 Konstantinos Mazarakos1
Konstantinos Mazarakos1 Ramesh Prasad
Ramesh Prasad Huan-Xiang Zhou
Huan-Xiang Zhou- 1Department of Physics, University of Illinois at Chicago, Chicago, IL, United States
- 2Department of Chemistry, University of Illinois at Chicago, Chicago, IL, United States
Phase separation of intrinsically disordered proteins (IDPs) is a phenomenon associated with many essential cellular processes, but a robust method to compute the binodal from molecular dynamics simulations of IDPs modeled at the all-atom level in explicit solvent is still elusive, due to the difficulty in preparing a suitable initial dense configuration and in achieving phase equilibration. Here we present SpiDec as such a method, based on spontaneous phase separation via spinodal decomposition that produces a dense slab when the system is initiated at a homogeneous, low density. After illustrating the method on four model systems, we apply SpiDec to a tetrapeptide modeled at the all-atom level and solvated in TIP3P water. The concentrations in the dense and dilute phases agree qualitatively with experimental results and point to binodals as a sensitive property for force-field parameterization. SpiDec may prove useful for the accurate determination of the phase equilibrium of IDPs.
Introduction
Biomolecular condensates formed via liquid-liquid phase separation drive much of biology, but accurate computation of the binodal, representing the equilibrium concentrations in the bulk and dense phases as a function of temperature, based on atomistic modeling of the components presents a significant challenge. A related issue is the mechanism leading to phase separation. Theories predict that, depending on the initial densities or compositions, phase separation occurs by two mechanisms (Figure 1). Inside the spinodal, the system is thermodynamically unstable and phase separation occurs spontaneously, in a process known as spinodal decomposition. This process is initiated by large-scale density fluctuations, leading to interconnected domains. Further condensation then leads to separated condensates. Between the binodal and spinodal, the system is metastable, and phase separation occurs by nucleation and growth. This mechanism is initiated by local density fluctuations, leading to the generation of nuclei. Further growth then produces stable condensates. The dividing line between these two mechanisms, i.e., the spinodal, is known for a number of theoretical models (Figure 1) and has been measured for both structured and disordered proteins (Thomson et al., 1987; Bracha et al., 2018). Both spinodal decomposition and nucleation have been observed for the formation of biomolecular condensates (Bracha et al., 2018; Kasinsky et al., 2021). It has been suggested that cells may want to keep component concentrations to a minimum required for phase separation, i.e., crossing the low-concentration branch of the binodal just enough into the left metastable region, and thus nucleation and growth is the favored mechanism (Zeng et al., 2020). However, in most theoretical models (Figure 1), the spinodal covers the bulk of the area under the binodal, and therefore random initial preparations have much higher chances for phase separation by spinodal decomposition than by nucleation and growth. Indeed, phase separation is observed instantaneously in many in vitro preparations; the rapid speed is consistent with spinodal decomposition.
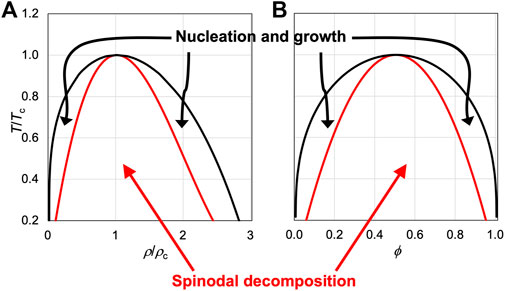
FIGURE 1. Binodals and spinodals of two model systems. (A) Van der Waals fluid, which satisfies the following equation of state:
In computer simulations of model systems, depending on the initial density, spinodal decomposition produces a variety of morphologies for the dense phase, including sphere, cylinder, slab, hollow cylinder, and hollow sphere (Binder et al., 2012; Díaz-Herrera et al., 2022). The morphologies are a reflection of the finite system size, where surface energy associated with interfacial tension becomes nonnegligible compared to the free energy within the dense phase. In essence, each shape optimizes the balance between these two terms of free energy at the given density. The slab morphology is of special interest because it represents the macroscopic form of the dense phase. More importantly, for this paper, the slab morphology is the basis of the method for computing binodals to be presented below.
Over the years, a variety of methods have been developed to compute the binodals of coarse-grained model systems (for a brief review and illustration, see Mazarakos et al., 2023). One of the earliest methods is based on preparing a dense slab at the center of an otherwise empty simulation box (Rao and Levesque, 1976). Molecular dynamics (MD) simulations then allow the dense and bulk phases to reach equilibration, and the densities of the two phases are then evaluated to build the binodal. This classical slab method has now been applied to a variety of coarse-grained models of biomolecular condensates, from spherical particles and homopolymers (as crude models of structured proteins and IDPs) (Mazarakos and Zhou, 2021; Mazarakos and Zhou, 2022) to residue-level models of IDPs (Das et al., 2018; Dignon et al., 2018; Statt et al., 2020) to multi-bead-per-residue models of dipeptides (Tang et al., 2021). An exciting recent development is the migration of this method to simulations of all-atom models of IDPs in explicit solvent, by first carrying out full simulations at the coarse-grained level and then mapping to atomistic systems (Zheng et al., 2020; Welsh et al., 2022). Still, the mapping is not a trivial task and, after mapping, simulations at the all-atom level may fail to allow exchange of protein molecules between the phases, let alone reaching phase equilibrium (Zheng et al., 2020). In comparison, Gibbs ensemble simulations (Panagiotopoulos, 1987), while proven useful for providing insights on biomolecular condensates (Nguemaha and Zhou, 2018; Ghosh et al., 2019) and complex coacervates (Lytle et al., 2016; Li et al., 2018), are more restrictive, in particular due to the difficulty in inserting a polymer chain into a dense solution. Recent implementation by field-theoretic simulations has widened the applicability of the Gibbs ensemble (McCarty et al., 2019), but application to all-atom models seems out of reach at present. Lastly the FMAP method, based on using the fast Fourier transform to evaluate intermolecular interaction energies, has been developed to calculate the binodals of structured proteins, but not IDPs, modeled at the all-atom level in implicit solvent (Qin and Zhou, 2016).
Here we present a method that we dub SpiDec, as an efficient alternative to the classical slab method. Instead of an initial dense slab, we start the system at a homogeneous, low density inside the spinodal. Spinodal decomposition then quickly brings the system to a slab morphology. We demonstrate this method both on four model systems and on a phase-separating tetrapeptide (Abbas et al., 2021) at the all-atom level in explicit solvent.
Computational methods
Model systems
We tested the SpiDec method on model systems composed of Lennard-Jones (LJ) particles, LJ chains, hydrophobic-hydrophilic (HP) chains, and patchy particles. The interaction potentials for LJ particles and LJ chains are the same as in our previous studies (Mazarakos and Zhou, 2021; Mazarakos and Zhou, 2022). Specifically, the LJ particles interact via the LJ potential,
but with a cutoff imposed at
The LJ chains consist of 10 beads. The beads interact via a force-shifted LJ potential:
with a cutoff
The interaction potential for patchy particles was the same as in our previous studies (Nguemaha and Zhou, 2018; Ghosh et al., 2019). Each particle has four equal-sized circular patches with centers located at the vertices of a tetrahedron. Each patch has a spanning polar angle of
where the range of interaction, λ, is fixed to 0.5σ;
Initialization for simulations
We studied LJ particles by both MD and Monte-Carlo (MC) simulations, LJ and HP chains by MD simulations, and patchy particles by MC simulation. The simulation boxes were rectangular, with side length Lx in two directions and Lz (≥Lx) in the third direction. To prepare for MD simulations, particles or chains were randomly inserted into the simulation box at density ρ0. For MC simulations, the particles were initially placed on a cubic lattice spanning the simulation box, again at an initial density ρ0. The values of ρ0 are given below. The periodic boundary condition was applied during the simulations.
Ranges in initial density for various dense-phase morphologies
We scanned the initial density to identify the dense-phase morphologies of a given system, with the temperature at the lowest value (0.65, 1.70, 1.05, and 0.61 for LJ particles, LJ chains, HP chains, and patchy particles, respectively). The initial density was scanned up to 0.8, in two series. In the first series, the particle numbers were fixed (1,000 for LJ particles and LJ and HP chains; 750 for patchy particles); the simulation boxes were cubic, and the side lengths were varied to span the range of initial densities, which was from 0.05 to 0.8 at increments of 0.05.
In the second series, done for LJ particles and LJ chains, Lx was fixed (10 for LJ particles and 13 for LJ chains), and Lz/Lx was varied from 1 to 5 in increments of 0.25 or 0.5. The initial densities were 0.01–0.12 in increments of 0.01, 0.125 to 0.3 in increments of 0.025, 0.3 to 0.8 (for LJ particles) or 0.7 (for LJ chains) in increments of 0.05. The corresponding particle (or bead) numbers ranged from 10 to 4,000 for LJ particles and from 20 to 7,690 for LJ chains. For a given Lz/Lx, at increasing initial densities, spinodal decomposition or lack thereof results in seven distinct cases: a low-density homogenous phase, five dense-phase morphologies, and a high-density homogenous phase. For the boundary between the case of a low-density homogenous phase and the case of a dense phase with a spherical morphology, we took the midpoint between the highest initial density that resulted in a low-density homogenous phase and the lowest initial density that resulted in a dense phase with a spherical morphology. Sometimes it was uncertain whether to identify the status resulted from an intermediate initial density as a low-density homogenous phase or a dense phase with a spherical morphology; we then assigned that initial density as the boundary value. A similar procedure was followed to identify the next boundary, i.e., between a dense phase with a spherical morphology and a dense phase with a cylindrical morphology. The process continued until the last boundary, i.e., between a dense phase with a hollow sphere and a high-density homogenous phase, was determined.
Molecular dynamics simulations for LJ particles and LJ and HP chains
These simulations were carried out using the HOOMD-blue package (version 2.5.0) on GPUs (Glaser et al., 2015). All the particles or beads have the same diameter
The initial densities were 0.3 for LJ particles and 0.25 for LJ and HP chains. Separate simulations were carried out over a range of temperatures. To investigate the effects of system size and Lz/Lx ratio, we chose N in the range of 1,000–10,000, and Lz/Lx from 1.25 to 33.3 for LJ particles, from 1.16 to 18.2 for LJ chains, and 1.5 to 5 for HP chains. For LJ particles, the simulation length was 10 million steps, but was extended to 100 or 200 million steps when multiple slabs took a long time to fuse into a single slab (at N ≥ 6,000 and Lz/Lx ≥ 20). The same was true for LJ chains, except that the longer simulations were 100–300 million steps and applied to more cases (N as low as 4,000 and Lz/Lx as low as 2). The simulation length was 100 million steps for HP chains. The time interval for saving snapshots was 1,000 time steps for simulations with a total length of 10 million steps and 10,000 time steps for longer simulations.
Monte Carlo simulations
For patchy particles, the initial density was 0.36, N ranged from 250 to 1,250, and Lz/Lx was 1.5, 3, and 5. MC simulations were run for 2 million steps, and up to 5 million steps for larger N and elongated simulation boxes. Snapshots were saved for analysis once every 2000 MC steps. Each MC step consisted of either a displacement or a rotation (with equal probability) for every particle. The displacement was randomly selected inside a cube centered at the original position and with a side length of 0.09
Time for phase separation via spinodal decomposition
In simulations where the dense phase had a slab morphology, we monitored the time that it took for slabs to emerge from spinodal decomposition. In each saved snapshot, the density profile along the normal direction of the slabs (typically the z direction) was calculated in slices with a default thickness of 1
When multiple slabs were formed, we also monitored the time,
Densities in the dense and bulk phases
In all the simulations where a slab morphology was formed and used to calculate equilibrium properties, averages were taken over the second half of the simulations. However, when
The density profile,
To obtain the densities,
where
Critical temperature
The binodal, comprising bulk- and dense-phase densities as a function of temperature, was fit to the following equations
where
Interfacial tension
The interfacial tension
where
Molecular dynamics simulations of phase-separating tetrapeptide
We also tested SpiDec on a peptide, consisting of two copies of a phenylalanine dipeptide crosslinked at the C-termini by a disulfide bond (denoted as FFssFF), that was recently shown to phase separate (Abbas et al., 2021). FFssFF was prepared in ChemDraw and saved in Protein Data Bank (PDB) format for force-field parametrization, which was done using Gaussian 16 at the HF/6-31G* level for atomic charges and using general Amber force field (GAFF) (Wang et al., 2004) for other parameters. The initial configuration of 64 copies of the peptide in a water box was prepared in two steps. First, eight copies were randomly inserted into a cubic box with a side length of 30 Å and solvated with TIP3P water (Jorgensen et al., 1983) using CHARMM-GUI (Jo et al., 2008). This system was relaxed by energy minimization (2000 steps of steepest descent and 3,000 steps of conjugate gradient) and a 100 ps MD simulation at constant NVT (ramping from 0 to 294 K in 40 ps and at 294 K for remaining 60 ps) with a 1 fs timestep. The box with only the peptides in the last snapshot was duplicated in each of the three orthogonal directions to build a system with 64 copies in a cubic box with a side length of 60 Å, which was solvated again with 4693 TIP3P water molecules. The latter system was also relaxed by energy minimization and 500 ps of constant-NVT simulation (ramping from 0 to 294 K in 40 ps and at 294 K for remaining 460 ps). To further condense the system, half of the water molecules were randomly removed and the system was again relaxed by the same procedure of energy minimization plus 500 ps of constant-NVT simulation. It was then equilibrated at constant NPT (294 K and 1 bar) and with a 2 fs timestep, for 8.6 µs until the peptides formed a single slab with normal in the z direction. The side length of the cubic box at this point was reduced to 51.72 Å. Long-range electrostatic interactions were treated by the particle mesh Ewald method (Essmann et al., 1995) with nonbonded cutoff at 8 Å. Temperature was regulated by the Langevin thermostat with a damping constant of 3 ps−1; pressure was regulated using the Berendsen barostat (Berendsen et al., 1984). All bonds connected with hydrogen atoms were constrained using the SHAKE algorithm (Ryckaert et al., 1977).
The single slab of 64 copies was finally placed in a rectangular box with Lz/Lx = 5. To model pH 7, 32 copies were randomly chosen to have a terminal amide protonated. The system was neutralized with 32 Cl− ions and solvated with 20,174 TIP3P water molecules; the total number of atoms was 66,986. Initial relaxation was done with energy minimization and 500 ps of constant NVT simulation with a 1 fs timestep, followed by 100 ns of equilibration at constant NPT with a 2 fs timestep. Finally constant-NVT production runs with a 2 fs timestep, at T = 294 K and 326 K, each for 2 µs. The box dimensions were 51.47 Å × 51.47 Å × 260.38 Å and 52.04 Å × 52.04 Å × 263.24 Å, respectively, at the two temperatures. The last snapshot of the 326 K simulation was ramped to 340 K or 360 K (in a 500 ps of constant NVT simulation) and production runs were carried out at these higher temperatures for 2 µs each. All MD simulations were carried out on GPUs using pmemd.cuda (Salomon-Ferrer et al., 2013). Snapshots were saved at 100 ps intervals for analysis.
Results
Variety of dense-phase morphologies from spinodal decomposition
In Figure 2A, we display the dense-phase morphologies of LJ particles in a cubic box at T = 0.65, obtained from MD simulations at increasing initial densities (ρ0). The dense phase appears as a sphere at ρ0 = 0.1, a cylinder at ρ0 = 0.2, a slab at ρ0 = 0.3, a hollow cylinder at ρ0 = 0.6, and a hollow sphere at ρ0 = 0.7. Similar observations are found for the other model systems, and are displayed in Figure 2B for LJ chains at T = 1.7 and in Supplementary Figure S1 for HP chains at T = 1.05.
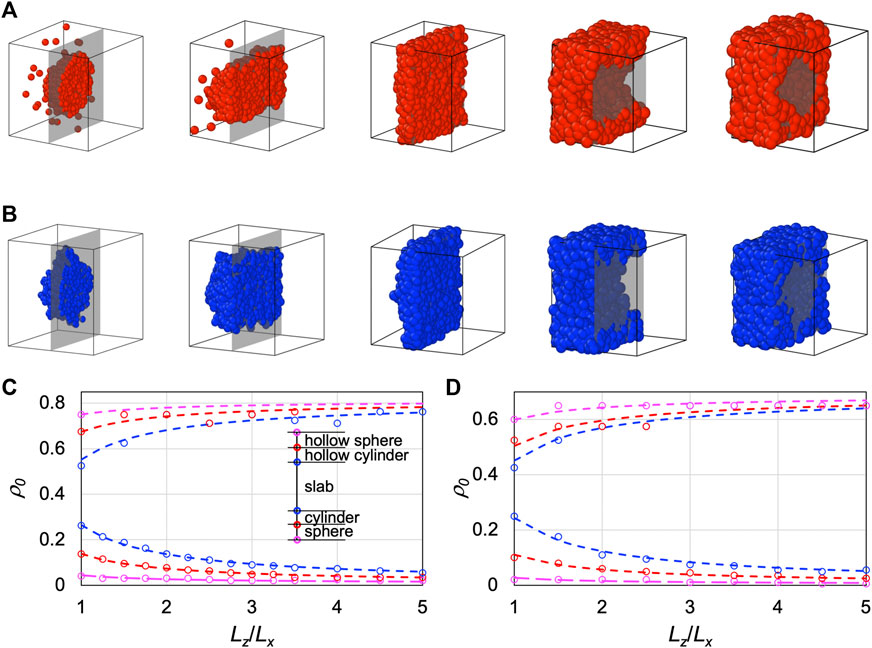
FIGURE 2. Morphologies of the dense phases of LJ particles and LJ chains over a range of initial densities inside the spinodal. (A) Morphologies for an LJ particle system of N = 1,000 particles in a cubic box at T = 0.65. The dense phase appears as a sphere, cylinder, slab, hollow cylinder, and hollow sphere at ρ0 = 0.1, 0.2, 0.3, 0.6, and 0.7 respectively. The initial densities were changed by varying the box side lengths, which are shown here not to scale. At each density, the cubic box is cut by a plane (rendered as gray when the background is empty), and only the half behind the cut is displayed. (B) Corresponding results for an LJ chain system (100 10-bead chains) at T = 1.7 and ρ0 = 0.1, 0.2, 0.3, 0.5, and 0.6. (C) Boundaries between different morphologies for LJ particles in rectangular boxes with different Lz/Lx ratios. The density ranges for different morphologies are illustrated in the inset. Lx = 10 and T = 0.65. (D) Corresponding results for an LJ chain system at Lx = 13 and T = 1.7.
We scanned the initial densities to identify the boundaries between different dense-phase morphologies. For example, for LJ particles in a cubic box (Lz/Lx = 1) at T = 0.65, the transitions from a single low-density phase to a spherical dense phase, from sphere to cylinder, from cylinder to slab, from slab to hollow cylinder, from hollow cylinder to hollow sphere, and from hollow sphere to a single high-density phase occur at initial densities of 0.04, 0.1375, 0.2625, 0.525, 0.675, and 0.75, respectively. Note that we checked the dense-phase morphologies on the very short timescale of phase separation via spinodal decomposition (see below for more details). On this timescale, phase separation via nucleation and growth would not have occurred. Therefore simulations from initial densities in the metastable regions, between the spinodal and binodal, would remain a single phase, and the boundaries with the two single phases effectively define the spinodal densities. In the foregoing example, the lowest and highest boundary values, 0.04 and 0.75, are the spinodal densities.
At increasing Lz/Lx, the range of initial densities for the slab morphology widens in both directions, squeezing the other inter-morphology boundaries at both ends of the range (Figure 2C). The dependences of the boundary density values on Lz/Lx fit well to a simple function,
where
Slab formation at different Lz/Lx ratios
The slab morphology allows easy determination of binodals (see Computational Methods). We thus pay special attention to this morphology. In Figures 3, 4, we display snapshots of slabs formed in simulations of the four model systems over a range of
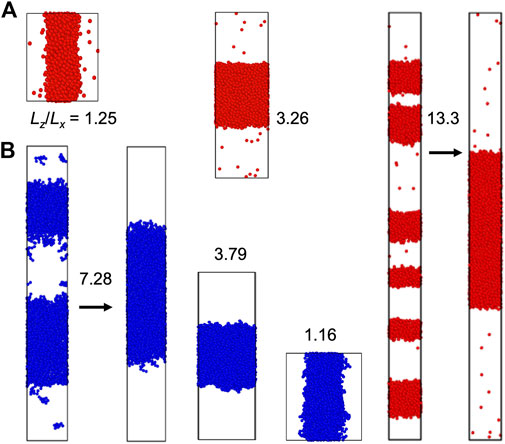
FIGURE 3. Slab formation at various Lz/Lx values. (A) LJ particle system at N = 4,000, T = 0.65, ρ0 = 0.3, and Lz/Lx = 1.25, 3.26, and 13.3. (B) LJ chain system at N = 4,000, T = 1.7, ρ0 = 0.25, and Lz/Lx = 1.16, 3.79, and 7.28. The z axis is along the vertical direction. For each system, the simulation boxes are drawn approximately to scale. Arrows indicate the fusion of multiple slabs into a single slab.
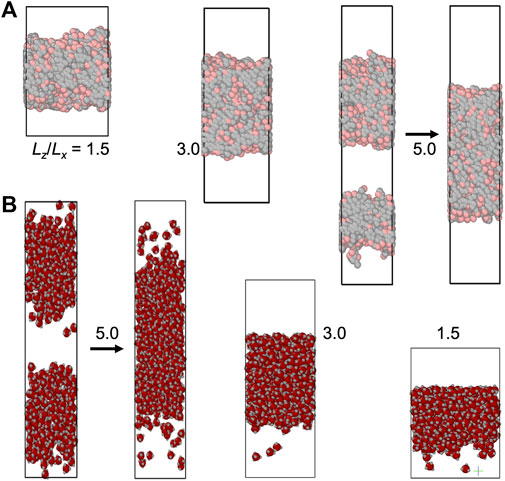
FIGURE 4. Slab formation at various Lz/Lx values. (A) HP chain system at N = 4,000, T = 1.05, ρ0 = 0.25, and Lz/Lx = 1.5, 3.0, and 5.0. (B) Patchy particle system at N = 500, T = 0.61, ρ0 = 0.36, and Lz/Lx = 1.5, 3.0, and 5.0. The z axis is along the vertical direction. For each system, the simulation boxes are drawn approximately to scale. Arrows indicate the fusion of multiple slabs into a single slab.
At
Timescales for phase separation via spinodal decomposition and for complete slab fusion
We devised a procedure to determine the time (
For the purpose of determining binodals, another important timescale is
Optimum in Lz/Lx and selection of ρ0 for binodal determination
So it appears that there is an optimum in
Slab formation also requires a correct choice for the initial density. Fortunately, at
Binodals of four model systems
Once a single slab is formed and equilibrated with the bulk phase at each temperature within a selected range, we can calculate the densities of the two phases as a function of temperature. The resulting binodals are shown in Figures 5A,B for the LJ particle and LJ chain systems at N = 4,000 and in Supplementary Figures S4A, S4B for the HP chain system at N = 4,000 and the patchy particle system at N = 500. For each system, we report binodals calculated at three
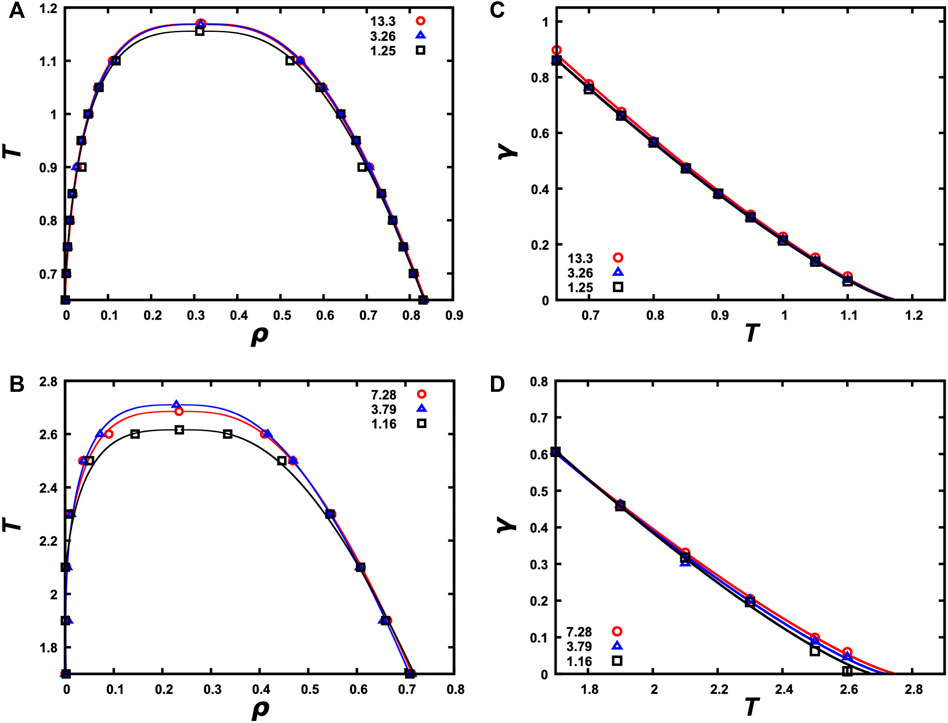
FIGURE 5. Binodals and interfacial tensions calculated from snapshots with a single slab. (A) Binodals of the LJ particle system at N = 4,000 and Lz/Lx = 1.25, 3.26, and 13.3. (B) Binodals of the LJ chain system at N = 4,000 and Lz/Lx = 1.16, 3.79, and 7.28. (C) Interfacial tension versus temperature for the LJ particle system at the three Lz/Lx ratios. (D) Interfacial tension versus temperature for LJ chain system at the three Lz/Lx ratios.
We also tested for possible effects of the spring constant on the binodal of the LJ chain system. As shown in Supplementary Figure S7A, the effects are very modest. The binodal remains essentially unchanged when the spring constant is reduced from 75,000
We also tested the effect of the HP chain sequence, specifically the positions of the two P beads. As shown in Supplementary Figure S7B, the positions of the P beads have a significant effect on the binodal. Tc has a significant increase when the two P beads are moved next to each other. P beads are repulsive to all beads, and the repulsion can be minimized when P beads are clustered together. When the two P beads within a chain are next to each other, P beads are much easier to cluster, and this increased clustering explains the increase in Tc.
Interfacial tensions of three model systems
The single-slab morphology generated by SpiDec also allows us to calculate the interfacial tension. The results are shown in Figures 5C,D for the LJ particle and LJ chain systems and in Supplementary Figure S4C for the HP chain system, all at N = 4,000. Again, as long as Lz/Lx is above 2, convergent results are obtained. We also obtained very similar interfacial tension for the LJ particle system from both MD and MC simulations (Supplementary Figure S6B).
Interfacial tension is a measure of the different extents of intermolecular interactions in the two phases on the opposite sides of an interface. As the temperature approaches Tc, the two phases become more and more similar. Correspondingly the interfacial tension decreases as T approaches Tc and finally vanishes at Tc, where the two phases become identical.
Application of SpiDec to a peptide system modeled at the all-atom level in explicit solvent
Finally we tested SpiDec on the tetrapeptide FFssFF (Figure 6A) that was recently shown to phase separate (Abbas et al., 2021). Starting from a random, loose configuration solvated in water, 64 copies of the peptide condensed into a single slab (Figure 6B). We subsequently solvated this single slab in an elongated box (Lz/Lx = 5). The copies equilibrated between the dense and bulk phases. We calculated the average concentrations (expressed as wt/wt, i.e., weight of peptide over weight of water) along the z axis, and fit the concentration profile to eq [6] to obtain the dense- and bulk-phase concentrations (Figure 6C). At 294 K, the two concentrations are 0.94 wt/wt and 0.0085 wt/wt, respectively. These values are each about 3-fold higher than the experimental counterparts (Abbas et al., 2021), reflecting the need for improved force-field parameterization. We also carried out simulations at 326, 340, and 360 K. At the elevated temperatures, the two densities move toward each other (Figure 6D), as expected for a system showing upper critical solution temperature.
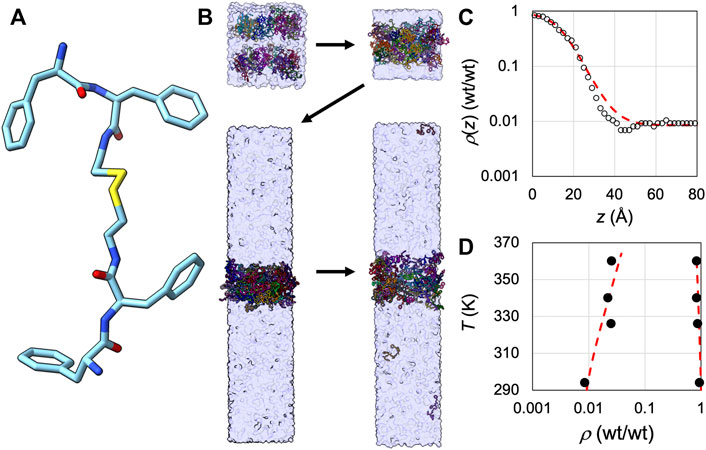
FIGURE 6. Application of SpiDec to a phase-separating peptide. (A) Structure of FFssFF. (B) The SpiDec simulation procedure. First, a random, loose configuration condensed into a single slab. Then the slab was solvated into an elongated box and the two phases reached equilibrium. The fourth snapshot shown was taken at 1.065 µs of a 2-µs simulation at 294 K. (C) Concentration profile (circles) at 294 K and fit to eq [6] (red curve). Concentrations were calculated by counting copy numbers in 2-Å slices along the z direction; the location of each copy was represented by the midpoint of the central disulfide bond. The conversion of concentrations to wt/wt used a molecular weight of 741.5 Da for the peptide and a density of 1 kg/L for water. (D) Dilute- and dense-phase concentrations at four temperatures, from 294 K to 360 K. The red curve shows a binodal to guide the eye.
Discussion
We have characterized the morphologies and timescales of spinodal decomposition in model systems and demonstrated that slab formation via spinodal decomposition can be used to calculate the binodal and interfacial tension. At different initial densities in a rectangular simulation box, spinodal decomposition produces distinct dense-phase morphologies, including sphere, cylinder, slab, hollow cylinder, and hollow sphere. The range of initial densities for slab formation is the widest among all the dense-phase morphologies, and this range further widens as Lz/Lx increases. At
In addition to computing the equilibrium properties (binodal and interfacial tension) of phase separation, SpiDec simulations can be adapted to study dynamic properties of related processes. In particular, the fusion between multiple slabs observed here corresponds to condensate fusion. Therefore SpiDec simulations at large
For the phase-separating tetrapeptide, our simulation results agree qualitatively with experimental data (Abbas et al., 2021), but quantitatively, the computed concentrations in the two phases are each about 3-fold higher than the experimental counterparts. Binodals are exquisitely sensitive to force fields, and now, with SpiDec, we will have the opportunity to use experimental binodals for force-field parameterization of IDPs, leading to accurate modeling of IDPs and their condensate properties.
The advantages of SpiDec can be summarized as follows. Whereas the classical slab method yields only phase equilibrium properties, SpiDec can also be used to study gelation and condensate fusion. For atomistic systems, the implementation of the slab method involved first carrying out a simulation at the coarse-grained level and then mapping a resulting dense slab to the atomistic level (Zheng et al., 2020; Welsh et al., 2022). The mapping is a complicated procedure, and in the subsequent atomistic simulation the protein molecules can easily get trapped in the dense slab. In contrast, as demonstrated here, SpiDec eliminates the need for an initial coarse-grained simulation and directly yields an atomistic system that exchanges between the phases. For systems without any prior knowledge of the binodal, it may be difficult to pick the right initial concentration and therefore one may need to start SpiDec simulations at several initial concentrations. We expect this difficulty to diminish as SpiDec is used on more systems.
Associated content
Supporting information
The following Supporting Information is available:
Seven additional figures (Supplementary Figures S1–S7) presenting the dense-phase morphologies of HP chains; the timescales for phase separation and for complete fusion of multiple slabs into a single slab, and illustration of their determination; binodal and interfacial tension of HP chains and binodal of patchy particles; the effects of system size and Lz/Lx ratio on the calculated critical temperature; comparison of the binodal and interfacial tension of LJ particles determined by MD and MC simulations; binodals of the LJ chain system with different spring constants and of the HP chain system with different sequences and Supplementary Movie S1 showing the phase separation of LJ particles via spinodal decomposition and the fusion of multiple slabs, and Supplementary Movie S2 showing arrested spinodal decomposition leading to gelation of HP chains at T = 0.2.
Data availability statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Author contributions
KM and H-XZ designed research; KM and RP carried out research and analyzed data. KM and H-XZ wrote the manuscript.
Funding
This work was supported by National Institutes of Health Grant GM118091.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2022.1021939/full#supplementary-material
Abbreviations
HP, hydrophobic-hydrophilic; IDP, intrinsically disordered protein; LJ, Lennard-Jones; MC, Monte Carlo; MD, molecular dynamics; WCA, Weeks-Chandler-Anderson.
References
Abbas, M., Lipiński, W. P., Nakashima, K. K., Huck, W. T. S., and Spruijt, E. (2021). A short peptide synthon for liquid–liquid phase separation. Nat. Chem. 13, 1046–1054. doi:10.1038/s41557-021-00788-x
Berendsen, H. J. C., Postma, J. P. M., Gunsteren, W. F. v., DiNola, A., and Haak, J. R. (1984). Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690. doi:10.1063/1.448118
Binder, K., Block, B. J., Virnau, P., and Tröster, A. (2012). Beyond the van der Waals loop: What can Be learned from simulating Lennard-Jones fluids inside the region of phase coexistence. Am. J. Phys. 80, 1099–1109. doi:10.1119/1.4754020
Bracha, D., Walls, M. T., Wei, M.-T., Zhu, L., Kurian, M., Avalos, J. L., et al. (2018). Mapping local and global liquid phase behavior in living cells using photo-oligomerizable seeds. Cell 175, 1467–1480. e13. doi:10.1016/j.cell.2018.10.048
Das, S., Amin, A. N., Lin, Y.-H., and Chan, H. S. (2018). Coarse-grained residue-based models of disordered protein condensates: Utility and limitations of simple charge pattern parameters. Phys. Chem. Chem. Phys. 20, 28558–28574. doi:10.1039/c8cp05095c
Díaz-Herrera, E., Cerón-García, E., Bryan Gutiérrez, A., and Chapela, G. A. (2022). Finite size effect on the existence of the liquid–vapour spinodal curve. Mol. Phys. 120, e1989071. doi:10.1080/00268976.2021.1989071
Dignon, G. L., Zheng, W., Kim, Y. C., Best, R. B., and Mittal, J. (2018). Sequence determinants of protein phase behavior from a coarse-grained model. PLoS Comput. Biol. 14, e1005941. doi:10.1371/journal.pcbi.1005941
Essmann, U., Perera, L., Berkowitz, M. L., Darden, T., Lee, H., and Pedersen, L. G. (1995). A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593. doi:10.1063/1.470117
Foffi, G., Michele, C. D., Sciortino, F., and Tartaglia, P. (2005). Arrested phase separation in a short-ranged attractive colloidal system: A numerical study. J. Chem. Phys. 122, 224903. doi:10.1063/1.1924704
Ghosh, A., Mazarakos, K., and Zhou, H. X. (2019). Three archetypical classes of macromolecular regulators of protein liquid-liquid phase separation. Proc. Natl. Acad. Sci. U. S. A. 116, 19474–19483. doi:10.1073/pnas.1907849116
Glaser, J., Nguyen, T. D., Anderson, J. A., Lui, P., Spiga, F., Millan, J. A., et al. (2015). Strong scaling of general-purpose molecular dynamics simulations on GPUs. Comput. Phys. Commun. 192, 97–107. doi:10.1016/j.cpc.2015.02.028
Jo, S., Kim, T., Iyer, V. G., and Im, W. (2008). CHARMM-GUI: A web-based graphical user interface for charmm. J. Comput. Chem. 29, 1859–1865. doi:10.1002/jcc.20945
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W., and Klein, M. L. (1983). Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935. doi:10.1063/1.445869
Kasinsky, H. E., Gowen, B. E., and Ausió, J. (2021). Spermiogenic chromatin condensation patterning in several hexapods may involve phase separation dynamics by spinodal decomposition or microemulsion inversion (nucleation). Tissue Cell 73, 101648. doi:10.1016/j.tice.2021.101648
Kern, N., and Frenkel, D. (2003). Fluid–fluid coexistence in colloidal systems with short-ranged strongly directional attraction. J. Chem. Phys. 118, 9882–9889. doi:10.1063/1.1569473
Kirkwood, J. G., and Buff, F. P. (1949). The statistical mechanical theory of surface tension. J. Chem. Phys. 17, 338–343. doi:10.1063/1.1747248
Li, L., Srivastava, S., Andreev, M., Marciel, A. B., de Pablo, J. J., and Tirrell, M. V. (2018). Phase behavior and salt partitioning in polyelectrolyte complex coacervates. Macromolecules 51, 2988–2995. doi:10.1021/acs.macromol.8b00238
Lytle, T. K., Radhakrishna, M., and Sing, C. E. (2016). High charge density coacervate assembly via hybrid Monte Carlo single chain in mean field theory. Macromolecules 49, 9693–9705. doi:10.1021/acs.macromol.6b02159
Mazarakos, K., Qin, S., and Zhou, H. X. (2023). Calculating binodals and interfacial tension of phase-separated condensates from molecular simulations, with finite-size corrections. Methods Mol. Biol. 2563, 1–35. doi:10.1007/978-1-0716-2663-4_1
Mazarakos, K., and Zhou, H.-X. (2022). Multiphase organization is a second phase transition within multi-component biomolecular condensates. J. Chem. Phys. 156, 191104. doi:10.1063/5.0088004
Mazarakos, K., and Zhou, H. X. (2021). Macromolecular regulators have matching effects on the phase equilibrium and interfacial tension of biomolecular condensates. Protein Sci. 30, 1360–1370. doi:10.1002/pro.4084
McCarty, J., Delaney, K. T., Danielsen, S. P. O., Fredrickson, G. H., and Shea, J. E. (2019). Complete phase diagram for liquid-liquid phase separation of intrinsically disordered proteins. J. Phys. Chem. Lett. 10, 1644–1652. doi:10.1021/acs.jpclett.9b00099
Nguemaha, V., and Zhou, H. X. (2018). Liquid-liquid phase separation of patchy particles illuminates diverse effects of regulatory components on protein droplet formation. Sci. Rep. 8, 6728. doi:10.1038/s41598-018-25132-1
Panagiotopoulos, A. Z. (1987). Direct determination of phase coexistence properties of fluids by Monte Carlo simulation in a new ensemble. Mol. Phys. 61, 813–826. doi:10.1080/00268978700101491
Qin, S., and Zhou, H. X. (2016). Fast method for computing chemical potentials and liquid-liquid phase equilibria of macromolecular solutions. J. Phys. Chem. B 120, 8164–8174. doi:10.1021/acs.jpcb.6b01607
Rao, M., and Levesque, D. (1976). Surface structure of a liquid film. J. Chem. Phys. 65, 3233–3236. doi:10.1063/1.433495
Ryckaert, J.-P., Ciccotti, G., and Berendsen, H. J. C. (1977). Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341. doi:10.1016/0021-9991(77)90098-5
Salomon-Ferrer, R., Götz, A. W., Poole, D., Le Grand, S., and Walker, R. C. (2013). Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J. Chem. Theory Comput. 9, 3878–3888. doi:10.1021/ct400314y
Statt, A., Casademunt, H., Brangwynne, C. P., and Panagiotopoulos, A. Z. (2020). Model for disordered proteins with strongly sequence-dependent liquid phase behavior. J. Chem. Phys. 152, 075101. doi:10.1063/1.5141095
Tang, Y., Bera, S., Yao, Y., Zeng, J., Lao, Z., Dong, X., et al. (2021). Prediction and characterization of liquid-liquid phase separation of minimalistic peptides. Cell Rep. Phys. Sci. 2, 100579. doi:10.1016/j.xcrp.2021.100579
Thomson, J. A., Schurtenberger, P., Thurston, G. M., and Benedek, G. B. (1987). Binary liquid phase separation and critical phenomena in a protein/water solution. Proc. Natl. Acad. Sci. U. S. A. 84, 7079–7083. doi:10.1073/pnas.84.20.7079
Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A., and Case, D. A. (2004). Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174. doi:10.1002/jcc.20035
Weeks, J. D., Chandler, D., and Andersen, H. C. (1971). Role of repulsive forces in determining the equilibrium structure of simple liquids. J. Chem. Phys. 54, 5237–5247. doi:10.1063/1.1674820
Welsh, T. J., Krainer, G., Espinosa, J. R., Joseph, J. A., Sridhar, A., Jahnel, M., et al. (2022). Surface electrostatics govern the emulsion stability of biomolecular condensates. Nano Lett. 22, 612–621. doi:10.1021/acs.nanolett.1c03138
Zeng, X., Holehouse, A. S., Chilkoti, A., Mittag, T., and Pappu, R. V. (2020). Connecting coil-to-globule transitions to full phase diagrams for intrinsically disordered proteins. Biophys. J. 119, 402–418. doi:10.1016/j.bpj.2020.06.014
Keywords: phase separation, biomolecular condensates, phase equilibrium, binodal, interfacial tension, spinodal decomposition
Citation: Mazarakos K, Prasad R and Zhou H-X (2022) SpiDec: Computing binodals and interfacial tension of biomolecular condensates from simulations of spinodal decomposition. Front. Mol. Biosci. 9:1021939. doi: 10.3389/fmolb.2022.1021939
Received: 17 August 2022; Accepted: 12 October 2022;
Published: 24 October 2022.
Edited by:
Arvind Ramanathan, Argonne National Laboratory (DOE), United StatesReviewed by:
Davit Potoyan, Iowa State University, United StatesJianhan Chen, University of Massachusetts Amherst, United States
Copyright © 2022 Mazarakos, Prasad and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huan-Xiang Zhou, aHpob3U0M0B1aWMuZWR1