 Chuixiong Wu
Chuixiong Wu Ruye Li†
Ruye Li† Kuang Yu
Kuang Yu- Tsinghua-Berkeley Shenzhen Institute, Tsinghua Shenzhen International Graduate School, Tsinghua University, Shenzhen, China
Molecular mechanics (MM) is a powerful tool to study the properties of molecular systems in the fields of biology and materials science. With the development of ab initio force field and the application of ab initio potential energy surface, the nuclear quantum effect (NQE) is becoming increasingly important for the robustness of the simulation. However, the state-of-the-art path-integral molecular dynamics simulation, which incorporates NQE in MM, is still too expensive to conduct for most biological and material systems. In this work, we analyze the locality of NQE, using both analytical and numerical approaches, and conclude that NQE is an extremely localized phenomenon in nonreactive molecular systems. Therefore, we can use localized machine learning (ML) models to predict quantum force corrections both accurately and efficiently. Using liquid water as example, we show that the ML facilitated centroid MD can reproduce the NQEs in both the thermodynamical and the dynamical properties, with a minimal increase in computational time compared to classical molecular dynamics. This simple approach thus largely decreases the computational cost of quantum simulations, making it really accessible to the studies of large-scale molecular systems.
Introduction
Molecular mechanics (MM) simulation is an extremely powerful tool in the studies of biomolecules and material systems. In most situations, MM simulations are conducted with Born-Oppenheimer approximation, with the atoms moving on an adiabatic electronic potential energy surface (PES). The motions of the atoms on PES are typically governed by classical statistical mechanics and Newtonian dynamics, so the nuclear quantum effects (NQEs) are often neglected. While many properties can be computed with satisfactory accuracy at classical level, it has been shown that the NQE can be quite essential in systems with light atoms (Wang et al., 2014; Markland and Ceriotti, 2018; Tuckerman and Ceperley, 2018). One important example is that NQE may significantly alter the strengths and the structures of the hydrogen bonding networks in aqueous solutions. Such effect brings profound impacts to the understandings of many important biological problems such as enzyme activities and DNA stabilities (Agarwal et al., 2002; Pérez et al., 2010). In materials science, it was also found that NQEs play nonnegligible roles in many problems such as guest molecule adsorptions and thermal transport simulations (Wahiduzzaman et al., 2014; Shulumba et al., 2017; Luo and Yu, 2020). Existing evidences show that NQE is not only a specific problem existing in a small number of systems, but a universal phenomenon that needs to be accounted for in most biological and materials simulations.
Conventionally, NQEs can be partially incorporated into classical MM through implicit ways. Ad hoc corrections can be used for some systems: for example, water molecules are often constrained to be rigid, simulating the freezing of the high-frequency intramolecular vibrations at room temperature. However, such techniques are highly specific to a certain type of molecule and can lead to significant errors as it does not treat all degrees of freedom consistently (Luo and Yu, 2020) Meanwhile, the force fields most widely used nowadays in molecular simulations (such as AMBER (Weiner et al., 1984), OPLS (Jorgensen et al., 1984, 1996), and CHARMM (Brooks and Karplus, 1983; MacKerell et al., 1995; MacKerell Jr et al., 2000) were developed empirically to reproduce the experimental results. Since the real world is quantum in nature, it can be argued that these empirical force field parameters are trained with quantum effects incorporated implicitly, legitimating their usages in classical MM simulations. However, we note that in the conventional force field training process, it is often difficult to fit both the small cluster ab initio data and the bulk experimental data simultaneously. Therefore, in order to obtain a balanced performance in bulk, one often needs to sacrifice the accuracy of the potential in the small clusters. Besides the long-range many-body interactions, the negligence of NQE in bulk simulations is potentially one of the major reasons causing this dilemma. Due to this problem, the conventional empirical force fields are often not reliable in the details of the PES, which limits its transferability and its predictive power. Therefore, in the past decade, it has becoming increasingly popular to construct accurate force fields from ab initio data directly without any experimental inputs (Lee et al., 1995; Martin et al., 1995, 95; McDaniel et al., 2012; Xu et al., 2018). For the systems that are affordable, ab initio MD (AIMD), including both Born-Oppenheimer MD (BOMD) and Car-Parinello MD (CPMD) (Car and Parrinello, 1985), are also frequently performed with the ab initio calculations being run on the fly to compute the PES. For these nonempirical MM simulations, NQE becomes an important issue with universal concerns and should be addressed explicitly in the simulation.
Most main stream NQE methods that are widely used in large-scale simulations are originated from Feynman’s path-integral formula (Feynman, 1998; Feynman et al., 2010). One simple technique is the Feynman-Hibbs correction (FHC) (Sesé, 1992, 1995; Guillot and Guissani, 1998; Feynman et al., 2010), in which atoms are smeared as in free-particle limit and the quantum correction is given by convoluting the PES with Gaussian distributions. In a pairwise additive potential, FHC also leads to a pairwise additive correction, which are only related to the high order derivatives of the pairwise interactions. Although being heavily approximated, FHC has been widely used in complicated simulations (e.g., Grand Canonical Monte Carlo simulations (Kowalczyk et al., 2005; Fischer et al., 2009; Durette et al., 2016; Ahmed et al., 2017, 2019)) due to its simplicity. However, FHC can be erroneous in many systems (as we will show in this work), and it can be difficult to implement for a general many-body potential. A more advanced method is the path-integral molecular dynamics (PIMD) (Berne and Thirumalai, 1986; Tuckerman and Martyna, 2000), which simulates a quantum particle using a classical “ring-polymer,” realizing the mathematical connection between the partition functions of the two systems. In order to describe the dynamics, PIMD is further succeeded by techniques such as ring-polymer molecular dynamics (RPMD) (Craig and Manolopoulos, 2004) and centroid molecular dynamics (CMD) (Cao and Voth, 1994a; 1994b; 1994c; 1994d; 1994e). All these methods can generate the exact quantum Boltzmann distribution when using large number of beads, but they are also much more expensive to run compared to classical MD. The large computational cost of PIMD roots in two facts: first, it utilizes multiple (typically 32–64) beads to represent one atom, so the number of energy and force evaluations it needs is much more than that of a classical simulation; and second, the hard spring potential between the neighboring beads requires a much smaller timestep for time integration. Tremendous efforts have been made to accelerate PIMD (including RPMD and CMD): for example, algorithms such as contraction (Markland and Manolopoulos, 2008b, 08, 2008a) or high-order PI(Pérez and Tuckerman, 2011; Kapil et al., 2016) reduces the number of beads effectively, and advance integrators were developed to allow the use of larger timesteps (Ceriotti et al., 2010). These efforts have made PIMD gradually enter the mainstream of MD simulations, but it is still much slower than the standard classical MD. Techniques such as contraction also relies on a subjective partition of the potential into fast and slow varying parts, which is sometimes not so straightforward to perform. Consequently, it is still relatively rare to see PIMD (and the related RPMD/CMD methods) being used on large systems (e.g., in biomolecule simulations) in conjunction with expensive many-body potentials (e.g., multipolar polarizable force fields, or even ab initio potentials).
In this work, using water as example, we will demonstrate how machine learning (ML) techniques can be utilized to accelerate the CMD simulation. CMD is an important category of path-integral method, which essentially coarse grains each ring-polymer into a single site located on the centroid of the polymer. The centroids move on an effective potential of mean force (PMF), which are conventionally sampled on-the-fly using explicit ring-polymers. The masses of the intra-polymer modes are set to be light, so the centroid and the intra-polymer motions are decoupled and the centroid motions can be considered as adiabatic. Rigorous adiabatic CMD usually requires a much smaller timestep, and the computation of the centroid PMF is also time consuming. However, noticing that the difference between the centroid PMF and the original PES (i.e., the quantum correction to the PES) is localized, it can be learned using localized ML models quite efficiently.
In the recent few years, we witness a rapid development of ML techniques in MM simulations. A variety of ML techniques, including BPNN (Behler and Parrinello, 2007), EANN (Zhang et al., 2019, 2021), SchNet (Schütt et al., 2018, 18), and DeepPotential (Zhang et al., 2018, 18), have been applied to develop accurate high-dimensional PES. All these models decompose the total energy into a sum of localized atomic energies, which are then predicted using neural networks with input features designed to describe the local environment of each atom. While such approaches have become increasingly popular in force field development, their applications on the description of NQE are relatively rare. Liu et al. developed the method called equilibrium continuity dynamics with the aim of calculating the quantum time correlation function in Wigner phase space, and accelerated this approach using ML techniques (Liu et al., 2021). However, to the best of our knowledge, the ML-facilitated path-integral methods (in particular, CMD) have not been thoroughly investigated. In this work, through analytical and numerical methods, we will rigorously quantify the locality of the CMD quantum force correction in molecular systems. Due to this locality, the CMD quantum force correction is actually much easier to model using ML methods compared to regular PES. We will show that a robust quantum force correction can be trained efficiently on very small cluster PIMD samples in molecular systems. Using this method, we can perform CMD simulations with a speed similar to the conventional classical MD, really making the quantum simulations accessible to the studies of large-scale biological and materials problems.
Methods
Path-Integral Based Methods
According to Feynman, the quantum partition function of a particle can be expressed in a path integral form, which, after discretization, is analogous to the classical partition function of a ring-polymer. For example, in the one-dimensional case (Tuckerman, 2010):
In here,
When the motion of the centroid and the intra-ring vibrations are decoupled, the centroid can be considered as moving on an adiabatic PMF (denoted as
Here, the action
In principle, one can conduct either MD or MC to sample the bead (
Another simple alternative was proposed by Feynman and Hibbs (Feynman, 1998), when they proved that in a variational sense, the optimal effective classical potential
It is interesting to observe that the Gaussian kernel features a width of
When the underlying potential is pairwise additive and spherically symmetric (i.e.,
Here,
Machine Learning Techniques and Model Locality
In this work, we will use the embedded atom neural network (EANN) technique to learn the quantum centroid force correction (Cao and Voth, 1994a; Feynman et al., 2010):
EANN, like many other popular ML methods, assumes that the total energy (in this work, the centroid PMF) can be written as a sum of atomic energies:
And each atomic energy
In this work, we will show that in molecular systems, the quantum centroid force correction (
Computational Details
In this work, we use water as our test example, with both q-TIP4P/F (Habershon et al., 2009) and AMOEBA (Ren and Ponder, 2003) force fields. The two force fields were selected due to the following reasons: 1) Both of them are not too expensive to run, so we can obtain the rigorous bulk PIMD benchmark data at a reasonable cost; 2) The two force fields represent two distinct categories of PES. The q-TIP4P/F force field is one of the conventional force fields with simple point charges and Lennard-Jones interactions, which are completely spherical symmetric and pairwise-additive. Meanwhile, AMOEBA features multipole moments and explicitly polarizable atoms, thus it resembles a more general many-body PES. Therefore, it is interesting to compare the performances of the localized ML model on both cases, in order to examine the generality of the method.
For each force field, we first performed a bulk (1,000 water) NPT PIMD simulation at the corresponding condition (1 bar, 300 K or 100 K), and randomly drew 2000 cluster configurations from the bulk trajectory. Each cluster was consisted by one central water molecule and seven nearest molecules, so we guarantee that the first solvation shell of the central water was fully included. These water octamers were then used to generate the training data: with centroid positions fixed, we ran 1 ns PIMD simulation to sample the bead distribution, so the averaged centroid force is converged within 0.2 kJ/(mol Å). To examine the width of the ring-polymer, we also performed 1 ns PIMD simulations on a smaller bulk system (with 216 water) with a density of 0.997 g/ml at different temperatures. All the sampling simulations mentioned above were conducted using the OpenMM 7.4.0 program, with modifications made to enable the fixed-centroid PIMD simulations. All sampling simulations were performed using 0.5 fs timestep and a Langevin thermostat with a 1.0 ps−1 friction constant. We used 32 beads at 300 K and 64 beads at 100 K, and the cutoff distance for the nonbonded interaction was set to 9 Å.
Once the averaged centroid forces were collected, their differences with the classical forces (i.e.,
The trained EANN model was then used in combination with the original force field to perform the machine learning CMD (ML-CMD) simulations. For validation, the simulation results were compared to the PIMD, RPMD, and the PA-CMD results, and all the production runs were conducted using the i-PI 2.2 program. In PA-CMD simulations, the lower limit of internal modes is set to 648 THz to ensure the adiabaticity between the centroid mode and the intra-ring modes, and the timestep was set to be 0.02 fs.
Results
Locality of the Quantum Force Correction
As we discussed in the theory section, the locality of the
In here,
and we can rewrite Eq. 9 as:
Therefore, the distribution of
And in here, the intrinsic vibrational frequency of the atom is:
Therefore, the total second moment of the distribution of
Define the dimensionless quantity
Then the second moment of the ring-polymer bead distribution is:
Apparently, the locality of the ring-polymer is determined by both the temperature and the curvature of the underlying potential. In the high temperature limit (
In Figure 1, using liquid water as example, we plot the widths of the H atoms given by the square root of Eq. 15, in conjunction with the values given by the FHC and the QHO formula, as well as the PIMD simulation results. The intrinsic frequency
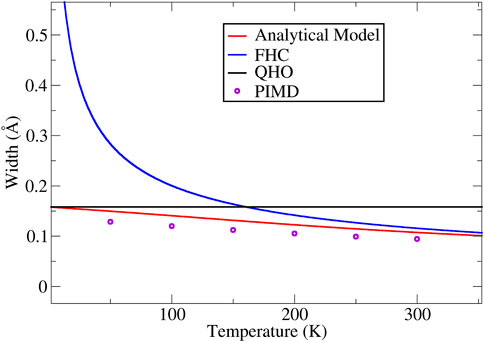
FIGURE 1. The width of the ring-polymer predicted by Eq. 15 at different temperatures (calculated using = 670 cm−1), plotted along the side with the FHC and QHO widths. The PIMD simulation results are also plotted in the figure using open circles.
Using the results of the bead distribution width, we can roughly estimate the magnitude of the quantum force correction at a certain range. Let us consider a pair of particles interacting with Coulombic force (the force with the slowest decay rate in molecular systems), and evaluate how the quantum correction to the magnitude of the force decays with increasing distance. For convenience, without writing down the charges explicitly, we assume that the magnitude of the classical force between the two particles is:
And let us assume the three-dimensional ring-polymer bead shifts for particle i and particle j are
Then, the averaged quantum force correction, truncated at second order, is:
Hence the relative correction is:
Considering
The largest force correction happens when the direction of
This inequality could be further improved, realizing that the correlation (
In this work, we use a cutoff distance of 4 Å for our ML model, and at this distance, Eq. 20 gives a relative force correction as small as 2% at all temperatures. Furthermore, both analytical and numerical analysis indicate that the correlation between the force corrections decays fast with respect to interatomic distance. Therefore, it is strongly indicated that the quantum force correction is an extremely localized phenomenon and can be tackled using a very small localized ML model.
It is noted that previous studies (Rognoni et al., 2021) show the vibrational modes of liquid water is highly nonlocal. While there are certainly nonlocal NQE existing in aqueous systems, such result is not necessarily contradictory to our findings in this work. In here, we focus on the locality of the centroid force correction: just like localized force can lead to nonlocal vibrational modes, local force correction can certainly give rise to global NQE in bulk systems.
Force Tests
Using the water octamer PIMD centroid forces as training data, we train the EANN model with a small cutoff of 4 Å, and test its validity in both bulk and surface water systems. We examine the accuracy of the EANN model on the quantum force corrections for both Q-TIP4P/F and AMOEBA potentials, and the test results are shown in Figure 2. Excellent agreement between the fitted and the reference data are achieved in all cases, showing the great success of the methodology. This result is in consistent with our analysis in the last section, showing that the quantum force correction is virtually fully determined by the structure of the first solvation shell. Water octamer clusters that contain one fully solvated water molecule and seven surface molecules is all it needs to obtain a model that is transferrable to both bulk and surface environments. Such extrapolation capability does not rely on the pairwise additivity of the underlying potential, and is extremely important for PES that is difficult to evaluate (such as AMEOBA).
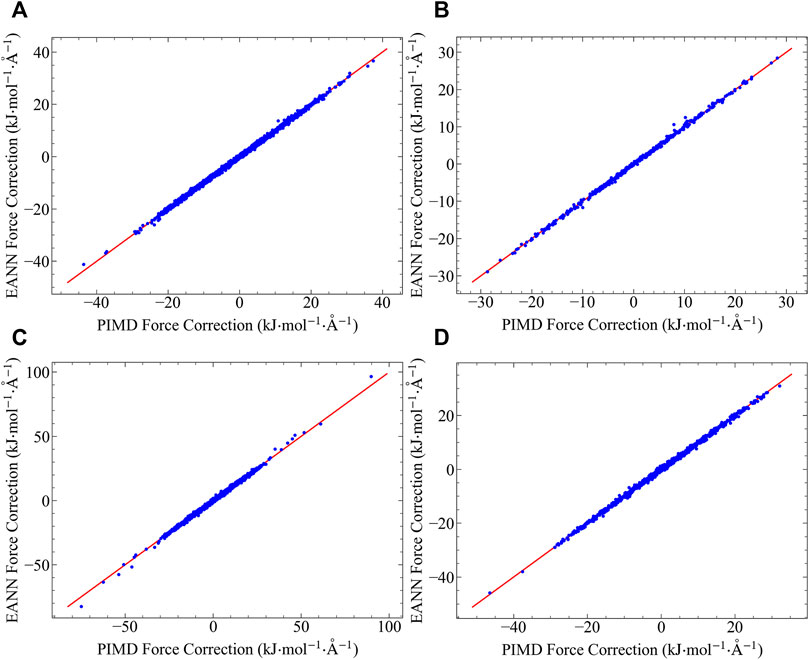
FIGURE 2. Quantum force corrections predicted by the EANN model, plotted against the reference data computed using PIMD: (A) Results in bulk water with q-TIP4P/F force field. (B) Results in surface water with q-TIP4P/F force field. (C) Results in bulk water with AMOEBA force field. (D) Results in surface water with AMOEBA force field.
Thermodynamic Properties
After examining the centroid force correction, we investigate the performance of the EANN model in the bulk ML-CMD simulations. We first investigate the centroid radial distribution functions (RDFs) for both O-O and O-H atom pairs, and the results are plotted in Figure 3. It is noted that the RDF reported here is not the regular RDF reported in literature, but the RDF computed by centroid positions. Although in principle, there is no essential technical difficulties to train the PMF of one bead (instead of the centroid) such that the real RDF can be obtained. Apparently, compared to pure classical MD, the machine learned quantum force correction reduces the error on the centroid RDFs significantly compared to the exact PIMD simulation. It is further observed that the NQE on the centroid RDFs are more significant in the AMOEBA simulations compared to the q-TIP4P/F simulations, and our correction is also more effective in AMOEBA. For both force fields, the quantum centroid RDF is generally less structured compared to the classical results, and such effect is accurately captured by the EANN model.
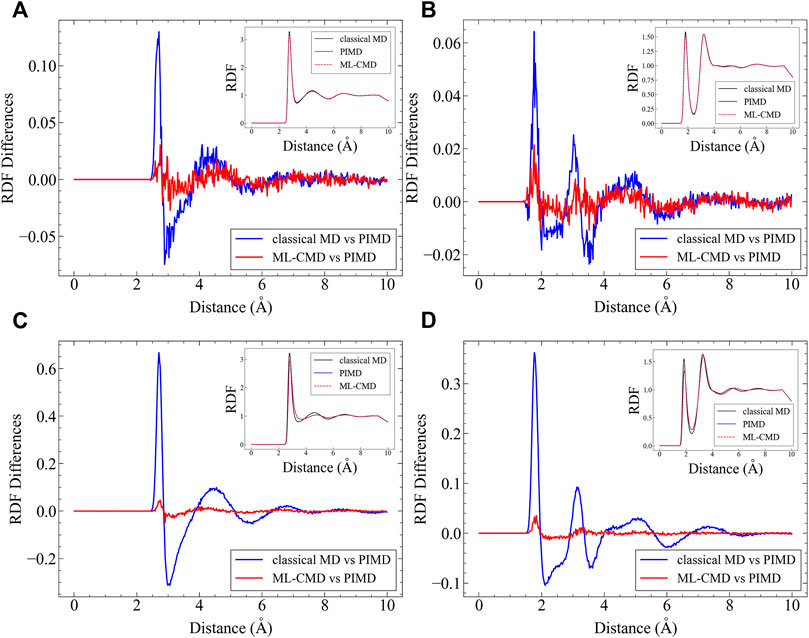
FIGURE 3. Comparison between different centroid RDFs. In each figure, the RDF differences between ML-CMD and PIMD are shown, in conjunction with the RDF differences between classical MD and PIMD. The corresponding RDFs are all plotted in the inset. (A) The O-O RDF in q-TIP4P/F simulations; (B) the O-H RDF in Sq-TIP4P/F simulations; (C) the O-O RDF in AMOEBA simulations; and (D) the O-H RDF in AMOEBA simulations.
Besides the liquid structure, we further examine the NQEs on the system pressure (or, equivalently, the system density), and the results are shown in Figure 4. In this part, we performed the reference PIMD simulations using the more efficient OpenMM program, while the ML-CMD was conducted using i-PI for convenience. To rule out the potential numerical differences caused by different programs, we computed the classical baselines using both codes. The classical pressures computed by both OpenMM and i-PI are in excellent agreement, proving that the different integrators and thermostat implementations in the two codes do not affect our simulation results significantly. With the classical baselines established, it is clearly shown in Figure 4 that ML-CMD reproduces the exact PIMD results with a reasonable accuracy. Small overestimations can be observed in both the low-density and the high-density ends, potentially because that the EANN model was trained at 0.998 g/ml, so the model error is slightly larger when the density deviates from this density. In summary, it is shown that in both the centroid RDF and the pressure tests, the ML force correction trained by only small cluster data can reproduce the bulk PIMD results accurately, proving the capability of our methodology in the studies of thermodynamic properties.
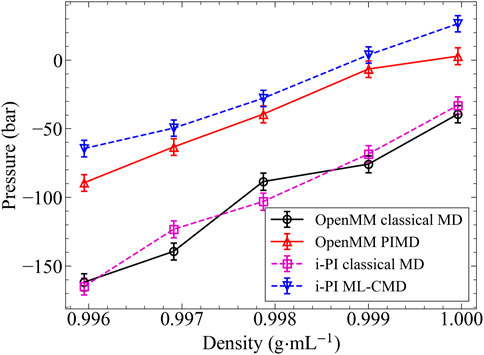
FIGURE 4. The pressure varies with density in 300 K. This is an alternative pressure plot because the barostat behave difference in two different software. The black solid line: classical MD simulated by OpenMM. The violet dashed line: classical MD simulated by i-PI. The red solid line: PIMD simulated by OpenMM. The dashed blue line: ML-CMD simulated by i-PI This model is trained in 0.998 g/ml.
Dynamic Property
As we discussed in the theoretical background section, the ML-CMD method is theoretically equivalent to adiabatic CMD, so it should also be able to capture the NQE in dynamic properties at CMD level of theory. We thus computed the self-diffusion constant of bulk water using classical MD, PA-CMD, and ML-CMD, the results of which are listed in Table 1. The effective mass used in PA-CMD is small enough such that the PA-CMD simulation we perform is a good approximation to the rigorous adiabatic CMD. Once again, the ML-CMD agrees with PA-CMD within the statistical uncertainty, validating our methodology.

TABLE 1. The self-diffusion constants from classical MD, PA-CMD, and ML-CMD simulations, respectively. The last digit in the parenthesis marks the uncertainty.
Computational Cost
To verify that ML-CMD indeed possesses significant advantages in computational efficiency, we compare the running speeds of classical MD, T-RPMD, PA-CMD, and ML-CMD in Table 2. AMOEBA force field is employed, and all simulations are run using i-PI, with one RTX 2080 graphic card, 8 CPU cores, and 80 GB of memory. The EANN model has not been implemented in CUDA platform, so this part of the calculation is performed on a separate CPU client, while all other calculations are run on GPU. Comparing ML-CMD with classical MD, we can see that even with a slow CPU implementation, the extra computational time due the EANN model is minor. Because of the extremely short cutoff distance (4 Å) we use, the EANN model is rather small and is very fast to evaluate. Meanwhile, both T-RPMD and PA-CMD are orders of magnitude slower than ML-CMD and classical MD, due to two major reasons. First, both simulations have to propagate the motions of 32 replicas of the system, which means at least 32 times slower due to more energy and force evaluations. Second, the timestep has to be smaller in both T-RPMD and CMD to maintain the stability of the simulations. We do acknowledge that a better integrator can be employed so a larger timestep is possible for at least T-RPMD, and techniques such as contraction can help to decrease the number of beads employed in force evaluation. All these techniques can alleviate, but not cure the heavy computational cost of path integral simulations. In comparison, ML-CMD is by far the simplest, and the most straightforward way to achieve the classical-like simulation speed, circumventing the complicated and highly specialized techniques used in PIMD.
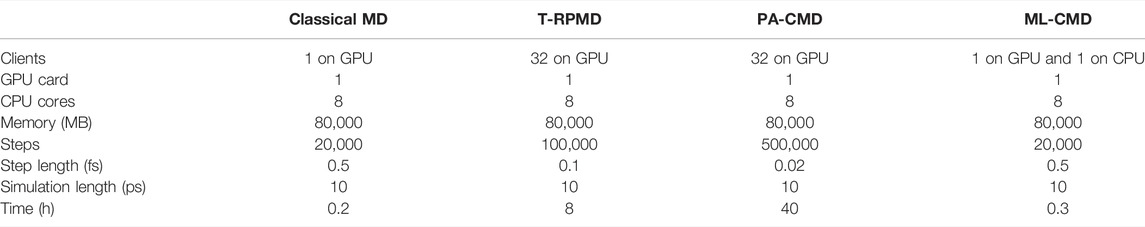
TABLE 2. The simulate resources and time costs of different methods.
Conclusion and Outlook
In this work, through both analytical and numerical approaches, we analyze the locality of the centroid force corrections in typical molecular systems such as bulk water. A general formula is given to quantify the locality, and it was found that at ambient condition, the NQE on the forces decays to less than 0.5% at the range of 4 Å. Exploiting this locality, we are able to train a reliable ML model to predict the NQE force correction, utilizing only small cluster training data. For a PES that is expensive to evaluate (e.g., polarizable force field or even ab initio potential), such locality is particularly important as the force correction in bulk system can be difficult, or even impossible to compute. Also due to the locality, the model is high transferrable to different physical environments, and applicable to both pairwise additive force fields and general many-body potentials. The ML model is formulated as a simple correction term to the classical forces and energies, thus is straightforward to implement and easy to use in complicated simulations such as GCMC. The ML-CMD simulation, which uses the ML quantum force corrections in combination with the classical forces, reproduces both the thermodynamics and the dynamics of adiabatic CMD. Thanks to the extreme locality of the model, the ML-CMD simulation runs as efficient as classical MD, which is much faster compared to both RPMD and CMD. The computational cost of ML-CMD is not significantly higher than FHC, while ML-CMD is formulated more rigorously and is more accurate compare to FHC. Therefore, the localized ML-CMD is considered to be an excellent approach to incorporate NQEs in the simulations of large biological and materials systems.
Meanwhile, we note that there are a few scenarios that the locality of the ML-CMD could be broken. One typical example is the proton tunneling under low temperature: Eq. 15 holds only when the curvature of the underlying PES is positive, which is not the case in the transition state region of a chemical reaction. At high temperature, the locality is still enforced by the FHC limit, but in low temperature, the locality of the proton at the transition region could be a problem. More detailed analysis is needed to quantify the necessary cutoff range of the ML model in these cases, which is left to future work. Nevertheless, in most nonreactive molecular system simulations (e.g., protein folding, drug molecule binding etc.), our method serves as a both robust and fast alternative to the rigorous PIMD and adiabatic CMD simulations.
Data Availability Statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Author Contributions
KY designed the research, CW and RL collected the data and performed the simulations, CW and KY analyzed the data and wrote the paper.
Funding
This work is supported by National Natural Science Foundation of China (Project Approval No. 22103048) and Guangdong Basic and Applied Basic Research Foundation (Grant No. 2019A1515110256) and Tsinghua SIGS International Corporation Fund (Grant Number HW2020009).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank Xinyan Wang for providing us his universal i-PI wrapper program and helpful discussions.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2022.851311/full#supplementary-material
References
Agarwal, P. K., Billeter, S. R., Rajagopalan, P. T. R., Benkovic, S. J., and Hammes-Schiffer, S. (2002). Network of Coupled Promoting Motions in Enzyme Catalysis. Proc. Natl. Acad. Sci. U.S.A. 99, 2794–2799. doi:10.1073/pnas.052005999
Ahmed, A., Liu, Y., Purewal, J., Tran, L. D., Wong-Foy, A. G., Veenstra, M., et al. (2017). Balancing Gravimetric and Volumetric Hydrogen Density in MOFs. Energy Environ. Sci. 10, 2459–2471. doi:10.1039/C7EE02477K
Ahmed, A., Seth, S., Purewal, J., Wong-Foy, A. G., Veenstra, M., Matzger, A. J., et al. (2019). Exceptional Hydrogen Storage Achieved by Screening Nearly Half a Million Metal-Organic Frameworks. Nat. Commun. 10, 1568. doi:10.1038/s41467-019-09365-w
Behler, J., and Parrinello, M. (2007). Generalized Neural-Network Representation of High-Dimensional Potential-Energy Surfaces. Phys. Rev. Lett. 98, 146401. doi:10.1103/PhysRevLett.98.146401
Berne, B. J., and Thirumalai, D. (1986). On the Simulation of Quantum Systems: Path Integral Methods. Annu. Rev. Phys. Chem. 37, 401–424. doi:10.1146/annurev.pc.37.100186.002153
Brooks, B., and Karplus, M. (1983). Harmonic Dynamics of Proteins: Normal Modes and Fluctuations in Bovine Pancreatic Trypsin Inhibitor. Proc. Natl. Acad. Sci. U.S.A. 80, 6571–6575. doi:10.1073/pnas.80.21.6571
Cao, J., and Voth, G. A. (1994a). The Formulation of Quantum Statistical Mechanics Based on the Feynman Path Centroid Density. I. Equilibrium Properties. J. Chem. Phys. 100, 5093–5105. doi:10.1063/1.467175
Cao, J., and Voth, G. A. (1994b). The Formulation of Quantum Statistical Mechanics Based on the Feynman Path Centroid Density. II. Dynamical Properties. J. Chem. Phys. 100, 5106–5117. doi:10.1063/1.467176
Cao, J., and Voth, G. A. (1994c). The Formulation of Quantum Statistical Mechanics Based on the Feynman Path Centroid Density. III. Phase Space Formalism and Analysis of Centroid Molecular Dynamics. J. Chem. Phys. 101, 6157–6167. doi:10.1063/1.468503
Cao, J., and Voth, G. A. (1994d). The Formulation of Quantum Statistical Mechanics Based on the Feynman Path Centroid Density. IV. Algorithms for Centroid Molecular Dynamics. J. Chem. Phys. 101, 6168–6183. doi:10.1063/1.468399
Cao, J., and Voth, G. A. (1994e). The Formulation of Quantum Statistical Mechanics Based on the Feynman Path Centroid Density. V. Quantum Instantaneous Normal Mode Theory of Liquids. J. Chem. Phys. 101, 6184–6192. doi:10.1063/1.468400
Car, R., and Parrinello, M. (1985). Unified Approach for Molecular Dynamics and Density-Functional Theory. Phys. Rev. Lett. 55, 2471–2474. doi:10.1103/PhysRevLett.55.2471
Ceriotti, M., Parrinello, M., Markland, T. E., and Manolopoulos, D. E. (2010). Efficient Stochastic Thermostatting of Path Integral Molecular Dynamics. J. Chem. Phys. 133, 124104. doi:10.1063/1.3489925
Craig, I. R., and Manolopoulos, D. E. (2004). Quantum Statistics and Classical Mechanics: Real Time Correlation Functions from Ring Polymer Molecular Dynamics. J. Chem. Phys. 121, 3368–3373. doi:10.1063/1.1777575
Durette, D., Bénard, P., Zacharia, R., and Chahine, R. (2016). Investigation of the Hydrogen Adsorbed Density inside the Pores of MOF-5 from Path Integral Grand Canonical Monte Carlo at Supercritical and Subcritical Temperature. Sci. Bull. 61, 594–600. doi:10.1007/s11434-016-1027-9
Feynman, R. P., Hibbs, A. R., and Styer, D. F. (2010). Quantum Mechanics and Path Integrals. Massachusetts, United States: Courier Corporation.
Feynman, R. P. (1998). Statistical Mechanics: A Set of Lectures. New York, United States: Avalon Publishing.
Fischer, M., Hoffmann, F., and Fröba, M. (2009). Preferred Hydrogen Adsorption Sites in Various MOFs-A Comparative Computational Study. ChemPhysChem 10, 2647–2657. doi:10.1002/cphc.200900459
Guillot, B., and Guissani, Y. (1998). Quantum Effects in Simulated Water by the Feynman-Hibbs Approach. J. Chem. Phys. 108, 10162–10174. doi:10.1063/1.476475
Habershon, S., Markland, T. E., and Manolopoulos, D. E. (2009). Competing Quantum Effects in the Dynamics of a Flexible Water Model. J. Chem. Phys. 131, 024501. doi:10.1063/1.3167790
Hone, T. D., Rossky, P. J., and Voth, G. A. (2006). A Comparative Study of Imaginary Time Path Integral Based Methods for Quantum Dynamics. J. Chem. Phys. 124, 154103. doi:10.1063/1.2186636
Jorgensen, W. L., Madura, J. D., and Swenson, C. J. (1984). Optimized Intermolecular Potential Functions for Liquid Hydrocarbons. J. Am. Chem. Soc. 106, 6638–6646. doi:10.1021/ja00334a030
Jorgensen, W. L., Maxwell, D. S., and Tirado-Rives, J. (1996). Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 118, 11225–11236. doi:10.1021/ja9621760
Kapil, V., Behler, J., and Ceriotti, M. (2016). High Order Path Integrals Made Easy. J. Chem. Phys. 145, 234103. doi:10.1063/1.4971438
Kowalczyk, P., Tanaka, H., Hołyst, R., Kaneko, K., Ohmori, T., and Miyamoto, J. (2005). Storage of Hydrogen at 303 K in Graphite Slitlike Pores from Grand Canonical Monte Carlo Simulation. J. Phys. Chem. B 109, 17174–17183. doi:10.1021/jp0529063
Lee, T. J., Martin, J. M. L., and Taylor, P. R. (1995). An Accurate Ab Initio Quartic Force Field and Vibrational Frequencies for CH4 and Isotopomers. J. Chem. Phys. 102, 254–261. doi:10.1063/1.469398
Liu, X., Zhang, L., and Liu, J. (2021). Machine Learning Phase Space Quantum Dynamics Approaches. J. Chem. Phys. 154, 184104. doi:10.1063/5.0046689
Luo, R., and Yu, K. (2020). Capturing the Nuclear Quantum Effects in Molecular Dynamics for Lattice Thermal Conductivity Calculations: Using Ice as Example. J. Chem. Phys. 153, 194105. doi:10.1063/5.0022013
MacKerell, A. D., Banavali, N., and Foloppe, N. (2000). Development and Current Status of the CHARMM Force Field for Nucleic Acids. Biopolymers 56, 257–265. doi:10.1002/1097-0282(2000)56:4<257::AID-BIP10029>3.0.CO;2-W
MacKerell, A. D., Wiorkiewicz-Kuczera, J., and Karplus, M. (1995). An All-Atom Empirical Energy Function for the Simulation of Nucleic Acids. J. Am. Chem. Soc. 117, 11946–11975. doi:10.1021/ja00153a017
Markland, T. E., and Ceriotti, M. (2018). Nuclear Quantum Effects Enter the Mainstream. Nat. Rev. Chem. 2, 1–14. doi:10.1038/s41570-017-0109
Markland, T. E., and Manolopoulos, D. E. (2008a). A Refined Ring Polymer Contraction Scheme for Systems with Electrostatic Interactions. Chem. Phys. Lett. 464, 256–261. doi:10.1016/j.cplett.2008.09.019
Markland, T. E., and Manolopoulos, D. E. (2008b). An Efficient Ring Polymer Contraction Scheme for Imaginary Time Path Integral Simulations. J. Chem. Phys. 129, 024105. doi:10.1063/1.2953308
Martin, J. M. L., Lee, T. J., Taylor, P. R., and François, J. P. (1995). The Anharmonic Force Field of Ethylene, C2H4, by Means of Accurate Ab Initio Calculations. J. Chem. Phys. 103, 2589–2602. doi:10.1063/1.469681
McDaniel, J. G., Yu, K., and Schmidt, J. R. (2012). Ab Initio, Physically Motivated Force Fields for CO2 Adsorption in Zeolitic Imidazolate Frameworks. J. Phys. Chem. C 116, 1892–1903. doi:10.1021/jp209335y
Pérez, A., Tuckerman, M. E., Hjalmarson, H. P., and von Lilienfeld, O. A. (2010). Enol Tautomers of Watson−Crick Base Pair Models Are Metastable Because of Nuclear Quantum Effects. J. Am. Chem. Soc. 132, 11510–11515. doi:10.1021/ja102004b
Pérez, A., and Tuckerman, M. E. (2011). Improving the Convergence of Closed and Open Path Integral Molecular Dynamics via Higher Order Trotter Factorization Schemes. J. Chem. Phys. 135, 064104. doi:10.1063/1.3609120
Ren, P., and Ponder, J. W. (2003). Polarizable Atomic Multipole Water Model for Molecular Mechanics Simulation. J. Phys. Chem. B 107, 5933–5947. doi:10.1021/jp027815+
Rognoni, A., Conte, R., and Ceotto, M. (2021). How Many Water Molecules Are Needed to Solvate One? Chem. Sci. 12, 2060–2064. doi:10.1039/D0SC05785A
Schütt, K. T., Sauceda, H. E., Kindermans, P.-J., Tkatchenko, A., and Müller, K.-R. (2018). SchNet - A Deep Learning Architecture for Molecules and Materials. J. Chem. Phys. 148, 241722. doi:10.1063/1.5019779
Sesé, L. M. (1992). A Quantum Monte Carlo Study of Liquid Lennard-Jones Methane, Path-Integral and Effective Potentials. Mol. Phys. 76, 1335–1346. doi:10.1080/00268979200102121
Sesé, L. M. (1995). Feynman-Hibbs Potentials and Path Integrals for Quantum Lennard-Jones Systems: Theory and Monte Carlo Simulations. Mol. Phys. 85, 931–947. doi:10.1080/00268979500101571
Shulumba, N., Hellman, O., and Minnich, A. J. (2017). Lattice Thermal Conductivity of Polyethylene Molecular Crystals from First-Principles Including Nuclear Quantum Effects. Phys. Rev. Lett. 119, 185901. doi:10.1103/PhysRevLett.119.185901
Tong, Y., Kampfrath, T., and Campen, R. K. (2016). Experimentally Probing the Libration of Interfacial Water: the Rotational Potential of Water Is Stiffer at the Air/water Interface Than in Bulk Liquid. Phys. Chem. Chem. Phys. 18, 18424–18430. doi:10.1039/C6CP01004K
Tuckerman, M., and Ceperley, D. (2018). Preface: Special Topic on Nuclear Quantum Effects. J. Chem. Phys. 148, 102001. doi:10.1063/1.5026714
Tuckerman, M. E., and Martyna, G. J. (2000). Understanding Modern Molecular Dynamics: Techniques and Applications. J. Phys. Chem. B 104, 159–178. doi:10.1021/jp992433y
Wahiduzzaman, M., Walther, C. F. J., and Heine, T. (2014). Hydrogen Adsorption in Metal-Organic Frameworks: The Role of Nuclear Quantum Effects. J. Chem. Phys. 141, 064708. doi:10.1063/1.4892670
Wang, L., Fried, S. D., Boxer, S. G., and Markland, T. E. (2014). Quantum Delocalization of Protons in the Hydrogen-Bond Network of an Enzyme Active Site. Proc. Natl. Acad. Sci. U.S.A. 111, 18454–18459. doi:10.1073/pnas.1417923111
Weiner, S. J., Kollman, P. A., Case, D. A., Singh, U. C., Ghio, C., Alagona, G., et al. (1984). A New Force Field for Molecular Mechanical Simulation of Nucleic Acids and Proteins. J. Am. Chem. Soc. 106, 765–784. doi:10.1021/ja00315a051
Xu, P., Guidez, E. B., Bertoni, C., and Gordon, M. S. (2018). Perspective:Ab Initioforce Field Methods Derived from Quantum Mechanics. J. Chem. Phys. 148, 090901. doi:10.1063/1.5009551
Zhang, L., Han, J., Wang, H., Car, R., and E, W. (2018). Deep Potential Molecular Dynamics: A Scalable Model with the Accuracy of Quantum Mechanics. Phys. Rev. Lett. 120, 143001. doi:10.1103/PhysRevLett.120.143001
Zhang, Y., Hu, C., and Jiang, B. (2019). Embedded Atom Neural Network Potentials: Efficient and Accurate Machine Learning with a Physically Inspired Representation. J. Phys. Chem. Lett. 10, 4962–4967. doi:10.1021/acs.jpclett.9b02037
Keywords: molecular dynamics, machine learning, nuclear quantum effects, path-integral molecular dynamics, centroid molecular dynamics
Citation: Wu C, Li R and Yu K (2022) Learning the Quantum Centroid Force Correction in Molecular Systems: A Localized Approach. Front. Mol. Biosci. 9:851311. doi: 10.3389/fmolb.2022.851311
Received: 09 January 2022; Accepted: 27 April 2022;
Published: 19 May 2022.
Edited by:
Kai Huang, Shenzhen Bay Laboratory, ChinaCopyright © 2022 Wu, Li and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kuang Yu, eXUua3VhbmdAc3oudHNpbmdodWEuZWR1LmNu
†These authors share first authorship