 Bai-Hui Zhang
Bai-Hui Zhang Fan Yin
Fan Yin Ya-Nan Qiao
Ya-Nan Qiao Shou-Dong Guo
Shou-Dong Guo- Institute of Lipid Metabolism and Atherosclerosis, Innovative Drug Research Centre, School of Pharmacy, Weifang Medical University, Weifang, China
Cardiovascular disease (CVD) is still the leading cause of death globally, and atherosclerosis is the main pathological basis of CVDs. Low-density lipoprotein cholesterol (LDL-C) is a strong causal factor of atherosclerosis. However, the first-line lipid-lowering drugs, statins, only reduce approximately 30% of the CVD risk. Of note, atherosclerotic CVD (ASCVD) cannot be eliminated in a great number of patients even their LDL-C levels meet the recommended clinical goals. Previously, whether the elevated plasma level of triglyceride is causally associated with ASCVD has been controversial. Recent genetic and epidemiological studies have demonstrated that triglyceride and triglyceride-rich lipoprotein (TGRL) are the main causal risk factors of the residual ASCVD. TGRLs and their metabolites can promote atherosclerosis via modulating inflammation, oxidative stress, and formation of foam cells. In this article, we will make a short review of TG and TGRL metabolism, display evidence of association between TG and ASCVD, summarize the atherogenic factors of TGRLs and their metabolites, and discuss the current findings and advances in TG-lowering therapies. This review provides information useful for the researchers in the field of CVD as well as for pharmacologists and clinicians.
Introduction
According to the WHO report in 2021, cardiovascular disease (CVD) is still the leading cause of death worldwide, and atherosclerotic CVD (ASCVD) is the most representative and dangerous one (Lin et al., 2021; WHO, 2021). Cholesterol carried by lipoproteins in blood is the major inducer of ASCVD. Among the lipoproteins, low-density lipoprotein (LDL) carries approximately 75% of total cholesterol (TC) that is carried by non-high density lipoprotein (HDL) particles (Jacobson et al., 2015). People with low LDL cholesterol (LDL-C) levels are less likely to develop CVD compared with those having average or high levels of LDL-C (Cohen et al., 2006; Ference et al., 2012). For instance, a long-term exposure to lower LDL-C is associated with a 54.5% reduction in the risk of coronary heart disease (CHD) for each mmol/L (38.7 mg/dl) reduction of LDL-C (Ference et al., 2012). The first line lipid-lowering drug, statins, can significantly reduce LDL-C levels, leading to a reduction in CVD events by 25%–40% (Michos et al., 2012; Silverman et al., 2016; Toth et al., 2019a). Moreover, the antibodies or siRNA of proprotein convertase subtilisin/kexin-type 9 (PCSK9) can further decrease LDL-C level as an add-on-statin therapy. However, patients-treated with statins in combination with PCSK9 inhibitors still experience ASCVD events even their LDL-C levels meet the clinical goals (Sampson et al., 2012; Nishikido and Ray, 2021; Su et al., 2021). Therefore, researchers are impelled to find novel strategies for treatment of residual ASCVD.
Given a low HDL cholesterol (HDL-C) level is a strong and independent risk factor associated with CVD events, enhancing HDL-C level has ever been expected to prevent and/or recover CVD (Catapano et al., 2016; Mach et al., 2020). Cholesteryl ester transport protein (CETP) inhibitors can significantly improve HDL-C levels and reverse cholesterol transport. However, these inhibitors are found to be useless in prevention of CVD events (Liu N et al., 2021). HDL dysfunction in patients with CVD may partially explain the failure of these CETP inhibitors (Sandesara et al., 2019). Recent studies suggested that although HDL-C is a useful risk biomarker, accumulating evidence from Mendelian randomization studies and other research have demonstrated that HDL-C is not a causal risk factor for ASCVD (Nordestgaard, 2016; Lincoff et al., 2017; Barter and Genest, 2019; Goyal et al., 2021). Therefore, researchers turn their attention to other targets for treatment of ASCVD.
Previously, whether the elevated plasma triglyceride (TG) levels are causally associated with ASCVD has been controversial. Genetic and epidemiological studies have demonstrated that TG and TG-rich lipoprotein (TGRL) are main causes of residual ASCVD (Do et al., 2013; Jørgensen et al., 2013; Thomsen et al., 2014; Nordestgaard, 2016; Generoso et al., 2019; Laufs et al., 2019; Matsunage et al., 2020; Farnier et al., 2021; Nordestgaard et al., 2021). For instance, approximately 26% adults in United State, including one-third of statin users, have a TG ≥ 150 mg/dl, and approximately 40% adults with diabetes have a TG ≥ 150 mg/dl despite statin use. These elevated TGs are associated with CVD risk even in patients with low LDL-C levels (Hoogeveen and Ballantyne, 2020; Toth et al., 2020). In the genome-wide association studies, the susceptibility sites for CHD are associated with genes involved in TG metabolism (Teslovich et al., 2010; Schunkert et al., 2011). Mendelian randomization studies also indicate that there is a causal relationship between TG metabolism and the risk of atherosclerosis (Johansen and Hegele, 2013; Jørgensen et al., 2013; Varbo et al., 2013; Thomsen et al., 2014; Si et al., 2021). The atherogenic effects of TG, TGRL, and TGRL metabolites are dependent on their roles in endothelial function, inflammation, oxidative stress, and formation of foam cells. In this review, we will make a short review of TG and TGRL metabolism, discuss the association between TG and ASCVD, summarize the atherogenic factors of TGRL, and outline the current advances in TG-lowering therapies and the targets with potential applications in TG modulation.
A Short Review of TG and TGRL
TG is the major storage form of fatty acid (FA) within cells and in circulation (Alves-Bezerra and Cohen, 2017; Duran and Pradhan, 2021). Liver is the central organ for metabolism of FAs that are originated from the plasma and/or hepatocellular de novo biosynthesis. FA synthesis is precisely controlled by a series of enzymes including sterol regulatory element binding protein (SREBP) 1c. When glucose is abundant, plasma insulin activates the endoplasmic reticulum (ER) membrane-bound transcription factor SREBP-1c, which can upregulate genes related to FA biosynthesis (Alves-Bezerra and Cohen, 2017). Within hepatocytes, FA is esterified to glycerol-3-phosphate (G3P) to generate TG. It is estimated that more than 90% of the total TG is synthesized by the G3P pathway in most mammalians (Alves-Bezerra and Cohen, 2017; Lee and Ridgway, 2020). The acylation of G3P is a rate-limiting step because G3P acyltransferase (GPAT) family members have the lowest specific activity within the enzymes involved in TG synthesis. GPAT1 is highly expressed in the liver, and deficiency of GPAT1 can reduce the plasma level of TG and secretion rate of VLDL (Hammond et al., 2005; Neschen et al., 2005). TG synthesis pathways and its metabolism in liver have been well-documented in the literature (Alves-Bezerra and Cohen, 2017; Lee and Ridgway, 2020; Castillo-Núñez et al., 2022). The assembly of TG is the primary way for the liver to store and export FA. However, only a small amount of FA is stored in the form of TG as lipid droplet because most of the FAs are either oxidized in the mitochondrion or packaged in the core of very low-density lipoprotein (VLDL) as TG and secreted into the blood.
As TG is a kind of nonpolar and hydrophobic molecule, it must be combined with related proteins and lipids to form lipoprotein particles during transportation in blood (Do et al., 2013; Castillo-Núñez et al., 2022). In the liver, a large amount of TGs are assembled with cholesterol, phospholipids, and apolipoprotein (apo) B100 (apoB100) into VLDL. In the small intestine, dietary TG is decomposed into FA and monoglyceride or diglyceride before being absorbed by enterocytes. These decomposition products are reassembled into CM with cholesterol, phospholipids, and apoB48 (Julve et al., 2016; Santos-Baez and Ginsberg, 2020). Next, CMs are released into the lymphatic system and enter the circulation, where they obtain other apolipoproteins including apoCII, apoCIII, and apoE (Rosenson et al., 2014; Nakajima and Tanaka, 2018a). Of importance, microsomal triglyceride transfer protein (MTP) plays a key role in the assembly of VLDL and CM via transporting the related lipids to apoB particles (Iqbal et al., 2020). Furthermore, recent studies have demonstrated that CETP increases the production of VLDL-TG in response to estrogen treatment via enhancing the expression of nuclear receptor and small heterodimer partner in female CETP transgenic mice (Palmisano et al., 2016; Palmisano et al., 2021). Therefore, some endogenous molecules, such as estrogen, mediate sex-specific modulation of TG metabolism. The secreted VLDL and CM particles transport FAs to muscle and adipose tissue for energy usage and/or storage via the blood flow (Duran and Pradhan, 2021; Castillo-Núñez et al., 2022).
In circulation, lipoprotein lipase (LPL) located at the surface of capillary lumen hydrolyzes TGs that are encapsulated in the core of CM and VLDL into FAs. LPL binds to its endothelial coenzyme, glycosylphosphatidylinositol-anchored HDL binding protein 1 (GPIHBP1), to provide a platform for lipolysis of apoB-containing lipoproteins on the surface of vascular endothelium (Davies et al., 2010; Young et al., 2019; Basu and Goldberg, 2020). Of note, the activity of LPL is highly regulated by several proteins, such as apoCII, apoCIII, apoE, and angiopoietin-like protein (ANGPTL)3, ANGPTL4, and ANGPTL8 (Rosenson et al., 2014; Wu et al., 2021). Along with TG hydrolysis, CM gradually turns into smaller and cholesterol-rich CM remnant (CMR), and VLDL becomes intermediate density lipoprotein (IDL), which is further catabolized to be LDL via hepatic TG lipase (HTGL) on the surface of hepatic sinusoidal endothelial cell cavity (Young and Zechner, 2013). TGRL remnants in circulation are cleared by liver receptors including LDL receptor (LDLR), LDLR-related protein (LRP)-1, scavenger receptor B type 1 (SR-B1), and heparan sulfate proteoglycan (HSPG) (Toth, 2016; Kockx and Kritharides, 2018; Christopoulou et al., 2019). For instance, small particles, such as LDL, are cleared through the binding of apoE and/or apoB to LDLR, while larger particles, such as CMR, are eliminated by the binding of apoE to HSPG and other potentially undefined hepatic receptors (Kockx and Kritharides, 2018; Christopoulou et al., 2019). The metabolism of TG and TGRL is summarized in Figure 1.
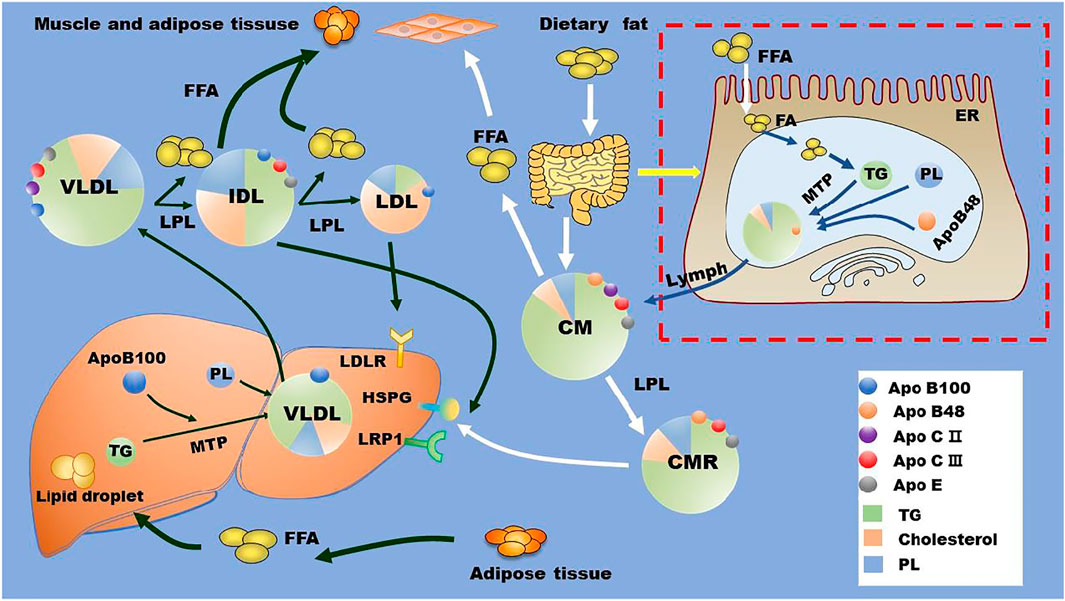
FIGURE 1. TG and TGRL metabolism. Dietary fat is metabolized by intestinal cells into FAs, which are reassembled with cholesterol, phospholipids, and apoB48 to form CMs. These CMs are released into blood via lymph. In the liver, exogenous and de novo synthesized FAs are assembled with cholesterol, phospholipids, and apoB100 to form VLDL with the assistant of MTP. In the circulation, LPL located at the surface of the capillary lumen hydrolyzes the TG in the core of TGRL (CM and VLDL) and promotes the production of TGRL remnants and free FAs. TGRL remnants are cleared by liver through receptors including HSPG, LDLR, LRP1, and other potentially unidentified receptors. Apo, apolipoprotein; CM, chylomicron; CMR, chylomicron residual; ER, endoplasmic reticulum; FA, fatty acid; FFA, free fatty acid; HSPG, heparan sulfate proteoglycan; IDL, intermediate density lipoprotein; LDL, low-density lipoprotein; LDLR, low-density lipoprotein receptor; LPL, lipoprotein lipase; LRP1, LDLR-related protein 1; MTP, microsomal triglyceride transfer protein; PL, phospholipids; TG, triglycerides; VLDL, very low-density lipoproteins.
Hypertriglyceridemia and Atherosclerosis
Generally, lipid analysis (such as TC and TG levels) is performed using overnight fasted blood samples. However, recent studies have demonstrated that non-fasting and fasting plasma samples show similar lipid profiles, and all these data can be used for prediction of CVD risk (Bansal et al., 2007; Nordestgaard et al., 2007; Jørgensen et al., 2013; Thomsen et al., 2014). On average, non-fasting plasma TG levels are approximately 0.3 mmol/L (27 mg/dl) higher than the corresponding fasting samples (Catapano et al., 2016; Mach et al., 2020). TG levels reach peak at 4–6 h after food intake. Therefore, people are in a state of non-fasting at most of the times within a day, and non-fasting lipid levels are more representative than those of fasting lipid profiles. Presently, the standard measurement of plasma TG is still performed under fasting conditions. Generally, the fasting TG concentration <1.7 mmol/L (150 mg/dl) is defined as normal, 1.7–11.4 mmol/L (150–1000 mg/dl) is defined as moderate HTG, and >11.4 mmol/L (1000 mg/dl) is defined as severe HTG (Parhofer and Laufs, 2019). Extreme HTG is rare and is defined as fasting TG concentration >20 mmol/L (∼1750 mg/dl) (Parhofer and Laufs, 2019; Mach et al., 2020). Of note, severe HTG is generally associated with pancreatitis (Laufs et al., 2019; Parhofer and Laufs, 2019). Although the threshold TG level of 1.7 mmol/L is accepted by all medical societies, moderate and severe HTG are differently defined by distinct medical societies. Such as, severe HTG is also defined as TG level >10 mmol/L (850 mg/dl) (Laufs et al., 2019).
Genetic studies indicate that HTG can be caused by both single gene and multiple gene variants (Dron and Hegele, 2020; Matsunage et al., 2020; Ginsberg et al., 2021; Tokgözoğlu and Libby, 2022). For instance, homozygous or biallelic variants in LPL, apoCII, apoCIII, apoAV, lipase maturation factor 1, GPIHBP1, and ANGPTLs are demonstrated to be correlated with HTG (Jørgensen et al., 2013; Rosenson et al., 2014; Dron and Hegele, 2020; Gill et al., 2021). TGRL is consisted of a TG, cholesterol ester, and cholesterol core that is surrounded by phospholipids and apolipoproteins. These apolipoproteins play important roles in CM assembly and degradation. Carriers of the rare non-synonymous mutation of apoAV have higher levels of plasma TG compared with those of non-carriers (Do et al., 2015). An E40K loss-of-function variant in the gene encoding ANGPTL4 is associated with substantially reduced plasma levels of TG in white persons (Folsom et al., 2008). Furthermore, GPIHBP1 deficiency develops severe plasma CM in mouse even on a low-fat diet (Beigneux et al., 2007). In apoA-IV knockout mice, larger CM particles are formed and the clearance of these larger CMs is significantly delayed in circulation compared with those of wild-type mice (Kohan et al., 2012). Mechanistically, apoA-IV may influence particle assembly and/or lipidation in the ER, thereby modulating CM size and secretion (Black, 2007). Furthermore, obese adolescents show higher levels of ANGPTL3 and apoCIII, which potentially inhibit LPL activity, leading to increased TGRL levels and residual atherosclerosis risk (Rodríguez-Mortera et al., 2020).
HTG is reported to affect 15–20% of the adult population and is associated with overweight, metabolic syndrome, and diabetes mellitus (Parhofer and Laufs, 2019). Of note, approximately 50% of patients with type 2 diabetes are accompanied with HTG (Parhofer and Laufs, 2019). Patients with mild to moderate HTG have a higher risk of atherosclerosis than people with normal TG (Crea, 2021; Nordestgaard et al., 2021; Tokgözoğlu and Libby, 2022). One study indicates that TG ≥ 150 and TG ≥ 200–499 mg/dl may enhance CVD risk by 25.0% and 34.9%, respectively (Toth et al., 2021). In patients with severe HTG, individuals with CMRs that are rich in TG also have an increased risk of atherosclerosis (Dron and Hegele, 2020). Accumulating epidemiological studies have indicated that plasma level of TG (both fasting and non-fasting) has a strong correlation with atherosclerosis, and elevated TG levels are an independent risk factor for ASCVD (Nordestgaard et al., 2007; Pirillo et al., 2014; Werner et al., 2014; Kockx and Kritharides, 2018; Shahid et al., 2018; Laufs et al., 2019). Some research support that non-fasting TG levels are more closely associated with incident CVD events than fasting TG levels (Bansal et al., 2007; Adiels et al., 2012). Of importance, each reduction of 88.5 mg/dl of TG level is associated with approximately 50% reduction in CVD risk (Jun et al., 2010). Therefore, lowering TG treatment can reduce the risk of ASCVD as that of lowering LDL-C (Ference et al., 2019). In 2019, European Society of Cardiology and European Atherosclerosis Society has clearly pointed out that TG ≥ 175 mg/dl is a risk factor of ASCVD events and TG-lowering therapy is recommended for residual ASCVD therapy (Mach et al., 2020).
TGRL and Atherosclerosis
TG is the main component of TGRL (CM and VLDL), and their remnants CMR and IDL (Duran and Pradhan, 2021; Castillo-Núñez et al., 2022). Therefore, plasma TG concentration is a biomarker for TGRL and their remnants in circulation. The methods used for isolation and quantification of TGRL remnants have been recently reviewed by distinct groups (Hoogeveen and Ballantyne, 2020; Duran and Pradhan, 2021). After meals, TGs are transported from the small intestine to bloodstream by CM particles, where they are converted to atherogenic CMRs by LPL in tissues (Ginsberg et al., 2021). Similarly, liver secreted VLDL particles are converted to IDL and then LDL by LPL and HTGL in circulation. TGRL and the hydrolyzed residuals including free FAs bind to leukocytes and endothelial cells in circulation, leading to a state of acute activation that is characterized by expression of integrins, generation of ROS, production of cytokines as well as a complement activation (DeVries et al., 2014). Recent studies have demonstrated that TGRL and their remnants are positively associated with atherosclerosis by up-regulating inflammation, oxidative stress, and foam cell formation as shown in Figure 2.
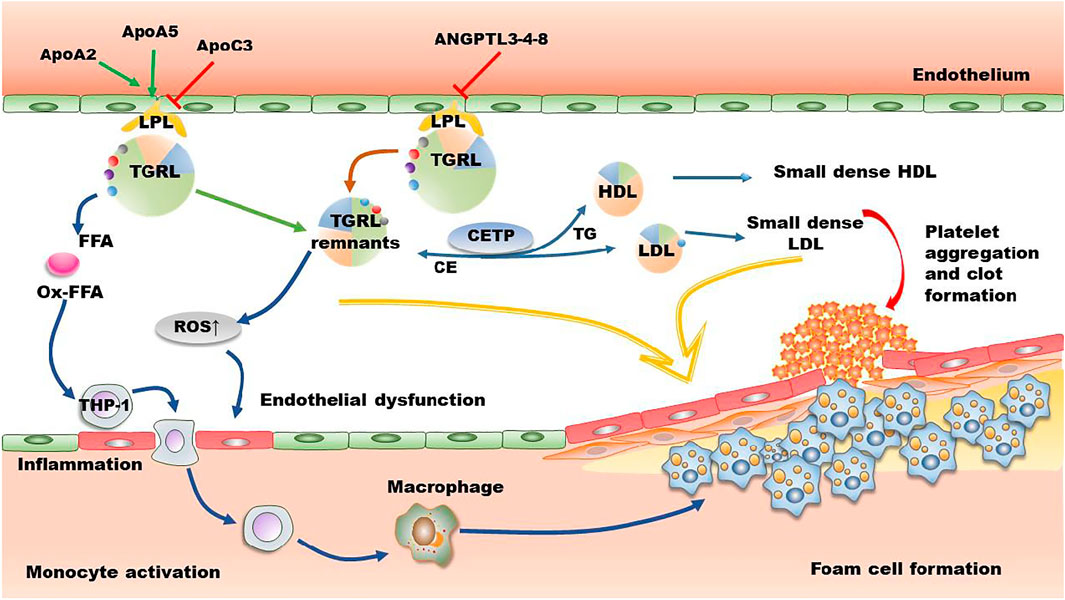
FIGURE 2. The mechanisms of TGRL on promoting atherosclerosis. In circulation, apoAII and apoAV enhance the activity of LPL, while apoAIII and ANGPTL3-4-8 suppress LPL-induced TGRL lipolysis. However, several studies demonstrate that apoAIII has no effect on the activity of LPL. The released FFA and the lipolysis process promote the production of ROS and secretion of inflammatory factors, leading to endothelial dysfunction and formation of foam cells. CETP-mediated lipid exchange between TGRL and HDL/LDL increases the production of small dense LDL and small dense HDL, further promoting deterioration of atherosclerosis. Furthermore, lipolytic products activate platelet and induce clot formation. ANGPTL, angiopoietin-like protein; Apo, apolipoprotein; CE, cholesteryl ester; CETP, cholesteryl ester transport protein; FFA, free fatty acid; HDL, high density lipoprotein; LDL, low-density lipoprotein; LPL, lipoprotein lipase; Ox-FFA, oxidized FFA; ROS, reactive oxygen species; TG, triglycerides; TGRL, triglyceride-rich lipoprotein.
TGRLs Activate Inflammation
Atherosclerosis is characterized as a chronic inflammatory disease. Of note, each mmol/L (39 mg/dl) increase of TGRL cholesterol is associated with a 37% increase of C-reactive protein level, suggesting TGRL cholesterol increases inflammatory response (Varbo et al., 2013). Furthermore, plasma levels of interleukin (IL)-6 and tumor necrosis factor-alpha (TNFα) are significantly higher in postprandial subjects than those in fasting state, suggesting that elevated levels of postprandial TGRL cholesterol are associated with inflammatory response, causing increased susceptibility for premature atherosclerosis (Twickler et al., 2003). TGRLs with high TG content up-regulate the level of TNF-α, thereby inducing the expression of vascular cell adhesion molecule (VCAM)-1 in human aortic endothelial cells and monocyte adhesion. On the contrary, TGRLs with low TG content have an atheroprotective effect by reducing VCAM-1 expression and monocyte recruitment (Gower et al., 2011; Wang et al., 2011; Sun et al., 2012). Postprandially released VLDL particles have an increased level of apoCIII (Wang et al., 2011; Sun et al., 2012), and these particles activate inflammation in endothelial cells by enhancing the protein kinase C (PKC)/NF-κB signaling pathway (Libby, 2007). Similarly, VLDL particles promote inflammation by activating the NF-κB signaling pathway in endothelial cells (Dichtl et al., 1999; den Hartigh et al., 2014). However, ApoCIII A allele at rs2070667 shows an inhibitory effect on polyunsaturated fatty acids (PUFA)-containing TGs and hepatic inflammation in nonalcoholic fatty liver disease (Xu et al., 2020).
Macrophage is a central link between lipid metabolism and inflammatory response. TG synthesis (lipid droplet formation) enhances macrophage inflammation (Castoldi et al., 2020). In vitro, VLDL enhances the expression of TNF-α, IL-1β, monocyte chemoattractant protein 1 (MCP-1), intercellular adhesion molecule-1 (ICAM-1), matrix metalloproteinase 3, and macrophage inflammatory protein 1-α. Mechanistically, VLDL activates mitogen-activated protein kinase (MAPK) signaling cascades including the phosphorylation of extracellular signal-regulated kinase (ERK) 1/2, c-Jun NH2-terminal kinase (JNK), and p38 MAPK (Jinno et al., 2011). VLDL particles further enhance the expression of TNF-α in macrophages that are induced by LPS via activating ERK1/2, MAPK kinase (MEK)1/2, and the transcription factor AP-1 rather than nuclear factor-κB (NF-κB) or peroxisome proliferator activated receptor (PPAR) γ (Stollenwerk et al., 2004). TGRL also induces inflammation by activating the inflammasome nucleotide binding domain like receptor family pyrrole domain containing protein 1 (NLRP1) (Bleda et al., 2016; Bleda et al., 2017). Furthermore, VLDL intensifies its pro-inflammatory effects by binding to LRP and activating the downstream p38 MAPK/NF-κB signaling pathway (Libby, 2007). Of note, ER stress and the unfolded protein response are also involved in TGRL-induced inflammation (Ozcan et al., 2004; Civelek et al., 2009; Wang et al., 2013). In addition, CMRs stimulate the expression of IL-1β via activating caspase-1 and NF-κB in THP-1 cells (Okumura et al., 2006). CMRs also activate human monocytes and enhance their migration in vitro, contributing to an inflammatory environment in the early stage of atherosclerosis (Bentley et al., 2011). The inflammation-associated hormone, growth and differentiation factor 15, is also involved in TGRL-mediated inflammation (Luan et al., 2019).
Accumulating evidence have demonstrated that TGRLs and their remnants increase endothelial inflammation and facilitate monocytes infiltration of the arterial wall. A previous study demonstrated that TGRL induces monocyte adhesion to vascular endothelial cells by sequentially activating the expression of PKC, RhoA, focal adhesion kinase, and integrins in vitro, suggesting a mechanism of TGRL remnants-mediated vascular inflammation during atherogenesis (Kawakami et al., 2002). TGRL remnants also induce the expression of TNF-α, VCAM-1, ICAM-1, E-selectin, and MCP-1 through modulation of lectin-like receptor for oxidized LDL (LOX-1) receptor and NF-κB-dependent nuclear transcription (Park et al., 2005). Furthermore, JNK and activating transcription factor 3 (ATF3) are involved in TGRL lipolysis products-induced vascular inflammation via upregulating the levels of IL-8 and E-selectin (Aung et al., 2013). Postprandial TGRL also stimulate inflammation via interferon regulatory factor-1, especially under shear stress in cultured endothelium (Sherrod DeVerse, et al., 2013). Oxylipids in TGRLs are found to promote endothelial inflammation following a high fat meal (Rajamani et al., 2019). Of importance, TGRL hydrolysis and the accumulation of intracellular TGs and free FAs, especially oxidized free FAs, play key roles in TGRL-mediated inflammation. Reductions in susceptibility of VLDL to LPL can attenuate the above inflammatory reactions (Saraswathi and Hasty, 2006; Jinno et al., 2011; Castillo-Núñez et al., 2022). FAs are transported into cells passively or actively by transporters including plasma membrane fatty acid-binding protein, fatty acid transport proteins, and cluster of differentiation 36 (Mallick and Duttaroy, 2022). Neutral and oxidized free FAs released during TGRL hydrolysis are found to induce endothelial inflammation and vascular apoptosis (Wang et al., 2009). The relationship between free FAs and inflammation has been reviewed recently by distinct groups (Mallick and Duttaroy, 2022; Panda et al., 2022; Ren et al., 2022). Some presently known inflammatory signaling pathways that are modulated by TGRL, TGRL remnants, and free FAs are summarized in Figure 3.
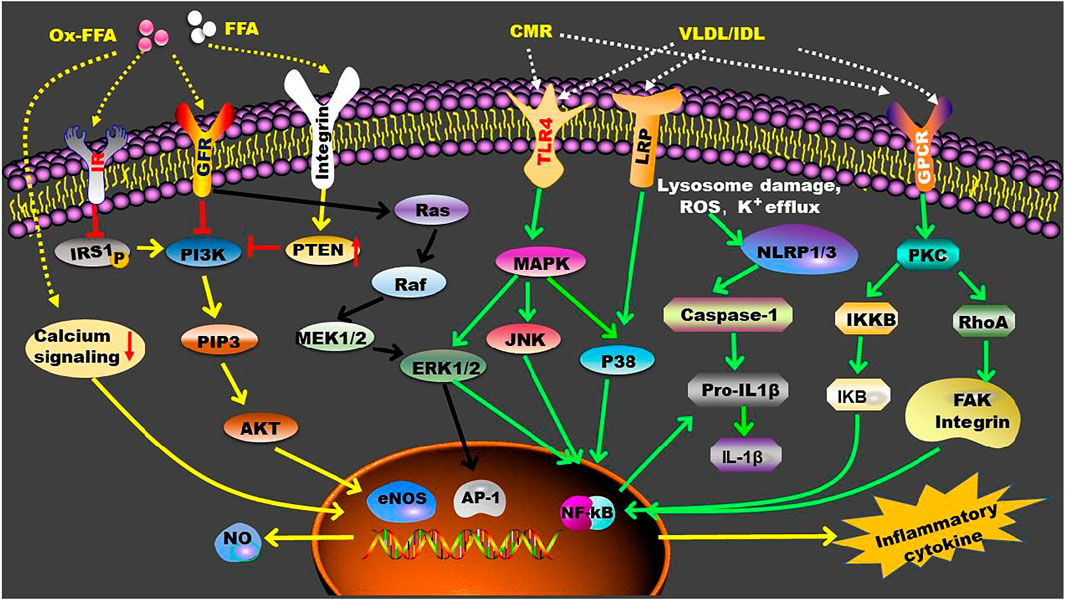
FIGURE 3. TGRL and TGRL metabolites-mediated inflammatory signaling pathways. In circulation, LPL converts TGRL to TGRL remnants, such as CMR and IDL, and promotes the production of FFA. These TGRL metabolites enhance inflammation by activating multiple receptors that located on the cell membrane. Furthermore, FFA can penetrate cell membrane and exert their functions intracellularly. AP-1, activating protein-1; eNOS, endothelial nitric oxide synthase; ERK1/2, extracellular signal-regulated kinase 1/2; FAK, focal adhesion kinase; FFA, free fatty acid; GFR, growth factor receptor; GPCR, G protein-coupled receptor; IKB, nuclear factor-kappa B inhibitor; IKKB, Inhibitor of kappa B kinase; IL-1β, interleukin-1β; IR, insulin receptor; IRS1, insulin receptor substrate 1; JNK, c-Jun NH2-terminal kinase; LRP, LDL receptor-related protein; MAPK, mitogen-activated protein kinase; MEK1/2, mitogen-activated protein kinase kinases 1/2; NF-kB, nuclear factor-kappa B; NLRP, nucleotide binding domain like receptor family pyrrole domain containing protein; NO, Nitric oxide; P38, P38 mitogen-activated protein kinase; PI3K, phosphoinositide 3-kinase; PIP3, phosphatidylinositol 3,4,5-trisphosphate; PKC, protein kinase C; PTEN, phosphatase and tensin homolog; RhoA, Ras homolog family member A; TRL-4, Toll-like receptor 4.
TGRLs Induce Oxidative Stress and Endothelial Cell Dysfunction
Oxidative stress plays an important role in the progression of atherosclerosis by interacting with inflammation, foam cell formation, and endothelial dysfunction. TGRL remnant cholesterol stimulates NAD(P)H oxidase–dependent superoxide formation and cytokine secretion in human umbilical vein endothelial cells by activation of LOX-1, leading to reduction in cell viability (Shin et al., 2004). Furthermore, TGRL remnant cholesterol upregulates endothelial expression of ICAM-1, VCAM-1, and tissue factor through a redox-sensitive mechanism, thus affecting the progression of atherosclerosis (Doi et al., 2000). CMRs also cause rapid and prolonged generation of ROS in monocytes, thereby influencing monocyte activation and migration (Bentley et al., 2011). Exposure of J774 macrophages to a pro-oxidizing state further promotes CMR-induced accumulation of intracellular lipids (Napolitano et al., 2001).
Endothelial dysfunction is one of the early pathological mechanisms of atherosclerosis. Free FAs released during lipolysis of TGRL can activate NADPH oxidase- and cytochrome P450-mediated ROS production within endothelial cells, causing oxidative stress and dysfunction of endothelial barrier (Wang et al., 2009). The mechanisms of action of FAs on modulation of endothelium function have been reviewed recently by Mallick and Duttaroy (2022). Furthermore, high levels of TGRL remnant cholesterol and TG have a strong correlation with endothelial vasomotor dysfunction. These TGRL remnants increase the susceptibility of coronary endothelium to oxidative stress, leading to inhibition of nitric oxide-mediated vascular dilation (Nakamura et al., 2005). Of note, postprandial elevated TGRL remnant cholesterol is associated with atherosclerotic progression even in normolipidemic subjects by affecting endothelial dysfunction, such as endothelium-dependent vasorelaxation (Inoue et al., 1998; Funada et al., 2002). TGRL lipolysis induces ROS production and alterations of lipid raft in morphology and protein components, such as LRP, nitric oxide synthase, and caveolin-1, leading to endothelial dysfunction (Wang et al., 2008). Furthermore, TGRL and their lipolytic products activate platelet aggregation and clot formation by suppressing fibrinolysis and promoting the assembly of prothombinase complex, the expression of plasminogen activator inhibitor-1 and its antigen, and the endothelial expression of tissue factor (Gianturco and Bradley, 1999; Toth, 2016; Reiner, 2017). Furthermore, TGs are found to increase the risk of atherosclerosis by increasing plasma viscosity, leading to impaired microcirculatory flow and enhanced interactions between atherogenic lipoproteins and endothelium (Rosenson et al., 2001).
TGRLs Promote Foam Cell Formation
Lipid accumulation in the subcutaneous space of endothelium is a key characteristic of atherosclerosis (Lin et al., 2021). Reductions in plasma TG levels are associated with reduced all-cause mortality in CVD patients potentially due to the low cholesterol content in TGRL and TGRL remnant particles. Indeed, TGRL remnant cholesterol is an independent risk factor for menopausal women with CHD (Feng X. et al., 2020). Another study indicates that the cholesterol levels of TGRL are a residual risk of future CVDs in patients with stable CHD and even in those with LDL-C < 70 mg/dl after statin therapy (Fujihara et al., 2019). Of note, the elevated postprandial VLDL residues are the main cause of the occurrence and development of atherosclerosis (Nakajima and Tanaka, 2018b). Furthermore, the reduced TGRL removal efficacy maybe a causal factor of the increased atherosclerosis in elderly people (Maranhão et al., 2020).
TGRLs carry approximately 25% of the TC carried by non-HDL particles. Due to the big size, the cholesterol content carried by each TGRL particle is approximately 5–20 times higher than that carried by each LDL particle (Toth, 2016; Ginsberg et al., 2021). Except for CM particles, TGRL and TGRL remnant particles, can penetrate blood vessel and be recognized and directly ingested by macrophages through the ligand apoE located at the surface of TGRL (Ohmura, 2019; Ginsberg et al., 2021). As is known, only modified LDL (such as oxidation) particles are ingested by macrophages. TGRLs are the only natural, unmodified lipoproteins that can cause rapid lipid accumulation in macrophages as revealed in vitro (Gianturco and Bradley, 1999; Peng and Wu, 2022). First, TGRLs are trapped in artery walls through the interaction between positively charged residues on apoB with negatively charged groups of proteoglycans located at the endothelium lining (Sandesara et al., 2019). In the subcutaneous space, TGRL particles are internalized by macrophages and smooth muscle cells, contributing to plaque formation and development (Padro et al., 2021). Therefore, TGRL is more pathogenic than LDL in causing atherosclerosis.
Indeed, both apoB48 and apoB100 are found in human aortic atherosclerotic plaques, indicating that TGRLs are involved in the formation of atherosclerotic plaque (Nakano et al., 2008; Behbodikhah et al., 2021). Furthermore, the majority of apoB proteins isolated from human atherosclerotic plaques are derived from VLDL and IDL, but not LDL, suggesting that VLDL and IDL play a key atherosclerotic role in the intima of the arteries (Rapp et al., 1994; Proctor and Mamo, 1998; Castillo-Núñez et al., 2022). Furthermore, a proportion of arterial plaque cholesterol is derived from VLDL and its residue IDL in patients with mild-to-moderate HTG (Gill et al., 2021). Of note, plasma VLDL cholesterol (VLDL-C) is an increased risk factor for major adverse CVD events, independent of the established risk factors such as LDL-C (Heidemann et al., 2021). It seems that the remnant cholesterol, but not TG, in TGRL particles is a causal factor of atherosclerosis (Jørgensen et al., 2013; Varbo and Nordestgaard, 2016; Dron and Hegele, 2020). However, TGs in TGRL particles assist the ingestion process of the cells, such as macrophages, involved in plaque foam cell formation. Unlike VLDL particles isolated from people with normal TG levels, VLDL particles obtained from patients with HTG have a high affinity for scavenger receptors, such as LDLR and VLDLR, that are specifically expressed by monocytes, macrophages, and endothelial cells (Gianturco et al., 1982; Takahashi, 2017).
CMRs also contribute to lipid accumulation in the atherosclerotic plaques. Unlike LDL particles, CMRs carrying dietary lipids induce the formation of foam cells without the oxidation process in circulation, and the uptake of CMRs by macrophages can induce intracellular accumulation of both TG and cholesterol. The rate of uptake and lipid accumulation is affected by the type of dietary fat in the granules (Botham et al., 2007; Morita, 2016). In the absence of LDLR or its ligand apoE, CMRs still contribute to lipid accumulation during atherosclerotic plaque formation (Fujioka et al., 1998; Bravo and Napolitano, 2007). For instance, apoB48 receptor is involved in CMR uptake and foam cell formation independent of the apoE-mediated pathway. Of note, apoB48 receptor can be used for further uptake of TGRL particles even when macrophages are accumulated with a large amount of lipids (Kawakami et al., 2005; Bermudez et al., 2012). In macrophages, CMR internalization results in rapid accumulation of cholesterol in lysosomes and cell death due to lysosomal instability, thereby promoting atherosclerosis deterioration (Wakita et al., 2015). Additionally, TG contained in TGRL particles can activate CETP, which promotes the exchange of core lipids between major lipoproteins, promoting the accumulation of residual cholesterol in TGRL remnants, thereby aggravating atherosclerosis (Brinton, 2015). For instance, CETP mediates the exchange of TG in TGRL with cholesterol in LDL, thereby promoting the production of small, dense LDL particles that are more atherosclerotic than LDL particles (Gianturco et al., 1982; Merkel, 2009). Similarly, TGRL promotes HDL remodeling and the formation of smaller, low-cholesterol HDL particles that are lack of atherosclerotic protection (Feng M. et al., 2020).
Strategies for Treatment of Hypertriglyceridemia
Except for statins, clinical drugs that can be used to treat HTG are fibrates, niacin, and omega-3 fatty acids (Preston Mason, 2019). Several previous reviews have summarized the effects of these clinically used drugs for TG-lowering (Sando and Knight, 2015; Simons, 2018; Feingold, 2021). Furthermore, our team demonstrated that exogenous supplement of N-acetylneuraminic acid can reduce TG by more than 60% in apoE(−/−) mice (Guo et al., 2016; Hou et al., 2019). Here, we make a short review about these clinically used drugs, and then focus on some targets with potential applications for TG-lowering.
Clinical Drugs
Statins
Statins are the standard therapy for many types of dyslipidemias because they can effectively decrease the endogenous biosynthesis of cholesterol via inhibiting 3-hydroxy-3-methyl glutaryl coenzyme A reductase and enhance the hepatic uptake of LDL particles by up-regulating LDLR through SREBP-2. In individuals with normal TG levels, statins have little effects on plasma VLDL. However, statins decrease VLDL and CM and their remnants via improving hepatic clearance in patients with HTG (Caslake and Packard, 2004). A previous study demonstrates that statin treatment decreases not only fasting TG but also postprandial TGs (Mora-Rodriguez et al., 2020). However, a survey in United State adults (9593 participants) indicates that the prevalence of TG < 150, 150–199, and ≥200 mg/dl is 75.3%, 12.8%, and 11.9% in adults without statin treatment; among statin users, the ratios are 68.4%, 16.2%, and 15.4%, respectively. Furthermore, the estimated mean 10-years ASCVD risk from TG < 150 to ≥500 mg/dl, ranges from 11.3% to 19.1% in statin users and 6.0%–15.6% in nonusers. These data suggest that statin treatment moderately elevates TG levels as well as ASCVD risk in patients with TG > 150 mg/dl (Fan et al., 2019). Another meta-analysis of randomized-controlled trials demonstrates that statins only reduce plasma TG by 8.4% in children and adolescents with familial hypercholesterolaemia (Anagnostis et al., 2020). Patients with increased CVD risk generally display high levels of plasma TGs and low levels of HDL-C, even after statin therapy (Larsson et al., 2014). Of importance, statin-treated patients with TG levels ≥ 150 mg/dl have even worse CVD risk than those with TG < 150 mg/dl (Toth et al., 2019a; Toth et al., 2019b). Collectively, statins show limited effects on TG-lowering in patients with dyslipidemia.
Fibrates
Mechanistically, fibrates exert their bioactivity primarily by activating PPARα (Chapman et al., 2010). Fibrate treatment reduces plasma TG levels by approximately 20%–70% (Katsiki et al., 2013; Wang et al., 2015). Compared with placebo, gemfibrozil (1.2 g/d) reduces TG level by 31% and increases HDL-C by 6% along with a reduction in the risk of CVD events by 4.4% in CHD patients (Rubins et al., 1999). Oral bezafibrate treatment (400 mg/d, twice) leads to a significant reduction in serum TG level (42.7%) as well as increases in HDL2-C, HDL3-C, and plasma content and activity of LPL. Bezafibrate may increase HDL3-C and HDL2-C by promoting TGRL catabolism and the conversion of HDL3 to HDL2, respectively (Sakuma et al., 2003). Fenofibrate or fenofibric acid can significantly reduce TG level in combination with statin (Huang et al., 2009; Ouwens et al., 2015). A meta-analysis demonstrates that fibrate reduces TG level by 46.5% in combination with statin. However, this combination increases risk of side effects (Choi et al., 2014). Pemafibrate (k-877), a selective PPARα modulator, is found to be superior to fenofibrate (106.6 mg/d) in TG lowering in patients with high TG (≥1.7 mmol/L and <5.7 mmol/L) and relatively low HDL-C levels at the dosage of 0.2–0.4 mg/d (Ishibashi et al., 2018). However, a meta-analysis suggests that pemafibrate reduces plasma TG levels and increases HDL-C similar as that of fenofibrate (Ida et al., 2019). In a multicenter, randomized, double-blind, phase IV study, fenofibrate significantly decreases TG levels from 269.8 to 145.5 mg/dl as an add-on-statin therapy, while statin monotherapy has no effect on TG levels (Park et al., 2021). A recent retrospective longitudinal study indicates that pemafibrate significantly reduces plasma TG by 43.8% and increases HDL-C by 10.8% in patients with dyslipidemia after 3 months treatment. Furthermore, this molecule improves liver function and serum levels of uric acid and hemoglobin A1c (Yanai et al., 2022).
Niacin
Niacin (nicotinic acid) decreases TG levels by up to 30%, and reduces LDL-C and lipoprotein (α)) levels to the greatest extent by 15% and 30%, respectively (Chapman et al., 2010). It inhibits the lipolysis of adipose tissue and reduces the flow of free FAs to the liver, leading to a reduction in hepatic synthesis of VLDL (Khetarpal et al., 2016). This molecule can successfully reduce TGs in patients with familial CM syndrome potentially by reducing the production of apoB48 and CM (Pang et al., 2016). Furthermore, niacin mimics the role of the physiological ligand β-hydroxybutyrate by interacting with the type 3 hydroxycarboxylic acid receptor (Chaudhry et al., 2018). In a clinical follow-up study, niacin treatment decreases TG level from 164 mg/dl to 122 mg/dl, reduces LDL-C from 74 mg/dl to 62 mg/dl, and increases HDL-C level from 35 mg/dl to 42 mg/dl. However, there is no incremental clinical benefit from the addition of niacin (1.5–2 g/d) to statin treatment (40–80 mg/d) during the 3 years of follow-up period in patients with ASCVD (Boden et al., 2011). A large randomized trial demonstrates that although the extended-release niacin treatment reduces TG levels by 33 mg/dl on average compared with placebo, the addition of niacin to statin therapy has no effect on reducing the risk of major vascular events. On the contrary, niacin is found to increase the risk of myopathy, especially in patient after simvastatin treatment (HPS2-THRIVE Collaborative Group, 2013). Therefore, the use of niacin in clinical practice is limited due to its useless for reducing CVD events and the high incidence of adverse reactions.
Omega-3 Fatty Acid
The beneficial roles and metabolism of PUFA have been recently review by several groups (Chen et al., 2022; Mallick and Duttaroy, 2022; Ren et al., 2022). Omega 3 FAs belong to the family of PUFA and play an important role in the formation and stability of cell membranes as well as cell physiology (Ganda et al., 2018). Omega-3 FAs, such as eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), can reduce TG levels, improve blood vessel function, and suppress inflammation, platelet aggregation, liver steatosis, and insulin resistance (Mozaffarian and Wu, 2011; Shahidi and Ambigaipalan, 2018). A previous review indicates that DHA and EPA treatment is associated with a net decrease in TG by 22.4% and 15.6%, respectively. Furthermore, DHA supplement is associated with more significant increases in LDL-C and HDL-C compared to that of EPA (Jacobson et al., 2012; Kotwal et al., 2012). These Omega 3 FAs affect lipid raft organization by disrupting acyl chain packing and molecular order within lipid rafts, thereby modulating protein lateral distribution and signaling (Shaikh, 2012). Of note, EPA and DHA have different effects on membrane bilayer width, membrane fluidity, and cholesterol crystalline domain formation, suggesting omegar-3 FAs with different structural characteristics may show distinct effects (Preston Mason et al., 2016). The TG-lowering mechanisms of action of these compounds are associated with increased FA degradation through peroxisome β-oxidation, reduced hepatic fat production, and enhanced TG clearance in circulation (Harris et al., 2008).
In a 12-weeks clinical trial, the EPA ethyl ester, AMR101, reduces TG level by 10.1% and 21.5% at the dosage of 2 and 4 g/d, respectively, in high-risk statin-treated patients with residually high TG (>200 and <500 mg/dl) (Ballantyne et al., 2012). In patients with fasting TG levels of 1.52–5.63 mmol/L and LDL-C levels of 1.06–2.59 mmol/L, 2 g/d of EPA ethyl (twice daily) significantly reduces the relative risk of ischemic events by 25% compared with the placebo group (Bhatt et al., 2019). Of note, EPA (1.8 g/d) combined with pitavastatin (4 mg/d) is found to reduce coronary plaque volume and reinforce plaque stabilization compared to pitavastatin monotherapy (Watanabe et al., 2017). Another study in Japan demonstrates that EPA (1.8 g/d) reduces CVD events by 19% in patients receiving statin therapy and decreases CVD events by 53% in patients with TG ≥ 150 mg/dl and HDL-C < 40 mg/dl, suggesting EPA is more effective in patient with abnormal TG and HDL-C levels (Saito et al., 2008). Icosapent ethyl is recommended for treatment of ASCVD patients with fasting TG between 139 and 499 mg/dl in combination with statin therapy (Orringer et al., 2019). However, omega-3 FA supplementation has no protective effects in patients with diabetes without evidence of CVD (Bowman et al., 2018). A meta-analysis involving 77917 high-risk individuals suggests that omegar-3 FAs have no significant association with fatal and nonfatal CHD or any major vascular events (Aung et al., 2018). Epanova, a mix of omega-3 free FAs, lowers plasma TG level by up to 31%. In 2018, the STRENGTH study was designed to check whether 4 g/day of epanova can reduce the incidence of CVD events in patients with HTG and low levels of HDL-C (Nicholls et al., 2018). This STRENGTH study terminated on 8 January 2020 demonstrates that there is no significant difference between omega-3 FA treatment and corn oil intervention (Nicholls et al., 2020; Nissen et al., 2021; Reyes-Soffer, 2021). Therefore, omega-3 FAs mix may also have limited application in clinical therapy for HTG.
Emerging Targets for Lowering Triglyceride
Mendelian randomization and genetic studies provide evidence of potential therapeutic targets for reducing TG and the risk of ASCVD. These potential targets include LPL and LPL-related proteins, such as apoCIII, apoCII, apoAV, ANGPTL4, and GPIHBP1, which cause alterations in TG levels and are related to the development of ASCVD. For instance, in a systematic review and meta-analysis, apoCIII is found to cause HTG and atherosclerosis (Wyler von Ballmoos et al., 2015; Rocha et al., 2017). The proatherosclerotic effects of GPIHBP1 deficiency are probably caused by the markedly elevated levels of CM/VLDL, which exacerbate atherosclerosis through increasing the formation of TGRL remnants and generation of proatherogenic lipid products (Vallerie and Bornfeldt, 2015). Furthermore, accumulating evidence have demonstrated that gut microbiota is also associated with TG metabolism.
Targeting LPL
LPL maintains TG homeostasis in blood and is the rate-limiting enzyme for the hydrolysis of TGs that are encapsulated in the core of TGRL particles (Wang and Eckel, 2009; Tada et al., 2018; Kumari et al., 2021). It has been reported that gain-of-function and loss-of-function gene mutations of LPL lead to the imbalance of plasma TG levels, thereby influencing CVD events (Rip et al., 2006; Wang and Eckel, 2009; Burnett et al., 2019). LPL S447X is a naturally occurring gain-of-function mutation (Rip et al., 2006). In 2012, alipogene tiparvovec (AAV1-LPLS447X) was approved in Europe for treatment of severe HTG and recurrent pancreatitis in patients with complete loss-of-function of LPL. This highly active recombinant LPL with S447X variant reduces the fasting TG level by >40% in half of the patients between 3 and 12 weeks (Gaudet et al., 2013). Furthermore, several interesting compounds are found to increase the activity of LPL. Among these agonists, 50F10 is found to stabilize LPL in vitro and successfully reduce postprandial HTG in apoAV(−/−) mice (Larsson et al., 2014). The agonist NO-1886 (ibrolipim) mainly increases the mRNA level of LPL, while the compound C10d primarily affects the hydrolysis activity of LPL (Larsson et al., 2014). Although a long-term administration of NO-1886 significantly inhibits the development of coronary atherosclerosis, this compound shows severe side effects. Except for lowering TG, the agonist C10d lowers TC, body fat, and fatty liver, suggesting its potential application in HTG treatment (Geldenhuys et al., 2017).
Targeting ApoCIII
ApoCIII is synthesized in the liver and intestine, and it is distributed in TGRL and HDL particles (Borén et al., 2020). This apolipoprotein is previously demonstrated to be an effective inhibitor of LPL (Christopoulou et al., 2019). However, several studies have shown that apoCIII has pleiotropic effects in regulating the metabolism of TGRL without affecting LPL (Gordts et al., 2016; Kovrov et al., 2022). ApoCIII mainly suppresses the hepatic clearance of TGRLs and their remnants through LDLR and LRP-1, thereby inducing HTG (Gordts et al., 2016; Christopoulou et al., 2019; Kegulian et al., 2019; Reyes-Soffer and Ginsberg, 2019). People with apoCIII loss-of-function mutations are associated with approximately 40% reductions in plasma TG and the risk of CVD (Pollin et al., 2008; Crosby et al., 2014; Jørgensen et al., 2014). Therefore, apoCIII is a therapeutic target for patients with severe HTG. Volanesorsen is an antisense oligonucleotide targeting apoCIII mRNA, and it is developed for treatment of familial CM syndrome (FCS), HTG, and familial partial lipodystrophy (Gouni-Berthold, 2017; Paik and Duggan, 2019). Based on the beneficial effects observed in the phase III study, volanesorsen was approved in the European Union for treatment of adult FCS patients in May 2019 (Hegele et al., 2018; Corbin et al., 2020; Gouni-Berthold et al., 2021; Lazarte and Hegele, 2021). The average TG level in FCS patients decreases by 77% after treatment using volanesorsen (300 mg) once a week for 3 months (Witztum et al., 2019). Subcutaneous injection of volanesorsen (300 mg) once a week for 3 months reduces the average TG level by 71.2% in patients that have applied conventional TG-lowering therapy but have a fasting TG > 500 mg/dl (Gouni-Berthold et al., 2021). However, this compound may cause side effects such as thrombocytopenia (Witztum et al., 2019).
Targeting ApoCII
ApoCII is a key cofactor for activation of LPL. A complete deficiency of apoCII causes the same phenotype, severe HTG, as LPL deficiency (Hegele et al., 2020). The apoCII mimetic peptide (C-II-a, 30 mg/kg) is found to reduce plasma TG level by 85% in apoE(−/−) mice (Amar et al., 2015). Intravenous injection of this short peptide (0.2–5 μmol/L) reverses HTG in apoCII(−/−) mice in a dose-dependent manner (Sakurai et al., 2016). C-II-a peptide is found to promote plasma clearance of TG-rich lipid emulsions and improve the following incorporation of FAs from these TG emulsions into specific peripheral tissues (Komatsu et al., 2019). However, this mimic peptide only acutely activates LPL due to its short half-life (1.33 h) (Sakurai et al., 2016). Recent studies have shown that apoCII mimic peptide D6PV is a novel compound for the treatment of HTG and apoCII deficiency (Wolska et al., 2020a; Wolska et al., 2020b). In apoCII(−/−) mice and human apoCIII-transgenic mice, this peptide consisted of 40-amino acid causes a rapid decrease in plasma TG and apoB by approximately 80% and 65%, respectively. Furthermore, it also works independent of LPL (Wolska et al., 2020a). Of importance, D6PV displays good TG-lowering bioactivity in nonhuman primates and shows an extended terminal half-life of 42–50 h (Wolska et al., 2020a). These data suggest that apoCII mimetic peptides have an attractive future for treatment of HTG.
Targeting ApoAⅤ
Genetic association studies have established a clear link between apoAⅤ variation and TG metabolism (Nilsson et al., 2011; Flores-Viveros et al., 2019). ApoAV variants affect not only TG concentration but also the distribution of lipoprotein subclasses, shifting them to atherogenic particles in high-risk subjects (Guardiola et al., 2015; Guardiola and Ribalta, 2017). A previous study demonstrates that three mutations including p.(Ser232_Leu235)del, p.Leu253Pro, and p.Asp332ValfsX4 are the direct cause of HTG by apoAⅤ (Mendoza-Barberá et al., 2013). Furthermore, there is a significant association between c.56C > G (rs3135506) apoAⅤ gene polymorphism and coronary artery disease in the Moroccan population (Morjane et al., 2020). Liver-derived apoAⅤ facilitates LPL-mediated TG hydrolysis in circulation. Furthermore, apoAⅤ is co-localized with perilipin through binding LRP-1, and it reduces intracellular TG concentration by suppressing adipogenesis-related factors in adipocytes (Su et al., 2020; Xu et al., 2020). ApoAV shows a protective effect against atherosclerosis in apoE2 gene knock-in and human apoAV transgenic mice via reducing TG and the residual particles rich in cholesterol esters, such as LDL and VLDL (Mansouri et al., 2008). Intravenous injection of wild-type apoAV reconstituted HDL significantly reduces TG by 60% in apoAV(−/−) mice, and this effect requires the functional GPIHBP1-LPL-apoAV axis (Shu et al., 2010). Furthermore, adenovirus overexpression of human apoAV reduces serum levels of TG and cholesterol in mice (Van der Vliet et al., 2002). However, apoAV is involved in fructose-induced metabolic dysregulation and is associated with hepatic steatosis. Furthermore, there is a significantly lower levels of hepatic TG in apoAV(−/−) mice compared with the control (Ress et al., 2020). Therefore, the working mechanisms of action of apoAV and the actual application for treatment of HTG by targeting apoAV need to be clarified in future.
Targeting ANGPTL
ANGPTL is a family of secreted glycoproteins consisting of eight members (ANGPTL1-8). These proteins, especially ANGPTL3, ANGPTL4, and ANGPTL8 are found to regulate the activity of LPL (Li et al., 2020; Kumari et al., 2021). In the following, we mainly describe several intensively studied ANGPTL members that have potential applications for treatment of HTG.
ANGPTL3
ANGPTL3 is a secreted protein mostly expressed in the liver. This protein increases the plasma levels of TG and LDL-C. However, loss-of-function variants in ANGPTL3 are associated with decreased plasma levels of TG, LDL-C, and HDL-C, as well as reduced ASCVD risk (Musunuru et al., 2012; Dewey et al., 2017). In mice, lack of ANGPTL3 reduces the plasma levels of TG, TC, and free FA, and increases the activity of LPL (Koishi et al., 2002; Fujimoto et al., 2006). In humans, evinacumab, an ANGPTL3 antibody, reduces fasting TG and LDL-C levels by approximately 76% and 23%, respectively, without affecting LDLR (Dewey et al., 2017). In an open-label study, 4 weeks evinacumab treatment reduces the plasma levels of TG, LDL-C, apoB, non-HDL-C, and HDL-C by 47%, 49%, 46%, 49%, and 36%, respectively, in patients with homozygous familial hypercholesterolemia (Gaudet et al., 2017). Evinacumab increases the fractional catabolic rate of IDL apoB and LDL apoB, suggesting this molecule may improve hepatic clearance of TGRL remnants (Reeskamp et al., 2021). The fully human monoclonal antibody (REGN1500) has a high affinity with ANGPTL3, and its effectiveness in reducing the plasma TG and LDL-C levels has been confirmed in monkeys and mice (Gusarova et al., 2015). Targeting ANGPTL3 mRNA by the antisense oligonucleotide, named as ANGPTL3-LRx, causes reductions in the levels of ANGPTL3 protein by 46.6%–84.5%, TG by 33.2%–63.1%, LDL-Cs by 1.3%–32.9%, VLDL-C by 27.9%–60.0%, non-HDL-C by10.0%–36.6%, apoB by 3.4%–25.7%, and apoCIII by18.9%–58.8% in mice (Graham et al., 2017). Gene editing through CRISPR-Cas9 technology has been established as a potential strategy for treatment of patients with atherosclerotic dyslipidemia. For instance, injection of BE3-ANGPTL3 causes reductions in TG, TC, and ANGPTL3 by 49%, 51%, and 19%, respectively, in LDLR(−/−) mice (Chadwick et al., 2018). Furthermore, a lipid nanoparticle delivery platform has recently been developed for targeted-delivery of CRISPR-Cas9-based genome editing of ANGPTL3. In this study, the reductions in ANGPTL3 mRNA and plasma TG level are stable for at least 100 days after a single administration (Qiu et al., 2021).
ANGPTL4
ANGPTL4 is mainly expressed in liver, adipose tissue, kidney, intestine, and heart. This protein plays an important role in lipid metabolism, especially in TG metabolism (Aryal et al., 2019). It mediates fasting-induced repression of LPL activity by promoting LPL unfolding, thereby enhancing degradation of LPL. However, this protein may show distinct bioactivity in distinct organs or cells. The biological functions of ANGPTL4 have been previously reviewed by Aryal et al. (2019) and Kersten (2021). ANGPTL4 derived from liver and adipose tissue primarily acts as an endocrine factor that regulates systemic lipid metabolism, while ANGPTL4 in macrophages reduces the formation of foam cells (Yang et al., 2020). Like ANGPTL3, loss-of-function mutations in ANGPTL4 are associated with low TG levels and reduced CVD risk (Dewey et al., 2016). ANGPTL4 monoclonal antibody (REGN1001) inhibits ANGPTL4 and reduces plasma levels of TG in mice and non-human primates (Dewey et al., 2016). Compared to wild-type mice, ANGPTL4(−/−) mice have lower levels of TG and TC due to the enhanced VLDL clearance and decreased VLDL production. Of note, the anti-ANGPTL4 monoclonal antibody, 14D12, is found to reduce TG by 50% in C57BL/6J mice. This antibody also reduces plasma levels of TG in LDLR(−/−), apoE(−/−), and db/db mice (Desai et al., 2007). Adipocyte-derived ANGPTL4 plays a key role in regulation of plasma TG in mice fed a regular chow diet, but this effect is diminished after a chronic high-fat diet feeding (Spitler et al., 2021). Of importance, clinical trials are needed for determining the actual effects of inhibiting ANGPTL4 on TG-lowering.
ANGPTL 8
ANGPTL8, also known as adipin/betatrophin, regulates LPL activity in the heart and skeletal muscle. This protein is also expressed in liver and adipose tissue (Zhang, 2016). Although ANGPTL8 has a functional LPL inhibitory motif, it only inhibits LPL and increases plasma TG levels in the presence of ANGPTL3 or possibly other ANGPTL family members in mice (Haller et al., 2017). Therefore, ANGPTL8 seems to increase plasma TG level by interacting with ANGPTL3. The fully human monoclonal antibody, REGN3776, can bind monkey and human ANGPTL8 with a high affinity, and reduces plasma TG by up-regulating LPL activity in humanized ANGPTL8 mice. In addition, blocking ANGPTL8 by this antibody reduces serum TG and increases serum HDL-C in cynomolgus monkeys with spontaneous HTG (Gusarova et al., 2017). An ANGPTL3-4-8 model has been provided for explaining the mechanisms of action of ANGPTLs in regulation of TG metabolism (Zhang, 2016). In brief, food intake induces the expression of ANGPTL8 in the liver and white adipose tissue. In the liver, ANGPTL8 activates ANGPTL3 and promotes the formation of ANGPTL3-8 complexes, which finally suppress the activity of LPL in circulation. In the white adipose tissue, ANGPTL8 promotes the formation of ANGPTL4-8 complexes, which enhance the activity of LPL locally. On the contrary, fasting inactivates the expression of ANGPTL8, thereby modulating LPL by an inverse way (Zhang and Zhang, 2022).
Other ANGPTLs
ANGPTL5 is primarily expressed in adipose tissue and heart. This protein is positively associated with obesity, type 2 diabetes, oxidized LDL, and especially glucose metabolism disorders, suggesting ANGPTL5 is involved in modulation of TG and glucose homeostasis (Alghanim et al., 2019; Hammad et al., 2020; Liu Y. Z et al., 2021). The plasma level of ANGPTL7 is also elevated in obese subjects and is positively associated with TG level, suggesting ANGPTL7 may be explored as a therapeutic target for modulating TG metabolism (Abu-Farha et al., 2017; Liu Y. Z et al., 2021). As reviewed previously, ANGPTL2 primarily derived from visceral fat is positively associated with inflammation and insulin resistance, while ANGPTL6 expressed in the liver is found to counteract obesity and insulin resistance by suppressing gluconeogenesis and enhancing energy expenditure (Kadomatsu et al., 2011). A recent study demonstrates that serum ANGPTL6 levels are a valuable predictor of metabolic syndrome (Namkung et al., 2019). Furthermore, ANGPTL6 is suggested to primarily maintain glucose homeostasis in response to hyperglycemia (Fan et al., 2020). Among the 8 ANGPTL members, ANGPTL1 shows a limited correlation with TG metabolism (Carbone et al., 2018). Although ANGPTL5-7 are associated with TG metabolism, the underlying mechanisms of action of these three ANGPTLs are still far from clear.
Targeting Gut Microbiota
In recent years, the relationship between gut microbiota and lipid metabolism has been paid more and more attentions. Cotillard et al. point out that reductions of microbial abundance in obese patients are related to the elevated levels of serum TC and TG (Cotillard et al., 2013). Similarly, another study suggests that individuals with low microbial gene counts have higher TG levels than individuals with high microbial gene counts (Le Chatelier et al., 2013). Compared with healthy volunteers, the phylum Bacteroides is decreased and the order Lactobacillus is increased in patients with coronary artery disease (Emoto et al., 2016). In apoE(−/−) mice, the relative abundances of Verrucomicrobia, Bacteroidaceae, Bacteroides, and Akkermansia are positively correlated with serum levels of TC, TG, HDL-C, and LDL-C. In addition, the relative abundance of Ruminococcaceae is positively correlated with HDL-C level, and the abundance of Rikenellaceae is negatively correlated with TG and LDL-C levels (Liu et al., 2020a).
Probiotics are live bacteria that colonize the gastrointestinal tract and endow beneficial effects for health. Some probiotics alleviate fat by modulating gut microbiota-short chain FA-hormone axis (Yadav et al., 2013). Of note, supplementation of Lactobacillus plantarum FRT10 is found to reduce the body weight, fat weight, and hepatic TG via upregulating the mRNA expression of PPARα and carnitine palmitoyltransferase-1α and down-regulating the mRNA expression of SREBP-1 and TG synthase diacylglycerol acyltransferase 1 in the liver of mice fed a high-fat diet (Cai et al., 2020). Furthermore, Lactobacillus plantarum FRT10 intervention is found to increase the abundance of Lactobacillus, Bifidobacterium, and Akkermansia, which could improve the imbalance of gut flora caused by a high-fat diet (Cai et al., 2020). Liraglutide is a glucagon-like peptide-1 (GLP-1) analog. This molecule significantly reduces hepatic TG content, insulin resistance, and serum LDL-C in db/db mice. Mechanistically, liraglutide significantly increases the abundance of Akkermansia, Romboutsia, and norank_f_Bacteroidales_S24-7_group, and decreases the abundance of Klebsiella, Anaerotruncus, Bacteroides, Lachnospiraceae_UCG-001, Lachnospiraceae_NK4A136_group, Ruminiclostridium, and Desulfovibrio (Liu et al., 2020b). Furthermore, oligofructose promotes satiety and reduces plasma TG in rats fed a high-fat diet by up-regulating the level of GLP-1 in the gut (Cani et al., 2005). Another study demonstrates that oligofructose increases the abundance of Bifidobacteria and lactobacilli in the gut of obese rats (Bomhof et al., 2014). Some polysaccharides may also exert their TG-lowering effects by regulating the gut microbiota (Yin et al., 2021; Li et al., 2022).
Future Directions
Statins only provide 25%–40% reductions in CVD risk, and high TG levels are closely associated with residual CVD risk. Cholesterol carried by TGRLs and their remnants is a causal factor of ASCVD except for LDL-C. The structural characteristics of TGRLs and their remnants, such as lipid content, apolipoprotein components, particle size, and retention time in circulation, determine the burden of atherosclerosis (Aguilar Salinas and John Chapman, 2020; Packard et al., 2020; Duran and Pradhan, 2021). Although there are several available methods for quantification of TGRLs and their remnants, more specific methods are needed to accurately determine and quantify the subclasses of these particles that are derived from different metabolic pathways. The recently developed omics technologies may assist the clarification of the component of these particles. These data will clarify what kinds of TGRLs and their remnants primarily determine the progression of atherosclerosis. Furthermore, the underlying mechanisms of action of the recently identified therapeutic targets, such as ANGPTLs, on modulating TG metabolism and even their own metabolism or interactions between each other are still far from clear.
Of importance, the presently used clinical TG-lowering drugs, such as fibrate, omega-3 FAs, and niacin, show equivocal effects or even futile in reduction of CVD risk by monotherapy or in combination with statin. The reasons need to be clarified in future. Although several novel compounds exhibit good TG-lowering and/or even CVD protective effects in animal models and/or clinical trials, their actual functions need to be verified in practice. Furthermore, pill burden of the patients with dyslipidemia is another question need to be resolved (Bittner, 2019; Toth et al., 2019b). Last but not the least, lifestyle changes, including reductions in carbohydrate (such as glucose, sucrose, and starch) intake, alcohol intake, smoking, and body weight, are the main measures to control HTG.
Author Contributions
B-HZ and FY performed reference collection and prepared the manuscript; Y-NQ and S-DG: Funding acquisition and editing.
Funding
This work was supported by the National Natural Science Foundation of China (82070469 and 81770463).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abu-Farha, M., Cherian, P., Al-Khairi, I., Madhu, D., Tiss, A., Warsam, S., et al. (2017). Plasma and Adipose Tissue Level of Angiopoietin-like 7 (ANGPTL7) Are Increased in Obesity and Reduced after Physical Exercise. PLoS. One. 12 (3), e0173024. doi:10.1371/journal.pone.0173024
Adiels, M., Matikainen, N., Westerbacka, J., Söderlund, S., Larsson, T., Olofsson, S.-O., et al. (2012). Postprandial Accumulation of Chylomicrons and Chylomicron Remnants Is Determined by the Clearance Capacity. Atherosclerosis 222 (1), 222–228. doi:10.1016/j.atherosclerosis.2012.02.001
Alghanim, G., Qaddoumi, M. G., Alhasawi, N., Cherian, P., Al-Khairi, I., Nizam, R., et al. (2019). Higher Levels of ANGPTL5 in the Circulation of Subjects with Obesity and Type 2 Diabetes Are Associated with Insulin Resistance. Front. Endocrinol. 10, 495. doi:10.3389/fendo.2019.00495
Alves‐Bezerra, M., and Cohen, D. E. (2017). Triglyceride Metabolism in the Liver. Compr. Physiol. 8 (1), 1–22. doi:10.1002/cphy.c170012
Amar, M. J. A., Sakurai, T., Sakurai-Ikuta, A., Sviridov, D., Freeman, L., Ahsan, L., et al. (2015). A Novel Apolipoprotein C-II Mimetic Peptide that Activates Lipoprotein Lipase and Decreases Serum Triglycerides in Apolipoprotein E-Knockout Mice. J. Pharmacol. Exp. Ther. 352 (2), 227–235. doi:10.1124/jpet.114.220418
Anagnostis, P., Vaitsi, K., Kleitsioti, P., Mantsiou, C., Pavlogiannis, K., Athyros, V. G., et al. (2020). Efficacy and Safety of Statin Use in Children and Adolescents with Familial Hypercholesterolaemia: a Systematic Review and Meta-Analysis of Randomized-Controlled Trials. Endocrine 69 (2), 249–261. doi:10.1007/s12020-020-02302-8
Aryal, B., Price, N. L., Suarez, Y., and Fernández-Hernando, C. (2019). ANGPTL4 in Metabolic and Cardiovascular Disease. Trends Mol. Med. 25 (8), 723–734. doi:10.1016/j.molmed.2019.05.010
Aung, H. H., Lame, M. W., Gohil, K., An, C.-I., Wilson, D. W., and Rutledge, J. C. (2013). Induction of ATF3 Gene Network by Triglyceride-Rich Lipoprotein Lipolysis Products Increases Vascular Apoptosis and Inflammation. Atvb 33 (9), 2088–2096. doi:10.1161/ATVBAHA.113.301375
Aung, T., Halsey, J., Kromhout, D., Gerstein, H. C., Marchioli, R., Tavazzi, L., et al. (2018). Associations of Omega-3 Fatty Acid Supplement Use with Cardiovascular Disease Risks. JAMA Cardiol. 3 (3), 225–234. doi:10.1001/jamacardio.2017.5205
Ballantyne, C. M., Bays, H. E., Kastelein, J. J., Stein, E., Isaacsohn, J. L., Braeckman, R. A., et al. (2012). Efficacy and Safety of Eicosapentaenoic Acid Ethyl Ester (AMR101) Therapy in Statin-Treated Patients with Persistent High Triglycerides (From the ANCHOR Study). Am. J. Cardiol. 110 (7), 984–992. doi:10.1016/j.amjcard.2012.05.031
Bansal, S., Buring, J. E., Rifai, N., Mora, S., Sacks, F. M., and Ridker, P. M. (2007). Fasting Compared with Nonfasting Triglycerides and Risk of Cardiovascular Events in Women. JAMA 298 (3), 309–316. doi:10.1001/jama.298.3.309
Barter, P., and Genest, J. (2019). HDL Cholesterol and ASCVD Risk Stratification: A Debate. Atherosclerosis 283, 7–12. doi:10.1016/j.atherosclerosis.2019.01.001
Basu, D., and Goldberg, I. J. (2020). Regulation of Lipoprotein Lipase-Mediated Lipolysis of Triglycerides. Curr. Opin. Lipidol. 31 (3), 154–160. doi:10.1097/MOL.0000000000000676
Behbodikhah, J., Ahmed, S., Elyasi, A., Kasselman, L. J., De Leon, J., Glass, A. D., et al. (2021). Apolipoprotein B and Cardiovascular Disease: Biomarker and Potential Therapeutic Target. Metabolites 11 (10), 690. doi:10.3390/metabo11100690
Beigneux, A. P., Davies, B. S. J., Gin, P., Weinstein, M. M., Farber, E., Qiao, X., et al. (2007). Glycosylphosphatidylinositol-anchored High-Density Lipoprotein-Binding Protein 1 Plays a Critical Role in the Lipolytic Processing of Chylomicrons. Cell Metab. 5 (4), 279–291. doi:10.1016/j.cmet.2007.02.002
Bentley, C., Hathaway, N., Widdows, J., Bejta, F., De Pascale, C., Avella, M., et al. (2011). Influence of Chylomicron Remnants on Human Monocyte Activation In Vitro. Nutr. Metabolism Cardiovasc. Dis. 21 (11), 871–878. doi:10.1016/j.numecd.2010.02.019
Bermudez, B., Lopez, S., Varela, L. M., Ortega, A., Pacheco, Y. M., Moreda, W., et al. (2012). Triglyceride-rich Lipoprotein Regulates APOB48 Receptor Gene Expression in Human THP-1 Monocytes and Macrophages. J. Nutr. 142 (2), 227–232. doi:10.3945/jn.111.149963
Bhatt, D. L., Steg, P. G., Miller, M., Brinton, E. A., Jacobson, T. A., Ketchum, S. B., et al. (2019). Cardiovascular Risk Reduction with Icosapent Ethyl for Hypertriglyceridemia. N. Engl. J. Med. 380 (1), 11–22. doi:10.1056/NEJMoa1812792
Bittner, V. (2019). Implications for REDUCE IT in Clinical Practice. Prog. Cardiovasc. Dis. 62 (5), 395–400. doi:10.1016/j.pcad.2019.11.003
Black, D. D. (2007). Development and Physiological Regulation of Intestinal Lipid Absorption. I. Development of Intestinal Lipid Absorption: Cellular Events in Chylomicron Assembly and Secretion. Am. J. Physiology-Gastrointestinal Liver Physiology 293 (3), G519–G524. doi:10.1152/ajpgi.00189.2007
Bleda, S., de Haro, J., and Acin, F. (2017). Nuclear Factor-Kappa B Role in NLRP1 Inflammasome Activation by Triglycerides and VLDL Cholesterol in Endothelial Cells. Int. J. Cardiol. 234, 104. doi:10.1016/j.ijcard.2016.12.161
Bleda, S., de Haro, J., Varela, C., Ferruelo, A., and Acin, F. (2016). Elevated Levels of Triglycerides and Vldl-Cholesterol Provoke Activation of Nlrp1 Inflammasome in Endothelial Cells. Int. J. Cardiol. 220, 52–55. doi:10.1016/j.ijcard.2016.06.193
Boden, W. E., Boden, W. E., Probstfield, J. L., Anderson, T., Chaitman, B. R., Desvignes-Nickens, P., et al. (2011). Niacin in Patients with Low HDL Cholesterol Levels Receiving Intensive Statin Therapy. N. Engl. J. Med. 365 (24), 2255–2267. doi:10.1056/NEJMoa1107579
Bomhof, M. R., Saha, D. C., Reid, D. T., Paul, H. A., and Reimer, R. A. (2014). Combined Effects of Oligofructose andBifidobacterium Animalison Gut Microbiota and Glycemia in Obese Rats. Obesity 22 (3), 763–771. doi:10.1002/oby.20632
Borén, J., Packard, C. J., and Taskinen, M.-R. (2020). The Roles of ApoC-III on the Metabolism of Triglyceride-Rich Lipoproteins in Humans. Front. Endocrinol. 11, 474. doi:10.3389/fendo.2020.00474
Botham, K. M., Moore, E. H., Pascale, C. D., and Bejta, F. (2007). The Induction of Macrophage Foam Cell Formation by Chylomicron Remnants. Biochem. Soc. Trans. 35 (Pt3), 454–458. doi:10.1042/BST0350454
Bowman, L., Mafham, M., Wallendszus, K., Stevens, W., Buck, G., Barton, J., et al. (2018). Effects of N−3 Fatty Acid Supplements in Diabetes Mellitus. N. Engl. J. Med. 379 (16), 1540–1550. doi:10.1056/NEJMoa1804989
Bravo, E., and Napolitano, M. (2007). Mechanisms Involved in Chylomicron Remnant Lipid Uptake by Macrophages. Biochem. Soc. Trans. 35 (Pt3), 459–463. doi:10.1042/BST0350459
Brinton, E. A. (2015). Management of Hypertriglyceridemia for Prevention of Atherosclerotic Cardiovascular Disease. Cardiol. Clin. 33 (2), 309–323. doi:10.1016/j.ccl.2015.02.007
Burnett, J. R., Hooper, A. J., and Hegele, R. A. (2019). Lipids and Cardiovascular Disease. Pathology 51 (2), 129–130. doi:10.1016/j.pathol.2018.12.001
Cai, H., Wen, Z., Li, X., Meng, K., and Yang, P. (2020). Lactobacillus Plantarum FRT10 Alleviated High-Fat Diet-Induced Obesity in Mice through Regulating the PPARα Signal Pathway and Gut Microbiota. Appl. Microbiol. Biotechnol. 104 (13), 5959–5972. doi:10.1007/s00253-020-10620-0
Cani, P. D., Neyrinck, A. M., Maton, N., and Delzenne, N. M. (2005). Oligofructose Promotes Satiety in Rats Fed a High-Fat Diet: Involvement of Glucagon-like Peptide-1. Obes. Res. 13 (6), 1000–1007. doi:10.1038/oby.2005.117
Carbone, C., Piro, G., Merz, V., Simionato, F., Santoro, R., Zecchetto, C., et al. (2018). Angiopoietin-like Proteins in Angiogenesis, Inflammation and Cancer. Ijms 19 (2), 431. doi:10.3390/ijms19020431
Caslake, M. J., and Packard, C. J. (2004). Phenotypes, Genotypes and Response to Statin Therapy. Curr. Opin. Lipidol. 15, 387–392. doi:10.1097/01.mol.0000137225.46654.4d
Castillo-Núñez, Y., Morales-Villegas, E., and Aguilar-Salinas, C. A. (2022). Triglyceride-rich Lipoproteins: Their Role in Atherosclerosis. Ric 74 (2), 61–70. doi:10.24875/RIC.21000416
Castoldi, A., Monteiro, L. B., van Teijlingen Bakker, N., Sanin, D. E., Rana, N., Corrado, M., et al. (2020). Triacylglycerol Synthesis Enhances Macrophage Inflammatory Function. Nat. Commun. 11 (1), 4107. doi:10.1038/s41467-020-17881-3
Catapano, A. L., Graham, I., De Backer, G., Wiklund, O., Chapman, M. J., Drexel, H., et al. (2016). 2016 ESC/EAS Guidelines for the Management of Dyslipidaemias. Eur. Heart J. 37 (39), 2999–3058. doi:10.1093/eurheartj/ehw272
Chadwick, A. C., Evitt, N. H., Lv, W., and Musunuru, K. (2018). Reduced Blood Lipid Levels with In Vivo CRISPR-Cas9 Base Editing of ANGPTL3. Circulation 137 (9), 975–977. doi:10.1161/CIRCULATIONAHA.117.031335
Chapman, M. J., Redfern, J. S., McGovern, M. E., and Giral, P. (2010). Niacin and Fibrates in Atherogenic Dyslipidemia: Pharmacotherapy to Reduce Cardiovascular Risk. Pharmacol. Ther. 126 (3), 314–345. doi:10.1016/j.pharmthera.2010.01.008
Chaudhry, R., Viljoen, A., and Wierzbicki, A. S. (2018). Pharmacological Treatment Options for Severe Hypertriglyceridemia and Familial Chylomicronemia Syndrome. Expert Rev. Clin. Pharmacol. 11 (6), 589–598. doi:10.1080/17512433.2018.1480368
Chen, J., Jayachandran, M., Bai, W., and Xu, B. (2022). A Critical Review on the Health Benefits of Fish Consumption and its Bioactive Constituents. Food Chem. 369, 130874. doi:10.1016/j.foodchem.2021.130874
Choi, H. D., Shin, W. G., Lee, J.-Y., and Kang, B. C. (2015). Safety and Efficacy of Fibrate-Statin Combination Therapy Compared to Fibrate Monotherapy in Patients with Dyslipidemia: A Meta-Analysis. Vasc. Pharmacol. 65-66, 23–30. doi:10.1016/j.vph.2014.11.002
Christopoulou, E., Tsimihodimos, V., Filippatos, T., and Elisaf, M. (2019). Apolipoprotein CIII and Diabetes. Is There a Link? Diabetes. Metab. Res. Rev. 35 (3), e3118. doi:10.1002/dmrr.3118
Civelek, M., Manduchi, E., Riley, R. J., Stoeckert, C. J., and Davies, P. F. (2009). Chronic Endoplasmic Reticulum Stress Activates Unfolded Protein Response in Arterial Endothelium in Regions of Susceptibility to Atherosclerosis. Circulation Res. 105 (5), 453–461. doi:10.1161/CIRCRESAHA.109.203711
Cohen, J. C., Boerwinkle, E., Mosley, T. H., and Hobbs, H. H. (2006). Sequence Variations inPCSK9,Low LDL, and Protection against Coronary Heart Disease. N. Engl. J. Med. 354 (12), 1264–1272. doi:10.1056/NEJMoa054013
Corbin, L. J., Hughes, D. A., Chetwynd, A. J., Taylor, A. E., Southam, A. D., Jankevics, A., et al. (2020). Metabolic Characterisation of Disturbances in the APOC3/triglyceride-Rich Lipoprotein Pathway through Sample-Based Recall by Genotype. Metabolomics 16 (6), 69. doi:10.1007/s11306-020-01689-9
Cotillard, A., Kennedy, S. P., Kong, L. C., Prifti, E., Pons, N., Le Chatelier, E., et al. (2013). Dietary Intervention Impact on Gut Microbial Gene Richness. Nature 500 (7464), 585–588. doi:10.1038/nature12480
Crea, F. (2021). An Update on Triglyceride-Rich Lipoproteins and Their Remnants in Atherosclerotic Cardiovascular Disease. Eur. Heart J. 42 (47), 4777–4780. doi:10.1093/eurheartj/ehab844
Crosby, J., Peloso, G. M., Auer, P. L., Crosslin, D. R., Stitziel, N. O., Lange, L. A., et al. (2014). Loss-of-Function Mutations inAPOC3,Triglycerides, and Coronary Disease. N. Engl. J. Med. 371 (1), 22–31. doi:10.1056/NEJMoa1307095
Davies, B. S. J., Beigneux, A. P., Barnes, R. H., Tu, Y., Gin, P., Weinstein, M. M., et al. (2010). GPIHBP1 Is Responsible for the Entry of Lipoprotein Lipase into Capillaries. Cell Metab. 12 (1), 42–52. doi:10.1016/j.cmet.2010.04.016
de Vries, M. A., Klop, B., Janssen, H. W., Njo, T. L., Westerman, E. M., and Castro Cabezas, M. (2014). Postprandial Inflammation: Targeting Glucose and Lipids. Adv. Exp. Med. Biol. 824, 161–170. doi:10.1007/978-3-319-07320-0_12
den Hartigh, L. J., Altman, R., Norman, J. E., and Rutledge, J. C. (2014). Postprandial VLDL Lipolysis Products Increase Monocyte Adhesion and Lipid Droplet Formation via Activation of ERK2 and NFκB. Am. J. Physiology-Heart Circulatory Physiology 306 (1), H109–H120. doi:10.1152/ajpheart.00137.2013
Desai, U., Lee, E.-C., Chung, K., Gao, C., Gay, J., Key, B., et al. (2007). Lipid-lowering Effects of Anti-angiopoietin-like 4 Antibody Recapitulate the Lipid Phenotype Found in Angiopoietin-like 4 Knockout Mice. Proc. Natl. Acad. Sci. U.S.A. 104 (28), 11766–11771. doi:10.1073/pnas.0705041104
DeVerse, J. S., Sandhu, A. S., Mendoza, N., Edwards, C. M., Sun, C., Simon, S. I., et al. (2013). Shear Stress Modulates VCAM-1 Expression in Response to TNF-α and Dietary Lipids via Interferon Regulatory Factor-1 in Cultured Endothelium. Am. J. Physiology-Heart Circulatory Physiology 305 (8), H1149–H1157. doi:10.1152/ajpheart.00311.2013
Dewey, F. E., Gusarova, V., Dunbar, R. L., O’Dushlaine, C., Schurmann, C., Gottesman, O., et al. (2017). Genetic and Pharmacologic Inactivation of ANGPTL3 and Cardiovascular Disease. N. Engl. J. Med. 377 (3), 211–221. doi:10.1056/NEJMoa1612790
Dewey, F. E., Gusarova, V., O’Dushlaine, C., Gottesman, O., Trejos, J., Hunt, C., et al. (2016). Inactivating Variants in ANGPTL4 and Risk of Coronary Artery Disease. N. Engl. J. Med. 374 (12), 1123–1133. doi:10.1056/NEJMoa1510926
Dichtl, W., Nilsson, L., Goncalves, I., Ares, M. P. S., Banfi, C., Calara, F., et al. (1999). Very Low-Density Lipoprotein Activates Nuclear Factor-Κb in Endothelial Cells. Circulation Res. 84 (9), 1085–1094. doi:10.1161/01.res.84.9.1085
Do, R., Willer, C. J., Schmidt, E. M., Sengupta, S., Gao, C., Peloso, G. M., et al. (2013). Common Variants Associated with Plasma Triglycerides and Risk for Coronary Artery Disease. Nat. Genet. 45 (11), 1345–1352. doi:10.1038/ng.2795
Do, R., Stitziel, N. O., Stitziel, N. O., Won, H.-H., Jørgensen, A. B., Duga, S., et al. (2015). Exome Sequencing Identifies Rare LDLR and APOA5 Alleles Conferring Risk for Myocardial Infarction. Nature 518 (7537), 102–106. doi:10.1038/nature13917
Doi, H., Kugiyama, K., Ohgushi, M., Sugiyama, S., Matsumura, T., Ohta, Y., et al. (1998). Remnants of Chylomicron and Very Low Density Lipoprotein Impair Endothelium-dependent Vasorelaxation. Atherosclerosis 137 (2), 341–349. doi:10.1016/s0021-9150(97)00291-8
Doi, H., Kugiyama, K., Oka, H., Sugiyama, S., Ogata, N., Koide, S.-i., et al. (2000). Remnant Lipoproteins Induce Proatherothrombogenic Molecules in Endothelial Cells through a Redox-Sensitive Mechanism. Circulation 102 (6), 670–676. doi:10.1161/01.cir.102.6.670
Dron, J. S., and Hegele, R. A. (2020). Genetics of Hypertriglyceridemia. Front. Endocrinol. 11, 455. doi:10.3389/fendo.2020.00455
Duran, E. K., and Pradhan, A. D. (2021). Triglyceride-rich Lipoprotein Remnants and Cardiovascular Disease. Clin. Chem. 67 (1), 183–196. doi:10.1093/clinchem/hvaa296
Emoto, T., Yamashita, T., Sasaki, N., Hirota, Y., Hayashi, T., So, A., et al. (2016). Analysis of Gut Microbiota in Coronary Artery Disease Patients: A Possible Link between Gut Microbiota and Coronary Artery Disease. Jat 23 (8), 908–921. doi:10.5551/jat.32672
Fan, K.-C., Wu, H.-T., Wei, J.-N., Chuang, L.-M., Hsu, C.-Y., Yen, I.-W., et al. (2020). Serum Angiopoietin-like Protein 6, Risk of Type 2 Diabetes, and Response to Hyperglycemia: A Prospective Cohort Study. J. Clin. Endocrinol. Metab. 105 (5), e1949–e1957. doi:10.1210/clinem/dgaa103
Fan, W., Philip, S., Granowitz, C., Toth, P. P., and Wong, N. D. (2019). Hypertriglyceridemia in Statin-Treated US Adults: The National Health and Nutrition Examination Survey. J. Clin. Lipidol. 13 (1), 100–108. doi:10.1016/j.jacl.2018.11.008
Farnier, M., Zeller, M., Masson, D., and Cottin, Y. (2021). Triglycerides and Risk of Atherosclerotic Cardiovascular Disease: An Update. Archives Cardiovasc. Dis. 114 (2), 132–139. doi:10.1016/j.acvd.2020.11.006
Feingold, K. R. (2021). “Triglyceride Lowering Drugs,” in Endotext [Internet] (South Dartmouth (MA): MDText.com, Inc.).
Feng, M., Darabi, M., Tubeuf, E., Canicio, A., Lhomme, M., Frisdal, E., et al. (2020a). Free Cholesterol Transfer to High-Density Lipoprotein (HDL) upon Triglyceride Lipolysis Underlies the U-Shape Relationship between HDL-Cholesterol and Cardiovascular Disease. Eur. J. Prev. Cardiol. 27 (15), 1606–1616. doi:10.1177/2047487319894114
Feng, X., Guo, Q., Zhou, S., Sun, T., Liu, Y., Zhou, Z., et al. (2020b). Could Remnant-like Particle Cholesterol Become a Risk Factor in Diabetic Menopausal Women with Coronary Artery Disease? A Cross-Sectional Study of Single Academic Center in China. Lipids Health. Dis. 19 (1), 44. doi:10.1186/s12944-020-01224-8
Ference, B. A., Kastelein, J. J. P., Ray, K. K., Ginsberg, H. N., Chapman, M. J., Packard, C. J., et al. (2019). Association of Triglyceride-Lowering LPL Variants and LDL-C-Lowering LDLR Variants with Risk of Coronary Heart Disease. JAMA 321, 364–373. doi:10.1001/jama.2018.20045
Ference, B. A., Yoo, W., Alesh, I., Mahajan, N., Mirowska, K. K., Mewada, A., et al. (2012). Effect of Long-Term Exposure to Lower Low-Density Lipoprotein Cholesterol Beginning Early in Life on the Risk of Coronary Heart Disease. J. Am. Coll. Cardiol. 60 (25), 2631–2639. doi:10.1016/j.jacc.2012.09.017
Flores-Viveros, K. L., Aguilar-Galarza, B. A., Ordóñez-Sánchez, M. L., Anaya-Loyola, M. A., Moreno-Celis, U., Vázquez-Cárdenas, P., et al. (2019). Contribution of Genetic, Biochemical and Environmental Factors on Insulin Resistance and Obesity in Mexican Young Adults. Obes. Res. Clin. Pract. 13 (6), 533–540. doi:10.1016/j.orcp.2019.10.012
Folsom, A. R., Peacock, J. M., Demerath, E., and Boerwinkle, E. (2008). Variation in ANGPTL4 and Risk of Coronary Heart Disease: the Atherosclerosis Risk in Communities Study. Metabolism 57 (11), 1591–1596. doi:10.1016/j.metabol.2008.06.016
Fujihara, Y., Nakamura, T., Horikoshi, T., Obata, J.-e., Fujioka, D., Watanabe, Y., et al. (2019). Remnant Lipoproteins Are Residual Risk Factor for Future Cardiovascular Events in Patients with Stable Coronary Artery Disease and On-Statin Low-Density Lipoprotein Cholesterol Levels <70 mg/dL. Circ. J. 83 (6), 1302–1308. doi:10.1253/circj.CJ-19-0047
Fujimoto, K., Koishi, R., Shimizugawa, T., and Ando, Y. (2006). Angptl3-null Mice Show Low Plasma Lipid Concentrations by Enhanced Lipoprotein Lipase Activity. Exp. Anim. 55 (1), 27–34. doi:10.1538/expanim.55.27
Fujioka, Y., Cooper, A. D., and Fong, L. G. (1998). Multiple Processes Are Involved in the Uptake of Chylomicron Remnants by Mouse Peritoneal Macrophages. J. Lipid Res. 39 (12), 2339–2349. doi:10.1016/s0022-2275(20)33313-7
Funada, J.-i., Sekiya, M., Hamada, M., and Hiwada, K. (2002). Postprandial Elevation of Remnant Lipoprotein Leads to Endothelial Dysfunction. Circ. J. 66 (2), 127–132. doi:10.1253/circj.66.127
Ganda, O. P., Bhatt, D. L., Mason, R. P., Miller, M., and Boden, W. E. (2018). Unmet Need for Adjunctive Dyslipidemia Therapy in Hypertriglyceridemia Management. J. Am. Coll. Cardiol. 72 (3), 330–343. doi:10.1016/j.jacc.2018.04.061
Gaudet, D., Gipe, D. A., Pordy, R., Ahmad, Z., Cuchel, M., Shah, P. K., et al. (2017). ANGPTL3 Inhibition in Homozygous Familial Hypercholesterolemia. N. Engl. J. Med. 377 (3), 296–297. doi:10.1056/NEJMc1705994
Gaudet, D., Méthot, J., Déry, S., Brisson, D., Essiembre, C., Tremblay, G., et al. (2013). Efficacy and Long-Term Safety of Alipogene Tiparvovec (AAV1-Lpls447x) Gene Therapy for Lipoprotein Lipase Deficiency: An Open-Label Trial. Gene Ther. 20 (4), 361–369. doi:10.1038/gt.2012.43
Geldenhuys, W. J., Caporoso, J., Leeper, T. C., Lee, Y.-K., Lin, L., Darvesh, A. S., et al. (2017). Structure-activity and In Vivo Evaluation of a Novel Lipoprotein Lipase (LPL) Activator. Bioorg. Med. Chem. Lett. 27 (2), 303–308. doi:10.1016/j.bmcl.2016.11.053
Generoso, G., Janovsky, C. C. P. S., and Bittencourt, M. S. (2019). Triglycerides and Triglyceride-Rich Lipoproteins in the Development and Progression of Atherosclerosis. Curr. Opin. Endocrinol. Diabetes Obes. 26 (2), 109–116. doi:10.1097/MED.0000000000000468
Gianturco, S. H., and Bradley, W. A. (1999). Pathophysiology of Triglyceride-Rich Lipoproteins in Atherothrombosis: Cellular Aspects. Clin. Cardiol. 22 (6 Suppl. l), II–7. doi:10.1002/clc.4960221403
Gianturco, S. H., Brown, F. B., Gotto, A. M., and Bradley, W. A. (1982). Receptor-mediated Uptake of Hypertriglyceridemic Very Low Density Lipoproteins by Normal Human Fibroblasts. J. Lipid Res. 23 (7), 984–993. doi:10.1016/s0022-2275(20)38070-6
Gill, P. K., Dron, J. S., and Hegele, R. A. (2021). Genetics of Hypertriglyceridemia and Atherosclerosis. Curr. Opin. Cardiol. 36 (3), 264–271. doi:10.1097/HCO.0000000000000839
Ginsberg, H. N., Packard, C. J., Chapman, M. J., Borén, J., Aguilar-Salinas, C. A., Averna, M., et al. (2021). Triglyceride-rich Lipoproteins and Their Remnants: Metabolic Insights, Role in Atherosclerotic Cardiovascular Disease, and Emerging Therapeutic Strategies-A Consensus Statement from the European Atherosclerosis Society. Eur. Heart J. 42 (47), 4791–4806. doi:10.1093/eurheartj/ehab551
Gordts, P. L. S. M., Nock, R., Son, N.-H., Ramms, B., Lew, I., Gonzales, J. C., et al. (2016). ApoC-III Inhibits Clearance of Triglyceride-Rich Lipoproteins through LDL Family Receptors. J. Clin. Invest. 126 (8), 2855–2866. doi:10.1172/JCI86610
Gouni-Berthold, I., Alexander, V. J., Yang, Q., Hurh, E., Steinhagen-Thiessen, E., Moriarty, P. M., et al. (2021). Efficacy and Safety of Volanesorsen in Patients with Multifactorial Chylomicronaemia (COMPASS): a Multicentre, Double-Blind, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet Diabetes & Endocrinol. 9 (5), 264–275. doi:10.1016/S2213-8587(21)00046-2
Gouni-Berthold, I. (2017). The Role of Antisense Oligonucleotide Therapy against Apolipoprotein-CIII in Hypertriglyceridemia. Atheroscler. Suppl. 30, 19–27. doi:10.1016/j.atherosclerosissup.2017.05.003
Gower, R. M., Wu, H., Foster, G. A., Devaraj, S., Jialal, I., Ballantyne, C. M., et al. (2011). CD11c/CD18 Expression Is Upregulated on Blood Monocytes during Hypertriglyceridemia and Enhances Adhesion to Vascular Cell Adhesion Molecule-1. Atvb 31, 160–166. doi:10.1161/atvbaha.110.215434
Goyal, S., Tanigawa, Y., Zhang, W., Chai, J.-F., Almeida, M., Sim, X., et al. (2021). APOC3 Genetic Variation, Serum Triglycerides, and Risk of Coronary Artery Disease in Asian Indians, Europeans, and Other Ethnic Groups. Lipids Health Dis. 20 (1), 113. doi:10.1186/s12944-021-01531-8
Graham, M. J., Lee, R. G., Brandt, T. A., Tai, L.-J., Fu, W., Peralta, R., et al. (2017). Cardiovascular and Metabolic Effects of ANGPTL3 Antisense Oligonucleotides. N. Engl. J. Med. 377 (3), 222–232. doi:10.1056/NEJMoa1701329
Guardiola, M., Cofán, M., de Castro-Oros, I., Cenarro, A., Plana, N., Talmud, P. J., et al. (2015). APOA5 Variants Predispose Hyperlipidemic Patients to Atherogenic Dyslipidemia and Subclinical Atherosclerosis. Atherosclerosis 240 (1), 98–104. doi:10.1016/j.atherosclerosis.2015.03.008
Guardiola, M., and Ribalta, J. (2017). Update on APOA5 Genetics: toward a Better Understanding of its Physiological Impact. Curr. Atheroscler. Rep. 19 (7), 30. doi:10.1007/s11883-017-0665-y
Guo, S., Tian, H., Dong, R., Yang, N., Zhang, Y., Yao, S., et al. (2016). Exogenous Supplement of N-Acetylneuraminic Acid Ameliorates Atherosclerosis in Apolipoprotein E-Deficient Mice. Atherosclerosis 251, 183–191. doi:10.1016/j.atherosclerosis
Gusarova, V., Alexa, C. A., Wang, Y., Rafique, A., Kim, J. H., Buckler, D., et al. (2015). ANGPTL3 Blockade with a Human Monoclonal Antibody Reduces Plasma Lipids in Dyslipidemic Mice and Monkeys. J. Lipid Res. 56 (7), 1308–1317. doi:10.1194/jlr.M054890
Gusarova, V., Banfi, S., Alexa-Braun, C. A., Shihanian, L. M., Mintah, I. J., Lee, J. S., et al. (2017). ANGPTL8 Blockade with a Monoclonal Antibody Promotes Triglyceride Clearance, Energy Expenditure, and Weight Loss in Mice. Endocrinology 158 (5), 1252–1259. doi:10.1210/en.2016-1894
Haller, J. F., Mintah, I. J., Shihanian, L. M., Stevis, P., Buckler, D., Alexa-Braun, C. A., et al. (2017). ANGPTL8 Requires ANGPTL3 to Inhibit Lipoprotein Lipase and Plasma Triglyceride Clearance. J. Lipid Res. 58 (6), 1166–1173. doi:10.1194/jlr.M075689
Hammad, M. M., Abu-Farha, M., Al-Taiar, A., Alam-Eldin, N., Al-Sabah, R., Shaban, L., et al. (2020). Correlation of Circulating ANGPTL5 Levels with Obesity, High Sensitivity C-Reactive Protein and Oxidized Low-Density Lipoprotein in Adolescents. Sci. Rep. 10 (1), 6330. doi:10.1038/s41598-020-63076-7
Hammond, L. E., Neschen, S., Romanelli, A. J., Cline, G. W., Ilkayeva, O. R., Shulman, G. I., et al. (2005). Mitochondrial Glycerol-3-Phosphate Acyltransferase-1 Is Essential inLiver for the Metabolism of ExcessAcyl-CoAs. J. Biol. Chem. 280 (27), 25629–25636. doi:10.1074/jbc.M503181200
Harris, W. S., Miller, M., Tighe, A. P., Davidson, M. H., and Schaefer, E. J. (2008). Omega-3 Fatty Acids and Coronary Heart Disease Risk: Clinical and Mechanistic Perspectives. Atherosclerosis 197, 12–24. doi:10.1016/j.atherosclerosis.2007.11.008
Hegele, R. A., Berberich, A. J., Ban, M. R., Wang, J., Digenio, A., Alexander, V. J., et al. (2018). Clinical and Biochemical Features of Different Molecular Etiologies of Familial Chylomicronemia. J. Clin. Lipidol. 12 (4), 920–927. doi:10.1016/j.jacl.2018.03.093
Hegele, R. A., Borén, J., Ginsberg, H. N., Arca, M., Averna, M., Binder, C. J., et al. (2020). Rare Dyslipidaemias, from Phenotype to Genotype to Management: a European Atherosclerosis Society Task Force Consensus Statement. Lancet Diabetes & Endocrinol. 8 (1), 50–67. doi:10.1016/S2213-8587(19)30264-5
Heidemann, B. E., Koopal, C., Bots, M. L., Asselbergs, F. W., Westerink, J., and Visseren, F. L. J. (2021). The Relation between VLDL-Cholesterol and Risk of Cardiovascular Events in Patients with Manifest Cardiovascular Disease. Int. J. Cardiol. 322, 251–257. doi:10.1016/j.ijcard.2020.08.030
Hoogeveen, R. C., and Ballantyne, C. M. (2020). Residual Cardiovascular Risk at Low LDL: Remnants, Lipoprotein(a), and Inflammation. Clin. Chem. 67 (1), 143–153. doi:10.1093/clinchem/hvaa252
Hou, P., Hu, S., Wang, J., Yang, Z., Yin, J., Zhou, G., et al. (2019). Exogenous Supplement of N-Acetylneuraminic Acid Improves Macrophage Reverse Cholesterol Transport in Apolipoprotein E-Deficient Mice. Lipids Health Dis. 18 (1), 24. doi:10.1186/s12944-019-0971-1
HPS2-THRIVE Collaborative Group (2013). HPS2-THRIVE Randomized Placebo-Controlled Trial in 25 673 High-Risk Patients of ER Niacin/laropiprant: Trial Design, Pre-specified Muscle and Liver Outcomes, and Reasons for Stopping Study Treatment. Eur. Heart. J. 34 (17), 1279–1291. doi:10.1093/eurheartj/eht055
Huang, X.-s., Zhao, S.-p., Bai, L., Hu, M., Zhao, W., and Zhang, Q. (2009). Atorvastatin and Fenofibrate Increase Apolipoprotein AV and Decrease Triglycerides by Up-Regulating Peroxisome Proliferator-Activated Receptor-α. Brit. J. Pharmacol. 158, 706–712. doi:10.1111/j.1476-5381.2009.00350.x
Ida, S., Kaneko, R., and Murata, K. (2019). Efficacy and Safety of Pemafibrate Administration in Patients with Dyslipidemia: a Systematic Review and Meta-Analysis. Cardiovasc. Diabetol. 18 (1), 38. doi:10.1186/s12933-019-0845-x
Inoue, T., Saniabadi, A. R., Matsunaga, R., Hoshi, K., Yaguchi, I., and Morooka, S. (1998). Impaired Endothelium-dependent Acetylcholine-Induced Coronary Artery Relaxation in Patients with High Serum Remnant Lipoprotein Particles. Atherosclerosis 139 (2), 363–367. doi:10.1016/s0021-9150(98)00098-7
Iqbal, J., Jahangir, Z., and Al-Qarni, A. A. (2020). Microsomal Triglyceride Transfer Protein: From Lipid Metabolism to Metabolic Diseases. Adv. Exp. Med. Biol. 1276, 37–52. doi:10.1007/978-981-15-6082-8_4
Ishibashi, S., Arai, H., Yokote, K., Araki, E., Suganami, H., and Yamashita, S. (2018). Efficacy and Safety of Pemafibrate (K-877), a Selective Peroxisome Proliferator-Activated Receptor α Modulator, in Patients with Dyslipidemia: Results from a 24-week, Randomized, Double Blind, Active-Controlled, Phase 3 Trial. J. Clin. Lipidol. 12 (1), 173–184. doi:10.1016/j.jacl.2017.10.006
Jacobson, T. A., Glickstein, S. B., Rowe, J. D., and Soni, P. N. (2012). Effects of Eicosapentaenoic Acid and Docosahexaenoic Acid on Low-Density Lipoprotein Cholesterol and Other Lipids: A Review. J. Clin. Lipidol. 6 (1), 5–18. doi:10.1016/j.jacl.2011.10.018
Jacobson, T. A., Ito, M. K., Maki, K. C., Orringer, C. E., Bays, H. E., Jones, P. H., et al. (2015). National Lipid Association Recommendations for Patient-Centered Management of Dyslipidemia: Part 1-Full Report. J. Clin. Lipidol. 9 (2), 129–169. doi:10.1016/j.jacl.2015.02.003
Jinno, Y., Nakakuki, M., Kawano, H., Notsu, T., Mizuguchi, K., and Imada, K. (2011). Eicosapentaenoic Acid Administration Attenuates the Pro-inflammatory Properties of VLDL by Decreasing its Susceptibility to Lipoprotein Lipase in Macrophages. Atherosclerosis 219 (2), 566–572. doi:10.1016/j.atherosclerosis.2011.09.046
Johansen, C. T., and Hegele, R. A. (2013). Using Mendelian Randomization to Determine Causative Factors in Cardiovascular Disease. J. Intern. Med. 273 (1), 44–47. doi:10.1111/j.1365-2796.2012.02586.x
Jørgensen, A. B., Frikke-Schmidt, R., Nordestgaard, B. G., and Tybjærg-Hansen, A. (2014). Loss-of-Function Mutations inAPOC3and Risk of Ischemic Vascular Disease. N. Engl. J. Med. 371 (1), 32–41. doi:10.1056/NEJMoa1308027
Jørgensen, A. B., Frikke-Schmidt, R., West, A. S., Grande, P., Nordestgaard, B. G., and Tybjærg-Hansen, A. (2013). Genetically Elevated Non-fasting Triglycerides and Calculated Remnant Cholesterol as Causal Risk Factors for Myocardial Infarction. Eur. Heart. J. 34 (24), 1826–1833. doi:10.1093/eurheartj/ehs431
Julve, J., Martín-Campos, J. M., Escolà-Gil, J. C., and Blanco-Vaca, F. (2016). Chylomicrons: Advances in Biology, Pathology, Laboratory Testing, and Therapeutics. Clin. Chim. Acta 455, 134–148. doi:10.1016/j.cca.2016.02.004
Jun, M., Foote, C., Lv, J., Neal, B., Patel, A., Nicholls, S. J., et al. (2010). Effects of Fibrates on Cardiovascular Outcomes: A Systematic Review and Meta-Analysis. Lancet 375, 1875–1884. doi:10.1016/S0140-6736(10)60656-3
Kadomatsu, T., Tabata, M., and Oike, Y. (2011). Angiopoietin-like Proteins: Emerging Targets for Treatment of Obesity and Related Metabolic Diseases. FEBS J. 278 (4), 559–564. doi:10.1111/j.1742-4658.2010.07979.x
Katsiki, N., Nikolic, D., Montalto, G., Banach, M., Mikhailidis, D. P., and Rizzo, M. (2013). The Role of Fibrate Treatment in Dyslipidemia: An Overview. Cpd 19 (17), 3124–3131. doi:10.2174/1381612811319170020
Kawakami, A., Tanaka, A., Nakajima, K., Shimokado, K., and Yoshida, M. (2002). Atorvastatin Attenuates Remnant Lipoprotein-Induced Monocyte Adhesion to Vascular Endothelium under Flow Conditions. Circulation Res. 91 (3), 263–271. doi:10.1161/01.res.0000028454.42385.8b
Kawakami, A., Tani, M., Chiba, T., Yui, K., Shinozaki, S., Nakajima, K., et al. (2005). Pitavastatin Inhibits Remnant Lipoprotein-Induced Macrophage Foam Cell Formation through ApoB48 Receptor-dependent Mechanism. Atvb 25 (2), 424–429. doi:10.1161/01.ATV.0000152632.48937.2d
Kegulian, N. C., Ramms, B., Horton, S., Trenchevska, O., Nedelkov, D., Graham, M. J., et al. (2019). ApoC-III Glycoforms Are Differentially Cleared by Hepatic TRL (Triglyceride-Rich Lipoprotein) Receptors. Atvb 39 (10), 2145–2156. doi:10.1161/ATVBAHA.119.312723
Kersten, S. (2021). Role and Mechanism of the Action of Angiopoietin-like Protein ANGPTL4 in Plasma Lipid Metabolism. J. Lipid Res. 62, 100150. doi:10.1016/j.jlr.2021.100150
Khetarpal, S. A., Qamar, A., Millar, J. S., and Rader, D. J. (2016). Targeting ApoC-III to Reduce Coronary Disease Risk. Curr. Atheroscler. Rep. 18 (9), 54. doi:10.1007/s11883-016-0609-y
Kockx, M., and Kritharides, L. (2018). Triglyceride-rich Lipoproteins. Cardiol. Clin. 36 (2), 265–275. doi:10.1016/j.ccl.2017.12.008
Kohan, A. B., Wang, F., Li, X., Bradshaw, S., Yang, Q., Caldwell, J. L., et al. (2012). Apolipoprotein A-IV Regulates Chylomicron Metabolism-Mechanism and Function. Am. J. Physiology-Gastrointestinal Liver Physiology 302 (6), G628–G636. doi:10.1152/ajpgi.00225.2011
Koishi, R., Ando, Y., Ono, M., Shimamura, M., Yasumo, H., Fujiwara, T., et al. (2002). Angptl3 Regulates Lipid Metabolism in Mice. Nat. Genet. 30 (2), 151–157. doi:10.1038/ng814
Komatsu, T., Sakurai, T., Wolska, A., Amar, M. J., Sakurai, A., Vaisman, B. L., et al. (2019). Apolipoprotein C-II Mimetic Peptide Promotes the Plasma Clearance of Triglyceride-Rich Lipid Emulsion and the Incorporation of Fatty Acids into Peripheral Tissues of Mice. J. Nutr. Metabolism 2019, 1–9. doi:10.1155/2019/7078241
Kotwal, S., Jun, M., Sullivan, D., Perkovic, V., and Neal, B. (2012). Omega 3 Fatty Acids and Cardiovascular Outcomes. Circ Cardiovasc. Qual. Outcomes 5 (6), 808–818. doi:10.1161/CIRCOUTCOMES.112.966168
Kovrov, O., Landfors, F., Saar-Kovrov, V., Näslund, U., and Olivecrona, G. (2022). Lipoprotein Size Is a Main Determinant for the Rate of Hydrolysis by Exogenous LPL in Human Plasma. J. Lipid Res. 63 (1), 100144. doi:10.1016/j.jlr.2021.100144
Kumari, A., Kristensen, K. K., Ploug, M., and Winther, A.-M. L. (2021). The Importance of Lipoprotein Lipase Regulation in Atherosclerosis. Biomedicines 9 (7), 782. doi:10.3390/biomedicines9070782
Larsson, M., Caraballo, R., Ericsson, M., Lookene, A., Enquist, P.-A., Elofsson, M., et al. (2014). Identification of a Small Molecule that Stabilizes Lipoprotein Lipase In Vitro and Lowers Triglycerides In Vivo. Biochem. Biophysical Res. Commun. 450 (2), 1063–1069. doi:10.1016/j.bbrc.2014.06.114
Laufs, U., Parhofer, K. G., Ginsberg, H. N., and Hegele, R. A. (2019). Clinical Review on Triglycerides. Eur. Heart. J. 41 (1), 99–109c. doi:10.1093/eurheartj/ehz785
Lazarte, J., and Hegele, R. A. (2021). Volanesorsen for Treatment of Familial Chylomicronemia Syndrome. Expert Rev. Cardiovasc. Ther. 19 (8), 685–693. doi:10.1080/14779072.2021.1955348
Le Chatelier., E., Nielsen, T., Qin, J., Prifti, E., Hildebrand, F., Falony, G., et al. (2013). Richness of Human Gut Microbiome Correlates with Metabolic Markers. Nature 500 (7464), 541–546. doi:10.1038/nature12506
Lee, J., and Ridgway, N. D. (2020). Substrate Channeling in the Glycerol-3-Phosphate Pathway Regulates the Synthesis, Storage and Secretion of Glycerolipids. Biochimica Biophysica Acta (BBA) - Mol. Cell Biol. Lipids 1865 (1), 158438. doi:10.1016/j.bbalip.2019.03.010
Prospective Studies Collaboration Lewington, S., Whitlock, G., Clarke, R., Sherliker, P., Emberson, J., Halsey, J., et al. (2007). Blood Cholesterol and Vascular Mortality by Age, Sex, and Blood Pressure: A Meta-Analysis of Individual Data from 61 Prospective Studies with 55,000 Vascular Deaths. Lancet 370 (9602), 1829–1839. doi:10.1016/S0140-6736(07)61778-4
Li, J., Li, L., Guo, D., Li, S., Zeng, Y., Liu, C., et al. (2020). Triglyceride Metabolism and Angiopoietin-like Proteins in Lipoprotein Lipase Regulation. Clin. Chim. Acta 503, 19–34. doi:10.1016/j.cca.2019.12.029
Li, Y., Miao, M., Yin, F., Shen, N., Yu, W.-Q., and Guo, S.-D. (2022). The Polysaccharide-Peptide Complex from Mushroom Cordyceps Militaris Ameliorates Atherosclerosis by Modulating the lncRNA-miRNA-mRNA axis. Food Funct. 13 (6), 3185–3197. doi:10.1039/d1fOo03285b10.1039/d1fo03285b
Libby, P. (2007). Fat Fuels the Flame. Circulation Res. 100 (3), 299–301. doi:10.1161/01.RES.0000259393.89870.58
Lin, P., Ji, H.-H., Li, Y.-J., and Guo, S.-D. (2021). Macrophage Plasticity and Atherosclerosis Therapy. Front. Mol. Biosci. 8, 679797. doi:10.3389/fmolb.2021.679797
Lincoff, A. M., Nicholls, S. J., Riesmeyer, J. S., Barter, P. J., Brewer, H. B., Fox, K. A. A., et al. (2017). Evacetrapib and Cardiovascular Outcomes in High-Risk Vascular Disease. N. Engl. J. Med. 376 (20), 1933–1942. doi:10.1056/NEJMoa1609581
Liu, N., Si, Y., Zhang, Y., Guo, S., and Qin, S. (2021). Human Cholesteryl Ester Transport Protein Transgene Promotes Macrophage Reverse Cholesterol Transport in C57BL/6 Mice and Phospholipid Transfer Protein Gene Knockout Mice. J. Physiol. Biochem. 77 (4), 683–694. doi:10.1007/s13105-021-00834-9
Liu, Q., Cai, B.-y., Zhu, L.-x., Xin, X., Wang, X., An, Z.-m., et al. (2020b). Liraglutide Modulates Gut Microbiome and Attenuates Nonalcoholic Fatty Liver in Db/db Mice. Life Sci. 261, 118457. doi:10.1016/j.lfs.2020.118457
Liu, Q., Li, Y., Song, X., Wang, J., He, Z., Zhu, J., et al. (2020a). Both gut Microbiota and Cytokines Act to Atherosclerosis in ApoE−/− Mice. Microb. Pathog. 138, 103827. doi:10.1016/j.micpath.2019.103827
Liu, Y.-Z., Zhang, C., Jiang, J.-F., Cheng, Z.-B., Zhou, Z.-Y., Tang, M.-Y., et al. (2021). Angiopoietin-like Proteins in Atherosclerosis. Clin. Chim. Acta 521, 19–24. doi:10.1016/j.cca.2021.06.024
Luan, H. H., Wang, A., Hilliard, B. K., Carvalho, F., Rosen, C. E., Ahasic, A. M., et al. (2019). GDF15 Is an Inflammation-Induced Central Mediator of Tissue Tolerance. Cell 178 (5), 1231–1244. doi:10.1016/j.cell.2019.07.033
Mach, F., Baigent, C., Catapano, A. L., Koskinas, K. C., Casula, M., Badimon, L., et al. (2020). 2019 ESC/EAS Guidelines for the Management of Dyslipidaemias: Lipid Modification to Reduce Cardiovascular Risk. Eur. Heart J. 41, 111–188. doi:10.1093/eurheartj/ehz455
Mallick, R., and Duttaroy, A. K. (2022). Modulation of Endothelium Function by Fatty Acids. Mol. Cell. Biochem. 477 (1), 15–38. doi:10.1007/s11010-021-04260-9
Mansouri, R. M., Baugé, E., Gervois, P., Fruchart-Najib, J., Fiévet, C., Staels, B., et al. (2008). Atheroprotective Effect of Human Apolipoprotein A5 in a Mouse Model of Mixed Dyslipidemia. Circulation Res. 103 (5), 450–453. doi:10.1161/CIRCRESAHA.108.179861
Maranhão, R. C., Pala, D., and Freitas, F. R. (2020). Lipoprotein Removal Mechanisms and Aging: Implications for the Cardiovascular Health of the Elderly. Curr. Opin. Endocrinol. Diabetes. Obes. 27 (2), 104–109. doi:10.1097/MED.0000000000000529
Mason, R. P., Jacob, R. F., Shrivastava, S., Sherratt, S. C. R., and Chattopadhyay, A. (2016). Eicosapentaenoic Acid Reduces Membrane Fluidity, Inhibits Cholesterol Domain Formation, and Normalizes Bilayer Width in Atherosclerotic-like Model Membranes. Biochimica Biophysica Acta (BBA) - Biomembr. 1858 (12), 3131–3140. doi:10.1016/j.bbamem.2016.10.002
Matsunaga, A., Nagashima, M., Yamagishi, H., and Saku, K. (2020). Variants of Lipid-Related Genes in Adult Japanese Patients with Severe Hypertriglyceridemia. Jat 27 (12), 1264–1277. doi:10.5551/jat.51540
Mendoza-Barberá, E., Julve, J., Nilsson, S. K., Lookene, A., Martín-Campos, J. M., Roig, R., et al. (2013). Structural and Functional Analysis of APOA5 Mutations Identified in Patients with Severe Hypertriglyceridemia. J. Lipid Res. 54 (3), 649–661. doi:10.1194/jlr.M031195
Merkel, M. (2009). Diabetische Dyslipoproteinämie: jenseits von LDL. Dtsch. Med. Wochenschr. 134 (20), 1067–1073. doi:10.1055/s-0029-1222571
Michos, E. D., Sibley, C. T., Baer, J. T., Blaha, M. J., and Blumenthal, R. S. (2012). Niacin and Statin Combination Therapy for Atherosclerosis Regression and Prevention of Cardiovascular Disease Events. J. Am. Coll. Cardiol. 59 (23), 2058–2064. doi:10.1016/j.jacc.2012.01.045
Mora‐Rodriguez, R., Ortega, J. F., Morales‐Palomo, F., Ramirez‐Jimenez, M., and Moreno‐Cabañas, A. (2020). Effects of Statin Therapy and Exercise on Postprandial Triglycerides in Overweight Individuals with Hypercholesterolaemia. Br. J. Clin. Pharmacol. 86 (6), 1089–1099. doi:10.1111/bcp.14217
Morita, S.-y. (2016). Metabolism and Modification of Apolipoprotein B-Containing Lipoproteins Involved in Dyslipidemia and Atherosclerosis. Biol. Pharm. Bull. 39 (1), 1–24. doi:10.1248/bpb.b15-00716
Morjane, I., Charoute, H., Ouatou, S., Elkhattabi, L., Benrahma, H., Saile, R., et al. (2020). Association of c.56C > G (Rs3135506) Apolipoprotein A5 Gene Polymorphism with Coronary Artery Disease in Moroccan Subjects: A Case-Control Study and an Updated Meta-Analysis. Cardiol. Res. Pract. 2020, 1–8. doi:10.1155/2020/5981971
Mozaffarian, D., and Wu, J. H. Y. (2011). Omega-3 Fatty Acids and Cardiovascular Disease. J. Am. Coll. Cardiol. 58 (20), 2047–2067. doi:10.1016/j.jacc.2011.06.063
Musunuru, K., Pirruccello, J. P., Do, R., Peloso, G. M., Guiducci, C., Sougnez, C., et al. (2010). Exome Sequencing,ANGPTL3Mutations, and Familial Combined Hypolipidemia. N. Engl. J. Med. 363 (23), 2220–2227. doi:10.1056/NEJMoa1002926
Nakajima, K., and Tanaka, A. (2018b). Atherogenic Postprandial Remnant Lipoproteins; VLDL Remnants as a Causal Factor in Atherosclerosis. Clin. Chim. Acta 478, 200–215. doi:10.1016/j.cca.2017.12.039
Nakajima, K., and Tanaka, A. (2018a). Postprandial Remnant Lipoproteins as Targets for the Prevention of Atherosclerosis. Curr. Opin. Endocrinol. Diabetes Obes. 25 (2), 108–117. doi:10.1097/MED.0000000000000393
Nakamura, T., Takano, H., Umetani, K., Kawabata, K.-i., Obata, J.-e., Kitta, Y., et al. (2005). Remnant Lipoproteinemia Is a Risk Factor for Endothelial Vasomotor Dysfunction and Coronary Artery Disease in Metabolic Syndrome. Atherosclerosis 181 (2), 321–327. doi:10.1016/j.atherosclerosis.2005.01.012
Nakano, T., Nakajima, K., Niimi, M., Fujita, M. Q., Nakajima, Y., Takeichi, S., et al. (2008). Detection of Apolipoproteins B-48 and B-100 Carrying Particles in Lipoprotein Fractions Extracted from Human Aortic Atherosclerotic Plaques in Sudden Cardiac Death Cases. Clin. Chim. Acta 390 (1-2), 38–43. doi:10.1016/j.cca.2007.12.012
Namkung, J., Sohn, J. H., Chang, J. S., Park, S.-W., Kim, J.-Y., Koh, S.-B., et al. (2019). Increased Serum Angiopoietin-like 6 Ahead of Metabolic Syndrome in a Prospective Cohort Study. Diabetes. Metab. J. 43 (4), 521–529. doi:10.4093/dmj.2018.0080
Napolitano, M., Rivabene, R., Avella, M., Botham, K. M., and Bravo, E. (2001). The Internal Redox Balance of the Cells Influences the Metabolism of Lipids of Dietary Origin by J774 Macrophages: Implications for Foam Cell Formation. J. Vasc. Res. 38 (4), 350–360. doi:10.1159/000051066
Neschen, S., Morino, K., Hammond, L. E., Zhang, D., Liu, Z.-X., Romanelli, A. J., et al. (2005). Prevention of Hepatic Steatosis and Hepatic Insulin Resistance in Mitochondrial Acyl-CoA:glycerol-Sn-3-Phosphate Acyltransferase 1 Knockout Mice. Cell Metab. 2 (1), 55–65. doi:10.1016/j.cmet.2005.06.006
Nicholls, S. J., Lincoff, A. M., Bash, D., Ballantyne, C. M., Barter, P. J., Davidson, M. H., et al. (2018). Assessment of Omega-3 Carboxylic Acids in Statin-Treated Patients with High Levels of Triglycerides and Low Levels of High-Density Lipoprotein Cholesterol: Rationale and Design of the STRENGTH Trial. Clin. Cardiol. 41 (10), 1281–1288. doi:10.1002/clc.23055
Nicholls, S. J., Lincoff, A. M., Garcia, M., Bash, D., Ballantyne, C. M., Barter, P. J., et al. (2020). Effect of High-Dose Omega-3 Fatty Acids vs Corn Oil on Major Adverse Cardiovascular Events in Patients at High Cardiovascular Risk. JAMA 324 (22), 2268–2280. doi:10.1001/jama.2020.22258
Nilsson, S. K., Heeren, J., Olivecrona, G., and Merkel, M. (2011). Apolipoprotein A-V; a Potent Triglyceride Reducer. Atherosclerosis 219 (1), 15–21. doi:10.1016/j.atherosclerosis.2011.07.019
Nishikido, T., and Ray, K. K. (2021). Targeting the Peptidase PCSK9 to Reduce Cardiovascular Risk: Implications for Basic Science and Upcoming Challenges. Br. J. Pharmacol. 178 (11), 2168–2185. doi:10.1111/bph.14851
Nissen, S. E., Lincoff, A. M., Wolski, K., Ballantyne, C. M., Kastelein, J. J. P., Ridker, P. M., et al. (2021). Association between Achieved ω-3 Fatty Acid Levels and Major Adverse Cardiovascular Outcomes in Patients with High Cardiovascular Risk. JAMA Cardiol. 6 (8), 910–918. doi:10.1001/jamacardio.2021.1157
Nordestgaard, B. G., Benn, M., Schnohr, P., and Tybjærg-Hansen, A. (2007). Nonfasting Triglycerides and Risk of Myocardial Infarction, Ischemic Heart Disease, and Death in Men and Women. JAMA 298 (3), 299–308. doi:10.1001/jama.298.3.299
Nordestgaard, B. G. (2016). Triglyceride-Rich Lipoproteins and Atherosclerotic Cardiovascular Disease. Circ. Res. 118 (4), 547–563. doi:10.1161/CIRCRESAHA.115.306249
Nordestgaard, L. T., Christoffersen, M., Afzal, S., Nordestgaard, B. G., Tybjærg-Hansen, A., and Frikke-Schmidt, R. (2021). Triglycerides as a Shared Risk Factor between Dementia and Atherosclerotic Cardiovascular Disease: A Study of 125 727 Individuals. Clin. Chem. 67 (1), 245–255. doi:10.1093/clinchem/hvaa269
Ohmura, H. (2019). Triglycerides as Residual Risk for Atherosclerotic Cardiovascular Disease. Circ. J. 83 (5), 969–970. doi:10.1253/circj.CJ-19-0239
Okumura, T., Fujioka, Y., Morimoto, S., Masai, M., Sakoda, T., Tsujino, T., et al. (2006). Chylomicron Remnants Stimulate Release of Interleukin-1.BETA. By THP-1 Cells. Jat 13 (1), 38–45. doi:10.5551/jat.13.38
Orringer, C. E., Jacobson, T. A., and Maki, K. C. (2019). National Lipid Association Scientific Statement on the Use of Icosapent Ethyl in Statin-Treated Patients with Elevated Triglycerides and High or Very-High ASCVD Risk. J. Clin. Lipidol. 13 (6), 860–872. doi:10.1016/j.jacl.2019.10.014
Ouwens, M. J. N. M., Nauta, J., Ansquer, J.-C., and Driessen, S. (2015). Systematic Literature Review and Meta-Analysis of Dual Therapy with Fenofibrate or Fenofibric Acid and a Statin versus a Double or Equivalent Dose of Statin Monotherapy. Curr. Med. Res. Opin. 31 (12), 2273–2285. doi:10.1185/03007995.2015.1098597
Özcan, U., Cao, Q., Yilmaz, E., Lee, A.-H., Iwakoshi, N. N., Özdelen, E., et al. (2004). Endoplasmic Reticulum Stress Links Obesity, Insulin Action, and Type 2 Diabetes. Science 306 (5695), 457–461. doi:10.1126/science.1103160
Packard, C. J., Boren, J., and Taskinen, M.-R. (2020). Causes and Consequences of Hypertriglyceridemia. Front. Endocrinol. 11, 252. doi:10.3389/fendo.2020.00252
Padro, T., Muñoz-Garcia, N., and Badimon, L. (2021). The Role of Triglycerides in the Origin and Progression of Atherosclerosis. Clínica Investig. Arterioscler. 33, 20–28. doi:10.1016/j.arteri.2021.02.007
Paik, J., and Duggan, S. (2019). Volanesorsen: First Global Approval. Drugs 79 (12), 1349–1354. doi:10.1007/s40265-019-01168-z
Palmisano, B. T., Le, T. D., Zhu, L., Lee, Y. K., and Stafford, J. M. (2016). Cholesteryl Ester Transfer Protein Alters Liver and Plasma Triglyceride Metabolism through Two Liver Networks in Female Mice. J. Lipid Res. 57 (8), 1541–1551. doi:10.1194/jlr.M069013
Palmisano, B. T., Zhu, L., Litts, B., Burman, A., Yu, S., Neuman, J. C., et al. (2021). Hepatocyte Small Heterodimer Partner Mediates Sex-specific Effects on Triglyceride Metabolism via Androgen Receptor in Male Mice. Metabolites 11 (5), 330. doi:10.3390/metabo11050330
Panda, C., Varadharaj, S., and Voruganti, V. S. (2022). PUFA, Genotypes and Risk for Cardiovascular Disease. Prostagl. Leukot. Essent. Fat. Acids 176, 102377. doi:10.1016/j.plefa.2021.102377
Pang, J., Chan, D. C., Hamilton, S. J., Tenneti, V. S., Watts, G. F., and Barrett, P. H. R. (2016). Effect of Niacin on Triglyceride-Rich Lipoprotein Apolipoprotein B-48 Kinetics in Statin-Treated Patients with Type 2 Diabetes. Diabetes. Obes. Metab. 18 (4), 384–391. doi:10.1111/dom.12622
Parhofer, K. G., and Laufs, U. (2019). The Diagnosis and Treatment of Hypertriglyceridemia. Dtsch. Arztebl. Int. 116 (49), 825–832. doi:10.3238/arztebl.2019.0825
Park, M. S., Youn, J.-C., Kim, E. J., Han, K. H., Lee, S. H., Kim, S. H., et al. (2021). Efficacy and Safety of Fenofibrate-Statin Combination Therapy in Patients with Inadequately Controlled Triglyceride Levels Despite Previous Statin Monotherapy: A Multicenter, Randomized, Double-Blind, Phase IV Study. Clin. Ther. 43, 1735–1747. doi:10.1016/j.clinthera.2021.08.005
Park, S. Y., Lee, J. H., Kim, Y. K., Kim, C. D., Rhim, B. Y., Lee, W. S., et al. (2005). Cilostazol Prevents Remnant Lipoprotein Particle-Induced Monocyte Adhesion to Endothelial Cells by Suppression of Adhesion Molecules and Monocyte Chemoattractant Protein-1 Expression via Lectin-like Receptor for Oxidized Low-Density Lipoprotein Receptor Activation. J. Pharmacol. Exp. Ther. 312 (3), 1241–1248. doi:10.1124/jpet.104.077826
Peng, X., and Wu, H. (2022). Inflammatory Links between Hypertriglyceridemia and Atherogenesis. Curr. Atheroscler. Rep. 24 (5), 297–306. doi:10.1007/s11883-022-01006-w
Pirillo, A., Norata, G. D., and Catapano, A. L. (2014). Postprandial Lipemia as a Cardiometabolic Risk Factor. Curr. Med. Res. Opin. 30 (8), 1489–1503. doi:10.1185/03007995.2014.909394
Pollin, T. I., Damcott, C. M., Shen, H., Ott, S. H., Shelton, J., Horenstein, R. B., et al. (2008). A Null Mutation in Human APOC3 Confers a Favorable Plasma Lipid Profile and Apparent Cardioprotection. Science 322 (5908), 1702–1705. doi:10.1126/science.1161524
Preston Mason, R. (2019). New Insights into Mechanisms of Action for Omega-3 Fatty Acids in Atherothrombotic Cardiovascular Disease. Curr. Atheroscler. Rep. 21 (1), 2. doi:10.1007/s11883-019-0762-1
Proctor, S. D., and L. Mamo, J. C. (1998). Retention of Fluorescent-Labelled Chylomicron Remnants within the Intima of the Arterial Wall - Evidence that Plaque Cholesterol May Be Derived from Post-prandial Lipoproteins. Eur. J. Clin. Investigation 28 (6), 497–503. doi:10.1046/j.1365-2362.1998.00317.x
Qiu, M., Glass, Z., Chen, J., Haas, M., Jin, X., Zhao, X., et al. (2021). Lipid Nanoparticle-Mediated Codelivery of Cas9 mRNA and Single-Guide RNA Achieves Liver-specific In Vivo Genome Editing of Angptl3. Proc. Natl. Acad. Sci. U.S.A. 118 (10), e2020401118. doi:10.1073/pnas.2020401118
Rajamani, A., Borkowski, K., Akre, S., Fernandez, A., Newman, J. W., Simon, S. I., et al. (2019). Oxylipins in Triglyceride-Rich Lipoproteins of Dyslipidemic Subjects Promote Endothelial Inflammation Following a High Fat Meal. Sci. Rep. 9 (1), 8655. doi:10.1038/s41598-019-45005-5
Rapp, J. H., Lespine, A., Hamilton, R. L., Colyvas, N., Chaumeton, A. H., Tweedie-Hardman, J., et al. (1994). Triglyceride-rich Lipoproteins Isolated by Selected-Affinity Anti-apolipoprotein B Immunosorption from Human Atherosclerotic Plaque. Arterioscler. Thromb. 14 (11), 1767–1774. doi:10.1161/01.atv.14.11.1767
Reeskamp, L. F., Millar, J. S., Wu, L., Jansen, H., van Harskamp, D., Schierbeek, H., et al. (2021). ANGPTL3 Inhibition with Evinacumab Results in Faster Clearance of IDL and LDL apoB in Patients with Homozygous Familial Hypercholesterolemia-Brief Report. Atvb 41 (5), 1753–1759. doi:10.1161/ATVBAHA.120.315204
Reiner, Ž. (2017). Hypertriglyceridaemia and Risk of Coronary Artery Disease. Nat. Rev. Cardiol. 14 (7), 401–411. doi:10.1038/nrcardio.2017.31
Ren, J., Ren, A., Deng, X., Huang, Z., Jiang, Z., Li, Z., et al. (2022). Long-chain Polyunsaturated Fatty Acids and Their Metabolites Regulate Inflammation in Age-Related Macular Degeneration. Jir 15, 865–880. doi:10.2147/JIR.S347231
Ress, C., Dobner, J., Rufinatscha, K., Staels, B., Hofer, M., Folie, S., et al. (2021). Apolipoprotein A5 Controls Fructose-Induced Metabolic Dysregulation in Mice. Nutr. Metabolism Cardiovasc. Dis. 31 (3), 972–978. doi:10.1016/j.numecd.2020.11.008
Reyes-Soffer, G., and Ginsberg, H. N. (2019). Life Is Complicated: So Is apoCIII. J. Lipid Res. 60 (8), 1347–1349. doi:10.1194/jlr.C119000214
Reyes-Soffer, G. (2021). Triglyceride-rich Lipoproteins and Atherosclerotic Cardiovascular Disease Risk: Current Status and Treatments. Curr. Opin. Endocrinol. Diatetes. Obes. 28 (2), 85–89. doi:10.1097/MED.0000000000000619
Rip, J., Nierman, M. C., Ross, C. J., Jukema, J. W., Hayden, M. R., Kastelein, J. J. P., et al. (2006). Lipoprotein Lipase S447X. Atvb 26 (6), 1236–1245. doi:10.1161/01.ATV.0000219283.10832.43
Rocha, N. A., East, C., Zhang, J., and McCullough, P. A. (2017). ApoCIII as a Cardiovascular Risk Factor and Modulation by the Novel Lipid-Lowering Agent Volanesorsen. Curr. Atheroscler. Rep. 19 (12), 62. doi:10.1007/s11883-017-0697-3
Rodríguez-Mortera, R., Caccavello, R., Garay-Sevilla, M. E., and Gugliucci, A. (2020). Higher ANGPTL3, apoC-III, and apoB48 Dyslipidemia, and Lower Lipoprotein Lipase Concentrations Are Associated with Dysfunctional Visceral Fat in Adolescents with Obesity. Clin. Chim. Acta 508, 61–68. doi:10.1016/j.cca.2020.05.014
Rosenson, R. S., Davidson, M. H., Hirsh, B. J., Kathiresan, S., and Gaudet, D. (2014). Genetics and Causality of Triglyceride-Rich Lipoproteins in Atherosclerotic Cardiovascular Disease. J. Am. Coll. Cardiol. 64 (23), 2525–2540. doi:10.1016/j.jacc.2014.09.042
Rosenson, R. S., Shott, S., Lu, L., and Tangney, C. C. (2001). Hypertriglyceridemia and Other Factors Associated with Plasma Viscosity. Am. J. Med. 110 (6), 488–492. doi:10.1016/s0002-9343(01)00643-x
Rubins, H. B., Robins, S. J., Collins, D., Fye, C. L., Anderson, J. W., Elam, M. B., et al. (1999). Gemfibrozil for the Secondary Prevention of Coronary Heart Disease in Men with Low Levels of High-Density Lipoprotein Cholesterol. N. Engl. J. Med. 341 (6), 410–418. doi:10.1056/NEJM199908053410604
Saito, Y., Yokoyama, M., Origasa, H., Matsuzaki, M., Matsuzawa, Y., Ishikawa, Y., et al. (2008). Effects of EPA on Coronary Artery Disease in Hypercholesterolemic Patients with Multiple Risk Factors: Sub-analysis of Primary Prevention Cases from the Japan EPA Lipid Intervention Study (JELIS). Atherosclerosis 200 (1), 135–140. doi:10.1016/j.atherosclerosis.2008.06.003
Sakuma, N., Ikeuchi, R., Hibino, T., Yoshida, T., Mukai, S., Akita, S., et al. (2003). Increased Serum Triglyceride Clearance and Elevated High-Density Lipoprotein 2 and 3 Cholesterol during Treatment of Primary Hypertriglyceridemia with Bezafibrate. Curr. Ther. Res. 64 (9), 697–706. doi:10.1016/j.curtheres.2003.10.002
Sakurai, T., Sakurai, A., Vaisman, B. L., Amar, M. J., Liu, C., Gordon, S. M., et al. (2016). Creation of Apolipoprotein C-II (ApoC-II) Mutant Mice and Correction of Their Hypertriglyceridemia with an ApoC-II Mimetic Peptide. J. Pharmacol. Exp. Ther. 356 (2), 341–353. doi:10.1124/jpet.115.229740
Salinas, C. A. A., and Chapman, M. J. (2020). Remnant Lipoproteins. Curr. Opin. Lipidol. 31 (3), 132–139. doi:10.1097/MOL.0000000000000682
Sampson, U. K., Fazio, S., and Linton, M. F. (2012). Residual Cardiovascular Risk Despite Optimal LDL Cholesterol Reduction with Statins: The Evidence, Etiology, and Therapeutic Challenges. Curr. Atheroscler. Rep. 14 (1), 1–10. doi:10.1007/s11883-011-0219-7
Sandesara, P. B., Virani, S. S., Fazio, S., and Shapiro, M. D. (2019). The Forgotten Lipids: Triglycerides, Remnant Cholesterol, and Atherosclerotic Cardiovascular Disease Risk. Endocr. Rev. 40 (2), 537–557. doi:10.1210/er.2018-00184
Sando, K. R., and Knight, M. (2015). Nonstatin Therapies for Management of Dyslipidemia: A Review. Clin. Ther. 37 (10), 2153–2179. doi:10.1016/j.clinthera.2015.09.001
Santos-Baez, L. S., and Ginsberg, H. N. (2020). Hypertriglyceridemia-causes, Significance, and Approaches to Therapy. Front. Endocrinol. 11, 616. doi:10.3389/fendo.2020.00616
Saraswathi, V., and Hasty, A. H. (2006). The Role of Lipolysis in Mediating the Proinflammatory Effects of Very Low Density Lipoproteins in Mouse Peritoneal Macrophages. J. Lipid Res. 47 (7), 1406–1415. doi:10.1194/jlr.M600159-JLR200
Schunkert, H., König, I. R., Kathiresan, S., Reilly, M. P., Assimes, T. L., Holm, H., et al. (2011). Large-scale Association Analysis Identifies 13 New Susceptibility Loci for Coronary Artery Disease. Nat. Genet. 43 (4), 333–338. doi:10.1038/ng.784
Shahid, S. U., Shabana, N. A., Rehman, A., and Humphries, S. (2018). GWAS Implicated Risk Variants in Different Genes Contribute Additively to Increase the Risk of Coronary Artery Disease (CAD) in the Pakistani Subjects. Lipids. Health. Dis. 17 (1), 89. doi:10.1186/s12944-018-0736-2
Shahidi, F., and Ambigaipalan, P. (2018). Omega-3 Polyunsaturated Fatty Acids and Their Health Benefits. Annu. Rev. Food Sci. Technol. 9, 345–381. doi:10.1146/annurev-food-111317-095850
Shaikh, S. R. (2012). Biophysical and Biochemical Mechanisms by Which Dietary N-3 Polyunsaturated Fatty Acids from Fish Oil Disrupt Membrane Lipid Rafts. J. Nutr. Biochem. 23 (2), 101–105. doi:10.1016/j.jnutbio.2011.07.001
Shin, H. K., Kim, Y. K., Kim, K. Y., Lee, J. H., and Hong, K. W. (2004). Remnant Lipoprotein Particles Induce Apoptosis in Endothelial Cells by NAD(P)H Oxidase-Mediated Production of Superoxide and Cytokines via Lectin-like Oxidized Low-Density Lipoprotein Receptor-1 Activation. Circulation 109 (8), 1022–1028. doi:10.1161/01.CIR.0000117403.64398.53
Shu, X., Nelbach, L., Weinstein, M. M., Burgess, B. L., Beckstead, J. A., Young, S. G., et al. (2010). Intravenous Injection of Apolipoprotein A-V Reconstituted High-Density Lipoprotein Decreases Hypertriglyceridemia in Apoav −/− Mice and Requires Glycosylphosphatidylinositol-Anchored High-Density Lipoprotein-Binding Protein 1. Atvb 30 (12), 2504–2509. doi:10.1161/ATVBAHA.110.210815
Si, S., Hou, L., Chen, X., Li, W., Liu, X., Liu, C., et al. (2022). Exploring the Causal Roles of Circulating Remnant Lipid Profile on Cardiovascular and Cerebrovascular Diseases: Mendelian Randomization Study. J. Epidemiol. 32, 205–214. doi:10.2188/jea.JE20200305
Silverman, M. G., Ference, B. A., Im, K., Wiviott, S. D., Giugliano, R. P., Grundy, S. M., et al. (2016). Association between Lowering LDL-C and Cardiovascular Risk Reduction Among Different Therapeutic Interventions. JAMA 316 (12), 1289–1297. doi:10.1001/jama.2016.13985
Simons, L. A. (2019). An Updated Review of Lipid‐modifying Therapy. Med. J. Aust. 211 (2), 87–92. doi:10.5694/mja2.50142
Spitler, K. M., Shetty, S. K., Cushing, E. M., Sylvers-Davie, K. L., and Davies, B. S. J. (2021). Regulation of Plasma Triglyceride Partitioning by Adipose-Derived ANGPTL4 in Mice. Sci. Rep. 11 (1), 7873. doi:10.1038/s41598-021-87020-5
Stollenwerk, M. M., Schiopu, A., Fredrikson, G. N., Dichtl, W., Nilsson, J., and Ares, M. P. S. (2005). Very Low Density Lipoprotein Potentiates Tumor Necrosis Factor-α Expression in Macrophages. Atherosclerosis 179 (2), 247–254. doi:10.1016/j.atherosclerosis.2004.12.002
Su, X., Cheng, Y., and Chang, D. (2021). Lipid-lowering Therapy: Guidelines to Precision Medicine. Clin. Chim. Acta 514, 66–73. doi:10.1016/j.cca.2020.12.019
Su, X., and Peng, D. (2020). The Exchangeable Apolipoproteins in Lipid Metabolism and Obesity. Clin. Chim. Acta 503, 128–135. doi:10.1016/j.cca.2020.01.015
Su, X., Weng, S., and Peng, D. (2020). New Insights into Apolipoprotein A5 and the Modulation of Human Adipose-Derived Mesenchymal Stem Cells Adipogenesis. Cmm 20 (2), 144–156. doi:10.2174/1566524019666190927155702
Sun, C., Alkhoury, K., Wang, Y. I., Foster, G. A., Radecke, C. E., Tam, K., et al. (2012). IRF-1 and miRNA126 Modulate VCAM-1 Expression in Response to a High-Fat Meal. Circ. Res. 111 (8), 1054–1064. doi:10.1161/CIRCRESAHA.112.270314
Tada, H., Nohara, A., and Kawashiri, M.-a. (2018). Serum Triglycerides and Atherosclerotic Cardiovascular Disease: Insights from Clinical and Genetic Studies. Nutrients 10 (11), 1789. doi:10.3390/nu10111789
Takahashi, S. (2017). Triglyceride Rich Lipoprotein -LPL-VLDL Receptor and Lp(a)-VLDL Receptor Pathways for Macrophage Foam Cell Formation. Jat 24 (6), 552–559. doi:10.5551/jat.RV17004
Teslovich, T. M., Musunuru, K., Smith, A. V., Edmondson, A. C., Stylianou, I. M., Koseki, M., et al. (2010). Biological, Clinical and Population Relevance of 95 Loci for Blood Lipids. Nature 466 (7307), 707–713. doi:10.1038/nature09270
Thomsen, M., Varbo, A., Tybjærg-Hansen, A., and Nordestgaard, B. G. (2014). Low Nonfasting Triglycerides and Reduced All-Cause Mortality: A Mendelian Randomization Study. Clin. Chem. 60 (5), 737–746. doi:10.1373/clinchem.2013.219881
Tokgözoğlu, L., and Libby, P. (2022). The Dawn of a New Era of Targeted Lipid-Lowering Therapies. Eur. Heart J. 00, 1–13. doi:10.1093/eurheartj/ehab841
Toth, P. P., Fazio, S., Wong, N. D., Hull, M., and Nichols, G. A. (2020). Risk of Cardiovascular Events in Patients with Hypertriglyceridaemia: A Review of Real‐world Evidence. Diabetes. Obes. Metab. 22 (3), 279–289. doi:10.1111/dom.13921
Toth, P. P., Hull, M., Granowitz, C., and Philip, S. (2021). Real-world Analyses of Patients with Elevated Atherosclerotic Cardiovascular Disease Risk from the Optum Research Database. Future Cardiol. 17 (4), 743–755. doi:10.2217/fca-2020-0123
Toth, P. P., Philip, S., Hull, M., and Granowitz, C. (2019b). Association of Elevated Triglycerides with Increased Cardiovascular Risk and Direct Costs in Statin-Treated Patients. Mayo Clin. Proc. 94 (9), 1670–1680. doi:10.1016/j.mayocp.2019.03.028
Toth, P. P., Philip, S., Hull, M., and Granowitz, C. (2019a). Elevated Triglycerides (≥150 mg/dL) and High Triglycerides (200-499 mg/dL) Are Significant Predictors of New Heart Failure Diagnosis: A Real-World Analysis of High-Risk Statin-Treated Patients. Vhrm 15, 533–538. doi:10.2147/VHRM.S221289
Toth, P. (2016). Triglyceride-rich Lipoproteins as a Causal Factor for Cardiovascular Disease. Vhrm 12, 171–183. doi:10.2147/VHRM.S104369
Twickler, T. B., Dallinga-Thie, G. M., Visseren, F. L. J., de Vries., W. R., Erkelens, D. W., and Koppeschaar, H. P. F. (2003). Induction of Postprandial Inflammatory Response in Adult Onset Growth Hormone Deficiency Is Related to Plasma Remnant-like Particle-Cholesterol Concentration. J. Clin. Endocrinol. Metabolism 88 (3), 1228–1233. doi:10.1210/jc.2002-020470
Vallerie, S. N., and Bornfeldt, K. E. (2015). Gpihbp1. Circ. Res. 116 (4), 560–562. doi:10.1161/CIRCRESAHA.115.305819
Van der Vliet, H. N., Schaap, F. G., Levels, J. H. M., Ottenhoff, R., Looije, N., Wesseling, J. G., et al. (2002). Adenoviral Overexpression of Apolipoprotein A-V Reduces Serum Levels of Triglycerides and Cholesterol in Mice. Biochem. Biophysical Res. Commun. 295 (5), 1156–1159. doi:10.1016/s0006-291x(02)00808-2
Varbo, A., Benn, M., Tybjærg-Hansen, A., and Nordestgaard, B. G. (2013). Elevated Remnant Cholesterol Causes Both Low-Grade Inflammation and Ischemic Heart Disease, whereas Elevated Low-Density Lipoprotein Cholesterol Causes Ischemic Heart Disease without Inflammation. Circulation 128 (12), 1298–1309. doi:10.1161/CIRCULATIONAHA.113.003008
Varbo, A., and Nordestgaard, B. G. (2016). Remnant Cholesterol and Triglyceride-Rich Lipoproteins in Atherosclerosis Progression and Cardiovascular Disease. Atvb 36 (11), 2133–2135. doi:10.1161/ATVBAHA.116.308305
Wakita, K., Morita, S.-y., Okamoto, N., Takata, E., Handa, T., and Nakano, M. (2015). Chylomicron Remnant Model Emulsions Induce Intracellular Cholesterol Accumulation and Cell Death Due to Lysosomal Destabilization. Biochimica Biophysica Acta (BBA) - Mol. Cell Biol. Lipids 1851 (5), 598–604. doi:10.1016/j.bbalip.2015.01.015
Wang, D., Liu, B., Tao, W., Hao, Z., and Liu, M. (2015). Fibrates for Secondary Prevention of Cardiovascular Disease and Stroke. Cochrane Database Syst. Rev. 2018 (10), CD009580. doi:10.1002/14651858.CD009580.pub2
Wang, H., and Eckel, R. H. (2009). Lipoprotein Lipase: from Gene to Obesity. Am. J. Physiology-Endocrinology Metabolism 297 (2), E271–E288. doi:10.1152/ajpendo.90920.2008
Wang, L., Gill, R., Pedersen, T. L., Higgins, L. J., Newman, J. W., and Rutledge, J. C. (2009). Triglyceride-rich Lipoprotein Lipolysis Releases Neutral and Oxidized FFAs that Induce Endothelial Cell Inflammation. J. Lipid Res. 50 (2), 204–213. doi:10.1194/jlr.M700505-JLR200
Wang, L., Sapuri-Butti, A. R., Aung, H. H., Parikh, A. N., and Rutledge, J. C. (2008). Triglyceride-rich Lipoprotein Lipolysis Increases Aggregation of Endothelial Cell Membrane Microdomains and Produces Reactive Oxygen Species. Am. J. Physiology-Heart Circulatory Physiology 295 (1), H237–H244. doi:10.1152/ajpheart.01366.2007
Wang, Y. I., Bettaieb, A., Sun, C., DeVerse, J. S., Radecke, C. E., Mathew, S., et al. (2013). Triglyceride-rich Lipoprotein Modulates Endothelial Vascular Cell Adhesion Molecule (VCAM)-1 Expression via Differential Regulation of Endoplasmic Reticulum Stress. PLoS. One. 8 (10), e78322. doi:10.1371/journal.pone.0078322
Wang, Y. I., Schulze, J., Raymond, N., Tomita, T., Tam, K., Simon, S. I., et al. (2011). Endothelial Inflammation Correlates with Subject Triglycerides and Waist Size after a High-Fat Meal. Am. J. Physiology-Heart Circulatory Physiology 300 (3), H784–H791. doi:10.1152/ajpheart.01036.2010
Watanabe, T., Ando, K., Daidoji, H., Otaki, Y., Sugawara, S., Matsui, M., et al. (2017). A Randomized Controlled Trial of Eicosapentaenoic Acid in Patients with Coronary Heart Disease on Statins. J. Cardiol. 70 (6), 537–544. doi:10.1016/j.jjcc.2017.07.007
Werner, C., Filmer, A., Fritsch, M., Groenewold, S., Gräber, S., Böhm, M., et al. (2014). Risk Prediction with Triglycerides in Patients with Stable Coronary Disease on Statin Treatment. Clin. Res. Cardiol. 103, 984–997. doi:10.1007/s00392-014-0740-0
WHO (2021). Cardiovascular Diseases (CVDs). Available at: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds (accessed sept 20, 2021).
Witztum, J. L., Gaudet, D., Freedman, S. D., Alexander, V. J., Digenio, A., Williams, K. R., et al. (2019). Volanesorsen and Triglyceride Levels in Familial Chylomicronemia Syndrome. N. Engl. J. Med. 381 (6), 531–542. doi:10.1056/NEJMoa1715944
Wolska, A., Lo, L., Sviridov, D. O., Pourmousa, M., Pryor, M., Ghosh, S. S., et al. (2020a). A Dual Apolipoprotein C-II Mimetic-Apolipoprotein C-III Antagonist Peptide Lowers Plasma Triglycerides. Sci. Transl. Med. 12 (528), eaaw7905. doi:10.1126/scitranslmed.aaw7905
Wolska, A., Reimund, M., and Remaley, A. T. (2020b). Apolipoprotein C-II. Curr. Opin. Lipidol. 31 (3), 147–153. doi:10.1097/MOL.0000000000000680
Wu, S. A., Kersten, S., and Qi, L. (2021). Lipoprotein Lipase and its Regulators: An Unfolding Story. Trends Endocrinol. Metabolism 32 (1), 48–61. doi:10.1016/j.tem.2020.11.005
Wyler von Ballmoos, M. C., Haring, B., and Sacks, F. M. (2015). The Risk of Cardiovascular Events with Increased Apolipoprotein CIII: A Systematic Review and Meta-Analysis. J. Clin. Lipidol. 9 (4), 498–510. doi:10.1016/j.jacl.2015.05.002
Xu, Q.-Y., Li, H., Cao, H.-X., Pan, Q., and Fan, J.-G. (2020). ApoC3 Rs2070667 Associates with Serum Triglyceride Profile and Hepatic Inflammation in Nonalcoholic Fatty Liver Disease. BioMed Res. Int. 2020, 1–9. doi:10.1155/2020/8869674
Yadav, H., Lee, J.-H., Lloyd, J., Walter, P., and Rane, S. G. (2013). Beneficial Metabolic Effects of a Probiotic via Butyrate-Induced GLP-1 Hormone Secretion. J. Biol. Chem. 288 (35), 25088–25097. doi:10.1074/jbc.M113.452516
Yanai, H., Katsuyama, H., and Hakoshima, M. (2022). Effects of a Novel Selective Peroxisome Proliferator-Activated Receptor α Modulator, Pemafibrate, on Metabolic Parameters: A Retrospective Longitudinal Study. Biomedicines 10, 401. doi:10.3390/biomedicines10020401
Yang, J., Li, X., and Xu, D. (2020). Research Progress on the Involvement of ANGPTL4 and Loss-Of-Function Variants in Lipid Metabolism and Coronary Heart Disease: Is the "Prime Time" of ANGPTL4-Targeted Therapy for Coronary Heart Disease Approaching? Cardiovasc. Drugs Ther. 35 (3), 467–477. doi:10.1007/s10557-020-07001-0
Yin, F., Lin, P., Yu, W.-Q., Shen, N., Li, Y., and Guo, S.-D. (2021). The Cordyceps Militaris-Derived Polysaccharide CM1 Alleviates Atherosclerosis in LDLR(-/-) Mice by Improving Hyperlipidemia. Front. Mol. Biosci. 8, 783807. doi:10.3389/fmolb.2021.783807
Young, S. G., Fong, L. G., Beigneux, A. P., Allan, C. M., He, C., Jiang, H., et al. (2019). GPIHBP1 and Lipoprotein Lipase, Partners in Plasma Triglyceride Metabolism. Cell Metab. 30 (1), 51–65. doi:10.1016/j.cmet.2019.05.023
Young, S. G., and Zechner, R. (2013). Biochemistry and Pathophysiology of Intravascular and Intracellular Lipolysis. Genes Dev. 27 (5), 459–484. doi:10.1101/gad.209296.112
Zhang, R. (2016). The ANGPTL3-4-8 Model, a Molecular Mechanism for Triglyceride Trafficking. Open Biol. 6 (4), 150272. doi:10.1098/rsob.150272
Zhang, R., and Zhang, K. (2022). An Updated ANGPTL3-4-8 Model as a Mechanism of Triglyceride Partitioning between Fat and Oxidative Tissues. Prog. Lipid Res. 85, 101140. doi:10.1016/j.plipres.2021.101140
Glossary
ANGPTL Angiopoietin-like protein
apo Apolipoprotein
ASCVD Atherosclerotic cardiovascular disease
ATF3 Activating transcription factor 3
CETP Cholesteryl ester transport protein
CHD Coronary heart disease
CM Chylomicron
CMR CM remnant
CVD Cardiovascular disease
DHA Docosahexaenoic acid
EPA Eicosapentaenoic acid
ER Endoplasmic reticulum
ERK Extracellular signal-regulated kinase
FA Fatty acid
FCS Familial CM syndrome
G3P Glycerol-3-phosphate
GLP-1 Glucagon-like peptide-1
GPAT G3P acyltransferase
GPIHBP1 Glycosylphosphatidylinositol-anchored HDL binding protein 1
HDL High density lipoprotein
HDL-C HDL cholesterol
HSPG Heparan sulfate proteoglycan
HTG Hypertriglyceridemia
HTGL Hepatic TG lipase
ICAM-1 Intercellular adhesion molecule-1
IDL Intermediate density lipoprotein
IL Interleukin
JNK c-Jun NH2-terminal kinase
LDL Low-density lipoprotein
LDL-C LDL cholesterol
LDLR LDL receptor
LOX-1 Lectin-like receptor for oxidized LDL
LPL Lipoprotein lipase
LRP-1 LDLR-related protein 1
MAPK Mitogen-activated protein kinase
MCP Monocyte chemoattractant protein
MEK MAPK kinase
MMP Matrix metalloproteinase
MTP Microsomal triglyceride transfer protein
NF-κB Nuclear factor-κB
NLRP1 Nucleotide binding domain like receptor family pyrrole domain containing protein 1
PCSK9 Proprotein convertase subtilisin/kexin-type 9
PKC Protein kinase C
PPAR Peroxisome proliferator activated receptor
PUFA Polyunsaturated fatty acids
SR-B1 Scavenger receptor B type 1
SREBP Sterol regulatory element binding protein
TG Triglyceride
TGRL TG-rich lipoprotein
TNF-α Tumor necrosis factor-alpha
VCAM Vascular cell adhesion molecule
VLDL Very low-density lipoprotein
VLDL-C VLDL cholesterol
Keywords: hypertriglyceridemia, cardiovascular disease, triglyceride-rich lipoprotein, residual risk, lipid-lowering
Citation: Zhang B-H, Yin F, Qiao Y-N and Guo S-D (2022) Triglyceride and Triglyceride-Rich Lipoproteins in Atherosclerosis. Front. Mol. Biosci. 9:909151. doi: 10.3389/fmolb.2022.909151
Received: 31 March 2022; Accepted: 06 May 2022;
Published: 25 May 2022.
Edited by:
Leming Sun, Northwestern Polytechnical University, ChinaReviewed by:
Guojun Zhao, The Sixth Affiliated Hospital of Guangzhou Medical University, ChinaJian Tu, Guilin Medical University, China
Copyright © 2022 Zhang, Yin, Qiao and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ya-Nan Qiao, MTgyNTMxNjQ2NTRAMTYzLmNvbQ==; Shou-Dong Guo, U0QtR1VPQGhvdG1haWwuY29t