 Fiorella Faienza
Fiorella Faienza Andrea Rasola2
Andrea Rasola2 Giuseppe Filomeni
Giuseppe Filomeni- 1Department of Biology, University of Rome Tor Vergata, Rome, Italy
- 2Department of Biomedical Sciences, University of Padova, Padova, Italy
- 3Redox Biology, Danish Cancer Society Research Center, Copenhagen, Denmark
- 4Center for Healthy Aging, University of Copenhagen, Copenhagen, Denmark
Nitric oxide in metabolism regulation
Nitric oxide (NO) is a gaseous signaling molecule able to modify protein structure and activity (Rizza and Filomeni, 2017). The addition of the NO moiety to a cysteine thiol is called S-nitrosylation, which is establishing as one of the major NO-induced protein posttranslational modifications involved in cell signaling (Fernando et al., 2019). In the presence of oxygen—and reactive oxygen species (ROS), mostly superoxide (O2-)—NO can also give rise to other biologically active compounds, such as peroxynitrite (ONOO−) (Fernando et al., 2019). Peroxynitrite is highly reactive and commonly recognized being detrimental to macromolecules, as it can cause irreversible modifications to lipids (peroxidation) and proteins (tyrosine nitration) (Radi, 2018).
Nitric oxide is endogenously generated by nitric oxide synthases (NOSs), a family of three isozymes (neuronal, endothelial, and inducible) whose subcellular localization and activation influence NO target selectivity (Benhar et al., 2009). NO and NO-derived species (e.g., N2O3, NO+) can directly react with thiol groups in proteins or in sulfhydryl-containing low molecular weight molecules, such as glutathione, free cysteine, and Coenzyme A (CoA). Otherwise, S-nitroso (SNO) groups can be generated via transnitrosylation, the transfer of an NO moiety between thiols and S-nitrosothiols (thiol/nitrosothiol exchange) (Nakamura et al., 2021). This mechanism is also the basis of protein denitrosylation, which is the process required to remove NO from S-nitrosothiols and regenerate protein thiol pools. Three are the denitrosylases so far characterized: i) S-nitrosoglutathione reductase (GSNOR); ii) S-nitroso-CoA reductase (ScoR) and iii) thioredoxin reductase (TrxR). All these enzymes do not directly react with S-nitrosylated proteins, but specifically recognize, and remove, the NO moiety from small S-nitrosothiols (S-nitrosoglutathione, S-nitroso-CoA and thioredoxin, respectively) generated by transnitrosylation with S-nitrosylated proteins (Rizza and Filomeni, 2017).
S-nitrosylation usually acts as an inhibitory mechanism of key metabolic enzymes in order to adapt metabolism to cell’s needs (Brown, 2007). Nitric oxide inhibits all electron transport chain (ETC) complexes (Poderoso et al., 2019). Cytochrome c oxidase (CcOX), the complex IV of ETC (Rich, 2017), represents the best-characterized mitochondrial target of NO. At low levels, NO inhibits complex IV competing with O2 for the binding to the active site, whereas, at high concentrations, the inhibition occurs via S-nitrosylation (Cleeter et al., 1994). The NO-mediated regulation of CcOX activity is closely related to the oxygen availability and finely tunes cellular respiration, as it controls the balance between O2 cellular uptake and consumption (Poderoso et al., 2019). As a side-effect of NO-mediated inhibition of mitochondrial complexes, ROS are produced, this leading to the production of peroxynitrite that further enhances NO inhibition on ETC complexes in a positive feedback loop (Brown, 2007).
NO also inhibits several tricarboxylic acid (TCA) cycle enzymes, such as aconitase, α-ketoglutarate dehydrogenase and succinate dehydrogenase (SDH) (Prime et al., 2009), as well as enzymes involved in fatty acid oxidation (FAO) (Piantadosi, 2012) and branched-chain amino acid metabolism (Coles et al., 2009). Likewise, pyruvate kinase M2 is inhibited by S-nitrosylation, redirecting glucose towards the pentose phosphate pathway (Lowenstein, 2019). Nitric oxide can also tune metabolism by targeting the chaperone tumor necrosis factor receptor-associated protein 1 (TRAP1) and the deacetylase sirtuin 3 (SIRT3) (Rizza et al., 2016; Kalous et al., 2021), which have been recently proposed to be mutually regulated in the mitochondria (Park et al., 2019).
Tumor necrosis factor receptor-associated protein 1
Tumor necrosis factor receptor-associated protein 1 in metabolism
TRAP1 is a mitochondrial chaperone belonging to heat shock protein 90 family (Hoter et al., 2018). It is considered a key molecule for the maintenance of mitochondrial homeostasis given its ability to: i) promote metabolism rewiring by sustaining a “Warburg-like”, aerobic glycolysis phenotype; ii) downregulate ROS production, iii) protect from stress-induced cell death (Hoter et al., 2018). TRAP1 works as a dimer in which each monomer is composed of three domains: 1) the N-terminal domain, responsible for ATP binding; 2) the middle domain, which participates in ATP binding site shaping and client allocation; 3) the C-terminal domain, indispensable for dimerization (Serapian et al., 2021). Interestingly, it has been recently demonstrated that TRAP1 is also able to generate tetramers (i.e., dimers of dimers) in association with changes in oxidative phosphorylation (OXPHOS) rate (Joshi et al., 2020; Liu et al., 2020).
Only few TRAP1 interactors, called clients, have been characterized to date (Sanchez-Martin et al., 2020b), even if emerging data indicate that TRAP1 can bind multiple mitochondrial components of bioenergetic circuits (Joshi et al., 2020; Cannino et al., 2022). Among the best defined TRAP1 clients is SDH, a key enzyme placed at the intersection of OXPHOS and TCA cycle. TRAP1 downregulates SDH activity (Sciacovelli et al., 2013), an effect reinforced by TRAP1 interaction with the mitochondrial fraction of the extracellular signal-regulated kinase (ERK) (Masgras et al., 2017a). Consequently, besides dampening OXPHOS rate, TRAP1-mediated inhibition of SDH activity results in succinate accumulation (Faienza et al., 2020b), an effect particularly relevant in cancer models characterized by hyperactivation of Ras/ERK signalling (Masgras et al., 2017b). Indeed, succinate is an oncometabolite that concurs to promote tumorigenesis by affecting the activity of α-ketoglutarate dependent hydroxylases (Islam et al., 2018). Among this huge class of enzymes, it is worth to mention: i) 5-methylcytosine hydroxylases and JmjC domain containing lysine demethylases, which broadly affect epigenetic regulation, and ii) prolyl hydroxylases, which leads to stabilization of the hypoxia inducible factor 1α (HIF-1α) and activation of a HIF1-mediated (pseudo)hypoxic transcription program, condition favoring neoplastic progression even under normoxia (Sanchez-Martin et al., 2020b; Masgras et al., 2021). TRAP1 is transcriptionally activated by HIF1, in a feed-forward loop between HIF1 and TRAP1, which sustains metabolic adaptations in cancer, as well as during embryonic development (Laquatra et al., 2021) and oxidative damage associated to ischemic conditions (Masgras et al., 2017b). The inhibitory interaction between TRAP1 and the mitochondrial fraction of the c-Src kinase, a CcOX activator (Yoshida et al., 2013), could also contribute to OXPHOS inhibition, in line with the antioxidant effect exerted by TRAP1 (Guzzo et al., 2014).
In agreement with its pro-neoplastic role, TRAP1 also inhibits cell death. Recent findings suggest that this could rely on TRAP1 interaction with F-ATP synthase: the OXPHOS complex that generates ATP and acts as the permeability transition pore (PTP) (Urbani et al., 2019), a mitochondrial channel whose opening commits cells to death (Masgras et al., 2021). Indeed, TRAP1 interacts with the F-ATP synthase subunit OSCP, blocking the PTP channel activity and outcompeting the PTP-activating interaction between OSCP and the prolyl isomerase cyclophilin D (Cannino et al., 2022).
Tumor necrosis factor receptor-associated protein 1 regulation by nitric oxide
TRAP1 is a target of S-nitrosylation, which affects protein stability and activity (Faienza et al., 2020b). This is particularly relevant in a GSNOR-deficient model of hepatocellular carcinoma (HCC) where GSNOR loss increases SDH activity and, in turn, confers high sensitivity to SDH-targeting drugs (Rizza et al., 2016). This mitochondrial alteration represents the Achilles’ heel of GSNOR-deficient HCC cells and is associated with TRAP1 S-nitrosylation at Cys501, which causes TRAP1 destabilization and proteasomal degradation (Rizza et al., 2016). S-nitrosylation of Cys501 also reduces TRAP1 ATPase activity via an allosteric mechanism, and cells expressing the non-nitrosylable TRAP1 mutant are less sensitive to apoptosis induction (Faienza et al., 2020a). This suggests that Cys501 nitrosylation inhibits TRAP1 ATPase activity, destabilizes its structure and directs TRAP1 to proteasomal degradation (Faienza et al., 2020b) or, as proposed (Elnatan et al., 2017), promotes a TRAP1 holdase activity that shields clients exposed to stress conditions in an ATP-independent way.
Sirtuin 3
Sirtuin 3 in metabolism
Sirtuins (SIRTs) are NAD+-dependent deacetylases with a catalytic site where four cysteine residues coordinate a Zinc ion (Zn2+-tetrathiolate) (Kalous et al., 2021). Since sirtuins need NAD+ to work, they are both sensors and regulators of the NAD+/NADH ratio, which affects metabolism and the redox status of cells (Elkhwanky and Hakkola, 2017). Seven sirtuins have been described in mammals, three of which (SIRT3, SIRT4, and SIRT5) are localized in mitochondria. SIRT3 is the main mitochondrial deacetylase, whereas SIRT4 is involved in ADP-ribosylation and SIRT5 in desuccinylation, demalonylation, and deglutarylation reactions (Singh et al., 2017a).
SIRT3 has been implicated in virtually all mitochondrial metabolic pathways (Rardin et al., 2013), as it controls the activity of a variety of mitochondrial enzymes involved in redox and bioenergetic homeostasis by regulating their acetylation status (Kalous et al., 2021). Mitochondrial translocation of SIRT3 is increased in response to several stress stimuli (Singh et al., 2017a).
The first mitochondrial proteins identified as SIRT3 targets were complex I and II of the ETC (Bong-Hyun et al., 2008), but further studies suggested that all ETC complexes can be regulated by SIRT3 (Bell et al., 2011). Numerous observations extended the number of SIRT3 substrates, which currently includes α-ketoglutarate dehydrogenase and malate dehydrogenase 2 Rardin al. (2013), whereas isocitrate dehydrogenase 2 and superoxide dismutase 2 (SOD2) are activated by SIRT3-dependent deacetylation (Zullo et al., 2022), leading to an increase in NADPH levels and in the antioxidant response (Yu et al., 2012). In further agreement with the antioxidant function of SIRT3, it is worth noting that its deacetylating activity is required for Forkhead box O3a-mediated transcription of both superoxide dismutase 2 and catalase (Singh et al., 2017a).
A large body of literature defines SIRT3 as a general booster of mitochondrial metabolism. Enzymes belonging to the urea cycle, FAO (Hirschey et al., 2010) and ketogenesis (Shimazu et al., 2010) are activated by SIRT3 deacetylation to keep metabolism running. Phenotypes observed in SIRT3 KO (Sir3−/−) mice support this general idea, showing: i) a general decrease of cellular respiration (Bong-Hyun et al., 2008; Singh et al., 2017a); ii) liver steatosis associated with increased levels of triglycerides and FAO intermediates (Hirschey et al., 2010; Rardin et al., 2013); iii) alterations in ketone body production (Elkhwanky and Hakkola, 2017).
Sirtuin 3 regulation by nitric oxide
Zn2+-tetrathiolate is a target of several oxidative post-translational modifications (Kalous et al., 2021). Among these, S-nitrosylation has been proposed to have a prominent role, and the S-nitrosylated form of Zn2+-tetrathiolate has a general inhibitory effect on sirtuins. However, in mitochondria, NO generated by NOS1 activates SIRT3, resulting in deacetylation-dependent SOD2 activation and down-regulation of ROS levels (Wang et al., 2019). Vice versa, massive NO production has been reported to inactivate SIRT3 via tyrosine nitration (Pérez et al., 2018). This argues for a Janus-faced (physio-pathological) effect for NO, depending on the fluxes applied, which is consistent with the crucial role of SIRT3 in the antioxidant defense of the cell (Kalous et al., 2020).
Tumor necrosis factor receptor-associated protein 1 and sirtuin 3 crosstalk
Tumor necrosis factor receptor-associated protein 1 and sirtuin 3 in cancer
An interaction between TRAP1 and SIRT3 has been reported in glioma stem cells (GSC) (Park et al., 2019), where the two proteins reciprocally sustain their activities: SIRT3-mediated deacetylation maintains TRAP1 chaperone activity, which in turn stabilizes SIRT3. Several OXPHOS components associate with TRAP1/SIRT3 complex, resulting in a high level of respiration matched by low ROS. However, in line with the metabolic plasticity that characterizes neoplastic cells, it is possible to envisage other metabolic effects of TRAP1 and SIRT3. For instance, in malignant cells related to the tumor-predisposing syndrome neurofibromatosis type 1 (NF1), the hyperactivation of Ras/ERK signaling, which occurs downstream to loss of neurofibromin, dampens the expression and activity of complex I, lowering both respiration and intracellular NAD+ levels. As a result, SIRT3 activity is reduced, supporting the hypothesis that SIRT3 has anti-neoplastic effects that synergize with TRAP1 inhibition (Masgras et al., 2022). Indeed, both SIRT3 induction and TRAP1 inhibition enhance SDH enzymatic activity to a similar extent and without any additive effect. Coherently, the allosteric TRAP1 inhibitor honokiol bis-dichloroacetate increases SDH activity at the same level of SIRT3 overexpression in NF1-related malignant peripheral nerve sheath tumor cells (Sanchez-Martin et al., 2020a). Even if these results are in contrast with the SIRT3-dependent activation of TRAP1 observed in GSC (Park et al., 2019), they provide a glimpse into a fascinating scenario in which SIRT3 and TRAP1 can harmonize the bioenergetic features of tumor cells with their needs and supplies, and provide a regulatory backbone that dynamically interacts with key metabolic components.
Nitric oxide in tumor necrosis factor receptor-associated protein 1 and Sirtuin 3 crosstalk
Nitric oxide allosterically inhibits TRAP1 ATPase activity, perturbing long-range structural communications and conformational changes (Faienza et al., 2020a). It also stimulates SIRT3 deacetylates activity (Wang et al., 2019) in a way resembling the effects of HDCA on both proteins (Sanchez-Martin et al., 2020a), and suggesting a common route that converges to regulate SDH activity. In conditions of NO toxicity (e.g., at very high concentrations), SDH is inhibited, most likely as a final effect of peroxynitrite-mediated protein damage and irreversible inactivation (Brown, 2007). However, in pathophysiological models of defective denitrosylation (e.g., in GSNOR-deficient systems), NO causes TRAP1 S-nitrosylation and degradation, which results in an increase in SDH activity (Rizza et al., 2016). This effect suggests that NO positively regulates SDH via S-nitrosylation through a dual mode, i.e., by inhibiting TRAP1 ATPase activity and activating SIRT3 (Figure 1). These opposite effects are mirrored by the dual role of NO in tumors, which finally results in driving metabolic adaptions of tumor cells. Notably, a switch between the chaperone and holdase-like activities of TRAP1—in which NO probably plays a role—may add a further level of complexity in the regulation of TRAP1/SIRT3 interaction (Figure 1).
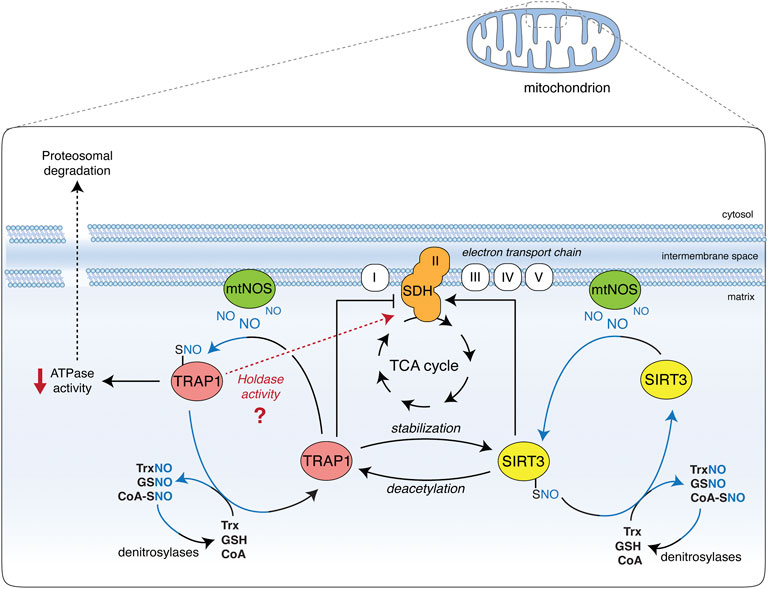
FIGURE 1. Effect of TRAP1 and SIRT3 S-nitrosylation on SDH. Nitric oxide (NO) is generated in the mitochondria by the mitochondrial portion of NOS2 (mtNOS). Besides nitration-induced irreversible damage (not shown), it causes the S-nitrosylation of TRAP1 and SIRT3 (TRAP1-SNO and SIRT3-SNO), which are in equilibrium with low molecular weight nitrosothiols (molecules and small proteins, such as GSNO, SNO-CoA, and TrxNO), the levels of which are controlled by denitrosylases (GSNOR, SCoR, and TrxR, respectively). S-nitrosylation increases SIRT3 activity, hence promoting succinate dehydrogenase (SDH) deacetylation and activation. SIRT3 has also been reported to deacetylate and activate TRAP1, which is crucial for SIRT3 protein stability. On the other hand, S-nitrosylation inhibits TRAP1 ATPase activity and promotes its degradation via the proteasome. Being TRAP1 a negative regulator of SDH, SDH activity is consequently increased. TRAP1 S-nitrosylation may also be responsible for a shift towards a holdase-like function of the protein (as recently proposed). This additional ability of TRAP1 could alternatively affect SDH stability and activity, and may help reconcile discrepancies between different studies on the effects of SIRT3 on SDH: if it takes place or not via TRAP1 inhibition or induction.
Conclusion
Although the role of NO in metabolism is well-established, most of the studies takes into account the direct inhibitory effect of NO on metabolic enzymes, and barely consider other layers of regulation. In this Opinion paper we have elaborated on, and proposed that NO-dependent regulation of metabolism could go beyond this direct effect and influence key mitochondrial regulators, i.e., TRAP1 and SIRT3, which are involved in cancer-associated increase of antioxidant response and metabolic rewiring (Singh et al., 2017a; Sanchez-Martin et al., 2020b). This is particularly relevant in neoplastic progression, where both TRAP1 and SIRT3 can simultaneously confer the ability to cope with oxidative stress and adapt to metabolic changes.
Author contributions
FF and GF conceived and designed the paper. All authors discussed and wrote the manuscript. All authors corrected and critically read the paper.
Funding
This work has been supported by grants from the Novo Nordisk Foundation (2018-0052550 to GF); Danish Cancer Society (KBVU R146-A9414 and R231-A13855 to GF); the Italian Association for Cancer Research, (IG 2017-20749 to AR and IG 2017-20719 to GF); Children’s Tumor Foundation Drug Discovery Initiative Registered Reports Award 2020-05-001 (to AR); PRIN MIUR grant 2020 9KYCH9 (to AR). FF is supported by an AIRC Fellowship for Italy. The laboratory in Copenhagen is part of the Center of Excellence in Autophagy, Recycling and Disease (CARD), funded by the Danish National Research Foundation (DNRF125).
Acknowledgments
The authors are grateful to Salvatore Rizza for kind help in drawing Figure 1, Laila Fisher for secretarial work, and Ludovica for having allowed the finalization of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
CoA, coenzyme A; CcOX, cytochrome c oxidase; ETC, electron transport chain; FAO, fatty acid oxidation; GSC, glioma stem cells; GSNOR, S-nitrosoglutathione reductase; HCC, hepatocellular carcinoma; HIF-1, hypoxia inducible factor 1; NO, nitric oxide; NF1, neurofibromatosis 1; OXPHOS, oxidative phosphorylation; PTP, permeability transition pore; ROS, reactive oxygen species; SDH, succinate dehydrogenase; SIRT, sirtuin; TCA, tricarboxylic acid; TRAP1, tumor necrosis factor receptor-associated protein 1.
References
Bell, E. L., Emerling, B. M., Ricoult, S. J. H., and Guarente, L. (2011). SirT3 suppresses hypoxia inducible factor 1α and tumor growth by inhibiting mitochondrial ROS production. Oncogene 30, 2986–2996. doi:10.1038/onc.2011.37
Benhar, M., Forrester, M. T., and Stamler, J. S. (2009). Protein denitrosylation: enzymatic mechanisms and cellular functions. Nat. Rev. Mol. Cell Biol. 10, 721–732. doi:10.1038/nrm2764
Bong-Hyun, A., Hyun-Seok, K., Shiwei, S., Hye, L. I., Jie, L., Athanassios, V., et al. (2008). A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. U. S. A. 105, 14447–14452. doi:10.1073/pnas.0803790105
Cannino, G., Urbani, A., Gaspari, M., Varano, M., Negro, A., Filippi, A., et al. (2022). TRAP1 and cyclophilin D compete at OSCP subunit to regulate enzymatic activity and permeability transition pore opening by F-ATP synthase. Cell Death Differ. 25. doi:10.1038/s41418-022-01020-0
Cleeter, M. W. J., Cooper, J. M., Darley-Usmar, V. M., Moncada, S., and Schapira, A. H. (1994). Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett. 345, 50–54. doi:10.1016/0014-5793(94)00424-2
Coles, S. J., Easton, P., Sharrod, H., Hutson, S. M., Hancock, J., Patel, V. B., et al. (2009). S-nitrosoglutathione inactivation of the mitochondrial and cytosolic BCAT proteins: S-nitrosation and S-thiolation. Biochemistry 48, 645–656. doi:10.1021/bi801805h
Elkhwanky, M.-S., and Hakkola, J. (2017). Extranuclear sirtuins and metabolic stress. Antioxid. Redox Signal. 28, 662–676. doi:10.1089/ars.2017.7270
Elnatan, D., Betegon, M., Liu, Y., Ramelot, T., Kennedy, M. A., Agard, D. A., et al. (2017). Symmetry broken and rebroken during the ATP hydrolysis cycle of the mitochondrial Hsp90 TRAP1. Elife 6, e25235. doi:10.7554/eLife.25235
Faienza, F., Lambrughi, M., Rizza, S., Pecorari, C., Giglio, P., Salamanca Viloria, J., et al. (2020a). S-nitrosylation affects TRAP1 structure and ATPase activity and modulates cell response to apoptotic stimuli. Biochem. Pharmacol. 176, 113869. doi:10.1016/j.bcp.2020.113869
Faienza, F., Rizza, S., Giglio, P., and Filomeni, G. (2020b). TRAP1: A metabolic hub linking aging pathophysiology to mitochondrial S-nitrosylation. Front. Physiol. 11, 340. doi:10.3389/fphys.2020.00340
Fernando, V., Zheng, X., Walia, Y., Sharma, V., Letson, J., Furuta, S., et al. (2019). S-nitrosylation: An emerging paradigm of redox signaling. Antioxidants 8, E404. doi:10.3390/antiox8090404
Guzzo, G., Sciacovelli, M., Bernardi, P., and Rasola, A. (2014). Inhibition of succinate dehydrogenase by the mitochondrial chaperone TRAP1 has anti-oxidant and anti-apoptotic effects on tumor cells. Oncotarget 5, 11897–11908. doi:10.18632/oncotarget.2472
Hirschey, M. D., Shimazu, T., Goetzman, E., Jing, E., Schwer, B., Lombard, D. B., et al. (2010). SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464, 121–125. doi:10.1038/nature08778
Hoter, A., El-Sabban, M. E., and Naim, H. Y. (2018). The HSP90 family: Structure, regulation, function, and implications in health and Disease. Int. J. Mol. Sci. 19, E2560. doi:10.3390/ijms19092560
Islam, Md. S., Leissing, T. M., Chowdhury, R., Hopkinson, R. J., and Schofield, C. J. (2018). 2-Oxoglutarate-Dependent oxygenases. Annu. Rev. Biochem. 87, 585–620. doi:10.1146/annurev-biochem-061516-044724
Joshi, A., Dai, L., Liu, Y., Lee, J., Ghahhari, N. M., Segala, G., et al. (2020). The mitochondrial HSP90 paralog TRAP1 forms an OXPHOS-regulated tetramer and is involved in mitochondrial metabolic homeostasis. BMC Biol. 18, 10. doi:10.1186/s12915-020-0740-7
Kalous, K. S., Wynia-Smith, S. L., Summers, S. B., and Smith, B. C. (2020). Human sirtuins are differentially sensitive to inhibition by nitrosating agents and other cysteine oxidants. J. Biol. Chem. 295, 8524–8536. doi:10.1074/jbc.RA119.011988
Kalous, K. S., Wynia-Smith, S. L., and Smith, B. C. (2021). Sirtuin oxidative post-translational modifications. Front. Physiol. 12, 763417. doi:10.3389/fphys.2021.763417
Laquatra, C., Sanchez-Martin, C., Dinarello, A., Cannino, G., Minervini, G., Moroni, E., et al. (2021). HIF1α-dependent induction of the mitochondrial chaperone TRAP1 regulates bioenergetic adaptations to hypoxia. Cell Death Dis. 12, 434. doi:10.1038/s41419-021-03716-6
Liu, Y., Sun, M., Elnatan, D., Larson, A. G., and Agard, D. A. (2020). Cryo-EM analysis of human mitochondrial Hsp90 in multiple tetrameric states. bioRxiv. doi:10.1101/2020.11.04.368837
Lowenstein, J. C. (2019). Metabolism reprogrammed by the nitric oxide signalling molecule. Nature 565, 33–34. doi:10.1038/d41586-018-07457-z
Masgras, I., Ciscato, F., Brunati, A. M., Tibaldi, E., Indraccolo, S., Curtarello, M., et al. (2017a). Absence of neurofibromin induces an oncogenic metabolic switch via mitochondrial ERK-mediated phosphorylation of the chaperone TRAP1. Cell Rep. 18, 659–672. doi:10.1016/j.celrep.2016.12.056
Masgras, I., Sanchez-Martin, C., Colombo, G., and Rasola, A. (2017b). The chaperone TRAP1 as a modulator of the mitochondrial adaptations in cancer cells. Front. Oncol. 7, 58. doi:10.3389/fonc.2017.00058
Masgras, I., Laquatra, C., Cannino, G., Serapian, S. A., Colombo, G., Rasola, A., et al. (2021). The molecular chaperone TRAP1 in cancer: From the basics of biology to pharmacological targeting. Semin. Cancer Biol. 76, 45–53. doi:10.1016/j.semcancer.2021.07.002
Masgras, I., Cannino, G., Ciscato, F., Sanchez-Martin, C., Darvishi, F. B., Scantamburlo, F., et al. (2022). Tumor growth of neurofibromin-deficient cells is driven by decreased respiration and hampered by NAD+ and SIRT3. Cell Death Differ. doi:10.1038/s41418-022-00991-4
Nakamura, T., Oh, C., Zhang, X., Tannenbaum, S. R., and Lipton, S. A. (2021). Protein transnitrosylation signaling networks contribute to inflammaging and neurodegenerative disorders. Antioxid. Redox Signal. 35, 531–550. doi:10.1089/ars.2021.0081
Park, H.-K., Hong, J.-H., Oh, Y. T., Kim, S. S., Yin, J., Lee, A.-J., et al. (2019). Interplay between TRAP1 and sirtuin-3 modulates mitochondrial respiration and oxidative stress to maintain stemness of glioma stem cells. Cancer Res. 79, 1369–1382. doi:10.1158/0008-5472.CAN-18-2558
Pérez, H., Finocchietto, P. V., Alippe, Y., Rebagliati, I., Elguero, M. E., Villalba, N., et al. (2018). p66Shc inactivation modifies RNS production, regulates Sirt3 activity, and improves mitochondrial homeostasis, delaying the aging process in mouse brain. Oxid. Med. Cell. Longev. 2018, 8561892. doi:10.1155/2018/8561892
Piantadosi, C. A. (2012). Regulation of mitochondrial processes by protein S-nitrosylation. Biochim. Biophys. Acta 1820, 712–721. doi:10.1016/j.bbagen.2011.03.008
Poderoso, J. J., Helfenberger, K., and Poderoso, C. (2019). The effect of nitric oxide on mitochondrial respiration. Nitric Oxide 88, 61–72. doi:10.1016/j.niox.2019.04.005
Prime, T. A., Blaikie, F. H., Evans, C., Nadtochiy, S. M., James, A. M., Dahm, C. C., et al. (2009). A mitochondria-targeted S-nitrosothiol modulates respiration, nitrosates thiols, and protects against ischemia-reperfusion injury. Proc. Natl. Acad. Sci. U. S. A. 106, 10764–10769. doi:10.1073/pnas.0903250106
Radi, R. (2018). Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. U. S. A. 115, 5839–5848. doi:10.1073/pnas.1804932115
Rardin, M. J., Newman, J. C., Held, J. M., Cusack, M. P., Sorensen, D. J., Li, B., et al. (2013). Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc. Natl. Acad. Sci. U. S. A. 110, 6601–6606. doi:10.1073/pnas.1302961110
Rich, P. R. (2017). Mitochondrial cytochrome c oxidase: catalysis, coupling and controversies. Biochem. Soc. Trans. 45, 813–829. doi:10.1042/BST20160139
Rizza, S., and Filomeni, G. (2017). Chronicles of a reductase: Biochemistry, genetics and physio-pathological role of GSNOR. Free Radic. Biol. Med. 110, 19–30. doi:10.1016/j.freeradbiomed.2017.05.014
Rizza, S., Montagna, C., Cardaci, S., Maiani, E., Di Giacomodi, G., Sanchez-Quiles, G. V., et al. (2016). S-nitrosylation of the mitochondrial chaperone TRAP1 sensitizes hepatocellular carcinoma cells to inhibitors of succinate dehydrogenase. Cancer Res. 76, 4170–4182. doi:10.1158/0008-5472.CAN-15-2637
Sanchez-Martin, C., Menon, D., Moroni, E., Ferraro, M., Masgras, I., Elsey, J., et al. (2020a). Honokiol bis-dichloroacetate is a selective allosteric inhibitor of the mitochondrial chaperone TRAP1. Antioxid. Redox Signal. 34, 505–516. doi:10.1089/ars.2019.7972
Sanchez-Martin, C., Serapian, S. A., Colombo, G., and Rasola, A. (2020b). Dynamically shaping chaperones. Allosteric modulators of HSP90 family as regulatory tools of cell metabolism in neoplastic progression. Front. Oncol. 10, 1177. doi:10.3389/fonc.2020.01177
Sciacovelli, M., Guzzo, G., Morello, V., Frezza, C., Zheng, L., Nannini, N., et al. (2013). The mitochondrial chaperone TRAP1 promotes neoplastic growth by inhibiting succinate dehydrogenase. Cell Metab. 17, 988–999. doi:10.1016/j.cmet.2013.04.019
Serapian, S. A., Moroni, E., Ferraro, M., and Colombo, G. (2021). Atomistic simulations of the mechanisms of the poorly catalytic mitochondrial chaperone Trap1: Insights into the effects of structural asymmetry on reactivity. ACS Catal. 11, 8605–8620. doi:10.1021/acscatal.1c00692
Shimazu, T., Hirschey, M. D., Hua, L., Dittenhafer-Reed, K. E., Schwer, B., Lombard, D. B., et al. (2010). SIRT3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metab. 12, 654–661. doi:10.1016/j.cmet.2010.11.003
Singh, C. K., Chhabra, G., Ndiaye, M. A., Garcia-Peterson, L. M., Mack, N. J., Ahmad, N., et al. (2017a). The role of sirtuins in antioxidant and redox signaling. Antioxid. Redox Signal. 28, 643–661. doi:10.1089/ars.2017.7290
Urbani, A., Giorgio, V., Carrer, A., Franchin, C., Arrigoni, G., Jiko, C., et al. (2019). Purified F-ATP synthase forms a Ca2+-dependent high-conductance channel matching the mitochondrial permeability transition pore. Nat. Commun. 10, 4341. doi:10.1038/s41467-019-12331-1
Wang, Q., Ye, S., Chen, X., Xu, P., Li, K., Zeng, S., et al. (2019). Mitochondrial NOS1 suppresses apoptosis in colon cancer cells through increasing SIRT3 activity. Biochem. Biophys. Res. Commun. 515, 517–523. doi:10.1016/j.bbrc.2019.05.114
Yoshida, S., Tsutsumi, S., Muhlebach, G., Sourbier, C., Lee, M.-J., Lee, S., et al. (2013). Molecular chaperone TRAP1 regulates a metabolic switch between mitochondrial respiration and aerobic glycolysis. Proc. Natl. Acad. Sci. U. S. A. 110, E1604–E1612. doi:10.1073/pnas.1220659110
Yu, W., Dittenhafer-Reed, K. E., and Denu, J. M. (2012). SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J. Biol. Chem. 287, 14078–14086. doi:10.1074/jbc.M112.355206
Keywords: nitric oxide, metabolism, mitochondria, SDH, nitrosylation
Citation: Faienza F, Rasola A and Filomeni G (2022) Nitric oxide-based regulation of metabolism: Hints from TRAP1 and SIRT3 crosstalk. Front. Mol. Biosci. 9:942729. doi: 10.3389/fmolb.2022.942729
Received: 12 May 2022; Accepted: 06 July 2022;
Published: 26 July 2022.
Edited by:
Gennaro Napolitano, Telethon Institute of Genetics and Medicine (TIGEM), ItalyReviewed by:
Khosrow Kashfi, City University of New York, United StatesStéphanie Plenchette, Université de Sciences Lettres de Paris, France
Copyright © 2022 Faienza, Rasola and Filomeni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fiorella Faienza, ZmlvcmVsbGEuZmFpZW56YUB1bmlyb21hMi5pdA==; Giuseppe Filomeni, Z2l1ZmlsQGNhbmNlci5kaw==, Zmlsb21lbmlAYmlvLnVuaXJvbWEyLml0