 Nathaniel Shannon
Nathaniel Shannon Randi Gravelle1
Randi Gravelle1 Brian Cunniff
Brian Cunniff- 1Department of Pathology and Laboratory Medicine, Redox Biology Program, University of Vermont Larner College of Medicine, Burlington, VT, United States
- 2University of Vermont Cancer Center, University of Vermont Larner College of Medicine, Burlington, VT, United States
Regulation of cell signaling cascades is critical in making sure the response is activated spatially and for a desired duration. Cell signaling cascades are spatially and temporally controlled through local protein phosphorylation events which are determined by the activation of specific kinases and/or inactivation of phosphatases to elicit a complete and thorough response. For example, A-kinase-anchoring proteins (AKAPs) contribute to the local regulated activity protein kinase A (PKA). The activity of kinases and phosphatases can also be regulated through redox-dependent cysteine modifications that mediate the activity of these proteins. A primary example of this is the activation of the epidermal growth factor receptor (EGFR) and the inactivation of the phosphatase and tensin homologue (PTEN) phosphatase by reactive oxygen species (ROS). Therefore, the local redox environment must play a critical role in the timing and magnitude of these events. Mitochondria are a primary source of ROS and energy (ATP) that contributes to redox-dependent signaling and ATP-dependent phosphorylation events, respectively. The strategic positioning of mitochondria within cells contributes to intracellular gradients of ROS and ATP, which have been shown to correlate with changes to protein redox and phosphorylation status driving downstream cellular processes. In this review, we will discuss the relationship between subcellular mitochondrial positioning and intracellular ROS and ATP gradients that support dynamic oxidation and phosphorylation signaling and resulting cellular effects, specifically associated with cell migration signaling.
Introduction
Two primary reversible post translational modifications, protein oxidation and phosphorylation, can elicit cooperative or divergent cell signaling responses affecting numerous cell processes including cell proliferation (Yao et al., 2019), cell migration (Hurd et al., 2012; Cao et al., 2015), transcription (Riedl and Egly, 2000; De Nigris et al., 2001; Al-Mehdi et al., 2012), stress response (Hamada et al., 2020), immune cell activation (Davidson et al., 2003; Gostner et al., 2013; Iwasaki et al., 2020) and more. These modifications directly impact protein structure and function, hence altering their downstream cell signaling cascades (Karasev et al., 2018; Fu et al., 2019). Mitochondria have emerged as an important source of ROS that contribute to redox signaling (Horn et al., 2017; Jezek et al., 2020) while being the primary source of cellular ATP required for cellular energy and protein phosphorylation. Mitochondria are dynamic organelles that vary in size, shape and location depending on cell type (normal and disease), energy status and metabolic demand for mitochondrial metabolites (Tilokani et al., 2018).
Mitochondria produce ROS and ATP at the electron transport chain (ETC) which takes place in the inner mitochondrial membrane (IMM). Electrons are passed from NADH and FADH2 through IMM bound protein complexes, with subsequent pumping of H+ ions to the intermembrane space (IMS). H+ ions are pumped from the IMS through the ATP synthase and into the mitochondrial matrix to generate ATP (Zhao et al., 2019). ROS generation occurs when the electrons from NADH/FADH2 leak out of the protein complex and bind with O2 to form superoxide (O2−) which can be enzymatically converted to H2O2 via the mitochondrial superoxide dismutase (SOD2) (Cadenas and Davies, 2000; Turrens, 2003). Approximately 0.2–2% of the electrons flowing through the ETC, under physiological conditions, can leak out to cause oxidation of proteins proximal to mitochondria (Cadenas and Davies, 2000; Turrens, 2003).
Not only can mitochondria produce ATP and ROS, but they can also regulate calcium (Ca2+) concentrations which also regulate mitochondrial function. A flux of mitochondrial Ca2+ causes activation of the dehydrogenases in the tricarboxylic acid (TCA) cycle, which are the rate limiting steps during oxidative phosphorylation; therefore, causing an increase in NADH which eventually feeds into the ETC (Duchen, 1992; Maechler and Wollheim, 2000; Rizzuto et al., 2000). The mitochondria can also associate with the endoplasmic reticulum (ER), which is involved in Ca2+ storage and release. Therefore, the interaction between mitochondria and the ER can lead to different Ca2+ associated pathways such as increased mitochondrial bioenergetics or even cell death (Carreras-Sureda et al., 2018; Marchi et al., 2018). The relationship between the mitochondria and Ca2+ signaling throughout the cell is extensive and not the central focus of this review.
The subcellular positioning of mitochondria, and the localized activity of mitochondria, drives intracellular gradients of ATP and ROS and therefore mitochondrial trafficking is necessary for localized accumulation of these molecules (Schuler et al., 2017; Alshaabi et al., 2021). A large body of research supports a key role for ROS-dependent redox signaling in regulating cell migration phenotypes (Hurd et al., 2012). Emerging research now shows the subcellular positioning of mitochondria also supports cell migration phenotypes (Desai et al., 2013; Altieri, 2017; Schuler et al., 2017), providing an interesting, yet unresolved, link between mitochondrial trafficking and redox signaling in cell migration. In this review we will discuss the relationship between mitochondrial positioning and the downstream signaling cascades elicited from localized mitochondrial ROS (mROS) and ATP with a focus on cell migration.
Sources of ROS and redox signaling
The oxidation of target proteins occurs through ROS, specially hydrogen peroxide (H2O2) reacting with a free thiol (-SH) to form a sulfenic acid (-SOH), typically on cysteine residues, but can also react with methionine, tryptophan, and tyrosine residues (Hoshi and Heinemann, 2001; van der Vliet et al., 2018) (Figure 1). Chemical cell signaling events are well characterized for the reversible oxidation of cysteine residues (Berndt et al., 2007; Garcia-Santamarina et al., 2014); however, far less is understood about the role of oxidation of methionine, tryptophan, and tyrosine residues. Specific, structurally distinct and solvent accessible cysteine residues are targets for oxidation by H2O2, and these modifications result in structural and functional changes in target proteins (Cecarini et al., 2007) (Figure 1). Similar to protein phosphorylation/dephosphorylation cascades, the reversible oxidation of specific cysteine residues modulates signaling pathways that govern all facets of cell physiology (Yanes et al., 2010). Physiochemical characteristics of oxidized cysteine residues in target proteins underlie the specificity and hierarchy of responses in redox signaling. ROS, like H2O2, regulate cellular physiology through direct oxidation of cysteine residues in target proteins, or via inactivation of resident scavenger/chaperone proteins (Nguyen and Sok, 2003; Dustin et al., 2020). Redox signaling through “redox-relays” utilizes the H2O2 reactive peroxiredoxin (PRX) family of enzymes as intermediates for transferring oxidation to target proteins through inter-disulfide exchange (Sobotta et al., 2015; Stocker et al., 2018a; Stocker et al., 2018b; Kim and Jang, 2019). The reversible oxidation of proteins plays a central role in regulating cell signaling cascades that govern all facets of cellular responses (Holmstrom and Finkel, 2014).
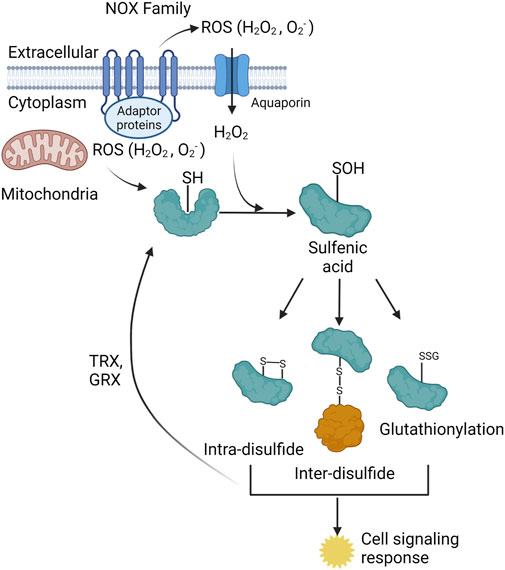
Figure 1. The two primary cellular sources of reactive oxygen species (ROS) are the NADPH oxidase (NOX) family of enzymes (NOX1-5, DUOX 1–2) and the mitochondrial electron transport chain (ETC). The NOX enzymes produces ROS (O2- and H2O2) towards the extracellular space, O2- is spontaneously or enzymatically (via SOD) dismutated to H2O2. H2O2is either transported through membrane channels or passed through the plasma membrane to elicit signaling in the cytoplasm. Mitochondria produce ROS into the mitochondrial matrix or the intermembrane space. Mitochondrial H2O2 can exit the mitochondria and signal in the cytoplasm. The ROS from both sources participate in redox-dependent signaling through oxidation of target cysteine residues on proteins. Cysteine sulfenic acids can form intra-disulfide bonds, inter-disulfide bonds, and become glutathionylated. All three of these species elicit several cell signaling responses within the cell. These three protein species can be converted back to the reduced thiol state via thioredoxin (TRX) and glutaredoxin (GRX) proteins.
Similar to oxidation, protein phosphorylation is a reversible post-translational modification that typically occurs on serine, threonine, and tyrosine residues (Rao et al., 2013). Phosphorylation of target proteins occurs when the gamma-phosphate of ATP is transferred to the hydroxyl group of an amino acid and this is accomplished by a set of proteins known as kinases (Endicott et al., 2012). The phosphate group can be removed by phosphatases, and this will return the residue back to the hydroxyl group, thus making this process reversible (Barford et al., 1998). In certain cell signaling cascades, these two reversible post translational modifications can converge to cooperatively promote signaling or compete to downregulate signaling (Chiarugi et al., 2003; Giannoni et al., 2005; Grintsevich et al., 2017; Londhe et al., 2020). Evolutionarily there are conserved cysteine residues proximal to a Ser/Thr/Tyr residue in various eukaryotic kinases that regulate activity, thus further demonstrating the dynamics between oxidation and phosphorylation (Byrne et al., 2020). A key example of this is the activation of kinases via oxidation of cysteine residues in the active site and the inactivation of protein tyrosine phosphatases via oxidation of active site cysteines (Ostman et al., 2011; Dustin et al., 2020), leading to prolonged phosphorylation of a target protein. A primary example is the oxidation of the epidermal growth factor receptor (EGFR) at Cys797 leading to enhanced tyrosine kinase activity (Paulsen et al., 2011). Inactivation of the phosphatase and tensin homologue (PTEN) phosphatase occurs during muscle differentiation when there is an increase in the oxidation of PTEN, leading to decreased activity which causes an upregulation of the PI3K/AKT/mTOR pathway since these target proteins are able to remain phosphorylated for a longer period of time (Kim et al., 2018). A critical gap in the understanding of control over dynamic oxidation/phosphorylating events is the source, location and duration of ROS governing these processes (Meng et al., 2002; Ostman et al., 2011; Londhe et al., 2020).
ROS can be generated from a variety of sources both externally and internally to the cell. Such internal sources are derived from NADPH oxidases (NOXs) as well as mitochondria via the electron transport chain (ETC). Cellular ROS has also been shown to be produced via the endoplasmic reticulum (ER) (Cao and Kaufman, 2014), peroxisomes (Sandalio et al., 2013), and various enzymatic reactions; however, the main sources of subcellular ROS are derived from the NOXs and mitochondria. The NOX family can be separated into two categories: NOXs and dual oxidases (DUOXs) both of which are membrane bound enzymes that typically extend from the cytosolic face to the extracellular space with ROS generation (superoxide (O2-) and H2O2) towards the exterior of the cell (Panday et al., 2015) (Figure 2). ROS generation by these enzymes is regulated by NADPH, protein cofactors, various stimuli, such as bacterial infection, calcium, and post-translation modifications (phosphorylation), to produce ROS, specifically O2− and H2O2 for the NOXs and H2O2 for the DUOXs (Panday et al., 2015). Subcellular localization of specific NOX isoforms has also been identified with NOX4 being localized to the mitochondria (Shanmugasundaram et al., 2017), nucleus, ER, and directly interacting with focal adhesions (FAs) (Block et al., 2009), as well as NOX2 being localized to the plasma membrane (Anilkumar et al., 2008). FAs are multiprotein segments of a cell responsible for cell attachment by connecting the cytoplasm to the extracellular matrix (ECM). Strategic localization to these subcellular compartments is shown to provide a burst of ROS needed for microbial killing and to inhibit local phosphatases, which contributes to cell migration or increased insulin signaling (Wu et al., 2005; Chen et al., 2008). During FA maturation, NOX4 has been shown to provide the ROS needed for the oxidation of two cysteine residues in actin which is critical in the binding of vinculin, a FA protein that links integrins to the actin cytoskeleton (Vukelic et al., 2018). DUOX specific H2O2 is also important for epithelial cell migration and rearrangement of the cytoskeleton, which will be discussed later in this review.
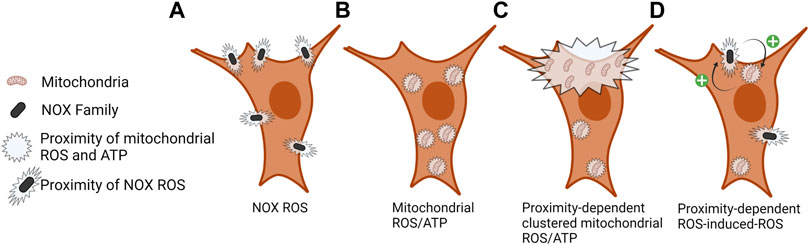
FIGURE 2. Local proximity of NOX ROS and mitochondrial ROS/ATP. (A) ROS is produced by NOX enzymes in the extracellular space proximal to the plasma membrane, signaling at the cytoplasmic face of the plasma membrane is dependent on local ROS concentration and ROS scavenging. (B) Mitochondrial ROS (mROS) and ATP are rapidly consumed at the site of production; therefore, the density of these mitochondrial outputs is localized to sites of mitochondrial density. (C) Clustering of mitochondria at subcellular sites contributes to a localized increase in ROS and ATP levels. (D) ROS-induced-ROS and mitochondrial–NOX crosstalk regulates the activity of each entity and the amount and duration of ROS production. Still unclear is how the proximity of NOX and mitochondria might regulate this process.
NOX enzymes have also been shown to contribute to a gradient of H2O2 in zebrafish tissues in response to injury. Following tail fin amputations, a 30 µm wide H2O2 gradient extending from the wound margin into the tissue has been observed (Niethammer et al., 2009; Jelcic et al., 2017). This NOX associated H2O2 gradient acts as a chemoattractant for inflammatory cell recruitment to aid in repair of the injury. More localized requirements for ROS have also been observed in the repair of the plasma membrane following plasma membrane injury (PMI), which will be discussed more later (Horn et al., 2020). Thus, NOX-dependent ROS gradients on both the micro and macro level contribute to the regulation of cell signaling cascades to aid in repair of tissues.
Unlike the NOXs which are membrane bound, the mitochondria are dynamic as they undergo cycles of fission and fusion, as well as are trafficked throughout the cytoplasm (Lopez-Domenech et al., 2018; Horn et al., 2020) (Figures 2, 3). Mitochondria provide a major cellular source of ROS via the ETC (Inoue et al., 2003). mROS are generated in the mitochondrial matrix and IMS by ETC complexes I and III, respectively, as a result of the single electron reduction of O2 to produce O2− which can be converted to H2O2 (Murphy, 2009). Manganese Superoxide Dismutase (MnSOD), located in the mitochondrial matrix, catalyzes the reaction of O2− to H2O2, thus changing the type of ROS, but not fully reducing it to H2O (Inoue et al., 2003). Complete reduction of H2O2 to H2O in the mitochondrial matrix is accomplished by mitochondrial glutathione peroxidase 4 (GPX4) (Handy et al., 2009) and peroxiredoxin 3 (PRX3) (Newick et al., 2012). Therefore, mROS diffusion out of the mitochondrial matrix will be dependent on the amount of ROS produced in time and space and the activity of resident ROS scavenging enzymes. mROS contribute to redox signaling through canonical cysteine oxidation of target proteins and through retrograde signaling to the nucleus (Tan and Finkel, 2020).
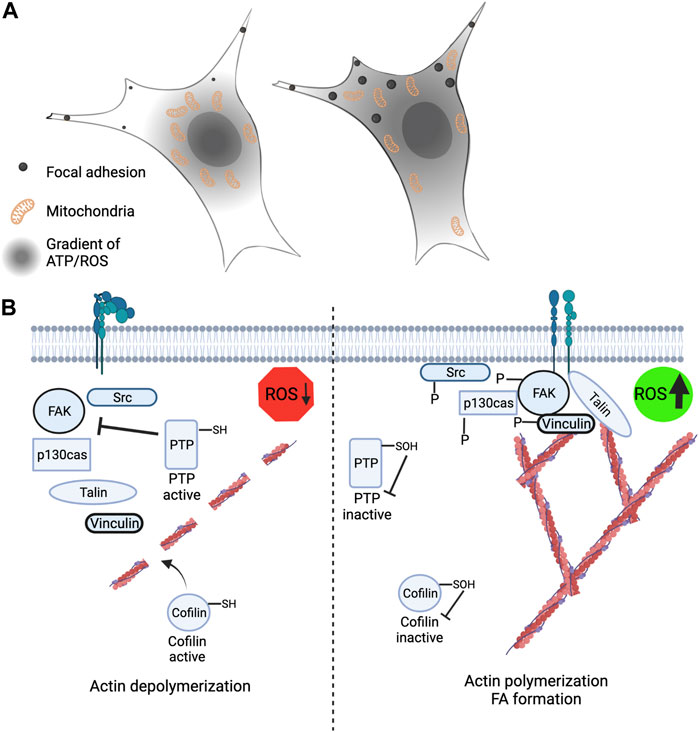
FIGURE 3. Leading edge mitochondria and redox signaling contribute to cytoskeleton rearrangement and cell migration. (A) (Left) Restriction of mitochondria to the perinuclear space leads to loss of peripheral ATP and ROS levels and correlates with smaller and less stable focal adhesions. (Right) Mitochondria that are strategically localized and recruited to the cell periphery have an extended gradient of ATP and mROS and is correlated with larger and more stable focal adhesions. (B) (Left) Low ROS levels at the cell periphery promotes actin severing through cofilin activation and increased protein tyrosine phosphatase activity leading to reduced phosphorylation of FAK, p130cas, vinculin and Src. (Right) Elevated ROS levels at the cell periphery promotes actin polymerization and branching through redox-dependent inactivation of cofilin. Inactivation of PTP’s by ROS promotes increased FAK, p130cas, vinculin and Src phosphorylation.
Both ATP and ROS are rapidly consumed at sites proximal to their source, largely due to the abundance of antioxidant enzymes present in the cell (Jones, 2010; Jelcic et al., 2017; Alshaabi et al., 2021) (Figure 2). An additional level of regulation is achieved through the compartmentalization of oxidant and antioxidant systems, allowing cells to utilize redox-dependent systems for physiological signaling and damage responses while protecting redox-sensitive cell compartments (Go and Jones, 2008; Pak et al., 2020). Recent studies in yeast described a mitochondria-to-cytosol H2O2 gradient where the mitochondrial H2O2 is rapidly consumed by the cytosolic antioxidant peroxiredoxin, thus the downstream signaling effects of mitochondrial H2O2 occurs proximal to its site of production (Carmona et al., 2019; de Cubas et al., 2021). Other studies show a strong correlation between mitochondrial matrix H2O2 levels and cellular growth rate (Morgan et al., 2016). Similar findings have also been described in mammalian cells and the subcellular localization of mitochondria has also been correlated with spatial cytosolic H2O2 levels (Alshaabi et al., 2021). These collective studies support an inside-out (mitochondrial-cytosolic) redox-signaling gradient from mitochondria.
In support of mitochondria H2O2 contributing to signaling in cancer cell metastasis, published reports have shown that mitochondria with experimentally decreased ETC function contributed to metastatic phenotypes; results showed an increase in migratory and invasive activity (Porporato et al., 2014). Their findings showed that in their “supermetastatic” and “superinvasive” cell lines there are defects in the TCA cycle characterized by increased succinate production. The unequal pairing of the TCA cycle with the ETC led to an increase in mROS; a notable increase in superoxide production was detected. The increase of succinate and superoxide suggests that more electrons could be transferred to ETC complex II by succinate, resulting in an overloaded ETC. Use of the mitochondria-targeted superoxide scavenger mitoTEMPO resulted in a decrease in tumor cell metastasis (Porporato et al., 2014). This work reinforces the suspected role of mitochondria in cancer aggressiveness and progression. Mutations in mitochondrial DNA (mtDNA) can result in ETC dysfunction, specifically relating to mutations in complex I where both mtDNA and nuclear DNA are required for its formation (Ishikawa et al., 2008). It is important to note that many carcinogenic chemicals are known to bind to mtDNA (Chen et al., 2004; Budnik et al., 2013). It was determined that mtDNA with mutations causing complex I dysfunction increased metastatic phenotypes in transformed cells but did not induce tumor formation in murine models (Ishikawa et al., 2008). Defective complex I function results in ROS accumulation in tumor cells. The mutations to complex I lead to the up-regulation of three genes with heavy implications in metastatic potential: MCL-1, HIF-1α, and VEGF (Ishikawa et al., 2008). The specific role and location of mitochondria in driving these supermetastatic processes is not clear, but likely local mitochondrial recruitment is required (Altieri, 2019).
Although not fully understood, crosstalk between mitochondria and NOX enzymes has been proposed by a mechanism termed “ROS-induced ROS release” (Zorov et al., 2000). ROS-dependent oxidation of mitochondrial ATP-sensitive potassium channels (Queliconi et al., 2011) and full enzymatic activity of NADPH oxidases is required for angiotensin II mediated mROS production (Doughan et al., 2008). Alternatively, mROS have been shown to activate NOX1 following serum withdrawal in human embryonic kidney 293T cells (Lee et al., 2006). Still missing from these studies is the role of subcellular mitochondrial positioning in mediating the initiation and execution of ROS-induced ROS release (Figure 2D). Better understanding the localization and abundance of mitochondria and subcellular ATP and ROS can lead to deciphering local cell signaling cascades in mediating mitochondria and NOX ROS-induced ROS release.
Mitochondrial trafficking dynamics
In mammalian cells mitochondria are strategically positioned throughout the cytoplasm to meet local energy requirements (Hollenbeck, 2005). This movement is orchestrated by the microtubule motor proteins kinesin and dynein (Fransson et al., 2006; Lopez-Domenech et al., 2018) and allows the mitochondria to move anterograde (to periphery) and retrograde (towards the nucleus), respectively. The actin cytoskeleton and myosin proteins also play a role in mitochondrial trafficking and anchoring, although this is believed to support short movements (Sheng, 2014). The microtubule motor proteins are linked to mitochondria via the TRAK1/2 (Milton) adapter proteins which connect to the outer mitochondrial membrane bound adaptor protein Miro1 or Miro2 (Debattisti et al., 2017; Li et al., 2021). When Miro1 is knocked out from many cell types this results in mitochondria becoming restricted around the nucleus compared to when Miro1 is present, and the mitochondria are strategically and dynamically re-localized throughout the cytoplasm (Ahmad et al., 2014; Schuler et al., 2017; Alshaabi et al., 2021) (Figure 3A). We recently have shown that the subcellular positioning of mitochondria by Miro1 directly impacts intracellular gradients of ATP and mROS (Schuler et al., 2017; Alshaabi et al., 2021) (Figure 3A). Additionally, disruption of the microtubule cytoskeleton with Taxol causes restriction of mitochondria around the nucleus comparable to Miro1 deletion and similar disruption to subcellular H2O2 gradients. Re-expression of Miro1 can rescue these gradient defects (Alshaabi et al., 2021). Loss of Miro2 does not elicit dramatic changes to mitochondrial trafficking in differentiated cells and therefore has been of less focus (Nguyen et al., 2014).
Another process that impacts mitochondrial trafficking is fission and fusion. To mitigate the effects of damaged mitochondria, a healthy and damaged mitochondrion may fuse together which can be trafficked to areas of the cell in high energy demand (Detmer and Chan, 2007). Mitochondria can also undergo fission which will cause one mitochondrion to split into two and this may support increased trafficking. The role of fission and fusion on mitochondrial trafficking is still unclear; however, fusion can be directly affected by AMP-activated protein kinase (AMPK), a cytoplasmic energy sensor. Therefore, mechanistically providing the cell with information when energy is low in various parts of the cell which in turn signals mitochondria to fragment and be transported to that area (Cunniff et al., 2016; Toyama et al., 2016). Overall, energy sensing plays a role in mitochondrial structure and location.
Mitochondria are also stopped and anchored at specific subcellular sites where mitochondrial functions are required. At sites of high energy demand in neurons mitochondria stop moving, partly by the protein syntaphilin which binds mitochondria to the microtubules (Kang et al., 2008). Mitochondrial movement is also halted in axons at sites of increased calcium (Ca2+) (Yi et al., 2004). EF-hands present in the Miro1 protein (Smith et al., 2020) are thought to play a role in this sensing, but there is also evidence that mitochondria can be halted at sites of increased Ca2+ when Miro1 is lost (Nguyen et al., 2014). ROS have also been shown to regulate the speed of mitochondrial trafficking, presumably through the p38 MAPK pathway (Debattisti et al., 2017). Mitochondria also respond to increased levels of extracellular glucose, where O-GlcNAc transferase (OGT) performs the glucose-dependent O-GlcNAcylation on key serine residues of the adaptor protein Milton which stops mitochondrial motility (Pekkurnaz et al., 2014). Similarly, by inhibiting the glucose transporter FGT-1 in Caenorhabditis elegans, there was a decreased mitochondrial recruitment to the basal membrane to help drive anchor cell (AC) invasion, which is responsible for the development of the reproductive system (Garde et al., 2022).
Another energy dependent process, the activation of AMPK, has been shown to contribute to the recruitment of mitochondria to the leading edge of migrating cells. When AMPK is selectively activated at the leading edge of the cell, mitochondria are trafficked to this specific area accompanied by increased ATP concentration and membrane ruffling, a direct readout of cell migration (Cunniff et al., 2016). Inhibition of mitochondrial activity with acute exposure to the complex I inhibitor, rotenone, blocked membrane ruffling. Local specific and temporal AMPK inactivation, using pharmacological and optogenetic approaches, caused decreased mitochondrial movement to the leading edge as well as decreased cell migration and invasion (Cunniff et al., 2016). These studies provide evidence that when the ATP: ADP ratio is spatially decreased, AMPK becomes activated to drive mitochondria to the site of interest to produce more ATP needed for various downstream signaling at the periphery of the cell. Mitochondrial fission through DRP1 activation is also mediated by AMPK activity in response to ETC inhibition (Toyama et al., 2016). Collectively, numerous metabolic dependent and independent processes converge to mediate the subcellular trafficking, anchoring, and severing of mitochondria to provide local mitochondrial byproducts to areas in demand.
Leading edge mitochondria can rearrange the cytoskeleton
As described above, subcellular H2O2 gradients have been shown to regulate cell signaling. Below we will discuss this in the context of phosphorylation dynamics, cytoskeleton remodeling and cell migration. ROS, in particular H2O2, can act on numerous signaling pathways controlling cell migration including receptor activation, kinase and phosphatase activity, FA dynamics, membrane reorganization and transcription factor activation (Hurd et al., 2012; Truong and Carroll, 2013) (Figure 3). During cell migration and invasion, the mitochondria have been found to localize to the leading edge of the cell to help drive cytoskeleton rearrangements (Madan et al., 2021). Anchor Cell (AC) invasion of the basement membrane (BM) in C. elegans requires mitochondrial recruitment to the invasive edge of the AC to drive invadopodia formation (Garde and Sherwood, 2021; Garde et al., 2022). Filamentous actin (F-actin) is responsible for the structure of the invadopodia and is increased by the presence of mitochondria at the invasive edge which provides a local source of ATP (Kelley et al., 2019). Localized ATP at the leading edge of the cell is necessary for the activation of the Arp2/3 complex which serves as a nucleation site for actin filaments. Arp2/3 is activated upon phosphorylation at Thr237/238 in Arp2 and this allows for increased lamellipodia at the leading edge of the cell through the branching of actin filaments (LeClaire et al., 2008). Therefore, the presence of mitochondria at the leading edge of the cell supports increased ATP concentrations to drive protein phosphorylation for the reconstruction of the cytoplasm.
As critical as phosphorylation events, protein oxidation plays a key role in the stability of actin filaments. Oxidation of actin filaments specifically in cell protrusions has recently been described using the ratiometric H2O2 biosensor HyPer7 fused to the actin binding peptide LifeAct. Using this probe, protrusions with elevated H2O2 levels were more stable compared to protrusions with lower H2O2 levels (Pak et al., 2020). This means that mitochondria can serve at least two purposes at the edge of the cell: 1) in providing the ATP needed for Arp2/3 activation for F-actin formation 2) in providing sufficient ROS needed to maintain F-actin stability. Similarly, mitochondria are required at the site of plasma membrane injury (PMI) to provide the necessary means for plasma membrane repair (PMR). At the site of PMI in mouse embryonic fibroblasts (MEFs), mitochondria fragment and this supports signaling to aid in repair, cells that lack the required machinery for mitochondrial fission (DRP1) fail to repair (Horn et al., 2020). The small GTPase, DRP1, oligomerizes around the mitochondrial outer membrane and is necessary for pinching of one mitochondrion into two via fission (Rosdah et al., 2020). The DRP1 adaptor protein MiD49 is involved in mitochondrial fission and when this is absent from the cell they fail to repair, and the mitochondria are not able to sustain increased calcium intake at the site of injury (Horn et al., 2020). Fragmented mitochondria cause an increase in F-actin abundance at the site of injury which aids in repairing the plasma membrane; however, unfragmented mitochondria fail to effectively heal the plasma membrane. Localized mROS production also contribute to plasma membrane repair through activation of RhoA and actin polymerization (Horn et al., 2017). These DRP1-dependent responses only occur proximal to the site of membrane damage. DRP1 is also upregulated in many cancer cells, including metastatic breast cancer cells (Zhao et al., 2013). DRP1-dependnet fission is thought to support fragmentation of mitochondria for subcellular transport (Giovarelli et al., 2020). Silencing of DRP1 in breast cancer cells decreases mitochondrial fission, cell migration and invasion (Zhao et al., 2013). Loss of DRP1 also accompanied a reduction in the number of mitochondria in the leading edge of these cells. DRP1 also supports the directional migration of breast cancer cells, supporting the movement of mitochondria to the anterior membrane in the direction of cell migration (Desai et al., 2013). Thus, mitochondrial fission and location are important in F-actin dynamics and cell migration.
Cell migration and invasion in vivo requires degradation and remodeling of the extracellular matrix (Bonnans et al., 2014). The primary set of enzymes known to degrade the extracellular matrix are the matrix metalloproteinases (MMPs) (Loffek et al., 2011). MMPs are also regulated via reversible oxidation and phosphorylation. Increasing intracellular H2O2 levels via MnSOD, the mitochondrial superoxide dismutase, as well as increasing mROS via rotenone and antimycin A increases the activity of the MMPs (Hazan et al., 2000). The expression levels of MMP-1 is increased by intracellular ROS concentrations; therefore, both the activity and expression levels are increased in the presence of elevated ROS (Shin et al., 2015). Cell migration and invasion are also correlated with the activity of MMPs in breast cancer cells (Ren et al., 2015). Oxidation activates MMPs; however, phosphorylation inactivates them, and it is believed that protein kinase C (PKC) is the kinase responsible for their inactivation (Sariahmetoglu et al., 2007; Williams and Coppolino, 2011). It is not fully understood if oxidation or phosphorylation is dominant when both species are present, thus these two post translational modifications do not crosstalk with each other, per se, but they do have opposing functions on MMPs. Therefore, mitochondria are critical in the regulation of MMPs which influence cell migration and invasion via reshaping the extracellular milieu.
mROS can alter localized phosphorylation status
The relationship between the positioning of mitochondria within the cell and the downstream effects on cell migration, invasion, and membrane repair are starting to be revealed; however, these processes are not fully understood (Cunniff et al., 2016; Schuler et al., 2017; Horn et al., 2020; Garde and Sherwood, 2021). Mitochondrial positioning directly maps to area of increased ATP as well as H2O2 which makes intracellular trafficking of these organelles critical for the function of the cell. When mitochondria are concentrated to the perinuclear area this causes a decrease in ATP and H2O2 concentrations in the cell periphery and an increased in perinuclear H2O2 levels; however, this is rescued when mitochondrial trafficking to the periphery is rescued (Schuler et al., 2017; Alshaabi et al., 2021).
Few relationships between mitochondrial positioning and the effects of their byproducts, ATP and H2O2, on proteins have yet to be fully understood. Two targets to have altered function based on Miro1-mediated mitochondrial positioning are vinculin, which is a cytoplasmic protein involved in the binding of actin in focal adhesions, and p130cas, which serves as a substrate for several tyrosine kinases (Peng et al., 2011) (Figure 3B). When mitochondria are restricted around the nucleus due to deletion of Miro1 (Miro1−/−) in MEFs there is decreased H2O2 in the cell periphery (Alshaabi et al., 2021) (Figure 3A). This correlates with lower vinculin and p130cas phosphorylation at tyrosine residues Y100 and Y410, respectively, residues critical for activity (Pellicena and Miller, 2001; Golji et al., 2012). When Miro1 is re-expressed via stable expression of Myc-tagged Miro1 in MEFs the mitochondria are redistributed throughout the cytoplasm causing an increase in H2O2 in the periphery, accompanied by increased phosphorylation of vinculin and p130cas (Alshaabi et al., 2021). Going alongside this, it has been shown that elevated H2O2 levels in metastatic bladder cancer cells increases the phosphorylation and membrane recruitment of p130cas through oxidation of the PTPN12 phosphatase, driving the metastatic phenotype (Hempel et al., 2013). Mitochondrial and NOX-dependent sources of ROS have both been implicated in regulation of these processes stated above, but due to the intimate crosstalk between mitochondria and NOX enzymes (Daiber, 2010), deciphering the precise contribution from each source has been challenging (Figure 2D).
Similarly, during cell migration, there is an increase in ROS in cell protrusions which is needed for the oxidation of cofilin at C139 and C147 (Cameron et al., 2015). Cofilin is a cytoplasmic protein that is responsible for the severing of F-actin. When oxidized at C139 and C147 cofilin becomes inactivated (Figure 3B). Oxidation resistant mutants of cofilin were shown to reduce breast cancer attachment, migration, and invasion (Cameron et al., 2015). Cofilin is also regulated via phosphorylation and when phosphorylated at S3 it renders the protein inactive (Agnew et al., 1995; Moriyama et al., 1996; Sumi et al., 1999). Since ATP and H2O2 are abundant in areas of high mitochondrial density (Schuler et al., 2017; Alshaabi et al., 2021) it is realistic that either or both molecules could regulate cofilin activity, however, it is unclear which molecule is preferentially utilized from mitochondria.
All the proteins listed above: vinculin, p130cas, and cofilin are all important in FA formation which aids in cell attachment and migration. Focal adhesion kinase (FAK) is a key kinase found in FA formations and it is known to be activated via phosphorylation; however, its phosphorylation is attenuated by inhibition of redox signaling in the cell periphery (Chiarugi et al., 2003) (Figure 3B). FAK dephosphorylation/inactivation can be positively regulated via integrin-induced ROS which inhibits low molecular weight protein tyrosine phosphatase (LMW-PTP), therefore keeping FAK activated for longer (Chiarugi et al., 2003; Scales and Parsons, 2011). Subcellular ROS has also been shown to activate FA proteins such as FAK, paxillin, and p130cas, which all are integral in FA maturation and cell adhesion (Gozin et al., 1998). Disruptions in the trafficking of mitochondria and changes in local H2O2 and ATP levels correlate with perturbations in FA dynamics (Schuler et al., 2017). Leading edge changes in mROS also contribute to Src and FAK signaling driving breast cancer cell migration. Downregulation of SIRT3 in breast cancer cells supports increased mROS signaling that increases Src-dependent phosphorylation of FAK (Tyr576/577) and p130Cas (Y410) at the leading-edge membrane (Lee et al., 2018). SIRT3 mediated changes in Src and FAK phosphorylation were also sensitive to addition of endogenous antioxidants. While performing scratch-migration assays, it was observed that SIRT3 levels were the lowest in cells at the leading edge of the scratch, compared to non-migrating cells at distal sites, indicating migrating cells downregulate SIRT3 expression to support increased mROS mediating Src and FAK phosphorylation (Lee et al., 2018).
FAK and Src activities are closely intertwined since they participate in overlapping signaling response. Oxidative stress, elicited by PI3 kinase, in Caco-2 colon epithelial cells, caused increased activity and phosphorylation of FAK at Y397, Y577, and Y925 as well as c-Src activity and phosphorylation at Y418 (Basuroy et al., 2010). This resulted in increased cell migration, but by expressing a dominant negative c-Src the oxidant induced cell migration was prevented; therefore, it was found that both oxidants and an active c-Src were needed to rapidly increase cell migration via FAK (Basuroy et al., 2010). Similarly in vascular endothelial cells, FAK is activated and phosphorylated in the presence of H2O2 in a time and dose dependent manner (Vepa et al., 1999). The increased FAK activity also corresponded with enhanced actin stress fibers because of cytoskeleton reorganization.
During cell attachment there is an integrin-induced release of ROS at the plasma membrane which oxidizes Src, therefore increasing Src activity by dephosphorylating Y527. Src activity has been linked to increased cell invasion and tumor onset; however, when antioxidants are used or an oxidant null Src (C245A and C487A) is expressed then Src activity decreases as well as cell invasion and tumor progression (Giannoni et al., 2005) (Figure 3B). Oxidation of Src via ATP-mediated activation of DUOX1-dependent H2O2 production increases Src activity which activates the epidermal growth factor receptor (EGFR) to activate downstream signaling pathways (Truong and Carroll, 2012; Heppner et al., 2016). DUOX-1 activity is also important for epithelial cell migration during repair via activation of EGFR (Gorissen et al., 2013). Still unclear is the role of mitochondria is these processes which presumably is important given the regulation of DUOX enzymes by ATP and Ca2+.
Gap in knowledge/Summary
The regulation of redox-dependent signaling by mitochondrial or NOX-dependent ROS production is well-established and new targets are continuously being uncovered. The crosstalk between these ROS sources, with distinct differences in subcellular localization, dynamics, substrates, and targets is still unclear. The dynamic nature of the mitochondria and the ability to produce both ATP and ROS at specific subcellular sites provides an additional layer of control to redox and phospho-signaling by mitochondria. The contribution of local mitochondrial populations and how disruption of the subcellular architecture of mitochondria may impact NOX activity is unclear. We hypothesize that disruption of intracellular ATP and ROS gradients via loss of Miro1 mediated mitochondrial positioning, or other mitochondrial disruptions, would alter NOX-dependent redox signaling and redox-dependent phosphorylation cascades. Critical gaps still exist regarding the role of localized mitochondria in regulating these signaling events during cell migration and other localized responses (ie. membrane repair). Herein, we have briefly summarized the literature that supports the subcellular trafficking of mitochondria in the regulation of redox and phospho-signaling events supporting cell migration, linking mitochondrial dynamics to the spatial and temporal control over redox and phospho-signaling cascades.
Author contributions
NS contributed to the writing, editing, figure creation and final editing of the manuscript. RG contributed to the writing and editing of the manuscript. BC contributed to the writing, editing and final approval of the manuscript.
Funding
This work was supported by development funds from the University of Vermont Department of Pathology and Laboratory Medicine (BC).
Acknowledgments
We apologize for any oversight and lack of inclusion of relevant literature.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Agnew, B. J., Minamide, L. S., and Bamburg, J. R. (1995). Reactivation of phosphorylated actin depolymerizing factor and identification of the regulatory site. J. Biol. Chem. 270 (29), 17582–17587. doi:10.1074/jbc.270.29.17582
Ahmad, T., Mukherjee, S., Pattnaik, B., Kumar, M., Singh, S., Kumar, M., et al. (2014). Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. 33 (9), 994–1010. doi:10.1002/embj.201386030
Al-Mehdi, A. B., Pastukh, V. M., Swiger, B. M., Reed, D. J., Patel, M. R., Bardwell, G. C., et al. (2012). Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci. Signal. 5 (231), ra47. doi:10.1126/scisignal.2002712
Alshaabi, H., Shannon, N., Gravelle, R., Milczarek, S., Messier, T., and Cunniff, B. (2021). Miro1-mediated mitochondrial positioning supports subcellular redox status. Redox Biol. 38, 101818. doi:10.1016/j.redox.2020.101818
Altieri, D. C. (2017). Mitochondria on the move: Emerging paradigms of organelle trafficking in tumour plasticity and metastasis. Br. J. Cancer 117 (3), 301–305. doi:10.1038/bjc.2017.201
Altieri, D. C. (2019). Mitochondrial dynamics and metastasis. Cell. Mol. Life Sci. 76 (5), 827–835. doi:10.1007/s00018-018-2961-2
Anilkumar, N., Weber, R., Zhang, M., Brewer, A., and Shah, A. M. (2008). Nox4 and nox2 NADPH oxidases mediate distinct cellular redox signaling responses to agonist stimulation. Arterioscler. Thromb. Vasc. Biol. 28 (7), 1347–1354. doi:10.1161/ATVBAHA.108.164277
Barford, D., Das, A. K., and Egloff, M. P. (1998). The structure and mechanism of protein phosphatases: Insights into catalysis and regulation. Annu. Rev. Biophys. Biomol. Struct. 27, 133–164. doi:10.1146/annurev.biophys.27.1.133
Basuroy, S., Dunagan, M., Sheth, P., Seth, A., and Rao, R. K. (2010). Hydrogen peroxide activates focal adhesion kinase and c-Src by a phosphatidylinositol 3 kinase-dependent mechanism and promotes cell migration in Caco-2 cell monolayers. Am. J. Physiol. Gastrointest. Liver Physiol. 299 (1), G186–G195. doi:10.1152/ajpgi.00368.2009
Berndt, C., Lillig, C. H., and Holmgren, A. (2007). Thiol-based mechanisms of the thioredoxin and glutaredoxin systems: Implications for diseases in the cardiovascular system. Am. J. Physiol. Heart Circ. Physiol. 292 (3), H1227–H1236. doi:10.1152/ajpheart.01162.2006
Block, K., Gorin, Y., and Abboud, H. E. (2009). Subcellular localization of Nox4 and regulation in diabetes. Proc. Natl. Acad. Sci. U. S. A. 106 (34), 14385–14390. doi:10.1073/pnas.0906805106
Bonnans, C., Chou, J., and Werb, Z. (2014). Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 15 (12), 786–801. doi:10.1038/nrm3904
Budnik, L. T., Kloth, S., Baur, X., Preisser, A. M., and Schwarzenbach, H. (2013). Circulating mitochondrial DNA as biomarker linking environmental chemical exposure to early preclinical lesions elevation of mtDNA in human serum after exposure to carcinogenic halo-alkane-based pesticides. PLoS One 8 (5), e64413. doi:10.1371/journal.pone.0064413
Byrne, D. P., Shrestha, S., Galler, M., Cao, M., Daly, L. A., Campbell, A. E., et al. (2020). Aurora A regulation by reversible cysteine oxidation reveals evolutionarily conserved redox control of Ser/Thr protein kinase activity. Sci. Signal. 13 (639), eaax2713. doi:10.1126/scisignal.aax2713
Cadenas, E., and Davies, K. J. (2000). Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 29 (3-4), 222–230. doi:10.1016/s0891-5849(00)00317-8
Cameron, J. M., Gabrielsen, M., Chim, Y. H., Munro, J., McGhee, E. J., Sumpton, D., et al. (2015). Polarized cell motility induces hydrogen peroxide to inhibit cofilin via cysteine oxidation. Curr. Biol. 25 (11), 1520–1525. doi:10.1016/j.cub.2015.04.020
Cao, S. S., and Kaufman, R. J. (2014). Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 21 (3), 396–413. doi:10.1089/ars.2014.5851
Cao, X., Kaneko, T., Li, J. S., Liu, A. D., Voss, C., and Li, S. S. C. (2015). Erratum: A phosphorylation switch controls the spatiotemporal activation of rho GTPases in directional cell migration. Nat. Commun. 6, 8320. doi:10.1038/ncomms9320
Carmona, M., de Cubas, L., Bautista, E., Moral-Blanch, M., Medrano-Fernandez, I., Sitia, R., et al. (2019). Monitoring cytosolic H2O2 fluctuations arising from altered plasma membrane gradients or from mitochondrial activity. Nat. Commun. 10 (1), 4526. doi:10.1038/s41467-019-12475-0
Carreras-Sureda, A., Pihan, P., and Hetz, C. (2018). Calcium signaling at the endoplasmic reticulum: Fine-tuning stress responses. Cell Calcium 70, 24–31. doi:10.1016/j.ceca.2017.08.004
Cecarini, V., Gee, J., Fioretti, E., Amici, M., Angeletti, M., Eleuteri, A. M., et al. (2007). Protein oxidation and cellular homeostasis: Emphasis on metabolism. Biochim. Biophys. Acta 1773 (2), 93–104. doi:10.1016/j.bbamcr.2006.08.039
Chen, J. Q., Eshete, M., Alworth, W. L., and Yager, J. D. (2004). Binding of MCF-7 cell mitochondrial proteins and recombinant human estrogen receptors alpha and beta to human mitochondrial DNA estrogen response elements. J. Cell. Biochem. 93 (2), 358–373. doi:10.1002/jcb.20178
Chen, K., Kirber, M. T., Xiao, H., Yang, Y., and Keaney, J. F. (2008). Regulation of ROS signal transduction by NADPH oxidase 4 localization. J. Cell Biol. 181 (7), 1129–1139. doi:10.1083/jcb.200709049
Chiarugi, P., Pani, G., Giannoni, E., Taddei, L., Colavitti, R., Raugei, G., et al. (2003). Reactive oxygen species as essential mediators of cell adhesion: The oxidative inhibition of a FAK tyrosine phosphatase is required for cell adhesion. J. Cell Biol. 161 (5), 933–944. doi:10.1083/jcb.200211118
Cunniff, B., McKenzie, A. J., Heintz, N. H., and Howe, A. K. (2016). AMPK activity regulates trafficking of mitochondria to the leading edge during cell migration and matrix invasion. Mol. Biol. Cell 27 (17), 2662–2674. doi:10.1091/mbc.E16-05-0286
Daiber, A. (2010). Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim. Biophys. Acta 1797 (6-7), 897–906. doi:10.1016/j.bbabio.2010.01.032
Davidson, D., Bakinowski, M., Thomas, M. L., Horejsi, V., and Veillette, A. (2003). Phosphorylation-dependent regulation of T-cell activation by PAG/Cbp, a lipid raft-associated transmembrane adaptor. Mol. Cell. Biol. 23 (6), 2017–2028. doi:10.1128/mcb.23.6.2017-2028.2003
de Cubas, L., Pak, V. V., Belousov, V. V., Ayte, J., and Hidalgo, E. (2021). The mitochondria-to-cytosol H2O2 gradient is caused by peroxiredoxin-dependent cytosolic scavenging. Antioxidants (Basel) 10 (5), 731. doi:10.3390/antiox10050731
De Nigris, F., Lerman, L. O., Condorelli, M., Lerman, A., and Napoli, C. (2001). Oxidation-sensitive transcription factors and molecular mechanisms in the arterial wall. Antioxid. Redox Signal. 3 (6), 1119–1130. doi:10.1089/152308601317203620
Debattisti, V., Gerencser, A. A., Saotome, M., Das, S., and Hajnoczky, G. (2017). ROS control mitochondrial motility through p38 and the motor adaptor miro/trak. Cell Rep. 21 (6), 1667–1680. doi:10.1016/j.celrep.2017.10.060
Desai, S. P., Bhatia, S. N., Toner, M., and Irimia, D. (2013). Mitochondrial localization and the persistent migration of epithelial cancer cells. Biophys. J. 104 (9), 2077–2088. doi:10.1016/j.bpj.2013.03.025
Detmer, S. A., and Chan, D. C. (2007). Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 8 (11), 870–879. doi:10.1038/nrm2275
Doughan, A. K., Harrison, D. G., and Dikalov, S. I. (2008). Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: Linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ. Res. 102 (4), 488–496. doi:10.1161/CIRCRESAHA.107.162800
Duchen, M. R. (1992). Ca(2+)-dependent changes in the mitochondrial energetics in single dissociated mouse sensory neurons. Biochem. J. 283 (Pt 1), 41–50. doi:10.1042/bj2830041
Dustin, C. M., Heppner, D. E., Lin, M. C. J., and van der Vliet, A. (2020). Redox regulation of tyrosine kinase signalling: More than meets the eye. J. Biochem. 167 (2), 151–163. doi:10.1093/jb/mvz085
Endicott, J. A., Noble, M. E., and Johnson, L. N. (2012). The structural basis for control of eukaryotic protein kinases. Annu. Rev. Biochem. 81, 587–613. doi:10.1146/annurev-biochem-052410-090317
Fransson, S., Ruusala, A., and Aspenstrom, P. (2006). The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem. Biophys. Res. Commun. 344 (2), 500–510. doi:10.1016/j.bbrc.2006.03.163
Fu, Q., Liu, R., Wang, H., Hua, C., Song, S., Zhou, G., et al. (2019). Effects of oxidation in vitro on structures and functions of myofibrillar protein from beef muscles. J. Agric. Food Chem. 67 (20), 5866–5873. doi:10.1021/acs.jafc.9b01239
Garcia-Santamarina, S., Boronat, S., and Hidalgo, E. (2014). Reversible cysteine oxidation in hydrogen peroxide sensing and signal transduction. Biochemistry 53 (16), 2560–2580. doi:10.1021/bi401700f
Garde, A., Kenny, I. W., Kelley, L. C., Chi, Q., Mutlu, A. S., Wang, M. C., et al. (2022). Localized glucose import, glycolytic processing, and mitochondria generate a focused ATP burst to power basement-membrane invasion. Dev. Cell 57 (6), 732–749 e7. doi:10.1016/j.devcel.2022.02.019
Garde, A., and Sherwood, D. R. (2021). Fueling cell invasion through extracellular matrix. Trends Cell Biol. 31 (6), 445–456. doi:10.1016/j.tcb.2021.01.006
Giannoni, E., Buricchi, F., Raugei, G., Ramponi, G., and Chiarugi, P. (2005). Intracellular reactive oxygen species activate Src tyrosine kinase during cell adhesion and anchorage-dependent cell growth. Mol. Cell. Biol. 25 (15), 6391–6403. doi:10.1128/MCB.25.15.6391-6403.2005
Giovarelli, M., Zecchini, S., Martini, E., Garre, M., Barozzi, S., Ripolone, M., et al. (2020). Drp1 overexpression induces desmin disassembling and drives kinesin-1 activation promoting mitochondrial trafficking in skeletal muscle. Cell Death Differ. 27 (8), 2383–2401. doi:10.1038/s41418-020-0510-7
Go, Y. M., and Jones, D. P. (2008). Redox compartmentalization in eukaryotic cells. Biochim. Biophys. Acta 1780 (11), 1273–1290. doi:10.1016/j.bbagen.2008.01.011
Golji, J., Wendorff, T., and Mofrad, M. R. (2012). Phosphorylation primes vinculin for activation. Biophys. J. 102 (9), 2022–2030. doi:10.1016/j.bpj.2012.01.062
Gorissen, S. H., Hristova, M., Habibovic, A., Sipsey, L. M., Spiess, P. C., Janssen-Heininger, Y. M. W., et al. (2013). Dual oxidase-1 is required for airway epithelial cell migration and bronchiolar reepithelialization after injury. Am. J. Respir. Cell Mol. Biol. 48 (3), 337–345. doi:10.1165/rcmb.2012-0393OC
Gostner, J. M., Becker, K., Fuchs, D., and Sucher, R. (2013). Redox regulation of the immune response. Redox Rep. 18 (3), 88–94. doi:10.1179/1351000213Y.0000000044
Gozin, A., Andrieu, V., Da Costa, L., RollEt-LabEllE, E., and Pasquier, C. (1998). Reactive oxygen species activate focal adhesion kinase, paxillin and p130cas tyrosine phosphorylation in endothelial cells. Free Radic. Biol. Med. 25 (9), 1021–1032. doi:10.1016/s0891-5849(98)00134-8
Grintsevich, E. E., Ge, P., Sawaya, M. R., Yesilyurt, H. G., Terman, J. R., Zhou, Z. H., et al. (2017). Catastrophic disassembly of actin filaments via Mical-mediated oxidation. Nat. Commun. 8 (1), 2183. doi:10.1038/s41467-017-02357-8
Hamada, Y., Furumoto, Y., Izutani, A., Taniuchi, S., Miyake, M., Oyadomari, M., et al. (2020). Nanosecond pulsed electric fields induce the integrated stress response via reactive oxygen species-mediated heme-regulated inhibitor (HRI) activation. PLoS One 15 (3), e0229948. doi:10.1371/journal.pone.0229948
Handy, D. E., Lubos, E., Yang, Y., Galbraith, J. D., Kelly, N., Zhang, Y. Y., et al. (2009). Glutathione peroxidase-1 regulates mitochondrial function to modulate redox-dependent cellular responses. J. Biol. Chem. 284 (18), 11913–11921. doi:10.1074/jbc.M900392200
Hazan, R. B., Phillips, G. R., Qiao, R. F., Norton, L., and Aaronson, S. A. (2000). Exogenous expression of N-cadherin in breast cancer cells induces cell migration, invasion, and metastasis. J. Cell Biol. 148 (4), 779–790. doi:10.1083/jcb.148.4.779
Hempel, N., Bartling, T. R., Mian, B., and Melendez, J. A. (2013). Acquisition of the metastatic phenotype is accompanied by H2O2-dependent activation of the p130Cas signaling complex. Mol. Cancer Res. 11 (3), 303–312. doi:10.1158/1541-7786.MCR-12-0478
Heppner, D. E., Hristova, M., Dustin, C. M., Danyal, K., Habibovic, A., and van der Vliet, A. (2016). The NADPH oxidases DUOX1 and NOX2 play distinct roles in redox regulation of epidermal growth factor receptor signaling. J. Biol. Chem. 291 (44), 23282–23293. doi:10.1074/jbc.M116.749028
Hollenbeck, P. J. (2005). Mitochondria and neurotransmission: Evacuating the synapse. Neuron 47 (3), 331–333. doi:10.1016/j.neuron.2005.07.017
Holmstrom, K. M., and Finkel, T. (2014). Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 15 (6), 411–421. doi:10.1038/nrm3801
Horn, A., Raavicharla, S., Shah, S., Cox, D., and Jaiswal, J. K. (2020). Mitochondrial fragmentation enables localized signaling required for cell repair. J. Cell Biol. 219 (5), e201909154. doi:10.1083/jcb.201909154
Horn, A., Van der Meulen, J. H., Defour, A., Hogarth, M., Sreetama, S. C., Reed, A., et al. (2017). Mitochondrial redox signaling enables repair of injured skeletal muscle cells. Sci. Signal. 10 (495), eaaj1978. doi:10.1126/scisignal.aaj1978
Hoshi, T., and Heinemann, S. (2001). Regulation of cell function by methionine oxidation and reduction. J. Physiol. 531 (Pt 1), 1–11. doi:10.1111/j.1469-7793.2001.0001j.x
Hurd, T. R., DeGennaro, M., and Lehmann, R. (2012). Redox regulation of cell migration and adhesion. Trends Cell Biol. 22 (2), 107–115. doi:10.1016/j.tcb.2011.11.002
Inoue, M., Sato, E. F., Nishikawa, M., Park, A. M., Kira, Y., Imada, I., et al. (2003). Mitochondrial generation of reactive oxygen species and its role in aerobic life. Curr. Med. Chem. 10 (23), 2495–2505. doi:10.2174/0929867033456477
Ishikawa, K., Takenaga, K., Akimoto, M., Koshikawa, N., Yamaguchi, A., Imanishi, H., et al. (2008). ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 320 (5876), 661–664. doi:10.1126/science.1156906
Iwasaki, Y., Takeshima, Y., and Fujio, K. (2020). Basic mechanism of immune system activation by mitochondria. Immunol. Med. 43 (4), 142–147. doi:10.1080/25785826.2020.1756609
Jelcic, M., Enyedi, B., Xavier, J. B., and Niethammer, P. (2017). Image-based measurement of H2O2 reaction-diffusion in wounded zebrafish larvae. Biophys. J. 112 (9), 2011–2018. doi:10.1016/j.bpj.2017.03.021
Jezek, P., Holendova, B., and Plecita-Hlavata, L. (2020). Redox signaling from mitochondria: Signal propagation and its targets. Biomolecules 10 (1), E93. doi:10.3390/biom10010093
Jones, D. P. (2010). Redox sensing: Orthogonal control in cell cycle and apoptosis signalling. J. Intern. Med. 268 (5), 432–448. doi:10.1111/j.1365-2796.2010.02268.x
Kang, J. S., Tian, J. H., Pan, P. Y., Zald, P., Li, C., Deng, C., et al. (2008). Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell 132 (1), 137–148. doi:10.1016/j.cell.2007.11.024
Karasev, D. A., Veselova, D. A., Veselovsky, A. V., Sobolev, B. N., Zgoda, V. G., and Archakov, A. I. (2018). Spatial features of proteins related to their phosphorylation and associated structural changes. Proteins 86 (1), 13–20. doi:10.1002/prot.25397
Kelley, L. C., Chi, Q., Caceres, R., Hastie, E., Schindler, A. J., Jiang, Y., et al. (2019). Adaptive F-actin polymerization and localized ATP production drive basement membrane invasion in the absence of MMPs. Dev. Cell 48 (3), 313–328. doi:10.1016/j.devcel.2018.12.018
Kim, J. H., Choi, T. G., Park, S., Yun, H. R., Nguyen, N. N. Y., Jo, Y. H., et al. (2018). Mitochondrial ROS-derived PTEN oxidation activates PI3K pathway for mTOR-induced myogenic autophagy. Cell Death Differ. 25 (11), 1921–1937. doi:10.1038/s41418-018-0165-9
Kim, Y., and Jang, H. H. (2019). Role of cytosolic 2-cys Prx1 and Prx2 in redox signaling. Antioxidants (Basel) 8 (6), E169. doi:10.3390/antiox8060169
LeClaire, L. L., Baumgartner, M., Iwasa, J. H., Mullins, R. D., and Barber, D. L. (2008). Phosphorylation of the Arp2/3 complex is necessary to nucleate actin filaments. J. Cell Biol. 182 (4), 647–654. doi:10.1083/jcb.200802145
Lee, J. J., van de Ven, R. A. H., Zaganjor, E., Ng, M. R., Barakat, A., Demmers, J. J. P. G., et al. (2018). Inhibition of epithelial cell migration and Src/FAK signaling by SIRT3. Proc. Natl. Acad. Sci. U. S. A. 115 (27), 7057–7062. doi:10.1073/pnas.1800440115
Lee, S. B., Bae, I. H., Bae, Y. S., and Um, H. D. (2006). Link between mitochondria and NADPH oxidase 1 isozyme for the sustained production of reactive oxygen species and cell death. J. Biol. Chem. 281 (47), 36228–36235. doi:10.1074/jbc.M606702200
Li, L., Conradson, D. M., Bharat, V., Kim, M. J., Hsieh, C. H., Minhas, P. S., et al. (2021). A mitochondrial membrane-bridging machinery mediates signal transduction of intramitochondrial oxidation. Nat. Metab. 3 (9), 1242–1258. doi:10.1038/s42255-021-00443-2
Loffek, S., Schilling, O., and Franzke, C. W. (2011). Series "matrix metalloproteinases in lung health and disease": Biological role of matrix metalloproteinases: A critical balance. Eur. Respir. J. 38 (1), 191–208. doi:10.1183/09031936.00146510
Londhe, A. D., Bergeron, A., Curley, S. M., Zhang, F., Rivera, K. D., Kannan, A., et al. (2020). Regulation of PTP1B activation through disruption of redox-complex formation. Nat. Chem. Biol. 16 (2), 122–125. doi:10.1038/s41589-019-0433-0
Lopez-Domenech, G., Covill-Cooke, C., Ivankovic, D., Halff, E. F., Sheehan, D. F., Norkett, R., et al. (2018). Miro proteins coordinate microtubule- and actin-dependent mitochondrial transport and distribution. EMBO J. 37 (3), 321–336. doi:10.15252/embj.201696380
Madan, S., Uttekar, B., Chowdhary, S., and Rikhy, R. (2021). Mitochondria lead the way: Mitochondrial dynamics and function in cellular movements in development and disease. Front. Cell Dev. Biol. 9, 781933. doi:10.3389/fcell.2021.781933
Maechler, P., and Wollheim, C. B. (2000). Mitochondrial signals in glucose-stimulated insulin secretion in the beta cell. J. Physiol. 529 Pt 1, 49–56. doi:10.1111/j.1469-7793.2000.00049.x
Marchi, S., Patergnani, S., Missiroli, S., Morciano, G., Rimessi, A., Wieckowski, M. R., et al. (2018). Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 69, 62–72. doi:10.1016/j.ceca.2017.05.003
Meng, T. C., Fukada, T., and Tonks, N. K. (2002). Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol. Cell 9 (2), 387–399. doi:10.1016/s1097-2765(02)00445-8
Morgan, B., Van Laer, K., Owusu, T. N. E., Ezerina, D., Pastor-Flores, D., Amponsah, P. S., et al. (2016). Real-time monitoring of basal H2O2 levels with peroxiredoxin-based probes. Nat. Chem. Biol. 12 (6), 437–443. doi:10.1038/nchembio.2067
Moriyama, K., Iida, K., and Yahara, I. (1996). Phosphorylation of Ser-3 of cofilin regulates its essential function on actin. Genes cells. 1 (1), 73–86. doi:10.1046/j.1365-2443.1996.05005.x
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem. J. 417 (1), 1–13. doi:10.1042/BJ20081386
Newick, K., Cunniff, B., Preston, K., Held, P., Arbiser, J., Pass, H., et al. (2012). Peroxiredoxin 3 is a redox-dependent target of thiostrepton in malignant mesothelioma cells. PLoS One 7 (6), e39404. doi:10.1371/journal.pone.0039404
Nguyen, S. D., and Sok, D. E. (2003). Oxidative inactivation of paraoxonase1, an antioxidant protein and its effect on antioxidant action. Free Radic. Res. 37 (12), 1319–1330. doi:10.1080/5760310001621351-1
Nguyen, T. T., Oh, S. S., Weaver, D., Lewandowska, A., Maxfield, D., Schuler, M. H., et al. (2014). Loss of Miro1-directed mitochondrial movement results in a novel murine model for neuron disease. Proc. Natl. Acad. Sci. U. S. A. 111 (35), E3631–E3640. doi:10.1073/pnas.1402449111
Niethammer, P., Grabher, C., Look, A. T., and Mitchison, T. J. (2009). A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature 459 (7249), 996–999. doi:10.1038/nature08119
Ostman, A., Frijhoff, J., Sandin, A., and Bohmer, F. D. (2011). Regulation of protein tyrosine phosphatases by reversible oxidation. J. Biochem. 150 (4), 345–356. doi:10.1093/jb/mvr104
Pak, V. V., Ezerina, D., Lyublinskaya, O. G., Pedre, B., Tyurin-Kuzmin, P. A., Mishina, N. M., et al. (2020). Ultrasensitive genetically encoded indicator for hydrogen peroxide identifies roles for the oxidant in cell migration and mitochondrial function. Cell Metab. 31 (3), 642–653. doi:10.1016/j.cmet.2020.02.003
Panday, A., Sahoo, M. K., Osorio, D., and Batra, S. (2015). NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 12 (1), 5–23. doi:10.1038/cmi.2014.89
Paulsen, C. E., Truong, T. H., Garcia, F. J., Homann, A., Gupta, V., Leonard, S. E., et al. (2011). Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat. Chem. Biol. 8 (1), 57–64. doi:10.1038/nchembio.736
Pekkurnaz, G., Trinidad, J. C., Wang, X., Kong, D., and Schwarz, T. L. (2014). Glucose regulates mitochondrial motility via Milton modification by O-GlcNAc transferase. Cell 158 (1), 54–68. doi:10.1016/j.cell.2014.06.007
Pellicena, P., and Miller, W. T. (2001). Processive phosphorylation of p130Cas by Src depends on SH3-polyproline interactions. J. Biol. Chem. 276 (30), 28190–28196. doi:10.1074/jbc.M100055200
Peng, X., Nelson, E. S., Maiers, J. L., and DeMali, K. A. (2011). New insights into vinculin function and regulation. Int. Rev. Cell Mol. Biol. 287, 191–231. doi:10.1016/B978-0-12-386043-9.00005-0
Porporato, P. E., Payen, V. L., Perez-Escuredo, J., De Saedeleer, C. J., Danhier, P., Copetti, T., et al. (2014). A mitochondrial switch promotes tumor metastasis. Cell Rep. 8 (3), 754–766. doi:10.1016/j.celrep.2014.06.043
Queliconi, B. B., Wojtovich, A. P., Nadtochiy, S. M., Kowaltowski, A. J., and Brookes, P. S. (2011). Redox regulation of the mitochondrial K(ATP) channel in cardioprotection. Biochim. Biophys. Acta 1813 (7), 1309–1315. doi:10.1016/j.bbamcr.2010.11.005
Rao, R., Xu, D., Thelen, J. J., and Miernyk, J. A. (2013). Circles within circles: Crosstalk between protein ser/thr/tyr-phosphorylation and met oxidation. BMC Bioinforma. 14 (Suppl. 14), S14. doi:10.1186/1471-2105-14-S14-S14
Ren, F., Tang, R., Zhang, X., Madushi, W. M., Luo, D., Dang, Y., et al. (2015). Overexpression of mmp family members functions as prognostic biomarker for breast cancer patients: A systematic review and meta-analysis. PLoS One 10 (8), e0135544. doi:10.1371/journal.pone.0135544
Riedl, T., and Egly, J. M. (2000). Phosphorylation in transcription: The CTD and more. Gene Expr. 9 (1-2), 3–13. doi:10.3727/000000001783992704
Rizzuto, R., Bernardi, P., and Pozzan, T. (2000). Mitochondria as all-round players of the calcium game. J. Physiol. 529 Pt 1, 37–47. doi:10.1111/j.1469-7793.2000.00037.x
Rosdah, A. A., Smiles, W. J., Oakhill, J. S., Scott, J. W., Langendorf, C. G., Delbridge, L. M. D., et al. (2020). New perspectives on the role of Drp1 isoforms in regulating mitochondrial pathophysiology. Pharmacol. Ther. 213, 107594. doi:10.1016/j.pharmthera.2020.107594
Sandalio, L. M., Rodriguez-Serrano, M., Romero-Puertas, M. C., and del Rio, L. A. (2013). Role of peroxisomes as a source of reactive oxygen species (ROS) signaling molecules. Subcell. Biochem. 69, 231–255. doi:10.1007/978-94-007-6889-5_13
Sariahmetoglu, M., Crawford, B. D., Leon, H., Sawicka, J., Li, L., Ballermann, B. J., et al. (2007). Regulation of matrix metalloproteinase-2 (MMP-2) activity by phosphorylation. FASEB J. 21 (10), 2486–2495. doi:10.1096/fj.06-7938com
Scales, T. M., and Parsons, M. (2011). Spatial and temporal regulation of integrin signalling during cell migration. Curr. Opin. Cell Biol. 23 (5), 562–568. doi:10.1016/j.ceb.2011.05.008
Schuler, M. H., Lewandowska, A., Caprio, G. D., Skillern, W., Upadhyayula, S., Kirchhausen, T., et al. (2017). Miro1-mediated mitochondrial positioning shapes intracellular energy gradients required for cell migration. Mol. Biol. Cell 28 (16), 2159–2169. doi:10.1091/mbc.E16-10-0741
Shanmugasundaram, K., Nayak, B. K., Friedrichs, W. E., Kaushik, D., Rodriguez, R., and Block, K. (2017). NOX4 functions as a mitochondrial energetic sensor coupling cancer metabolic reprogramming to drug resistance. Nat. Commun. 8 (1), 997. doi:10.1038/s41467-017-01106-1
Sheng, Z. H. (2014). Mitochondrial trafficking and anchoring in neurons: New insight and implications. J. Cell Biol. 204 (7), 1087–1098. doi:10.1083/jcb.201312123
Shin, D. H., Dier, U., Melendez, J. A., and Hempel, N. (2015). Regulation of MMP-1 expression in response to hypoxia is dependent on the intracellular redox status of metastatic bladder cancer cells. Biochim. Biophys. Acta 1852 (12), 2593–2602. doi:10.1016/j.bbadis.2015.09.001
Smith, K. P., Focia, P. J., Chakravarthy, S., Landahl, E. C., Klosowiak, J. L., Rice, S. E., et al. (2020). Insight into human Miro1/2 domain organization based on the structure of its N-terminal GTPase. J. Struct. Biol. 212 (3), 107656. doi:10.1016/j.jsb.2020.107656
Sobotta, M. C., Liou, W., Stocker, S., Talwar, D., Oehler, M., Ruppert, T., et al. (2015). Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 11 (1), 64–70. doi:10.1038/nchembio.1695
Stocker, S., Maurer, M., Ruppert, T., and Dick, T. P. (2018). A role for 2-Cys peroxiredoxins in facilitating cytosolic protein thiol oxidation. Nat. Chem. Biol. 14 (2), 148–155. doi:10.1038/nchembio.2536
Stocker, S., Van Laer, K., Mijuskovic, A., and Dick, T. P. (2018). The conundrum of hydrogen peroxide signaling and the emerging role of peroxiredoxins as redox relay hubs. Antioxid. Redox Signal. 28 (7), 558–573. doi:10.1089/ars.2017.7162
Sumi, T., Matsumoto, K., Takai, Y., and Nakamura, T. (1999). Cofilin phosphorylation and actin cytoskeletal dynamics regulated by rho- and Cdc42-activated LIM-kinase 2. J. Cell Biol. 147 (7), 1519–1532. doi:10.1083/jcb.147.7.1519
Tan, J. X., and Finkel, T. (2020). Mitochondria as intracellular signaling platforms in health and disease. J. Cell Biol. 219 (5), e202002179. doi:10.1083/jcb.202002179
Tilokani, L., Nagashima, S., Paupe, V., and Prudent, J. (2018). Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 62 (3), 341–360. doi:10.1042/EBC20170104
Toyama, E. Q., Herzig, S., Courchet, J., Lewis, T. L., Loson, O. C., Hellberg, K., et al. (2016). Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 351 (6270), 275–281. doi:10.1126/science.aab4138
Truong, T. H., and Carroll, K. S. (2012). Redox regulation of epidermal growth factor receptor signaling through cysteine oxidation. Biochemistry 51 (50), 9954–9965. doi:10.1021/bi301441e
Truong, T. H., and Carroll, K. S. (2013). Redox regulation of protein kinases. Crit. Rev. Biochem. Mol. Biol. 48 (4), 332–356. doi:10.3109/10409238.2013.790873
Turrens, J. F. (2003). Mitochondrial formation of reactive oxygen species. J. Physiol. 552 (Pt 2), 335–344. doi:10.1113/jphysiol.2003.049478
van der Vliet, A., Janssen-Heininger, Y. M. W., and Anathy, V. (2018). Oxidative stress in chronic lung disease: From mitochondrial dysfunction to dysregulated redox signaling. Mol. Asp. Med. 63, 59–69. doi:10.1016/j.mam.2018.08.001
Vepa, S., Scribner, W. M., Parinandi, N. L., English, D., Garcia, J. G., and Natarajan, V. (1999). Hydrogen peroxide stimulates tyrosine phosphorylation of focal adhesion kinase in vascular endothelial cells. Am. J. Physiol. 277 (1), L150–L158. doi:10.1152/ajplung.1999.277.1.L150
Vukelic, S., Xu, Q., Seidel-Rogol, B., Faidley, E. A., Dikalova, A. E., Hilenski, L. L., et al. (2018). NOX4 (NADPH oxidase 4) and Poldip2 (polymerase delta-interacting protein 2) induce filamentous actin oxidation and promote its interaction with vinculin during integrin-mediated cell adhesion. Arterioscler. Thromb. Vasc. Biol. 38 (10), 2423–2434. doi:10.1161/ATVBAHA.118.311668
Williams, K. C., and Coppolino, M. G. (2011). Phosphorylation of membrane type 1-matrix metalloproteinase (MT1-MMP) and its vesicle-associated membrane protein 7 (VAMP7)-dependent trafficking facilitate cell invasion and migration. J. Biol. Chem. 286 (50), 43405–43416. doi:10.1074/jbc.M111.297069
Wu, R. F., Xu, Y. C., Ma, Z., Nwariaku, F. E., Sarosi, G. A., and Terada, L. S. (2005). Subcellular targeting of oxidants during endothelial cell migration. J. Cell Biol. 171 (5), 893–904. doi:10.1083/jcb.200507004
Yanes, O., Clark, J., Wong, D. M., Patti, G. J., Sanchez-Ruiz, A., Benton, H. P., et al. (2010). Metabolic oxidation regulates embryonic stem cell differentiation. Nat. Chem. Biol. 6 (6), 411–417. doi:10.1038/nchembio.364
Yao, C. H., Wang, R., Wang, Y., Kung, C. P., Weber, J. D., and Patti, G. J. (2019). Mitochondrial fusion supports increased oxidative phosphorylation during cell proliferation. Elife 8, e41351. doi:10.7554/eLife.41351
Yi, M., Weaver, D., and Hajnoczky, G. (2004). Control of mitochondrial motility and distribution by the calcium signal: A homeostatic circuit. J. Cell Biol. 167 (4), 661–672. doi:10.1083/jcb.200406038
Zhao, J., Zhang, M., Yu, , Xie, Y., Huang, Y., Wolff, D. W., et al. (2013). Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 32 (40), 4814–4824. doi:10.1038/onc.2012.494
Zhao, R. Z., Jiang, S., Zhang, L., and Yu, Z. B. (2019). Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 44 (1), 3–15. doi:10.3892/ijmm.2019.4188
Zorov, D. B., Filburn, C. R., Klotz, L. O., Zweier, J. L., and Sollott, S. J. (2000). Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 192 (7), 1001–1014. doi:10.1084/jem.192.7.1001
Keywords: redox signaling, phosphorylation, cell migration, mitochondrial trafficking, focal adhesion (FA)
Citation: Shannon N, Gravelle R and Cunniff B (2022) Mitochondrial trafficking and redox/phosphorylation signaling supporting cell migration phenotypes. Front. Mol. Biosci. 9:925755. doi: 10.3389/fmolb.2022.925755
Received: 21 April 2022; Accepted: 05 July 2022;
Published: 22 July 2022.
Edited by:
Benoit Boivin, State University of New York, United StatesReviewed by:
Mikael Molin, Chalmers University of Technology, SwedenNelli Mnatsakanyan, The Pennsylvania State University, United States
Copyright © 2022 Shannon, Gravelle and Cunniff. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brian Cunniff, QnJpYW4uQ3VubmlmZkB1dm0uZWR1