
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Biosci. , 14 November 2022
Sec. Cellular Biochemistry
Volume 9 - 2022 | https://doi.org/10.3389/fmolb.2022.1058602
This article is part of the Research Topic Prion-Like Transmission of Pathogenic Proteins in Neurodegenerative Diseases: Structural and Molecular Bases View all 5 articles
 Natallia Makarava1,2Elizaveta Katorcha1,2Jennifer Chen-Yu Chang1,2
Natallia Makarava1,2Elizaveta Katorcha1,2Jennifer Chen-Yu Chang1,2 Joseph T. Y. Lau3
Joseph T. Y. Lau3 Ilia V. Baskakov1,2*
Ilia V. Baskakov1,2*Prion diseases are a group of fatal neurodegenerative diseases caused by misfolding of the normal cellular form of the prion protein or PrPC, into a disease-associated self-replicating state or PrPSc. PrPC and PrPSc are posttranslationally modified with N-linked glycans, in which the terminal positions occupied by sialic acids residues are attached to galactose predominantly via α2-6 linkages. The sialylation status of PrPSc is an important determinant of prion disease pathogenesis, as it dictates the rate of prion replication and controls the fate of prions in an organism. The current study tests whether a knockout of ST6Gal1, one of the two mammalian sialyltransferases that catalyze the sialylation of glycans via α2-6 linkages, reduces the sialylation status of PrPSc and alters prion disease pathogenesis. We found that a global knockout of ST6Gal1 in mice significantly reduces the α2-6 sialylation of the brain parenchyma, as determined by staining with Sambucus Nigra agglutinin. However, the sialylation of PrPSc remained stable and the incubation time to disease increased only modestly in ST6Gal1 knockout mice (ST6Gal1-KO). A lack of significant changes in the PrPSc sialylation status and prion pathogenesis is attributed to the redundancy in sialylation and, in particular, the plausible involvement of a second member of the sialyltransferase family that sialylate via α2-6 linkages, ST6Gal2.
Prion diseases are a group of fatal neurodegenerative diseases in humans and animals that can arise spontaneously or via transmission (Prusiner, 2001). Prions or PrPSc are transmissible and spread from host-to-host or cell-to-cell by recruiting and converting the cellular form of the prion protein or PrPC into a disease-associated PrPSc state (Prusiner, 1997; Legname et al., 2004). Prions elicit multiple disease phenotypes characterized by different clinical symptoms, cell tropism, affected brain regions, deposition patterns and incubation times to disease (Collinge and Clarke, 2007). The diversity of disease phenotypes within the same host has been attributed to the ability of PrPC to acquire multiple, structurally distinct, self-replicating PrPSc states referred to as prion strains (Bessen and Marsh, 1992; Safar et al., 1998; Peretz et al., 2001; Ayers et al., 2011; Gonzalez-Montalban et al., 2011; Klimova et al., 2015; Morales et al., 2016). The fact that prion strains are structurally different has been well documented (Caughey et al., 2022; Manka et al., 2022), yet how prion strains elicit multiple, strain-specific disease phenotypes has not been answered (Baskakov, 2021).
PrPC is posttranslationally modified with one or two N-linked glycans and a GPI anchor (Stahl et al., 1987; Endo et al., 1989). At the terminal positions of the N-linked glycans are sialic acid residues that are attached to galactose predominantly via α2-6 or, less often, via α2-3 linkages (Turk et al., 1988; Endo et al., 1989; Stimson et al., 1999; Katorcha and Baskakov, 2017). Each of the two glycans can carry up to five sialic acid residues (Endo et al., 1989; Rudd et al., 1999). According to mass-spectroscopy analysis, the variation in structure and composition of N-linked glycans give rise to more than 400 different PrPC sialoglycoforms (Endo et al., 1989; Stimson et al., 1999). Traditional mass-spectroscopy largely underestimates the sialylation status of glycans due to the stripping of sialic residues by acidic solvents, so the actual diversity is expected to be greater. As judged from biochemical analysis, up to 90% of the terminal positions of PrPSc N-glycans are sialylated (Katorcha and Baskakov, 2018). Upon conversion of PrPC into PrPSc, the sialylated glycans and GPI are carried over, giving rise to sialylated PrPSc (Bolton et al., 1985; Stahl et al., 1993; Rudd et al., 1999).
Among hundreds of PrPC sialoglycoforms expressed in a brain, prion strains recruit sialoglycoforms selectively, according to the strain-specific structure of PrPSc and the fitness of individual PrPC sialoglycoforms (Katorcha et al., 2015b; Baskakov and Katorcha, 2016). Previously, we proposed that as a result of selective recruitment, strain-specific patterns of carbohydrate groups are formed on PrPSc surfaces (Baskakov et al., 2018). Among solvent-exposed groups, terminal sialic acids and galactose residues are functionally most important, as they elicit response of innate immune cells (Srivastava et al., 2018). For instance, terminal galactose residues are sensed by myeloid cells and elicit an ‘eat-me’ response. Consistent with the above hypothesis, the sialylation status of the N-glycans of PrPSc was found to dictate the outcomes of prion infection (Katorcha et al., 2014; Srivastava et al., 2017). Partially desialylated PrPSc, i.e. PrPSc with increased amounts of exposed galactose, failed to induce prion disease upon intracranial or intraperitoneal administration (Katorcha et al., 2014; Srivastava et al., 2017). Moreover, prion infectivity could be switched off and on in a reversible manner via removing and reinstalling the sialylation of PrPSc, respectively (Katorcha et al., 2016a). Finally, desialylation of PrPSc altered its trafficking in the periphery, redirecting PrPSc away from secondary lymphoid organs to the liver (Srivastava et al., 2017).
Considering the import role of sialylation in dictating disease outcomes, modulating the sialylation status of PrPSc might offer new strategies for developing therapeutic intervention against prion diseases. Moreover, learning how carbohydrates dictate strain-specific pathology will help to establish the relationship between strain-specific structure and disease phenotype in a predictable manner. In mammals, sialylation of glycans takes place in the trans-Golgi and is executed by twenty sialyltransferases (STs) (Audry et al., 2011). STs are classified into four families according to the type of linkages synthesized (α2-3, α2-6, α2-8 or α2-9) and the selectivity towards N- or O-linked glycans (Takashima, 2008; Audry et al., 2011). Among the twenty STs, three enzymes (ST3Gal3, ST3Gal4 and ST3Gal6) sialylate N-linked glycans via an α2-3 linkage, whereas only two, ST6Gal1 and ST6Gal2, sialylate via an α2-6 linkage (Takashima, 2008; Audry et al., 2011). In PrPSc, an α2-6 linkage is predominant (Endo et al., 1989; Katorcha and Baskakov, 2017), so we decided to target the ST6 family for manipulating the sialylation status of PrPC and PrPSc. Between the two members of the ST6 family, ST6Gal1 is believed to be the main enzyme responsible for α2-6 sialylation in mammals. In adults, it is expressed ubiquitously in almost all tissues, including the brain (Manzi et al., 2000; Martin et al., 2002). Numerous reports document the involvement of ST6GAL1-mediated sialylation in pathologies relating to inflammation, immune responses, hematopoietic regulation, and cancer (e.g. recently reviewed in (Gc et al., 2022). Very little is known of ST6GAL2 and its catalytic properties. In fact, nothing is known about the physiologic roles of ST6GAL2, and its expression appears to be restricted to a brain, lung and spleen (Krzewinski-Recchi et al., 2003; Laporte et al., 2009; Lehoux et al., 2010; Ohmi et al., 2021).
The current study tested whether a deficiency of ST6Gal1 reduces the sialylation status of PrPSc and alters prion disease pathogenesis. We found that a global knockout of ST6Gal1 significantly reduces α2-6 sialylation of the brain parenchyma, as judged from staining with Sambucus Nigra agglutinin (SNA). However, the sialylation of PrPSc remained stable and the incubation time to disease showed only a modest increase in ST6Gal1 knockout mice (ST6Gal1-KO). Lack of significant changes is attributed to the redundancy in sialylation of PrPC and, in particular, the involvement of ST6GAL2. Our data indirectly points to the involvement of ST6GAL2, the only other known sialyltransferase to be able to construct the α2,6-Sia linkage in PrP, in the pathogenic progression of prion disease.
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The animal protocol was approved by the Institutional Animal Care and Use Committee of the University of Maryland, Baltimore (Assurance Number A32000-01; Permit Number: 0215002).
The ST6Gal1-KO mouse has a globally inactivated ST6Gal1 gene and was originally produced by Marth and co-workers (Hennet et al., 1998). The strain was backcrossed >10 generations into the C57Bl/6 background. Age-matched C57Bl/6 animals were used as controls.
ST6Gal1-KO and C57BL/6 mice (females and males) were inoculated intracerebrally into the left hemisphere ∼2 mm to the left of the midline and ∼2 mm anterior to a line drawn between the ears with 20 μL of 1% 22L brain homogenate under isoflurane anesthesia. Inoculum is delivered slowly by a 26 G needle inserted to a depth of approximately 3 mm. Signs of neurological disease were detected between 127–138 days post inoculation and consisted of hind-limb clasp, ataxia, and weight loss. Within 13–26 days after the first clinical signs, mice became unable to walk on a beam, developed kyphosis and became lethargic. Mice were considered terminally ill when they were unable to rear and/or lost 20% of their weight. At this point they were euthanized by CO2 asphyxiation and decapitation.
10% (wt/vol) homogenates in PBS from brains and spleens were prepared using glass/Teflon homogenizers as described previously (Makarava et al., 2012). For analysis of brain-derived PrPSc, an aliquot of 10% homogenate was diluted with nine volumes of 1% Triton X-100 in PBS, sonicated for 30 s inside a Misonix S-4000 microplate horn (Qsonica), and treated with 10 μg/ml of proteinase K (New England BioLabs) for 1 h at 37°C. For analysis of spleen-derived PrPSc, 10% homogenate was supplemented with 1% Triton X-100, sonicated as above, and treated with 20 μg/ml of proteinase K for 1 h at 37°C. Resulting brain and spleen samples were supplemented with 4xSDS loading buffer and heated for 10 min in a boiling water bath. Samples were loaded onto NuPAGE 12% Bis-Tris gels, transferred to PVDF membranes, and probed with anti-prion antibodies ab3531 (Abcam, Waltham, MA). Western blot signals were visualized using the FlourChem M imaging system (Protein Simple, San Jose, CA). For generating glycoform profiles and calculating total PrPSc amount, densitometry analysis of 1D blots was performed using the “Lane profile” and “Multiplex Band Analysis” functions in AlphaView software (Protein Simple), respectively.
For 2D electrophoresis of brain-derived materials, an aliquot of 10% (wt/vol) homogenate was diluted with nine volumes of 1% (vol/vol) Triton X-100 in PBS, sonicated as above, and treated with 25 μg/ml of proteinase K for 30 min at 37 °C. For 2D electrophoresis of spleen-derived material, 250 µL of 10% (wt/vol) homogenate was diluted 1:1 with PBS, aliquotted into 0.2-ml thin-wall PCR tubes, sonicated as above, and combined into one tube, which was subjected to a 30-min 16,000 g centrifugation at 4°C. The pellet was resuspended in 25 µL of 1% (wt/vol) Triton X-100 in PBS, treated with 20 μg/ml of proteinase K for 30 min at 37 °C, then supplemented with 4xSDS loading buffer, heated for 10 min in a boiling water bath, and processed for 2D electrophoresis. 25 µL of the resulting samples were solubilized for 1 h at room temperature in 200 µL of solubilization buffer (8 M Urea, 2% (wt/vol) CHAPS, 5 mM TBP, 20 mM TrisHCl pH 8.0), then alkylated by adding 7 µL of 0.5 M iodoacetamide and incubated for 1 h at room temperature in the dark. Then, 1150 µL of ice-cold methanol was added and samples were incubated for 2 h at −20°C. After 16,000 g centrifugation at 4°C, the supernatant was discarded, and the pellet was re-solubilized in 160 µL rehydration buffer (7 M urea, 2 M thiourea, 1% (wt/vol) DTT, 1% (wt/vol) CHAPS, 1% (wt/vol) Triton X-100, 1% (vol/vol) ampholyte, and a trace amount of Bromophenol Blue). Fixed immobilized pre-cast IPG strips (cat. # ZM0018, Life Technologies, Carlsbad, CA) with a linear pH gradient 3–10 were rehydrated in 155 µL of the resulting mixture overnight at room temperature inside IPG Runner cassettes (cat. # ZM0008, Life Technologies). Isoelectrofocusing (first dimension separation) was performed at room temperature with increasing voltage (175 V for 15 min, then 175–2,000 V linear gradient for 45 min, then 2,000 V for 30 min) on a Life Technologies Zoom Dual Power Supply using an XCell SureLock Mini-Cell Electrophoresis System (cat. # EI0001, Life Technologies). The IPG strips were then equilibrated for 15 min consecutively in 1) 6 M Urea, 20% (vol/vol) glycerol, 2% SDS, 375 mM Tris-HCl, pH 8.8, 130 mM DTT, then 2) 6 M Urea, 20% (vol/vol) glycerol, 2% SDS, 375 mM Tris-HCl, pH 8.8, 135 mM iodoacetamide, and loaded on 4–12% Bis-Tris ZOOM SDS-PAGE pre-cast gels (cat. # NP0330BOX, Life Technologies). For the second dimension, SDS-PAGE was performed for 1 h at 170 V. Immunoblotting was performed with ab3531 (Abcam).
Formalin-fixed brain halves divided at the midline (left hemisphere) were treated in formic acid (95%) to deactivate prion infectivity before being embedded in paraffin. 4 µm sections mounted on slides were processed for hematoxylin-eosin (H&E) staining and immunohistochemistry. To expose epitopes, slides were subjected to 20 min hydrated autoclaving at 121°C in trisodium citrate buffer, pH 6.0 with 0.05% Tween 20. For detection of disease-associated PrP, a 5 min treatment with 88% formic acid was used following autoclaving. PrP was stained with anti-prion antibody SAF-84 (Cayman Chemical, Ann Arbor, MI). Rabbit anti-Iba1 (Wako, Richmond, VA) was used to stain microglia. Detection was performed using 3,3’ Diaminobenzidine (DAB) Quanto chromogen and substrate (VWR, Radnor, PA).
For SNA staining, 4 µm brain sections mounted on slides were subjected to the standard rehydration procedure, then submerged in 10 mM tri-sodium citrate buffer, pH 6.0, boiled for 5 min by microwaving at 20% power, and cooled for 1 h before proceeding with lectin staining. Incubation in 3% hydrogen peroxide in methanol for 20 min was used to remove endogenous peroxidase activity. After a 5 min wash in running water, slides were incubated for 1 h with 5 μg/ml biotin-labeled elderberry bark lectin (SNA, Vector laboratories, Burlingame, CA) diluted in lectin buffer, pH 7.6 (50 mM Tris, 150 mM NaCl, 1 mM MgCl2, 0.75 mM CaCl2). Following three 5 min washes in lectin buffer, the slides were incubated for 30 min in 5 μg/ml horse radish peroxidase-labeled streptavidin (Thermo Fisher scientific, Waltham, MA), then again washed three times with lectin buffer, and developed with DAB Quanto chromogen and substrate (VWR, Radnor, PA).
Brains were divided at the midline, and the right hemispheres were used to dissect cortex, hippocampus, midbrain and thalamus regions. 10% (wt/vol) homogenates were prepared within 1.5 ml tubes in Trizol (Thermo Fisher Scientific, Waltham, MA, United States), using RNase-free disposable pestles (Fisher scientific, Hampton, NH). Total RNA was isolated using the Aurum Total RNA Mini Kit (Bio-Rad, Hercules, CA, United States) and subjected to DNase I digestion to remove contaminating genomic DNA. Total RNA was dissolved in elution buffer and stored at −80°C. An absorbance 260/280 value of ∼2.0, determined using a NanoDrop ND-1000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, United States) proved RNA purity. Reverse transcription was performed using 1 μg of extracted RNA and an iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, United States). RT-qPCR was performed in triplicate from one ST6Gal1-KO and one C57Bl/6 (WT) brain using the SsoAdvanced universal SYBR Green Supermix (Bio-Rad, Hercules, CA, United States) with Bio-Rad designed and validated primers for ST6Gal1 (qMmuCID0009827). Alternative ST6Gal1 primers designed for exon 2 were as follows: forward 5′- ACACCACTGAATGGGAGGGT - 3′, and reverse 5′- CAGAGATCCTGCAGAAGACACG - 3’. The housekeeping gene TBP (Bio-Rad, qMmuCID0040542) was used for normalization. The PCR protocol consisted of a treatment at 95 °C for 2 min followed by 40 amplification cycles with the following steps: 95 °C for 5 s, and 60 °C for 30 s. The RT-qPCR was performed with a CFX96 Touch Real-Time PCR Detection System (Bio-Rad, Hercules, CA, United States), and analyzed with Bio-Rad CFX Manager software (Bio-Rad, Hercules, CA, United States).
In mice, the highest level of ST6Gal1 expression was found in the spleen (Takashima et al., 2002). In the current study, spleens were used to confirm the global knockout of ST6Gal1. As expected, ST6Gal1was found in control C56Bl/6 mice (WT) but was lacking in ST6Gal1-KO mice (Figure 1A). The level of ST6Gal1 expression in the brains of WT mice was below the detection limits of the Western blot (data not shown). To confirm the knockout of ST6Gal1 in animal brains, we used RT-qPCR with two sets of ST6Gal1 primers, including primers to the exon 2. ST6Gal1-KO mice were generated by deleting the exon 2 that resulted in a loss of over 50% of the ST6Gal1 coding sequence including cytoplasmic, transmembrane, and considerable catalytic domain sequences (Hennet et al., 1998). As expected, RT-qPCR with primers to exon 2 detected expression of ST6Gal1 in multiple brain areas of WT mice, but not in ST6Gal-KO mice (Figure 1B).
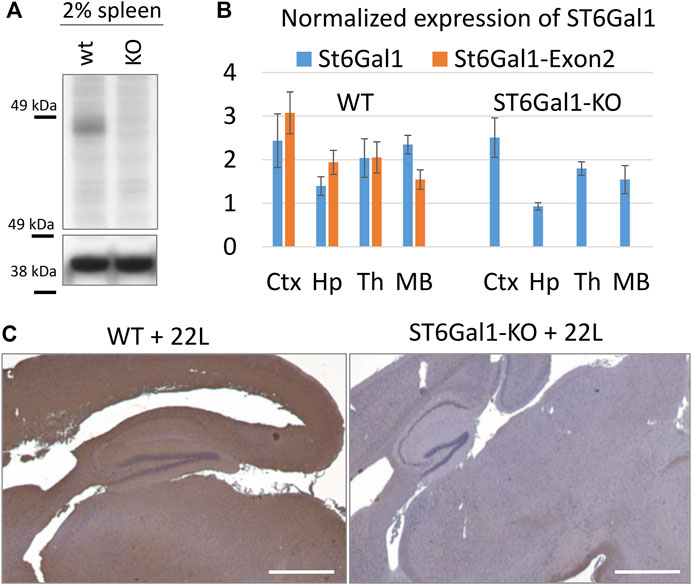
FIGURE 1. The global knockout of ST6Gal1. (A) Western blot of 2% spleen tissues from ST6Gal1-KO (KO) and control C57Bl/6 (WT) mice stained with anti-ST6Gal1 antibody (#AF5924, R&D Systems, Minneapolis, MN). (B) RT-qPCR analysis of ST6Gal1 expression using Bio-Rad primers (qMmuCID0009827) or primers to Exon 2 of the gene in cortex (Ctx), hippocampus (Hp), thalamus (Th) and midbrain (MB) from ST6Gal1-KO (KO) and control C57Bl/6 (WT) mice. Each sample was analyzed in triplicate. The mean and standard deviation are shown. TBP was used as a housekeeping gene. (C) Staining of 22L-infected brains of ST6Gal1-KO (KO) and C57Bl/6 (WT) mice, euthanized at 146 and 145 days post-inoculation (dpi), respectively, using Sambucus Nigra lectin (SNA). Scale bar = 1 mm.
To test whether the knockout of ST6Gal1 reduces sialylation of the brain parenchyma, brain slices were stained with SNA lectin that detects terminal α2-6 sialylation of both N- and O-linked glycans. Consistent with previous studies (Martin et al., 2002), ST6Gal1-KO mice showed significant loss in α2-6 sialylation of the brain parenchyma across multiple brain regions including thalamus, hippocampus and cortex in comparison to control WT mice (Figure 1C).
To test whether the knockout of ST6Gal1 modifies prion disease pathogenesis, ST6Gal1-KO and control WT mice were inoculated intracerebrally with 1% scrapie brain homogenate of 22L, a mouse-adapted prion strain. ST6Gal1-KO and control WT groups succumbed to the diseases at 155.3 ± 4.9 and 150.3 ± 4.1 days post-inoculation, respectively (Figure 2A). While ST6Gal1-KO mice showed slightly longer incubation times relative to the control group, the difference in their survival curves was bordering statistical significance (Figure 2A). No statistically significant difference was found between the two groups with respect to the amounts of PK-resistant PrPSc in their brains (Figure 2B). The knockout of ST6Gal1 did not change the ratios of un-, mono-, and diglycosylated glycoforms within PrPSc (Figure 2C). As reported previously (Katorcha et al., 2015b), in the 22L mouse adapted strain, the diglycosylated glycoform is predominant, whereas the unglycosylated is the least abundant PrPSc glycoform. In both groups, PrPSc spread to spleen showing large variations between individual animals with respect to its amounts, which is expected for the 22L strain inoculated intracranially (Figure 2B). In spleen, the amounts of PrPSc was lower in ST6Gal1-KO group relative to WT group (Figure 2B). Histopathological analysis showed the main pathological hallmarks of prion disease including PrPSc deposition, spongiosis and reactive microglia in both ST6Gal1-KO and control WT mice (Figure 3).
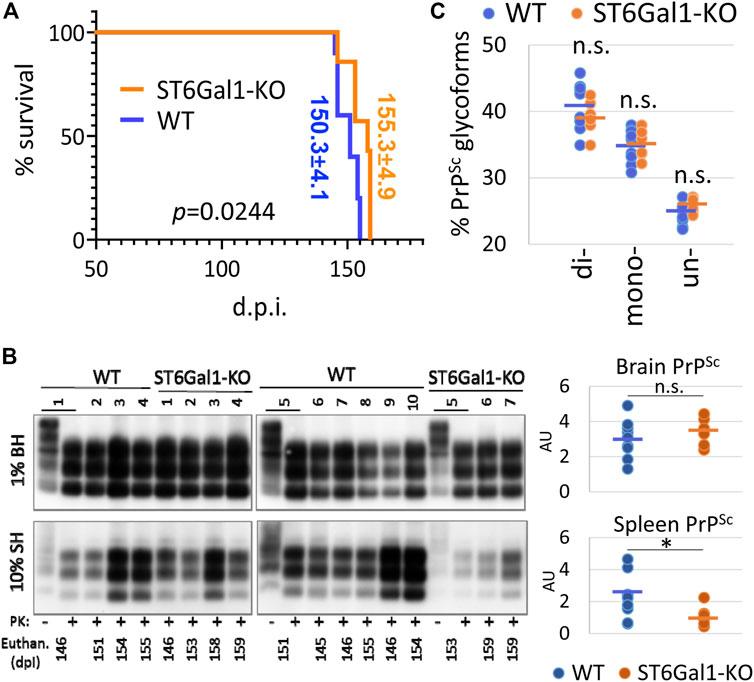
FIGURE 2. A modest impact of ST6Gal1 knockout on the incubation time to disease. (A) Kaplan-Meier survival plot for ST6Gal1-KO and C57Bl/6 (WT) mice inoculated IC with 1% scrapie brain homogenate of the 22L strain. p value was calculated using Mantel-Cox test for comparison of survival curves. (B) Western blot analysis and quantification of PrPSc in brain homogenates (BH) and spleen homogenates (SH) of ST6Gal1-KO and C57Bl/6 (WT) mice inoculated with 22L. Samples were treated with PK. Untreated (-PK) samples were used as a reference, and were loaded as a 10-fold dilution. Western blots were stained with the ab3531 antibody. PrPSc amount in brains and spleens were compared using unpaired t-tests and presented as individual animals and means (n = 10 WT, n = 7 ST6Gal1-KO, *p < 0.05). (C) The percentage of di-, mono- and unglycosylated brain-derived PrPSc from ST6Gal1-KO and C57Bl/6 (WT) mice inoculated with 22L. Data presented as individual animals and means (n = 10 WT and n = 7 ST6Gal1-KO).
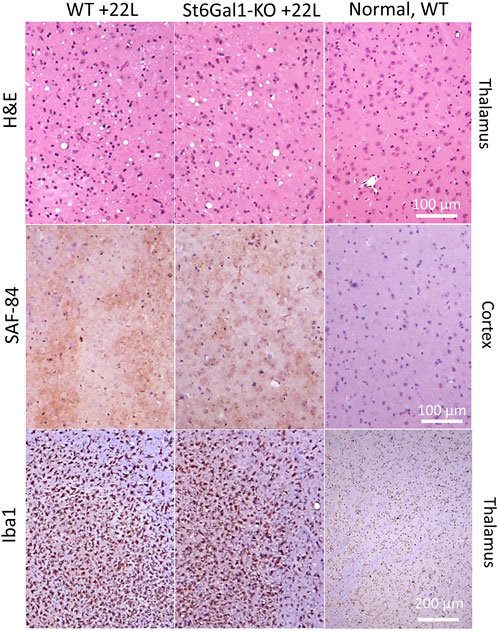
FIGURE 3. Histopathological analysis of 22L-infected mouse brains. Vacuolation revealed with hematoxylin-eosin (H&E) staining, deposition of PrPSc stained with SAF-84 antibody, and reactive microglia stained for Iba1 in ST6Gal1-KO and C57Bl/6 (WT) mice inoculated IC with 22L and euthanized at 146 and 145 dpi, respectively. Staining of normal C57Bl/6 (WT) euthanized at 212 days of age is shown as references (right panels).
To test whether the knockout of ST6Gal1 reduces the sialylation status of PrPSc, brain- and spleen-derived material was analyzed using two-dimensional (2D) gel electrophoresis. Prior to 2D analysis, the samples were treated with PK and then denatured, so that the sialylation status of individual PrP molecules could be visualized as a distribution of charge isoforms in the horizontal dimension of 2D. Each sialic acid residue adds one negative charge and hypersialylated PrP molecules move toward an acidic pH, whereas hyposialylated PrP molecules move toward a basic pH in the horizontal dimension of 2D gels (Figure 4). Three horizontal rows of charged isoforms from bottom to top correspond to the non-, mono- and diglycosylated PrP molecules.
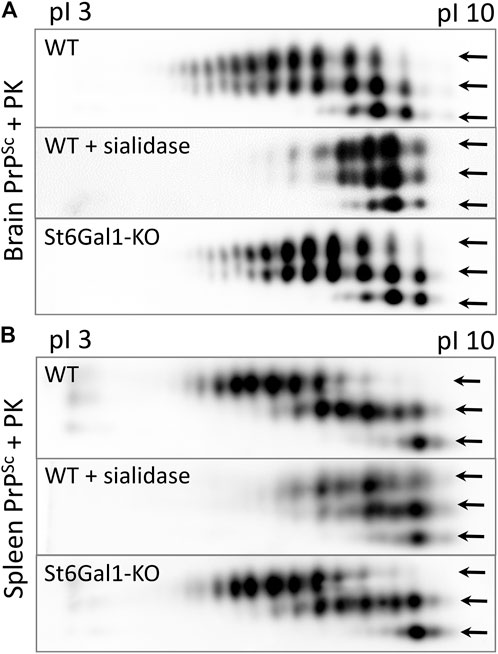
FIGURE 4. Analysis of the sialylation status of PrPSc. Representative 2D Western blot of brain-derived PrPSc from ST6Gal1-KO and C57Bl/6 (WT) mice euthanized at 146 and 145 dpi, respectively (A), and spleen-derived PrPSc from ST6Gal1-KO and C57Bl/6 (WT) mice euthanized at 158 and 154 dpi, respectively (B). Mice were inoculated IC with 1% scrapie brain homogenate of the 22L strain. Middle panels show brain- and spleen-derived PrPSc from WT mice desialylated using sialidase Arthrobacter ureafaciens. Prior to 2D blot, brain and spleen materials were treated with 20 μg/mL PK. Anti-PrP antibody ab3531 was used for immunodetection. Arrows point at di-, mono- and non-glycosylated glycoforms.
Consistent with previous studies, non-glycosylated PrP molecules showed multiple charge isoforms (all of which were at basic pH) that were attributed to the structural heterogeneity of GPI anchors (Katorcha et al., 2016b). No notable differences between ST6Gal1-KO and control group brains with respect to the distribution of PrP sialoglycoforms can be found in brain- or spleen-derived PrPSc (Figures 4A,B). As reported in previous studies (Srivastava et al., 2015), the sialylation pattern of brain-derived PrPSc was different from that of spleen-derived PrPSc (Figures 4A,B). As expected, treatment with sialidase Arthrobacter ureafaciens (AU), that has broad substrate specificity, shifted the distribution of PrP sialoglycoforms to basic pH (Figures 4A,B). These results illustrate that the global knockout of ST6Gal1 did not change the sialylation pattern of PrPSc in the brain or spleen.
Considering the important role of sialylation in dictating the rate of prion replication and their fate in an organism, manipulation of the sialylation status of PrPSc via altering the steady-state sialylation of its precursor PrPC might offer a novel strategy for developing therapeutic intervention against prion diseases. Sialylation of glycoproteins is controlled by two groups of enzymes: sialyltransferases (STs) and neuraminidases or sialidases (Audry et al., 2011; Miyagi and Yamaguchi, 2012). Previously, we found that a knockout of Neu1, Neu3, Neu4, or both Neu3 and Neu4, which are all expressed in the brain, does not affect the steady-state levels of PrPC sialylation in mice (Katorcha et al., 2015a). This fact suggests that desialylated PrPC molecules do not contribute to the steady-state level of PrPC sialoglycoforms, presumably, due to degradation. In the absence of detectable changes in PrPC sialylation in Neu’s knockout models, targeting STs seemed to offer a more effective strategy for manipulating the sialylation levels of PrPC and PrPSc. In support of this notion, the sialylation levels of PrPC in cultured N2a cells were suppressed to a large degree by the administration of a general inhibitor of STs, 3Fax-Neu5Ac (Katorcha et al., 2015a). However, because 3Fax-Neu5Ac targets a broad spectrum of STs and blocks global sialylation (Burkart et al., 2000; Lairson et al., 2008), it causes irreversible kidney dysfunction in animals which leads to rapid death (Macauley et al., 2014). In the absence of inhibitors that selectively target individual STs (Wang et al., 2016; Szabo and Skropeta, 2017), we decided to test the effect of a knockout of ST6Gal1.
Western blotting of spleen tissues from the ST6Gal1-KO cohort confirmed a global knockout of ST6Gal1, while RT-qPCR documented the lack of ST6Gal1 expression in brain tissue. Consistent with previous studies (Martin et al., 2002), SNA staining revealed significantly lower levels of α2-6 sialylation of the brain parenchyma in ST6Gal1-KO mice relative to that of control WT animals. However, despite a global knockout of the ST6Gal1 gene, the sialylation status of brain- and spleen-derived PrPSc did not show notable changes. In spleen-derived PrPSc, dyglycosylated PrP molecules shifted toward acidic pH relative to those of monoglycosylated PrP molecules in both WT and ST6Gal1 mice (Figure 4B), suggesting that spleen-derived PrPSc is fully sialylated. Such sialylation pattern is consistent with very high levels of sialyltransferase activity in secondary lymphoid organs. In contrast, in brain-derived PrPSc of WT and ST6Gal1, the pattern of charged isoforms of diglycosylated PrP molecules was very similar to that of monoglycosylated PrPs (Figure 4A). Such pattern suggests that in a brain, diglycosylated PrP molecules are only partially sialylated and on average have a similar number of sialic acid residues per PrP molecule as monoglycosylated PrPs, despite the fact that diglycosylated PrPs have two glycans, while monoglycosylated PrPs have only one glycan. Nevertheless, similar sialylation levels of PrPSc in ST6Gal1-KO and WT mice could be due to 1) the possibility that sialylation is conducted primarily by the ST6Gal2 enzyme, 2) redundancy in activities of ST6Gal1 and ST6Gal2 enzymes, and/or 3) redundancy between the ST6 and ST3 families of STs.
As the last member of STs discovered, ST6Gal2 has not been characterized as thoroughly as ST6Gal1 (Krzewinski-Recchi et al., 2003). ST6Gal2 seems to exhibit a more narrow enzymatic substrate specificity in comparison to ST6Gal1 (Takashima et al., 2002; Takashima et al., 2003). While the ST6Gal1 gene is expressed in almost all tissues, the expression of ST6Gal2 shows a restricted tissue-specific pattern and is primarily limited to the brain and spleen (Krzewinski-Recchi et al., 2003; Laporte et al., 2009; Lehoux et al., 2010; Ohmi et al., 2021). A direct comparison of expression using RT-qPCR revealed that in an adult mouse brain, ST6Gal1 and ST6Gal2 genes are expressed at similar levels (Ohmi et al., 2021). However, ST6Gal1 mRNAs were found across different cell types in mice including neurons, astrocytes and microglia, whereas the expression of ST6Gal2 gene was detected primarily in astrocytes and neurons. Notably, the expression of STI6Gal2 in primary epithelial cultures was found to be upregulated by treatment with IL-6, suggesting that STI6Gal2 responds to pro-inflammatory conditions (Laporte et al., 2009; Lehoux et al., 2010). In a mouse brain, region-specific expression of the ST6Gal2 gene and the region-specific sialylation status of PrPSc showed the same ranking order (Makarava et al., 2020a), raising the possibility that ST6Gal2 is responsible for the sialylation. It would be interesting to test whether a knockout of ST6Gal2 or a double knockout of ST6Gal1 and ST6Gal2 affect prion disease pathogenesis.
It is not surprising that in the absence of notable effects on PrPSc sialylation, the knockout of ST6Gal1 caused only a modest delay in the incubation time to terminal disease. Because in brain ST6Gal1 is expressed across different cell types, the delay could be attributed to a perturbation of the homeostatic states of neurons and glia or changes in their response to prion infection. For example, ST6Gal1 was found to promote long-term cell activation through two pro-inflammatory pathways, NFκB and JAK/STAT3 (Holdbrooks et al., 2020). STAT3 is a transcription factor which is believed to be a master regulator of astrocyte reactivity (Ben Haim et al., 2015; Ceyzériat et al., 2018; Reichenbach et al., 2019). Because reactive astrocytes associated with prion disease are detrimental to neurons (Kushwaha et al., 2021; Makarava et al., 2021), the delay in the incubation time could be attributed to an attenuation of a STAT3-mediated activation of astrocytes in ST6Gal1-KO mice. Several families of receptors including siglecs, selectins, galectins, complement system components and asialoglycoprotein receptors recognize terminal sialic acid or galactose residue, and are involved in innate immunity (Linnartz et al., 2012; Rabinovich and Croci, 2012). As a result of a significant reduction in α2-6 sialylation of the brain parenchyma in ST6Gal1-KO mice, one should expect a perturbation of multiple signaling pathways involved in innate immunity (Zhuo and Bellis, 2011). Alternatively, deficiency in ST6Gal1 could affect the incubation time by altering homeostatic and/or reactive states of peripheral immune cells involved in prion disease pathogenesis (Nasirikenari et al., 2006; Liu et al., 2011). ST6Gal1 was found to regulate monocyte-macrophage development and survival (Rusiniak et al., 2022). As such, a ST6Gal1 deficiency could attenuate pro-inflammatory toxic effects of monocytes infiltrating prion-infected brains. Lower amounts of PrPSc in spleens of ST6Gal1-KO mice relative to WT mice is consistent with the idea that ST6Gal1 regulates function of peripheral immune cells important for prion trafficking in periphery and/or prion replication in secondary lymphoid organs.
Understanding the role of sialylation might be important for gaining insight into the etiology of sporadic CJD. There is a decline in the total sialic acid content as well as cell surface sialylation with normal aging (Svennerholm et al., 1997). Previously, we showed that the rate of PrPSc replication is accelerated by PrPC with low sialylation status (Katorcha et al., 2014; Katorcha et al., 2015b). It is not known whether a possible increase in the proportion of hyposialylated PrPC with aging might facilitate the spontaneous conversion of PrPC into PrPSc and increase the rate of sporadic CJD (Baskakov and Katorcha, 2016). Consistent with this hypothesis is a correlation between the region-specific sialylation status of PrPSc and the vulnerability of brain regions to prion infection (Makarava et al., 2020a). The thalamus displays prion deposition and chronic neuroinflammation prior to cortex and hippocampus and, by the terminal stage of the disease, the thalamus is affected more severely than any other brain region (Karapetyan et al., 2009; Sandberg et al., 2014; Carroll et al., 2016; Makarava et al., 2019; Makarava et al., 2020b; Makarava et al., 2021). Regardless of prion strain, thalamic PrPSc is sialylated less than cortex and hippocampus PrPSc (Makarava et al., 2020a). Lower levels of sialylation promotes faster prion replication and triggers profound neuroinflammation (Katorcha et al., 2015b; Srivastava et al., 2018). As such, testing a knockout of ST6Gal2 in future studies might provide new insight into etiology of sporadic CJD.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The animal study was reviewed and approved by Institutional Animal Care and Use Committee of the University of Maryland, Baltimore (Assurance Number A32000-01).
IB and NM conceived the idea and designed the experiments; JL provided ST6Gal1 KO mice; NM, EK, and JC performed experiments; NM analyzed the data; IB wrote the manuscript. All authors read and approved the final manuscript.
Financial support for this study was provided by National Institute of Health (Grants R01 NS045585 and AI128925) to IB.
We thank Daryl Butler for his assistance in handling animals, Regina Savtchenko for her assistance in running Western blots, and Kara Molesworth for editing the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Audry, M., Jeanneau, C., Imberty, A., Harduin-Lepers, A., Delannoy, P., and Breton, C. (2011). Current trends in the structure-activity relationships of sialyltransferases. Glycobiology 21 (6), 716–726. doi:10.1093/glycob/cwq189
Ayers, J. L., Schutt, C. R., Shikiya, R. A., Aguzzi, A., Kincaid, A. E., and Bartz, J. C. (2011). The strain-encoded relationship between PrP replication, stability and processing in neurons is predictive of the incubation period of disease. PLoS Pathog. 7 (3), e1001317. doi:10.1371/journal.ppat.1001317
Baskakov, I. V. (2021). From posttranslational modifications to disease phenotype: a substrate selection hypothesis in neurodegenerative diseases. Int. J. Mol. Sci. 22 (2), E901. doi:10.3390/ijms22020901
Baskakov, I. V., Katorcha, E., and Makarava, N. (2018). Prion strain-specific structure and pathology: a view from the perspective of glycobiology. Viruses 10 (12), 723. doi:10.3390/v10120723
Baskakov, I. V., and Katorcha, E. (2016). Multifaceted role of sialylation in prion diseases. Front. Neurosci. 10 (1), e358. doi:10.3389/fnins.2016.00358
Ben Haim, L., Ceyzériat, K., Carrillo-de Sauvage, M. A., Aubry, F., Auregan, G., Guillermier, M., et al. (2015). The JAK/STAT3 pathway is a common inducer of astrocyte reactivity in Alzheimer's and Huntington's diseases. J. Neurosci. 35 (6), 2817–2829. doi:10.1523/jneurosci.3516-14.2015
Bessen, R. A., and Marsh, R. F. (1992). Identification of two biologically distinct strains of transmissible mink encephalopathy in hamsters. J. Gen. Virol. 73 (2), 329–334. doi:10.1099/0022-1317-73-2-329
Bolton, D. C., Meyer, R. K., and Prusiner, S. B. (1985). Scrapie PrP 27-30 is a sialoglycoprotein. J. Virol. 53 (2), 596–606. doi:10.1128/JVI.53.2.596-606.1985
Burkart, M. D., Vincent, S. P., Duffels, A., Murray, B. W., Ley, S. V., and Wong, C. H. (2000). Chemo-enzymatic synthesis of fluorinated sugar nucleotide: useful mechanistic probes for glycosyltransferases. Bioorg. Med. Chem. 8, 1937–1946. doi:10.1016/s0968-0896(00)00139-5
Carroll, J. A., Striebel, J. F., Rangel, A., Woods, T., Phillips, K., Peterson, K. E., et al. (2016). Prion strain differences in accumulation of PrPSc on neurons and glia are associated with similar expression profiles of neuroinflammatory genes: Comparison of three prion strains. PLoS Pathog. 12 (4), e1005551. doi:10.1371/journal.ppat.1005551
Caughey, B., Standke, H. G., Artikis, E., Hoyt, F., and Kraus, A. (2022). Pathogenic prion structures at high resolution. PLoS Pathog. 18 (6), e1010594. doi:10.1371/journal.ppat.1010594
Ceyzériat, K., Ben Haim, L., Denizot, A., Pommier, D., Matos, M., Guillemaud, O., et al. (2018). Modulation of astrocyte reactivity improves functional deficits in mouse models of Alzheimer's disease. Acta Neuropathol. Commun. 6 (1), 104. doi:10.1186/s40478-018-0606-1
Collinge, J., and Clarke, A. R. (2007). A general model of prion strains and their pathogenicity. Science 318 (5852), 930–936. doi:10.1126/science.1138718
Endo, T., Groth, D., Prusiner, S. B., and Kobata, A. (1989). Diversity of oligosaccharide structures linked to asparagines of the scrapie prion protein. Biochemistry 28 (21), 8380–8388. doi:10.1021/bi00447a017
Gc, S., Bellis, S. L., and Hjelmeland, A. B. (2022). ST6Gal1: Oncogenic signaling pathways and targets. Front. Mol. Biosci. 9, 962908. doi:10.3389/fmolb.2022.962908
Gonzalez-Montalban, N., Makarava, N., Savtchenko, R., and Baskakov, I. V. (2011). Relationship between conformational stability and amplification efficiency of prions. Biochemistry 50 (37), 7933–7940. doi:10.1021/bi200950v
Hennet, T., Chui, D., Paulson, J. C., and Marth, J. D. (1998). Immune regulation by the ST6Gal sialyltransferase. Proc. Natl. Acad. Sci. U. S. A. 95 (8), 4504–4509. doi:10.1073/pnas.95.8.4504
Holdbrooks, A. T., Ankenbauer, K. E., Hwang, J., and Bellis, S. L. (2020). Regulation of inflammatory signaling by the ST6Gal-I sialyltransferase. PLoS One 15 (11), e0241850. doi:10.1371/journal.pone.0241850
Karapetyan, Y. E., Saa, P., Mahal, S. P., Sferrazza, G. F., Sherman, A., Sales, N., et al. (2009). Prion strain discrimination based on rapid in vivo amplification and analysis by the cell panel assay. PLoS One 4 (5), e5730. doi:10.1371/journal.pone.0005730
Katorcha, E., and Baskakov, I. V. (2017). Analyses of N-linked glycans of PrPSc revealed predominantly 2, 6-linked sialic acid residues. FEBS J. 284 (21), 3727–3738. doi:10.1111/febs.14268
Katorcha, E., and Baskakov, I. V. (2018). Analysis of covalent modifications of amyloidogenic proteins using two-dimensional electrophoresis: Prion protein and its sialylation. Methods Mol. Biol. 1779 (241-255), 241–255. doi:10.1007/978-1-4939-7816-8_15
Katorcha, E., Daus, M. L., Gonzalez-Montalban, N., Makarava, N., Lasch, P., Beekes, M., et al. (2016a). Reversible off and on switching of prion infectivity via removing and reinstalling prion sialylation. Sci. Rep. 6, 33119. doi:10.1038/srep33119
Katorcha, E., Klimova, N., Makarava, N., Savtchenko, R., Pan, X., Annunziata, I., et al. (2015a). Loss of cellular sialidases does not affect the sialylation status of the prion protein but increases the amounts of its proteolytic fragment C1. PLoS One 10 (11), e0143218. doi:10.1371/journal.pone.0143218
Katorcha, E., Makarava, N., Savtchenko, R., and Baskakov, I. V. (2015b). Sialylation of the prion protein glycans controls prion replication rate and glycoform ratio. Sci. Rep. 5 (1), 16912. doi:10.1038/srep16912
Katorcha, E., Makarava, N., Savtchenko, R., D'Azzo, A., and Baskakov, I. V. (2014). Sialylation of prion protein controls the rate of prion amplification, the cross-species barrier, the ratio of PrPSc glycoform and prion infectivity. PLoS Pathog. 10 (9), e1004366. doi:10.1371/journal.ppat.1004366
Katorcha, E., Srivastava, S., Klimova, N., and Baskakov, I. V. (2016b). Sialylation of glycosylphosphatidylinositol (GPI) anchors of mammalian prions is regulated in a host-tissue-and cell-specific manner. J. Biol. Chem. 291 (33), 17009–17019. doi:10.1074/jbc.M116.732040
Klimova, N., Makarava, N., and Baskakov, I. V. (2015). The diversity and relationship of prion protein self-replicating states. Virus Res. 207 (1), 113–119. doi:10.1016/j.virusres.2014.10.002
Krzewinski-Recchi, M. A., Julien, S., Juliant, S., Teintenier-Lelièvre, M., Samyn-Petit, B., Montiel, M. D., et al. (2003). Identification and functional expression of a second human beta-galactoside alpha2, 6-sialyltransferase, ST6Gal II. Eur. J. Biochem. 270 (5), 950–961. doi:10.1046/j.1432-1033.2003.03458.x
Kushwaha, R., Sinha, A., Makarava, N., Molesworth, K., and Baskakov, I. V. (2021). Non-cell autonomous astrocyte-mediated neuronal toxicity in prion diseases. Acta Neuropathol. Commun. 9 (1), 22. doi:10.1186/s40478-021-01123-8
Lairson, L. L., Henrissat, B., Davies, G. J., and Withers, S. G. (2008). Glycosyltransferases: structures, functions, and mechanisms. Annu. Rev. Biochem. 77, 521–555. doi:10.1146/annurev.biochem.76.061005.092322
Laporte, B., Gonzalez-Hilarion, S., Maftah, A., and Petit, J.-M. (2009). The second bovine beta-galactoside-alpha2, 6-sialyltransferase (ST6Gal II): genomic organization and stimulation of its in vitro expression by IL-6 in bovine mammary epithelial cells. Glycobiology 19 (10), 1082–1093. doi:10.1093/glycob/cwp094
Legname, G., Baskakov, I. V., Nguyen, H. O. B., Riesner, D., Cohen, F. E., DeArmond, S. J., et al. (2004). Synthetic mammalian prions. Science 305 (5684), 673–676. doi:10.1126/science.1100195
Lehoux, S., Groux-Degroote, S., Cazet, A., Dhaenens, C.-M., Maurage, C.-A., Caillet-Boudin, M.-L., et al. (2010). Transcriptional regulation of the human ST6GAL2 gene in cerebral cortex and neuronal cells. Glycoconj. J. 27, 99–114. doi:10.1007/s10719-009-9260-y
Linnartz, B., Bodea, L.-G., and Neumann, H. (2012). Microglial carbohydrate-binding receptors for neural repair. Cell Tissue Res. 349, 215–227. doi:10.1007/s00441-012-1342-7
Liu, Z., Swindall, A. F., Kesterson, R. A., Schoeb, T. R., Bullard, D. C., and Bellis, S. L. (2011). ST6Gal-I regulates macrophage apoptosis via α2-6 sialylation of the TNFR1 death receptor. J. Biol. Chem. 286 (45), 39654–39662. doi:10.1074/jbc.M111.276063
Macauley, M. S., Arlian, B. M., Rillahan, C. D., Pang, P. C., Bortell, N., Marcondes, M. C., et al. (2014). Systemic blockade of sialylation in mice with a global inhibitor of sialyltransferases. J. Biol. Chem. 289 (35149-35158), 35149–35158. doi:10.1074/jbc.M114.606517
Makarava, N., Chang, J. C.-Y., and Baskakov, I. V. (2020a). Region-specific sialylation pattern of prion strains provides novel insight into prion neurotropism. Int. J. Mol. Sci. 21 (3), 828. doi:10.3390/ijms21030828
Makarava, N., Chang, J. C.-Y., Kushwaha, R., and Baskakov, I. V. (2019). Region-specific response of astrocytes to prion infection. Front. Neurosci. 13 (1), e1048. doi:10.3389/fnins.2019.01048
Makarava, N., Chang, J. C.-Y., Molesworth, K., and Baskakov, I. V. (2020b). Region-specific glial homeostatic signature in prion diseases is replaced by a uniform neuroinflammation signature, common for brain regions and prion strains with different cell tropism. Neurobiol. Dis. 137 (1), e104783. doi:10.1016/j.nbd.2020.104783
Makarava, N., Kovacs, G. G., Savtchenko, R., Alexeeva, I., Budka, H., Rohwer, R. G., et al. (2012). Stabilization of a prion strain of synthetic origin requires multiple serial passages. J. Biol. Chem. 287 (36), 30205–30214. doi:10.1074/jbc.M112.392985
Makarava, N., Mychko, O., Chang, J. C.-Y., Molesworth, K., and Baskakov, I. V. (2021). The degree of astrocyte activation is predictive of the incubation time to prion disease. Acta Neuropathol. Commun. 9 (1), 87. doi:10.1186/s40478-021-01192-9
Manka, S. W., Wenborn, A., Collinge, J., and Wadsworth, J. D. F. (2022). Prion strains viewed through the lens of cryo-EM. Cell Tissue Res. doi:10.1007/s00441-022-03676-z
Manzi, A. E., Norgard-Sumnicht, K., Argade, S., Marth, J. D., van Halbeek, H., and Varki, A. (2000). Exploring the glycan repertoire of genetically modified mice by isolation and profiling of the major glycan classes and nano-NMR analysis of glycan mixtures. Glycobiology 10 (7), 669–689. doi:10.1093/glycob/10.7.669
Martin, L. T., Marth, J. D., Varki, A., and Varki, N. M. (2002). Genetically altered mice with different sialyltransferase deficiencies show tissue-specific alterations in sialylation and sialic acid 9-O-acetylation. J. Biol. Chem. 277 (36), 32930–32938. doi:10.1074/jbc.M203362200
Miyagi, T., and Yamaguchi, K. (2012). Mammalian sialidases: physiological and pathological roles in cellular functions. Glycobiology 22 (7), 880–896. doi:10.1093/glycob/cws057
Morales, R., Hu, P. P., Duran-Aniotz, C., Moda, F., Diaz-Espinoza, R., Chen, B., et al. (2016). Strain-dependent profile of misfolded prion protein aggregates. Sci. Rep. 6 (1), e20526. doi:10.1038/srep20526
Nasirikenari, M., Segal, B. H., Ostberg, J. R., Urbasic, A., and Lau, J. T. (2006). Altered granulopoietic profile and exaggerated acute neutrophilic inflammation in mice with targeted deficiency in the sialyltransferase ST6Gal I. Blood 108 (10), 3397–3405. doi:10.1182/blood-2006-04-014779
Ohmi, Y., Nishikaze, T., Kitaura, Y., Ito, T., Yamamoto, S., Sugiyama, F., et al. (2021). Majority of alpha2, 6-sialylated glycans in the adult mouse brain exist in O-glycans: SALSA-MS analysis for knockout mice of alpha2, 6-sialyltransferase genes. Glycobiology 31 (5), 557–570. doi:10.1093/glycob/cwaa105
Peretz, D., Scott, M., Groth, D., Williamson, A., Burton, D., Cohen, F. E., et al. (2001). Strain-specified relative conformational stability of the scrapie prion protein. Protein Sci. 10 (4), 854–863. doi:10.1110/ps.39201
Prusiner, S. B. (1997). Prion diseases and the BSE crisis. Science 278 (5336), 245–251. doi:10.1126/science.278.5336.245
Prusiner, S. B. (2001). Shattuck lecture -- neurodegenerative diseases and prions. N. Engl. J. Med. 344, 1516–1526. doi:10.1056/NEJM200105173442006
Rabinovich, G. A., and Croci, D. O. (2012). Regulatory circuits mediated by lectin-glycan interactions in autoimmunity and cancer. Immunity 36, 322–335. doi:10.1016/j.immuni.2012.03.004
Reichenbach, N., Delekate, A., Plescher, M., Schmitt, F., Krauss, S., Blank, N., et al. (2019). Inhibition of Stat3-mediated astrogliosis ameliorates pathology in an Alzheimer's disease model. EMBO Mol. Med. 11 (2), e9665. doi:10.15252/emmm.201809665
Rudd, P. M., Endo, T., Colominas, C., Groth, D., Wheeler, S. F., Harvey, D. J., et al. (1999). Glycosylation differences between the normal and pathogenic prion protein isoforms. Proc. Natl. Acad. Sci. U. S. A. 96, 13044–13049. doi:10.1073/pnas.96.23.13044
Rusiniak, M. E., Punch, P. R., Hait, N. C., Maiti, A., Burns, R. T., Chapla, D., et al. (2022). Extracellular ST6GAL1 regulates monocyte-macrophage development and survival. Glycobiology 32 (8), 701–711. doi:10.1093/glycob/cwac032
Safar, J., Wille, H., Itri, V., Groth, D., Serban, H., Torchia, M., et al. (1998). Eight prion strains have PrP Sc molecules with different conformations. Nat. Med. 4 (10), 1157–1165. doi:10.1038/2654
Sandberg, M. K., Al-Doujaily, H., Sharps, B., De Oliveira, M. W., Schmidt, C., Richard-Londt, A., et al. (2014). Prion neuropathology follows the accumulation of alternate prion protein isoforms after infective titre has peaked. Nat. Commun. 5, e4347. doi:10.1038/ncomms5347
Srivastava, S., Katorcha, E., Daus, M. L., Lasch, P., Beekes, M., and Baskakov, I. V. (2017). Sialylation controls prion fate in vivo. J. Biol. Chem. 292 (6), 2359–2368. doi:10.1074/jbc.M116.768010
Srivastava, S., Katorcha, E., Makarava, N., Barrett, J. P., Loane, D. J., and Baskakov, I. V. (2018). Inflammatory response of microglia to prions is controlled by sialylation of PrPSc. Sci. Rep. 8 (1), e11326. doi:10.1038/s41598-018-29720-z
Srivastava, S., Makarava, N., Katorcha, E., Savtchenko, R., Brossmer, R., and Baskakov, I. V. (2015). Post-conversion sialylation of prions in lymphoid tissues. Proc. Natl. Acad. Sci. U. S. A. 112 (48), E6654–E6662. doi:10.1073/pnas.1517993112
Stahl, N., Baldwin, M. A., Teplow, D. B., Hood, L., Gibson, B. W., Burlingame, A. L., et al. (1993). Structural studies of the scrapie prion protein using mass spectrometry and amino acid sequencing. Biochemistry 32, 1991–2002. doi:10.1021/bi00059a016
Stahl, N., Borchelt, D. R., Hsiao, K., and Prusiner, S. B. (1987). Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 51 (2), 229–240. doi:10.1016/0092-8674(87)90150-4
Stimson, E., Hope, J., Chong, A., and Burlingame, A. L. (1999). Site-specific characterization of the N-linked glycans of murine prion protein by high-performance liquid chromatography/electrospray mass spectrometry and exoglycosidase digestions. Biochemistry 38, 4885–4895. doi:10.1021/bi982330q
Svennerholm, L., Bostrom, K., and Jungbjer, B. (1997). Changes in weight and compositions of major membrane components of human brain during the span of adult human life of Swedes. Acta Neuropathol. 94, 345–352. doi:10.1007/s004010050717
Szabo, R., and Skropeta, D. (2017). Advancement of sialyltransferase inhibitors: Therapeutic challenges and opportunities. Med. Res. Rev. 37 (2), 219–270. doi:10.1002/med.21407
Takashima, S. (2008). Characterization of mouse sialyltransferase genes: their evolution and diversity. Biosci. Biotechnol. Biochem. 72, 1155–1167. doi:10.1271/bbb.80025
Takashima, S., Truji, S., and Tsujimoto, M. (2002). Characterization of the second type of human beta-galactoside alpha 2, 6-sialyltransferase (ST6Gal II), which sialylates Galbeta 1, 4GlcNAc structures on oligosaccharides preferentially. Genomic analysis of human sialyltransferase genes. J. Biol. Chem. 277 (48), 45719–45728. doi:10.1074/jbc.M206808200
Takashima, S., Tsuji, S., and Tsujimoto, M. (2003). Comparison of the enzymatic properties of mouse beta-galactoside alpha2, 6-sialyltransferases, ST6Gal I and II. J. Biochem. 134 (2), 287–296. doi:10.1093/jb/mvg142
Turk, E., Teplow, D. B., Hood, L. E., and Prusiner, S. B. (1988). Purification and properties of the cellular and scrapie hamster prion proteins. Eur. J. Biochem. 176 (1), 21–30. doi:10.1111/j.1432-1033.1988.tb14246.x
Wang, L., Liu, Y., Wu, L., and Sun, X. L. (2016). Sialyltransferase inhibition and recent advances. Biochim. Biophys. Acta 1864 (1), 143–153. doi:10.1016/j.bbapap.2015.07.007
Keywords: prion, prion diseases, N-glycosylation, sialic acid, sialyltransferases, ST6GAL1, ST6GAL2, mouse
Citation: Makarava N, Katorcha E, Chang JC-Y, Lau JTY and Baskakov IV (2022) Deficiency in ST6GAL1, one of the two α2,6-sialyltransferases, has only a minor effect on the pathogenesis of prion disease. Front. Mol. Biosci. 9:1058602. doi: 10.3389/fmolb.2022.1058602
Received: 30 September 2022; Accepted: 31 October 2022;
Published: 14 November 2022.
Edited by:
Fei Wang, University of Texas Health Science Center at Houston, United StatesReviewed by:
Kazuchika Nishitsuji, Wakayama Medical University, JapanCopyright © 2022 Makarava, Katorcha, Chang, Lau and Baskakov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ilia V. Baskakov, QmFza2Frb3ZAc29tLnVtYXJ5bGFuZC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.