 Shivangi Gupta
Shivangi Gupta Jerome Baudry
Jerome Baudry Vineetha Menon1*
Vineetha Menon1*- 1Department of Computer Science, The University of Alabama in Huntsville, Huntsville, AL, United States
- 2Department of Biological Sciences, The University of Alabama in Huntsville, Huntsville, AL, United States
This research introduces new machine learning and deep learning approaches, collectively referred to as Big Data analytics techniques that are unique to address the protein conformational selection mechanism for protein:ligands complexes. The novel Big Data analytics techniques presented in this work enables efficient data processing of a large number of protein:ligand complexes, and provides better identification of specific protein properties that are responsible for a high probability of correct prediction of protein:ligand binding. The GPCR proteins ADORA2A (Adenosine A2a Receptor), ADRB2 (Adrenoceptor Beta 2), OPRD1 (Opioid receptor Delta 1) and OPRK1 (Opioid Receptor Kappa 1) are examined in this study using Big Data analytics techniques, which can efficiently process a huge ensemble of protein conformations, and significantly enhance the prediction of binding protein conformation (i.e., the protein conformations that will be selected by the ligands for binding) about 10–38 times better than its random selection counterpart for protein conformation selection. In addition to providing a Big Data approach to the conformational selection mechanism, this also opens the door to the systematic identification of such “binding conformations” for proteins. The physico-chemical features that are useful in predicting the “binding conformations” are largely, but not entirely, shared among the test proteins, indicating that the biophysical properties that drive the conformation selection mechanism may, to an extent, be protein-specific for the protein properties used in this work.
1 Introduction
The prediction of which small molecules, e.g., substrates or modulators, are more likely than other small molecules to bind to a specific protein, is one the most formidable challenges of contemporary biology, chemical biology and pharmacology. Only a small fraction of the large number of small organic molecules present in living organisms will, in most cases, bind to a specific protein. There is a considerable amount of work that aims at improving the biophysical approaches to predicting such protein:ligand interactions.
As exemplified in the current special issue of Frontiers, the dynamics of the protein target is increasingly taken into account in such predictive approaches. Indeed, a protein cycles through multiple conformations, a few of which will be bound by its ligands, as conceptualized in the “conformational selection” mechanism of ligand binding. Virtual docking (Amaro et al., 2018) that aims at predicting if a given small chemical binds to a given protein, usually considers only one protein conformation in an “induced fit” mechanism. Advances beyond a simple induced fit mechanisms have been proposed, such as submitting the protein:ligands complexes to molecular dynamics simulations after docking (Seelinger and de Groot, 2010), which identifies binding modes of known ligands close to that of their experimental co-crystallized structures, or generating an ensemble of holo structures from experimental structures deposited in the PDB for a given protein target (Aggarwal et al., 2021). This present work, continuing in that direction, aims at using the information contained in molecular dynamics simulations of a single protein target structure prior to any docking.
In principle the “binding” protein conformations will correspond to the free energy minima of the (protein + ligand) complex free energy hypersurface. In our research, we are looking into whether we can identify these rare apo-conformations that possess this capacity to bind their ligands, while the vast majority of the other apo-protein conformations do not. This paper describes our Big Data analytics work toward such characterization of what properties of an apo-protein conformation more likely lead to conformational selection.
The data we used here has been obtained using supermassive “ensemble docking” from proteins’ molecular dynamics simulations, and is described in (Evangelista et al., 2016). The data corresponds to about 1.5 millions of protein conformation and protein:ligand complex structures and their associated docking scores.
Big Data analytics provides an efficient approach to analyzing such a large amount of data, and also addresses the class imbalance problem (Abd Elrahman and Abraham, 2013), which is a result of imbalanced groups or sub-categories present in the data, where the majority class or larger group of data consists of non-binding protein conformations and it overshadows the minority class or smaller data group, which comprises the data-of-interest i.e., the binding protein conformations. In our prior work (Akondi et al., 2019; Gupta et al., 2022; Sripriya Akondi et al., 2022), a novel two-stage sampling-based classifier framework was proposed with the primary goal of addressing the class imbalance problem and maximizing the detection of potential binding protein conformations as conventional machine learning (ML) algorithms are ill-equipped to deal with the issue of class imbalance during the data-learning phase. This paper extends on our previous work by presenting additional improvements to our two-stage sampling-based classification approach (Gupta et al., 2022) using deep learning techniques and four different feature selection methods in conjunction with an Enrichment ratio framework.
2 Materials and methods
2.1 Dataset description
As described in our previous work (Gupta et al., 2022), Molecular Dynamics (MD) simulations of four proteins, namely, ADORA2A (Adenosine A2a Receptor), ADRB2 (Adrenoceptor Beta 2), OPRD1 (Delta Opioid Receptor) and OPRK1 (Opioid Receptor Kappa 1) were used to study the efficacy of our proposed method. The conformations of these four proteins have been well-studied, and the protein conformations that: a) will bind to ligands (binding conformations) and b) will not bind to ligands (non-binding conformations), are known and have been previously documented and published (Evangelista et al., 2016).
ADORA2A: This dataset has 50 attributes and consists of 2,998 protein conformations among which 851 protein conformations are “binding” and 2,147 protein conformations that are “non-binding”. Here the imbalance ratio is 3:1 i.e., for every datasample belonging to minority class (binding conformations) there are three data samples belonging to the majority class (non-binding conformations).
ADRB2: This dataset has 51 attributes and consists of 2,565 protein conformations among which 156 are binding and 2,411 protein conformations are non-binding. Here the imbalance ratio is 16:1 i.e., for every datasample belonging to minority class (binding conformations) there are 16 data samples belonging to the majority class (non-binding conformations).
OPRD1: This dataset has 51 attributes and consists of 3,004 protein conformations among which 72 protein conformations are binding and 2,932 protein conformations are non-binding. Here the imbalance ratio is 41:1 i.e., for every datasample belonging to minority class (binding conformations) there are 41 data samples belonging to the majority class (non-binding conformations).
OPRK1: This dataset has 50 attributes and consists of 2,998 protein conformations among which 138 protein conformations are binding and 2,862 protein conformations are non-binding. Here the imbalance ratio is 20:1 i.e., for every data sample belonging to minority class (binding conformations) there are 20 data samples belonging to the majority class (non-binding conformations).
Tables describing the protein attributes/features/descriptors for ADORA2A, ADRB2, OPRD1, and OPRK1 datasets can be found in our previous work (Gupta et al., 2022). ADRB2 and OPRD1 have one additional feature - pro_pl_seq (Sequence based pI) in comparison to ADORA2A and OPRK1. The molecular descriptors were calculated using the protein descriptors from the program MOE (Akondi et al., 2019; Chemical Computing Group, 2019; Gupta et al., 2022; Sripriya Akondi et al., 2022).
2.1.1 Analysis of variance
Analysis of variance (ANOVA) is a statistical analysis method used here to calculate the linear relationship between the various protein features and to select the important protein features that correspond to the highest F-values (Johnson and Synovec, 2002). The top “x” features with the greatest F-values were selected in this case, where the x features to be retained is determined experimentally by the user. Thus, ANOVA technique allows for selection of the primary physio-chemical protein properties that essay a critical role in protein:ligand interaction and conformation selection.
2.1.2 Mutual information
Mutual Information (MI) (Macedo et al., 2019) is a measure of the amount of information that can be inferred about a variable U through the use of the other given random variable V. The mutual information I (U; V) for random variables U and V can be defined as follows (Guyon and Elisseeff, 2003; Gupta et al., 2022):
where
• p(u,v) is the joint probability density function.
• p(u) is the probability density function
In Eq. 1, if the MI value I is 1, then U and V are dependent on each other, i.e., protein features share similar information. If the MI value I is 0, then U and V are independent of each other i.e., no common (in other words unique) information between the features. The MI in physio-chemical properties are calculated as follows:
• First, calculate the MI value for all properties to determine how dependent the physio-chemical features vectors are and understand the common information contained in all the protein features.
• Then, sort the protein features according to their highest MI values. The top “x” protein features with the greatest MI values are retained, where x is user defined.
2.1.3 Recurrence quantification analysis
Recurrence Quantification Analysis (RQA) is a non-linear data-analysis method that is used to study the dynamical systems (Eckmann et al., 1987). The first step in the recurrence analysis is to quantify the repeating patterns of a dynamic system. One of the variables generated by the quantification of the recurrences is Entropy (ENT), which is the probability distribution p(j) of the diagonal line on the RQA plot and is defined as:
where M is the number of points on the state space trajectory and j is the length of the diagonal line in the RQA plot. We investigate the RQA-based entropy measure’s link to the probability of detecting potential binding conformations in terms of time-space evolution of protein conformations.
2.1.4 Spearman correlation coefficient
Spearman correlation coefficient is a statistical measure (Hauke and Kossowski, 2011) of the strength and direction of the monotonic relationship between each protein feature and target variable. The correlation coefficient for each feature is obtained by applying the formula as defined below:
where u is the feature vector and
2.1.5 Extreme gradient boosting
Extreme Gradient Boosting (XGBoost) is a tree ensemble boosting approach that merges a number of weak classifiers into a single strong classifier (Chen and Guestrin, 2016). Starting with a base learner, the strong learner is trained iteratively for best classification or prediction performance. Given a dataset X with m samples and n protein descriptors, let (
where
where
where D is the number of leaves, w is the weight of each leaf,
2.1.6 K-Means clustering
K-Means clustering is an unsupervised machine learning algorithm (Oyelade et al., 2010) that is used to understand the data patterns in the input data by grouping the instances in the dataset that are similar into different clusters. K-Means clustering is often used to produce compact clusters with minimum intra-cluster distances and maximum inter-cluster distances (Oyelade et al., 2010). This goal is achieved by splitting the data into a number of clusters “k” that the user specifies (Wilkin and Huang, 2007). Here we employ the K-Means clustering algorithm to under sample the data points from the majority class samples i.e., non-binding protein conformations as demonstrated in our prior work (Akondi et al., 2019).
2.1.7 Generative adversarial networks
Generative adversarial networks (GAN) is an unsupervised learning method that involves learning regularities or patterns in the input data to produce new examples that mimic the original dataset. The GAN technique uses two artificial models, the discriminator and generator, which compete for data learning (Jo and Kim, 2022). The discriminator focuses on discriminating or distinguishing between the original and synthetic data, whereas the generator tries to create synthetic data that is comparable to the real data. The loss function of GAN (Jo and Kim, 2022) is defined as:
where.
•
•
• u is the real input data
• v is the noise input to the generator neural network
• F(u) is the output probability of the generator
• D(v) is the sample generated by the generator neural network
Here the GAN is used to oversample or replicate the minority class in the dataset to alleviate the class imbalance problem and in turn maximize the prediction of the potential binding protein conformations.
2.1.8 Convolutional neural networks
Convolutional neural network (CNN) is a supervised deep learning technique (Hossain and Sajib, 2019) that has emerged as the most widely used artificial neural network in many computer vision applications, including texture recognition (Cimpoi et al., 2016), remote sensing scene classification (Hu et al., 2015; Penatti et al., 2015) and structure-based protein analysis (Torng and Altman, 2017). Architectural design of a CNN consists of several convolutional, pooling and dropout layers followed by one or more fully-connected layers (FC) (Sultana et al., 2018). Figure 1 describes the architecture of the CNN used in our work.
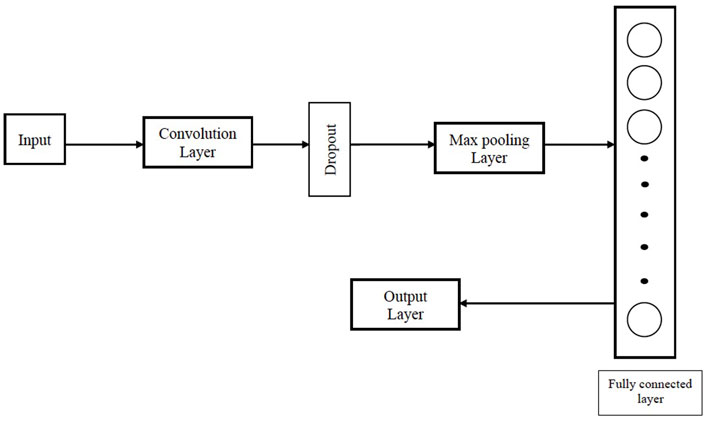
FIGURE 1. Architecture of the CNN used in our proposed Big Data analytics based AI/ML protein conformation selection/prediction framework.
The architectural design of the CNN in our work consists of a convolutional layer followed by dropout to reduce overfitting, a max pooling layer, a fully connected layer and an output layer. Rectified linear unit (ReLU) is used as the activation function for the convolution layer and fully connected layer. Binary cross-entropy L is used as the loss function for the CNN.
2.1.9 Recurrent neural networks
Recurrent neural network (RNN) is a class of neural networks which is used to detect patterns in a sequence of data (Ho and Wookey, 2020). In our work, the RNN architecture consists of two long-short term memory (LSTM) (Schmidt, 2019) layers with dropout followed by a dense layer with dropout and an output layer. The LSTM unit introduces a gate mechanism to select whether to retain or discard specific information in the existing memory. If the LSTM unit recognizes a pivotal protein descriptor from an input sequence early on, then it captures any potential long-distance dependencies between the protein descriptor and target value. Figure 2 describes the architecture of the RNN used in our work. Rectified linear unit (ReLU) is used as the activation function for the LSTM unit and dense layer, sigmoid function is used as the activation function in the output layer and binary cross-entropy as the loss function.
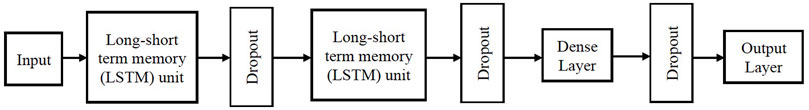
FIGURE 2. Architecture of the RNN used in our proposed Big Data analytics based AI/ML protein conformation selection/prediction framework.
2.1.10 Evaluation metrics
The confusion matrix and its derived evaluation parameters such as classification accuracy, sensitivity, specificity, etc., are some of the most commonly used ML evaluation metrics to validate a classification or prediction performance of ML algorithms. In this case of binary classification between binding and non-binding protein conformations, the confusion matrix has four categories of classification results as follows:
• True Positive (TP): When the classifier accurately predicts “binding,” indicating that the ligand and target protein did bind (Right predictions of class 1)
• True Negative (TN): When the classifier accurately predicts “non-binding,” indicating that the ligand and target protein did not bind (Right predictions of class 0)
• False Negative (FN): When the classifier inaccurately predicted “non-binding,” but the ligand and target protein did bind (Wrong predictions of class 0)
• False Positive (FP): When the classifier inaccurately predicted “binding,” but the ligand and target protein did not bind (Wrong predictions of class 1)
Here class 0 refers to the non-binding protein conformations (majority class) and class 1 denotes the binding protein conformations (minority class).
Accuracy of an AI/ML framework is calculated as the sum of correctly predicted binding and non-binding protein conformations divided by the total number of conformations in the data set. It is defined as:
Sensitivity is the ability of the AI/ML framework to correctly predict binding protein conformations. It is calculated as the number of correctly predicted binding protein conformations divided by the total number of binding protein conformations in the data set as defined below:
Eqs 8, 9 are used for performance evaluation of the proposed AI/ML protein conformation selection/prediction framework.
2.1.11 Enrichment ratio framework
The enrichment was calculated using the TP and FN predictions from the Big Data analytics based AI/ML protein conformation selection/prediction framework, described in Section 2.2. The base enrichment ratio is calculated to measure the effectiveness of general predictive performance in the absence of the ML protein conformation selection framework as in our prior work (Gupta et al., 2022). For accurate base enrichment ratio we performed subset data selection on previously calculated and published anticipated protein:ligand interactions energies in (Evangelista et al., 2016). The assumption is that the computed protein:ligand interaction energies are quantitatively valid, i.e., a “preferred” binding conformation would be the one in which the protein binds the ligand stronger (i.e., with lower interaction energies) than other alternative conformations. Thus, the base enrichment was calculated from (Evangelista et al., 2016) by dividing the number of binding conformations by the total number of conformations. Eq. 10 calculates the base enrichment detected during the test phase if the ML algorithm is not implemented. We select different subsets of the TP and FN values in order to calculate the ML prediction framework enrichment ratios in Eq. 10. The values returned by both Eqs 10, 11 were then used to calculate the final enrichment ratio returned by each of the four filters (A,B,C,D) defined in Eq. 12.
The final enrichment ratios for proteins ADORA2A, ADRB2, OPRD1, and OPRK1 were calculated using four different filters (A,B,C,D) and have been described and published in our previous work (Gupta et al., 2022). The proposed enrichment ratio framework used is depicted in Supplementary Figure SI-1 (Gupta et al., 2022).
2.2 The proposed Big Data analytics based AI/ML protein conformation selection/prediction framework
In this work, we combine the feature selection techniques discussed in Section 2.1 with the improved two-stage sampling based classification approach (Gupta et al., 2022) using deep learning techniques. The steps given below describe the new improved methodology and is illustrated in Figure 3:
• The first step in the methodology is to input the dataset and then apply the ML feature selection methods: i) Analysis of variance (ANOVA), ii) Mutual Information (MI), iii) Recurrence Quantification Analysis (RQA), and iv) Spearman correlation to select the important protein features from each of the methods respectively.
• We then obtain a feature ranking score for all features based on the common consensus of all the feature selection methods. Only the subset of protein features that are selected by all four feature selection methods are chosen to create a new dataset.
• Both the original dataset and a new dataset that is more biased towards samples in class 0 are sent as inputs to the XGBoost classifier. Samples of class 0 (TN) and class 1 (TP) are recorded as classification results 1.
• In order to create a new training dataset, the GAN algorithm was applied to both the original dataset and the new modified dataset.
• K-Means clustering (Akondi et al., 2019) is used on the XGBoost classifier’s classification results, class 0 samples are undersampled, and the intended class 1 samples are oversampled. This step increases the detection rate of class 1 samples or binding protein conformations to address the class imbalance issue. The new training dataset has the same size as the initial training dataset in order to maintain consistency.
• Supervised classification using deep learning methodologies: CNN and RNN are applied to the newly created training dataset. Both classifiers are used to identify the binding and non-binding conformations in the new training dataset. The results of both classifiers are recorded.
• As a final step, the TP (binding conformations), and FN (binding conformations but are incorrectly predicted as non-binding conformations) by the AI/ML protein conformation prediction framework (CNN and RNN) are employed in the Enrichment ratio framework to calculate the Enrichment ratios. The outcomes of the framework for enrichment ratios are recorded.
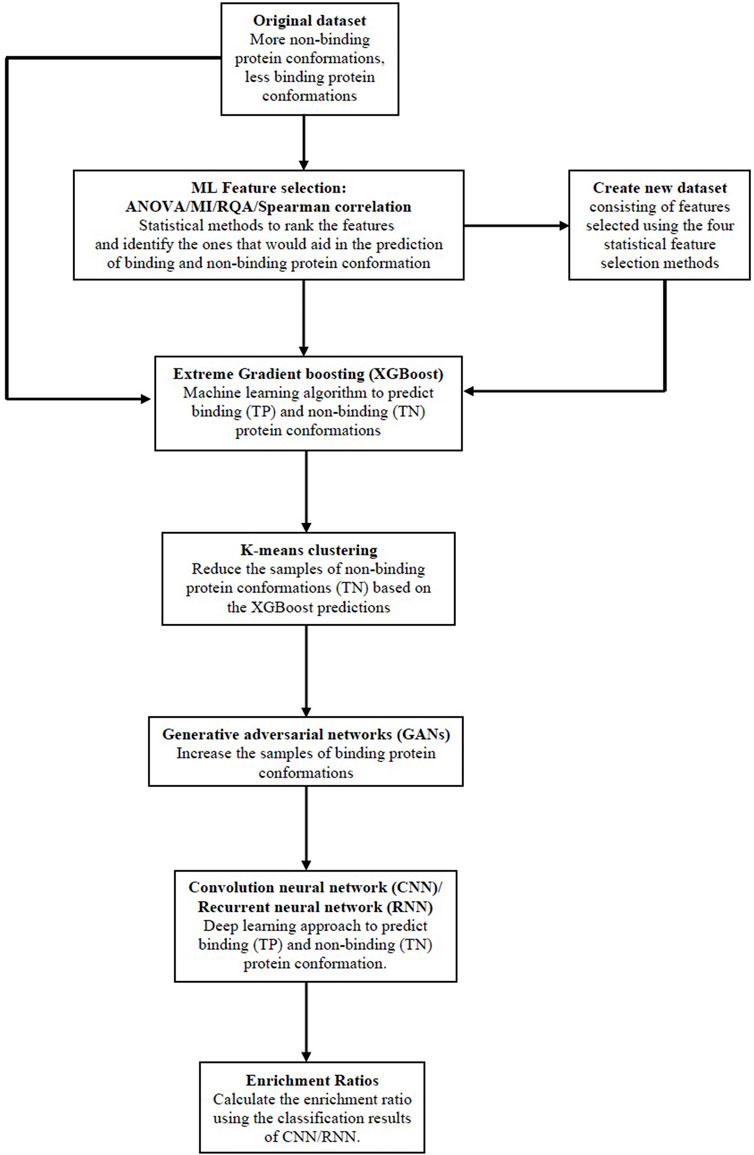
FIGURE 3. The proposed Big Data analytics based AI/ML protein conformation selection/prediction framework.
3 Results
The overview of enrichment ratios for ADORA2A that were determined using the predicted binding conformations from the AI/ML framework is shown in Table 1. As indicated in Supplementary Table SI-1 through Supplementary Table SI-5, the AI/ML framework was evaluated on the remaining 70% of the dataset after being trained on 30% of it. It can be observed that the data selection filter A of the Enrichment ratio framework gave the maximum enrichment ratio of 7.1 using XGBoost + GANs–RNN framework predictions.
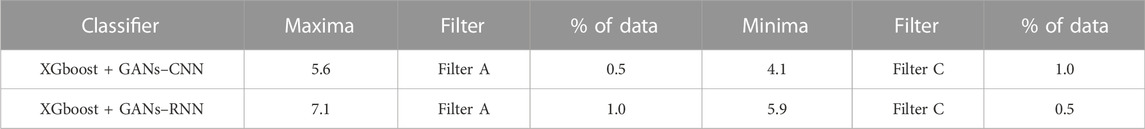
TABLE 1. Enrichment Ratios of ADORA2A on the original dataset with no feature selection with training size of 30%.
The list of protein descriptors that the four ML feature selection techniques determined to be significant is shown in Table 2 and it can be observed that 11 of the 50 features were chosen. Table 3 gives the overview of the enrichment ratios that were calculated using the features listed in Table 2. It can be observed that data selection filter A of Enrichment ratio framework gave the maximum enrichment ratio of 10.2 using XGBoost + GANs–CNN framework predictions.

TABLE 2. 11 features out of 50 were selected having a feature score of four using the feature scoring table for ADORA2A.

TABLE 3. Enrichment Ratios of ADORA2A on the dataset consisting of features as shown in Table 2 with training size of 30%.
The three common protein descriptors for the proteins ADORA2A, OPRK1, and OPRD1 that were determined to be significant by the four ML feature selection methods are listed in Supplementary Table SI-6. A summary of the enrichment ratios that were estimated using the characteristics indicated in Supplementary Table SI-6 is provided in Supplementary Table SI-7. It can be observed that employing data selection filter A, the XGBoost + GANs–CNN framework predictions provided the maximum enrichment ratio of 8.2.
The overview of enrichment ratios for the ADRB2 binding conformations predicted by the AI/ML framework is shown in Table 4. On 30% of the dataset, the AI/ML framework was trained, and on the remaining 70%, it was tested. It can be observed that employing data selection filter C, the XGBoost + GANs–RNN framework predictions provided the maximum enrichment ratio of 13.8.
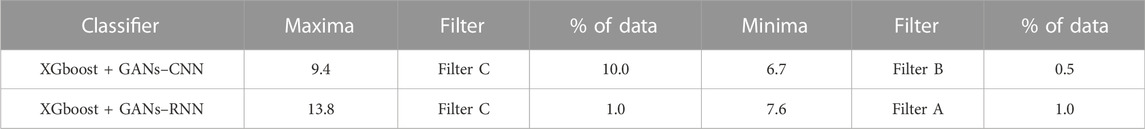
TABLE 4. Enrichment Ratios of ADRB2 on the original dataset with no feature selection with training size of 30%.
The list of protein descriptors that the three out of four ML feature selection techniques determined to be significant is shown in Table 5. It can be seen from the table that 8 of the 51 features were chosen. Table 6 provides an overview of the enrichment ratios that were computed using the features listed in Table 5. It can be observed that employing data selection filter D, the XGBoost + GANs–RNN framework predictions provided the maximum enrichment ratio of 24.2.

TABLE 5. 8 features out of 51 were selected having a feature score of three using the feature scoring table for ADRB2.
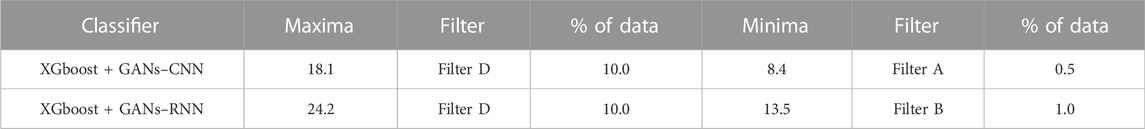
TABLE 6. Enrichment Ratios of ADRB2 on the dataset consisting of features as shown in Table 5 with training size of 30%.
The summary of enrichment ratios for OPRD1 that were determined using the predicted binding conformations from the AI/ML framework is shown in Table 7. On 30% of the dataset, the AI/ML framework was trained, and on the remaining 70%, it was tested. It can be observed that utilizing data selection filter B, the XGBoost + GANs–RNN framework predictions produced an enrichment ratio of up to 37.
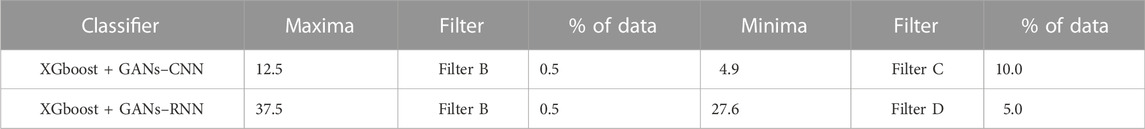
TABLE 7. Enrichment Ratios of OPRD1 on the original dataset with no feature selection with training size of 30%.
The list of protein descriptors that the four ML feature selection techniques determined to be significant is shown in Table 8. It can be seen that 12 of the 51 features were chosen, and Table 9 provides an overview of the enrichment ratios that were computed using the features listed in Table 8. It can be observed that employing data selection filter B, the XGBoost + GANs–RNN framework predictions provided the maximum enrichment ratio of 37.5.
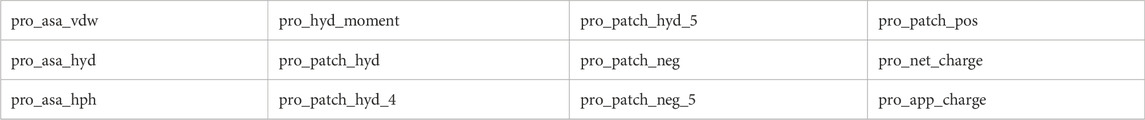
TABLE 8. 12 features out of 51 were selected having a feature score of four using the feature scoring table for OPRD1.
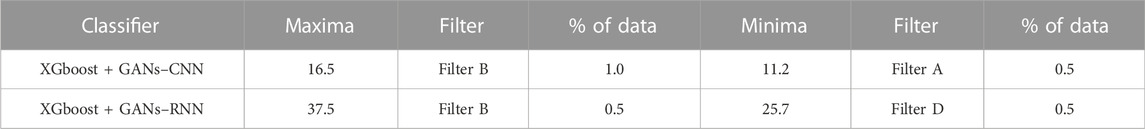
TABLE 9. Enrichment Ratios of OPRD1 on the dataset consisting of features as shown in Table 8 with training size of 30%.
Supplementary Table SI-8 gives the overview of enrichment ratios that were calculated using the features that were listed in Supplementary Table SI-6. It can be seen that both XGBoost + GANs–CNN and XGBoost + GANs–RNN framework predictions gave the same enrichment ratio of 37.5, using data selection filter B.
A summary of the enrichment ratios for OPRK1 that were determined using the predicted binding conformations from the AI/ML framework is shown in Table 10. On 30% of the dataset, the AI/ML framework was trained, and on the remaining 70%, it was tested. It can be seen that employing data selection filter A, the XGBoost + GANs–RNN framework predictions provided the maximum enrichment ratio of 27.6.

TABLE 10. Enrichment Ratios of OPRK1 on the original dataset with no feature selection with training size of 30%.
The list of protein descriptors that the four ML feature selection techniques determined to be significant is shown in Table 11. It can be seen that 5 of the 50 features were chosen, and Table 12 provides an overview of the enrichment ratios that were computed using the features listed in Table 11. It can be seen that both XGBoost + GANs–CNN and XGBoost + GANs–RNN frameworks gave the same enrichment ratio of 27.6, using data selection filter A.

TABLE 11. 5 features out of 50 were selected having a feature score of four using the feature scoring table for OPRK1.
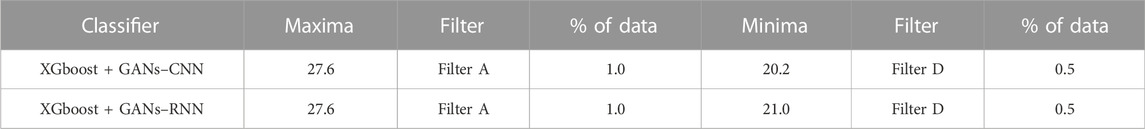
TABLE 12. Enrichment Ratios of OPRK1 on the dataset consisting of features as shown in Table 11 with training size of 30%.
An overview of the enrichment ratios that were calculated using the descriptors listed in Supplementary Table SI-6 is provided in Supplementary Table SI-9. It can be seen that employing data selection filter A, the XGBoost + GANs–RNN framework predictions provided the maximum enrichment ratio of 30.1.
4 Discussion
The Big Data analytics research outcomes in this study suggest that four proteins ADORA2A, ADRB2, OPRK1, and OPRD1, and their binding conformations considered in this work do possess similar global properties that can be leveraged to predict whether they will be more likely to bind their ligands than other conformations. The enrichment factors obtained with the best approaches are about 10 to about 40 times better than what would be available with a random selection of protein conformations for docking. For three out of the four targets of interest here (i.e., ADORA2A, OPRK1, and OPRD1), the physico-chemical features that are most associated with a high propensity to be selected for binding by the ligands are the water accessible surface area (MOE descriptor pro_asa_vdw), the hydrophobic surface area (MOE descriptor pro_asa_hyd) and the hydrophobicity moment (MOE descriptor pro_hyd_moment). That these properties, which are global and not limited to the binding sites, are common to the important descriptor of all proteins point to a dual role of exposure to solvent and hydrophobicity as globally driving the capacity of proteins to bind, or not, their ligands. Note that this work is not a structure-activity relationship studies, i.e., we do not at this point give a range of values for these proteins that would be associated with ligand binding and a range of values that would be associated with non-ligand binding.
The fourth protein target that was used here, ADRB2, can also be analyzed by deep learning approaches to identify the ligand binding conformations about 24 times better than a random selection of conformations. Yet, that one protein target yields different physico-chemical features than the other three proteins used here, although the general role of surface hydrophobicity and electrostatics (negatively-charged regions, precisely) is conserved. We do not yet know if this difference observed between ADRB2 and the other proteins is a result of different actual physicochemical mechanisms involved in ligand binding, or if this is an artifact of the data and of specific issues with class imbalance from the MD trajectories of this target.
Nonetheless, the fact that the apo-proteins’ global physico chemicals properties may—to an extent—predict the ligand-binding character of conformations is remarkable. Naturally, this does not mean that only global protein properties are “holding” the keys to the conformational selection mechanisms. This work will have to be continued and repeated with features that are specific to the binding sites’ conformations rather than describing the global protein structure.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
Conceptualization, SG, JB, and VM; methodology, SG; software, SG; validation, SG, JB, and VM; formal analysis, SG, JB, and VM; investigation, SG; resources, SG and JB; data curation, SG and JB; writing—original draft preparation, SG, JB, and VM; writing—review and editing, JB and VM; visualization, SG; supervision, JB and VM; project administration, JB and VM; funding acquisition, VM. All authors have read and agreed to the published version of the manuscript.
Funding
Funding to VM and JB was provided by The University of Alabama.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2022.953984/full#supplementary-material
References
Abd Elrahman, S. M., and Abraham, A. (2013). A review of class imbalance problem. J. Netw. Innov. Comput. 1, 332–340. doi:10.20943/01201706.4351
Aggarwal, R., Gupta, A., and Priyakumar, U. D. (2021). APObind: A dataset of ligand unbound protein conformations for machine learning applications in de novo drug design. Available at: https://arxiv.org/abs/2108.09926 (Accessed 08 25, 2021).
Akondi, V. S., Menon, V., Baudry, J., and Whittle, J. (2019). “Novel K-means clustering-based undersampling and feature selection for drug discovery applications,” in Proceedings of the 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), San Diego, CA, USA, 18–21 November 2019, 2771–2778.
Amaro, R. E., Baudry, J., Chodera, J., Demir, Ö., McCammon, J. A., Miao, Y., et al. (2018). Ensemble docking in drug discovery. Biophys. J. 114, 2271–2278. doi:10.1016/j.bpj.2018.02.038
Babajide Mustapha, I., and Saeed, F. (2016). Bioactive molecule prediction using extreme gradient boosting. Molecules 21, 983. doi:10.3390/molecules21080983
Chemical Computing Group (2019). Molecular operating environment (MOE). Available at: https://www.chemcomp.com/Products.htm.
Chung, J., Gülçehre, Ç., Cho, K., and Bengio, Y. (2014). Empirical evaluation of gated recurrent neural networks on sequence modeling. ArXiv, abs/1412.3555.
Cimpoi, M., Maji, S., Kokkinos, I., and Vedaldi, A. (2016). Deep filter banks for texture recognition, description, and segmentation. IJCV 118 (1), 65–94. doi:10.1007/s11263-015-0872-3
Eckmann, J.-P., Kamphorst, S. O., and Ruelle, D. (1987). Recurrence plots of dynamical systems. Europhys. Lett. 4, 973–977. doi:10.1209/0295-5075/4/9/004
Evangelista, W., Weir, R. L., Ellingson, S. R., Harris, J. B., Kapoor, K., Smith, J. C., et al. (2016). Ensemble-based docking: From hit discovery to metabolism and toxicity predictions. Bioorg. Med. Chem. 24, 4928–4935. doi:10.1016/j.bmc.2016.07.064
Gupta, S., Baudry, J., and Menon, V. (2022). Using big data analytics to “back engineer” protein conformational selection mechanisms. Molecules 27 (8), 2509. doi:10.3390/molecules27082509
Guyon, I., and Elisseeff, A. (2003). An introduction to variable and feature selection. J. Mach. Learn. Res. 3, 1157–1182.
Hauke, J., and Kossowski, T. (2011). Comparison of values of pearson's and spearman's correlation coefficients on the same sets of data. Quaest. Geogr. 30, 87–93. doi:10.2478/v10117-011-0021-1
Ho, Y., and Wookey, S. (2020). The real-world-weight cross-entropy loss function: Modeling the costs of mislabeling. IEEE Access 8, 4806–4813. doi:10.1109/ACCESS.2019.2962617
Hossain, M. A., and Sajib, M. (2019). Classification of image using convolutional neural network (CNN). Glob. J. Comput. Sci. Technol. 19, 13–18. doi:10.34257/GJCSTDVOL19IS2PG13
Hu, F., Xia, G.-S., Hu, J., and Zhang, L. (2015). Transferring deep convolutional neural networks for the classification of high- resolution remote sensing imagery. Remote Sens. 7 (11), 14680–14707. doi:10.3390/rs71114680
Jo, W., and Kim, D. (2022). Obgan: Minority oversampling near borderline with generative adversarial networks. Expert Syst. Appl. 197, 116694. doi:10.1016/j.eswa.2022.116694
Johnson, K. J., and Synovec, E. R. (2002). Pattern recognition of jet fuels: Comprehensive GCGC with ANOVA-based feature selection and principal component analysis. Chemom. Intell. Lab. Syst. 60, 225–237. doi:10.1016/s0169-7439(01)00198-8
Macedo, F., Oliveira, M. R., Pacheco, A., and Valadas, R. (2019). Theoretical foundations of forward feature selection methods based on mutual information. Neurocomputing 325, 67–89. doi:10.1016/j.neucom.2018.09.077
Oyelade, O. J., Oladipupo, O. O., and Obagbuwa, I. C. (2010) Application of k means clustering algorithm for prediction of students academic performance. arXiv preprint arXiv:1002.2425, 2010.
Penatti, O., Nogueira, K., and Santos, J. (2015). “Do deep features generalize from everyday objects to remote sensing and aerial scenes domains?,” in 2015 IEEE Conference on Computer Vision and Pattern Recognition Workshops (CVPRW), Boston, MA, USA, June 7 2015 to June 12 2015.
Seelinger, D., and de Groot, B. L. (2010). Conformational transitions upon ligand binding: Holo-structure prediction from apo conformations. PLOS Comput. Biol. 6 (1), e1000634. doi:10.1371/journal.pcbi.1000634
Sripriya Akondi, V., Menon, V., Baudry, J., and Whittle, J. (2022). Novel big data-driven machine learning models for drug discovery application. Molecules 27, 594. doi:10.3390/molecules27030594
Sultana, F., Sufian, A., and Dutta, P. (2018). “Advancements in image classification using convolutional neural network,” in 2018 Fourth International Conference on Research in Computational Intelligence and Communication Networks (ICRCICN), Kolkata, India, 22-23 Nov. 2018, 122–129. doi:10.1109/ICRCICN.2018.8718718
Torng, W., and Altman, R. B. (2017). 3D deep convolutional neural networks for amino acid environment similarity analysis. BMC Bioinforma. 18, 302. doi:10.1186/s12859-017-1702-0
Keywords: protein conformation selection, Big Data, deep learning, machine learning, feature selection, drug discovery
Citation: Gupta S, Baudry J and Menon V (2023) Big Data analytics for improved prediction of ligand binding and conformational selection. Front. Mol. Biosci. 9:953984. doi: 10.3389/fmolb.2022.953984
Received: 26 May 2022; Accepted: 16 December 2022;
Published: 12 January 2023.
Edited by:
J. Andrew McCammon, University of California, San Diego, United StatesReviewed by:
Mithun Radhakrishna, Indian Institute of Technology Gandhinagar, IndiaQin Xu, Shanghai Jiao Tong University, China
Copyright © 2023 Gupta, Baudry and Menon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vineetha Menon, dmluZWV0aGEubWVub25AdWFoLmVkdQ==; Jerome Baudry, amVyb21lLmJhdWRyeUB1YWguZWR1