 Megha K. Thakkar
Megha K. Thakkar Jamie Lee
Jamie Lee Stefan Meyer
Stefan Meyer Vivian Y. Chang
Vivian Y. Chang- 1Department of Pediatrics, Division of Pediatric Hematology-Oncology, University of California, Los Angeles, Los Angeles, CA, United States
- 2Division of Cancer Studies, University of Manchester, Manchester, United Kingdom
- 3Department of Pediatric Hematology Oncology, Royal Manchester Children’s Hospital and Christie NHS Foundation Trust, Manchester, United Kingdom
- 4Childrens Discovery and Innovation Institute, UCLA, Los Angeles, CA, United States
- 5Jonsson Comprehensive Cancer Center, UCLA, Los Angeles, CA, United States
Named the “caretakers” of the genome, RecQ helicases function in several pathways to maintain genomic stability and repair DNA. This highly conserved family of enzymes consist of five different proteins in humans: RECQL1, BLM, WRN, RECQL4, and RECQL5. Biallelic germline mutations in BLM, WRN, and RECQL4 have been linked to rare cancer-predisposing syndromes. Emerging research has also implicated somatic alterations in RecQ helicases in a variety of cancers, including hematological malignancies, breast cancer, osteosarcoma, amongst others. These alterations in RecQ helicases, particularly overexpression, may lead to increased resistance of cancer cells to conventional chemotherapy. Downregulation of these proteins may allow for increased sensitivity to chemotherapy, and, therefore, may be important therapeutic targets. Here we provide a comprehensive review of our current understanding of the role of RecQ DNA helicases in cancer and discuss the potential therapeutic opportunities in targeting these helicases.
1 Introduction
The highly conserved RecQ family of proteins play fundamental roles in the maintenance of genomic stability (Hickson, 2003; Croteau et al., 2014). Impaired function in these RecQ proteins widely impacts health and has been associated with cancer and aging (Ellis et al., 1995; Yu et al., 1996; Kitao et al., 1999; Kyng et al., 2003; Orren, 2006; Kudlow et al., 2007). There are at least five human RecQ helicase proteins, RECQL1, BLM, WRN, RECQL4, and RECQL5. Biallelic, loss-of-function germline pathogenic variants in three of them, BLM, WRN, and RECQL4, have been linked to rare cancer-predisposing syndromes. More recently, mutations in RECQL1 have been identified with the novel genome instability disorder called RECON (RECql ONe) syndrome though cancers have not yet been described in these patients (Abu-Libdeh et al., 2022).
The PanCancer Atlas with 10,967 samples of different cancer types shows that these RecQ helicase genes are somatically mutated in 0.9–1.9% of cases (Figure 1) (Cerami et al., 2012; Gao et al., 2013). These missense, truncating, inframe, splice, and structural variants/fusions have been identified across these genes with no clear somatic hot spots. Of note, RECQL5 has no known driver mutations, although there are 164 variants of uncertain significance. Despite the relative paucity of somatic mutations, the RecQ helicase family has significant roles in cancer. This review aims to provide a summary of the RecQ protein family structure and function, and the various roles in cancer, with a focus on somatic alterations that drive tumorigenesis and potential therapeutic opportunities through small molecules or a synthetic lethal approach (Table 1).
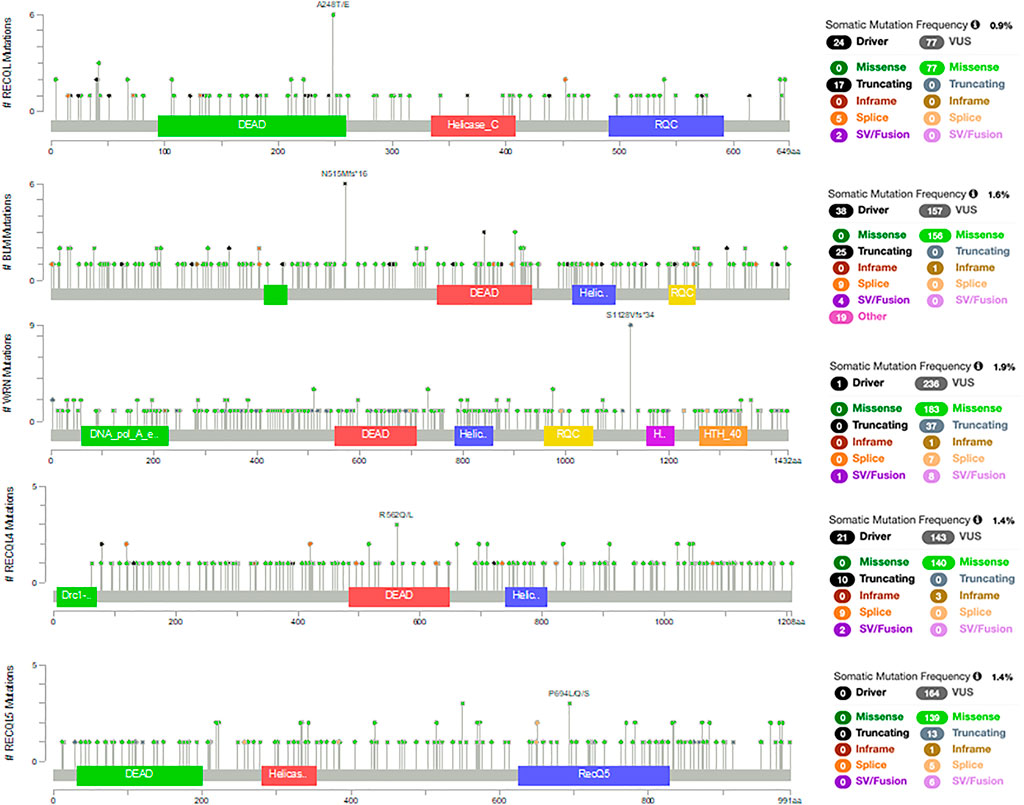
FIGURE 1. Somatic mutations in RecQ helicases from the PanCancer Atlas with 10,967 samples of different cancer types (Cerami et al., 2012; Gao et al., 2013).
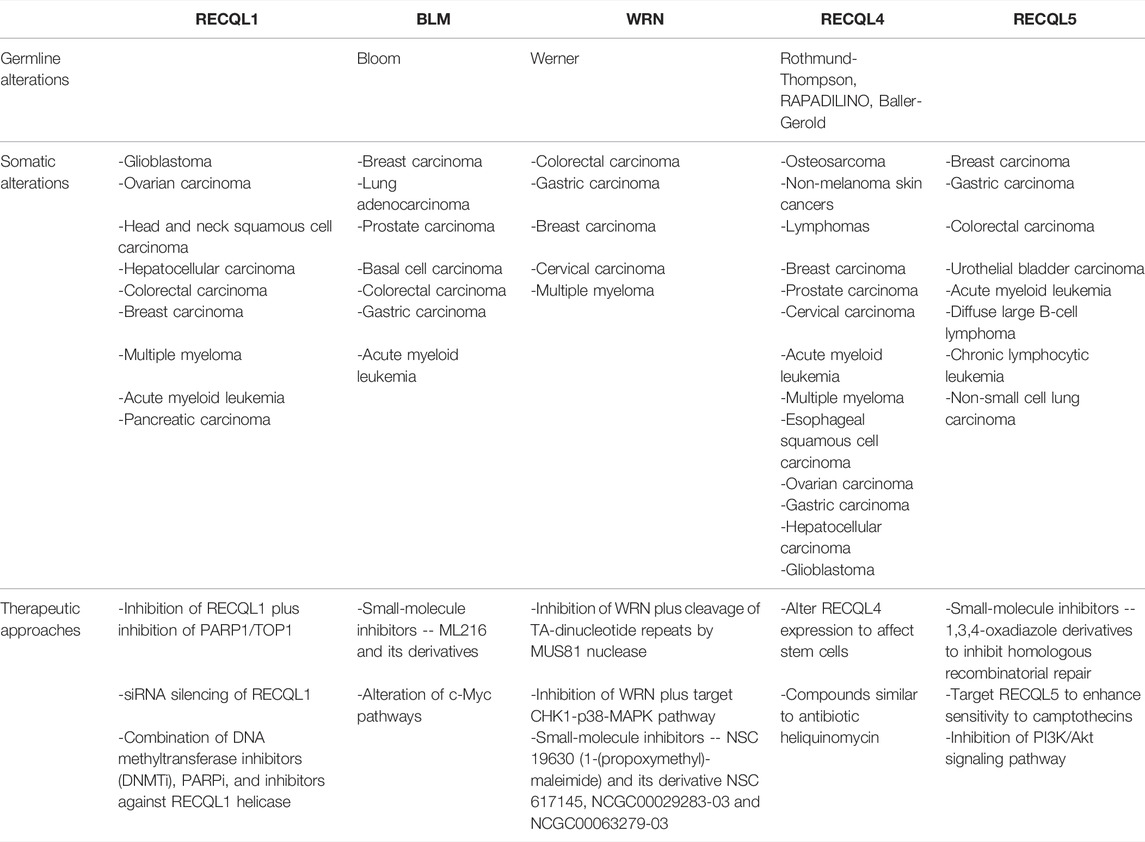
TABLE 1. Summary of germline and somatic alterations in RecQ helicases with potential anti-cancer therapeutic opportunities.
1.1 RecQ Protein Structure
Sequence analyses have identified three domains conserved across bacteria and eukaryotes, the helicase, RecQ carboxy-terminal (RQC), and helicase and RNase D C-terminal (HRDC) domains that interact with many proteins involved in DNA replication, recombination, and repair (Morozov et al., 1997). The core functional unit is comprised of the ATPase-dependent RecQ helicase domain and the RQC. The helicase domain has a single strand DNA sensor element and a DNA-binding region that utilizes ATP hydrolysis to catalyze unwinding of complementary of DNA in a 3′ to 5’ direction. The RQC domain is less conserved across the RecQ family and is only partially present in RECQL4 and RECQL5. It is thought to regulate contact with DNA and be involved in oligomerization status of the helicase (Soultanas and Wigley, 2001; Singleton et al., 2007; Pike et al., 2009; Lucic et al., 2011). The HRDC domain is the least conserved region of the RecQ helicase family, being present only in BLM and WRN, as well as bacterial and yeast homologues (Morozov et al., 1997). It mediates DNA binding activity of the helicase and RQC core but appears to be dispensable for enzyme function both in vitro and in vivo (Mullen et al., 2000; Mullen et al., 2001; Bernstein and Keck, 2003; Janscak et al., 2003).
WRN is unique amongst the RecQ family proteins because in addition to the helicase domain, it also has 3′ to 5’ exonuclease domain (Huang et al., 1998; Choi et al., 2007). This domain is located in the N terminus and becomes activated upon binding of DNA, independently of its helicase domain (Choi et al., 2007). WRN helicase interacts with DNA2 nuclease to degrade reversed replication forks and stimulate replication restart, while the exonuclease activity may play in a role in protection of the stalled fork (Thangavel et al., 2015; Datta et al., 2021).
Remarkably, the R4ZBD domain of the RECQL4 structure is different from RQC domains in other RecQ helicases in that it contains a new C-terminal domain (Kaiser et al., 2017). This unique domain consists of a zinc-binding motif and characteristic winged-helix domains that do not act in expected DNA binding or helicase activities. Rather, it may be involved in protein stability, and mutations may lead to cancers and specific RECQL4-associated syndromes (Kaiser et al., 2017).
1.2 RecQ Functions in DNA Repair Pathways
DNA is constantly being damaged by a variety of endogenous and exogenous factors, which then trigger different cellular pathways to repair damaged DNA lesions (Lindahl, 1993; Friedberg et al., 2006). When DNA damage remains unresolved, this results in genomic instability with accumulation of mutations and chromosomal rearrangements, which are then associated with cell death, accelerated aging, and cancer (White and Vijg, 2016; Tubbs and Nussenzweig, 2017; d'Adda di Fagagna, 2008). Each RecQ family protein is involved in multiple aspects of DNA repair, both shared and unique.
1.3 Homologous Recombination
Homologous recombination (HR) repairs double-strand breaks (DSB) using a homologous DNA template, primarily restricted to the S and G2 phases of cell cycle. HR also plays an important role in DNA replication and telomere maintenance. Alterations to the HR pathway are known to cause genomic instability and drive cancer, as well as affect sensitivity of cancers to chemotherapy (Thomas and Capecchi, 1987; Scully et al., 1997; Moynahan et al., 1999; Canto et al., 2019).
BLM is involved in HR through its interactions with EXO1 and DNA2 (Nimonkar et al., 2008; Nimonkar et al., 2011; Sturzenegger et al., 2014), resecting DSB and generating 3′ single-stranded DNA that can then be bound by replication protein A (RPA) and RAD51 (Gravel et al., 2008; Nimonkar et al., 2008; Nimonkar et al., 2011). RAD51 searches for a homologous DNA template, leads strand invasion and creates a “D-loop” structure where two strands of double-stranded DNA are separated. Once DNA is resynthesized, BLM then processes these two strands of double-stranded DNA (dsDNA), Holliday junctions, and restores separate DNA duplexes, in a complex with Topoisomerase Iiα and RMI1/2 (Wu and Hickson, 2003; Singh et al., 2008). BLM also builds up at stalled replication forks and interacts with FANCM and FANCC to dissolve double Holliday junctions (Davalos and Campisi, 2003; Wu and Hickson, 2003; Singh et al., 2008; Moder et al., 2017). This process normally limits genetic exchange from occurring between homologous sequences during HR, which is the basis for the increased sister chromatid exchange observed in cells from patients with Bloom syndrome (Wu and Hickson, 2003; Wang et al., 2015a). The interaction between TopBP1 and BLM has been suggested to play a role in suppressing sister chromatid exchange by maintaining BLM in the S phase cycle (Singh et al., 2008). Additionally, BLM and RECQL5 both play an anti-recombinase role by disrupting RAD51 nucleoprotein filaments from resected DNA and preventing RAD51-mediated D-loop formation thereby regulating HR (Hu et al., 2007; Patel et al., 2017). RECQL5 inhibits the exonuclease activity of MRE11, part of the MRE11-RAD50-NBS1 (MRN) complex. The MRN complex senses DNA DSBs and recruits RECQL5 to sites needing DNA repair (Zheng et al., 2009) This suggests that RECQL5 potentially controls errors in HR, thereby acting as a crucial tumor suppressor (Hu et al., 2007; Zheng et al., 2009). In addition to promoting NHEJ, RECQL4 is also involved in homologous recombination (HR)-dependent DNA DSB repair. This occurs when RECQL4 interactions initiate 5’ DNA end resection (Lu et al., 2016; Lu et al., 2017).
1.4 Nonhomologous End-Joining
Nonhomologous end-joining (NHEJ) is a template-independent DSB repair process that is more error-prone than homologous recombination but is active across phases of the cell cycle. Cells that lack various components of the NHEJ pathway display increased genomic rearrangements (Karanjawala et al., 1999). NHEJ itself can also induce genomic arrangements by ligating two junctions of DSBs that are not compatible, resulting in insertions or deletions (Rothkamm et al., 2001; Ghezraoui et al., 2014). Therefore, NHEJ has a dual role in suppressing and promoting cancer (Sishc and Davis, 2017).
The five human RecQ helicases are involved in DSB repair. WRN promotes canonical NHEJ by interacting with Ku, the loading protein that recruits other NHEJ proteins, which in turn stimulates WRN exonuclease activity (Li and Comai, 2000; Karmakar et al., 2002a; Kim et al., 2020). DNA-PKcs, another important protein involved in NHEJ, phosphorylates WRN, inhibiting helicase activity and mediating the re-localization of WRN to the nucleolus (Karmakar et al., 2002b; Kusumoto-Matsuo et al., 2010; Kusumoto-Matsuo et al., 2014). BLM helicase has recently been shown to be required for recruitment of NHEJ component, XRCC4, to chromatin in the G1 phase and also negatively regulate NHEJ during S-phase (Tripathi et al., 2018). Both RECQL1 and RECQL4 interact with the Ku70/80 subunit of the DNA-PK complex. This interaction is important to modulate DNA double-strand break repair via nonhomologous end-joining (NHEJ) (Parvathaneni et al., 2013; Shamanna et al., 2014; Lu et al., 2017).
1.5 Base Excision Repair
Base excision repair (BER) is the predominant pathway utilized to repair endogenous DNA damage, such as deaminations, alkylations, oxidative damage, and abasic single base damage (Mitra et al., 1997; Zharkov, 2008; Dianov and Hubscher, 2013). BER is initiated by DNA glycosylases, which demonstrate specificity for damaged DNA bases and conduct specific mechanisms to excise these lesions. The next step of the BER pathway consists of recognition of the AP site by AP endonuclease 1 (APE-1), which cleaves the abasic site, then eventually for DNA polymerase ß and DNA ligase to complete BER via repair and replacement of a single damaged DNA nucleotide (Carter and Parsons, 2016).
Key protein interactions overlap between SSB repair and the BER pathway (Popuri et al., 2013). BLM and WRN interact with many proteins involved in BER and single-strand break repair (SSBR) pathways. WRN interacts with APE-1. WRN and BLM stimulate several BER proteins including DNA polymerase β, FEN-1, and NEIL1 (Brosh et al., 2001; Harrigan et al., 2003; Sharma et al., 2004). RECQL4 interacts with several proteins to promote BER, including FEN1, APE1, and PARP1, where these protein interactions are essential to conduct oxidative damage (Luong and Bernstein, 2021). RECQL5 interacts with FEN1 and PCNA proteins, involved in BER (Popuri et al., 2013).
1.6 Telomere Maintenance
Telomeres, which protect the ends of chromosomes, have an important function in maintaining genomic integrity. Telomere length decreases with age as a result of normal processes that lead to cellular senescence and organismal aging. DNA damage, repeated cell division, and variants in the telomere protective complex can cause shortening and dysfunction of telomeres, leading to cell death, senescence, or additional genomic instability (Bejarano et al., 2019; McNally et al., 2019). Interestingly, both shorter and longer telomere lengths have been associated with cancer predisposition and cancer-associated mortality (Telomeres Mendelian Randomization et al., 2017; McNally et al., 2019; Samavat et al., 2019; Protsenko et al., 2020; Schmutz et al., 2020).
Patients with Werner Syndrome have features of accelerated aging and cells from these patients display shortened telomere lengths (Ouellette et al., 2000; Wyllie et al., 2000; Chang et al., 2004; Crabbe et al., 2007). Consistent with this aging phenotype, it has been demonstrated that the WRN helicase is necessary for replication of G-rich telomeric DNA (Crabbe et al., 2004). WRN and BLM are also involved in the alternative lengthening of telomeres (ALT) pathway that is independent of telomerase, by localizing to telomeric DNA and using HR to lengthen telomeres (Bryan et al., 1995; Yankiwski et al., 2000; Johnson et al., 2001). Similar to WRN and BLM, RECQL4 has also been linked with telomere maintenance by supporting telomeric D-loop resolution through its interaction with shelterin proteins as well as through WRN stimulation (Ghosh et al., 2012; Kaiser et al., 2017).
1.7 Replication Stress
During DNA replication, dsDNA that are separated into two branching single strands form a replication fork. Stalled replication forks lead to genomic instability and chromosomal rearrangements which are associated with cancer. RecQ helicases interact with replication forks in several ways by unwinding DNA, assisting with branch migration, and strand annealing (Bachrati and Hickson, 2008; Bohr, 2008; Vindigni et al., 2010). BLM can promote fork reversal and restart, and suppress firing (Davies et al., 2007; Machwe et al., 2011a; Machwe et al., 2011b; Bugreev et al., 2011). WRN is also required after replication arrest via MUS81 endonuclease activity (Franchitto et al., 2008). RECQL1 is involved in reverse branch migration, while RECQL5 disrupts RAD51 filaments on stalled replication forks by also interacting with endonuclease MUS81 (Urban et al., 2016; Di Marco et al., 2017; Chappidi et al., 2020).
2 Overview of DNA RecQ Helicases in Cancer
2.1 RECQL1
2.1.1 Germline Variants in RECQL1
Missense biallelic mutations in RECQL1 gene are associated with the novel RECql One (RECON) syndrome. Remarkably, although individuals with RECON syndrome display overlapping features with the other RecQ-associated genetic disorders, the overall clinical phenotype is distinct. The characteristic features include progeroid facies, skin photosensitivity, xeroderma, pinched nose, and elongated thumbs (Abu-Libdeh et al., 2022). Studies recently demonstrated that these mutations affect the ability to repair DNA damage and perform successful DNA replication once exposed to topoisomerase inhibitors. More studies are needed to determine if there is an association with cancer development (Abu-Libdeh et al., 2022).
There are conflicting studies investigating germline variants in RECQL1 as a cancer susceptibility gene. Germline missense, truncating, and splice site variants in RECQL1 have been enriched in participants with breast cancer in five different ethnic groups (Cybulski et al., 2015; Sun et al., 2015; Kwong et al., 2016; Sun et al., 2017; Tervasmaki et al., 2018). However, there have been a few subsequent studies with conflicting evidence, such that RECQL1 appears to be a moderate breast cancer risk gene that needs further study (Bogdanova et al., 2017; Li et al., 2018a; Nguyen-Dumont et al., 2018).
One study evaluated 13 single nucleotide polymorphisms (SNPs) in DNA repair genes, including RECQL1, in patients with pancreatic adenocarcinoma (Li et al., 2006). A polymorphism in RECQL1 A159C had the strongest effect on survival and strongly interacted with other genotypes in RAD54L, XRCC1, and ATM. Although the functional impact of this polymorphism is unknown, it is possible that deficient RECQL1 may lead to an aggressive tumor subtype through increased genomic instability (Li et al., 2006).
2.2 RECQL1 and Cancer
RECQL1 is highly expressed in proliferating cells and has been shown to be upregulated in multiple cancer cell lines, as part of a common cancer signature identified through cancer microarray data (Kawabe et al., 2000; Xu et al., 2007; Futami et al., 2008a). Overexpression of RECQL1 has been found in glioblastoma, ovarian cancer, and head and neck squamous cell carcinoma (Arai et al., 2011; Mendoza-Maldonado et al., 2011; Sharma, 2011).
Hypopharyngeal carcinomas, an aggressive subtype of head and neck squamous cell carcinoma, highly express RECQL1 and WRN proteins (Arai et al., 2011). Cell lines and clinical samples of hypopharyngeal carcinoma showed increased ɣH2AX, a marker of proliferation and activated DNA damage response. Tumor cell growth was significantly inhibited by RECQL1-or WRN-silencing by siRNA. Furthermore, in vivo combination treatment with cisplatin and siRNA silencing of RECQL1 or WRN led to increased DNA damage, apoptosis, and mitotic catastrophe.
High expression of RECQL1 has been significantly associated with poor overall survival in breast cancer patients (Gyorffy et al., 2010). In 595 breast cancer patient samples found in the Cancer Genome Atlas (TCGA) database, RECQL1 mRNA expression was correlated with several other genes, including CDK6, CENPA, and TGFBI, suggesting that RECQL1 may regulate expression of other critical genes involved in cancer cell migration, invasion, and metastasis (Li et al., 2014). Zhu et al. investigated a large sample size of breast cancer patients with various tumor characteristics. They found that increased expression of RECQL1 was significantly associated with reduced relapse-free survival and post-progression survival in all breast cancers but improved overall survival in patients with basal-like breast cancer and mutant-p53-type breast cancer (Wang et al., 2009).
RECQL1 protein levels are elevated in ovarian cancer and correlate with histological type and high proliferative potential (Sanada et al., 2013). There was, however, no association between RECQL1 expression and overall survival. When ten ovarian cancer cell lines of various histologic subtypes were subject to siRNA silencing of RECQL1, cancer cells decreased in number through mitotic cell death, compared to two normal cell lines. These results suggest a potential anticancer effect of RECQL1 silencing (Futami et al., 2008b; Sanada et al., 2013).
Additionally, RECQL1 protein expression was identified in the majority of hepatocellular carcinoma (HCC) samples by immunohistochemistry and correlated with histological grade, portal vein invasion, and tumor size >2 cm (Futami et al., 2010). Silencing of RECQL1 using siRNA induced mitotic catastrophe, leading to cell death in various HCC cell lines and demonstrating anticancer activity in vivo. This has been proposed as a potential therapeutic approach against HCC.
RECQL1, WRN, and RECQL5 mRNA expression have been found to be lower in primary colorectal carcinoma (CRC) samples compared to normal colonic mucosa, while BLM and RECQL4 mRNA levels are increased (Lao et al., 2013). When evaluating molecular subtypes of CRC, increased BLM expression was the only RecQ gene correlated with CpG island methylator phenotype (CIMP) status. CRC with microsatellite instability (MSI) had significantly lower RECQL1 and RECQL5 expression compared to normal colonic tissue. There were, however, no differences in protein expression of any of the RecQ helicases by immunohistochemistry between localized disease compared to advanced stage disease (Lao et al., 2013).
The role of RECQL1 in replication stress is highlighted in multiple myeloma. In a study by Viziteu et al. focusing on multiple myeloma cells, RECQL1 overexpression was significantly higher in primary myeloma cells from newly diagnosed patients compared to normal bone marrow plasma cells (Viziteu et al., 2016). Additionally, overexpression of RECQL1 was correlated with poorer prognosis, while depletion led to apoptosis and arrest of cell growth in multiple myeloma cells (MMC) (Viziteu et al., 2017). Forced expression of RECQL1 in human myeloma cell lines correlated with an increased resistance to melphalan and bortezomib-induced cell death. Additionally, it was found that RECQL1 depletion induced cytotoxicity in MMCs by PARP inhibitors. This data demonstrates the crucial role of RECQL1 in not only protecting multiple myeloma cells against DNA damage and replicative stress but also against chemotherapeutic agents (Viziteu et al., 2016). As DNA methyltransferase (DNMT) inhibitors can lead to decreased expression of RECQL1, MMCs can potentially be targeted using combination therapy of DNMT inhibitors and chemotherapy and/or PARP inhibitors (Viziteu et al., 2017).
2.3 BLM
2.3.1 Germline Variants in BLM Cause Bloom Syndrome
Bloom Syndrome (BSyn) is an autosomal recessive disease caused by variants in the BLM gene, located at the 15q26.1 locus (German et al., 1994). Patients with BSyn typically present with short stature, immunodeficiency, skin photosensitivity, and susceptibility to cancer (Mojumdar, 2020). Chromosomes in cells from patients with BSyn display increased frequency of sister chromatid exchanges secondary to the role of BLM in homologous recombination described above. This is thought to be the underlying mechanism for their increased risk of cancer (Ababou, 2021). They develop a wide spectrum of cancer types, with hematologic and gastrointestinal malignancies being most frequent, at younger ages than the general population.
Heterozygous carriers of BLM variants have not been extensively studied but there are a few studies showing mixed results with regard to cancer risk (Cleary et al., 2003; Baris et al., 2007; Antczak et al., 2013; Laitman et al., 2016; Schayek et al., 2017). One study examined patients with metastatic prostate cancer who underwent germline genetic testing and found that truncating BLM variants occurred at a higher frequency in patients with metastatic prostate cancer than control populations (Ledet et al., 2020). Several studies in patients with colon cancer have shown an enrichment of deleterious BLM variants (Gruber et al., 2002; de Voer et al., 2015). An elegant study examining gene-environment links demonstrated that germline BLM variants increase susceptibility to asbestos-related carcinogenesis, leading to mesothelioma, a link first identified in patients with mesothelioma and confirmed in a mouse model (Bononi et al., 2020). Smaller studies trying to link germline variants in BLM and breast cancer have yielded mixed results (Thompson et al., 2012; Kluzniak et al., 2019).
Case-control studies have shown associations between polymorphisms in BLM and breast cancer (Wirtenberger et al., 2006; Broberg et al., 2009; Ding et al., 2009; Sassi et al., 2013). One study investigated polymorphisms in WRN, RMI1, and BLM in a large population-based case-control study of colorectal cancer patients and controls and found an association of BLM P868L with rectal cancer risk (Frank et al., 2010). One haplotype overlapping with BLM was shown to be significantly associated with prostate cancer susceptibility (Wang et al., 2015b). Further studies are needed to better understand these polymorphisms in BLM and their influence on cancer risk and prognosis.
2.4 BLM and Cancer
Studies have shown that overexpression of BLM, both mRNA and protein, has significant prognostic value in breast cancer (Arora et al., 2015; Zhu et al., 2018). In the previously mentioned study by Zhu et al., increased expression of BLM was associated with reduced distant metastasis-free survival in all patients (Zhu et al., 2018).
An integrated genomic approach analyzing two cohorts of triple-negative breast cancers resistant to cisplatin therapy, showed that BLM had increased DNA copy number and gene expression in cases that responded to cisplatin chemotherapy (Birkbak et al., 2018). Using lentiviral overexpression of BLM in cell lines, it has been shown that increased BLM expression promotes increased sensitivity to cisplatin. The mechanism of this finding seems to be through increased DNA damage seen in BLM overexpressing cells, potentially due to predominating anti-HR effects of BLM at high levels (Birkbak et al., 2018).
One recent study on lung adenocarcinoma validated a cancer stem cell-related biomarker by an mRNA stemness index (mRNAsi) that was significantly higher in patients with lung cancer than controls (Zhao et al., 2020). Lung cancer patients with higher mRNAsi also had higher stage cancers and worse overall survival. BLM was found to be differentially expressed amongst lung cancer patients and low expression of BLM was significantly correlated with better overall survival (Zhao et al., 2020). A second study profiling the mutational landscape of lung cancer identified BLM as being one of nine recurrently mutated genes (Zhou et al., 2020). Interestingly, mutations in BLM were found in higher frequently in male patients compared to female patients. A subset of patients with BLM mutations had no clinically actionable targets such as KRAS, ERBB2, MET, RET, BRAF and PIK3CA, leaving gaps for future novel therapies (Zhou et al., 2020).
BLM mRNA and protein expression are upregulated in prostate cancer cells compared to controls (Qian et al., 2017). Inhibition of BLM in vitro reduces cell proliferation and increases apoptosis, while having no effect on prostate cancer cell migration and invasion. One study investigated the role of BLM in prostate cancer progression (Chen et al., 2019). Using CRISPR/Cas9, they showed that BLM deletion in prostate cancer cells inhibits cell proliferation by downregulating pAKT and pRAS, which leads to increased reactive oxygen species production (Chen et al., 2019).
When mice with a hypomorphic Blm allele (Blmtm3Brd) were crossed with a Ptch1+/− mouse model of basal cell nevus syndrome, there was significantly increased formation of basal cell carcinoma (BCC) and rhabdomyosarcoma (RMS) (Aszterbaum et al., 1999; Luo et al., 2000; Davari et al., 2010). Although tumor histology and DNA copy number were not significantly different, mice with Blm deficiency in the background of Ptch heterozygosity had decreased survival. Similarly, mutant Blm increases intestinal tumor formation in Apc+/min mice without affecting tumor histology or chromosomal aberrations (Goss et al., 2002; Traverso et al., 2003; Snijders et al., 2008). These studies therefore demonstrated that BLM deficiency increases tumor formation in a tissue-specific pattern.
Patients with BSyn have presented with numerous colonic adenomas, similar to attenuated familial adenomatous polyposis (Lowy et al., 2001). Some adenomas from patients with BSyn harbor somatic mutations in APC and display microsatellite instability (Calin et al., 2000; Lowy et al., 2001). Somatic frameshift mutations in BLM have been identified in colon cancers and gastric cancers with high microsatellite instability (Calin et al., 2000; Calin et al., 2001). Additionally, it has been shown that BLM expression, by both mRNA and protein levels, is increased in colon cancer samples, along with RECQL4, while RECQL1 and RECQL5 expression are significantly decreased (Lao et al., 2013).
The majority of colon cancer samples and cell lines have overexpression of c-Myc at the RNA and protein levels (Finley et al., 1989; He et al., 1998). Significant overlap between tumors that over-express c-Myc and show loss of BLM has been observed (Chandra et al., 2013).
2.5 WRN
2.5.1 Germline Variants in WRN Cause Werner Syndrome
Werner Syndrome is an autosomal recessive disorder caused by mutations in the WRN gene, located on chromosome 8p11. It is associated with short stature, skin atrophy, and premature aging comorbidities, including premature atherosclerosis, stroke, myocardial infarction, cataracts, diabetes mellitus, and osteoporosis (Lauper et al., 2013). These patients have an increased risk of cancers, particularly sarcomas, thyroid carcinoma, meningioma, and hematologic malignancies (Lauper et al., 2013).
A case-control study looking at single-nucleotide polymorphisms in WRN identified WRN c.4330T > C to be associated with increased susceptibility to esophageal carcinoma (Li et al., 2012). There have been additional small reports of p.C1367R being associated with increased risk of breast cancer but also protective against non-Hodgkin lymphoma and soft tissue sarcoma (Shen et al., 2006; Nakayama et al., 2008; Zins et al., 2015).
WRN Leu1074Phe was evaluated for prostate cancer risk and found to potentially increase risk in patients younger than 72 years of age (Wang et al., 2011a). This same polymorphism was associated with an increased risk of breast cancer, earlier age at menarche, as well as an association with onset of cardiovascular disease (Castro et al., 2000; Wang et al., 2009).
2.6 WRN and Cancer
Loss of heterozygosity involving the WRN loci at chromosome 8p11.2-p12 occurs frequently in many different cancers, pointing to its role as a tumor suppressor gene (Chughtai et al., 1999; Armes et al., 2004). It has been shown that the WRN promoter is commonly hypermethylated, thereby leading to silencing of WRN expression, in different human cancer cell lines (Agrelo et al., 2006). Indeed, in colorectal cancer, one study demonstrated that epigenetic silencing of WRN was associated with improved survival in patients treated with a topoisomerase inhibitor, while another study was not able to validate these findings (Agrelo et al., 2006; Bosch et al., 2016). Gastric cancer samples have also been shown to have WRN promoter methylation. Patients with WRN methylation and methylation of heparan sulfate 6-O-endosulfatase (SULF2) had increased sensitivity to chemotherapy irinotecan but no significant clinical or pathologic correlations were identified (Wang et al., 2013).
Colon cancer samples with somatic variants in WRN were more likely to be from right-sided cancers and were associated with increased tumor mutation burden and microsatellite instability (MSI) (Zimmer et al., 2020). WRN has been found to have a synthetic lethal interaction with MSI, making it an interesting therapeutic target to be discussed further below (Kategaya et al., 2019; Picco et al., 2021). In addition, WRN-mutated colon cancer has a characteristic immunologic profile with higher PD-L1 expression in contrast to WRN wildtype which could impact response to immunotherapy (Zimmer et al., 2020).
Breast cancer cell lines have also demonstrated hypermethylation of WRN. Expression of WRN in a breast cancer cell line appears to inhibit tumor growth in athymic nude mice (Agrelo et al., 2006). In patients with breast cancer, methylation status of WRN was significantly correlated with expression (Li et al., 2016) and an independent study demonstrated that increased expression of WRN mRNA and protein were associated with improved overall survival (OS) and relapse-free survival (RFS) (Shamanna et al., 2016; Zhu et al., 2018).
Primary cervical cancer samples and cervical cancer cell lines were analyzed for WRN hypermethylation and found in 33.3% of patients and 33.3% of cancer cell lines (Masuda et al., 2012). Treatment with siRNA for WRN increased sensitivity of cancer cells to CPT-11, a topoisomerase I inhibitor. Decreased WRN mRNA expression negatively correlated with cervical cancer progression and WRN protein regulates the life cycle of viral carcinogen human papillomavirus 16 (HPV-16), linked with causing cervical and oropharyngeal cancers (James et al., 2020).
2.7 RECQL4
2.7.1 Germline variants in RECQL4 cause Rothmund-Thompson, RAPADILINO, and Baller-Gerold Syndromes
Rothmund-Thompson Syndrome (RTS) has two clinical subtypes, RTS I and RTS II. RTS I is characterized by poikiloderma, ectodermal dysplasia and juvenile cataracts, and a subset of these patients harbor variants in ANAPC1 (Zirn et al., 2021). RTS II, on the other hand, is caused by autosomal recessive variants in the RECQL4 gene located at the 8q24.3 locus (Wang et al., 2003; Mo et al., 2018; Lu et al., 2020) and is characterized by poikiloderma, congenital bone defects, and an increased risk of osteosarcoma and skin cancers, squamous and basal cell carcinomas (Lu et al., 2020). The developmental defects, including radial ray defects, classically seen in RTS II correlating with the presence of RECQL4 variants suggest that RECQL4 likely plays a crucial role in both normal skeletal development and oncogenesis, in addition to other major signaling pathways such as Wnt, Hedgehog, and Notch (Wang et al., 2003; Hu et al., 2005; Glass and Karsenty, 2007; Cleton-Jansen et al., 2009; Kansara et al., 2009; Tao et al., 2010; Vijayakumar et al., 2011; Belyea et al., 2012; Tao et al., 2014; Lu et al., 2020).
A recent study examined the prevalence of cancer risk in patients carrying monoallelic pathogenic variants in the RECQL4 gene (Martin-Giacalone et al., 2022). Despite some data suggesting that heterozygous germline variants in RECQL4 increase risk of osteosarcoma, in this analysis of an international registry of RTS II patients and their family members, investigators found that RTS II family members with heterozygous germline pathogenic variant in RECQL4 did not have an increased risk of developing cancer, compared to the age-adjusted population estimate per the Surveillance, Epidemiology, and End Results (SEER) program (Maciaszek et al., 2019; Martin-Giacalone et al., 2022).
Other syndromes caused by variants in RECQL4 include RAPADILINO syndrome and Baller-Gerold syndrome. In RAPADILINO syndrome, patients present with skeletal defects, including radial ray and limb deformities, small stature, palatal defects, and absent patella. These patients are susceptible to developing lymphoma and osteosarcoma (Wang et al., 2003). Patients with Baller-Gerold syndrome are typically characterized by craniosynostosis, poikiloderma, and radial ray defects. These patients are predisposed to developing lymphoma (Wang et al., 2003).
2.8 RECQL4 and Cancer
Patients with pathogenic variants in RECQL4 are more susceptible to developing OS, thereby making RTS II a good model that can be used to understand OS and develop targeted therapy. Although OS risk is increased in patients with RTS, RECQL4 variants have not been found in sporadic OS, suggesting that it may be targeted directly in germline but not somatic variants (Wang et al., 2003; Lu et al., 2020).
A previous study indicated that altered RecQ enzyme expression correlated with prognostic outcome in hematologic malignancies (Viziteu et al., 2016). Patients with AML with abnormal karyotype were found to have low RECQL4 and BLM expression, which correlated with adverse outcomes. On the other hand, overexpression of RECQL4 indicated a better prognosis. In multiple myeloma patients, overexpression of RECQL1, WRN, and RECQL4 were associated with poor prognosis (Viziteu et al., 2016).
Upregulated RECQL4 expression has been found to correlate with increased tumor aggressiveness in human prostate cancer cells (Su et al., 2010; Mo et al., 2018). A study demonstrated that RECQL4 inhibition in prostate cancer cells led to significant reduction in invasive growth in vitro and tumorigenic potential in vivo (Su et al., 2010; Mo et al., 2018). The RECQL4 harboring chromosome region (8q24.3) became amplified, and the level of amplification corresponded with tumor aggressiveness. This correlation may be beneficial as a prognostic tool in metastatic prostate cancer (Mo et al., 2018).
Tumors with increased RECQL4 expression may be resistant to radiation therapy (Jiang et al., 2009). After mitochondria in Raji cells were exposed to radiation, proteomic analysis revealed that RECQL4 along with GAPDH, MK167, and ATAD3B proteins can contribute as biomarkers for radiation resistance (Jiang et al., 2009). While it is unclear whether RECQL4 expression in cancer cells is related to radiation resistance, a preliminary study showed that RECQL4 inhibition enhanced radiation sensitivity of prostate cancer cells (Jiang et al., 2009; Mo et al., 2018).
Similar to prostate cancer cells, breast cancer cells show overexpression of RECQL4 (Fang et al., 2013; Mo et al., 2018) A study demonstrated that the majority of the breast cancer cells exhibited gene amplification via increased 8q24 (Fang et al., 2013). Studies reported that RECQL4 is not only a metastasis-promoting gene but also a prognostic indicator in breast cancer, as elevated levels of RECQL4 gene amplification, mRNA, and protein demonstrated considerably increased tumor aggressiveness (Thomassen et al., 2009; Santarpia et al., 2013; Arora et al., 2016a; Mo et al., 2018; Zhu et al., 2018). This association between overexpression and cancer progression highlights the potential clinical benefit of targeting RECQL4 in breast cancer.
A study analyzed five human lines with esophageal squamous cell carcinoma (ESCC) and found that RECQL4 expression was significantly increased in tumor tissues in contrast to non-tumor tissues (Lyu et al., 2021). Additionally, RECQL4 overexpression was associated with worse survival outcomes, including poor tumor differentiation, lymph node invasion, and metastatic disease. When RECQL4 was depleted, the cells were arrested in G0/G1 phase, and cell senescence occurred. Depletion also led to enhanced DNA damage, production of reactive oxygen species and impaired DNA damage response via the phosphorylation or activation of the kinases ATM, ATR, CHK1, and CHK2 (Lyu et al., 2021).
When ovarian cancer tissues were examined, upregulation of RECQL4 positively correlated with enhanced cell proliferation and invasion relating to potential worse survival (Guo et al., 2020). RECQL4 knockout caused cell cycle arrest as well as apoptosis. RECQL4 depletion led to increased sensitivity of the ovarian cancer cells to cisplatin and olaparib, a PARP inhibitor, which suggests that RECQL4 may be a critical component in the resistance of ovarian malignant cells to cisplatin. MAFB, involved in cell differentiation and oncogenesis, is a downstream effector of RECQL4 and expression of MAFB was associated with RECQL4 expression. MAFB silencing led to reduced cell viability, proliferation, and invasion. Furthermore, miR-10a-5p, a tumor suppressor, negatively regulates RECQL4 expression, thus suggesting its role in RECQL4 overexpression and, subsequently, oncogenic effect (Guo et al., 2020).
Similar to esophageal and ovarian cancers, RECQL4 mRNA expression was also increased in human gastric cancer and hepatocellular carcinoma cells, correlating with poor prognosis. Gastric cancer cells with enhanced RECQL4 expression correlated with more extensive invasion, as compared with normal gastric mucosa cells (Chen et al., 2018). Overexpression of RECQL4 in more than half of the seven gastric cancer cell lines analyzed, were not only associated with poor prognosis but also with enhanced resistance to cisplatin via the downstream AKT-YB1-MDR1 signaling pathway (Mo et al., 2016). In the setting of elevated endogenous RECQL4, silencing of ectopic RECQL4 in cisplatin-resistant gastric cancer cells led to reduced activation of this pathway and subsequent resensitization to cisplatin (Mo et al., 2016). In hepatocellular carcinoma cells, RECQL4 overexpression positively correlated with significantly shorter disease-free survival (DFS) and overall survival (OS) times compared with tissue showing lower RECQL4 expression (Li et al., 2018b). These cells demonstrated features of poor prognosis, including elevated a-fetoprotein (AFP) levels and higher staging (Li et al., 2018b).
Overexpression of RECQL4 mRNA and protein levels were also associated with poor survival outcomes in glioblastoma, while silencing of the gene led to significant chemosensitivity. Absence of RECQL4 in glioma cell lines demonstrated increased sensitivity to temozolomide via increased apoptotic proteins (Krol et al., 2020). This data highlights that targeting RECQL4 may potentially improve prognosis of a variety of cancers.
2.9 RECQL5
2.9.1 Germline Variants in RECQL5
Although there are no known syndromes associated with germline variants in RECQL5, there are a few studies that suggest that RECQL5 could be a cancer susceptibility gene. In a Spanish study of 700 families with breast and ovarian cancer who were negative for variants in BRCA1/2, there were deleterious or likely deleterious variants in RECQL5, which was enriched compared to controls (Tavera-Tapia et al., 2019). Polymorphisms in RECQL5 were associated with breast cancer, osteosarcoma, and laryngeal in a Chinese population (He et al., 2014; Qi and Zhou, 2014; Zhi et al., 2014).
2.10 RECQL5 and Cancer
RECQL5 deficiency is associated with genomic instability and thought to lead to cancer. RECQL5 is overexpressed in human urothelial carcinoma of the bladder (UCC) tissue compared to control normal bladder tissue and is associated with negative outcomes (Patterson et al., 2016). RECQL5 depletion in both UCC and normal bladder cells cause a significant decrease in cell survival in only the malignant cells. This differential effect of RECQL5 depletion on UCC cells compared to normal, suggests a role for RECQL5 targeted therapy (Patterson et al., 2016).
While overexpression of RECQL5 in UCC led to poor prognosis, low expression in human gastric carcinoma (GC) samples correlated with worse overall survival (Lin et al., 2020). RECQL5 may be a prognostic indicator in GC, particularly relating to extent of disease invasion and aggressive histology.
Increased RECQL5 mRNA expression was observed in breast cancer cells and associated with poor prognosis (Arora et al., 2016b). High RECQL5 levels correlated with worse phenotypes, including high histological grade, increased HER2 expression, and ER positivity. High RECQL5 protein expression combined with low RAD51 nuclear protein levels also correlated with worse survival outcomes (Arora et al., 2016b).
In the previously mentioned study investigating altered RecQ expression and prognostic value, overexpression of BLM, RECQL1, and RECQL5 in AML patients with normal karyotype were associated with poor prognosis (Viziteu et al., 2016). RECQL5 overexpression was also found in several other hematologic malignancies, such as diffuse large B cell lymphoma and chronic lymphocytic leukemia, correlated with poor outcomes (Viziteu et al., 2016).
Overexpression of RECQL5 has been found in two subtypes of non-small cell lung cancer (NSCLC), lung adenocarcinoma (LUAD) and lung squamous carcinoma (LUSC), along with NSCLC cell lines (Xia et al., 2021). Upon further analysis, it was found that RECQL5 depletion led to not only inhibition of invasion and migration of NSCLC cells but also suppression of lung metastasis. It also inhibited epithelial-mesenchymal transition (EMT), which is a function of metastasis by malignant cells (Valastyan and Weinberg, 2011; Xia et al., 2021).
3 Exploiting RecQ Helicases for Therapeutic Opportunity
To our knowledge, there are no approved or known clinical trials for therapies that directly target RecQ helicases (Futami and Furuichi, 2014; Hengel et al., 2017; Sommers et al., 2019). However, there are ongoing efforts to identify small-molecule inhibitors against RECQL1, BLM, and WRN (Futami and Furuichi, 2014; Hengel et al., 2017; Sommers et al., 2019). As the structure and biochemical interactions of these current biomolecular small-molecule inhibitors are further studied, a deeper understanding of their inhibitory effects in DNA repair pathways may lead to the development of successful targeted therapies.
Most traditional chemotherapy and radiation approaches to treat cancer cause DSBs or other DNA lesions that lead to subsequent cell death. When certain genes involved in DNA repair are altered through mutations or epigenetic silencing, cancer cells may rely on other repair pathways for survival. Synthetic lethality is a model where inhibition of two or more involved pathways can trigger cell death. For example, loss of BRCA1 or BRCA2 results in decreased HR, forcing cancer cells to utilize Poly (ADP-ribose) polymerases (PARP), an enzyme that facilitates repair of single-strand breaks and BER (Ame et al., 2004). When PARP is suppressed and single-strand breaks accumulate, stalled replication forks degrade into DSBs, which are then not able to be repaired in the setting of BRCA-deficient cancer cells. Thus, this synthetic lethality model has led to the development of multiple PARP inhibitors which are now approved to treat several different cancers, most effective in patients with germline BRCA alterations (Bryant et al., 2005; Farmer et al., 2005; Audeh et al., 2010; Tutt et al., 2010; Gelmon et al., 2011; Kaye et al., 2012; Mateo et al., 2016; de Bono et al., 2017).
3.1 RECQL1
Similar to PARP inhibitors, topoisomerase I (TOP1) inhibitors, including camptothecin (CPT), prevent DNA repair through the development of replication-mediated DSBs, and are used for the treatment of several metastatic cancers (Fox et al., 2003; Pommier et al., 2003; Pommier et al., 2006; Berti et al., 2013). TOP1 inhibitors trap the TOP1 cleavage complex, a catalytic intermediate consisting of TOP1 covalently linked to supercoiled DNA, which are then transformed into replication-induced DSBs. Additionally, low doses of TOP1 inhibitors stall replication, further impairing DNA repair (Fox et al., 2003; Pommier et al., 2003; Pommier et al., 2006; Berti et al., 2013). One of the unique features of RECQL1 compared to the rest of the RecQ enzyme family is that it is able to drive recovery of DNA replication forks after they undergo structural reversal that results from replication stress secondary to TOP1 inhibition. One study showed that PARP1 is required to regulate RECQL1 activity in this state and it, therefore, stabilizes regressed forks (Berti et al., 2013). PARP1 suppression combined with TOP1 inhibition, even at low doses, can lead to replication-mediation DSBs (Fox et al., 2003; Pommier et al., 2003; Pommier et al., 2006; Berti et al., 2013). This may imply that when both PARP1 and TOP1 are inhibited in cells depleted of homologous recombination, further inhibition by RECQL1 may potentiate the outcome by TOP1 inhibition and significantly impair DNA repair (Pommier et al., 2006).
As previously mentioned, siRNA silencing of RECQL1 has been shown to kill various HCC, ovarian cancer cell lines, and hypopharyngeal carcinoma and have in vivo activity in mice (Futami et al., 2010; Arai et al., 2011; Sanada et al., 2013). This approach could be effective against other cancers as well. The mechanism of anticancer activity was via mitotic catastrophe, which was even enhanced with the addition of chemotherapeutic agents (Futami et al., 2008b; Futami and Furuichi, 2014).
The study by Viziteu et al. demonstrated that RECQL1 depletion sensitizes multiple myeloma cells to PARPi-induced apoptosis (Viziteu et al., 2016). The data suggests that future therapeutic targets against multiple myeloma can potentially combine DNA methyltransferase inhibitors (DNMTi), PARPi, and inhibitors against RECQL1 helicase to both downregulate RECQL1 activity in replication stress and minimize resistance to chemotherapeutic agents (Viziteu et al., 2017).
3.2 BLM
The first small-molecule inhibitor against BLM is ML216 (1-(4-fluoro-3-trifluoromethyl)phenyl)-3-(5-(pyridine-4-yl)-1,3,4-thiadiazol-2-yl) urea). ML216 and its derivatives inhibit BLM by interfering in the BLM-ssDNA network and impairing cellular proliferation though they do not appear to be highly specific against BLM (Hengel et al., 2017). RNAi-mediated shRNA gene silencing allowed for depletion of BLM, which led to inhibition of specific osteosarcoma cells, implicating its potential use in osteosarcoma (Mao et al., 2010; Futami and Furuichi, 2014).
One study demonstrated that BLM enhances c-Myc turnover by interacting with ligase Fbw7 and promoting its degradation (Chandra et al., 2013). In a mouse xenograft model, BLM inhibits c-Myc dependent colon cancer initiation (Chandra et al., 2013). This opens the possibility of additional pathways that could be attractive therapeutic targets.
3.3 WRN
WRN has been identified as being an attractive synthetic lethal target in the setting of microsatellite unstable cancers (Chan et al., 2019; Kategaya et al., 2019; Lieb et al., 2019). Microsatellite instability (MSI) resulting from DNA mismatch repair deficiency leads to several different cancers, including gastrointestinal cancers and gynecologic cancers (Gurin et al., 1999; Boland and Goel, 2010). Decreased WRN causes catastrophic DNA damage in MSI cells, leading to cell cycle arrest and apoptosis (Chan et al., 2019; Kategaya et al., 2019; Lieb et al., 2019). One mechanism for this synthetic lethality is through accumulation of DSBs at TA-dinucleotide repeats (van Wietmarschen et al., 2020). These TA-dinucleotide repeats are prone to stalls in replication forks, which requires unwinding by the WRN helicase. Without WRN, these TA-dinucleotide repeats are cleaved by MUS81 nuclease, leading to massive genomic instability (van Wietmarschen et al., 2020). This could form the basis of WRN-directed cancer therapy.
With regard to ionizing radiation therapy, WRN-deficient cells are dependent on CHK1 mediated homologous recombination repair (Gupta et al., 2021). It has been shown that the CHK1-p38-MAPK pathway is important in the setting of WRN-deficient cells. Thus, targeting of CHK1 in the setting of WRN deficiency, results in radiosensitivity, as shown in melanoma tumors in vivo (Gupta et al., 2021).
NSC 19630 (1-(propoxymethyl)-maleimide) and its derivative NSC 617145 are the first human DNA small-molecule inhibitors against WRN helicase. Studies have shown that these inhibitors of WRN helicase trap WRN on DNA as well as lead to the accumulation of stalled replication forks and apoptosis, compromising cellular proliferation (Hengel et al., 2017). Treatment with NSC 19630 was able to induce apoptosis in human T-cell leukemia virus type 1 (HTLV-1)-transformed T-cell leukemia/lymphoma-derived cell lines (Moles et al., 2016). This data, in addition to the prevalence of hematologic neoplasms in Werner Syndrome patients, suggests the beneficial impact of inhibiting WRN helicase in leukemia cells in particular.
A large-scale high-throughput screen of about 350,000 small molecules identified other small-molecule inhibitors of WRN helicase (Sommers et al., 2019). NCGC00029283-03 and NCGC00063279-03 are partially reversible inhibitors, which make them suitable drug candidates as irreversible inhibitors may function nonspecifically (Sommers et al., 2019). Both showed favorable chemical properties and biological activity unlike other identified molecules, and highlight the potential role of WRN helicase inhibition (Sommers et al., 2019).
3.4 RECQL4
Cancer stem cells possess high proliferative capacity and resistance to DNA damage and cell death via constitutive checkpoint inhibitor and repair mechanisms (Skvortsova et al., 2015; Abad et al., 2020; Balajee, 2021). The authors of a review explained that RECQL4 expression may be important for stemness, as demonstrated by a positive association between RECQL4 and some stem cell markers, including Myc and CD133 (Balajee, 2021). They proposed that since RECQL4 expression may contribute to the unlimited proliferative potential and survival of neoplastic stem cells, RECQL4 is a potential target that would address these DNA damage-resistant cells. Studies show higher RECQL4 expression was observed in glioblastoma stem cells, whose development was impaired upon RECQL4 inhibition (Krol et al., 2020; Balajee, 2021). Additionally, RECQL4 expression significantly correlates with its downstream target, MAFB, a transcription factor whose inhibition affects aggressiveness of ovarian cancer cells as well as osteosarcoma stem cells (Chen et al., 2020; Guo et al., 2020; Balajee, 2021).
Previous studies also found that the antibiotic heliquinomycin suppressed the replication effects of many DNA helicases, including RECQL4 (Sugiyama et al., 2012; Balajee, 2021). Future therapies can potentially use compounds like heliquinomycin to inhibit RECQL4-induced replication of cancer cells.
3.5 RECQL5
Investigators developed a small-molecule inhibitor, compound 4a, which is a 1,3,4-oxadiazole derivative that specifically targeted RECQL5-positive breast cancers (Chakraborty et al., 2021). By securing the RECQL5-RAD51 interaction, this compound suppressed homologous recombinatorial repair, induced DNA double-stranded breaks, and inhibited tumorigenesis. This process led to preferential killing of RECQL5-expressing cancer cells. In addition to targeting RECQL5 specifically, the compound was also designed to evade neoadjuvant/adjuvant mediated chemoresistance often encountered in breast carcinoma (Chakraborty et al., 2021).
Colorectal carcinomas are often treated with camptothecins (CPTs), including irinotecan and topotecan, which are type I topoisomerase inhibitors. However, tumors may develop resistance against these therapies (Strumberg et al., 2000; Furuta et al., 2003; Wang et al., 2011b). A study investigated whether RECQL5 plays a role in CPT resistance in colorectal carcinoma. When two colorectal cancer cell lines were depleted of RECQL5, CPT sensitivity was remarkedly increased as compared to parental lines containing RECQL5. Additionally, CPTs successfully treated xenograft tumors derived from one of the RECQL5-deficient neoplastic cell lines though not cells consisting of RECQL5. This data suggested that RECQL5 likely contributes to CPT resistance in colorectal carcinoma and may be used as directed targeted therapy to treat colorectal malignant disease (Wang et al., 2011b).
The PI3K/Akt signaling pathway is one studied way in which neoplasms can proliferate and induce EMT as well as cause resistance to platinum-based treatments (Valastyan and Weinberg, 2011; Xia et al., 2021). When Akt inhibitor LY294002 was added, the effects of RECQL5 overexpression were reversed, overall leading to reduced proliferation and metastasis via EMT. RECQL5 knockdown also enhanced apoptosis of cisplatin-resistant cells upon cisplatin treatment (Xia et al., 2021). This study also suggests the therapeutic implications of targeting RECQL5 in non-small cell lung cancer.
4 Conclusion
RecQ helicases are a highly conserved family of enzymes involved in several pathways to maintain genomic stability. Of the five different proteins in humans, RECQL1, BLM, WRN, RECL4, and RECQL5, three are associated with germline alterations associated with an increased risk of cancer. This review has focused on somatic alterations and how loss of RecQ helicase function and upregulated expression have been shown to correlate with tumorigenesis. We also review novel strategies in which RecQ helicases can be exploited for anti-cancer therapies either through direct targeting via small molecules or synthetic lethality. Further studies are needed to better understand the mechanisms of the role of RecQ helicases in cancer which will have broad implications beyond cancer, including in the fields of DNA repair, cancer susceptibility, and aging.
Author Contributions
MT, JL, SM, and VC all contributed to the literature review and writing of this manuscript. VC conceived and designed the structure of the review.
Funding
VC is supported by NIH K08HL138305.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ababou, M. (2021). Bloom Syndrome and the Underlying Causes of Genetic Instability. Mol. Genet. Metab. 133 (1), 35–48. doi:10.1016/j.ymgme.2021.03.003
Abad, E., Graifer, D., and Lyakhovich, A. (2020). DNA Damage Response and Resistance of Cancer Stem Cells. Cancer Lett. 474, 106–117. doi:10.1016/j.canlet.2020.01.008
Abu-Libdeh, B., Jhujh, S. S., Dhar, S., Sommers, J. A., Datta, A., Longo, G. M., et al. (2022). RECON Syndrome Is a Genome Instability Disorder Caused by Mutations in the DNA Helicase RECQL1. J. Clin. Invest. 132 (5), e147301. doi:10.1172/JCI147301
Agrelo, R., Cheng, W. H., Setien, F., Ropero, S., Espada, J., Fraga, M. F., et al. (2006). Epigenetic Inactivation of the Premature Aging Werner Syndrome Gene in Human Cancer. Proc. Natl. Acad. Sci. U. S. A. 103 (23), 8822–8827. doi:10.1073/pnas.0600645103
Ame, J. C., Spenlehauer, C., and de Murcia, G. (2004). The PARP Superfamily. Bioessays 26 (8), 882–893. doi:10.1002/bies.20085
Antczak, A., Kluzniak, W., Wokolorczyk, D., Kashyap, A., Jakubowska, A., Gronwald, J., et al. (2013). A Common Nonsense Mutation of the BLM Gene and Prostate Cancer Risk and Survival. Gene 532 (2), 173–176. doi:10.1016/j.gene.2013.09.079
Arai, A., Chano, T., Futami, K., Furuichi, Y., Ikebuchi, K., Inui, T., et al. (2011). RECQL1 and WRN Proteins Are Potential Therapeutic Targets in Head and Neck Squamous Cell Carcinoma. Cancer Res. 71 (13), 4598–4607. doi:10.1158/0008-5472.can-11-0320
Armes, J. E., Hammet, F., de Silva, M., Ciciulla, J., Ramus, S. J., Soo, W. K., et al. (2004). Candidate Tumor-Suppressor Genes on Chromosome Arm 8p in Early-Onset and High-Grade Breast Cancers. Oncogene 23 (33), 5697–5702. doi:10.1038/sj.onc.1207740
Arora, A., Abdel-Fatah, T. M., Agarwal, D., Doherty, R., Croteau, D. L., Moseley, P. M., et al. (2016). Clinicopathological and Prognostic Significance of RECQL5 Helicase Expression in Breast Cancers. Carcinogenesis 37 (1), 63–71. doi:10.1093/carcin/bgv163
Arora, A., Abdel-Fatah, T. M., Agarwal, D., Doherty, R., Moseley, P. M., Aleskandarany, M. A., et al. (2015). Transcriptomic and Protein Expression Analysis Reveals Clinicopathological Significance of Bloom Syndrome Helicase (BLM) in Breast Cancer. Mol. Cancer Ther. 14 (4), 1057–1065. doi:10.1158/1535-7163.mct-14-0939
Arora, A., Agarwal, D., Abdel-Fatah, T. M., Lu, H., Croteau, D. L., Moseley, P., et al. (2016). RECQL4 Helicase Has Oncogenic Potential in Sporadic Breast Cancers. J. Pathol. 238 (4), 495–501. doi:10.1002/path.4681
Aszterbaum, M., Epstein, J., Oro, A., Douglas, V., LeBoit, P. E., Scott, M. P., et al. (1999). Ultraviolet and Ionizing Radiation Enhance the Growth of BCCs and Trichoblastomas in Patched Heterozygous Knockout Mice. Nat. Med. 5 (11), 1285–1291. doi:10.1038/15242
Audeh, M. W., Carmichael, J., Penson, R. T., Friedlander, M., Powell, B., Bell-McGuinn, K. M., et al. (2010). Oral poly(ADP-Ribose) Polymerase Inhibitor Olaparib in Patients with BRCA1 or BRCA2 Mutations and Recurrent Ovarian Cancer: a Proof-Of-Concept Trial. Lancet 376 (9737), 245–251. doi:10.1016/s0140-6736(10)60893-8
Bachrati, C. Z., and Hickson, I. D. (2008). RecQ Helicases: Guardian Angels of the DNA Replication Fork. Chromosoma 117 (3), 219–233. doi:10.1007/s00412-007-0142-4
Balajee, A. S. (2021). Human RecQL4 as a Novel Molecular Target for Cancer Therapy. Cytogenet Genome Res. 161 (6-7), 305–327. doi:10.1159/000516568
Baris, H. N., Kedar, I., Halpern, G. J., Shohat, T., Magal, N., Ludman, M. D., et al. (2007). Prevalence of Breast and Colorectal Cancer in Ashkenazi Jewish Carriers of Fanconi Anemia and Bloom Syndrome. Isr. Med. Assoc. J. 9 (12), 847–850.
Bejarano, L., Bosso, G., Louzame, J., Serrano, R., Gomez-Casero, E., Martinez-Torrecuadrada, J., et al. (2019). Multiple Cancer Pathways Regulate Telomere Protection. EMBO Mol. Med. 11 (7), e10292. doi:10.15252/emmm.201910292
Belyea, B., Kephart, J. G., Blum, J., Kirsch, D. G., and Linardic, C. M. (2012). Embryonic Signaling Pathways and Rhabdomyosarcoma: Contributions to Cancer Development and Opportunities for Therapeutic Targeting. Sarcoma 2012, 406239. doi:10.1155/2012/406239
Bernstein, D. A., and Keck, J. L. (2003). Domain Mapping of Escherichia coli RecQ Defines the Roles of Conserved N- and C-Terminal Regions in the RecQ Family. Nucleic Acids Res. 31 (11), 2778–2785. doi:10.1093/nar/gkg376
Berti, M., Ray Chaudhuri, A., Thangavel, S., Gomathinayagam, S., Kenig, S., Vujanovic, M., et al. (2013). Human RECQ1 Promotes Restart of Replication Forks Reversed by DNA Topoisomerase I Inhibition. Nat. Struct. Mol. Biol. 20 (3), 347–354. doi:10.1038/nsmb.2501
Birkbak, N. J., Li, Y., Pathania, S., Greene-Colozzi, A., Dreze, M., Bowman-Colin, C., et al. (2018). Overexpression of BLM Promotes DNA Damage and Increased Sensitivity to Platinum Salts in Triple-Negative Breast and Serous Ovarian Cancers. Ann. Oncol. 29 (4), 903–909. doi:10.1093/annonc/mdy049
Bogdanova, N., Pfeifer, K., Schurmann, P., Antonenkova, N., Siggelkow, W., Christiansen, H., et al. (2017). Analysis of a RECQL Splicing Mutation, c.1667_1667+3delAGTA, in Breast Cancer Patients and Controls from Central Europe. Fam. Cancer 16 (2), 181–186. doi:10.1007/s10689-016-9944-y
Bohr, V. A. (2008). Rising from the RecQ-Age: the Role of Human RecQ Helicases in Genome Maintenance. Trends Biochem. Sci. 33 (12), 609–620. doi:10.1016/j.tibs.2008.09.003
Boland, C. R., and Goel, A. (2010). Microsatellite Instability in Colorectal Cancer. Gastroenterology 138 (6), 2073–2087 e3. doi:10.1053/j.gastro.2009.12.064
Bononi, A., Goto, K., Ak, G., Yoshikawa, Y., Emi, M., Pastorino, S., et al. (2020). Heterozygous Germline BLM Mutations Increase Susceptibility to Asbestos and Mesothelioma. Proc. Natl. Acad. Sci. U. S. A. 117 (52), 33466–33473. doi:10.1073/pnas.2019652117
Bosch, L. J., Luo, Y., Lao, V. V., Snaebjornsson, P., Trooskens, G., Vlassenbroeck, I., et al. (2016). WRN Promoter CpG Island Hypermethylation Does Not Predict More Favorable Outcomes for Patients with Metastatic Colorectal Cancer Treated with Irinotecan-Based Therapy. Clin. Cancer Res. 22 (18), 4612–4622. doi:10.1158/1078-0432.ccr-15-2703
Broberg, K., Huynh, E., Schlawicke Engstrom, K., Bjork, J., Albin, M., Ingvar, C., et al. (2009). Association between Polymorphisms in RMI1, TOP3A, and BLM and Risk of Cancer, a Case-Control Study. BMC Cancer 9, 140. doi:10.1186/1471-2407-9-140
Brosh, R. M., von Kobbe, C., Sommers, J. A., Karmakar, P., Opresko, P. L., Piotrowski, J., et al. (2001). Werner Syndrome Protein Interacts with Human Flap Endonuclease 1 and Stimulates its Cleavage Activity. EMBO J. 20 (20), 5791–5801. doi:10.1093/emboj/20.20.5791
Bryan, T. M., Englezou, A., Gupta, J., Bacchetti, S., and Reddel, R. R. (1995). Telomere Elongation in Immortal Human Cells without Detectable Telomerase Activity. EMBO J. 14 (17), 4240–4248. doi:10.1002/j.1460-2075.1995.tb00098.x
Bryant, H. E., Schultz, N., Thomas, H. D., Parker, K. M., Flower, D., Lopez, E., et al. (2005). Specific Killing of BRCA2-Deficient Tumours with Inhibitors of poly(ADP-Ribose) Polymerase. Nature 434 (7035), 913–917. doi:10.1038/nature03443
Bugreev, D. V., Rossi, M. J., and Mazin, A. V. (2011). Cooperation of RAD51 and RAD54 in Regression of a Model Replication Fork. Nucleic Acids Res. 39 (6), 2153–2164. doi:10.1093/nar/gkq1139
Calin, G., Ranzani, G. N., Amadori, D., Herlea, V., Matei, I., Barbanti-Brodano, G., et al. (2001). Somatic Frameshift Mutations in the Bloom Syndrome BLM Gene Are Frequent in Sporadic Gastric Carcinomas with Microsatellite Mutator Phenotype. BMC Genet. 2, 14. doi:10.1186/1471-2156-2-14
Calin, G. A., Gafa, R., Tibiletti, M. G., Herlea, V., Becheanu, G., Cavazzini, L., et al. (2000). Genetic Progression in Microsatellite Instability High (MSI-H) Colon Cancers Correlates with Clinico-Pathological Parameters: A Study of the TGRbetaRII, BAX, hMSH3, hMSH6, IGFIIR and BLM Genes. Int. J. Cancer 89 (3), 230–235. doi:10.1002/1097-0215(20000520)89:3<230::aid-ijc4>3.0.co;2-j
Canto, L. M. D., Cury, S. S., Barros-Filho, M. C., Kupper, B. E. C., Begnami, M., Scapulatempo-Neto, C., et al. (2019). Locally Advanced Rectal Cancer Transcriptomic-Based Secretome Analysis Reveals Novel Biomarkers Useful to Identify Patients According to Neoadjuvant Chemoradiotherapy Response. Sci. Rep. 9 (1), 8702. doi:10.1038/s41598-019-45151-w
Carter, R. J., and Parsons, J. L. (2016). Base Excision Repair, a Pathway Regulated by Posttranslational Modifications. Mol. Cell. Biol. 36 (10), 1426–1437. doi:10.1128/mcb.00030-16
Castro, E., Edland, S. D., Lee, L., Ogburn, C. E., Deeb, S. S., Brown, G., et al. (2000). Polymorphisms at the Werner Locus: II. 1074Leu/Phe, 1367Cys/Arg, Longevity, and Atherosclerosis. Am. J. Med. Genet. 95 (4), 374–380. doi:10.1002/1096-8628(20001211)95:4<374::aid-ajmg14>3.0.co;2-4
Cerami, E., Gao, J., Dogrusoz, U., Gross, B. E., Sumer, S. O., Aksoy, B. A., et al. (2012). The cBio Cancer Genomics Portal: an Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2 (5), 401–404. doi:10.1158/2159-8290.cd-12-0095
Chakraborty, S., Dutta, K., Gupta, P., Das, A., Das, A., Ghosh, S. K., et al. (2021). Targeting RECQL5 Functions, by a Small Molecule, Selectively Kills Breast Cancer In Vitro and In Vivo. J. Med. Chem. 64 (3), 1524–1544. doi:10.1021/acs.jmedchem.0c01692
Chan, E. M., Shibue, T., McFarland, J. M., Gaeta, B., Ghandi, M., Dumont, N., et al. (2019). WRN Helicase Is a Synthetic Lethal Target in Microsatellite Unstable Cancers. Nature 568 (7753), 551–556. doi:10.1038/s41586-019-1102-x
Chandra, S., Priyadarshini, R., Madhavan, V., Tikoo, S., Hussain, M., Mudgal, R., et al. (2013). Enhancement of C-Myc Degradation by BLM Helicase Leads to Delayed Tumor Initiation. J. Cell. Sci. 126 (Pt 16), 3782–3795. doi:10.1242/jcs.124719
Chang, S., Multani, A. S., Cabrera, N. G., Naylor, M. L., Laud, P., Lombard, D., et al. (2004). Essential Role of Limiting Telomeres in the Pathogenesis of Werner Syndrome. Nat. Genet. 36 (8), 877–882. doi:10.1038/ng1389
Chappidi, N., Nascakova, Z., Boleslavska, B., Zellweger, R., Isik, E., Andrs, M., et al. (2020). Fork Cleavage-Religation Cycle and Active Transcription Mediate Replication Restart after Fork Stalling at Co-transcriptional R-Loops. Mol. Cell. 77 (3), 528–541 e8. doi:10.1016/j.molcel.2019.10.026
Chen, H., Yuan, K., Wang, X., Wang, H., Wu, Q., Wu, X., et al. (2018). Overexpression of RECQL4 Is Associated with Poor Prognosis in Patients with Gastric Cancer. Oncol. Lett. 16 (4), 5419–5425. doi:10.3892/ol.2018.9318
Chen, K., Xu, H., and Zhao, J. (2019). Bloom Syndrome Protein Activates AKT and PRAS40 in Prostate Cancer Cells. Oxid. Med. Cell. Longev. 2019, 3685817. doi:10.1155/2019/3685817
Chen, Y., Wang, T., Huang, M., Liu, Q., Hu, C., Wang, B., et al. (2020). MAFB Promotes Cancer Stemness and Tumorigenesis in Osteosarcoma through a Sox9-Mediated Positive Feedback Loop. Cancer Res. 80 (12), 2472–2483. doi:10.1158/0008-5472.CAN-19-1764
Choi, J. M., Kang, S. Y., Bae, W. J., Jin, K. S., Ree, M., and Cho, Y. (2007). Probing the Roles of Active Site Residues in the 3'-5' Exonuclease of the Werner Syndrome Protein. J. Biol. Chem. 282 (13), 9941–9951. doi:10.1074/jbc.m609657200
Chughtai, S. A., Crundwell, M. C., Cruickshank, N. R., Affie, E., Armstrong, S., Knowles, M. A., et al. (1999). Two Novel Regions of Interstitial Deletion on Chromosome 8p in Colorectal Cancer. Oncogene 18 (3), 657–665. doi:10.1038/sj.onc.1202340
Cleary, S. P., Zhang, W., Di Nicola, N., Aronson, M., Aube, J., Steinman, A., et al. (2003). Heterozygosity for the BLM(Ash) Mutation and Cancer Risk. Cancer Res. 63 (8), 1769–1771.
Cleton-Jansen, A. M., Anninga, J. K., Briaire-de Bruijn, I. H., Romeo, S., Oosting, J., Egeler, R. M., et al. (2009). Profiling of High-Grade Central Osteosarcoma and its Putative Progenitor Cells Identifies Tumourigenic Pathways. Br. J. Cancer 101 (12), 2064. doi:10.1038/sj.bjc.6605482
Crabbe, L., Jauch, A., Naeger, C. M., Holtgreve-Grez, H., and Karlseder, J. (2007). Telomere Dysfunction as a Cause of Genomic Instability in Werner Syndrome. Proc. Natl. Acad. Sci. U. S. A. 104 (7), 2205–2210. doi:10.1073/pnas.0609410104
Crabbe, L., Verdun, R. E., Haggblom, C. I., and Karlseder, J. (2004). Defective Telomere Lagging Strand Synthesis in Cells Lacking WRN Helicase Activity. Science 306 (5703), 1951–1953. doi:10.1126/science.1103619
Croteau, D. L., Popuri, V., Opresko, P. L., and Bohr, V. A. (2014). Human RecQ Helicases in DNA Repair, Recombination, and Replication. Annu. Rev. Biochem. 83, 519–552. doi:10.1146/annurev-biochem-060713-035428
Cybulski, C., Carrot-Zhang, J., Kluzniak, W., Rivera, B., Kashyap, A., Wokolorczyk, D., et al. (2015). Germline RECQL Mutations Are Associated with Breast Cancer Susceptibility. Nat. Genet. 47 (6), 643–646. doi:10.1038/ng.3284
d'Adda di Fagagna, F. (2008). Living on a Break: Cellular Senescence as a DNA-Damage Response. Nat. Rev. Cancer 8 (7), 512–522. doi:10.1038/nrc2440
Datta, A., Biswas, K., Sommers, J. A., Thompson, H., Awate, S., Nicolae, C. M., et al. (2021). WRN Helicase Safeguards Deprotected Replication Forks in BRCA2-Mutated Cancer Cells. Nat. Commun. 12 (1), 6561. doi:10.1038/s41467-021-26811-w
Davalos, A. R., and Campisi, J. (2003). Bloom Syndrome Cells Undergo P53-dependent Apoptosis and Delayed Assembly of BRCA1 and NBS1 Repair Complexes at Stalled Replication Forks. J. Cell. Biol. 162 (7), 1197–1209. doi:10.1083/jcb.200304016
Davari, P., Hebert, J. L., Albertson, D. G., Huey, B., Roy, R., Mancianti, M. L., et al. (2010). Loss of Blm Enhances Basal Cell Carcinoma and Rhabdomyosarcoma Tumorigenesis in Ptch1+/- Mice. Carcinogenesis 31 (6), 968–973. doi:10.1093/carcin/bgp309
Davies, S. L., North, P. S., and Hickson, I. D. (2007). Role for BLM in Replication-Fork Restart and Suppression of Origin Firing after Replicative Stress. Nat. Struct. Mol. Biol. 14 (7), 677–679. doi:10.1038/nsmb1267
de Bono, J., Ramanathan, R. K., Mina, L., Chugh, R., Glaspy, J., Rafii, S., et al. (2017). Phase I, Dose-Escalation, Two-Part Trial of the PARP Inhibitor Talazoparib in Patients with Advanced Germline BRCA1/2 Mutations and Selected Sporadic Cancers. Cancer Discov. 7 (6), 620–629. doi:10.1158/2159-8290.cd-16-1250
de Voer, R. M., Hahn, M. M., Mensenkamp, A. R., Hoischen, A., Gilissen, C., Henkes, A., et al. (2015). Deleterious Germline BLM Mutations and the Risk for Early-Onset Colorectal Cancer. Sci. Rep. 5, 14060. doi:10.1038/srep14060
Di Marco, S., Hasanova, Z., Kanagaraj, R., Chappidi, N., Altmannova, V., Menon, S., et al. (2017). RECQ5 Helicase Cooperates with MUS81 Endonuclease in Processing Stalled Replication Forks at Common Fragile Sites during Mitosis. Mol. Cell. 66 (5), 658–671 e8. doi:10.1016/j.molcel.2017.05.006
Dianov, G. L., and Hubscher, U. (2013). Mammalian Base Excision Repair: the Forgotten Archangel. Nucleic Acids Res. 41 (6), 3483–3490. doi:10.1093/nar/gkt076
Ding, S. L., Yu, J. C., Chen, S. T., Hsu, G. C., Kuo, S. J., Lin, Y. H., et al. (2009). Genetic Variants of BLM Interact with RAD51 to Increase Breast Cancer Susceptibility. Carcinogenesis 30 (1), 43–49. doi:10.1093/carcin/bgn233
Ellis, N. A., Groden, J., Ye, T.-Z., Straughen, J., Lennon, D. J., and Ciocci, S. (1995). The Bloom's Syndrome Gene Product Is Homologous to RecQ Helicases. Cell. 83 (4), 655–666. doi:10.1016/0092-8674(95)90105-1
Fang, H., Nie, L., Chi, Z., Liu, J., Guo, D., Lu, X., et al. (2013). RecQL4 Helicase Amplification Is Involved in Human Breast Tumorigenesis. PLoS One 8 (7), e69600. doi:10.1371/journal.pone.0069600
Farmer, H., McCabe, N., Lord, C. J., Tutt, A. N., Johnson, D. A., Richardson, T. B., et al. (2005). Targeting the DNA Repair Defect in BRCA Mutant Cells as a Therapeutic Strategy. Nature 434 (7035), 917–921. doi:10.1038/nature03445
Finley, G. G., Schulz, N. T., Hill, S. A., Geiser, J. R., Pipas, J. M., and Meisler, A. I. (1989). Expression of the Myc Gene Family in Different Stages of Human Colorectal Cancer. Oncogene 4 (8), 963–971.
Fox, B. M., Xiao, X., Antony, S., Kohlhagen, G., Pommier, Y., Staker, B. L., et al. (2003). Design, Synthesis, and Biological Evaluation of Cytotoxic 11-alkenylindenoisoquinoline Topoisomerase I Inhibitors and Indenoisoquinoline-Camptothecin Hybrids. J. Med. Chem. 46 (15), 3275–3282. doi:10.1021/jm0300476
Franchitto, A., Pirzio, L. M., Prosperi, E., Sapora, O., Bignami, M., and Pichierri, P. (2008). Replication Fork Stalling in WRN-Deficient Cells Is Overcome by Prompt Activation of a MUS81-dependent Pathway. J. Cell. Biol. 183 (2), 241–252. doi:10.1083/jcb.200803173
Frank, B., Hoffmeister, M., Klopp, N., Illig, T., Chang-Claude, J., and Brenner, H. (2010). Colorectal Cancer and Polymorphisms in DNA Repair Genes WRN, RMI1 and BLM. Carcinogenesis 31 (3), 442–445. doi:10.1093/carcin/bgp293
Friedberg, E. C., Aguilera, A., Gellert, M., Hanawalt, P. C., Hays, J. B., Lehmann, A. R., et al. (2006). DNA Repair: from Molecular Mechanism to Human Disease. DNA Repair (Amst) 5 (8), 986–996. doi:10.1016/j.dnarep.2006.05.005
Furuta, T., Takemura, H., Liao, Z. Y., Aune, G. J., Redon, C., Sedelnikova, O. A., et al. (2003). Phosphorylation of Histone H2AX and Activation of Mre11, Rad50, and Nbs1 in Response to Replication-dependent DNA Double-Strand Breaks Induced by Mammalian DNA Topoisomerase I Cleavage Complexes. J. Biol. Chem. 278 (22), 20303–20312. doi:10.1074/jbc.m300198200
Futami, K., and Furuichi, Y. (2014). RECQL1 and WRN DNA Repair Helicases: Potential Therapeutic Targets and Proliferative Markers against Cancers. Front. Genet. 5, 441. doi:10.3389/fgene.2014.00441
Futami, K., Kumagai, E., Makino, H., Goto, H., Takagi, M., Shimamoto, A., et al. (2008). Induction of Mitotic Cell Death in Cancer Cells by Small Interference RNA Suppressing the Expression of RecQL1 Helicase. Cancer Sci. 99 (1), 71–80. doi:10.1111/j.1349-7006.2007.00647.x
Futami, K., Kumagai, E., Makino, H., Sato, A., Takagi, M., Shimamoto, A., et al. (2008). Anticancer Activity of RecQL1 Helicase siRNA in Mouse Xenograft Models. Cancer Sci. 99 (6), 1227–1236. doi:10.1111/j.1349-7006.2008.00794.x
Futami, K., Ogasawara, S., Goto, H., Yano, H., and Furuichi, Y. (2010). RecQL1 DNA Repair Helicase: A Potential Tumor Marker and Therapeutic Target against Hepatocellular Carcinoma. Int. J. Mol. Med. 25 (4), 537–545. doi:10.3892/ijmm_00000375
Gao, J., Aksoy, B. A., Dogrusoz, U., Dresdner, G., Gross, B., Sumer, S. O., et al. (2013). Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal 6 (269), pl1. doi:10.1126/scisignal.2004088
Gelmon, K. A., Tischkowitz, M., Mackay, H., Swenerton, K., Robidoux, A., Tonkin, K., et al. (2011). Olaparib in Patients with Recurrent High-Grade Serous or Poorly Differentiated Ovarian Carcinoma or Triple-Negative Breast Cancer: a Phase 2, Multicentre, Open-Label, Non-randomised Study. Lancet Oncol. 12 (9), 852–861. doi:10.1016/s1470-2045(11)70214-5
German, J., Roe, A. M., Leppert, M. F., and Ellis, N. A. (1994). Bloom Syndrome: an Analysis of Consanguineous Families Assigns the Locus Mutated to Chromosome Band 15q26.1. Proc. Natl. Acad. Sci. U. S. A. 91 (14), 6669–6673. doi:10.1073/pnas.91.14.6669
Ghezraoui, H., Piganeau, M., Renouf, B., Renaud, J. B., Sallmyr, A., Ruis, B., et al. (2014). Chromosomal Translocations in Human Cells Are Generated by Canonical Nonhomologous End-Joining. Mol. Cell. 55 (6), 829–842. doi:10.1016/j.molcel.2014.08.002
Ghosh, A. K., Rossi, M. L., Singh, D. K., Dunn, C., Ramamoorthy, M., Croteau, D. L., et al. (2012). RECQL4, the Protein Mutated in Rothmund-Thomson Syndrome, Functions in Telomere Maintenance. J. Biol. Chem. 287 (1), 196–209. doi:10.1074/jbc.m111.295063
Glass, D. A., and Karsenty, G. (2007). In Vivo analysis of Wnt Signaling in Bone. Endocrinology 148 (6), 2630–2634. doi:10.1210/en.2006-1372
Goss, K. H., Risinger, M. A., Kordich, J. J., Sanz, M. M., Straughen, J. E., Slovek, L. E., et al. (2002). Enhanced Tumor Formation in Mice Heterozygous for Blm Mutation. Science 297 (5589), 2051–2053. doi:10.1126/science.1074340
Gravel, S., Chapman, J. R., Magill, C., and Jackson, S. P. (2008). DNA Helicases Sgs1 and BLM Promote DNA Double-Strand Break Resection. Genes. Dev. 22 (20), 2767–2772. doi:10.1101/gad.503108
Gruber, S. B., Ellis, N. A., Scott, K. K., Almog, R., Kolachana, P., Bonner, J. D., et al. (2002). BLM Heterozygosity and the Risk of Colorectal Cancer. Science 297 (5589), 2013. doi:10.1126/science.1074399
Guo, L., Li, Y., Zhao, C., Peng, J., Song, K., Chen, L., et al. (2020). RECQL4, Negatively Regulated by miR-10a-5p, Facilitates Cell Proliferation and Invasion via MAFB in Ovarian Cancer. Front. Oncol. 10, 524128. doi:10.3389/fonc.2020.524128
Gupta, P., Saha, B., Chattopadhyay, S., and Patro, B. S. (2021). Pharmacological Targeting of Differential DNA Repair, Radio-Sensitizes WRN-Deficient Cancer Cells In Vitro and In Vivo. Biochem. Pharmacol. 186, 114450. doi:10.1016/j.bcp.2021.114450
Gurin, C. C., Federici, M. G., Kang, L., and Boyd, J. (1999). Causes and Consequences of Microsatellite Instability in Endometrial Carcinoma. Cancer Res. 59 (2), 462–466.
Gyorffy, B., Lanczky, A., Eklund, A. C., Denkert, C., Budczies, J., Li, Q., et al. (2010). An Online Survival Analysis Tool to Rapidly Assess the Effect of 22,277 Genes on Breast Cancer Prognosis Using Microarray Data of 1,809 Patients. Breast Cancer Res. Treat. 123 (3), 725–731. doi:10.1007/s10549-009-0674-9
Harrigan, J. A., Opresko, P. L., von Kobbe, C., Kedar, P. S., Prasad, R., Wilson, S. H., et al. (2003). The Werner Syndrome Protein Stimulates DNA Polymerase Beta Strand Displacement Synthesis via its Helicase Activity. J. Biol. Chem. 278 (25), 22686–22695. doi:10.1074/jbc.m213103200
He, T. C., Sparks, A. B., Rago, C., Hermeking, H., Zawel, L., da Costa, L. T., et al. (1998). Identification of C-MYC as a Target of the APC Pathway. Science 281 (5382), 1509–1512. doi:10.1126/science.281.5382.1509
He, Y. J., Qiao, Z. Y., Gao, B., Zhang, X. H., and Wen, Y. Y. (2014). Association between RECQL5 Genetic Polymorphisms and Susceptibility to Breast Cancer. Tumour Biol. 35 (12), 12201–12204. doi:10.1007/s13277-014-2528-2
Hengel, S. R., Spies, M. A., and Spies, M. (2017). Small-Molecule Inhibitors Targeting DNA Repair and DNA Repair Deficiency in Research and Cancer Therapy. Cell. Chem. Biol. 24 (9), 1101–1119. doi:10.1016/j.chembiol.2017.08.027
Hickson, I. D. (2003). RecQ Helicases: Caretakers of the Genome. Nat. Rev. Cancer 3 (3), 169–178. doi:10.1038/nrc1012
Hu, H., Hilton, M. J., Tu, X., Yu, K., Ornitz, D. M., and Long, F. (2005). Sequential Roles of Hedgehog and Wnt Signaling in Osteoblast Development. Development 132 (1), 49–60. doi:10.1242/dev.01564
Hu, Y., Raynard, S., Sehorn, M. G., Lu, X., Bussen, W., Zheng, L., et al. (2007). RECQL5/Recql5 Helicase Regulates Homologous Recombination and Suppresses Tumor Formation via Disruption of Rad51 Presynaptic Filaments. Genes. Dev. 21 (23), 3073–3084. doi:10.1101/gad.1609107
Huang, S., Li, B., Gray, M. D., Oshima, J., Mian, I. S., and Campisi, J. (1998). The Premature Ageing Syndrome Protein, WRN, Is a 3'-->5' Exonuclease. Nat. Genet. 20 (2), 114–116. doi:10.1038/2410
James, C. D., Das, D., Morgan, E. L., Otoa, R., Macdonald, A., and Morgan, I. M. (2020). Werner Syndrome Protein (WRN) Regulates Cell Proliferation and the Human Papillomavirus 16 Life Cycle during Epithelial Differentiation. mSphere 5 (5). doi:10.1128/mSphere.00858-20
Janscak, P., Garcia, P. L., Hamburger, F., Makuta, Y., Shiraishi, K., Imai, Y., et al. (2003). Characterization and Mutational Analysis of the RecQ Core of the Bloom Syndrome Protein. J. Mol. Biol. 330 (1), 29–42. doi:10.1016/s0022-2836(03)00534-5
Jiang, Y., Liu, X., Fang, X., and Wang, X. (2009). Proteomic Analysis of Mitochondria in Raji Cells Following Exposure to Radiation: Implications for Radiotherapy Response. Protein Pept. Lett. 16 (11), 1350–1359. doi:10.2174/092986609789353646
Johnson, F. B., Marciniak, R. A., McVey, M., Stewart, S. A., Hahn, W. C., and Guarente, L. (2001). The Saccharomyces cerevisiae WRN Homolog Sgs1p Participates in Telomere Maintenance in Cells Lacking Telomerase. EMBO J. 20 (4), 905–913. doi:10.1093/emboj/20.4.905
Kaiser, S., Sauer, F., and Kisker, C. (2017). The Structural and Functional Characterization of Human RecQ4 Reveals Insights into its Helicase Mechanism. Nat. Commun. 8, 15907. doi:10.1038/ncomms15907
Kansara, M., Tsang, M., Kodjabachian, L., Sims, N. A., Trivett, M. K., Ehrich, M., et al. (2009). Wnt Inhibitory Factor 1 Is Epigenetically Silenced in Human Osteosarcoma, and Targeted Disruption Accelerates Osteosarcomagenesis in Mice. J. Clin. Invest. 119 (4), 837–851. doi:10.1172/jci37175
Karanjawala, Z. E., Grawunder, U., Hsieh, C. L., and Lieber, M. R. (1999). The Nonhomologous DNA End Joining Pathway Is Important for Chromosome Stability in Primary Fibroblasts. Curr. Biol. 9 (24), 1501–1504. doi:10.1016/s0960-9822(00)80123-2
Karmakar, P., Piotrowski, J., Brosh, R. M., Sommers, J. A., Miller, S. P., Cheng, W. H., et al. (2002). Werner Protein Is a Target of DNA-dependent Protein Kinase In Vivo and In Vitro, and its Catalytic Activities Are Regulated by Phosphorylation. J. Biol. Chem. 277 (21), 18291–18302. doi:10.1074/jbc.m111523200
Karmakar, P., Snowden, C. M., Ramsden, D. A., and Bohr, V. A. (2002). Ku Heterodimer Binds to Both Ends of the Werner Protein and Functional Interaction Occurs at the Werner N-Terminus. Nucleic Acids Res. 30 (16), 3583–3591. doi:10.1093/nar/gkf482
Kategaya, L., Perumal, S. K., Hager, J. H., and Belmont, L. D. (2019). Werner Syndrome Helicase Is Required for the Survival of Cancer Cells with Microsatellite Instability. iScience 13, 488–497. doi:10.1016/j.isci.2019.02.006
Kawabe, T., Tsuyama, N., Kitao, S., Nishikawa, K., Shimamoto, A., Shiratori, M., et al. (2000). Differential Regulation of Human RecQ Family Helicases in Cell Transformation and Cell Cycle. Oncogene 19 (41), 4764–4772. doi:10.1038/sj.onc.1203841
Kaye, S. B., Lubinski, J., Matulonis, U., Ang, J. E., Gourley, C., Karlan, B. Y., et al. (2012). Phase II, Open-Label, Randomized, Multicenter Study Comparing the Efficacy and Safety of Olaparib, a Poly (ADP-Ribose) Polymerase Inhibitor, and Pegylated Liposomal Doxorubicin in Patients with BRCA1 or BRCA2 Mutations and Recurrent Ovarian Cancer. J. Clin. Oncol. 30 (4), 372–379. doi:10.1200/jco.2011.36.9215
Kim, K., Min, J., Kirby, T. W., Gabel, S. A., Pedersen, L. C., and London, R. E. (2020). Ligand binding characteristics of the Ku80 von Willebrand domain. DNA Repair (Amst) 85, 102739. doi:10.1016/j.dnarep.2019.102739
Kitao, S., Shimamoto, A., Goto, M., Miller, R. W., Smithson, W. A., Lindor, N. M., et al. (1999). Mutations in RECQL4 Cause a Subset of Cases of Rothmund-Thomson Syndrome. Nat. Genet. 22 (1), 82–84. doi:10.1038/8788
Kluzniak, W., Wokolorczyk, D., Rusak, B., Huzarski, T., Kashyap, A., Stempa, K., et al. (2019). Inherited Variants in BLM and the Risk and Clinical Characteristics of Breast Cancer. Cancers (Basel) 11 (10), 1548. doi:10.3390/cancers11101548
Krol, S. K., Kaczmarczyk, A., Wojnicki, K., Wojtas, B., Gielniewski, B., Grajkowska, W., et al. (2020). Aberrantly Expressed RECQL4 Helicase Supports Proliferation and Drug Resistance of Human Glioma Cells and Glioma Stem Cells. Cancers (Basel) 12 (10). doi:10.3390/cancers12102919
Kudlow, B. A., Kennedy, B. K., and Monnat, R. J. (2007). Werner and Hutchinson-Gilford Progeria Syndromes: Mechanistic Basis of Human Progeroid Diseases. Nat. Rev. Mol. Cell. Biol. 8 (5), 394–404. doi:10.1038/nrm2161
Kusumoto-Matsuo, R., Ghosh, D., Karmakar, P., May, A., Ramsden, D., and Bohr, V. A. (2014). Serines 440 and 467 in the Werner Syndrome Protein Are Phosphorylated by DNA-PK and Affects its Dynamics in Response to DNA Double Strand Breaks. Aging (Albany NY) 6 (1), 70–81. doi:10.18632/aging.100629
Kusumoto-Matsuo, R., Opresko, P. L., Ramsden, D., Tahara, H., and Bohr, V. A. (2010). Cooperation of DNA-PKcs and WRN Helicase in the Maintenance of Telomeric D-Loops. Aging (Albany NY) 2 (5), 274–284. doi:10.18632/aging.100141
Kwong, A., Shin, V. Y., Cheuk, I. W. Y., Chen, J., Au, C. H., Ho, D. N., et al. (2016). Germline RECQL Mutations in High Risk Chinese Breast Cancer Patients. Breast Cancer Res. Treat. 157 (2), 211–215. doi:10.1007/s10549-016-3784-1
Kyng, K. J., May, A., Kolvraa, S., and Bohr, V. A. (2003). Gene Expression Profiling in Werner Syndrome Closely Resembles that of Normal Aging. Proc. Natl. Acad. Sci. U. S. A. 100 (21), 12259–12264. doi:10.1073/pnas.2130723100
Laitman, Y., Boker-Keinan, L., Berkenstadt, M., Liphsitz, I., Weissglas-Volkov, D., Ries-Levavi, L., et al. (2016). The Risk for Developing Cancer in Israeli ATM, BLM, and FANCC Heterozygous Mutation Carriers. Cancer Genet. 209 (3), 70–74. doi:10.1016/j.cancergen.2015.12.006
Lao, V. V., Welcsh, P., Luo, Y., Carter, K. T., Dzieciatkowski, S., Dintzis, S., et al. (2013). Altered RECQ Helicase Expression in Sporadic Primary Colorectal Cancers. Transl. Oncol. 6 (4), 458–469. doi:10.1593/tlo.13238
Lauper, J. M., Krause, A., Vaughan, T. L., and Monnat, R. J. (2013). Spectrum and Risk of Neoplasia in Werner Syndrome: a Systematic Review. PLoS One 8 (4), e59709. doi:10.1371/journal.pone.0059709
Ledet, E. M., Antonarakis, E. S., Isaacs, W. B., Lotan, T. L., Pritchard, C., and Sartor, A. O. (2020). Germline BLM Mutations and Metastatic Prostate Cancer. Prostate 80 (2), 235–237. doi:10.1002/pros.23924
Li, B., and Comai, L. (2000). Functional Interaction between Ku and the Werner Syndrome Protein in DNA End Processing. J. Biol. Chem. 275 (37), 28349–28352. doi:10.1074/jbc.c000289200
Li, D., Frazier, M., Evans, D. B., Hess, K. R., Crane, C. H., Jiao, L., et al. (2006). Single Nucleotide Polymorphisms of RecQ1, RAD54L, and ATM Genes Are Associated with Reduced Survival of Pancreatic Cancer. J. Clin. Oncol. 24 (11), 1720–1728. doi:10.1200/jco.2005.04.4206
Li, J., Jin, J., Liao, M., Dang, W., Chen, X., Wu, Y., et al. (2018). Upregulation of RECQL4 Expression Predicts Poor Prognosis in Hepatocellular Carcinoma. Oncol. Lett. 15 (4), 4248–4254. doi:10.3892/ol.2018.7860
Li, N., Rowley, S. M., Goode, D. L., Amarasinghe, K. C., McInerny, S., Devereux, L., et al. (2018). Mutations in RECQL Are Not Associated with Breast Cancer Risk in an Australian Population. Nat. Genet. 50 (10), 1346–1348. doi:10.1038/s41588-018-0206-9
Li, T., Suo, Q., He, D., Du, W., Yang, M., Fan, X., et al. (2012). Esophageal Cancer Risk Is Associated with Polymorphisms of DNA Repair Genes MSH2 and WRN in Chinese Population. J. Thorac. Oncol. 7 (2), 448–452. doi:10.1097/jto.0b013e31823c487a
Li, X. L., Lu, X., Parvathaneni, S., Bilke, S., Zhang, H., Thangavel, S., et al. (2014). Identification of RECQ1-Regulated Transcriptome Uncovers a Role of RECQ1 in Regulation of Cancer Cell Migration and Invasion. Cell. Cycle 13 (15), 2431–2445. doi:10.4161/cc.29419
Li, Z., Heng, J., Yan, J., Guo, X., Tang, L., Chen, M., et al. (2016). Integrated Analysis of Gene Expression and Methylation Profiles of 48 Candidate Genes in Breast Cancer Patients. Breast Cancer Res. Treat. 160 (2), 371–383. doi:10.1007/s10549-016-4004-8
Lieb, S., Blaha-Ostermann, S., Kamper, E., Rippka, J., Schwarz, C., Ehrenhofer-Wolfer, K., et al. (2019). Werner Syndrome Helicase Is a Selective Vulnerability of Microsatellite Instability-High Tumor Cells. Elife, 8. doi:10.7554/eLife.43333
Lin, Y., Chen, H., Wang, X., Xiang, J., Wang, H., and Peng, J. (2020). Mining the Role of RECQL5 in Gastric Cancer and Seeking Potential Regulatory Network by Bioinformatics Analysis. Exp. Mol. Pathol. 115, 104477. doi:10.1016/j.yexmp.2020.104477
Lindahl, T. (1993). Instability and Decay of the Primary Structure of DNA. Nature 362 (6422), 709–715. doi:10.1038/362709a0
Lowy, A. M., Kordich, J. J., Gismondi, V., Varesco, L., Blough, R. I., and Groden, J. (2001). Numerous Colonic Adenomas in an Individual with Bloom's Syndrome. Gastroenterology 121 (2), 435–439. doi:10.1053/gast.2001.26259
Lu, H., Shamanna, R. A., de Freitas, J. K., Okur, M., Khadka, P., Kulikowicz, T., et al. (2017). Cell Cycle-dependent Phosphorylation Regulates RECQL4 Pathway Choice and Ubiquitination in DNA Double-Strand Break Repair. Nat. Commun. 8 (1), 2039. doi:10.1038/s41467-017-02146-3
Lu, H., Shamanna, R. A., Keijzers, G., Anand, R., Rasmussen, L. J., Cejka, P., et al. (2016). RECQL4 Promotes DNA End Resection in Repair of DNA Double-Strand Breaks. Cell. Rep. 16 (1), 161–173. doi:10.1016/j.celrep.2016.05.079
Lu, L., Jin, W., and Wang, L. L. (2020). RECQ DNA Helicases and Osteosarcoma. Adv. Exp. Med. Biol. 1258, 37–54. doi:10.1007/978-3-030-43085-6_3
Lucic, B., Zhang, Y., King, O., Mendoza-Maldonado, R., Berti, M., Niesen, F. H., et al. (2011). A Prominent Beta-Hairpin Structure in the Winged-Helix Domain of RECQ1 Is Required for DNA Unwinding and Oligomer Formation. Nucleic Acids Res. 39 (5), 1703–1717. doi:10.1093/nar/gkq1031
Luo, G., Santoro, I. M., McDaniel, L. D., Nishijima, I., Mills, M., Youssoufian, H., et al. (2000). Cancer Predisposition Caused by Elevated Mitotic Recombination in Bloom Mice. Nat. Genet. 26 (4), 424–429. doi:10.1038/82548
Luong, T. T., and Bernstein, K. A. (2021). Role and Regulation of the RECQL4 Family during Genomic Integrity Maintenance. Genes. (Basel) 12 (12), 1919. doi:10.3390/genes12121919
Lyu, G., Su, P., Hao, X., Chen, S., Ren, S., Zhao, Z., et al. (2021). RECQL4 Regulates DNA Damage Response and Redox Homeostasis in Esophageal Cancer. Cancer Biol. Med. 18 (1), 120–138. doi:10.20892/j.issn.2095-3941.2020.0105
Machwe, A., Karale, R., Xu, X., Liu, Y., and Orren, D. K. (2011). The Werner and Bloom Syndrome Proteins Help Resolve Replication Blockage by Converting (Regressed) Holliday Junctions to Functional Replication Forks. Biochemistry 50 (32), 6774–6788. doi:10.1021/bi2001054
Machwe, A., Lozada, E., Wold, M. S., Li, G. M., and Orren, D. K. (2011). Molecular Cooperation between the Werner Syndrome Protein and Replication Protein A in Relation to Replication Fork Blockage. J. Biol. Chem. 286 (5), 3497–3508. doi:10.1074/jbc.m110.105411
Maciaszek, J. L., Oak, N., Chen, W., Hamilton, K. V., McGee, R. B., Nuccio, R., et al. (2019). Enrichment of Heterozygous Germline RECQL4 Loss-Of-Function Variants in Pediatric Osteosarcoma. Cold Spring Harb. Mol. Case Stud. 5 (5). doi:10.1101/mcs.a004218
Mao, F. J., Sidorova, J. M., Lauper, J. M., Emond, M. J., and Monnat, R. J. (2010). The Human WRN and BLM RecQ Helicases Differentially Regulate Cell Proliferation and Survival after Chemotherapeutic DNA Damage. Cancer Res. 70 (16), 6548–6555. doi:10.1158/0008-5472.CAN-10-0475
Martin-Giacalone, B. A., Rideau, T-T., Scheurer, M. E., Lupo, P. J., and Wang, L. L. (2022). Cancer Risk Among RECQL4 Heterozygotes. Cancer Genet. 263, 107–110. doi:10.1016/j.cancergen.2022.02.001
Masuda, K., Banno, K., Yanokura, M., Tsuji, K., Kobayashi, Y., Kisu, I., et al. (2012). Association of Epigenetic Inactivation of the WRN Gene with Anticancer Drug Sensitivity in Cervical Cancer Cells. Oncol. Rep. 28 (4), 1146–1152. doi:10.3892/or.2012.1912
Mateo, J., Moreno, V., Gupta, A., Kaye, S. B., Dean, E., Middleton, M. R., et al. (2016). An Adaptive Study to Determine the Optimal Dose of the Tablet Formulation of the PARP Inhibitor Olaparib. Target Oncol. 11 (3), 401–415. doi:10.1007/s11523-016-0435-8
McNally, E. J., Luncsford, P. J., and Armanios, M. (2019). Long Telomeres and Cancer Risk: the Price of Cellular Immortality. J. Clin. Invest. 129 (9), 3474–3481. doi:10.1172/jci120851
Mendoza-Maldonado, R., Faoro, V., Bajpai, S., Berti, M., Odreman, F., Vindigni, M., et al. (2011). The Human RECQ1 Helicase Is Highly Expressed in Glioblastoma and Plays an Important Role in Tumor Cell Proliferation. Mol. Cancer 10, 83. doi:10.1186/1476-4598-10-83
Mitra, S., Hazra, T. K., Roy, R., Ikeda, S., Biswas, T., Lock, J., et al. (1997). Complexities of DNA Base Excision Repair in Mammalian Cells. Mol. Cells 7 (3), 305–312.
Mo, D., Fang, H., Niu, K., Liu, J., Wu, M., Li, S., et al. (2016). Human Helicase RECQL4 Drives Cisplatin Resistance in Gastric Cancer by Activating an AKT-YB1-MDR1 Signaling Pathway. Cancer Res. 76 (10), 3057–3066. doi:10.1158/0008-5472.can-15-2361
Mo, D., Zhao, Y., and Balajee, A. S. (2018). Human RecQL4 Helicase Plays Multifaceted Roles in the Genomic Stability of Normal and Cancer Cells. Cancer Lett. 413, 1–10. doi:10.1016/j.canlet.2017.10.021
Moder, M., Velimezi, G., Owusu, M., Mazouzi, A., Wiedner, M., Ferreira da Silva, J., et al. (2017). Parallel Genome-wide Screens Identify Synthetic Viable Interactions between the BLM Helicase Complex and Fanconi Anemia. Nat. Commun. 8 (1), 1238. doi:10.1038/s41467-017-01439-x
Mojumdar, A. (2020). Mutations in Conserved Functional Domains of Human RecQ Helicases Are Associated with Diseases and Cancer: A Review. Biophys. Chem. 265, 106433. doi:10.1016/j.bpc.2020.106433
Moles, R., Bai, X. T., Chaib-Mezrag, H., and Nicot, C. (2016). WRN-targeted Therapy Using Inhibitors NSC 19630 and NSC 617145 Induce Apoptosis in HTLV-1-Transformed Adult T-Cell Leukemia Cells. J. Hematol. Oncol. 9 (1), 121. doi:10.1186/s13045-016-0352-4
Morozov, V., Mushegian, A. R., Koonin, E. V., and Bork, P. (1997). A Putative Nucleic Acid-Binding Domain in Bloom's and Werner's Syndrome Helicases. Trends Biochem. Sci. 22 (11), 417–418. doi:10.1016/s0968-0004(97)01128-6
Moynahan, M. E., Chiu, J. W., Koller, B. H., and Jasin, M. (1999). Brca1 Controls Homology-Directed DNA Repair. Mol. Cell. 4 (4), 511–518. doi:10.1016/s1097-2765(00)80202-6
Mullen, J. R., Kaliraman, V., and Brill, S. J. (2000). Bipartite Structure of the SGS1 DNA Helicase in Saccharomyces cerevisiae. Genetics 154 (3), 1101–1114. doi:10.1093/genetics/154.3.1101
Mullen, J. R., Kaliraman, V., Ibrahim, S. S., and Brill, S. J. (2001). Requirement for Three Novel Protein Complexes in the Absence of the Sgs1 DNA Helicase in Saccharomyces cerevisiae. Genetics 157 (1), 103–118. doi:10.1093/genetics/157.1.103
Nakayama, R., Sato, Y., Masutani, M., Ogino, H., Nakatani, F., Chuman, H., et al. (2008). Association of a Missense Single Nucleotide Polymorphism, Cys1367Arg of the WRN Gene, with the Risk of Bone and Soft Tissue Sarcomas in Japan. Cancer Sci. 99 (2), 333–339. doi:10.1111/j.1349-7006.2007.00692.x
Nguyen-Dumont, T., Myszka, A., Karpinski, P., Sasiadek, M. M., Akopyan, H., Hammet, F., et al. (2018). FANCM and RECQL Genetic Variants and Breast Cancer Susceptibility: Relevance to South Poland and West Ukraine. BMC Med. Genet. 19 (1), 12. doi:10.1186/s12881-018-0524-x
Nimonkar, A. V., Genschel, J., Kinoshita, E., Polaczek, P., Campbell, J. L., Wyman, C., et al. (2011). BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN Constitute Two DNA End Resection Machineries for Human DNA Break Repair. Genes. Dev. 25 (4), 350–362. doi:10.1101/gad.2003811
Nimonkar, A. V., Ozsoy, A. Z., Genschel, J., Modrich, P., and Kowalczykowski, S. C. (2008). Human Exonuclease 1 and BLM Helicase Interact to Resect DNA and Initiate DNA Repair. Proc. Natl. Acad. Sci. U. S. A. 105 (44), 16906–16911. doi:10.1073/pnas.0809380105
Orren, D. K. (2006). Werner Syndrome: Molecular Insights into the Relationships between Defective DNA Metabolism, Genomic Instability, Cancer and Aging. Front. Biosci. 11, 2657–2671. doi:10.2741/1999
Ouellette, M. M., McDaniel, L. D., Wright, W. E., Shay, J. W., and Schultz, R. A. (2000). The Establishment of Telomerase-Immortalized Cell Lines Representing Human Chromosome Instability Syndromes. Hum. Mol. Genet. 9 (3), 403–411. doi:10.1093/hmg/9.3.403
Parvathaneni, S., Stortchevoi, A., Sommers, J. A., Brosh, R. M., and Sharma, S. (2013). Human RECQ1 Interacts with Ku70/80 and Modulates DNA End-Joining of Double-Strand Breaks. PLoS One 8 (5), e62481. doi:10.1371/journal.pone.0062481
Patel, D. S., Misenko, S. M., Her, J., and Bunting, S. F. (2017). BLM Helicase Regulates DNA Repair by Counteracting RAD51 Loading at DNA Double-Strand Break Sites. J. Cell. Biol. 216 (11), 3521–3534. doi:10.1083/jcb.201703144
Patterson, K., Arya, L., Bottomley, S., Morgan, S., Cox, A., Catto, J., et al. (2016). Altered RECQL5 Expression in Urothelial Bladder Carcinoma Increases Cellular Proliferation and Makes RECQL5 Helicase Activity a Novel Target for Chemotherapy. Oncotarget 7 (46), 76140–76150. doi:10.18632/oncotarget.12683
Picco, G., Cattaneo, C. M., van Vliet, E. J., Crisafulli, G., Rospo, G., Consonni, S., et al. (2021). Werner Helicase Is a Synthetic-Lethal Vulnerability in Mismatch Repair-Deficient Colorectal Cancer Refractory to Targeted Therapies, Chemotherapy, and Immunotherapy. Cancer Discov. 11 (8), 1923–1937. doi:10.1158/2159-8290.cd-20-1508
Pike, A. C., Shrestha, B., Popuri, V., Burgess-Brown, N., Muzzolini, L., Costantini, S., et al. (2009). Structure of the Human RECQ1 Helicase Reveals a Putative Strand-Separation Pin. Proc. Natl. Acad. Sci. U. S. A. 106 (4), 1039–1044. doi:10.1073/pnas.0806908106
Pommier, Y., Barcelo, J. M., Rao, V. A., Sordet, O., Jobson, A. G., Thibaut, L., et al. (2006). Repair of Topoisomerase I-Mediated DNA Damage. Prog. Nucleic Acid. Res. Mol. Biol. 81, 179–229. doi:10.1016/s0079-6603(06)81005-6
Pommier, Y., Redon, C., Rao, V. A., Seiler, J. A., Sordet, O., Takemura, H., et al. (2003). Repair of and Checkpoint Response to Topoisomerase I-Mediated DNA Damage. Mutat. Res. 532 (1-2), 173–203. doi:10.1016/j.mrfmmm.2003.08.016
Popuri, V., Tadokoro, T., Croteau, D. L., and Bohr, V. A. (2013). Human RECQL5: Guarding the Crossroads of DNA Replication and Transcription and Providing Backup Capability. Crit. Rev. Biochem. Mol. Biol. 48 (3), 289–299. doi:10.3109/10409238.2013.792770
Protsenko, E., Rehkopf, D., Prather, A. A., Epel, E., and Lin, J. (2020). Are Long Telomeres Better Than Short? Relative Contributions of Genetically Predicted Telomere Length to Neoplastic and Non-neoplastic Disease Risk and Population Health Burden. PLoS One 15 (10), e0240185. doi:10.1371/journal.pone.0240185
Qi, Y., and Zhou, X. (2014). Haplotype Analysis of RECQL5 Gene and Laryngeal Cancer. Tumour Biol. 35 (3), 2669–2673. doi:10.1007/s13277-013-1351-5
Qian, X., Feng, S., Xie, D., Feng, D., Jiang, Y., and Zhang, X. (2017). RecQ Helicase BLM Regulates Prostate Cancer Cell Proliferation and Apoptosis. Oncol. Lett. 14 (4), 4206–4212. doi:10.3892/ol.2017.6704
Rothkamm, K., Kuhne, M., Jeggo, P. A., and Lobrich, M. (2001). Radiation-induced Genomic Rearrangements Formed by Nonhomologous End-Joining of DNA Double-Strand Breaks. Cancer Res. 61 (10), 3886–3893.
Samavat, H., Xun, X., Jin, A., Wang, R., Koh, W. P., and Yuan, J. M. (2019). Association between Prediagnostic Leukocyte Telomere Length and Breast Cancer Risk: the Singapore Chinese Health Study. Breast Cancer Res. 21 (1), 50. doi:10.1186/s13058-019-1133-0
Sanada, S., Futami, K., Terada, A., Yonemoto, K., Ogasawara, S., Akiba, J., et al. (2013). RECQL1 DNA Repair Helicase: a Potential Therapeutic Target and a Proliferative Marker against Ovarian Cancer. PLoS One 8 (8), e72820. doi:10.1371/journal.pone.0072820
Santarpia, L., Iwamoto, T., Di Leo, A., Hayashi, N., Bottai, G., Stampfer, M., et al. (2013). DNA Repair Gene Patterns as Prognostic and Predictive Factors in Molecular Breast Cancer Subtypes. Oncologist 18 (10), 1063–1073. doi:10.1634/theoncologist.2013-0163
Sassi, A., Popielarski, M., Synowiec, E., Morawiec, Z., and Wozniak, K. (2013). BLM and RAD51 Genes Polymorphism and Susceptibility to Breast Cancer. Pathol. Oncol. Res. 19 (3), 451–459. doi:10.1007/s12253-013-9602-8
Schayek, H., Laitman, Y., Katz, L. H., Pras, E., Ries-Levavi, L., Barak, F., et al. (2017). Colorectal and Endometrial Cancer Risk and Age at Diagnosis in BLMAsh Mutation Carriers. Isr. Med. Assoc. J. 19 (6), 365–367.
Schmutz, I., Mensenkamp, A. R., Takai, K. K., Haadsma, M., Spruijt, L., de Voer, R. M., et al. (2020). TINF2 Is a Haploinsufficient Tumor Suppressor that Limits Telomere Length. Elife, 9. doi:10.7554/eLife.61235
Scully, R., Chen, J., Ochs, R. L., Keegan, K., Hoekstra, M., Feunteun, J., et al. (1997). Dynamic Changes of BRCA1 Subnuclear Location and Phosphorylation State Are Initiated by DNA Damage. Cell. 90 (3), 425–435. doi:10.1016/s0092-8674(00)80503-6
Shamanna, R. A., Lu, H., Croteau, D. L., Arora, A., Agarwal, D., Ball, G., et al. (2016). Camptothecin Targets WRN Protein: Mechanism and Relevance in Clinical Breast Cancer. Oncotarget 7 (12), 13269–13284. doi:10.18632/oncotarget.7906
Shamanna, R. A., Singh, D. K., Lu, H., Mirey, G., Keijzers, G., Salles, B., et al. (2014). RECQ Helicase RECQL4 Participates in Non-homologous End Joining and Interacts with the Ku Complex. Carcinogenesis 35 (11), 2415–2424. doi:10.1093/carcin/bgu137
Sharma, S., Sommers, J. A., Wu, L., Bohr, V. A., Hickson, I. D., and Brosh, R. M. (2004). Stimulation of Flap Endonuclease-1 by the Bloom's Syndrome Protein. J. Biol. Chem. 279 (11), 9847–9856. doi:10.1074/jbc.m309898200
Sharma, S. (2011). Non-B DNA Secondary Structures and Their Resolution by RecQ Helicases. J. Nucleic Acids 2011, 724215. doi:10.4061/2011/724215
Shen, M., Zheng, T., Lan, Q., Zhang, Y., Zahm, S. H., Wang, S. S., et al. (2006). Polymorphisms in DNA Repair Genes and Risk of Non-hodgkin Lymphoma Among Women in Connecticut. Hum. Genet. 119 (6), 659–668. doi:10.1007/s00439-006-0177-2
Singh, T. R., Ali, A. M., Busygina, V., Raynard, S., Fan, Q., Du, C. H., et al. (2008). BLAP18/RMI2, a Novel OB-Fold-Containing Protein, Is an Essential Component of the Bloom Helicase-Double Holliday Junction Dissolvasome. Genes. Dev. 22 (20), 2856–2868. doi:10.1101/gad.1725108
Singleton, M. R., Dillingham, M. S., and Wigley, D. B. (2007). Structure and Mechanism of Helicases and Nucleic Acid Translocases. Annu. Rev. Biochem. 76, 23–50. doi:10.1146/annurev.biochem.76.052305.115300
Sishc, B. J., and Davis, A. J. (2017). The Role of the Core Non-homologous End Joining Factors in Carcinogenesis and Cancer. Cancers (Basel) 9 (7), 81. doi:10.3390/cancers9070081
Skvortsova, I., Debbage, P., Kumar, V., and Skvortsov, S. (2015). Radiation Resistance: Cancer Stem Cells (CSCs) and Their Enigmatic Pro-survival Signaling. Semin. Cancer Biol. 35, 39–44. doi:10.1016/j.semcancer.2015.09.009
Snijders, A. M., Hermsen, M. A., Baughman, J., Buffart, T. E., Huey, B., Gajduskova, P., et al. (2008). Acquired Genomic Aberrations Associated with Methotrexate Resistance Vary with Background Genomic Instability. Genes. Chromosom. Cancer 47 (1), 71–83. doi:10.1002/gcc.20509
Sommers, J. A., Kulikowicz, T., Croteau, D. L., Dexheimer, T., Dorjsuren, D., Jadhav, A., et al. (2019). A High-Throughput Screen to Identify Novel Small Molecule Inhibitors of the Werner Syndrome Helicase-Nuclease (WRN). PLoS One 14 (1), e0210525. doi:10.1371/journal.pone.0210525
Soultanas, P., and Wigley, D. B. (2001). Unwinding the 'Gordian Knot' of Helicase Action. Trends Biochem. Sci. 26 (1), 47–54. doi:10.1016/s0968-0004(00)01734-5
Strumberg, D., Pilon, A. A., Smith, M., Hickey, R., Malkas, L., and Pommier, Y. (2000). Conversion of Topoisomerase I Cleavage Complexes on the Leading Strand of Ribosomal DNA into 5'-phosphorylated DNA Double-Strand Breaks by Replication Runoff. Mol. Cell. Biol. 20 (11), 3977–3987. doi:10.1128/mcb.20.11.3977-3987.2000
Sturzenegger, A., Burdova, K., Kanagaraj, R., Levikova, M., Pinto, C., Cejka, P., et al. (2014). DNA2 Cooperates with the WRN and BLM RecQ Helicases to Mediate Long-Range DNA End Resection in Human Cells. J. Biol. Chem. 289 (39), 27314–27326. doi:10.1074/jbc.m114.578823
Su, Y., Meador, J. A., Calaf, G. M., Proietti De-Santis, L., Zhao, Y., Bohr, V. A., et al. (2010). Human RecQL4 Helicase Plays Critical Roles in Prostate Carcinogenesis. Cancer Res. 70 (22), 9207–9217. doi:10.1158/0008-5472.can-10-1743
Sugiyama, T., Chino, M., Tsurimoto, T., Nozaki, N., and Ishimi, Y. (2012). Interaction of Heliquinomycin with Single-Stranded DNA Inhibits MCM4/6/7 Helicase. J. Biochem. 151 (2), 129–137. doi:10.1093/jb/mvr130
Sun, J., Meng, H., Yao, L., Lv, M., Bai, J., Zhang, J., et al. (2017). Germline Mutations in Cancer Susceptibility Genes in a Large Series of Unselected Breast Cancer Patients. Clin. Cancer Res. 23 (20), 6113–6119. doi:10.1158/1078-0432.ccr-16-3227
Sun, J., Wang, Y., Xia, Y., Xu, Y., Ouyang, T., Li, J., et al. (2015). Mutations in RECQL Gene Are Associated with Predisposition to Breast Cancer. PLoS Genet. 11 (5), e1005228. doi:10.1371/journal.pgen.1005228
Tao, J., Chen, S., and Lee, B. (2010). Alteration of Notch Signaling in Skeletal Development and Disease. Ann. N. Y. Acad. Sci. 1192, 257–268. doi:10.1111/j.1749-6632.2009.05307.x
Tao, J., Jiang, M. M., Jiang, L., Salvo, J. S., Zeng, H. C., Dawson, B., et al. (2014). Notch Activation as a Driver of Osteogenic Sarcoma. Cancer Cell. 26 (3), 390–401. doi:10.1016/j.ccr.2014.07.023
Tavera-Tapia, A., de la Hoya, M., Calvete, O., Martin-Gimeno, P., Fernandez, V., Macias, J. A., et al. (2019). RECQL5: Another DNA Helicase Potentially Involved in Hereditary Breast Cancer Susceptibility. Hum. Mutat. 40 (5), 566–577. doi:10.1002/humu.23732
Telomeres Mendelian Randomization, C., Haycock, P. C., Burgess, S., Nounu, A., Zheng, J., Okoli, G. N., et al. (2017). Association between Telomere Length and Risk of Cancer and Non-neoplastic Diseases: A Mendelian Randomization Study. JAMA Oncol. 3 (5), 636–651. doi:10.1001/jamaoncol.2016.5945
Tervasmaki, A., Mantere, T., Hartikainen, J. M., Kauppila, S., Lee, H. M., Koivuluoma, S., et al. (2018). Rare Missense Mutations in RECQL and POLG Associate with Inherited Predisposition to Breast Cancer. Int. J. Cancer 142 (11), 2286–2292. doi:10.1002/ijc.31259
Thangavel, S., Berti, M., Levikova, M., Pinto, C., Gomathinayagam, S., Vujanovic, M., et al. (2015). DNA2 Drives Processing and Restart of Reversed Replication Forks in Human Cells. J. Cell. Biol. 208 (5), 545–562. doi:10.1083/jcb.201406100
Thomas, K. R., and Capecchi, M. R. (1987). Site-directed Mutagenesis by Gene Targeting in Mouse Embryo-Derived Stem Cells. Cell. 51 (3), 503–512. doi:10.1016/0092-8674(87)90646-5
Thomassen, M., Tan, Q., and Kruse, T. A. (2009). Gene Expression Meta-Analysis Identifies Chromosomal Regions and Candidate Genes Involved in Breast Cancer Metastasis. Breast Cancer Res. Treat. 113 (2), 239–249. doi:10.1007/s10549-008-9927-2
Thompson, E. R., Doyle, M. A., Ryland, G. L., Rowley, S. M., Choong, D. Y., Tothill, R. W., et al. (2012). Exome Sequencing Identifies Rare Deleterious Mutations in DNA Repair Genes FANCC and BLM as Potential Breast Cancer Susceptibility Alleles. PLoS Genet. 8 (9), e1002894. doi:10.1371/journal.pgen.1002894
Traverso, G., Bettegowda, C., Kraus, J., Speicher, M. R., Kinzler, K. W., Vogelstein, B., et al. (2003). Hyper-recombination and Genetic Instability in BLM-Deficient Epithelial Cells. Cancer Res. 63 (24), 8578–8581.
Tripathi, V., Agarwal, H., Priya, S., Batra, H., Modi, P., Pandey, M., et al. (2018). MRN Complex-dependent Recruitment of Ubiquitylated BLM Helicase to DSBs Negatively Regulates DNA Repair Pathways. Nat. Commun. 9 (1), 1016. doi:10.1038/s41467-018-03393-8
Tubbs, A., and Nussenzweig, A. (2017). Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell. 168 (4), 644–656. doi:10.1016/j.cell.2017.01.002
Tutt, A., Robson, M., Garber, J. E., Domchek, S. M., Audeh, M. W., Weitzel, J. N., et al. (2010). Oral poly(ADP-Ribose) Polymerase Inhibitor Olaparib in Patients with BRCA1 or BRCA2 Mutations and Advanced Breast Cancer: a Proof-Of-Concept Trial. Lancet 376 (9737), 235–244. doi:10.1016/s0140-6736(10)60892-6
Urban, V., Dobrovolna, J., Huhn, D., Fryzelkova, J., Bartek, J., and Janscak, P. (2016). RECQ5 Helicase Promotes Resolution of Conflicts between Replication and Transcription in Human Cells. J. Cell. Biol. 214 (4), 401–415. doi:10.1083/jcb.201507099
Valastyan, S., and Weinberg, R. A. (2011). Tumor Metastasis: Molecular Insights and Evolving Paradigms. Cell. 147 (2), 275–292. doi:10.1016/j.cell.2011.09.024
van Wietmarschen, N., Sridharan, S., Nathan, W. J., Tubbs, A., Chan, E. M., Callen, E., et al. (2020). Repeat Expansions Confer WRN Dependence in Microsatellite-Unstable Cancers. Nature 586 (7828), 292–298. doi:10.1038/s41586-020-2769-8
Vijayakumar, S., Liu, G., Rus, I. A., Yao, S., Chen, Y., Akiri, G., et al. (2011). High-frequency Canonical Wnt Activation in Multiple Sarcoma Subtypes Drives Proliferation through a TCF/beta-catenin Target Gene, CDC25A. Cancer Cell. 19 (5), 601–612. doi:10.1016/j.ccr.2011.03.010
Vindigni, A., Marino, F., and Gileadi, O. (2010). Probing the Structural Basis of RecQ Helicase Function. Biophys. Chem. 149 (3), 67–77. doi:10.1016/j.bpc.2010.03.012
Viziteu, E., Kassambara, A., Pasero, P., Klein, B., and Moreaux, J. (2016). RECQ Helicases Are Deregulated in Hematological Malignancies in Association with a Prognostic Value. Biomark. Res. 4, 3. doi:10.1186/s40364-016-0057-4
Viziteu, E., Klein, B., Basbous, J., Lin, Y. L., Hirtz, C., Gourzones, C., et al. (2017). RECQ1 Helicase Is Involved in Replication Stress Survival and Drug Resistance in Multiple Myeloma. Leukemia 31 (10), 2104–2113. doi:10.1038/leu.2017.54
Wang, J., Chen, J., and Gong, Z. (2015). TopBP1 Stabilizes BLM Protein to Suppress Sister Chromatid Exchange. Mol. Cell. 57 (6), 955–956. doi:10.1016/j.molcel.2015.02.011
Wang, L., Kaku, H., Huang, P., Xu, K., Yang, K., Zhang, J., et al. (2011). Single Nucleotide Polymorphism WRN Leu1074Phe Is Associated with Prostate Cancer Susceptibility in Chinese Subjects. Acta Med. Okayama 65 (5), 315–323.
Wang, L., Xie, L., Wang, J., Shen, J., and Liu, B. (2013). Correlation between the Methylation of SULF2 and WRN Promoter and the Irinotecan Chemosensitivity in Gastric Cancer. BMC Gastroenterol. 13, 173. doi:10.1186/1471-230x-13-173
Wang, L. L., Gannavarapu, A., Kozinetz, C. A., Levy, M. L., Lewis, R. A., Chintagumpala, M. M., et al. (2003). Association between Osteosarcoma and Deleterious Mutations in the RECQL4 Gene in Rothmund-Thomson Syndrome. J. Natl. Cancer Inst. 95 (9), 669–674. doi:10.1093/jnci/95.9.669
Wang, Q., Lv, H., Lv, W., Shi, M., Zhang, M., Luan, M., et al. (2015). Genome-wide Haplotype Association Study Identifies BLM as a Risk Gene for Prostate Cancer in Chinese Population. Tumour Biol. 36 (4), 2703–2707. doi:10.1007/s13277-014-2893-x
Wang, X., Lu, X., Zhou, G., Lou, H., and Luo, G. (2011). RECQL5 Is an Important Determinant for Camptothecin Tolerance in Human Colorectal Cancer Cells. Biosci. Rep. 31 (5), 363–369. doi:10.1042/bsr20100108
Wang, Z., Xu, Y., Tang, J., Ma, H., Qin, J., Lu, C., et al. (2009). A Polymorphism in Werner Syndrome Gene Is Associated with Breast Cancer Susceptibility in Chinese Women. Breast Cancer Res. Treat. 118 (1), 169–175. doi:10.1007/s10549-009-0327-z
White, R. R., and Vijg, J. (2016). Do DNA Double-Strand Breaks Drive Aging? Mol. Cell. 63 (5), 729–738. doi:10.1016/j.molcel.2016.08.004
Wirtenberger, M., Frank, B., Hemminki, K., Klaes, R., Schmutzler, R. K., Wappenschmidt, B., et al. (2006). Interaction of Werner and Bloom Syndrome Genes with P53 in Familial Breast Cancer. Carcinogenesis 27 (8), 1655–1660. doi:10.1093/carcin/bgi374
Wu, L., and Hickson, I. D. (2003). The Bloom's Syndrome Helicase Suppresses Crossing over during Homologous Recombination. Nature 426 (6968), 870–874. doi:10.1038/nature02253
Wyllie, F. S., Jones, C. J., Skinner, J. W., Haughton, M. F., Wallis, C., Wynford-Thomas, D., et al. (2000). Telomerase Prevents the Accelerated Cell Ageing of Werner Syndrome Fibroblasts. Nat. Genet. 24 (1), 16–17. doi:10.1038/71630
Xia, H. W., Zhang, Z. Q., Yuan, J., and Niu, Q. L. (2021). Human RECQL5 Promotes Metastasis and Resistance to Cisplatin in Non-small Cell Lung Cancer. Life Sci. 265, 118768. doi:10.1016/j.lfs.2020.118768
Xu, L., Geman, D., and Winslow, R. L. (2007). Large-scale Integration of Cancer Microarray Data Identifies a Robust Common Cancer Signature. BMC Bioinforma. 8, 275. doi:10.1186/1471-2105-8-275
Yankiwski, V., Marciniak, R. A., Guarente, L., and Neff, N. F. (2000). Nuclear Structure in Normal and Bloom Syndrome Cells. Proc. Natl. Acad. Sci. U. S. A. 97 (10), 5214–5219. doi:10.1073/pnas.090525897
Yu, C. E., Oshima, J., Fu, Y. H., Wijsman, E. M., Hisama, F., Alisch, R., et al. (1996). Positional Cloning of the Werner's Syndrome Gene. Science 272 (5259), 258–262. doi:10.1126/science.272.5259.258
Zhao, M., Chen, Z., Zheng, Y., Liang, J., Hu, Z., Bian, Y., et al. (2020). Identification of Cancer Stem Cell-Related Biomarkers in Lung Adenocarcinoma by Stemness Index and Weighted Correlation Network Analysis. J. Cancer Res. Clin. Oncol. 146 (6), 1463–1472. doi:10.1007/s00432-020-03194-x
Zharkov, D. O. (2008). Base Excision DNA Repair. Cell. Mol. Life Sci. 65 (10), 1544–1565. doi:10.1007/s00018-008-7543-2
Zheng, L., Kanagaraj, R., Mihaljevic, B., Schwendener, S., Sartori, A. A., Gerrits, B., et al. (2009). MRE11 Complex Links RECQ5 Helicase to Sites of DNA Damage. Nucleic Acids Res. 37 (8), 2645–2657. doi:10.1093/nar/gkp147
Zhi, L. Q., Ma, W., Zhang, H., Zeng, S. X., and Chen, B. (2014). Association of RECQL5 Gene Polymorphisms and Osteosarcoma in a Chinese Han Population. Tumour Biol. 35 (4), 3255–3259. doi:10.1007/s13277-013-1425-4
Zhou, X., Xu, X., Tian, Z., Xu, W. Y., and Cui, Y. (2020). Mutational Profiling of Lung Adenocarcinoma in China Detected by Next-Generation Sequencing. J. Cancer Res. Clin. Oncol. 146 (9), 2277–2287. doi:10.1007/s00432-020-03284-w
Zhu, X., Chen, H., Yang, Y., Xu, C., Zhou, J., Zhou, J., et al. (2018). Distinct Prognosis of mRNA Expression of the Five RecQ DNA-Helicase Family Members - RECQL, BLM, WRN, RECQL4, and RECQL5-In Patients with Breast Cancer. Cancer Manag. Res. 10, 6649–6668. doi:10.2147/cmar.s185769
Zimmer, K., Puccini, A., Xiu, J., Baca, Y., Spizzo, G., Lenz, H. J., et al. (2020). WRN-mutated Colorectal Cancer Is Characterized by a Distinct Genetic Phenotype. Cancers (Basel) 12 (5), 1319. doi:10.3390/cancers12051319
Zins, K., Frech, B., Taubenschuss, E., Schneeberger, C., Abraham, D., and Schreiber, M. (2015). Association of the Rs1346044 Polymorphism of the Werner Syndrome Gene RECQL2 with Increased Risk and Premature Onset of Breast Cancer. Int. J. Mol. Sci. 16 (12), 29643–29653. doi:10.3390/ijms161226192
Keywords: RecQ helicases, RECQ1, BLM, WRN, Recql4, Recql5, cancer
Citation: Thakkar MK, Lee J, Meyer S and Chang VY (2022) RecQ Helicase Somatic Alterations in Cancer. Front. Mol. Biosci. 9:887758. doi: 10.3389/fmolb.2022.887758
Received: 01 March 2022; Accepted: 23 May 2022;
Published: 15 June 2022.
Edited by:
Francesca M. Pisani, National Research Council (CNR), ItalyReviewed by:
Yuliang Wu, University of Saskatchewan, CanadaHuiming Lu, University of Texas Southwestern Medical Center, United States
Copyright © 2022 Thakkar, Lee, Meyer and Chang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vivian Y. Chang, dmNoYW5nQG1lZG5ldC51Y2xhLmVkdQ==