
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Biosci. , 28 April 2022
Sec. Molecular Diagnostics and Therapeutics
Volume 9 - 2022 | https://doi.org/10.3389/fmolb.2022.865494
This article is part of the Research Topic Methods and Applications in Molecular Diagnostics View all 9 articles
 Ewelina Bukowska-Olech1*†
Ewelina Bukowska-Olech1*† Anna Sowińska-Seidler1†‡
Anna Sowińska-Seidler1†‡ Dawid Larysz2,3†‡
Dawid Larysz2,3†‡ Paweł Gawliński4†‡
Paweł Gawliński4†‡ Grzegorz Koczyk5,6†‡
Grzegorz Koczyk5,6†‡ Delfina Popiel5†
Delfina Popiel5† Lidia Gurba-Bryśkiewicz7†Anna Materna-Kiryluk1,5†Zuzanna Adamek8
Lidia Gurba-Bryśkiewicz7†Anna Materna-Kiryluk1,5†Zuzanna Adamek8 Aleksandra Szczepankiewicz9†Paweł Dominiak8‡Filip Glista8‡Karolina Matuszewska1,5†
Aleksandra Szczepankiewicz9†Paweł Dominiak8‡Filip Glista8‡Karolina Matuszewska1,5† Aleksander Jamsheer1,5*†
Aleksander Jamsheer1,5*†Background: Craniosynostosis (CS) represents a highly heterogeneous genetic condition whose genetic background has not been yet revealed. The abnormality occurs either in isolated form or syndromic, as an element of hundreds of different inborn syndromes. Consequently, CS may often represent a challenging diagnostic issue.
Methods: We investigated a three-tiered approach (karyotyping, Sanger sequencing, followed by custom gene panel/chromosomal microarray analysis, and exome sequencing), coupled with prioritization of variants based on dysmorphological assessment and description in terms of human phenotype ontology. In addition, we have also performed a statistical analysis of the obtained clinical data using the nonparametric test χ2.
Results: We achieved a 43% diagnostic success rate and have demonstrated the complexity of mutations’ type harbored by the patients, which were either chromosomal aberrations, copy number variations, or point mutations. The majority of pathogenic variants were found in the well-known CS genes, however, variants found in genes associated with chromatinopathies or RASopathies are of particular interest.
Conclusion: We have critically summarized and then optimised a cost-effective diagnostic algorithm, which may be helpful in a daily diagnostic routine and future clinical research of various CS types. Moreover, we have pinpointed the possible underestimated co-occurrence of CS and intellectual disability, suggesting it may be overlooked when intellectual disability constitutes a primary clinical complaint. On the other hand, in any case of already detected syndromic CS and intellectual disability, the possible occurrence of clinical features suggestive for chromatinopathies or RASopathies should also be considered.
Premature fusion of calvarial sutures, i.e., craniosynostosis (CS), represents a highly heterogeneous neurocranium malformation (Morriss-Kay and Wilkie, 2005). The disease can be classified following at least three criteria: the origin, the number of affected sutures, and the occurrence of additional clinical features. Regarding the etiology, CS is distinguished as a primary condition, i.e., genetic; or secondary, if it arises from mechanical, metabolic, or hormonal defects. The assessment of affected sutures’ numbers allows recognising single or compound CS, and finally, a syndromic CS is described when additional features other than secondary to premature suture fusion occur. Otherwise, CS should be classified as an isolated condition (Wilkie et al., 2007; Johnson and Wilkie, 2011; Lattanzi et al., 2017).
The neonatal skull comprises six calvarial sutures—one metopic, one sagittal, two coronal, and two lambdoid, closing physiologically from 3 months to late 50 years of postnatal life. Calvarial sutures are fibrous junctions, which allow the skull to grow and develop during the expansion of the brain and permit skull compression during delivery (Baer, 1954; Opperman, 2000; Rice, 2008). Thus, the lack of physiological sutural patency impedes the allometric cranial growth, resulting in cranial deformities and, frequently, increased intracranial pressure, i.e., craniostenosis. Consequently, affected patients present with facial dysmorphism, cortex lesion, seizures, intellectual disability, visual and hearing impairments, or breathing difficulties that are all secondary to CS (Renier et al., 1982; Thompson et al., 1995; Tubbs et al., 2001; Gupta et al., 2003; Mathijssen and Arnaud, 2007; Chieffo et al., 2010).
CS affects approximately one in 2,500 births and burdens public health due to the requirement of extensive surgical treatment in the first year of life and multi-level specialist medical care in the subsequent postnatal periods (Wilkie et al., 2010; Lattanzi et al., 2017). Despite recent advancements in genetic diagnostics, the pathogenesis of CS remains still unknown or partially understood. The large cohort screenings reveal genetic etiology in barely 21–62% of all recruited cases, depending on the size of the study, ethnicity of the population, and range of the molecular analysis. Conversely, about 40–80% of CS cases remain molecularly unresolved (Roscioli et al., 2013; Paumard-Hernandez et al., 2015; Timberlake et al., 2016; Lee et al., 2018; Topa et al., 2020). In this paper, we have presented the study results encompassing 166 individuals in whom we had applied a three-step diagnostic algorithm to identify different mutation types. In addition, we have also performed a statistical analysis of the clinical data we had obtained.
All procedures involving human participants were performed under the Helsinki Declaration. The Institutional Review Board of Poznan University of Medical Sciences granted ethics approval (no. 742/17). All patients agreed to participate in this study, and written informed consent for participation and publishing the information and images in an online open-access publication was obtained from all participants and the parents of minors before genetic testing.
The patients were recruited provided they were born from pregnancies without exposure to environmental factors potentially causative for CS. Our cohort consisted of 166 individuals (33 patients belonging to 18 families and 133 sporadic patients), of whom 85 were males and 81 females. All patients underwent dysmorphological assessment, which allowed us to section off the two subgroups—isolated (if CS was accompanied only by the secondary defects directly resulting from CS) and syndromic (if CS was accompanied by additional defects, not resulting from CS).
STATISTICA (version 13.3) TIBCO software was used for data analysis. The statistical significance of the phenotypic diversity of the cohort and the differences between the frequency of identified genetic modification in the different phenotypic groups was tested using the nonparametric test χ2.
We extracted genomic DNA from peripheral blood leukocytes drawn into EDTA-coated tubes using either the manual salting-out method or automated extraction using the MagCore® HF16 Automated Nucleic Acid Extractor (RBC Bioscience Corp.). Whole blood for lymphocyte culture was drawn into heparin-coated tubes. The study has been divided into three tiers. Tier 1 included karyotyping and screening of the most frequent mutations located in exon no. 7 of FGFR1 (NM_023110.3), exons no. 7 and 8 of FGFR2 (NM_000141.5), and exon no. 7 of FGFR3 (NM_000142.5), and the entire coding sequence of TWIST1 (NM_000474.4). In tier 2, chromosomal microarray analysis and targeted next-generation sequencing (NGS) of custom gene panel were performed. Exome sequencing (ES) was applied in Tier 3.
We performed molecular screening for all recruited patients (n = 166) utilizing targeted PCR followed by Sanger sequencing. We tested the occurrence of the most frequent, recurrent mutations located in exon 7 of FGFR1 (NM_023110.3), exons 7 and 8 of FGFR2 (NM_000141.5), and exon 7 of FGFR3 (NM_000142.5), and the entire coding sequence of TWIST1 (NM_000474.4). Specific primers for amplification were designed using the online available Primer3 tool v. 0.4.0. For the detailed list of primers, see Supplementary Table S1. PCR products were sequenced using dye-terminator chemistry (kit v.3, ABI 3130XL) and run on automated sequencer Applied Biosystems Prism 3700 DNA Analyzer.
Whole blood lymphocyte culture was performed following the standard protocol. Next, we used the Giemsa-banding (GTG) technique at 550 band resolution per haploid genome.
Patients suspected of harboring pathogenic copy number variations (CNVs) were tested using (CMA). Depending on the research stages’, different CMA formats were used. Firstly, the assay was performed employing a high resolution 1.4 NimbleGen oligonucleotide array comparative genomic hybridization (aCGH; Roche NimbleGen) according to standard protocols provided by the manufacturer. Results were analysed with Deva software (Roche Nimblegen) using the ADM2 segmentation algorithm (Sowińska-Seidler et al., 2018). The chromosomal profile was visualized using SignalMap software (NimbleGen Systems Inc.). Next, we applied SurePrint G3 Human CGH Microarray 8 × 60 k, 4 × 180 k, 1 × 1M arrays (Agilent Technologies). The hybridization signals were detected with SureScan Dx Microarray Scanner (Agilent Technologies) and visualized with the use of Agilent CytoGenomics software (Agilent Technologies) (Bukowska-Olech et al., 2020b). The pathogenicity of CNVs was evaluated using the following tools and available online databases-Cytoscape 3.7.1, ClinGen, DECIPHER, database of Genomic Variants (DGV), Mouse Genome Informatics (MGI), or UCSC Genome Browser applying tracks such as Conservation, VistaEnhancers, ENCODE Regulation or HiC.
A cohort with negative results was subjected to targeted next-generation sequencing of a custom 225.709 kb in size gene panel (Agilent Technologies). Captured and indexed libraries were sequenced on the previously described Ion Torrent S5 sequencing system. Variants identified by TorrentSuite, as first described by Bukowska-Olech et al., were further analyzed using an extended custom pipeline (Bukowska-Olech et al., 2020c). For prioritizing candidate variants in probably causative genes, local Phen2Gene installation was used. For final SNV/indel prioritization, the updated pipeline combined Exomiser 12.1.0 with ANNOVAR (all non-commercially available databases, downloaded on 16th December 2020), Ensembl/VEP 102.0, and CADD 1.6 Exomiser default phenotype scoring was supplemented with alternate scoring where the original formula used Phen2Gene scores instead (Zhao et al., 2020). Two prioritizers (OMIM and HIPHIVE) were used with Exomiser. For both Phen2Gene and Exomiser, each sample was labeled with Human Phenotype Ontology terms assigned according to clinical notes (manual curation) (Supplementary Table S2). Common (AF>0.001 in population frequency datasets) and benign (as per strong ClinVar support) alleles were dropped out for reporting. The final pathogenicity of detected variants was analysed in line with the American College of Medical Genetics (ACMG) classification (Richards et al., 2015). Confirmation and segregation studies were performed applying PCR followed by Sanger sequencing as described in section PCR and Sanger sequencing. A list of primers was summarized in Supplementary Table S1.
The coding region and flanking intronic regions were enriched using a custom-designed in-solution exome enrichment (TWIST bioscience, San Francisco, United States) and were sequenced using the Illumina NovaSeq system (Illumina, San Diego, United States). Sequencing reads were demultiplexed using Illumina bcl2fastq2. The removal of the adapter was performed with Skewer. The trimmed reads were mapped to the human reference genome (hg19) using the Burrows-Wheeler Aligner, and variants were called using in-house software. First, Only SNVs and small indels in the coding regions and the flanking intronic regions (±8 bp) with a minor allele frequency (MAF) < 1.5% were evaluated. Second, the known disease-causing variants (according to Human Gene Mutation database; HGMD) were also evaluated in up to ±30 bp of flanking regions and up to 5% MAF. Downstream analysis was carried out using pipeline described for Tier 2 above. As before, the variant evaluation was based on the ACMG guidelines for interpreting sequence variants. Confirmation and segregation studies were performed applying PCR followed by Sanger sequencing as described in section PCR and Sanger sequencing. A list of primers was summarized in Supplementary Table S1.
We summarized all differentiating phenotypic features in our cohort in Table 1 and Supplementary Table S3. Patients involved in this study were more frequently sporadic (81%) than familial cases (19%)–χ2 (1;166) = 62.67; p < 0.001. We have not revealed any differences between the occurrence of isolated (56%) and syndromic forms of CS (44%)–χ2 (1; 146) = 2.22; p = 0.14. However, we have reported more frequently single CS (66%) than multiple CS (34%)–χ2 (1;144) = 14.69; p < 0.001. Next, we have also shown that the most often affected suture in isolated CS was coronal (53%), followed by metopic (33%), sagittal (15%), and lambdoid (0%)–χ2 (3;95) = 51.54; p < 0.001.

TABLE 1. The phenotypic characterization of the cohort of 166 patients affected with craniosynostosis.
Our diagnostic success was 43% (n = 72). The following paragraphs described the detailed results, entitled Tier 1–3, which were also summarized in Tables 2–4. Mutations located within the FGFR1, FGFR2, FGFR3 genes, and in the TWIST1 gene constitute the most often occurring alterations (73%)–χ2 (1;71) = 14.43; p < 0.001. This diagnostic indicator occurred significantly statistic (χ2 (1;52) = 4.92; p < 0.05) more often in a female group of patients (48%, FGFR2 was the most frequently affected–19%) than male–25% (the most common pathogenic variants were located in the TWIST1 gene–12%).
Regarding sex, we have not revealed any relevant diagnostic success rate changes, which were made in 62% among female patients and 38% among male patients–χ2 (1;71) = 3.61; p = 0.06. Next, we have shown that more often, we could diagnose the patients affected with multiple CS (65%) than single CS (35%)–χ2 (1;49) = 4.50; p = 0.03. Detailed results were summarized in Supplementary Table S4.
PCR followed by Sanger sequencing of the most frequent mutations located within the FGFR1, FGFR2, FGFR3 genes and screening of the entire TWIST1 coding sequence allowed us to diagnose 43 patients from 32 different families. Out of them, three patients carried the same alteration in the 7th exon of FGFR1 gene, 15 individuals presented with one from 10 mutations in the FGFR2 gene, 10 patients harbored one recurrent variant in the FGFR3, whereas 15 patients harbored nine distinct variants in the TWIST1 gene (Table 2). Karyotyping revealed three heterozygous deletions: one in locus 7q32.3-q35, second in locus 9p, and third in locus 18q21.32-q23 (Supplementary Table S5). The first two were additionally resized using 4 × 180 k Agilent CMA, while the third was by ES.
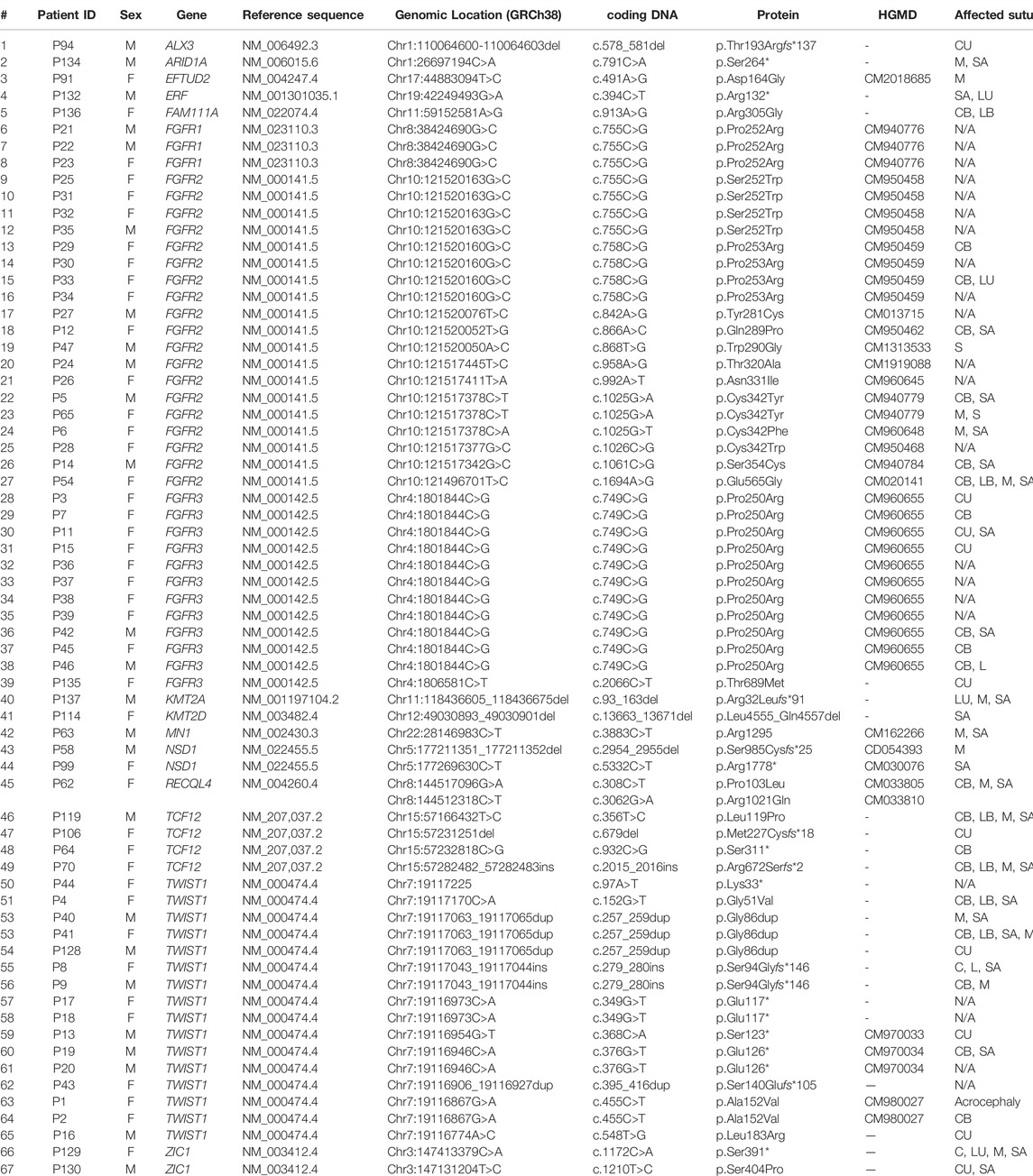
TABLE 2. The list of point variants found in the cohort of 166 patients affected with craniosynostosis. HGMD, Human Gene Mutation database (accession date: October 2021); B, bilateral; C, coronal; I, isolated; L, lambdoid; M, metopic; SA, sagittal; U, unilateral.
We have detected four CNVs in four individuals using CMA, i.e., three duplications in locus 1q22-q23.1, locus 2p21 encompassing solely the SIX2 gene, locus 17p13.3, and one deletion in locus 5q35.3, which included exons 18–21 in the NSD1 gene (Table 3; Supplementary Table S5) (Sowińska-Seidler et al., 2018). Targeted NGS of a custom gene panel allowed us to establish the molecular diagnosis in the subsequent 14 sporadic patients (15 variants) (Table 2; Supplementary Table S6). We have found 15 following heterozygous variants, from which 9 were not reported in HGMD–c.578_581del p.Thr193Argfs*137 in the ALX3 gene (variant of unknown significance, VUS), c.491A>G p. Asp164Gly in the EFTUD2 gene, c.394C>T p.Arg132* in the ERF gene (linked to Craniosynostosis 4), c.868T>G p.Trp290Gly (HGMD no: CM1313533), c.1025G>A p.Cys342Tyr (HGMD no: CM940779), c.1694A>G p.Glu565Gly (HGMD no: CM020141) in the FGFR2 gene (HGMD no: CM020141), and c.2066C>T p.Thr689Met in the FGFR3 gene, c.356T>C p.Leu119Pro, c.679del p.Met227Cysfs*18, c.932C>G p.Ser311*, c.2015_2016ins p.Arg672Serfs*2 in the TCF12 gene (linked to Craniosynostosis 3), c.1172C>A p.Ser391*, c.1210T>C p.Ser404Pro (VUS) in the ZIC1 gene (linked to Craniosynostosis 6) and two alterations in compound heterozygosity c.308C>T p.Pro103Leu (HGMD no: CM033805), and c.3062G>A p.Arg1021Gln (HGMD no: CM033810) located within the RECQL4 gene (linked to Rothmund-Thomson, Baller-Gerold, and RAPADILINO syndromes).

TABLE 3. The list of de novo aberrations and copy number variations (CNVs) found in the cohort of 166 patients affected by syndromic craniosynostosis. ISCN, International System for Human Cytogenetic Nomenclature; N/A, not applicable. P140 was diagnosed with Sotos syndrome, P142 with 17p13.3 microduplication syndrome class I.
Finally, applying ES, we revealed seven heterozygous variants in the subsequent seven patients–c.791C>A p.Ser264* in the ARID1A gene (linked to Coffin-Siris type 2 syndrome), c.93_163del p.Arg32Leufs*91 in the KMT2A gene (linked to Wiedemann-Steiner syndrome), c.13663_13671del p.Leu4555_Gln4557del in the KMT2D gene (linked to Kabuki type 1 syndrome), c.3883C>T p.Arg1295* in the MN1 gene (linked to MN1 C-terminal truncation syndrome; MCTT syndrome, and CEBALID syndrome), c.2954_2955del p.Ser985Cysfs*25, and c.5332C>T p.Arg1778* in the NSD1 gene (linked to Sotos type 1 syndrome), and c.913A>G p.Arg305Gly in FAM111A (linked to Gracile bone dysplasia, and Kenny-Caffey syndrome type 2) (Table 2; Supplementary Table S6). Those variants were not reported in the medical literature, except for mutations detected in the NSD1 gene–p.Ser985Cysfs*25 (HGMD no: CD054393), and p.Arg1778* (HGMD no: CM030076).
Craniosynostosis represents a highly heterogeneous medical condition whose etiology has not been yet fully elucidated. The results obtained from cohorts screened worldwide showed that the molecular background could be indicated in merely 21%, to propitiously 62% of patients (Roscioli et al., 2013; Paumard-Hernandez et al., 2015; Timberlake et al., 2016; Lee et al., 2018; Topa et al., 2020). Positive genetic testing is mainly achieved among subgroups with syndromic CS. The reported germline mutations are usually classified as point mutations, however, chromosomal aberrations, copy number variations, minor exonic deletions/duplication, or biallelic inheritance were also reported in the medical literature (Timberlake et al., 2016; Goos and Mathijssen, 2019; Yilmaz et al., 2019).
In this study, we have reported 166 individuals affected with different forms of CS in whom we had applied a three-step diagnostic algorithm (Tier 1–3) (Figure 1). To our best knowledge, this is the first large CS patients’ screening in which multi-leveled methods, including chromosomal aberrations and CNVs detection, were applied. The proposed approach allowed us to identify an exact genetic cause in around 43% of all CS patients. Since our multi-leveled molecular diagnostic strategy of CS patients is unique and previously unreported, we were unable to directly compare all the results obtained here with previous similar research studies. This is because all reports that have been submitted so far, were aimed at identifying only point mutations via targeted Sanger sequencing, targeted gene panel NGS or ES. Similar to other researchers analyzing these mutations type occurrence, we have shown that causative variants in the FGFR1, FGFR2, FGFR3, TWIST1, and TCF12 genes account for most common cause of CS (Roscioli et al., 2013; Paumard-Hernandez et al., 2015; Lee et al., 2018; Topa et al., 2020).
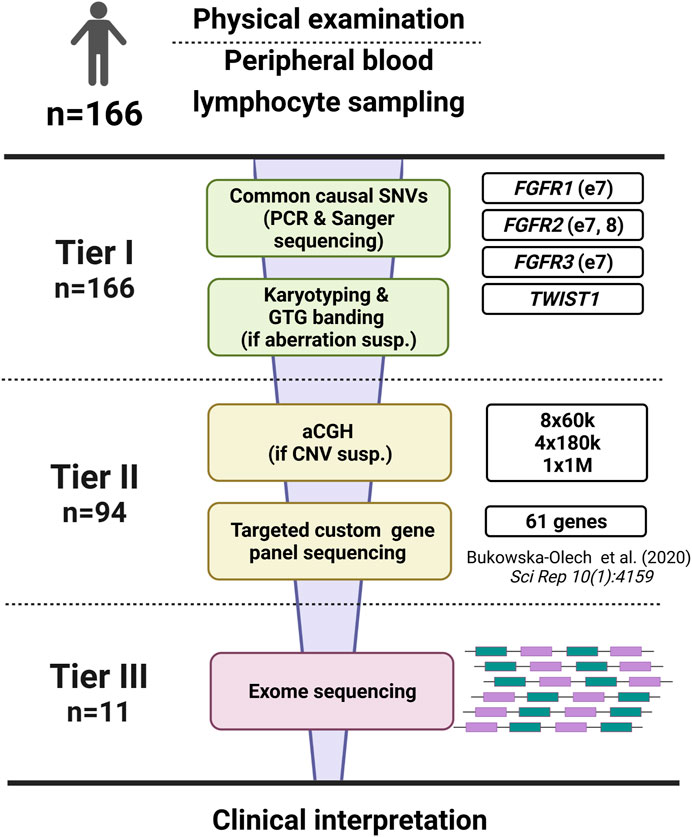
FIGURE 1. The scheme of the diagnostic algorithm applied in our study regarding 166 patients affected with craniosynostosis. The algorithm was divided into three tiers.
Notably, FGFR1, FGFR2, FGFR3 variants mainly occur in hot-spot positions and, along with TWIST1 gene mutations, were analyzed in Tier 1. Undeniably, this step was crucial in the CS testing algorithm and cost-effective compared to other genetic methods (73% of all diagnoses;–χ2 (1;71) = 14.43; p < 0.001). On the other hand, four novel variants in the TCF12 gene and additional FGFR2, FGFR3 alterations (other than hot-spots) were found using custom gene panel NGS (Table 2). No other gene included in the applied custom gene panel housed more than one pathogenic variant. Interestingly, based on the medical literature, the EFNB1 gene is usually reported as the sixth most commonly affected gene in CS (Paumard-Hernandez et al., 2015; Miller et al., 2017; Lee et al., 2018). However, we postulate that a very characteristic disease, i.e., craniofrontonasal dysplasia (CFND) resulting from pathogenic variants of the EFNB1, should be considered a standalone genetic disorder in which features other than CS guide the proper diagnosis (Bukowska-Olech et al., 2021). Moreover, CFND represents a classic viscerocranium defect, whereas CS is a neurocranium abnormality. Hence, we have excluded all individuals suggestive of CFND and consequently have not reported EFNB1 mutations in this study.
Next, we have revealed that six patients with syndromic CS carried pathogenic variants in genes involved in epigenetic regulations such as ARID1A (P134), KMT2A (P137), KMT2D (P114), NSD1 (P58, P99, P166) (Table 2), resulting in Coffin-Siris syndrome type 2, Wiedemann-Steiner syndrome, Kabuki syndrome type 1, and Sotos syndrome type 1, respectively. Such Mendelian disorders, i.e., those resulting from disruptions of epigenetic processes, were termed chromatinopathies. They are all characterized by intellectual disability, immune deficiencies, or skeletal anomalies. However, CS was rarely described among them (Zollino et al., 2017). Occasionally, CS has been reported only in Kabuki syndrome and Sotos syndrome thus far (Tatton-Brown et al., 2005; Martínez-Lage et al., 2010; Topa et al., 2017). The second group of Mendelian diseases in which CS has been noted are RASopathies resulting from the dysregulation of RAS/MAPK pathway. Retrospectively, we have described here one patient carrying CNVs in which BRAF gene deletion occurred (P141) (Bukowska-Olech et al., 2020b). Similar to our finding, other researchers have also highlighted the co-occurrence of both CS and RASopathy, resulting from mutations in other components of RAS/MAPK pathway (Kratz et al., 2009; Takenouchi et al., 2014; Ueda et al., 2017).
Custom genes panel allowed us to detect novel pathogenic variants–p.Arg132* in the ERF gene (P129), and p.Ser391* in the ZIC1 gene (P131), resulting in Craniosynostosis 4 and Craniosynostosis 6, respectively (Twigg et al., 2013, 2015). Both ERF and ZIC1 are newly recognized CS-related genes, however, only a few cases carrying variants in those two have been reported (Twigg et al., 2015; Miller et al., 2017; Glass et al., 2019). In addition, we have evaluated one variant in the ZIC1 gene as VUS p.Ser404Pro (P130) since it was present in the patient’s healthy father. However, this alteration was absent from the gnomAD v3.1.2 database (accession date: 3 December 2021). Finally, ES revealed two additional alterations in individuals with syndromic CS–p.Arg1295* in the MN1 (P63), which was recently discovered, and subsequently linked to CS, and p.Arg305Gly in FAM111A (P136), resulting in Kenny-Caffey syndrome type 2, in which CS represents an unseen clinical feature (Table 2) (Mak et al., 2020).
Importantly, we have noted an apparent gap regarding screening for chromosomal aberrations or CNVs among CS patients (Lattanzi et al., 2012; Poot, 2019). To our knowledge, no major CS groups were analyzed via karyotyping or CMA, therefore most research data describing microscopic chromosomal changes or submicroscopic CNVs causative for CS were published as single case reports (Villa et al., 2007; Marques et al., 2015). Besides, only a few chromosomal aberrations and CNVs known thus far represent recurrent changes underlying CS (e.g., deletions in 7p21, 9p22-p24, and 11q23-q24 or duplication in 5q33.3), however none of them was present in our cohort (Reardon et al., 1993; Shiihara et al., 2004; Poot, 2019). All genomic losses or gains reported in this research were not commonly associated with CS (Budisteanu et al., 2010; Dilzell et al., 2015). Hence, we could not recommend additional loci to be screened regarding the cohort of syndromic CS, especially those associated with intellectual disability. Here, karyotyping followed by GTG banding allowed us to detect two intrachromosomal deletions 7q32.3-q35, and 18q21.32-q23, both resized to chr7:131837067-144607071, and chr18:620405559-80247644, respectively. Next, using CMA, we have detected four CNVs including three duplications–1q22-q23.1 (chr1:155961428-157217426), 2p21 (chr2:44990857-45008348), 17p13.3 (chr17:847955-1641601), and one deletion 5q35.3 (chr5:177277901-177283748) from which the largest encompassed 1.3 Mb, whereas the smallest 5.8 kb (Sowińska-Seidler et al., 2018; Bukowska-Olech et al., 2020a). The detailed mutations description following the International System for Human Cytogenomic Nomenclature (ISCN) was listed in Table 3; for a list of genomic mutations’ content, see Supplementary Table S5. Notably, in most chromosomal aberrations or CNVs identified here, CS occurred as an additional phenotype. The above findings suggest that karyotyping and CMA cannot be replaced by targeted chromosomal testing when applied in a cohort of syndromic CS associated with an intellectual disability or developmental delay.
Regarding the results presented in this study, we would like to point to the possible underestimated co-occurrence of CS and intellectual disability. Our clinical experience suggests that CS may be overlooked when intellectual disability constitutes a primary clinical complaint. Hence, we recommend calvarial sutures’ evaluation in patients with intellectual disability. On the other hand, in any case of already detected syndromic CS and intellectual disability, the possible occurrence of clinical features suggestive for either chromatinopathies or RASopathies should also be considered (Kratz et al., 2009; Cao et al., 2017; Zollino et al., 2017; Davis et al., 2019).
Undeniably, the molecular diagnosis of CS should distinguish its isolated or syndromic form, which presence determines the subsequent diagnostic steps. In addition, syndromic CS should be classified as a disorder associated with intellectual disability or a disorder without intellectual disability (Figure 2). Targeted PCR and Sanger sequencing of FGFR1, FGFR2, FGFR3, TWIST1, and TCF12 genes resulted in the highest diagnostic rate in our cohort of craniosynostosis patients strongly recommend analysing those genes first (isolated CS and syndromic CS without intellectual disability). Because of many advantages of NGS-based methods, including mosaicism detection, screening those genes using targeted genes panel via NGS would be optimal. However, based on our results, we suggest applying a custom genes panel limited to the fewer genes, such as FGFR1, FGFR2, FGFR3, TWIST1, TCF12, ERF, ZIC1, RECQL4, and NSD1 (Siitonen et al., 2009; Sharma et al., 2013; Twigg et al., 2013, Twigg et al., 2015; Twigg and Wilkie, 2015; Miller et al., 2017; Kutkowska-Kaźmierczak et al., 2018; Lee et al., 2018; Bukowska-Olech et al., 2020c; Topa et al., 2020). In the case of syndromic CS and intellectual disability, the genetic investigation should start from chromosomal aberrations or CNVs detection. Lastly, when ES data bioinformatic analysis is performed, genes associated with RASopathies and chromatinopathies should be considered.
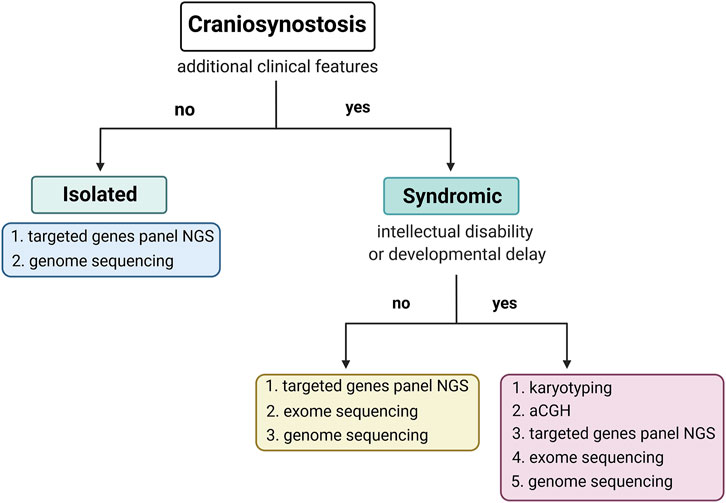
FIGURE 2. The scheme of the proposed diagnostic algorithm was prepared based on results obtained from the current study.
The heterogeneity of CS is enormous, resulting from either anatomical variability of the disorder, in which different types and number of sutures can be affected, or epidemiological aspects, including isolated presentation of CS or occurrence of various accompanying symptoms (Kutkowska-Kazmierczak et al., 2018). It has also been shown that CS may be detected in at least 180 different syndromes, which often are rare and in which CS is not a pathognomic feature. Besides, some researchers have also documented or postulated the association between CS and two-locus inheritance. Consequently, molecular causes of the disease seem to be complex in some CS individuals and, as presented in this study, the pathogenic variant or affected gene may be restricted to only one individual (Sharma et al., 2013; Flaherty et al., 2016; Timberlake et al., 2016, Timberlake et al., 2018; Wilkie et al., 2007; Timberlake and Persing, 2018). However, in many CS patients, including our cohort, genetic causes of observed phenotypes remain unrevealed. One may suspect the presence of deep-intronic and regulatory variants, polygenic inheritance, or even epigenetic influences. Another explanation may be the technical limitations of currently available diagnostic methods. It has been shown, for example, that the application of long-read sequencing in NGS-based methods may clarify the genetic background in many unresolved cases (Fujimoto et al., 2021; Hiatt et al., 2021; Miller et al., 2021; Rastegar and Yasui, 2021). Considering the above, the next steps that we should consider to implement for diagnosis of our unsolved cases include whole-genome sequencing, RNA-seq, whole-genome bisulfite sequencing, or long-read ES.
To conclude, our research may constitute a significant source of epidemiological information as we have presented precise phenotypic and genetic data derived from 166 consecutive CS patients of Caucasian origin. We yielded a 43% diagnostic success rate using the presented approach, highlighting the high occurrence of pathogenic variants within “classic” CS genes, i.e., FGFR1, FGFR2, FGFR3, TWIST1, and TCF12. Moreover, we have critically summarized the applied diagnostic methods (Figure 1) and proposed the optimized, cost-effective diagnostic algorithm, which may be helpful in a daily diagnostic routine of various CS’ types (Figure 2).
Database of Genomic Variants (http://dgv.tcag.ca/dgv/app/home).
DECIPHER (https://www.deciphergenomics.org/browser).
Human Gene Mutation database (http://www.hgmd.cf.ac.uk/ac/index.php).
Human Phenotype Ontology (https://hpo.jax.org/app/).
Online Mendelian Inheritance in Man (https://www.omim.org).
Primer3 tool (http://bioinfo.ut.ee/primer3-0.4.0/).
UCSC Genome Browser (https://genome.ucsc.edu/index.html).
Varsome (https://varsome.com/).
The datasets generated during and/or analyzed during the current study are available from the corresponding authors on reasonable request.
The studies involving human participants were reviewed and approved by the Institutional Review Board of Poznan University of Medical Sciences (no. 742/17). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
EB-O: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, validation, visualization, writing—original draft; DL: data curation, investigation, resources; AS-S, PG, and GK: formal analysis, methodology, validation, visualization, DP, LG-B, AS: methodology; AM-K: data curation, investigation, ZA: data curation, visualization, PD, FG: data curation; KM: investigation; AJ: conceptualization, data curation, investigation, review and editing.
This work was supported by the Polish National Science Centre grant, Poland UMO-2016/23/N/NZ5/02577 to EB-O; AJ was supported by the grant from the Polish National Science Centre UMO-2016/22/E/NZ5/00270.
LG-B was employed by Celon Pharma S.A.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We are grateful to the patients and family members for participating in this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2022.865494/full#supplementary-material
Baer, M. J. (1954). Patterns of Growth of the Skull as Revealed by Vital Staining. Hum. Biol. 26, 80–126.
Budisteanu, M., Arghir, A., Chirieac, S. M., Tutulan-Cunita, A., and Lungeanu, A. (2010). 18q Deletion Syndrome - A Case Report. Maedica (Bucur) 5, 135–138.
Bukowska‐Olech, E., Dmitrzak‐Węglarz, M., Larysz, D., Wojciechowicz, B., Simon, D., Walczak‐Sztulpa, J., et al. (2020a). Compound Craniosynostosis, Intellectual Disability, and Noonan‐Like Facial Dysmorphism Associated with 7q32.3‐q35 Deletion. Birth Defects Res. 112, 740–748. doi:10.1002/bdr2.1744
Bukowska‐Olech, E., Dmitrzak‐Węglarz, M., Larysz, D., Wojciechowicz, B., Simon, D., Walczak‐Sztulpa, J., et al. (2020b). Compound Craniosynostosis, Intellectual Disability, and Noonan‐Like Facial Dysmorphism Associated with 7q32.3‐q35 Deletion. Birth Defects Res. 112, 740–748. doi:10.1002/bdr2.1744
Bukowska-Olech, E., Gawliński, P., Jakubiuk-Tomaszuk, A., Jędrzejowska, M., Obersztyn, E., Piechota, M., et al. (2021). Clinical and Molecular Characterization of Craniofrontonasal Syndrome: New Symptoms and Novel Pathogenic Variants in the EFNB1 Gene. Orphanet J. Rare Dis. 16, 286. doi:10.1186/s13023-021-01914-1
Bukowska-Olech, E., Popiel, D., Koczyk, G., Sowińska-Seidler, A., Socha, M., Wojciechowicz, B., et al. (2020c). Adapting SureSelect Enrichment Protocol to the Ion Torrent S5 Platform in Molecular Diagnostics of Craniosynostosis. Sci. Rep. 10, 4159. doi:10.1038/s41598-020-61048-5
Cao, H., Alrejaye, N., Klein, O. D., Goodwin, A. F., and Oberoi, S. (2017). A Review of Craniofacial and Dental Findings of the RASopathies. Orthod. Craniofac. Res. 20 (Suppl. 1), 32–38. doi:10.1111/ocr.12144
Chieffo, D., Tamburrini, G., Massimi, L., Di Giovanni, S., Giansanti, C., Caldarelli, M., et al. (2010). Long-Term Neuropsychological Development in Single-Suture Craniosynostosis Treated Early. Ped 5, 232–237. doi:10.3171/2009.10.PEDS09231
Davis, A. A., Zuccoli, G., Haredy, M. M., Losee, J., Pollack, I. F., Madan-Khetarpal, S., et al. (2019). RASopathy in Patients with Isolated Sagittal Synostosis. Glob. Pediatr. Health 6, 2333794X1984677–2333794X19846774. doi:10.1177/2333794X19846774
Dilzell, K., Darcy, D., Sum, J., and Wallerstein, R. (20152015). Deletion of 7q33-Q35 in a Patient with Intellectual Disability and Dysmorphic Features: Further Characterization of 7q Interstitial Deletion Syndrome. Case Rep. Genet. 2015, 1–5. doi:10.1155/2015/131852
Fujimoto, A., Wong, J. H., Yoshii, Y., Akiyama, S., Tanaka, A., Yagi, H., et al. (2021). Whole-Genome Sequencing with Long Reads Reveals Complex Structure and Origin of Structural Variation in Human Genetic Variations and Somatic Mutations in Cancer. Genome Med. 13, 65. doi:10.1186/s13073-021-00883-1
Glass, G. E., O'Hara, J., Canham, N., Cilliers, D., Dunaway, D., Fenwick, A. L., et al. (2019). ERF-Related Craniosynostosis: The Phenotypic and Developmental Profile of a New Craniosynostosis Syndrome. Am. J. Med. Genet. 179, 615–627. doi:10.1002/ajmg.a.61073
Goos, J. A. C., and Mathijssen, I. M. J. (2019). Genetic Causes of Craniosynostosis: An Update. Mol. Syndromol 10, 6–23. doi:10.1159/000492266
Gupta, P. C., Foster, J., Crowe, S., Papay, F. A., Luciano, M., and Traboulsi, E. I. (2003). Ophthalmologic Findings in Patients with Nonsyndromic Plagiocephaly. J. Craniofac. Surg. 14, 529–532. doi:10.1097/00001665-200307000-00026
Hiatt, S. M., Lawlor, J. M. J., Handley, L. H., Ramaker, R. C., Rogers, B. B., Partridge, E. C., et al. (2021). Long-Read Genome Sequencing for the Molecular Diagnosis of Neurodevelopmental Disorders. Hum. Genet. Genomics Adv. 2, 100023. doi:10.1016/j.xhgg.2021.100023
Johnson, D., and Wilkie, A. O. M. (2011). Craniosynostosis. Eur. J. Hum. Genet. 19, 369–376. doi:10.1038/ejhg.2010.235
Kratz, C. P., Zampino, G., Kriek, M., Kant, S. G., Leoni, C., Pantaleoni, F., et al. (2009). Craniosynostosis in Patients with Noonan Syndrome Caused by Germline KRAS Mutations. Am. J. Med. Genet. 149A, 1036–1040. doi:10.1002/ajmg.a.32786
Kutkowska-Kaźmierczak, A., Gos, M., and Obersztyn, E. (2018). Craniosynostosis as a Clinical and Diagnostic Problem: Molecular Pathology and Genetic Counseling. J. Appl. Genet. 59, 133–147. doi:10.1007/s13353-017-0423-4
Lattanzi, W., Barba, M., Di Pietro, L., and Boyadjiev, S. A. (2017). Genetic Advances in Craniosynostosis. Am. J. Med. Genet. 173, 1406–1429. doi:10.1002/ajmg.a.38159
Lattanzi, W., Bukvic, N., Barba, M., Tamburrini, G., Bernardini, C., Michetti, F., et al. (2012). Genetic Basis of Single-Suture Synostoses: Genes, Chromosomes and Clinical Implications. Childs Nerv Syst. 28, 1301–1310. doi:10.1007/s00381-012-1781-1
Lee, E., Le, T., Zhu, Y., Elakis, G., Turner, A., Lo, W., et al. (2018). A Craniosynostosis Massively Parallel Sequencing Panel Study in 309 Australian and New Zealand Patients: Findings and Recommendations. Genet. Med. 20, 1061–1068. doi:10.1038/gim.2017.214
Mak, C. C. Y., Doherty, D., Lin, A. E., Vegas, N., Cho, M. T., Viot, G., et al. (2020). MN1 C-Terminal Truncation Syndrome Is a Novel Neurodevelopmental and Craniofacial Disorder with Partial Rhombencephalosynapsis. Brain. 143, 55–68. doi:10.1093/brain/awz379
Marques, F., Heredia, R., de Oliveira, C., Cardoso, M. T., Mazzeu, J., and Pogue, R. (2015). Partial Trisomy 17q and Partial Monosomy 20q in a Boy with Craniosynostosis. Am. J. Med. Genet. 167, 412–416. doi:10.1002/ajmg.a.36844
Martínez-Lage, J. F., Felipe-Murcia, M., Navarro, E. G., Almagro, M.-J., López-Guerrero, A. L., and Pérez-Espejo, M. A. (2010). Craniosynostosis in Kabuki Syndrome. Ped 6, 198–201. doi:10.3171/2010.5.PEDS09286
Mathijssen, I. M. J., and Arnaud, E. (2007). Benchmarking for Craniosynostosis. J. Craniofac. Surg. 18, 436–442. doi:10.1097/scs.0b013e31802d4c6c
Miller, D. E., Sulovari, A., Wang, T., Loucks, H., Hoekzema, K., Munson, K. M., et al. (2021). Targeted Long-Read Sequencing Identifies Missing Disease-Causing Variation. Am. J. Hum. Genet. 108, 1436–1449. doi:10.1016/j.ajhg.2021.06.006
Miller, K. A., Twigg, S. R. F., McGowan, S. J., Phipps, J. M., Fenwick, A. L., Johnson, D., et al. (2017). Diagnostic Value of Exome and Whole Genome Sequencing in Craniosynostosis. J. Med. Genet. 54, 260–268. doi:10.1136/jmedgenet-2016-104215
Morriss-Kay, G. M., and Wilkie, A. O. M. (2005). Growth of the Normal Skull Vault and its Alteration in Craniosynostosis: Insights From Human Genetics and Experimental Studies. J. Anat. 207, 637–653. doi:10.1111/j.1469-7580.2005.00475.x
Opperman, L. A. (2000). Cranial Sutures as Intramembranous Bone Growth Sites. Dev. Dyn. 219, 472–485. doi:10.1002/1097-0177(2000)9999:9999<::aid-dvdy1073>3.0.co;2-f
Paumard-Hernández, B., Berges-Soria, J., Barroso, E., Rivera-Pedroza, C. I., Pérez-Carrizosa, V., Benito-Sanz, S., et al. (2015). Expanding the Mutation Spectrum in 182 Spanish Probands with Craniosynostosis: Identification and Characterization of Novel TCF12 Variants. Eur. J. Hum. Genet. 23, 907–914. doi:10.1038/ejhg.2014.205
Poot, M. (2019). Structural Genome Variations Related to Craniosynostosis. Mol. Syndromol. 10, 24–39. doi:10.1159/000490480
Rastegar, M., and Yasui, D. H. (2021). Editorial: Epigenetic Mechanisms and Their Involvement in Rare Diseases. Front. Genet. 12, 755076. doi:10.3389/fgene.2021.755076
Reardon, W., McManus, S. P., Summers, D., and Winter, R. M. (1993). Cytogenetic Evidence that the Saethre-Chotzen Gene Maps to 7p21.2. Am. J. Med. Genet. 47, 633–636. doi:10.1002/ajmg.1320470510
Renier, D., Sainte-Rose, C., Marchac, D., and Hirsch, J.-F. (1982). Intracranial Pressure in Craniostenosis. J. Neurosurg. 57, 370–377. doi:10.3171/jns.1982.57.3.0370
Rice, D. P. (2008). Developmental Anatomy of Craniofacial Sutures. Front. Oral Biol. 12, 1–21. doi:10.1159/000115028
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Roscioli, T., Elakis, G., Cox, T. C., Moon, D. J., Venselaar, H., Turner, A. M., et al. (2013). Genotype and Clinical Care Correlations in Craniosynostosis: Findings from a Cohort of 630 Australian and New Zealand Patients. Am. J. Med. Genet. 163, 259–270. doi:10.1002/ajmg.c.31378
Sharma, V. P., Fenwick, A. L., Fenwick, A. L., Brockop, M. S., McGowan, S. J., Goos, J. A. C., et al. (2013). Mutations in TCF12, Encoding a Basic Helix-Loop-Helix Partner of TWIST1, Are a Frequent Cause of Coronal Craniosynostosis. Nat. Genet. 45, 304–307. doi:10.1038/ng.2531
Shiihara, T., Kato, M., Kimura, T., Hayasaka, K., Yamamori, S., and Ogata, T. (2004). Craniosynostosis with Extra Copy of MSX2 in a Patient with Partial 5q-Trisomy. Am. J. Med. Genet. 128A, 214–216. doi:10.1002/ajmg.a.20552
Siitonen, H. A., Sotkasiira, J., Biervliet, M., Benmansour, A., Capri, Y., Cormier-Daire, V., et al. (2009). The Mutation Spectrum in RECQL4 Diseases. Eur. J. Hum. Genet. 17, 151–158. doi:10.1038/ejhg.2008.154
Sowińska-Seidler, A., Olech, E. M., Socha, M., Larysz, D., and Jamsheer, A. (2018). Novel 1q22-q23.1 Duplication in a Patient with Lambdoid and Metopic Craniosynostosis, Muscular Hypotonia, and Psychomotor Retardation. J. Appl. Genet. 59, 281–289. doi:10.1007/s13353-018-0447-4
Takenouchi, T., Sakamoto, Y., Miwa, T., Torii, C., Kosaki, R., Kishi, K., et al. (2014). Severe Craniosynostosis with Noonan Syndrome Phenotype Associated with SHOC2 Mutation: Clinical Evidence of Crosslink Between FGFR and RAS Signaling Pathways. Am. J. Med. Genet. 164, 2869–2872. doi:10.1002/ajmg.a.36705
Tatton-Brown, K., Douglas, J., Coleman, K., Baujat, G., Cole, T. R. P., Das, S., et al. (2005). Genotype-Phenotype Associations in Sotos Syndrome: An Analysis of 266 Individuals with NSD1 Aberrations. Am. J. Hum. Genet. 77, 193–204. doi:10.1086/432082
Thompson, D. N. P., Malcolm, G. P., Jones, B. M., Harkness, W. J., and Hayward, R. D. (1995). Intracranial Pressure in Single-Suture Craniosynostosis. Pediatr. Neurosurg. 22, 235–240. doi:10.1159/000120907
Timberlake, A. T., Choi, J., Zaidi, S., Lu, Q., Nelson-Williams, C., Brooks, E. D., et al. (2016). Two Locus Inheritance of Non-Syndromic Midline Craniosynostosis via Rare SMAD6 and Common BMP2 Alleles. eLife 5, e20125. doi:10.7554/eLife.20125
Topa, A., Rohlin, A., Andersson, M. K., Fehr, A., Lovmar, L., Stenman, G., et al. (2020). NGS Targeted Screening of 100 Scandinavian Patients with Coronal Synostosis. Am. J. Med. Genet. 182, 348–356. doi:10.1002/ajmg.a.61427
Topa, A., Samuelsson, L., Lovmar, L., Stenman, G., and Kölby, L. (2017). On the Significance of Craniosynostosis in a Case of Kabuki Syndrome with a Concomitant KMT2D Mutation and 3.2 Mbp De Novo 10q22.3q23.1 Deletion. Am. J. Med. Genet. 173, 2219–2225. doi:10.1002/ajmg.a.38296
Tubbs, R. S., Elton, S., Blount, J. P., and Oakes, W. J. (2001). Preliminary Observations on the Association Between Simple Metopic Ridging in Children without Trigonocephaly and the Chiari I Malformation. Pediatr. Neurosurg. 35, 136–139. doi:10.1159/000050407
Twigg, S. R. F., Forecki, J., Goos, J. A. C., Richardson, I. C. A., Hoogeboom, A. J. M., van den Ouweland, A. M. W., et al. (2015). Gain-of-Function Mutations in ZIC1 Are Associated with Coronal Craniosynostosis and Learning Disability. Am. J. Hum. Genet. 97, 378–388. doi:10.1016/j.ajhg.2015.07.007
Twigg, S. R. F., Vorgia, E., McGowan, S. J., Peraki, I., Fenwick, A. L., Sharma, V. P., et al. (2013). Reduced Dosage of ERF Causes Complex Craniosynostosis in Humans and Mice and Links ERK1/2 Signaling to Regulation of Osteogenesis. Nat. Genet. 45, 308–313. doi:10.1038/ng.2539
Twigg, S. R. F., and Wilkie, A. O. M. (2015). A Genetic-Pathophysiological Framework for Craniosynostosis. Am. J. Hum. Genet. 97, 359–377. doi:10.1016/j.ajhg.2015.07.006
Ueda, K., Yaoita, M., Niihori, T., Aoki, Y., and Okamoto, N. (2017). Craniosynostosis in Patients with RASopathies: Accumulating Clinical Evidence for Expanding the Phenotype. Am. J. Med. Genet. 173, 2346–2352. doi:10.1002/ajmg.a.38337
Villa, O., Del Campo, M., Salido, M., Gener, B., Astier, L., Del Valle, J., et al. (2007). Small Supernumerary Marker Chromosome Causing Partial Trisomy 6p in a Child with Craniosynostosis. Am. J. Med. Genet. 143A, 1108–1113. doi:10.1002/ajmg.a.31709
Wilkie, A. O. M., Bochukova, E. G., Hansen, R. M. S., Taylor, I. B., Rannan-Eliya, S. V., Byren, J. C., et al. (2007). Clinical Dividends from the Molecular Genetic Diagnosis of Craniosynostosis. Am. J. Med. Genet. 143A, 1941–1949. doi:10.1002/ajmg.a.31905
Wilkie, A. O. M., Byren, J. C., Hurst, J. A., Jayamohan, J., Johnson, D., Knight, S. J. L., et al. (2010). Prevalence and Complications of Single-Gene and Chromosomal Disorders in Craniosynostosis. Pediatrics 126, e391–e400. doi:10.1542/peds.2009-3491
Yilmaz, E., Mihci, E., Nur, B., Alper, Ö. M., and Taçoy, Ş. (2019). Recent Advances in Craniosynostosis. Pediatr. Neurol. 99, 7–15. doi:10.1016/j.pediatrneurol.2019.01.018
Zhao, M., Havrilla, J. M., Fang, L., Chen, Y., Peng, J., Liu, C., et al. (2020). Phen2Gene: Rapid Phenotype-Driven Gene Prioritization for Rare Diseases. NAR Genomics and Bioinformatics 2, lqaa032. doi:10.1093/nargab/lqaa032
Zollino, M., Lattante, S., Orteschi, D., Frangella, S., Doronzio, P. N., Contaldo, I., et al. (2017). Syndromic Craniosynostosis Can Define New Candidate Genes for Suture Development or Result from the Non-Specifc Effects of Pleiotropic Genes: Rasopathies and Chromatinopathies as Examples. Front. Neurosci. 11, 1–8. doi:10.3389/fnins.2017.00587
Keywords: calvarial sutures, craniosynostosis, next-generation sequencing, chromosomal microarray analysis, cohort screening
Citation: Bukowska-Olech E, Sowińska-Seidler A, Larysz D, Gawliński P, Koczyk G, Popiel D, Gurba-Bryśkiewicz L, Materna-Kiryluk A, Adamek Z, Szczepankiewicz A, Dominiak P, Glista F, Matuszewska K and Jamsheer A (2022) Results from Genetic Studies in Patients Affected with Craniosynostosis: Clinical and Molecular Aspects. Front. Mol. Biosci. 9:865494. doi: 10.3389/fmolb.2022.865494
Received: 29 January 2022; Accepted: 21 March 2022;
Published: 28 April 2022.
Edited by:
Amit Prasad, Indian Institute of Technology Mandi, IndiaReviewed by:
Muhammad Abdul Rehman Rashid, Government College University, PakistanCopyright © 2022 Bukowska-Olech, Sowińska-Seidler, Larysz, Gawliński, Koczyk, Popiel, Gurba-Bryśkiewicz, Materna-Kiryluk, Adamek, Szczepankiewicz, Dominiak, Glista, Matuszewska and Jamsheer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ewelina Bukowska-Olech, ZXdlLm9sZWNoQGdtYWlsLmNvbQ==; Aleksander Jamsheer, amFtc2hlZXJAd3AucGw=
†ORCID: Ewelina Bukowska-Olech, orcid.org/0000-0003-0509-1696; Dawid Larysz, orcid.org/0000-0001-7520-9106; Anna Sowińska-Seidler, orcid.org/0000-0002-2493-898X; Paweł Gawliński, orcid.org/0000-0002-3672-5834; Grzegorz Koczyk, orcid.org/0000-0002-5414-4689; Delfina Popiel, orcid.org/0000-0002-6820-0338; Lidia Gurba-Bryśkiewicz, orcid.org/0000-0002-7807-6641; Anna Materna-Kiryluk, orcid.org/0000-0002-2889-5892; Aleksandra Szczepankiewicz, orcid.org/0000-0003-3379-9106; Karolina Matuszewska, orcid.org/0000-0001-7520-9106; Aleksander Jamsheer, orcid.org/0000-0003-4058-3901
‡These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.