 Oliver Brylski1,2†
Oliver Brylski1,2† Puja Shrestha1†
Puja Shrestha1† Philip J. House3Patricia Gnutt2
Philip J. House3Patricia Gnutt2 Jonathan Wolf Mueller3,4*
Jonathan Wolf Mueller3,4* Simon Ebbinghaus1,2*
Simon Ebbinghaus1,2*- 1Institute of Physical and Theoretical Chemistry, TU Braunschweig, Braunschweig, Germany
- 2Institute of Physical Chemistry II, Ruhr University, Bochum, Germany
- 3Institute of Metabolism and Systems Research (IMSR), University of Birmingham, Birmingham, United Kingdom
- 4Centre for Endocrinology, Diabetes and Metabolism (CEDAM), Birmingham Health Partners, Birmingham, United Kingdom
Cellular sulfation pathways rely on the activated sulfate 3′-phosphoadenosine-5′-phosphosulfate (PAPS). In humans, PAPS is exclusively provided by the two PAPS synthases PAPSS1 and PAPSS2. Mutations found in the PAPSS2 gene result in severe disease states such as bone dysplasia, androgen excess and polycystic ovary syndrome. The APS kinase domain of PAPSS2 catalyzes the rate-limiting step in PAPS biosynthesis. In this study, we show that clinically described disease mutations located in the naturally fragile APS kinase domain are associated either with its destabilization and aggregation or its deactivation. Our findings provide novel insights into possible molecular mechanisms that could give rise to disease phenotypes associated with sulfation pathway genes.
Introduction
Sulfation is a highly important biological process where a sulfate moiety from the activated sulfate donor 3′-phosphoadenosine-5′-phosphosulfate (PAPS) is transferred onto acceptor molecules. Adding the negatively charged sulfate group to a hydroxyl-group induces significant changes in the chemical properties of the acceptor molecule with a major impact on their function. It is the sheer variety of sulfated metabolites that makes sulfation impactful on numerous biological systems. Sulfotransferases use activated sulfate to modify proteins, glycans and other biomolecules like steroid hormones (Klassen and Boles, 1997; Strott, 2002; Mueller et al., 2015).
Active PAPS synthase enzymes generate active sulfate in the form of PAPS. In humans, there are two isoforms PAPSS1 and PAPSS2 (van den Boom et al., 2012). Nevertheless, disease-related protein variants have been exclusively reported for PAPSS2.
In a variety of human genetics studies, a total of 65 individuals with various inactivating alleles of the human PAPSS2 gene have been described (Ahmad et al., 1998; Haque et al., 1998; Noordam et al., 2009; Miyake et al., 2012; Iida et al., 2013; Tüysüz et al., 2013; Oostdijk et al., 2015; Handa et al., 2016; Bownass et al., 2019; Eltan et al., 2019; Perez-Garcia et al., 2021). These have been analyzed and summarized recently (Baranowski et al., 2018; Brylski et al., 2019; Paganini et al., 2020). An additional PAPSS2 variant is known for brachymorphic mice (Kurima et al., 1998).
Recently, several studies investigated the essential steps in sulfation pathways, whose malfunction is correlated with disease symptoms (Mueller et al., 2015; Foster and Mueller, 2018). Among these symptoms are bone and cartilage dysplasia (Oostdijk et al., 2015), as well as androgen excess and polycystic ovary syndrome (PCOS) (Noordam et al., 2009; Oostdijk et al., 2015), all caused by point-mutations in the PAPSS2 gene encoding for the PAPS synthase 2 enzyme. These point mutations diminish the enzyme activity and they are mainly located within the kinase domain of PAPSS2 (Kurima et al., 1998; Noordam et al., 2009; Iida et al., 2013).
It is evident that PAPSS2 plays a vital role in skeletal development as well as steroid hormones regulation (Kurima et al., 1998; Noordam et al., 2009; Iida et al., 2013). Kurima and coworkers linked the mutation G78R within the nucleotide kinase domain of the PAPSS2 isoform, with the bone phenotype seen in the brachymorphic mouse (bm) (Kurima et al., 1998). G78R is located in the adenosine-5′-phosphosulfate (APS) kinase domain, close to the ligand-binding site. The mutation causes catalytic inactivation and hence lowered intracellular PAPS availability. As a consequence, bm mice show reduced postnatal growth that was ascribed to under-sulfation of the extracellular matrix; they also show abnormal hepatic detoxification and prolonged bleeding times (Kurima et al., 1998). One of the most prominent roles of sulfation is the modification of glycosaminoglycans (GAGs) by Golgi-residing carbohydrate sulfotransferases (Gesteira et al., 2021). Sulfated GAGs play a vital role in cell signaling to regulate many biochemical processes like cell growth and proliferation, promotion of cell adhesion, anticoagulation and wound repair (Raman et al., 2005; Prydz, 2015). In brachymorphic cartilage, GAGs are found at normal level but significantly under-sulfated, affecting the formation of connective tissue, such as, cartilage (Kurima et al., 1998; Cho et al., 2004).
More recently, Noordam and coworkers reported a case study of a girl with premature pubarche, hyperandrogenic anovulation, very low level of dehydro-epiandrosterone sulfate (DHEAS) and high level of androgen. The steroid sulfation defect of this patient was associated with a T48R mutation found in the APS kinase domain. Due to this mutation, PAPS synthesis is affected, leading to incompetent DHEA inactivation, with the latter resulting in increased levels of androgens causing PCOS-like phenotypes (Noordam et al., 2009). In 2013, Iida and coworkers reported more PAPSS2 mutations (C43Y, L76Q, E183K, V540D) out of which three, C43Y, L76Q and E183K, were found in the APS kinase domain. C43Y and L76Q cause loss of function leading to brachyolmia and abnormal androgen metabolism (Iida et al., 2013).
Eukaryotic cells express another PAPS synthase gene, PAPSS1, that shares 78% identity at the level of amino acid sequence (van den Boom et al., 2012). However, this protein isoform cannot compensate for the loss of the other (Mueller et al., 2018). This lack of compensation raises the question of whether the two isoforms impact differently on subsets of sulfation pathways. Subcellular localization sequences (Schröder et al., 2012) were identified in both PAPS synthases and dimer formation (Sekulic et al., 2007; Grum et al., 2010; Brylski et al., 2019) was observed, both features proposed to be crucial for proper localization and activity of the enzyme. In addition to these physiological aspects, in vitro biophysical studies focused on the stability of PAPS synthases revealed that isoforms of this enzyme are only marginally stable as recombinant proteins (van den Boom et al., 2012). However, PAPS synthase proteins can be stabilized by preferential binding of their substrates to the APS kinase domain, namely PAPS, adenosine diphosphate (ADP) and APS (van den Boom et al., 2012; Mueller and Shafqat, 2013).
Using a recently developed folding sensor of the APS kinase domain of the human PAPS synthase PAPSS2 (Brylski et al., 2021), we investigate how clinically reported single-point-mutations change the in-cell stability of the APS kinase domain and if destabilization could lead to aggregation and thus loss of metabolic activity.
Materials and Methods
Construction of PAPSS2 and APSK37 Variants
The pEGFP-C1-PAPSS2 plasmid encoding human full-length PAPSS2b C-terminally fused to an EGFP fluorescent protein (Schröder et al., 2012), was used for DpnI-based site-directed mutagenesis and subsequently for cell counting experiments. To generate the APSK37 sensor, the APS kinase domain of PAPSS2 was PCR-subcloned into a modified pDream2.1 vector with an N-terminal AcGFP1 and a C-terminal mCherry (Ebbinghaus et al., 2010). The APSK enzyme was truncated between the two isoleucine residues I220 and I221, within the flexible linker that connects the kinase and the sulfurylase domains (Harjes et al., 2005). Furthermore, the flexible and disordered N-terminal region, which is known to assist in the dimerization of the protein (Sekulic et al., 2007; Grum et al., 2010), was truncated by 37 amino acids (Δ37). Further, DpnI-based site-directed mutagenesis was used to introduce different disease related point mutations (G78R, L76Q, C43Y and T48R). All constructs were verified by Sanger DNA sequencing.
Cell Culture and Plasmid Transfection
HeLa cells were grown at 5% CO2 at 37°C in DMEM supplemented with 10% FBS, 100 U/ml penicillin and 0.1 mg/ml streptomycin. Cells were passaged at a 1:4 or 1:6 ratios at 80–90% confluence, using trypsin digestion. For transfection, cells were seeded in six-well plates (Sarstedt). Using Lipofectamine 3000 (Thermo Fisher), cells were transfected according to the manufacturers protocol. Concisely, a mixture of 125 µl Opti-MEM (Thermo Fisher) with 2 µg of the respective plasmid DNA and 4 µl P3000 reagents was prepared. After 5 min of incubation, the mixture was transferred to another solution containing 125 µl Opti-MEM supplemented with 4 µl Lipofectamine3000 reagent. Cells were incubated for 6 h after the addition of transfection mixture to the cellular growth medium at 5% CO2, 37°C. The cells were passaged using trypsin digestion and seeded in 35 mm glass bottom dishes (Fluorodish, World Precision Instruments). Cells were grown for 2 days at standard cell culture conditions before imaging.
Sample Preparation
Fast Relaxation Imaging (FReI) was performed with transfected cells grown on 35 mm glass bottom dishes (Fluorodish, World Precision Instruments). Cells were washed with Dulbecco’s Phosphate Buffered Saline (DPBS) (Sigma-Aldrich) after removing the growth medium. 30 µL Leibovitz’s L15 medium supplemented with 30% FBS were sealed between a glass cover slip (Menzel #1.0) with a 120 µm thick imaging spacer (Sigma-Aldrich) and a glass bottom dish with cells.
Fast Relaxation Imaging Measurements
FReI is a combination of wide field fluorescence microscopy with millisecond temperature jumps induced by an IR diode laser (m2k-Laser, 2200 nm). The technique was previously described (Ebbinghaus et al., 2010; Gnutt et al., 2019a). Shortly, fluorescent light was split by a dichroic beam splitter into donor and acceptor signal that was recorded using CCD cameras while the sample is rapidly heated by an IR laser. The temperature sensitive dye Rhodamine B (Sigma Aldrich) was used for the calibration of temperature jumps (Vöpel et al., 2015; Gao et al., 2016; Büning et al., 2017). The heat profile used in this study showed an average temperature increase of 2.2°C per jump at intervals of 50 s, covering a range from 23.0 to 58.2°C in 16 steps. Image acquisition was performed at one frame per second (fps) with LED exposure times typically between 50 and 200 ns. Data was recorded using AxioVision software and the images were processed and analyzed using ImageJ (National Institute of Health, United States) and further evaluated using self-written MatLab (Mathworks) codes and GraphPad Prism 6 (GraphPad).
For data analysis, fluorescence intensities were averaged throughout the cytoplasmic region for each channel individually (Dhar et al., 2011). Further, background subtraction was performed for the individual channels and the ratio of the donor and acceptor channel (D/A) calculated. The changes of D/A ratio upon temperature jump yield information about the associated conformational change. An increase in D/A refers a decrease in FRET that may be attributed to protein unfolding. To analyze the kinetics of protein unfolding, the individual channel intensities were used as D-αA according to (Dhar et al., 2011). To determine the melting point (TM) of the protein, the thermodynamic model introduced as Better thermodynamics from kinetics (Girdhar et al., 2011) was used:
Where, ∂g1 is pre-factor of the linear Taylor approximation of the two-state populations. ΔT is the amplitude of the temperature jump (set to 2.2°C) and A0 and mA are fitting parameters of the underlying baseline (with mA set to 0).
HEK293 Cell Culture and Wide Field Microscopy
HEK293 cells were cultured in DMEM with high glucose (Gibco, United Kingdom), supplemented with 10% fetal FBS and penicillin/streptomycin at 1%. Cells were passaged at 80–90% confluence, using trypsin digestion. Regular checks ensured that all cells were mycoplasma-free. Cells were seeded 1:8 or 1:10 in culture flasks or maintaining stocks or at 200,000 cells per well into six-well plates with microscopic slides in them. Transfection of these HEK293 cells 24 h after seeding on cover slips was performed using the XtremeGENE HP DNA transfection reagent (Roche, United Kingdom), according to manufacturer’s instructions. Cells were left growing for 24 or 48 h, then washed with ice-cold PBS and incubated with ice-cold methanol, followed by three further washing steps using PBS. Finally, cover slips with cells were mounted on microscopic slides, using fluorescence mounting media, and fixed with nail varnish. The slides were anonymized to enable blind, non-biased analysis. The slides were then viewed under a wide-field fluorescent microscope and scored at least three different sections using a ×20 objective. Cells were imaged with a ×20 objective. The number of speckles per cell was scored in large numbers of cells, in a blinded fashion. As a control, fluorescence intensity was ranked as well (low/medium/high). No correlation was found between fluorescence intensity and number of speckles, suggesting that protein over-expression levels were not linked to the observed patterns of speckles. Significant changes of population of non-speckled cells have been determined using two-way ANOVA with a post-hoc Holm-Sidak’s test correcting for multiple comparisons.
Results
Mutations in APSK37 Reveal Distinct Folding Stabilities
We analyzed the effect of disease-related mutations on the folding stability and aggregation of the APS kinase domain of the bifunctional PAPSS2 protein, using our recently established APSK folding sensor (APSK37) (Brylski et al., 2021). The sensor reports intramolecular FRET between the N-terminal AcGFP1 and the C-terminal mCherry fusion proteins (see Materials and Methods for details).
We analyzed PAPS synthase disease point mutations located in the APS kinase domain to understand whether the phenotypes seen clinically correlate with misfolded, destabilized or inactive protein. Therefore, we created the variants G78R, L76Q, T48R and C43Y within APSK37 (Oostdijk et al., 2015), expressed them in HeLa cells and studied their in-cell protein stability in comparison to the wt protein using Fast Relaxation Imaging (FReI). In FReI, a rapid perturbation of temperature is applied by absorption of infrared light (IR) by the sample (Ebbinghaus et al., 2010; Vöpel et al., 2017; Gnutt et al., 2019b) (Figures 1A,B). Dual-color imaging allows to measure changes in donor-to-acceptor intensity ratio (D/A) (Figure 1C) that display unfolding kinetics and thermodynamics of the protein in the cell (see Materials and Methods for details) (Figure 1D).
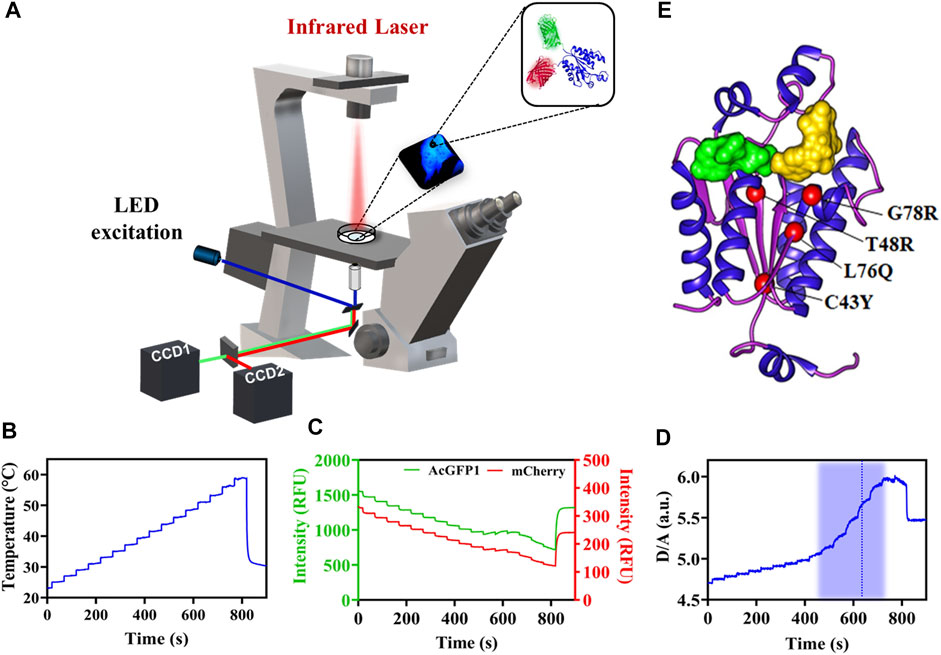
FIGURE 1. In-cell thermal unfolding of APSK37 wt using Fast Relaxation Imaging. (A) Schematic representation of the Fast Relaxation Imaging setup. (B) Induced temperature profile calculated by the calibration procedure published in Büning et al., 2017. (C) Change in fluorescence according to the temperature profile in B of the APSK37 AcGFP1 FRET donor (D) and the mCherry FRET acceptor (A). (D) D/A ratio calculated from the intensity data in (C). The thermal unfolding region is shaded in blue and the calculated TM is displayed by a dash blue line. (E) Crystal structure of the APSK domain of PAPSS2 (PDB:2AX4). Studied mutations are shown as red spheres. ADP/ATP (green) and APS/PAPS (yellow) binding sites are indicated by the substrates surface representation.
A structural analysis of the surface exposure of the disease-related mutants revealed that C43Y, T48R and L76Q are deeply buried in the protein core (solvent accessible surface area (SASA) ≤ 1 Å2) compared to G78R (Kabsch and Sander, 1983; Brylski et al., 2019); all of them located in close proximity in the central beta-sheet of the APSK (Figure 1E).
For APSK37 wt, we observed an increase of the normalized D/A ratio upon IR-laser heating and decrease after returning to the starting temperature (Figure 2A). This behavior is also evident for G78R mutant (Figure 2B), which exhibits an apparent two-state folding behavior as the respective unfolding kinetics can be fitted by a single exponential function. Plotting and fitting the respective amplitudes against temperature (Figures 2C,D) allowed the determination of a TM = 46.1 ± 2.1°C which is similar to wt (48.0 ± 1.7°C) (Figure 2E). Additionally, no significant differences were found with respect to the modified standard state free energies of folding ΔGf0’, suggesting that this mutation does not affect the stability of the protein (Figure 2F).
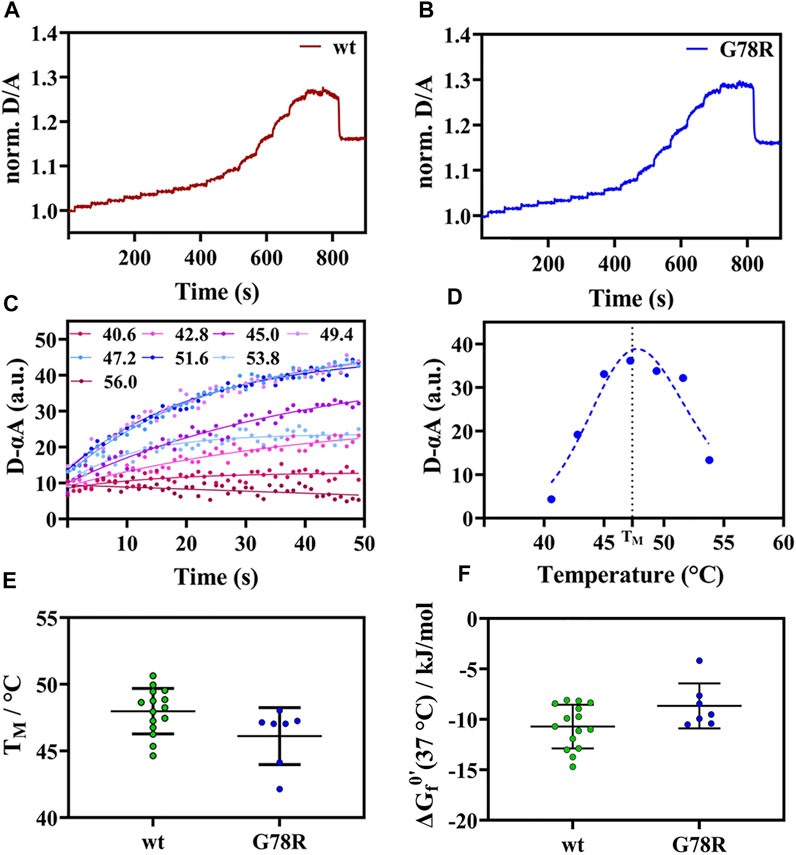
FIGURE 2. Thermal unfolding curve APSK37 wt and mutants (G78R). (A) Exemplary temperature induced thermal unfolding curves of wt (data shown from (Brylski et al., 2021)). (B) Exemplary temperature induced thermal unfolding curves of G78R. (C) Exponential unfolding curves of single temperature jumps from panel (B) showing the relaxations kinetics at the respective temperatures. (D) Kinetic amplitudes as a function of temperature to determine the Tm (dashed line). (E) Thermal stability comparison of the mutant G78R with APSK37 wt: Melting points of APSK37 (green) and melting point of G78R (blue) derived from FReI measurrement showing average ± s.d. (F) Folding free energy ∆Gf0’ for both APSK37 wt and G78R mutant with mean ± s.d. There are no statistically significant differences between wt and G78R. Significance were tested via one-way ANOVA with a post-hoc Holm-Sidak test correcting for multiple comparisons (no significant changes observed).
For the mutations C43Y, T48R and L76Q, we did not detect any unfolding transitions, impeding the determination of TM. We rather observed a strong decrease in the D/A ratio (Figures 3A–C) that can be attributed to an increase in FRET by intermolecular energy transfer due to self-association (Ebbinghaus et al., 2010; Büning et al., 2017).
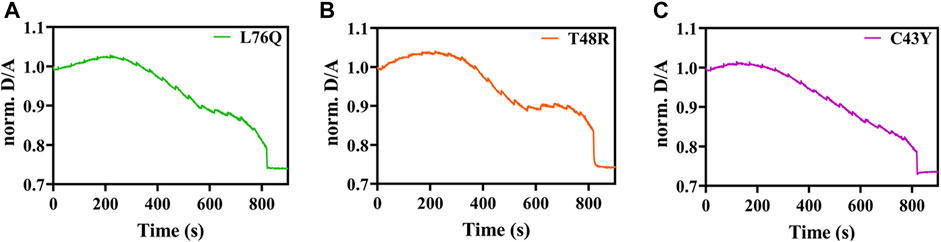
FIGURE 3. Exemplary temperature-induced thermal unfolding curves of the L76Q, T48RM, and C43Y mutants are visible in Figures 3A–C, respectively.
Self-Association and Aggregation of C43Y, T48R and L76Q
We then investigated if the self-association events observed in FReI result in the formation of microscopically visible aggregates. We monitored their formation by wide-field fluorescence microscopy of EGFP-labeled full-length human PAPS synthase 2 carrying the disease mutants, expressed in HEK293 cells along with PAPSS 2 wt proteins. The cellular distribution pattern of the protein and degree of aggregate formation was scored by classifying individual cells according to the number of speckles that were visible inside each cell (Figure 4A). Figure 4B illustrates the cellular distribution pattern of the protein and the degree of aggregate formation for different mutants compared to PAPSS2 wt. The mutants C43Y, T48R and L76Q caused a higher number of aggregates compared to PAPSS2 wt. The level of aggregate formation of the G78R mutation is not significantly different from wt. Thus, the results show that the self-association of the C43Y, T48R and L76Q APSK37 proteins measured by FReI is in accordance with protein aggregation of the respective mutants in the full-length PAPSS2 protein. On the other hand, the G78R mutant is stable and does not lead to aggregation both in APSK37 and PAPSS2 proteins.
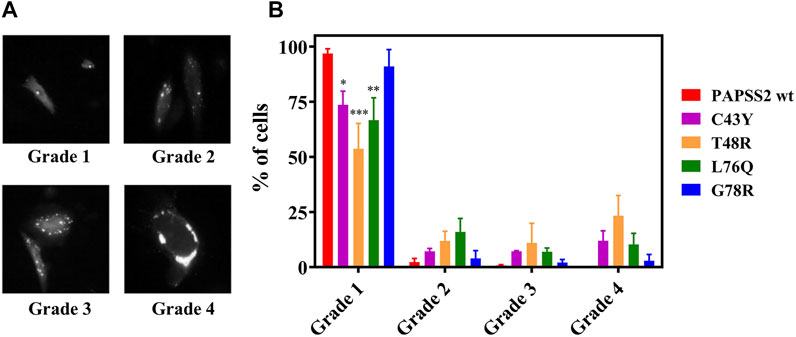
FIGURE 4. Expression and distribution of recombinant human PAPSS in HEK293 cells. (A) Exemplary fluorescence image of HEK293 cells showing fluorescence spots classified according to three categories. (B) Even distribution; no speckles: grade 1, 1–3 speckles: grade 2, 4–10 speckles: grade 3 and more than 10 speckles or large clumps: grade 4. Data is presented as average ± s.e.m. Cells were counted from four different slides (N = 4) with n > 200 cells in total for each protein variant. Asterisks indicate significant differences of the fraction of cells showing no speckles compared to PAPSS2 wt (*p < 0.05, ***p < 0.001). Additional statistical significance within grade 1 was found between G78R and T48R(***) or G78R and L76Q(*).
Discussion
Generally, proteins with disease-related mutations either show a loss of catalytic function, a gain of toxic function (Winklhofer et al., 2008; De Baets et al., 2015) or a significant loss in stability of the protein leading to misfolding and aggregation (Waters, 2001; Denny et al., 2013; Gandhi et al., 2019). For PAPSS2, many studies have shown that disease-related gene defects cause different forms of bone and cartilage malformation (Kurima et al., 1998; Iida et al., 2013), as a consequence of under-sulfation of extracellular matrix. Dysregulation of steroid metabolism causing an increase in androgen activation is further associated with diseases like PCOS and premature pubarche (Noordam et al., 2009).
The results of this study show that PAPSS2 disease-related mutations cause a destabilization and aggregation of the enzyme in cellular environments for the mutants C43Y, T48R, L76Q. The G78R mutation however shows a folding stability that is comparable to wt, preventing aggregation. Regarding catalysis, Kurima and coworkers have reported that the G78R variant has very little residual APS kinase activity, but the ATP sulfurylase activity was comparable to wt (Kurima et al., 1998). Conformational changes of the APS binding site upon mutation, modifying the interaction between the ATP γ-phosphate group, the magnesium ion and the DGDN-loop can be a potential reason for the catalytic inhibition APS kinase (Kurima et al., 1998). The mutation may not disrupt the native fold, however, a catalytic conversion, for example due to a loss in flexibility within the DGDN-loop, may not be possible anymore.
The mutations C43Y, T48R, and L76Q destabilize APSK37 and lead to aggregation of both APSK37 and full-length PAPS synthase. All three mutations reside in the central β-sheet region of the protein (Figure 1E), suggesting that this is a sensitive region that maintains the native fold and prevents self-association and aggregation. Changes in the protein’s native structure, protein-protein interactions and many other sequential and parallel events can lead to misfolded/unfolded conformations, resulting in aggregation. Protein aggregation is often linked with various pathologies, including neurodegenerative diseases, such as Alzheimer’s, Parkinson’s and Huntington’s. These disease-related aggregates are generally sub-divided into loss-of-function and gain-of- toxic function effects (Ross and Poirier, 2004; Wang, 2005; Soto and Pritzkow, 2018). Indeed, the three above-mentioned PAPSS mutations were previously classified as “missense mutants,” causing its loss of function (Noordam et al., 2009; Iida et al., 2013). Missense mutations in nuclear deubiquitinase BAP1 were previously shown to induce destabilization and aggregation of this enzyme, with the latter being suggested as the main cause of its functional loss (Bhattacharya et al., 2016). In fact, this hypothesis is further supported by our previous studies (Brylski et al., 2021) using an alanine scanning mutagenesis of the substrate binding site of APSK37 in HeLa cells. The results revealed a large range of different in-cell stabilities for the single point mutations (ΔGf0 = −10.7 to +13.8 kJ/mol).
Our results suggest two distinct possible disease mechanisms, one related to misfolding and aggregation, and the other one related to inhibition of catalytic function. However, whether these processes are causal for the different pathologies needs to be elucidated in future studies.
Conclusion
So far, many sulfotransferase-related mutations are known to be associated with the sulfation pathway but a lot less is reported for PAPSS. Our results report that PAPSS2 disease-related mutations cause misfolding and aggregation (L76Q, T48R, and C43Y), and inhibition of the catalytic function (G78R). Even though our study showed that the three missense mutants (L76Q, T48R and C43Y) lead to aggregation, the molecular details of this process remains to be explored, particularly putative cytotoxic effects of amyloid formation. Therapeutic approaches against the rare diseases that are associated with these mutations may thus be different, encompassing supplementation of lacking compounds (Klinge et al., 2018) or inhibitors to reduce aggregation (Vöpel et al., 2017).
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author Contributions
JWM and SE, designed the study; OB, PS, PJH, and PG, acquired data. OB, PS, PJH, PG, JWM, and SE, analyzed and interpreted data. OB and JWM, drafted the work. OB, PS, JWM, and SE, revised it critically for important intellectual content. All authors approved the final version of the manuscript and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding
We acknowledge funding from the Cluster of Excellence RESOLV (EXC 1069) and the Human Frontier Science Program (Research Grant RGP0022/2017). OB was supported by the Graduate School of Solvation Science (Ruhr University Bochum). JWM was supported by the European Commission Marie Curie Fellowship SUPA-HD (625451).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ahmad, M., Ul Haque, M. F., Ahmad, W., Abbas, H., Haque, S., Krakow, D., et al. (1998). Distinct, Autosomal Recessive Form of Spondyloepimetaphyseal Dysplasia Segregating in an Inbred Pakistani kindred. Am. J. Med. Genet. 78, 468–473. doi:10.1002/(sici)1096-8628(19980806)78:5<468:aid-ajmg13>3.0.co;2-d
Baranowski, E. S., Arlt, W., and Idkowiak, J. (2018). Monogenic Disorders of Adrenal Steroidogenesis. Horm. Res. Paediatr. 89, 292–310. doi:10.1159/000488034
Bhattacharya, S., Hanpude, P., and Maiti, T. K. (2016). Cancer Associated Missense Mutations in BAP1 Catalytic Domain Induce Amyloidogenic Aggregation: A New Insight in Enzymatic Inactivation. Sci. Rep. 5, 18462. doi:10.1038/srep18462
Bownass, L., Abbs, S., Armstrong, R., Baujat, G., Behzadi, G., Berentsen, R. D., et al. (2019). PAPSS2 ‐related Brachyolmia: Clinical and Radiological Phenotype in 18 New Cases. Am. J. Med. Genet. 179, 1884–1894. doi:10.1002/ajmg.a.61282
Brylski, O., Ebbinghaus, S., and Mueller, J. W. (2019). Melting Down Protein Stability: PAPS Synthase 2 in Patients and in a Cellular Environment. Front. Mol. Biosci. 6, 31. doi:10.3389/fmolb.2019.00031
Brylski, O., Shrestha, P., Gnutt, P., Gnutt, D., Mueller, J. W., and Ebbinghaus, S. (2021). Cellular ATP Levels Determine the Stability of a Nucleotide Kinase. Front. Mol. Biosci. 8, 790304. doi:10.3389/fmolb.2021.790304
Büning, S., Sharma, A., Vachharajani, S., Newcombe, E., Ormsby, A., Gao, M., et al. (2017). Conformational Dynamics and Self-Association of Intrinsically Disordered Huntingtin Exon 1 in Cells. Phys. Chem. Chem. Phys. 19, 10738–10747. doi:10.1039/C6CP08167C
Cho, Y. R., Lee, S. J., Jeon, H. B., Park, Z. Y., Chun, J.-S., and Yoo, Y. J. (2004). Under-sulfation by PAPS Synthetase Inhibition Modulates the Expression of ECM Molecules during Chondrogenesis. Biochem. Biophysical Res. Commun. 323, 769–775. doi:10.1016/j.bbrc.2004.08.173
De Baets, G., Van Doorn, L., Rousseau, F., and Schymkowitz, J. (2015). Increased Aggregation Is More Frequently Associated to Human Disease-Associated Mutations Than to Neutral Polymorphisms. PLOS Comput. Biol. 11, e1004374. doi:10.1371/journal.pcbi.1004374
Denny, R. A., Gavrin, L. K., and Saiah, E. (2013). Recent Developments in Targeting Protein Misfolding Diseases. Bioorg. Med. Chem. Lett. 23, 1935–1944. doi:10.1016/j.bmcl.2013.01.089
Dhar, A., Girdhar, K., Singh, D., Gelman, H., Ebbinghaus, S., and Gruebele, M. (2011). Protein Stability and Folding Kinetics in the Nucleus and Endoplasmic Reticulum of Eucaryotic Cells. Biophysical J. 101, 421–430. doi:10.1016/j.bpj.2011.05.071
Ebbinghaus, S., Dhar, A., McDonald, J. D., and Gruebele, M. (2010). Protein Folding Stability and Dynamics Imaged in a Living Cell. Nat. Methods 7, 319–323. doi:10.1038/nmeth.1435
Eltan, M., Yavas Abali, Z., Arslan Ates, E., Kirkgoz, T., Kaygusuz, S. B., Türkyılmaz, A., et al. (2019). Low DHEAS Concentration in a Girl Presenting with Short Stature and Premature Pubarche: A Novel PAPSS2 Gene Mutation. Horm. Res. Paediatr. 92, 262–268. doi:10.1159/000502114
Foster, P. A., and Mueller, J. W. (2018). SULFATION PATHWAYS: Insights into Steroid Sulfation and Desulfation Pathways. J. Mol. Endocrinol. 61, T271–T283. doi:10.1530/JME-18-0086
Gandhi, J., Antonelli, A. C., Afridi, A., Vatsia, S., Joshi, G., Romanov, V., et al. (2019). Protein Misfolding and Aggregation in Neurodegenerative Diseases: a Review of Pathogeneses, Novel Detection Strategies, and Potential Therapeutics. Rev. Neurosci. 30, 339–358. doi:10.1515/revneuro-2016-0035
Gao, M., Gnutt, D., Orban, A., Appel, B., Righetti, F., Winter, R., et al. (2016). RNA Hairpin Folding in the Crowded Cell. Angew. Chem. Int. Ed. 55, 3224–3228. doi:10.1002/anie.201510847
Gesteira, T. F., Marforio, T. D., Mueller, J. W., Calvaresi, M., and Coulson-Thomas, V. J. (2021). Structural Determinants of Substrate Recognition and Catalysis by Heparan Sulfate Sulfotransferases. ACS Catal. 11, 10974–10987. doi:10.1021/acscatal.1c03088
Girdhar, K., Scott, G., Chemla, Y. R., and Gruebele, M. (2011). Better Biomolecule Thermodynamics from Kinetics. J. Chem. Phys. 135, 015102. doi:10.1063/1.3607605
Gnutt, D., Sistemich, L., and Ebbinghaus, S. (2019a). Protein Folding Modulation in Cells Subject to Differentiation and Stress. Front. Mol. Biosci. 6, 38. doi:10.3389/fmolb.2019.00038
Gnutt, D., Timr, S., Ahlers, J., König, B., Manderfeld, E., Heyden, M., et al. (2019b). Stability Effect of Quinary Interactions Reversed by Single Point Mutations. J. Am. Chem. Soc. 141, 4660–4669. doi:10.1021/jacs.8b13025
Grum, D., van den Boom, J., Neumann, D., Matena, A., Link, N. M., and Mueller, J. W. (2010). A Heterodimer of Human 3′-Phospho-Adenosine-5′-Phosphosulphate (PAPS) Synthases Is a New Sulphate Activating Complex. Biochem. Biophysical Res. Commun. 395, 420–425. doi:10.1016/j.bbrc.2010.04.039
Handa, A., Tham, E., Wang, Z., Horemuzova, E., and Grigelioniene, G. (2016). Autosomal Recessive Brachyolmia: Early Radiological Findings. Skeletal Radiol. 45, 1557–1560. doi:10.1007/s00256-016-2458-8
Haque, M. F. u., King, L. M., Krakow, D., Cantor, R. M., Rusiniak, M. E., Swank, R. T., et al. (1998). Mutations in Orthologous Genes in Human Spondyloepimetaphyseal Dysplasia and the Brachymorphic Mouse. Nat. Genet. 20, 157–162. doi:10.1038/2458
Harjes, S., Bayer, P., and Scheidig, A. J. (2005). The Crystal Structure of Human PAPS Synthetase 1 Reveals Asymmetry in Substrate Binding. J. Mol. Biol. 347, 623–635. doi:10.1016/j.jmb.2005.01.005
Iida, A., Simsek-Kiper, P. Ö., Mizumoto, S., Hoshino, T., Elcioglu, N., Horemuzova, E., et al. (2013). Clinical and Radiographic Features of the Autosomal Recessive Form of Brachyolmia Caused byPAPSS2Mutations. Hum. Mutat. 34, 1381–1386. doi:10.1002/humu.22377
Kabsch, W., and Sander, C. (1983). Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers 22, 2577–2637. doi:10.1002/bip.360221211
Klassen, C. D., and Boles, J. W. (1997). Sulfation and Sulfotransferases 5: the Importance of 3’-phosphoadenosine 5’-phosphosulfate (PAPS) in the Regulation of Sulfation. FASEB J. 11, 404–418. doi:10.1096/fasebj.11.6.9194521
Klinge, C. M., Clark, B. J., and Prough, R. A. (2018). “Dehydroepiandrosterone Research: Past, Current, and Future,” in Vitamins and Hormones (Cambridge, MA: Elsevier), 108.1–28. doi:10.1016/bs.vh.2018.02.002
Kurima, K., Warman, M. L., Krishnan, S., Domowicz, M., Krueger, R. C., Deyrup, A., et al. (1998). A Member of a Family of Sulfate-Activating Enzymes Causes Murine Brachymorphism. Proc. Natl. Acad. Sci. 95, 8681–8685. doi:10.1073/pnas.95.15.8681
Miyake, N., Elcioglu, N. H., Iida, A., Isguven, P., Dai, J., Murakami, N., et al. (2012). PAPSS2mutations Cause Autosomal Recessive Brachyolmia. J. Med. Genet. 49, 533–538. doi:10.1136/jmedgenet-2012-101039
Mueller, J. W., Gilligan, L. C., Idkowiak, J., Arlt, W., and Foster, P. A. (2015). The Regulation of Steroid Action by Sulfation and Desulfation. Endocr. Rev. 36, 526–563. doi:10.1210/er.2015-1036
Mueller, J. W., Idkowiak, J., Gesteira, T. F., Vallet, C., Hardman, R., van den Boom, J., et al. (2018). Human DHEA Sulfation Requires Direct Interaction between PAPS Synthase 2 and DHEA Sulfotransferase SULT2A1. J. Biol. Chem. 293, 9724–9735. doi:10.1074/jbc.RA118.002248
Mueller, J. W., and Shafqat, N. (2013). Adenosine-5′-phosphosulfate - a Multifaceted Modulator of Bifunctional 3′-Phospho-Adenosine-5′-Phosphosulfate Synthases and Related Enzymes. FEBS J. 280, 3050–3057. doi:10.1111/febs.12252
Noordam, C., Dhir, V., McNelis, J. C., Schlereth, F., Hanley, N. A., Krone, N., et al. (2009). Inactivating PAPSS2 Mutations in a Patient with Premature Pubarche. N. Engl. J. Med. 360, 2310–2318. doi:10.1056/NEJMoa0810489
Oostdijk, W., Idkowiak, J., Mueller, J. W., House, P. J., Taylor, A. E., O'Reilly, M. W., et al. (2015). PAPSS2 Deficiency Causes Androgen Excess via Impaired DHEA Sulfation-In Vitro and In Vivo Studies in a Family Harboring Two Novel PAPSS2 Mutations. J. Clin. Endocrinol. Metab. 100, E672–E680. doi:10.1210/jc.2014-3556
Paganini, C., Gramegna Tota, C., Superti-Furga, A., and Rossi, A. (2020). Skeletal Dysplasias Caused by Sulfation Defects. Ijms 21, 2710. doi:10.3390/ijms21082710
Perez-Garcia, E. M., Whalen, P., and Gurtunca, N. (2021). Novel Inactivating Homozygous PAPSS2 Mutation in Two Siblings with Disproportionate Short Stature. AACE Clin. Case Rep.. doi:10.1016/j.aace.2021.11.003 https://www.aaceclinicalcasereports.com/article/S2376-0605(21)00121-8/fulltext.
Prydz , K. (2015). Determinants of Glycosaminoglycan (GAG) Structure. Biomolecules 5, 2003–2022. doi:10.3390/biom5032003
Raman, R., Sasisekharan, V., and Sasisekharan, R. (2005). Structural Insights into Biological Roles of Protein-Glycosaminoglycan Interactions. Chem. Biol. 12, 267–277. doi:10.1016/j.chembiol.2004.11.020
Ross, C. A., and Poirier, M. A. (2004). Protein Aggregation and Neurodegenerative Disease. Nat. Med. 10, S10–S17. doi:10.1038/nm1066
Schröder, E., Gebel, L., Eremeev, A. A., Morgner, J., Grum, D., Knauer, S. K., et al. (2012). Human PAPS Synthase Isoforms Are Dynamically Regulated Enzymes with Access to Nucleus and Cytoplasm. PLoS ONE 7, e29559. doi:10.1371/journal.pone.0029559
Sekulic, N., Dietrich, K., Paarmann, I., Ort, S., Konrad, M., and Lavie, A. (2007). Elucidation of the Active Conformation of the APS-Kinase Domain of Human PAPS Synthetase 1. J. Mol. Biol. 367, 488–500. doi:10.1016/j.jmb.2007.01.025
Soto, C., and Pritzkow, S. (2018). Protein Misfolding, Aggregation, and Conformational Strains in Neurodegenerative Diseases. Nat. Neurosci. 21, 1332–1340. doi:10.1038/s41593-018-0235-9
Strott, C. A. (2002). Sulfonation and Molecular Action. Endocr. Rev. 23, 703–732. doi:10.1210/er.2001-0040
Tüysüz, B., Yılmaz, S., Gül, E., Kolb, L., Bilguvar, K., Evliyaoğlu, O., et al. (2013). Spondyloepimetaphyseal Dysplasia Pakistani Type: Expansion of the Phenotype. Am. J. Med. Genet. 161, 1300–1308. doi:10.1002/ajmg.a.35906
van den Boom, J., Heider, D., Martin, S. R., Pastore, A., and Mueller, J. W. (2012). 3′-Phosphoadenosine 5′-Phosphosulfate (PAPS) Synthases, Naturally Fragile Enzymes Specifically Stabilized by Nucleotide Binding. J. Biol. Chem. 287, 17645–17655. doi:10.1074/jbc.M111.325498
Vöpel, T., Bravo-Rodriguez, K., Mittal, S., Vachharajani, S., Gnutt, D., Sharma, A., et al. (2017). Inhibition of Huntingtin Exon-1 Aggregation by the Molecular Tweezer CLR01. J. Am. Chem. Soc. 139, 5640–5643. doi:10.1021/jacs.6b11039
Vöpel, T., Scholz, R., Davico, L., Groß, M., Büning, S., Kareth, S., et al. (2015). Infrared Laser Triggered Release of Bioactive Compounds from Single Hard Shell Microcapsules. Chem. Commun. 51, 6913–6916. doi:10.1039/C4CC09745A
Wang, W. (2005). Protein Aggregation and its Inhibition in Biopharmaceutics. Int. J. Pharmaceutics 289, 1–30. doi:10.1016/j.ijpharm.2004.11.014
Waters, P. J. (2001). Degradation of Mutant Proteins, Underlying “Loss of Function” Phenotypes, Plays a Major Role in Genetic Disease. Curr. Issues Mol. Biol. 3 (3), 57–65. doi:10.21775/cimb.003.057
Keywords: PAPS synthase, sulfation pathways, in-cell spectroscopy, protein folding, stability and aggregation
Citation: Brylski O, Shrestha P, House PJ, Gnutt P, Mueller JW and Ebbinghaus S (2022) Disease-Related Protein Variants of the Highly Conserved Enzyme PAPSS2 Show Marginal Stability and Aggregation in Cells. Front. Mol. Biosci. 9:860387. doi: 10.3389/fmolb.2022.860387
Received: 22 January 2022; Accepted: 28 February 2022;
Published: 08 April 2022.
Edited by:
Giuseppe Calamita, University of Bari Aldo Moro, ItalyReviewed by:
Annarita Di Mise, University of Bari Aldo Moro, ItalyAntonio Rossi, University of Pavia, Italy
Copyright © 2022 Brylski, Shrestha, House, Gnutt, Mueller and Ebbinghaus. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonathan Wolf Mueller, ai53Lm11ZWxsZXJAYmhhbS5hYy51aw==; Simon Ebbinghaus, cy5lYmJpbmdoYXVzQHR1LWJyYXVuc2Nod2VpZy5kZQ==
†These authors have contributed equally to this work