 G Pepe
G Pepe R Appierdo1
R Appierdo1 F Ballesio
F Ballesio M Helmer-Citterich
M Helmer-Citterich PF Gherardini
PF Gherardini- 1Department of Biology, University of Rome “Tor Vergata”, Rome, Italy
- 2PhD Program in Cellular and Molecular Biology, Department of Biology, University of Rome “Tor Vergata”, Rome, Italy
Understanding how RNAs interact with proteins, RNAs, or other molecules remains a challenge of main interest in biology, given the importance of these complexes in both normal and pathological cellular processes. Since experimental datasets are starting to be available for hundreds of functional interactions between RNAs and other biomolecules, several machine learning and deep learning algorithms have been proposed for predicting RNA-RNA or RNA-protein interactions. However, most of these approaches were evaluated on a single dataset, making performance comparisons difficult. With this review, we aim to summarize recent computational methods, developed in this broad research area, highlighting feature encoding and machine learning strategies adopted. Given the magnitude of the effect that dataset size and quality have on performance, we explored the characteristics of these datasets. Additionally, we discuss multiple approaches to generate datasets of negative examples for training. Finally, we describe the best-performing methods to predict interactions between proteins and specific classes of RNA molecules, such as circular RNAs (circRNAs) and long non-coding RNAs (lncRNAs), and methods to predict RNA-RNA or RNA-RBP interactions independently of the RNA type.
Introduction
The involvement of RNAs in a wide range of biological processes, such as transcription, translation, neurogenesis, and the biogenesis and function of non-coding RNAs (ncRNAs) has been discussed in multiple studies (Newman et al., 2015; Turner and Díaz-Muñoz, 2018; Peng et al., 2022). Basic cellular physiology is critically dependant on RNA-Protein interactions (RPIs), as exemplified by their role in RNA splicing, transcription efficiency, stabilization and termination (Kelaini et al., 2021), in triggering RNA release from the transcription complex (Van Assche et al., 2015), and in regulating RNA degradation (Gilbertson et al., 2018). RNAs interact with RNA-Binding Proteins (RBPs) through sequence and structural motifs (Dominguez et al., 2018). Adinolfi et al. (Adinolfi et al., 2019) identified several RNA binding motifs by analyzing PAR-CLIP, eCLIP and HITS-CLIP experiments. Starting from these motifs, Guarracino et al. developed a web server for the identification of enriched structure or sequence motifs in a pool of RNAs which returns putative interacting RBPs (Guarracino et al., 2021). Altered functionality of RBPs and subsequent disruption of RNA-RBPs regulatory networks are commonly observed in human genetic diseases, neurodegenerative diseases and multiple cancer types (Pereira et al., 2017; Gebauer et al., 2021; Schieweck et al., 2021). Besides interacting with proteins, RNAs can also interact with each other, giving rise to complex regulatory networks that control cellular physiology in health and disease (e.g. mRNA regulation exerted by miRNA) (Chen et al., 2019; Pepe et al., 2022b; Wang et al., 2022). Moreover, RNAs influence each others’ expression level by competing for a limited pool of microRNAs (miRNAs) (Seitz, 2009; Poliseno et al., 2010), as postulated by the “competitive endogenous RNA” (ceRNA) theory (Salmena et al., 2011). The interaction between viral DNA or RNA genomes and host miRNAs is involved in immune system evasion and viral replication (Qiao et al., 2019). Accordingly, the role of exogenous DNA or RNA in viral infection has been extensively studied, highlighting how viral genomes can act as “sponges” for specific host miRNAs. This mechanism has been described for Hepatitis C Virus (Luna et al., 2015) and Epstein-Barr Virus (Riley et al., 2012) and it has also been suggested for SARS-CoV-2 (Pepe et al., 2022a).
Given the importance that RNA interactions play in fundamental cellular processes, cancer, and other diseases, several methods for studying the physical interactions between RNA and proteins have been developed (Ferrè et al., 2015). These in vitro or in vivo methods can be classified into two main categories: i) RNA-centric methods used to study proteins associated with a specific RNA; ii) protein-centric methods used to identify RNAs interacting with a specific protein (Ramanathan et al., 2019). Despite the large number of RNA interactions identified thanks to these methods, experimental validation is still expensive and time-consuming and computational approaches remain an active area of research.
In this review, we aim to elucidate recent advances in RNA interaction predictions, focusing on state-of-the-art methods currently used for the prediction of RNA-RNA or RNA-RBP interactions. The development of these methods is critically dependent on the quality and characteristics of datasets of known interactions. Accordingly, we will also review publicly available sources of RNA interaction data.
Overview of databases
A crucial element in the development of RNA-protein interaction prediction models is the retrieval of datasets containing known interacting pairs to be used for ML models’ training. The present section will therefore survey two fundamental aspects in this respect. Firstly, we describe the main features of the most widely employed datasets for RNA-protein interaction prediction. Indeed, during the last decade, various datasets have been constructed and released to pursue this task. Such datasets, typically, rely on information maintained in databases or obtained through literature-mining operations and they involve interactions supported by experimental evidence. Subsequently in this section, a second crucial aspect is pointed out. Since machine learning methods for binary classification need to be trained on datasets containing a balanced number of samples from both classes to be predicted, in the case of RPI prediction this translates into disposing of datasets containing RNA-protein pairs that are known to interact (which will henceforth be referred to as “positive dataset”) as well as non-interacting RNA-protein pairs (which will henceforth be referred to as “negative dataset”). We reported an overview of the major methods employed for the construction of RPI negative sets as well as a summary table reporting assumptions and outlines of such strategies.
Publicly available datasets of RNA interactions
Datasets currently considered as benchmarks for training, cross-validating or testing RPI prediction models include RPI369 and RPI2241 (Muppirala et al., 2011), RPI488 (Pan et al., 2016), and RPI1807 (Suresh et al., 2015). These are structure-based datasets which incorporate interaction pairs obtained from RNA-protein complexes whose structures have been deposited in the PDB (Velankar et al., 2021). Another commonly used dataset is NPInter2.0 (Yuan et al., 2014), which contains interactions derived from literature-mining and other databases.
The RPI2241 and RPI369 datasets were obtained from PRIDB (Lewis et al., 2011), a database of protein-RNA interfaces derived from PDB complexes (Burley et al., 2021). A total of 943 complexes from PRIDB (9,689 protein chains and 2,074 RNA chains) were initially selected. A final dataset consisting of 2241 experimentally validated RNA-protein interacting pairs (952 protein chains and 443 RNA chains) was derived, by redundancy reduction (discarding similar interaction on the basis of sequence identity) and sequence length filtering. When the RPI2241 dataset was constructed, a sizable fraction of all the RNA-protein complexes in the PDB corresponded to ribosomal structures, leading to a strong bias towards ribosomal RPIs. Accordingly, a second dataset, RPI369, was generated from RPI2241 by removing all RPIs that contained ribosomal proteins or ribosomal RNAs. Moreover, to generate a balanced dataset of non-interacting RNA-protein pairs, the RNAs and proteins from the original 943 complexes were randomly paired and pairs similar to known interactions were further discarded.
The RPI488 dataset is a structure-based dataset, derived from PDB complexes and specifically incorporating lncRNA-protein interactions. In order to generate the dataset, 18 ncRNA-protein complexes were downloaded from the PDB and 726 lncRNA-protein pairs were collected from them. In order to derive both a positive and a negative dataset, a distance cutoff of 5Å was used. Also, redundant sequences (sequence identity greater than 90% for both protein and lncRNA sequences) were excluded by using CD-HIT (Fu et al., 2012). Following redundancy reduction, the final RPI488 dataset contains 488 protein-lncRNA pairs (243 interacting pairs and 245 non-interacting ones).
The RPI1807 dataset was derived by integrating the Nucleic Acid Database (NDB) (Coimbatore Narayanan et al., 2014) and the PRIDB. A total of 1560 RPI complexes available in NDB were selected and, for 1336 of them, atomic interactions were extracted from PRIDB, thus obtaining 13,163 protein and 2715 RNA chains. The procedure for constructing the dataset included sequence length filtering and redundancy removal according to sequence similarity. In order to obtain both positive and negative sets, the selected non-redundant pairs were further analyzed for atomic interactions with a distance threshold (3.40 Å). This threshold was used to distinguish strongly interacting protein-RNA pairs (positive set) from weakly interacting protein-RNA pairs (negative set). The final RPI1807 dataset consists of 1807 positive pairs and 1436 negative pairs.
The overlap between RPI datasets is reported in Figure 1. This overlap could be greater than that obtained by simply intersecting the RNA-protein pairs since a redundancy reduction was applied to each one of the RPI datasets. In each of the RPI datasets, RNA-protein pairs were clustered and only one pair was chosen as representative; this could influence the overlap between the four datasets.
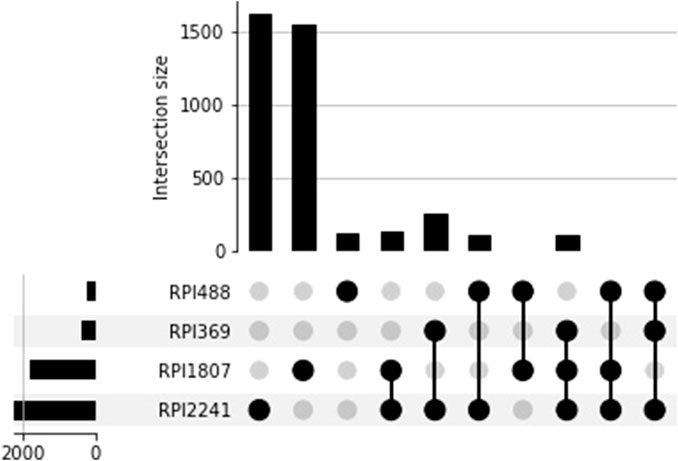
FIGURE 1. Overlap between the four RPI datasets generated from PDB RNA-protein complexes.
NPInter2.0 is a database that integrates experimentally-validated functional interactions between ncRNAs and other biomolecules (RNAs, proteins and DNAs), collected both from literature mining and from multiple databases. Although newer releases of the database exist (up to NPInter v4.0), NPInter v2.0 is the most widely used dataset for the development of prediction models. The dataset contains a total of 201,107 ncRNA interactions from 18 organisms, excluding interactions involving tRNAs and rRNAs.
Interactions were derived from manual annotation of articles published between 2008 and 2013 and include both experimentally-validated interactions as well as binding sites identified by genome-wide techniques (Yuan et al., 2014). The authors also integrated data from other resources, mainly the LncRNADisease database (Chen et al., 2013), and finally performed a redundancy reduction procedure within the dataset.
Several datasets have been derived from NPInter2.0 by selecting subsets of interactions with characteristics of interest. More specifically, the most widely used non-structure-based dataset for the development and testing of RPI prediction models is a subset of this database (namely NPInter10412) first assembled by Suresh et al, (2015), and subsequently used in numerous other works (Li et al., 2021; Wang et al., 2021; Zhao et al., 2021). NPInter10412 contains 10,412 ncRNA-protein interactions, distributed among the different species as illustrated in Table 1.
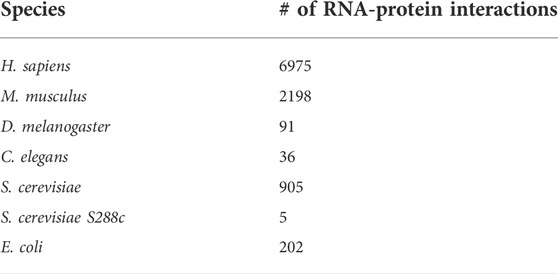
TABLE 1. Number of RNA-protein interactions by species in the NPInter10412 dataset.
A fourth release of NPInter was published in 2019 that increases the amount of high-throughput interactomes available data. NPInter4.0 (Teng et al., 2019) includes 600,000 new ncRNA interactions, particularly ncRNA–DNA interactions obtained via the ChIRP-seq technique, as well as interactions involving circular RNAs. Additionally, disease associations were added to the database.
Lastly, RNAInter4.0 is a recent resource that integrates experimentally validated and computationally predicted RNA interactions from literature-mining and databases (Kang et al., 2022). It provides information about different types of interactions in different taxa. Tables 2 and 3 summarize RNAInter’s content.

TABLE 2. Number of interactions of each type in the RNAInter database.

TABLE 3. Number of interactions in the 8 taxa in the RNAInter database. An overview of the main publicly available datasets for RNA-protein interaction prediction is given in Table 4.

TABLE 4. Publicly available datasets for RNA-protein interaction prediction.
Ultimately, despite remarkable advances in experimental techniques, the development of large and reliable RPI datasets is still the main bottleneck for training ML models. Hence, we would also like to stress the importance of redundancy control within data, since its presence may cause a leakage of information between training and test set during model training, resulting in untruthful prediction performance.
Strategies for the construction of a negative dataset
The lack of reliable datasets of non-interacting RNA-protein pairs is a major concern in the development of computational methods for RPI prediction. Indeed, it is not trivial to conclusively state that a given protein does not interact with a given RNA molecule (absence of evidence does not constitute evidence of absence). Indeed, various papers have demonstrated the critical effect of negative dataset composition on the performance of Machine Learning and Deep Learning models (Muppirala et al., 2011; Pan et al., 2016; Peng et al., 2019). Additionally, having balanced positive and negative sets is crucial to avoid overfitting on one class.
The most often used (Muppirala et al., 2011; Pan et al., 2016; Yi et al., 2020) method to construct a dataset of non-interacting pairs is to randomly pair RNAs and proteins in the positive set, followed by discarding the thus obtained pairs that showed high sequence similarity to the interacting ones, while retaining the others.
An interesting, albeit not widely used, technique to construct negative samples is the FIRE (FInding Reliable nEgative samples) method (Cheng et al., 2017). The core idea of this method relies on the following observation: given an experimentally-validated interaction between protein p1 and RNA r, and given another protein p2, the more similar p2 is to p1, the higher the likelihood that r interacts with p2. Thus, for each positive RPI (p1, r) the p2 protein that is most dissimilar to p1 is selected; if (p2, r) is not an experimentally-validated RPI, then it is selected as a negative RPI. The innovation introduced in this work lies in the way the similarity between each pair of proteins was computed, by taking into account functional annotations and protein domains information in addition to sequence similarity.
An additional approach that circumvents the requirement to create a negative dataset is PU learning, a binary classification method that can be applied when only positive (P) and unlabeled (U) data are available. For example, PRIPU trains a biased SVM on only positive and unlabelled examples (Cheng et al., 2015, 2017).
Some of the most often employed strategies for the construction of a negative dataset are listed in Table 5.

TABLE 5. Strategies for the construction of a negative dataset for RNA-protein interaction prediction.
Computational methods for RNA-protein interaction discovery
If on the one hand, the choice of the right training dataset is critical, on the other hand the choice of the right algorithm for RNA-RBP interaction prediction is also important, considering that some predictors were developed for a specific class of RNAs, such as lncRNA or circRNA. We will therefore review the latest methods for RNA-RBP interaction prediction. Some methods predict interactions between proteins and a specific class of RNA molecules, such as circRNAs (Yang et al., 2021; Niu et al., 2022) and lncRNAs (Ge et al., 2016; Zhao et al., 2018; Xie et al., 2019; Zhou et al., 2021). Others were developed to predict RNA-RBP interactions independently from the RNA type (Akbaripour-Elahabad et al., 2016; Yi et al., 2018; Zhan et al., 2018; Wang et al., 2019, 2021; Zhang et al., 2020).
LPI-deepGBDT: An artificial intelligence algorithm for the prediction of long non-coding RNA-protein interactions
Long non-coding RNAs (lncRNAs) are a class of RNA molecules that have attracted strong interest in recent years due to their abundance and their role in many physiological and pathological processes (Kornienko et al., 2016). Since many of the functions performed by lncRNAs require their interaction with proteins (LPIs), and most of lncRNAs are of unknown function, identifying new LPIs is a very important task. Most of the methods developed for this task are based on hand-crafted features, which is a process that requires time, domain knowledge and is based on strong assumptions. We describe the LPI-deepGBDT algorithm (Table 6), which uses a feed-forward deep architecture based on gradient boosting decision trees (Zhou et al., 2021). In this work three human and two plant LPI datasets, derived from the NPInter database, were used as training for the classifier. These datasets were processed using several filters, similar to previous works (Li et al., 2015; Zheng et al., 2017; Zheng et al., 2017; Zhang et al., 2018; Bai et al., 2019). Multiple features of lncRNAs and proteins were calculated from their sequences using Pyfeat (Muhammod et al., 2019) and BioProt (Márquez and Castro Amaya, 2019). The dimensionality of the feature space was then reduced using PCA, and protein and RNA features were concatenated to obtain a matrix of features representing the interaction pairs. This matrix was used as input to the classifier, which consisted of a multi-layered deep framework based on a gradient boosting model. The authors compared their model with five state-of-the-art LPI prediction methods, namely LPI-BLS, LPI-CatBoost, PLIPCOM, LPI-SKF and LPI-HNM (Yang et al., 2016; Deng et al., 2018; Fan and Zhang, 2019; Wekesa et al., 2020; Zhou et al., 2020), using six measurements: precision, recall, accuracy, F1-score, AUC and AUPR, and obtaining better average performances. Furthermore, the LPI-deepGBDT algorithm was successfully applied to the identification of potential protein partners for a specific lncRNA and, given a specific protein, to infer its potential interacting lncRNAs. The authors highlight that one of the main drivers of performance improvement for this method is the integration of biological features.
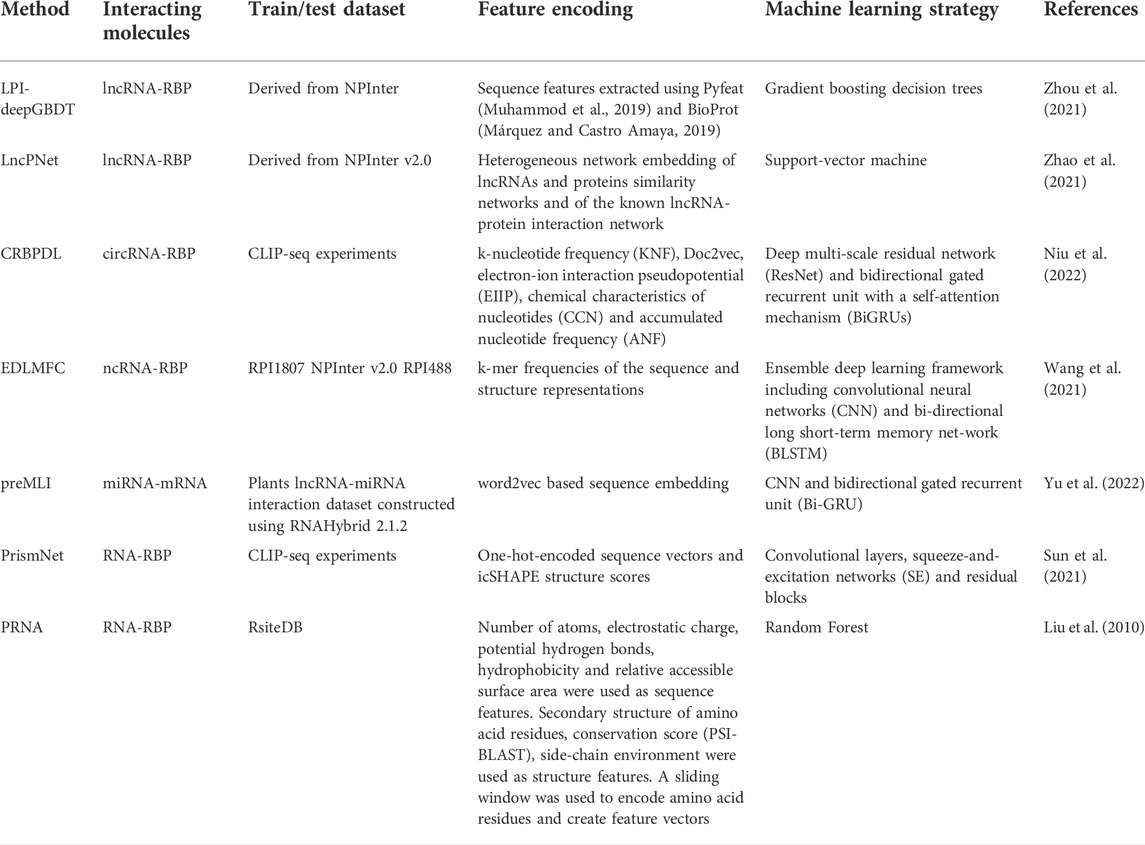
TABLE 6. Description of the train/test datasets, feature encoding and machine learning strategy for each of the described methods.
LncPNet: A human long non-coding RNA-protein interactions predictor
Most models are developed to predict lncRNA-protein interactions irrespective of the species, which can result in the introduction of noise and negatively affect performance.
To address this and other limitations, Zhao et al, (2021) introduced a new predictor model called LncPNet (Table 6). This method is designed to exclusively predict human lncRNA-protein interactions. Moreover, protein and lncRNA features are automatically generated using a network embedding. For this study, human lncRNA-protein interactions were selected from NPInter v2.0 resulting in 7523 experimentally validated pairs, including 3052 lncRNAs and 212 proteins. LncRNAs and proteins lacking sequence information were removed, thus obtaining a dataset of 4578 interactions between 2009 lncRNAs and 78 proteins. The negative dataset was built using the subcellular localization method (see Table 5). This method is based on a heterogeneous network of lncRNA-protein which is constructed using: i) lncRNA-lncRNA and protein-protein similarity; ii) known lncRNA-protein association. The similarity between lncRNAs and proteins is both calculated by Jaccard similarity and BLAST similarity. Subsequently the metapath2vec (Dong et al., 2017) method is used for network embedding and dimensionality reduction. LncRNA-protein interactions are represented as vectors of dimensionality 1 x 256 and those vectors are used to train a Support Vector Machine in order to predict whether an lncRNA interacts with a protein. Comparison with other state-of-the-art methods shows that LncPNet achieves better performances in terms of accuracy, F1-score and MCC.
CRBPDL: A deep learning approach for the prediction of circular RNA-RBP interactions
Circular RNAs or circRNAs are non-coding RNA molecules which can bind RBPs and are involved in multiple regulatory processes (Zang et al., 2020). CRBPDL (Table 6) (Niu et al., 2022) is a recently developed method that uses a deep learning approach (also used in other studies, e.g. Pan and Shen, 2018; Zhang et al., 2019; Yang et al., 2021) to predict interactions between circRNAs and proteins. The main improvement of CRBPDL is in the feature encoding step, which is critical for prediction performance. CRBPDL uses five different coding schemes (k-nucleotide frequency, Doc2vec, electron-ion interaction pseudopotential, nucleotide chemical properties, and cumulative nucleotide frequency) for the construction of a feature matrix. The method then uses a deep neural network architecture in order to extract local and global context information and subsequently train the model with a self-attention mechanism checking the robustness of the method. The deep neural network architecture is composed by a ResNet (a deep multi-scale residual network) and a BiGRUs (bidirectional gated recurrent unit) with the final integration of AdaBoost algorithm in order to improve the prediction performances. The authors trained and benchmarked CRBPDL using a circRNAs-RBPs interaction dataset derived from the CircInteractome database (Dudekula et al., 2016), consisting of interactions from 37 CLIP-seq experiments, consistently obtaining better performances when compared with existing methods. CRBPDL encodes different types of information about the sequence of circRNA: the dinucleotide and trinucleotide composition frequency (KNF), the free electron energy (EEIP), and also chemical informations about the nucleotides that compose circRNA sequences. For long-term context dependencies Doc2vec, used as encoding scheme, demonstrated to give a great contribution to the feature representation. CRBPDL was also tested on 31 datasets of linear RNA-RBP interactions, obtaining an average AUC of 0.91, which is significantly higher than the AUCs of other methods (ICIRCRBP-DHN (Yang et al., 2021), CRIP (Zhang et al., 2019), iDeepS (Pan et al., 2018), and CIRCSLNN (Ju et al., 2019)). CRBPDL is available on Github (https://github.com/nmt315320/CRBPDL).
EDLMFC: An ensemble deep learning framework for the prediction of non-coding RNA-RBP interactions
In this section, we discuss a class of ncRNA-RBP interaction predictors not designed for a specific RNA type. A recent computational method developed in this field, called EDLMFC, uses an Ensemble Deep Learning framework with Multi-scale Features Combination (Table 6) (Wang et al., 2021). EDLMFC was trained on ncRNA-RBP interaction pairs derived from the RPI1807, NPInter v2.0, and RPI488 datasets and uses different types of features as input such as the primary sequence and the secondary and tertiary structure of ncRNAs and proteins. Using a greater number of features was shown to increase prediction performance compared with single features. This method combines two different techniques: i) a convolutional neural network (CNN); ii) a bi-directional long short-term memory network (BLSTM). The first one is a deep learning-based method which is used to extract high-level information from the features and the second one is a recurrent neural network method which learns long-range dependencies between features, mainly on sequential data. Finally, a three-layer, fully connected, layer is able to predict ncRNA-protein interactions. In a five-fold cross-validation experiment, EDLMFC obtained better performance than RPITER (Peng et al., 2019), IPMiner (Pan et al., 2016), and CFRP (Dai et al., 2019). Moreover, independent tests demonstrated that EDLMFC can be effectively used to predict potential ncRNA-protein interactions in different organisms.
PRNA: Binding site features enable improvement RNA-protein interaction prediction
For the prediction of RNA-RBP interactions, several methods have been developed in order to find the potential binding sites in RNA or in RBP sequences. One of them is from Liu et al, (2010) (Table 6). In this work the authors highlighted the importance of both sequence and structure features in RNA-binding proteins, that simultaneously contribute towards the recognition of a specific RNA sequence site. In order to determine in a more comprehensive way the interacting sites in protein sequences, the authors suggested a parameter to consider interaction propensity of an amino acid. This variable represents a measure of mutual dependence of a triplet of amino acids in proteins where the central amino acid binds a nucleotide on the RNA sequence. Then this feature is encoded in a vector of other hybrid features to describe exhaustively the amino acids in the protein sequence. The method was trained using a dataset of protein-RNA complexes obtained from RsiteDB and used to predict RNA binding residues in proteins given the previous set of features, using Random Forest (RF), that with a sliding window of 5 amino acids on the protein sequence predicts the possible site of a binding event. The result in terms of AUC is of 0.905 with a ACC of 81.4% indicating a good performance if compared to other methods (RNABindR (Terribilini et al., 2007), BindN (Wang and Brown, 2006), RNAProB (Cheng et al., 2008), PPRint (Kumar et al., 2008)). In this paper the idea emerges that by integrating the information carried by the neighborhood of an amino acid with other features of the protein sequence and structure analyzed, we can substantially improve the prediction of RNA-RBP interactions by finding the binding sites. A concept well developed also in a recent work of Niu et al. in which instead of focusing on the binding protein sequence, the RNA sequence is fundamental.
PrismNet: A deep learning algorithm to predict RPIs that uses in vivo RNA structures
One of the most important factors determining the interaction between RNAs and proteins is the RNA secondary structure (Taliaferro et al., 2016). Therefore, leveraging this feature in prediction models can significantly increase their performance. Although there are different methods for the prediction of RNA secondary structure (Seetin and Mathews, 2012), computational methods based exclusively on the primary sequence do not take into account the dynamic nature of these structures. Indeed, RNA secondary structures are extremely dynamic and can change depending on various factors such as the interaction with chaperones and other RBPs. All these factors, ultimately, vary depending on the cellular conditions in vivo (Lewis et al., 2017). PrismNet is an RNA-protein prediction method that leverages experimental data on RNA secondary structures, being capable, in this way, to take into account their dynamism (Table 6). This method is based on secondary structure information obtained via in vivo click selective 2′-hydroxyl acylation and profiling experiments (icSHAPE) (Flynn et al., 2016) that were carried out in 7 cell types (i.e. K562, HepG2, HEK293, HEK 293T, HeLa, H9, and mES) in which RNA structures were profiled transcriptome-wide. This data was integrated with RBPs binding sites data from CLIP experiments in the same cell types. To construct the model input, the structure scores derived from the icSHAPE experiments were encoded as a one-dimensional vector and the sequence was represented as a four-dimensional one-hot-encoded vector. The deep learning model consists of a series of convolutional layers, while squeeze-and-excitation networks were used to recalibrate the convolutional channels and residual blocks to capture the joint sequence and structural determinants of RBP binding. The authors compared their model with other computational methods including RCK (Orenstein et al., 2016), GraphProt (Maticzka et al., 2014; Orenstein et al., 2016) and DeepBind (Alipanahi et al., 2015), using the binding sites obtained from the CLIP-seq datasets for each RBP, and obtaining better performance in terms of AUC and AUPRC. Furthermore, by training their model using different combinations of inputs, they observed that the model trained using both the sequence and the experimentally determined RNA secondary structures outperformed other models, demonstrating that experimental information on the RNA secondary structure in vivo is critical to the performance improvement.
Computational methods for RNA-RNA interactions prediction
RNAs can also interact with other RNAs and several studies have shown these interactions to be crucially involved in the regulation of gene transcription, cell metabolism, and other key cellular functions (Deogharia and Gurha, 2021; Singh et al., 2022; Wang et al., 2022). Despite the fact that a large number of RNA-RNA interactions have been experimentally validated, many more have yet to be identified. Therefore, several computational methods have been developed for the prediction of RNA-RNA interaction, many of which are based on sequence complementarity (Kang et al., 2020, 2021; Yang et al., 2020). In the last 5 years, these methods have been gradually revolutionized by the introduction of deep learning approaches borrowed from the field of natural language processing. PreMLI is one of the latest methods in this field, it was published in early 2022 by Yu and collaborators (Table 6) (Yu et al., 2022), and, currently, it achieves better overall performance compared with other existing methods. This method was specifically built to predict miRNA-lncRNA interactions and relies exclusively on RNA sequence information. PreMLI was trained using a plant lncRNA-miRNA interaction dataset, constructed using RNAHybrid 2.1.2. The approach consists of three steps: i) in the pre-training phase the RNA sequences are used as input for rna2vec training in order to obtain a weight matrix that better describes the RNA sequence and can be used as the input to the next step; ii) deep feature mining approaches, based on Convolutional Neural Network, Bidirectional Gated Recurrent Unit, and attention layers are used to obtain additional potential features; iii) in the last step the two feature vectors are connected as input to the prediction layer. The authors demonstrate how the pre-training and the deep feature mining phases improve prediction performance and, furthermore, they show how this method performs better than already existing advanced RNA-RNA interaction predictors in terms of sensitivity, specificity, and AUC. Although the pre-training step improves the model performance, it also increases the computational time required for the entire prediction process. Moreover, this method is optimized for the prediction of miRNA-lncRNA interactions in plants. In order to extend its use to other types of RNA-RNA interactions or other organisms the model needs to be trained on an appropriate specific dataset and the hyperparameters need to be adjusted.
Conclusion
In the last few years several studies have explored the RNA interactions landscape, given the crucial role that RNA-RBPs and RNA-RNA networks play in cell biology. Despite the advances made so far, novel experimental methods for the identification of binding sites (such as HITS-CLIP and PAR-CLIP) are still time-consuming and cost-intensive. That is why computational approaches represent a complementary strategy to guide experimental work. In this review, we provide an overview of the most recent prediction methods. We summarize recent advances in the algorithms developed to solve specific tasks, such as circRNA- or lncRNA-RBPs interaction predictions or, more generally RNA-RBPs interactions. Besides, we highlight how the development of a larger dataset of interactions is crucial to increase performance. Lastly, despite the fact that many methods rely only on sequence information, among the ones analyzed, those that obtain the best performances tend to include a variety of different biological features. Performance comparison of the described methods shows how the inclusion of structure information contributes to improving the accuracy and efficiency of the models. Only one of the described methods uses both RNAs and proteins structural information as input features for the predictive model because if, on the one hand, a large number of reliable protein structures is available, on the other hand, RNA structures are mainly obtained through computational prediction. RNA structure uncertainty could add noise to the model, resulting in untruthful prediction performances. The prediction of protein structure has reached satisfactory levels of performance thanks to the development of AlphaFold (Jumper et al., 2021). Conversely, RNA structure prediction still lags far behind. One of the main limitations is the paucity of known RNA structures that can be used for model training. To address this issue a new deep learning model called Atomic Rotationally Equivariant Scorer (ARES) has been developed (Townshend et al., 2021). ARES achieves good performances in the prediction of RNA structures, based on a training dataset of only 18 experimentally determined RNA structures. While this is a useful development, further work is needed in this area. Ultimately, as demonstrated by the methods described in this review, the availability of high-quality RNA structure predictions could greatly improve the inference of RNA-RBP and RNA-RNA interactions. Moreover, the advances in RNA secondary structure determination methods, that takes into account the information from biochemical assay like icSHAPE-seq (Flynn et al., 2016), could improve the confidence of such information as a feature for prediction models, likely leading to an improvement of their performance.
Author contributions
GP, RA, CC, FB performed the literature research and wrote the paper; PFG, MH-C reviewed the manuscript writing; GP, PFG, MH-C supervised the study.
Funding
AIRC project [to MHC] (grant number IG 23539) funded by the European Union – NextGenerationEU: National Center for Gene Therapy and Drugs based on RNA Technology.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adinolfi, M., Pietrosanto, M., Parca, L., Ausiello, G., Ferrè, F., and Helmer-Citterich, M. (2019). Discovering sequence and structure landscapes in RNA interaction motifs. Nucleic Acids Res. 47, 4958–4969. doi:10.1093/nar/gkz250
Akbaripour-Elahabad, M., Zahiri, J., Rafeh, R., Eslami, M., and Azari, M. (2016). rpiCOOL: A tool for in silico RNA–protein interaction detection using random forest. J. Theor. Biol. 402, 1–8. doi:10.1016/j.jtbi.2016.04.025
Alipanahi, B., Delong, A., Weirauch, M. T., and Frey, B. J. (2015). Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 33, 831–838. doi:10.1038/nbt.3300
Bai, Y., Dai, X., Ye, T., Zhang, P., Yan, X., Gong, X., et al. (2019). PlncRNADB: A repository of plant lncRNAs and lncRNA-RBP protein interactions. Curr. Bioinform. 14, 621–627. doi:10.2174/1574893614666190131161002
Burley, S. K., Bhikadiya, C., Bi, C., Bittrich, S., Chen, L., Crichlow, G. V., et al. (2021). RCSB protein data bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 49, D437–D451. doi:10.1093/nar/gkaa1038
Chen, G., Wang, Z., Wang, D., Qiu, C., Liu, M., Chen, X., et al. (2013). LncRNADisease: A database for long-non-coding RNA-associated diseases. Nucleic Acids Res. 41, D983–D986. doi:10.1093/nar/gks1099
Chen, Q., Meng, X., Liao, Q., and Chen, M. (2019). Versatile interactions and bioinformatics analysis of noncoding RNAs. Brief. Bioinform. 20, 1781–1794. doi:10.1093/bib/bby050
Cheng, C.-W., Su, E. C.-Y., Hwang, J.-K., Sung, T.-Y., and Hsu, W.-L. (2008). Predicting RNA-binding sites of proteins using support vector machines and evolutionary information. BMC Bioinforma. 9, S6. doi:10.1186/1471-2105-9-S12-S6
Cheng, Z., Huang, K., Wang, Y., Liu, H., Guan, J., and Zhou, S. (2017). Selecting high-quality negative samples for effectively predicting protein-RNA interactions. BMC Syst. Biol. 11, 9. doi:10.1186/s12918-017-0390-8
Cheng, Z., Zhou, S., and Guan, J. (2015). Computationally predicting protein-RNA interactions using only positive and unlabeled examples. J. Bioinform. Comput. Biol. 13, 1541005. doi:10.1142/s021972001541005x
Coimbatore Narayanan, B., Westbrook, J., Ghosh, S., Petrov, A. I., Sweeney, B., Zirbel, C. L., et al. (2014). The nucleic acid database: New features and capabilities. Nucleic Acids Res. 42, D114–D122. doi:10.1093/nar/gkt980
Dai, Q., Guo, M., Duan, X., Teng, Z., and Fu, Y. (2019). Construction of complex features for computational predicting ncRNA-protein interaction. Front. Genet. 10, 18. doi:10.3389/fgene.2019.00018
Deng, L., Wang, J., Xiao, Y., Wang, Z., and Liu, H. (2018). Accurate prediction of protein-lncRNA interactions by diffusion and HeteSim features across heterogeneous network. BMC Bioinforma. 19, 370. doi:10.1186/s12859-018-2390-0
Deogharia, M., and Gurha, P. (2021). The “guiding” principles of noncoding RNA function. New Jersey, United States: Wiley Interdiscip. Rev. RNA, e1704.
Dominguez, D., Freese, P., Alexis, M. S., Su, A., Hochman, M., Palden, T., et al. (2018). Sequence, structure, and context preferences of human RNA binding proteins. Mol. Cell. 70, 854–867. e9. doi:10.1016/j.molcel.2018.05.001
Dong, Y., Chawla, N. V., and Swami, A. (2017). metapath2vec. Proc. 23rd ACM SIGKDD Int. Conf. Knowl. Discov. Data Min. doi:10.1145/3097983.3098036
Dudekula, D. B., Panda, A. C., Grammatikakis, I., De, S., Abdelmohsen, K., and Gorospe, M. (2016). CircInteractome: A web tool for exploring circular RNAs and their interacting proteins and microRNAs. RNA Biol. 13, 34–42. doi:10.1080/15476286.2015.1128065
Fan, X.-N., and Zhang, S.-W. (2019). LPI-BLS: Predicting lncRNA–protein interactions with a broad learning system-based stacked ensemble classifier. Neurocomputing 370, 88–93. doi:10.1016/j.neucom.2019.08.084
Ferrè, F., Colantoni, A., and Helmer-Citterich, M. (2015). Revealing protein–lncRNA interaction. Brief. Bioinform. 17, 106–116. doi:10.1093/bib/bbv031
Flynn, R. A., Zhang, Q. C., Spitale, R. C., Lee, B., Mumbach, M. R., and Chang, H. Y. (2016). Transcriptome-wide interrogation of RNA secondary structure in living cells with icSHAPE. Nat. Protoc. 11, 273–290. doi:10.1038/nprot.2016.011
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi:10.1093/bioinformatics/bts565
Ge, M., Li, A., and Wang, M. (2016). A bipartite network-based method for prediction of long non-coding RNA-protein interactions. Genomics Proteomics Bioinforma. 14, 62–71. doi:10.1016/j.gpb.2016.01.004
Gebauer, F., Schwarzl, T., Valcárcel, J., and Hentze, M. W. (2021). RNA-binding proteins in human genetic disease. Nat. Rev. Genet. 22, 185–198. doi:10.1038/s41576-020-00302-y
Gilbertson, S., Federspiel, J. D., Hartenian, E., Cristea, I. M., and Glaunsinger, B. (2018). Changes in mRNA abundance drive shuttling of RNA binding proteins, linking cytoplasmic RNA degradation to transcription. Elife 7, e37663. doi:10.7554/eLife.37663
Guarracino, A., Pepe, G., Ballesio, F., Adinolfi, M., Pietrosanto, M., Sangiovanni, E., et al. (2021). Brio: A web server for RNA sequence and structure motif scan. Nucleic Acids Res. 49, W67–W71. doi:10.1093/nar/gkab400
Ju, Y., Yuan, L., Yang, Y., and Zhao, H. (2019). CircSLNN: Identifying RBP-binding sites on circRNAs via sequence labeling neural networks. Front. Genet. 10, 1184. doi:10.3389/fgene.2019.01184
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. doi:10.1038/s41586-021-03819-2
Kang, J., Tang, Q., He, J., Li, L., Yang, N., Yu, S., et al. (2022). RNAInter v4.0: RNA interactome repository with redefined confidence scoring system and improved accessibility. Nucleic Acids Res. 50, D326–D332. doi:10.1093/nar/gkab997
Kang, Q., Meng, J., Cui, J., Luan, Y., and Chen, M. (2020). PmliPred: A method based on hybrid model and fuzzy decision for plant miRNA–lncRNA interaction prediction. Bioinformatics 36, 2986–2992. doi:10.1093/bioinformatics/btaa074
Kang, Q., Meng, J., Shi, W., and Luan, Y. (2021). Ensemble deep learning based on multi-level information enhancement and greedy fuzzy decision for plant miRNA-lncRNA interaction prediction. Interdiscip. Sci. 13, 603–614. doi:10.1007/s12539-021-00434-7
Kelaini, S., Chan, C., Cornelius, V. A., and Margariti, A. (2021). RNA-binding proteins hold key roles in function, dysfunction, and disease. Biology 10, 366. doi:10.3390/biology10050366
Kornienko, A. E., Dotter, C. P., Guenzl, P. M., Gisslinger, H., Gisslinger, B., Cleary, C., et al. (2016). Long non-coding RNAs display higher natural expression variation than protein-coding genes in healthy humans. Genome Biol. 17, 14. doi:10.1186/s13059-016-0873-8
Kumar, M., Gromiha, M. M., and Raghava, G. P. S. (2008). Prediction of RNA binding sites in a protein using SVM and PSSM profile. Proteins 71, 189–194. doi:10.1002/prot.21677
Lewis, B. A., Walia, R. R., Terribilini, M., Ferguson, J., Zheng, C., Honavar, V., et al. (2011). Pridb: A protein-RNA interface database. Nucleic Acids Res. 39, D277–D282. doi:10.1093/nar/gkq1108
Lewis, C. J. T., Pan, T., and Kalsotra, A. (2017). RNA modifications and structures cooperate to guide RNA-protein interactions. Nat. Rev. Mol. Cell. Biol. 18, 202–210. doi:10.1038/nrm.2016.163
Li, A., Ge, M., Zhang, Y., Peng, C., and Wang, M. (2015). Predicting long noncoding RNA and protein interactions using heterogeneous network model. Biomed. Res. Int. 2015, 671950. doi:10.1155/2015/671950
Li, Y., Sun, H., Feng, S., Zhang, Q., Han, S., and Du, W. (2021). Capsule-LPI: A LncRNA-protein interaction predicting tool based on a capsule network. BMC Bioinforma. 22, 246. doi:10.1186/s12859-021-04171-y
Liu, Z.-P., Wu, L.-Y., Wang, Y., Zhang, X.-S., and Chen, L. (2010). Prediction of protein-RNA binding sites by a random forest method with combined features. Bioinformatics 26, 1616–1622. doi:10.1093/bioinformatics/btq253
Luna, J. M., Scheel, T. K. H., Danino, T., Shaw, K. S., Mele, A., Fak, J. J., et al. (2015). Hepatitis C virus RNA functionally sequesters miR-122. Cell. 160, 1099–1110. doi:10.1016/j.cell.2015.02.025
Márquez, B. G., and Castro Amaya, J. (2019). BIOPROT contenedor autónomo de Residuos biológicos. Rev. Colomb. De. Tecnol. De. Av. (RCTA) 1, 33. doi:10.24054/16927257.v33.n33.2019.3330
Maticzka, D., Lange, S. J., Costa, F., and Backofen, R. (2014). GraphProt: Modeling binding preferences of RNA-binding proteins. Genome Biol. 15, R17. doi:10.1186/gb-2014-15-1-r17
Muhammod, R., Ahmed, S., Md Farid, D., Shatabda, S., Sharma, A., and Dehzangi, A. (2019). PyFeat: A python-based effective feature generation tool for DNA, RNA and protein sequences. Bioinformatics 35, 3831–3833. doi:10.1093/bioinformatics/btz165
Muppirala, U. K., Honavar, V. G., and Dobbs, D. (2011). Predicting RNA-protein interactions using only sequence information. BMC Bioinforma. 12, 489. doi:10.1186/1471-2105-12-489
Newman, R., McHugh, J., and Turner, M. (2015). RNA binding proteins as regulators of immune cell biology. Clin. Exp. Immunol. 183, 37–49. doi:10.1111/cei.12684
Niu, M., Zou, Q., and Lin, C. (2022). Crbpdl: Identification of circRNA-RBP interaction sites using an ensemble neural network approach. PLoS Comput. Biol. 18, e1009798. doi:10.1371/journal.pcbi.1009798
Orenstein, Y., Wang, Y., and Berger, B. (2016). Rck: Accurate and efficient inference of sequence- and structure-based protein-RNA binding models from RNAcompete data. Bioinformatics 32, i351–i359. doi:10.1093/bioinformatics/btw259
Pan, X., Fan, Y.-X., Yan, J., and Shen, H.-B. (2016). IPMiner: Hidden ncRNA-protein interaction sequential pattern mining with stacked autoencoder for accurate computational prediction. BMC Genomics 17, 582. doi:10.1186/s12864-016-2931-8
Pan, X., Rijnbeek, P., Yan, J., and Shen, H.-B. (2018). Prediction of RNA-protein sequence and structure binding preferences using deep convolutional and recurrent neural networks. BMC Genomics 19, 511. doi:10.1186/s12864-018-4889-1
Pan, X., and Shen, H.-B. (2018). Predicting RNA-protein binding sites and motifs through combining local and global deep convolutional neural networks. Bioinformatics 34, 3427–3436. doi:10.1093/bioinformatics/bty364
Peng, C., Han, S., Zhang, H., and Li, Y. (2019). Rpiter: A hierarchical deep learning framework for ncRNA−Protein interaction prediction. Int. J. Mol. Sci. 20, E1070. doi:10.3390/ijms20051070
Peng, S., Guo, D., Guo, Y., Zhao, H., Mei, J., Han, Y., et al. (2022). CONSTITUTIVE EXPRESSER OF PATHOGENESIS-RELATED GENES 5 is an RNA-binding protein controlling plant immunity via an RNA processing complex. Plant Cell. 34, 1724–1744. doi:10.1093/plcell/koac037
Pepe, G., Guarracino, A., Ballesio, F., Parca, L., Ausiello, G., and Helmer-Citterich, M. (2022a). Evaluation of potential miRNA sponge effects of SARS genomes in human. Noncoding. RNA Res. 7, 48–53. doi:10.1016/j.ncrna.2022.01.003
Pepe, G., Parca, L., Viviani, L., Ausiello, G., and Helmer-Citterich, M. (2022b). Variation in the co-expression profile highlights a loss of miRNA-mRNA regulation in multiple cancer types. Noncoding. RNA Res. 7, 98–105. doi:10.1016/j.ncrna.2022.03.003
Pereira, B., Billaud, M., and Almeida, R. (2017). RNA-binding proteins in cancer: Old players and new actors. Trends Cancer 3, 506–528. doi:10.1016/j.trecan.2017.05.003
Poliseno, L., Salmena, L., Zhang, J., Carver, B., Haveman, W. J., and Pandolfi, P. P. (2010). A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 465, 1033–1038. doi:10.1038/nature09144
Qiao, Y., Zhao, X., Liu, J., and Yang, W. (2019). Epstein-Barr virus circRNAome as host miRNA sponge regulates virus infection, cell cycle, and oncogenesis. Bioengineered 10, 593–603. doi:10.1080/21655979.2019.1679698
Ramanathan, M., Porter, D. F., and Khavari, P. A. (2019). Methods to study RNA-protein interactions. Nat. Methods 16, 225–234. doi:10.1038/s41592-019-0330-1
Riley, K. J., Rabinowitz, G. S., Yario, T. A., Luna, J. M., Darnell, R. B., and Steitz, J. A. (2012). EBV and human microRNAs co-target oncogenic and apoptotic viral and human genes during latency. EMBO J. 31, 2207–2221. doi:10.1038/emboj.2012.63
Salmena, L., Poliseno, L., Tay, Y., Kats, L., and Pandolfi, P. P. (2011). A ceRNA hypothesis: The rosetta stone of a hidden RNA language? Cell. 146, 353–358. doi:10.1016/j.cell.2011.07.014
Schieweck, R., Ninkovic, J., and Kiebler, M. A. (2021). RNA-binding proteins balance brain function in health and disease. Physiol. Rev. 101, 1309–1370. doi:10.1152/physrev.00047.2019
Seetin, M. G., and Mathews, D. H. (2012). RNA structure prediction: An overview of methods. Methods Mol. Biol. 905, 99–122. doi:10.1007/978-1-61779-949-5_8
Seitz, H. (2009). Redefining microRNA targets. Curr. Biol. 19, 870–873. doi:10.1016/j.cub.2009.03.059
Singh, S., Shyamal, S., and Panda, A. C. (2022). Detecting RNA-RNA interactome. New Jersey, United States: Wiley Interdiscip. Rev. RNA, e1715.
Sun, L., Xu, K., Huang, W., Yang, Y. T., Li, P., Tang, L., et al. (2021). Predicting dynamic cellular protein-RNA interactions by deep learning using in vivo RNA structures. Cell Res. 31, 495–516.
Suresh, V., Liu, L., Adjeroh, D., and Zhou, X. (2015). RPI-pred: Predicting ncRNA-protein interaction using sequence and structural information. Nucleic Acids Res. 43, 1370–1379. doi:10.1093/nar/gkv020
Taliaferro, J. M., Lambert, N. J., Sudmant, P. H., Dominguez, D., Merkin, J. J., Alexis, M. S., et al. (2016). RNA sequence context effects measured in vitro predict in vivo protein binding and regulation. Mol. Cell. 64, 294–306. doi:10.1016/j.molcel.2016.08.035
Teng, X., Chen, X., Xue, H., Tang, Y., Zhang, P., Kang, Q., et al. (2019). NPInter v4.0: An integrated database of ncRNA interactions. Nucleic Acids Res. 48, D160–D165. doi:10.1093/nar/gkz969
Terribilini, M., Sander, J. D., Lee, J.-H., Zaback, P., Jernigan, R. L., Honavar, V., et al. (2007). RNABindR: A server for analyzing and predicting RNA-binding sites in proteins. Nucleic Acids Res. 35, W578–W584. doi:10.1093/nar/gkm294
Townshend, R. J. L., Eismann, S., Watkins, A. M., Rangan, R., Karelina, M., Das, R., et al. (2021). Geometric deep learning of RNA structure. Science 373, 1047–1051. doi:10.1126/science.abe5650
Turner, M., and Díaz-Muñoz, M. D. (2018). RNA-binding proteins control gene expression and cell fate in the immune system. Nat. Immunol. 19, 120–129. doi:10.1038/s41590-017-0028-4
Van Assche, E., Van Puyvelde, S., Vanderleyden, J., and Steenackers, H. P. (2015). RNA-binding proteins involved in post-transcriptional regulation in bacteria. Front. Microbiol. 6, 141 doi:10.3389/fmicb.2015.00141
Velankar, S., Burley, S. K., Kurisu, G., Hoch, J. C., and Markley, J. L. (2021). The protein data bank archive. Methods Mol. Biol. 2305, 3–21. doi:10.1007/978-1-0716-1406-8_1
Wang, D., Ye, R., Cai, Z., and Xue, Y. (2022). Emerging roles of RNA-RNA interactions in transcriptional regulation. New Jersey, United States: Wiley Interdiscip. Rev. RNA, e1712.
Wang, J., Zhao, Y., Gong, W., Liu, Y., Wang, M., Huang, X., et al. (2021). Edlmfc: An ensemble deep learning framework with multi-scale features combination for ncRNA–protein interaction prediction. BMC Bioinforma. 22, 133. doi:10.1186/s12859-021-04069-9
Wang, L., and Brown, S. J. (2006). BindN: A web-based tool for efficient prediction of DNA and RNA binding sites in amino acid sequences. Nucleic Acids Res. 34, W243–W248. doi:10.1093/nar/gkl298
Wang, L., Yan, X., Liu, M.-L., Song, K.-J., Sun, X.-F., and Pan, W.-W. (2019). Prediction of RNA-protein interactions by combining deep convolutional neural network with feature selection ensemble method. J. Theor. Biol. 461, 230–238. doi:10.1016/j.jtbi.2018.10.029
Wekesa, J. S., Meng, J., and Luan, Y. (2020). Multi-feature fusion for deep learning to predict plant lncRNA-protein interaction. Genomics 112, 2928–2936. doi:10.1016/j.ygeno.2020.05.005
Xie, G., Wu, C., Sun, Y., Fan, Z., and Liu, J. (2019). LPI-IBNRA: Long non-coding RNA-protein interaction prediction based on improved bipartite network recommender algorithm. Front. Genet. 10, 343. doi:10.3389/fgene.2019.00343
Yang, J., Li, A., Ge, M., and Wang, M. (2016). Relevance search for predicting lncRNA–protein interactions based on heterogeneous network. Neurocomputing 206, 81–88. doi:10.1016/j.neucom.2015.11.109
Yang, S., Wang, Y., Lin, Y., Shao, D., He, K., and Huang, L. (2020). LncMirNet: Predicting LncRNA–miRNA interaction based on deep learning of ribonucleic acid sequences. Molecules 25, 4372. doi:10.3390/molecules25194372
Yang, Y., Hou, Z., Ma, Z., Li, X., and Wong, K.-C. (2021). iCircRBP-DHN: identification of circRNA-RBP interaction sites using deep hierarchical network. Brief. Bioinform. 22, bbaa274. doi:10.1093/bib/bbaa274
Yi, H.-C., You, Z.-H., Huang, D.-S., Li, X., Jiang, T.-H., and Li, L.-P. (2018). A deep learning framework for robust and accurate prediction of ncRNA-protein interactions using evolutionary information. Mol. Ther. Nucleic Acids 11, 337–344. doi:10.1016/j.omtn.2018.03.001
Yi, H.-C., You, Z.-H., Cheng, L., Zhou, X., Jiang, T.-H., Li, X., et al. (2020). Learning distributed representations of RNA and protein sequences and its application for predicting lncRNA-protein interactions. Comput. Struct. Biotechnol. J. 18, 20–26. doi:10.1016/j.csbj.2019.11.004
Yu, X., Jiang, L., Jin, S., Zeng, X., and Liu, X. (2022). preMLI: a pre-trained method to uncover microRNA–lncRNA potential interactions. Brief. Bioinform. 23, bbab470. doi:10.1093/bib/bbab470
Yuan, J., Wu, W., Xie, C., Zhao, G., Zhao, Y., and Chen, R. (2014). NPInter v2.0: An updated database of ncRNA interactions. Nucleic Acids Res. 42, D104–D108. doi:10.1093/nar/gkt1057
Zang, J., Lu, D., and Xu, A. (2020). The interaction of circRNAs and RNA binding proteins: An important part of circRNA maintenance and function. J. Neurosci. Res. 98, 87–97. doi:10.1002/jnr.24356
Zhan, Z.-H., You, Z.-H., Li, L.-P., Zhou, Y., and Yi, H.-C. (2018). Accurate prediction of ncRNA-protein interactions from the integration of sequence and evolutionary information. Front. Genet. 9, 458. doi:10.3389/fgene.2018.00458
Zhang, K., Pan, X., Yang, Y., and Shen, H.-B. (2019). Crip: Predicting circRNA-RBP-binding sites using a codon-based encoding and hybrid deep neural networks. RNA 25, 1604–1615. doi:10.1261/rna.070565.119
Zhang, S.-W., Zhang, X.-X., Fan, X.-N., and Li, W.-N. (2020). LPI-CNNCP: Prediction of lncRNA-protein interactions by using convolutional neural network with the copy-padding trick. Anal. Biochem. 601, 113767. doi:10.1016/j.ab.2020.113767
Zhang, W., Qu, Q., Zhang, Y., and Wang, W. (2018). The linear neighborhood propagation method for predicting long non-coding RNA–protein interactions. Neurocomputing 273, 526–534. doi:10.1016/j.neucom.2017.07.065
Zhao, G., Li, P., Qiao, X., Han, X., and Liu, Z.-P. (2021). Predicting lncRNA-protein interactions by heterogenous network embedding. Front. Genet. 12, 814073. doi:10.3389/fgene.2021.814073
Zhao, Q., Yu, H., Ming, Z., Hu, H., Ren, G., and Liu, H. (2018). The bipartite network projection-recommended algorithm for predicting long non-coding RNA-protein interactions. Mol. Ther. Nucleic Acids 13, 464–471. doi:10.1016/j.omtn.2018.09.020
Zheng, X., Wang, Y., Tian, K., Zhou, J., Guan, J., Luo, L., et al. (2017). Fusing multiple protein-protein similarity networks to effectively predict lncRNA-protein interactions. BMC Bioinforma. 18, 420. doi:10.1186/s12859-017-1819-1
Zhou, L., Wang, Z., Tian, X., and Peng, L. (2021). LPI-deepGBDT: A multiple-layer deep framework based on gradient boosting decision trees for lncRNA–protein interaction identification. BMC Bioinforma. 22, 479. doi:10.1186/s12859-021-04399-8
Keywords: RNA, RNA interaction predictors, natural language processing, deep learning, machine learning, embedding, RNA sequence, RNA secondary structure
Citation: Pepe G, Appierdo R, Carrino C, Ballesio F, Helmer-Citterich M and Gherardini P (2022) Artificial intelligence methods enhance the discovery of RNA interactions. Front. Mol. Biosci. 9:1000205. doi: 10.3389/fmolb.2022.1000205
Received: 21 July 2022; Accepted: 20 September 2022;
Published: 07 October 2022.
Edited by:
Gian Gaetano Tartaglia, Italian Institute of Technology, ItalyReviewed by:
Zhi-Ping Liu, Shandong University, ChinaZilong Zhang, Hainan University, China
Alessio Colantoni, Italian Institute of Technology, Italy
Copyright © 2022 Pepe, Appierdo, Carrino, Ballesio, Helmer-Citterich and Gherardini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: G Pepe, Z2VyYXJkby5wZXBlQHVuaXJvbWEyLml0; M Helmer-Citterich, Y2l0dGVyaWNoQHVuaXJvbWEyLml0