 Jinshi Zhang1†
Jinshi Zhang1† Qiang He
Qiang He Juan Jin
Juan Jin
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Biosci. , 30 June 2022
Sec. Molecular Diagnostics and Therapeutics
Volume 9 - 2022 | https://doi.org/10.3389/fmolb.2022.951275
This article is part of the Research Topic Ferroptosis in Cancer and Beyond View all 34 articles
Ferroptosis is a novel cell death method discovered in recent years. It is usually accompanied by massive accumulations of iron and lipid peroxidation during cell death. Recent studies have shown that ferroptosis is closely associated with the pathophysiological processes of many diseases, such as tumors, neurological diseases, localized ischemia-reperfusion injury, kidney injury, and hematological diseases. How to intervene in the incidence and development of associated diseases by regulating the ferroptosis of cells has become a hot topic of research. This article provides a review of the role of ferroptosis in the pathogenesis and potential treatment of acute kidney injury.
Acute kidney injury (AKI), is a disease that can have a variety of causes, including ischemia, nephrotoxic drugs, and urinary tract obstruction (Wang and Tao, 2015). AKI has high morbidity and mortality rates in hospitalized patients, and yet research on the therapeutic options for AKI prevention and treatment, other than hemodialysis, has been slow. Therefore, new therapeutic options are urgently needed to prevent AKI, as well as to promote kidney repair after AKI onset. In 2012, Dixon et al. (2012) proposed a new concept of cell death, known as ferroptosis, which was subsequently proven to be closely associated with the pathophysiological processes of many diseases (Alvarez et al., 2017; Tang et al., 2021). A recent study has shed light on the role of iron homeostasis in the pathogenesis of AKI and its therapeutic potential (Swaminathan, 2018). This article reviews the current research on the regulatory mechanisms, research progress, and therapeutic potential of ferroptosis in AKI.
As one of the most important essential trace elements, iron is involved in a wide variety of metabolic processes in the body. In 2003, Dolma et al. (Yao et al., 2021) discovered a novel erastin compound that is selectively lethal to tumor cells with the RAS gene mutation, but this causes cell death in a manner that is different from conventional apoptosis without nuclear morphological changes. Neither DNA fragmentation, cysteine-containing aspartate protein hydrolase (caspase) activation, nor caspase inhibitors inhibit this mode of cell death. Subsequently, Yang et al. (Heintzman et al., 2022) and Yagoda et al. (2007) found that this mode of cell death could be inhibited by iron chelators and discovered another compound, RSL3, which could also cause this mode of cell death. In 2012, Dixon et al. (2012) officially named this mode of cell death “ferroptosis,” or “iron death,” based on its characteristics. Unlike other regulated cell deaths like apoptosis and autophagy, ferroptosis has the following characteristics: 1) Morphologically, it is primarily characterized by cell membrane rupture and blebbing, reduction in mitochondrial volume, increase in membrane density, and reduction or even disappearance of mitochondrial cristae. It does not have the morphological features of apoptosis, such as cell shrinkage, chromatin condensation, skeleton disassembly, or apoptotic vesicle formation (Mou et al., 2019); 2) Unlike apoptosis, ferroptosis occurs without the activation of caspase 3, and the process cannot be reversed by caspase inhibitors; 3) Ferroptosis cannot be interrupted by inhibitors of apoptosis, pyroptosis, or cellular autophagy, but can be inhibited by iron chelators and antioxidants; 4) Ferroptosis can be induced or inhibited through several metabolic pathways (Figure 1), it is characterized by the aggregation of iron and reactive oxygen species (ROS), both of which are thought to be central to ferroptosis, and the inhibition of cystine/glutamate antiporter (cystine/glutamate antiporter system, or System Xc-) and glutathione peroxidase 4 (GPX4) activity, by reducing cystine uptake, depleting glutathione (GSH), and releasing molecules, such as arachidonic acid (Doll and Conrad, 2017); 5) Genetically abnormal expression of several genes can occur, especially of those related to iron metabolism, such as the transferrin receptor (TfR), divalent metal transporter 1 (DMT1), ferritin heavy chain 1 (FTH1), and nuclear receptor coactivator 4 (NCOA4) (Wang et al., 2018). Caspase activation and autophagic lysosome formation are specific markers of apoptosis and autophagy, respectively. Although there are no recognized specific markers for ferroptosis, acyl-coenzyme A synthetase long chain family member 4 (ACSL4) causes the earliest upregulation of adrenaline in non-lethal ferroptosis in various AKI models, which suggests that this molecule is a reliable biomarker for ferroptosis detection. Whether these different modes of cell death can be integrated into a complete regulatory network requires further exploration (Doll et al., 2017).
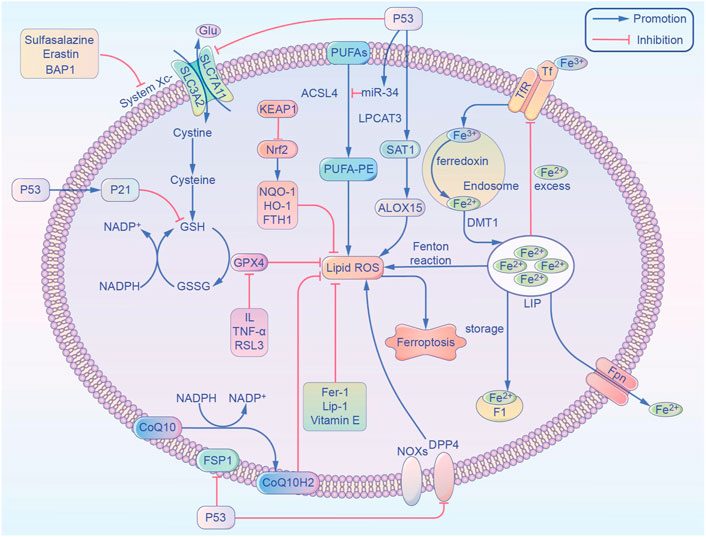
FIGURE 1. Overview of the metabolic routes contributing to ferroptosis. This non-exhaustive list includes (1) iron-Fenton reaction. (2) GPX4 antioxidant activity. (3) Lipid metabolism pathway. ACSL4, acyl-coenzyme A synthetase long chain family member 4; ALOX15, arachidonate 15-lipoxygenase; BAP1, BRCA1 associated protein 1; CoQ10, coenzyme Q10; CoQ10 H2, ubiquinol; DPP4, dipeptidyl-peptidase-4; DMT1, divalent metal transporter 1; FTH1, ferritin heavy chain 1; Fer-1, ferrostatin-1; FSP1, ferroptosis suppressor protein 1; Fpn, ferroportin; GPX4, glutathione peroxidase 4; GSH, glutathione; GSSG, oxidized glutathione; Glu, glutamic acid; HO-1, heme oxygenase-1; IL, interleukin; Lip-1, liproxstatin-1; LPCAT3, lysophosphatidylcholine acyltransferase 3; LIP, labile iron pool; NADPH, nicotinamide adenine dinucleotide phosphate; Nrf2, nuclear factor E2 related factor 2; NOX1, NADPH Oxidase 1; NQO-1, NAD(P)H quinone dehydrogenase 1; PUFAs, polyunsaturated fatty acids; ROS, reactive oxygen species; SAT1, spermidine/spermine N1-acetyltransferase 1; System Xc-, glutamate-cystine/antiporter; SLC7A11, solute carrier family 7 member 11; SLC3A2, solute carrier family 3 member 2; TNF-α, tumor necrosis factor-α; TfR, transferrin receptor; Tf, transferrin.
Iron homeostasis has an important influence on the incidence of ferroptosis, and iron is present in the circulation as the trivalent iron (Fe3+) bound to transferrin. Fe3+ is converted to Fe2+ by the membrane protein transferrin receptor 1 and divalent met al transport protein 1 and is released into the cytoplasmic labile iron pool (LIP). Excess iron is stored as ferritin, and the ferritin heavy chain (FTH1) has iron oxidase activity that catalyzes the conversion of Fe2+ into Fe3+, allowing iron to be safely incorporated into the ferritin shell and therefore reduce free iron levels. Hydrogen peroxide reacts with Fe2+ to produce hydroxyl radicals with strong oxidizing properties, and this is known as the Fenton reaction. Iron overload causes abnormalities in the mitochondrial oxidative phosphorylation pathway, producing ATP along with large amounts of ROS, oxidizing polyunsaturated fatty acids (PUFA) on cell and organelle membranes, forming lipid peroxides, and directly or indirectly disrupting cellular structure and function. In AKI due to IRI, ROS production is increased during reperfusion and induces ferroptosis in renal tubular epithelial cells. In AKI, plasma catalytic iron concentrations were significantly increased and correlated with extensive injury caused by cisplatin, ischemia-reperfusion, aminoglycosides, rhabdomyolysis, and hemoglobinuria, and both higher plasma catalytic iron levels and lower hepcidin concentrations were associated with increased mortality in patients with AKI (Leaf et al., 2019).
The glutamate-cystine/antiporter (System Xc-) is a non-sodium-dependent cystine/glutamate reverse transporter protein encoded by the solute carrier family 7 member 11 (SLC7A11), consisting of the catalytic subunit SLC7A11 and the chaperone subunit solute carrier family 3 member 2 (SLC3A2). SLC7A11 is also commonly used to refer to the Xc- system. The Xc-system exchanges glutamate and cystine intracellularly and extracellularly in a 1:1 ratio, and the intracellular cystine is reduced to cysteine. The methionine amino acids generate S-adenosylmethionine under enzymatic action, and cysteine is also produced after demethylation and deadenosylation. Cysteine binds glutamate and glycine to form GSH, which is an intracellular antioxidant that is used in intracellular enzymatic and non-enzymatic antioxidant reactions to maintain hydrogen peroxide levels within the physiological range. GPX4 is a GSH-dependent enzyme with an active center, composed of selenoproteins. Selenoproteins contain selenocysteine that reduces peroxide accumulation by reducing lipid-peroxide (L-OOH) to the corresponding harmless alcohol (L-OH) using GSH and thiol-containing compounds, thereby inhibiting lipoxygenase (LOX) activity and phospholipid/cardiolipid oxidation events. Mevalonate is used as a raw material for the synthesis of isoglutarate pyrophosphate (IPP) from acetyl coenzyme A (CoA), which is an essential signal for the maturation of selenocysteine tRNAs and the synthesis of active GPX4 (Moon et al., 2019). Erastin and sulfasalazine inhibit System Xc- function, resulting in insufficient cystine uptake, decreased intracellular antioxidant capacity, and lipid accumulation, and thereby, inducing cellular ferroptosis. AKI morbidity and mortality were significantly increased in GPX4 knockout mice, and during IRI there was a significant decrease in GSH levels, reduced GPX4 activity, increased iron accumulation and lipid peroxidation, and upregulated expression of proteins and genes associated with iron-sensitivity in kidney tissue (Ma et al., 2021).
PUFA contains arachidonic acid (AA) or its derivative adrenergic acid (AdA), and the pentose phosphate pathway directly dehydrogenates and decarboxylates glucose oxidatively to produce nicotinamide adenine dinucleotide phosphate (NADPH) as a reducing agent involved in the synthesis of fatty acids. AA and ACSL4 are esterified into phosphatidylethanolamine (PE), which in turn is formed by LOX through association with recombinant human phosphatidylethanolamine binding protein 1 to form the complex 15-LOX/phosphatidylethanolamine binding protein 1 (PEBP1). This undergoes metamorphic regulation to provide the signal sn2-15-HpETE-PE site that promotes ferroptosis and is ultimately oxidized to phospholipid hydroperoxides (PE-AA/AdA-OOH) and ROS. This is where 15-LO2 is highly expressed in renal tubular epithelial cells, and both 15-LO1 and 15-LO2 are involved in ischemic acute kidney injury (Anthonymuthu et al., 2018). ROS generate destructive hydroxyl radicals through the Fenton reaction, which react rapidly with neighboring molecules or peroxidize with cellular lipid components to generate large amounts of lipid radicals, resulting in thinning of PUFA-rich cell membranes and plasma membranes and irreversible damage to structure and function. Nuclear factor E2-related factor 2 (Nrf2) is considered an important regulator of the antioxidant system, and its downstream target genes are involved in maintaining redox homeostasis. These include GSH synthase, GPX4, and the oxidative stress sensor molecule kelch-like ECH-associated protein 1 (KEAP1), which reduces Nrf2 activity by ubiquitination. In AKI, methylpadoxolone activates Nrf2 by inhibiting KEAP1’s ubiquitin activity to activate Nrf2. This results in improved glomerular filtration rate. Renal lipid peroxidation was found in FA-AKI, and mice pretreated with ferritin-1 had improved renal function and reduced tissue damage (Shimada et al., 2016).
P53 has become one of the most extensively studied genes since its discovery in 1979. (Liu and Gu, 2021). Apart from its effects on common forms of cell death, p53 is well known for its key role in ferroptosis. (Tang et al., 2019). In the first study to investigate the role of p53 in ferroptosis, we found that p53 promotes ferroptosis by decreasing the expression of SLC7A11 (Jiang et al., 2015). In addition, p53 can regulate ferroptosis and lipid peroxidation by virtue of its target gene spermidine/sper-mine N1-acetyltransferase (SAT1) whose effect is demonstrated by ALOX15 upregulation after SAT1 induction (Gao et al., 2015). Recently, p53 has been discovered to delay the ferroptosis by up-regulating the expression of its downstream target p21 (Tarangelo et al., 2018). Furthermore, in p53-deficient cells, the dipeptidyl-peptidase-4 (DPP4) interacts with NADPH Oxidase 1(NOX1), thus forming a NOX1-DPP4 complex that mediates plasma membrane lipid peroxidation and ferroptosis (Gao et al., 2019). ACSL4 has been reported as both a reliable biomarker and a pivotal contributor to ferroptosis. Recent studies have confirmed that ACSL4 levels are post-transcriptionally downregulated by p53-activated miR-34 (Chang et al., 2007; Jiang et al., 2020). And there is a possibility that the p53/miR-34/ACSL4 axis represses ferroptosis by limiting lipid peroxidation. Controversial results of p53 are quite common in different ferroptosis studies, which may result from “different cell types,” “duality of p53 functions” or “different interventions” (Liu and Gu, 2022).
Erastin can bind to VDAC2 and VDAC3 in the mitochondrial voltage-dependent anion channel (VDAC) to alter cell membrane permeability and ion selectivity, causing mitochondrial dysfunction and the release of oxidative substances, which ultimately lead to ferroptosis (Yagoda et al., 2007). Heme oxygenase-1 (HO-1) is an important source of intracellular iron, and Adedoyin et al. (2018) found that HO-1 can act as a protective enzyme to inhibit erastin-induced occurrence of ferroptosis in proximal tubular epithelial cells in acute kidney injury. In contrast, in fibrosarcoma cells, Kwon et al. (2015) found that inhibition of erastin-induced ferroptosis was alleviated, and this contradictory result suggests that the exact relationship between HO-1 and ferroptosis requires further exploration. Kinolfin, a class of oral, small molecule, gold-containing compounds for the treatment of RA, was recently found to cause ferroptosis through high doses of inhibition of thioredoxin reductase (TXNRD) activity, which led to lipid peroxidation and ferroptosis, and suggested that TXNRD is a key factor in ferroptosis (Yang et al., 2020). Autophagy-related protein Beclin 1 can form a complex with SLC7A11 to promote ferroptosis, and this process requires AMP-activated protein kinase (AMPK) to mediate phosphorylation of the S90/93/96 sites of Beclin 1 (BECN1) (Song et al., 2018). Bersuker et al. (2019) found that coenzyme Q10 (CoQ10) captures lipid reactive oxygen species through its reduced form, while ferroptosis suppressor protein 1 (FSP1) catalyzes the regeneration of CoQ10 via NADH to prevent the release of lipid peroxides, and that FSP/CoQ10 can inhibit ferroptosis independently of the glutathione peroxidase 4 (CPX4) pathway.
As the mechanism of ferroptosis is being studied, many specific ferroptosis inhibitors have been discovered, such as the novel compounds ferrostatin-1 (Fer-1), liproxstatin-1 (Lip-1), and vitamin E, in addition to iron chelators. It is important to clarify the mechanism of ferroptosis and its regulation to provide new research ideas and therapeutic options for diseases related to ferroptosis, and the inhibitors.
The etiology of rhabdomyolysis syndrome is complex, with studies pointing to more than 190 acquired causes and more than 40 genetically related causes. Common causes include strenuous exercise, direct trauma, metabolic myopathy, toxic chemicals, physical or biological agents, and genetic factors (Thévenod et al., 2020). Previous studies suggest that the main mechanisms of rhabdomyolysis-induced AKI are: 1) Accumulation of myoglobin (Mb) in the kidney after massive damage to skeletal muscles that blocks the distal renal tubules. This is the central mechanism leading to renal damage; 2) Leakage of bodily fluids after muscle damage, insufficient blood volume, activation of renin-angiotensin, sympathetic nervous system, release of plasma antidiuretic hormone, other inflammatory mediators, such as endothelin-1 (ET1), tumor necrosis factor-α (TNF-α), which promote vasoconstriction; 3) Nitric oxide in the renal microcirculation is scavenged by Mb resulting in insufficient vasodilator levels. Increased vasoconstrictor and decreased vasodilator levels lead to the constriction of renal arteries, causing inadequate blood supply to the kidneys. Some recent studies suggest that ferroptosis plays a more important role in Mb-induced AKI than previously thought (Tang et al., 2021). In rhabdomyolysis, Mb is released in large amounts beyond the binding capacity of α2-globulin, and Mb is filtered out of the glomerulus into the proximal tubule, where it is eventually broken down into free iron ions and ferrous hemoglobin. Transferrin receptors and divalent metal ion transporters transfer extracellular free iron ions into the cell. Fe3+ entering the cell is converted to Fe2+, and part of Fe2+ is stored in ferritin, while the other part is transported extracellularly by membrane iron transport proteins to be rebound by Hb or Mb (Tang et al., 2021). If there were too much Mb in the renal tubules, it would lead to Fe2+ overload in the renal tubular cells in the presence of insufficient intracellular ATP, hypotension, and hypoperfusion. Overloaded Fe2+ will then induce direct damage to proximal tubular lipid peroxidation through the Fenton reaction, leading to acute tubular necrosis or renal failure (Bosch et al., 2009; Zeng and Tomlinson, 2021), where ferritin heavy chain 1 (FTH1) is of importance (Xie et al., 2016a). Zarjou et al. (2013) found that FTH1 knockout mice had higher mortality rates and more severe kidney injury than wild-type mice in a rhabdomyolysis-induced AKI model, which shows the protective effect of FTH1 against tubular injury and the role of iron ions in AKI. A study by Guerrero-Hue et al. (2019) found that Fer-1, a small molecule inhibitor of ferroptosis, could inhibit Mb-induced AKI, a finding that demonstrates the key role played by ferroptosis in this process. Skouta et al. (Tang et al., 2021) also found that Fer-1 effectively inhibited hydroxyquinoline and ferrous ammonium sulfate-induced cell necrosis in a rhabdomyolysis-induced in vitro ferroptosis model. In summary, the literature show that ferroptosis is one of the important mechanisms causing rhabdomyolysis-induced AKI.
Ferroptosis and AKI due to ischemia reperfusion injury (IRI) is a condition in which the degree of tissue damage increases rapidly after restoration of blood flow to cells that have suffered a period of ischemia, resulting in a clinical condition called reperfusion syndrome. This process exacerbates tissue injury by initiating an inflammatory cascade of responses. The cascade mainly covers ROS, cytokines, chemokines, and leukocyte activation. (Nieuwenhuijs-Moeke et al., 2020; Messerer et al., 2021). IRI is also an important factor in contributing to AKI. The main pathophysiological mechanisms of renal IRI include inflammation, oxidative stress and lipid peroxidation, mitochondrial dysfunction, and activation of the renin-angiotensin system (Franzin et al., 2021). A recent study suggests that ferroptosis may be one of the main drivers of renal IRI (Tonnus and Linkermann, 2017). In a mouse model of IRI, application of ferroptosis small molecule inhibitors protected mice from AKI and other organ damage (Tonnus et al., 2021). In clinical practice, renal IRI is usually the leading cause of AKI after cardiac surgery. The pathogenesis of postoperative AKI in the heart is complex and multifactorial, and these mechanisms of injury might play different roles at different times and may also act synergistically, with the release of free iron being a key part (Han et al., 2021). Due to the prolonged exposure of blood cells to the mechanical extracorporeal circulation system during various manipulations, such surgical procedures may lead to the destruction of red blood cells, resulting in the entry of free Hb into the circulation (van Swelm et al., 2020). It has been shown that free Hb levels in the blood increase to several times the physiological level during extracorporeal circulation and remain so until several hours after the procedure (Lee et al., 2021). The role of Hb in the induction of AKI is similar to that of Mb, with tubular cell iron overload being the main cause of eventual acute renal failure or tubular necrosis (Tang et al., 2021); studies have shown that free Hb levels are a significant independent risk factor for renal impairment after cardiac surgery (Leaf et al., 2019). Hemodynamic instability might occur during the transition from complete extracorporeal circulatory support to the patient’s own circulation, and this state of hemodynamic instability likely leads to systemic hypoperfusion, particularly ischemia and hypoxia of the kidneys (Haase et al., 2010). During ischemia, free iron levels in the kidney are increased by lower pH and dissociation of protein-bound iron. An increase in free iron might activate cellular ferroptosis, ultimately leading to tubular necrosis (van Swelm et al., 2020). Choi et al. (2019) proposed that lower intraoperative levels of iron-binding protein indirectly reflect impaired processing of catalytic iron by the body during extracorporeal circulation, leading to renal injury. This result emphasizes the importance of iron homeostasis in ischemia-reperfusion injury and suggests that iron homeostasis is a potential therapeutic target in kidney injury associated with cardiac surgery or in AKI induced by ischemia-reperfusion (Choi et al., 2019). In addition, studies have found that mechanical ventilation is an independent risk factor for the development of AKI after cardiac surgery. In an IRI mouse model, prolonged mechanical ventilation led to a gradual decrease in GPX4 levels, increased renal lipid peroxidation, and a gradual decrease in blood GSH and renal homogenate GSH levels. This suggests that prolonged mechanical ventilation leads to blood GSH depletion and induces renal ferroptosis via the GSH⁃GPX4 axis (Zhou et al., 2020a). In a model of hypoxic injury to renal tubular epithelial cells, application of small molecule inhibitors of ferroptosis effectively attenuated hypoxic damage to renal tubular epithelial cells, which suggests that ferroptosis may be an earlier mode of cell death in hypoxic renal tubular epithelial cells and that hypoxia plays a critical role in the development of ischemia-reperfusion AKI (Li et al., 2021a). Reperfusion injury to other organs during extracorporeal circulation may also bring additional iron to the kidney, and iron released from the upstream necrotic kidney unit might further descend to the next kidney unit, further exacerbating oxidative stress and inducing tissue damage (Martines et al., 2013). Linkermann et al. (Tonnus et al., 2021) found that ferroptosis is a key mechanism for sequential renal tubular cell death after ischemic AKI, and that triggering ferroptosis by inhibiting the cysteine glutamate transporter and applying the ferroptosis activator erastin to reduce intracellular GSH could cause proximal tubular cell cascade cell death. The use of Fer-1 not only inhibited ferroptosis in vitro, but also attenuated ischemia-reperfusion injury in mouse kidneys. Therefore, it can be inferred that ferroptosis is one of the important mechanisms of ischemia-reperfusion AKI. Huang et al. (2019) inhibited the expression of augmenter of liver regeneration (ALR) in an in vitro model of IRI-induced AKI and found that the level of cellular ferroptosis was increased, along with an increase in ROS and significant mitochondrial damage. In addition, the inhibition of System Xc⁃ with erastin promoted cellular ferroptosis and silenced ALR expression, which suggests that ischemia-reperfusion-induced AKI is mediated by ALR and that this process is associated with the glutathione⁃glutathione peroxidase (GSH⁃GPx) system. It was recently shown that IRI induced the upregulation of miR-182-5p and miR378a-3p and further downregulated GPX4 and SLC7A11, which also induced ferroptosis in AKI (Ding et al., 2020).
Cisplatin is an important chemotherapeutic agent for the treatment of many solid tumors, but its clinical application is limited by serious adverse effects, especially nephrotoxicity. Baliga et al. (Sharma and Leaf, 2019) reported in 1998 on a cisplatin-induced cytotoxicity in vitro model and a cisplatin-induced acute renal failure in vivo model, which showed that iron was detectable in bleomycin released into the medium after exposure to cisplatin, which, a significant increase in iron that could catalyze free radical reactions. They also found that iron chelators significantly reduced cisplatin induced cytotoxicity, suggesting an important role for iron, but the exact mechanism could not be elucidated. Ferritin plays a central role in iron metabolism, and it was found that in the proximal tubule, FTH1 knockout mice had more severe renal injury after cisplatin administration than the control group, which is a finding that underscored the protective role of FTH1 in AKI (van Swelm et al., 2020). Inositol dioxygenase is a proximal renal tubular enzyme, whose overexpression was found by Deng et al. (2019) to exacerbate cisplatin-induced redox damage in cells with AKI. They also found that it promotes ferroptosis through ferritin phagocytosis and lipid peroxidation, and that it might also inhibit GPX4 activity and intracellular GSH concentration by downregulating the “ferroptosis termination system.” In rodents, folic acid could lead to the development of AKI, and certain doses of folic acid could form crystals in the kidney lumen, and high doses could also be directly toxic to the renal tubular epithelium. Martin⁃Sanchez et al. (2017) confirmed the presence of lipid peroxidation and GSH downregulation, which are typical features of ferroptosis, in a folic acid-induced AKI mouse model. In addition, the ferroptosis inhibitor Fer-1 reduced oxidative stress, decreased tubular cell death, and attenuated tissue injury by inhibiting the upregulation of chemokines and cytokines, such as interleukin 33 (IL-33), and by inhibiting macrophage infiltration and downregulating the protective factor Klotho. Caspase inhibitors, however, had no nephroprotective effect (Martin-Sanchez et al., 2017). It was also found that receptor⁃interacting protein kinase 3 (RIPK3) and mixed lineage domain⁃like protein kinase (MLKL) were inhibited. However, the use of RIPK1 inhibitors or RIPK3 and MLKL gene defects did not prevent kidney injury, which suggests that ferroptosis was the predominant cell death pathway in folic acid-induced AKI (Martin-Sanchez et al., 2017). One recent study from China found that higher p53 activation in tubular cells of folic acid-induced AKI model, while Α-lipoic acid (ALA) supplementation blocked the activity of p53 and inhibit ferroptosis (Li et al., 2021b). Thus, p53-mediated ferroptosis in tubular epithelial cell may be a target of treatment of AKI. FG⁃4592 is an inhibitor of hypoxia⁃inducible factor (HIF) precursor hydroxylase, and Li et al. (2020) recently found in a folic acid-induced AKI model that renal function was significantly improved in FG⁃4592 pretreated mice, while the levels of iron, malondialdehyde, and 4-hydroxynonenal in tissues were reduced. In addition, there was upregulation of HIF-1α expression, activation of nuclear factor E2 related factor 2 (Nrf2), and high expression of downstream proteins, including hemeoxygenase 1 (HO1), GPX4, System Xc⁃, and membrane iron transport protein. Further signaling pathway studies suggested that Nrf2 activation is regulated by protein kinase B/glycogen synthase kinase-3β (PKB/GSK⁃3β), which suggests that PKB/GSK⁃3β is a potential therapeutic target for ferroptosis.
Increased iron accumulation and lipid peroxidation in the renal tissue of severe acute pancreatitis rats showed that these changes were accompanied by decreased GPX4 activity and up-regulation of ferroptosis-related proteins and genes. This demonstrates that ferroptosis is associated with severe acute pancreatitis-induced AKI, and lipase I (LIPI) could inhibit ferroptosis, and both reduce renal damage and improve renal function (Ma et al., 2021).
Recent studies demonstrated the importance of ferroptosis in AKI and many studies have shown that ferroptosis can be successfully modulated in different kinds of AKI. A variety of ferroptosis inducers and inhibitors have been administrated in those studies (Figure 2). Several studies have shown that ferroptosis is a promising therapeutic target, especially in diseases dominated by tubular necrosis (Linkermann, 2016). GPX4 is an antioxidant enzyme that catalyzes the reduction of lipid peroxides at the expense of reducing GSH, and thereby preventing oxidative damage and ferroptosis. If GPX4 is absent, it will trigger the accumulation of lipid peroxidation products, which eventually leads to the death of mice due to acute renal failure (Demuynck et al., 2021). Small molecule ferroptosis inhibitors (lipoxygenases), which completely block lipid peroxidation, were able to increase the survival rate of GPX4-deficient mice by 35% (Kang et al., 2021). A novel ferroptosis inhibitor1 that is independent of the classical GPX4 signaling pathway was also recently identified. It uses reduced NADPH to reduce CoQ10 to CoQ10 H2 in the cell membrane which reducing cell membrane lipid peroxidation and thereby inhibiting ferroptosis (Bersuker et al., 2019; Doll et al., 2019). Another study found that thiazolidinediones (e.g., rosiglitazone) reduced mortality in GPX4-deficient mice by inhibiting ACSL4, and demonstrated their potential benefit for AKI (Doll et al., 2017; Kagan et al., 2017). Fer-1 can prevent membrane lipid damage through redox reactions, thereby inhibiting cellular ferroptosis (Xie et al., 2016b; Martin-Sanchez et al., 2017; Zilka et al., 2017; Zhou et al., 2020b). Linkermann et al. (Tonnus et al., 2021) found that the use of Fer-1 not only blocked erastin-induced ferroptosis in vitro, but also prevented renal IRI in mice. It was also found that desferrioxamine could be used to obtain the less cytotoxic compound 3-Hydroxy-Adamant-1-yl by binding to iron adamantine derivatives, which attenuated AKI-causing rhabdomyolysis in rats by inhibiting lipid peroxidation, reducing the ferrous form of Mb, and inhibiting ferroptosis (Groebler et al., 2011; Yu et al., 2017). Pannexin1 (Panx1) is an ATP release pathway family protein. Su et al. (2019) found in a mouse model of ischemia-reperfusion-induced AKI that silencing Panx1 expression significantly attenuated lipid peroxidation and iron accumulation, mainly through mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) signaling. MAPK/ERK signaling activated ferritin and regulated NCAO4-mediated iron phagocytosis and HO1 expression, which provided potential therapeutic targets for AKI management (Su et al., 2019). Deferiprone has been used in patients with high ferrous load from repeated transfusions, but there have been no studies related to the application of deferiprone for the prevention or treatment of AKI due to rhabdomyolysis (Szymonik et al., 2021). Baicalein, an important active ingredient in the Chinese herb Scutellaria baicalensis, attenuated myocardial ischemia-reperfusion-induced AKI by regulating the B-cell lymphoma-2 (BCL2), extracellular signal-regulated kinase 1/2, and BCL2 associated X (BAX) activation of protein kinase B (PKB) (Lai et al., 2016; Szymonik et al., 2021). It has been shown that baicalein can also enhance cellular resistance to ferroptosis by limiting iron accumulation and lipid peroxidation in cells, which makes it a promising therapeutic agent for ferroptosis-related tissue damage (Xie et al., 2016b).
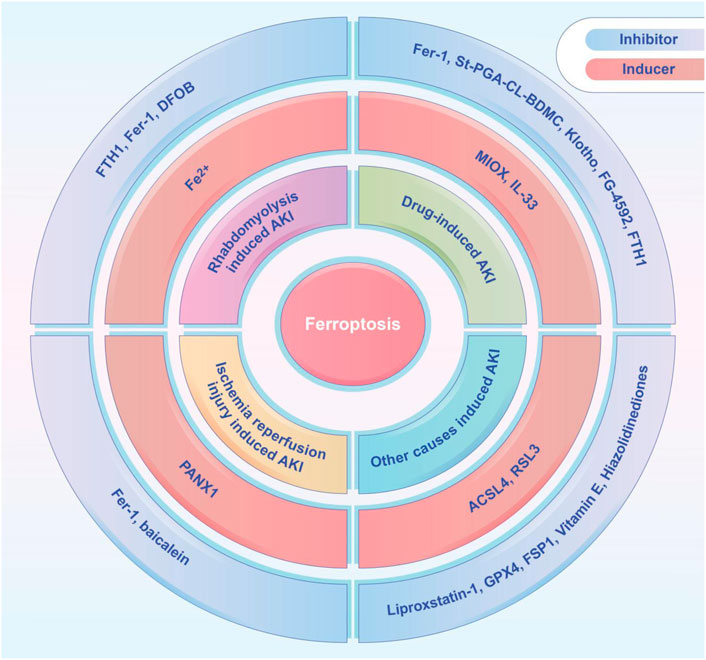
FIGURE 2. The ferroptosis inducers and inhibitors tested in animal models of AKI, including rhabdomyolysis induced AKI, drug-induced AKI, ischemia-reperfusion induced AKI, and other causes induced AKI. ACSL4, acyl-coenzyme A synthetase long chain family member 4; DFOB, desferrioxamine B; FTH1, ferritin heavy chain 1; Fer-1, ferrostatin-1; FG-4592, roxadustat; FSP1, ferroptosis suppressor protein 1; GPX4, glutathione peroxidase 4; IL-33, interleukin 33; MIOX, myo-inositol oxygenase; PANX1, pannexin1; St-PGA-CL-BDMC, star-shaped polyglutamate conjugate of bisdemethoxycurcumin.
In a mouse model of folic acid-induced AKI, Córdoba-David et al. (2020) proposed that stellate polyglutamate⁃curcumin coupling inhibited nuclear factor kappa B (NF-κB) activation and downregulated ferroptosis marker expression, while preserving the renal expression of Klotho, ultimately exerting a reno-protective effect. In addition, rhabdomyolysis-induced renal dysfunction and histological damage could also be reduced by curcumin treatment, with the involvement of HO1 (Guerrero-Hue et al., 2019). Li et al. (2021b) confirmed that ALA also could be used as an anti-ferroptosis agent to reduce iron overload in folic acid-induced AKI through upregulation of Ferritin and ferroportin (Fpn). Additionally, LA has the ability to increase the expression of system xCT, thus increasing the synthesis of GSH and enhancing the activity of GPX4. Roxadustat, an inhibitor of the hypoxia-inducible factor prolyl hydroxylase, is protective against folic acid-induced AKI, and studies have shown improved renal function, inhibition of iron accumulation and lipid peroxidation, and increased levels of antioxidant enzymes and GSH in pretreated mice as compared to untreated mice (Li et al., 2020). Mishima et al. (2020) found in a mouse model of cisplatin-induced AKI that cytochrome P450 (CYP450) substrates, such as isoproterenol and rifampin, attenuated tissue damage and cell death, by scavenging lipid peroxidation radicals. However, due to non-specific ferroptosis markers, multiple cell death pathways might exist in the injury model, and the inhibitory effect of drugs on ferroptosis was not directly confirmed. Some studies have tentatively shown that vitamin D receptor (VDR) activation is protective against cisplatin-induced AKI, and that paricalcitol reduces lipid peroxidation, 4-hydroxynonenal, and malondialdehyde, functionally and histologically attenuating cisplatin-induced AKI (Mishima et al., 2020).
Ferroptosis is a recently discovered, iron-dependent, non-apoptotic cell death mechanism. This paper outlines the mechanisms underlying its incidence, its characteristics, and its general preliminary understanding. In AKI, it has been possible to clarify that ferroptosis is one of the important causes of cell death. The application of small molecule ferroptosis inhibitors to inhibit ferroptosis is expected to be a new strategy in the treatment of AKI. It is applied in AKI particularly in cases of rhabdomyolysis or ischemia-reperfusion AKI, which is mainly caused by heme and non-matrix iron. Although the majority of these studies targeting small molecule ferroptosis inhibitors have been conducted in mice models of AKI or in vitro experiments, we believe that an in-depth exploration of the role played by ferroptosis in the process of AKI and the rational use of ferroptosis in the regulation of AKI would provide new perspectives and new strategies for AKI treatment.
JZ contributed to the organization of the preparation, writing, reading, correction and submission of the manuscript, as well as to the writing of the Abstract and Chapters 1, 2, and 4; BW and SY contributed to the writing of Chapter 3 and 5; JJ and QH contributed to the editing and reading of all manuscripts.
This research was supported by the Construction of Key Projects by Zhejiang Provincial Ministry (Project No. WKJ-ZJ-2017), the Zhejiang Province Chinese Medicine Modernization Program (Project No. 2020ZX001), the Key Project of Scientific Research Foundation of Chinese Medicine (2022ZZ002), the Key project of Zhejiang Science and Technology Department (2022C03118), the Huadong Medicine Joint Funds of the Zhejiang Provincial Natural Science Foundation of China (Grant No. LHDMZ22H050001), the Key project of Basic Scientific Research Operating Funds of Hangzhou Medical College (KYZD202002) and the Clinical and Experimental Research of YSHS Granule.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adedoyin, O., Boddu, R., Traylor, A., Lever, J. M., Bolisetty, S., George, J. F., et al. (2018). Heme Oxygenase-1 Mitigates Ferroptosis in Renal Proximal Tubule Cells. Am. J. Physiology-Renal PhysiologyRenal physiology 314, F702–F714. doi:10.1152/ajprenal.00044.2017
Alvarez, S. W., Sviderskiy, V. O., Terzi, E. M., Papagiannakopoulos, T., Moreira, A. L., Adams, S., et al. (2017). NFS1 Undergoes Positive Selection in Lung Tumours and Protects Cells from Ferroptosis. Nature 551, 639–643. doi:10.1038/nature24637
Anthonymuthu, T. S., Kenny, E. M., Shrivastava, I., Tyurina, Y. Y., Hier, Z. E., Ting, H.-C., et al. (2018). Empowerment of 15-Lipoxygenase Catalytic Competence in Selective Oxidation of Membrane ETE-PE to Ferroptotic Death Signals, HpETE-PE. J. Am. Chem. Soc. 140, 17835–17839. doi:10.1021/jacs.8b09913
Bersuker, K., Hendricks, J. M., Li, Z., Magtanong, L., Ford, B., Tang, P. H., et al. (2019). The CoQ Oxidoreductase FSP1 Acts Parallel to GPX4 to Inhibit Ferroptosis. Nature 575, 688–692. doi:10.1038/s41586-019-1705-2
Bosch, X., Poch, E., and Grau, J. M. (2009). Rhabdomyolysis and Acute Kidney Injury. N. Engl. J. Med. 361, 62–72. doi:10.1056/nejmra0801327
Chang, T.-C., Wentzel, E. A., Kent, O. A., Ramachandran, K., Mullendore, M., Lee, K. H., et al. (2007). Transactivation of miR-34a by P53 Broadly Influences Gene Expression and Promotes Apoptosis. Mol. Cell 26, 745–752. doi:10.1016/j.molcel.2007.05.010
Choi, N., Whitlock, R., Klassen, J., Zappitelli, M., Arora, R. C., Rigatto, C., et al. (2019). Early Intraoperative Iron-Binding Proteins Are Associated with Acute Kidney Injury after Cardiac Surgery. J. Thorac. Cardiovasc. Surg. 157, 287–297. e2. doi:10.1016/j.jtcvs.2018.06.091
Córdoba-David, G., Duro-Castano, A., Castelo-Branco, R. C., González-Guerrero, C., Cannata, P., Sanz, A. B., et al. (2020). Effective Nephroprotection Against Acute Kidney Injury with a Star-Shaped Polyglutamate-Curcuminoid Conjugate. Sci. Rep. 10, 2056. doi:10.1038/s41598-020-58974-9
Demuynck, R., Efimova, I., Naessens, F., and Krysko, D. (2021). Immunogenic Ferroptosis and where to Find it? J. Immunother. cancer 9, e003430. doi:10.1136/jitc-2021-003430
Deng, F., Sharma, I., Dai, Y., Yang, M., and Kanwar, Y. S. (2019). Myo-inositol Oxygenase Expression Profile Modulates Pathogenic Ferroptosis in the Renal Proximal Tubule. J. Clin. investigation 129, 5033–5049. doi:10.1172/jci129903
Ding, C., Ding, X., Zheng, J., Wang, B., Li, Y., Xiang, H., et al. (2020). miR-182-5p and miR-378a-3p Regulate Ferroptosis in I/R-induced Renal Injury. Cell Death Dis. 11, 929. doi:10.1038/s41419-020-03135-z
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: an Iron-dependent Form of Nonapoptotic Cell Death. Cell 149, 1060–1072. doi:10.1016/j.cell.2012.03.042
Doll, S., and Conrad, M. (2017). Iron and Ferroptosis: A Still Ill‐defined Liaison. IUBMB life 69, 423–434. doi:10.1002/iub.1616
Doll, S., Freitas, F. P., Shah, R., Aldrovandi, M., da Silva, M. C., Ingold, I., et al. (2019). FSP1 Is a Glutathione-independent Ferroptosis Suppressor. Nature 575, 693–698. doi:10.1038/s41586-019-1707-0
Doll, S., Proneth, B., Tyurina, Y. Y., Panzilius, E., Kobayashi, S., Ingold, I., et al. (2017). ACSL4 Dictates Ferroptosis Sensitivity by Shaping Cellular Lipid Composition. Nat. Chem. Biol. 13, 91–98. doi:10.1038/nchembio.2239
Franzin, R., Stasi, A., Fiorentino, M., Simone, S., Oberbauer, R., Castellano, G., et al. (2021). Renal Delivery of Pharmacologic Agents During Machine Perfusion to Prevent Ischaemia-Reperfusion Injury: From Murine Model to Clinical Trials. Front. Immunol. 12, 673562. doi:10.3389/fimmu.2021.673562
Gao, M., Yi, J., Zhu, J., Minikes, A. M., Monian, P., Thompson, C. B., et al. (2019). Role of Mitochondria in Ferroptosis. Mol. Cell 73, 354–e3. doi:10.1016/j.molcel.2018.10.042
Gao, M., Monian, P., Quadri, N., Ramasamy, R., and Jiang, X. (2015). Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 59, 298–308. doi:10.1016/j.molcel.2015.06.011
Groebler, L. K., Liu, J., Shanu, A., Codd, R., and Witting, P. K. (2011). Comparing the Potential Renal Protective Activity of Desferrioxamine B and the Novel Chelator Desferrioxamine B-N-(3-hydroxyadamant-1-yl)carboxamide in a Cell Model of Myoglobinuria. Biochem. J. 435, 669–677. doi:10.1042/bj20101728
Guerrero-Hue, M., García-Caballero, C., Palomino-Antolín, A., Rubio-Navarro, A., Vázquez-Carballo, C., Herencia, C., et al. (2019). Curcumin Reduces Renal Damage Associated with Rhabdomyolysis by Decreasing Ferroptosis-Mediated Cell Death. FASEB J. 33, 8961–8975. doi:10.1096/fj.201900077R
Haase, M., Bellomo, R., and Haase-Fielitz, A. (2010). Novel Biomarkers, Oxidative Stress, and the Role of Labile Iron Toxicity in Cardiopulmonary Bypass-Associated Acute Kidney Injury. J. Am. Coll. Cardiol. 55, 2024–2033. doi:10.1016/j.jacc.2009.12.046
Han, H., Wen, Z., Wang, J., Zhang, P., Gong, Q., Ge, S., et al. (2021). Prediction of Short-Term Mortality with Renal Replacement Therapy in Patients with Cardiac Surgery-Associated Acute Kidney Injury. Front. Cardiovasc. Med. 8, 738947. doi:10.3389/fcvm.2021.738947
Heintzman, D., Fisher, E., and Rathmell, J. (2022). Microenvironmental Influences on T Cell Immunity in Cancer and Inflammation. Cell. Mol. Immunol. 19, 316. doi:10.1038/s41423-021-00833-2
Huang, L. l., Liao, X. h., Sun, H., Jiang, X., Liu, Q., and Zhang, L. (2019). Augmenter of Liver Regeneration Protects the Kidney from Ischaemia‐reperfusion Injury in Ferroptosis. J. Cell Mol. Med. 23, 4153–4164. doi:10.1111/jcmm.14302
Jiang, L., Kon, N., Li, T., Wang, S.-J., Su, T., Hibshoosh, H., et al. (2015). Ferroptosis as a P53-Mediated Activity during Tumour Suppression. Nature 520, 57–62. doi:10.1038/nature14344
Jiang, X., Guo, S., Zhang, Y., Zhao, Y., Li, X., Jia, Y., et al. (2020). LncRNA NEAT1 Promotes Docetaxel Resistance in Prostate Cancer by Regulating ACSL4 via Sponging miR-34a-5p and miR-204-5p. Cell. Signal. 65, 109422. doi:10.1016/j.cellsig.2019.109422
Kagan, V. E., Mao, G., Qu, F., Angeli, J. P. F., Doll, S., Croix, C. S., et al. (2017). Oxidized Arachidonic and Adrenic PEs Navigate Cells to Ferroptosis. Nat. Chem. Biol. 13, 81–90. doi:10.1038/nchembio.2238
Kang, Y. P., Mockabee-Macias, A., Jiang, C., Falzone, A., Prieto-Farigua, N., Stone, E., et al. (2021). Non-canonical Glutamate-Cysteine Ligase Activity Protects against Ferroptosis. Cell metab. 33, 174–189. e7. doi:10.1016/j.cmet.2020.12.007
Kwon, M.-Y., Park, E., Lee, S.-J., and Chung, S. W. (2015). Heme Oxygenase-1 Accelerates Erastin-Induced Ferroptotic Cell Death. Oncotarget 6, 24393–24403. doi:10.18632/oncotarget.5162
Lai, C. C., Huang, P. H., Yang, A. H., Chiang, S. C., Tang, C. Y., Tseng, K. W., et al. (2016). Baicalein, a Component of Scutellaria Baicalensis, Attenuates Kidney Injury Induced by Myocardial Ischemia and Reperfusion. Planta Med. 82, 181–189. doi:10.1055/s-0035-1558114
Leaf, D. E., Rajapurkar, M., Lele, S. S., Mukhopadhyay, B., Boerger, E. A. S., Mc Causland, F. R., et al. (2019). Iron, Hepcidin, and Death in Human AKI. J. Am. Soc. Nephrol. 30, 493–504. doi:10.1681/asn.2018100979
Lee, T., Lee, C., Chen, J., Fan, P., Tu, Y., Yen, C., et al. (2021). Assessment of Cardiopulmonary Bypass Duration Improves Novel Biomarker Detection for Predicting Postoperative Acute Kidney Injury after Cardiovascular Surgery. J. Clin. Med. 10, 2741. doi:10.3390/jcm10132741
Li, J.-y., Liu, S.-q., Yao, R.-q., Tian, Y.-p., and Yao, Y.-m. (2021). A Novel Insight into the Fate of Cardiomyocytes in Ischemia-Reperfusion Injury: From Iron Metabolism to Ferroptosis. Front. Cell Dev. Biol. 9, 799499. doi:10.3389/fcell.2021.799499
Li, X., Zou, Y., Xing, J., Fu, Y. Y., Wang, K. Y., Wan, P. Z., et al. (2020). Pretreatment with Roxadustat (FG-4592) Attenuates Folic Acid-Induced Kidney Injury through Antiferroptosis via Akt/GSK-3β/Nrf2 Pathway. Oxid. Med. Cell Longev. 2020, 6286984. doi:10.1155/2020/6286984
Li, X., Zou, Y., Fu, Y.-Y., Xing, J., Wang, K.-Y., Wan, P.-Z., et al. (2021). A-Lipoic Acid Alleviates Folic Acid-Induced Renal Damage through Inhibition of Ferroptosis. Front. Physiol. 12, 680544. doi:10.3389/fphys.2021.680544
Linkermann, A. (2016). Nonapoptotic Cell Death in Acute Kidney Injury and Transplantation. Kidney Int. 89, 46–57. doi:10.1016/j.kint.2015.10.008
Liu, Y., and Gu, W. (2022). p53 in Ferroptosis Regulation: the New Weapon for the Old Guardian. Cell Death Differ. 29, 895–910. doi:10.1038/s41418-022-00943-y
Liu, Y., and Gu, W. (2021). The Complexity of P53-Mediated Metabolic Regulation in Tumor Suppression. Semin. Cancer Biol. S1044-579X (21), 00060–00062. doi:10.1016/j.semcancer.2021.03.010
Ma, D., Li, C., Jiang, P., Jiang, Y., Wang, J., and Zhang, D. (2021). Inhibition of Ferroptosis Attenuates Acute Kidney Injury in Rats with Severe Acute Pancreatitis. Dig. Dis. Sci. 66, 483–492. doi:10.1007/s10620-020-06225-2
Martin-Sanchez, D., Ruiz-Andres, O., Poveda, J., Carrasco, S., Cannata-Ortiz, P., Sanchez-Niño, M. D., et al. (2017). Ferroptosis, but Not Necroptosis, Is Important in Nephrotoxic Folic Acid-Induced AKI. J. Am. Soc. Nephrol. 28, 218–229. doi:10.1681/asn.2015121376
Martines, A. M. F., Masereeuw, R., Tjalsma, H., Hoenderop, J. G., Wetzels, J. F. M., and Swinkels, D. W. (2013). Iron Metabolism in the Pathogenesis of Iron-Induced Kidney Injury. Nat. Rev. Nephrol. 9, 385–398. doi:10.1038/nrneph.2013.98
Messerer, D. A. C., Halbgebauer, R., Nilsson, B., Pavenstädt, H., Radermacher, P., and Huber-Lang, M. (2021). Immunopathophysiology of Trauma-Related Acute Kidney Injury. Nat. Rev. Nephrol. 17, 91–111. doi:10.1038/s41581-020-00344-9
Mishima, E., Sato, E., Ito, J., Yamada, K.-i., Suzuki, C., Oikawa, Y., et al. (2020). Drugs Repurposed as Antiferroptosis Agents Suppress Organ Damage, Including AKI, by Functioning as Lipid Peroxyl Radical Scavengers. J. Am. Soc. Nephrol. 31, 280–296. doi:10.1681/asn.2019060570
Moon, S.-H., Huang, C.-H., Houlihan, S. L., Regunath, K., Freed-Pastor, W. A., Morris, J. P., et al. (2019). p53 Represses the Mevalonate Pathway to Mediate Tumor Suppression. Cell 176, 564–580. e19. doi:10.1016/j.cell.2018.11.011
Mou, Y., Wang, J., Wu, J., He, D., Zhang, C., Duan, C., et al. (2019). Ferroptosis, a New Form of Cell Death: Opportunities and Challenges in Cancer. J. Hematol. Oncol. 12, 34. doi:10.1186/s13045-019-0720-y
Nieuwenhuijs-Moeke, G. J., Pischke, S. E., Berger, S. P., Sanders, J. S. F., Pol, R. A., Struys, M. M. R. F., et al. (2020). Ischemia and Reperfusion Injury in Kidney Transplantation: Relevant Mechanisms in Injury and Repair. J. Clin. Med. 9, 253. doi:10.3390/jcm9010253
Sharma, S., and Leaf, D. E. (2019). Iron Chelation as a Potential Therapeutic Strategy for AKI Prevention. J. Am. Soc. Nephrol. 30, 2060–2071. doi:10.1681/asn.2019060595
Shimada, K., Skouta, R., Kaplan, A., Yang, W. S., Hayano, M., Dixon, S. J., et al. (2016). Global Survey of Cell Death Mechanisms Reveals Metabolic Regulation of Ferroptosis. Nat. Chem. Biol. 12, 497–503. doi:10.1038/nchembio.2079
Song, X., Zhu, S., Chen, P., Hou, W., Wen, Q., Liu, J., et al. (2018). AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System Xc- Activity. Curr. Biol. 28, 2388–2399. e5. doi:10.1016/j.cub.2018.05.094
Su, L., Jiang, X., Yang, C., Zhang, J., Chen, B., Li, Y., et al. (2019). Pannexin 1 Mediates Ferroptosis that Contributes to Renal Ischemia/reperfusion Injury. J. Biol. Chem. 294, 19395–19404. doi:10.1074/jbc.ra119.010949
Swaminathan, S. (2018). Iron Homeostasis Pathways as Therapeutic Targets in Acute Kidney Injury. Nephron 140, 156–159. doi:10.1159/000490808
Szymonik, J., Wala, K., Górnicki, T., Saczko, J., Pencakowski, B., and Kulbacka, J. (2021). The Impact of Iron Chelators on the Biology of Cancer Stem Cells. Int. J. Mol. Sci. 23, 89. doi:10.3390/ijms23010089
Tang, C., Ma, Z., Zhu, J., Liu, Z., Liu, Y., Liu, Y., et al. (2019). P53 in Kidney Injury and Repair: Mechanism and Therapeutic Potentials. Pharmacol. Ther. 195, 5–12. doi:10.1016/j.pharmthera.2018.10.013
Tang, D., Chen, X., Kang, R., and Kroemer, G. (2021). Ferroptosis: Molecular Mechanisms and Health Implications. Cell Res. 31, 107–125. doi:10.1038/s41422-020-00441-1
Tarangelo, A., Magtanong, L., Bieging-Rolett, K. T., Li, Y., Ye, J., Attardi, L. D., et al. (2018). p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep. 22, 569–575. doi:10.1016/j.celrep.2017.12.077
Thévenod, F., Lee, W.-K., and Garrick, M. D. (2020). Iron and Cadmium Entry into Renal Mitochondria: Physiological and Toxicological Implications. Front. Cell Dev. Biol. 8, 848. doi:10.3389/fcell.2020.00848
Tonnus, W., and Linkermann, A. (2017). The In Vivo Evidence for Regulated Necrosis. Immunol. Rev. 277, 128–149. doi:10.1111/imr.12551
Tonnus, W., Meyer, C., Steinebach, C., Belavgeni, A., von Mässenhausen, A., Gonzalez, N. Z., et al. (2021). Dysfunction of the Key Ferroptosis-Surveilling Systems Hypersensitizes Mice to Tubular Necrosis during Acute Kidney Injury. Nat. Commun. 12, 4402. doi:10.1038/s41467-021-24712-6
van Swelm, R. P. L., Wetzels, J. F. M., and Swinkels, D. W. (2020). The Multifaceted Role of Iron in Renal Health and Disease. Nat. Rev. NephrolNephrology 16, 77–98. doi:10.1038/s41581-019-0197-5
Wang, S., Luo, J., Zhang, Z., Dong, D., Shen, Y., Fang, Y., et al. (2018). Iron and Magnetic: New Research Direction of the Ferroptosis-Based Cancer Therapy. Am. J. Cancer Res. 8, 1933–1946.
Wang, Y., and Tao, Y. (2015). Research Progress on Regulatory T Cells in Acute Kidney Injury. J. Immunol. Res. 2015, 174164. doi:10.1155/2015/174164
Xie, Y., Hou, W., Song, X., Yu, Y., Huang, J., Sun, X., et al. (2016). Ferroptosis: Process and Function. Cell Death Differ. 23, 369–379. doi:10.1038/cdd.2015.158
Xie, Y., Song, X., Sun, X., Huang, J., Zhong, M., Lotze, M. T., et al. (2016). Identification of Baicalein as a Ferroptosis Inhibitor by Natural Product Library Screening. Biochem. biophysical Res. Commun. 473, 775–780. doi:10.1016/j.bbrc.2016.03.052
Yagoda, N., von Rechenberg, M., Zaganjor, E., Bauer, A. J., Yang, W. S., Fridman, D. J., et al. (2007). RAS-RAF-MEK-dependent Oxidative Cell Death Involving Voltage-dependent Anion Channels. Nature 447, 864–868. doi:10.1038/nature05859
Yang, L., Wang, H., Yang, X., Wu, Q., An, P., Jin, X., et al. (2020). Auranofin Mitigates Systemic Iron Overload and Induces Ferroptosis via Distinct Mechanisms. Sig Transduct. Target Ther. 5, 138. doi:10.1038/s41392-020-00253-0
Yao, X., Li, W., Fang, D., Xiao, C., Wu, X., Li, M., et al. (2021). Emerging Roles of Energy Metabolism in Ferroptosis Regulation of Tumor Cells. Adv. Sci. Weinh 8, e2100997. Baden-Wurttemberg, Germany. doi:10.1002/advs.202100997
Yu, H., Guo, P., Xie, X., Wang, Y., and Chen, G. (2017). Ferroptosis, a New Form of Cell Death, and its Relationships with Tumourous Diseases. J. Cell. Mol. Med. 21, 648–657. doi:10.1111/jcmm.13008
Zarjou, A., Bolisetty, S., Joseph, R., Traylor, A., Apostolov, E. O., Arosio, P., et al. (2013). Proximal Tubule H-Ferritin Mediates Iron Trafficking in Acute Kidney Injury. J. Clin. Invest. 123, 4423–4434. doi:10.1172/jci67867
Zeng, W., and Tomlinson, B. (2021). Causes and Outcome of Rhabdomyolysis in Patients Admitted to Medical Wards in the Prince of Wales Hospital. Ann. Transl. Med. 9, 1329. doi:10.21037/atm-21-3660
Zhou, F., Yang, Y., Luo, L., Chen, Y., Luo, Q., and Chen, J. (2020). Impact of Prolonged Mechanical Ventilation on Ferroptosis in Renal Ischemia/Reperfusion Injury in Rats. Biomed. Res. Int. 2020, 6097516. doi:10.1155/2020/6097516
Zhou, R.-P., Chen, Y., Wei, X., Yu, B., Xiong, Z.-G., Lu, C., et al. (2020). Novel Insights into Ferroptosis: Implications for Age-Related Diseases. Theranostics 10, 11976–11997. doi:10.7150/thno.50663
Keywords: acute kidney injury, ferroptosis, mechanism, treatment, regulators
Citation: Zhang J, Wang B, Yuan S, He Q and Jin J (2022) The Role of Ferroptosis in Acute Kidney Injury. Front. Mol. Biosci. 9:951275. doi: 10.3389/fmolb.2022.951275
Received: 23 May 2022; Accepted: 13 June 2022;
Published: 30 June 2022.
Edited by:
Xin Wang, National Institutes of Health (NIH), United StatesReviewed by:
Gang Wang, Weill Cornell Medicine, United StatesCopyright © 2022 Zhang, Wang, Yuan, He and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan Jin, bGFuZ18wMThAMTYzLmNvbQ==; Qiang He, cWlhbmdoZTE5NzNAMTI2LmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.