 Joseph D. Kaszubowski
Joseph D. Kaszubowski Michael A. Trakselis
Michael A. Trakselis- Department of Chemistry and Biochemistry, Baylor University, Waco, TX, United States
High fidelity (HiFi) DNA polymerases (Pols) perform the bulk of DNA synthesis required to duplicate genomes in all forms of life. Their structural features, enzymatic mechanisms, and inherent properties are well-described over several decades of research. HiFi Pols are so accurate that they become stalled at sites of DNA damage or lesions that are not one of the four canonical DNA bases. Once stalled, the replisome becomes compromised and vulnerable to further DNA damage. One mechanism to relieve stalling is to recruit a translesion synthesis (TLS) Pol to rapidly synthesize over and past the damage. These TLS Pols have good specificities for the lesion but are less accurate when synthesizing opposite undamaged DNA, and so, mechanisms are needed to limit TLS Pol synthesis and recruit back a HiFi Pol to reestablish the replisome. The overall TLS process can be complicated with several cellular Pols, multifaceted protein contacts, and variable nucleotide incorporation kinetics all contributing to several discrete substitution (or template hand-off) steps. In this review, we highlight the mechanistic differences between distributive equilibrium exchange events and concerted contact-dependent switching by DNA Pols for insertion, extension, and resumption of high-fidelity synthesis beyond the lesion.
Introduction
DNA damage can come from a variety of endogenous and exogenous sources and persist within the genome into the synthesis (S) phase of the cell cycle (Hakem, 2008). During S-phase, high fidelity (HiFi) polymerases (Pols) duplicate complementary DNA strands with a rapid rate of synthesis and a low rate of error. However, upon encountering DNA base damage within a template strand, HiFi Pols are unable to continue DNA synthesis and are stalled in place. Specialized Pols capable of bypassing the template-strand lesion, known as translesion synthesis (TLS) Pols, are recruited to the site of the damage, substituted in for the HiFi Pol, and then used to synthesize over and past the lesion (Figure 1A). TLS Pols have evolved to ensure error-free synthesis across preferred lesions but have overall lower fidelity than HiFi Pols (Waters et al., 2009; Goodman and Woodgate, 2013; Sale, 2013; Fujii and Fuchs, 2020). Following successful insertion of one or more nucleotides opposite (or past) the lesion, TLS Pols must dissociate from the DNA template and substitute back to the replicative Pol to resume high fidelity genomic synthesis (Trakselis et al., 2017).
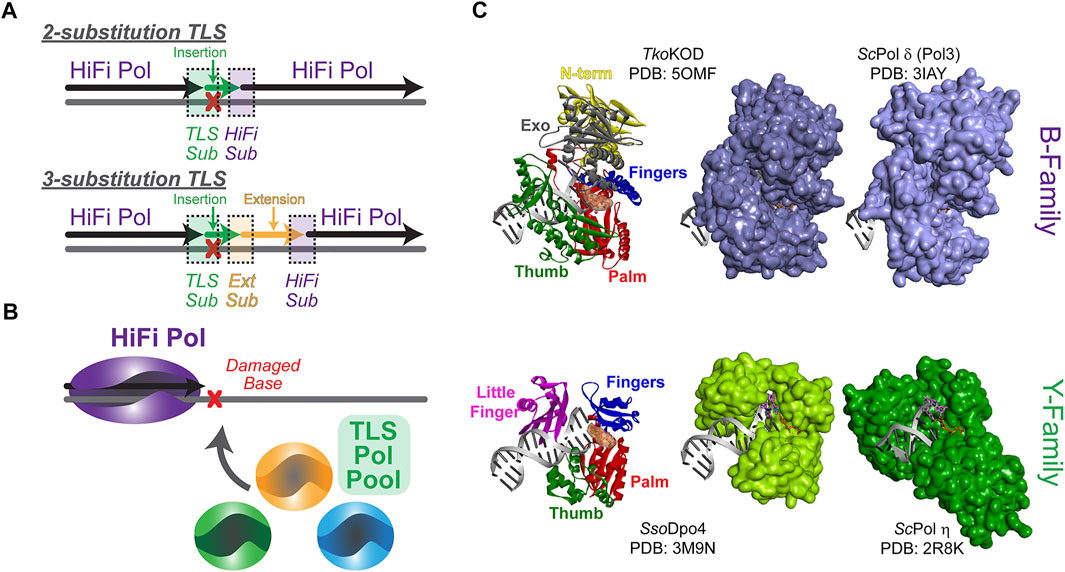
FIGURE 1. Steps required for the overall translesion synthesis (TLS) process. (A) A high fidelity (HiFi) polymerase (Pol) will stall at a lesion requiring two- or three-substitution TLS for bypass. In a two-substitution mechanism, the first TLS substitution (sub) will recruit an inserter TLS Pol (green) for translesion DNA insertion (green) followed by substitution back to a HiFi Pol (purple). In a three-substitution mechanism, an extender (Ext) TLS Pol is substituted after insertion and is required for limited downstream extension (orange) prior to substitution back to the HiFi Pol. (B) TLS substitution will be influenced by the available TLS Pol Pool as well as the specificity for a particular lesion. (C) Compares the polymerase domain structure (N-term—yellow, Exo—grey, Palm—red, Fingers—blue, Thumb—green, and Little Finger—pink) for the more closed structures of HiFi B-family Pols KOD (Thermococcus kodakarensis) (Kropp et al., 2017) and Pol δ (Saccharomyces cerevisiae) (Swan et al., 2009) with the more open active sites of TLS Y-family Pols SsoDpo4 (Wong et al., 2010) and Pol η (Saccharomyces cerevisiae) (Alt et al., 2007) in complex with DNA, all arranged in the same orientation.
Although relatively simple in description, there are many intricate elements of the TLS mechanism. Previously, TLS has been described by a “one-polymerase” or “two-polymerase” process; however, the number and subunit composition of Pols found at TLS sites is expanding, and while some Pols have direct synthesis activities, others act only as scaffolds for assemblies. Therefore, the diversity of TLS processes would be better defined by a “two-substitution” or “three-substitution” mechanism (Figure 1A), focusing primarily on the steps required for synthesis instead of individual Pols involved. Insertion opposite a lesion requires that an appropriately selected TLS “inserter” Pol is substituted for the HiFi Pol. After TLS insertion, there can be direct substitution back to the HiFi Pol to resume synthesis (i.e., two-substitution) or subsequently an intermediary “extender” Pol can synthesize downstream of the lesion prior to substitution back to the HiFi Pol in a three-substitution mechanism. There are several mechanistic facets that regulate this process to help select TLS Pols from the cellular pool (Figure 1B) and restrict their activities after TLS for the benefit of maintaining a stable genome. Although many of these basic principles have been better characterized in prokaryotic systems, where a more limited set of Pols are available, this review will focus primarily on defining the TLS process in more complex eukaryotic systems, culminating with mechanistic hypotheses for limiting lower fidelity TLS Pol synthesis downstream of the lesion to favor resumption of high-fidelity synthesis.
Translesion Synthesis Pol Open Active Site
All DNA Pols resemble a right hand, with a structure consisting minimally of palm, finger, and thumb domains, comprising the catalytic core (Figure 1C). This conformation allows for binding of the DNA substrate while simultaneously inserting nucleotides during DNA synthesis. HiFi Pols also include a second exonuclease (Exo) active site that is utilized to proofread insertions and ensure fidelity. While HiFi Pols of the B-family have smaller and more closed active sites to ensure efficient and accurate DNA replication opposite undamaged template strands, TLS Pols of the Y-family have larger and more open active sites that allow insertion of one or more nucleotides opposite a lesion. Y-family TLS Pols generally have small finger domains and have an additional little finger (LF) domain that attenuates lesion bypass abilities and processivities (Boudsocq et al., 2004; Pata, 2010). Varying distances between the LF and the catalytic core, creating a structural gap, add specificity to the size of lesions that each TLS Pol can bypass (Yang and Gao, 2018). As a result of these structural differences, TLS Pols are capable “inserters” opposite specific lesions but are inherently less accurate and can quickly generate errors in the nascent DNA strand when synthesizing opposite undamaged DNA (Vaisman and Woodgate, 2017). Although these Pols have tolerant active sites to accommodate bulky lesions, mutagenesis even during TLS is fairly common (Volkova et al., 2020). Therefore, it is paramount to cellular survival that synthesis by certain TLS Pols be restricted only to base insertion opposite DNA lesions to prevent misincorporations downstream. HiFi Pols with more discriminative active sites must promptly resume replicating DNA to minimize inaccurate synthesis. To facilitate the end of a TLS event, the DNA replisome needs to exchange or switch back to the HiFi Pol to proceed with faithful DNA replication.
Translesion Synthesis Pols and Lesion Specificity
DNA damage can lead to genomic instability and carcinogenesis (Hoeijmakers, 2009; Shilkin et al., 2020). TLS serves as a DNA damage tolerance mechanism by allowing DNA replication to persevere in the presence of DNA lesions. By allowing DNA synthesis to continue upon the encountering of a template-strand lesion, TLS Pols reduce the risk of cellular apoptosis and prevent occurrence of double-strand breaks from excessive replisome stalling (Chatterjee and Walker, 2017).
Before discussing the plethora of Pols capable of performing TLS, it is important to understand the types of lesions that are encountered as well as their sources. For DNA-damaging agents that have been prevalent for long periods of time, it is possible that TLS Pols have evolved to specifically bypass the resulting lesions (Livneh et al., 2010). For example, eukaryotic TLS Pol η is capable of perfectly bypassing thymine cyclobutane pyrimidine dimers (CPDs) following UV radiation from sunlight with correctly base-paired adenines (Johnson et al., 2000b; Biertumpfel et al., 2010) (Figure 1C). Separately, archaeal DNA polymerase 4 (Dpo4) can synthesize nascent DNA beyond 7,8-dihydro-8-oxoguanine (8-oxoG) lesions generated by reactive oxygen species, as well as many additional lesions (Yang, 2014; Cranford et al., 2020; Jung and Lee, 2020) (Figure 1C).
Relatively newer sources of exogenous damage, such as platinating agents used in cancer treatment, have not yielded an evolutionary adaptation in a specific TLS Pol and are instead bypassed by the most suitable (or a combination of multiple) Pol(s). In organisms with multiple available TLS Pols, specificity to lesions is a determining factor for which Pol will perform the bypass (Yang, 2014). Human cells contain four Y-family TLS Pols: Pols η, ι, κ, and Rev1, each with varying active site dimensions and unique bypass capabilities. Pols η, ι, and κ have all been shown to act as the “inserter” Pol opposite a variety of lesions, with the large active site of Pol κ allowing bypass of the bulkiest lesions (Vaisman et al., 2001; Kusumoto et al., 2002; Albertella et al., 2005a; Alt et al., 2007; Yoon et al., 2009; Phi et al., 2019; Jung et al., 2020; Vaisman and Woodgate, 2020; Ghodke et al., 2021). Human Pol ν, an A-family Pol, has also demonstrated an ability to act as an “inserter” TLS Pol opposite lesions (Takata et al., 2006; Gowda and Spratt, 2016; Du et al., 2020). While structurally similar to other Y-family Pols, Rev1 acts less frequently as an “inserter” and more often as a scaffold to facilitate the binding of other Pols through conserved Rev1-interacting regions (RIRs) (Qin et al., 2013; Boehm et al., 2016b; Pustovalova et al., 2016). Recent studies have shown that Pol λ, an X-family Pol, can also perform this scaffolding role during TLS (Yoon et al., 2021). The varying active site structures, activities, and potential contacts of TLS Pols regulate Pol selection for lesion bypass.
Polymerase Holoenzyme Complexes With PCNA
A polymerase holoenzyme complex is assembled to provide some stability when bound to DNA that presents biochemically as processivity. Processivity can be defined by an equation that quantifies the probability of a Pol continuing at a specific site based on the kpol for nucleotide incorporation relative to koff for dissociation from the DNA template (Hippel et al., 1994). Several mechanisms have evolved to increase processivities of HiFi Pols and limit processivities of TLS Pols to ensure genome stability. For Pols, the trimeric clamp protein (proliferating cell nuclear antigen, PCNA, in archaea and eukaryotes) encircles duplex DNA and provides a topological link to the DNA, effectively reducing koff (Trakselis and Benkovic, 2001; Boehm et al., 2016a).
The binding affinity of the Pol itself for DNA can also impact the processivity. Interestingly, HiFi Pols generally have a higher affinity for PCNA than DNA, while TLS Pols have a higher affinity for DNA than for PCNA (Lin et al., 2012; Hedglin et al., 2016a). This implies that binding to clamp proteins is often required for high fidelity and processive DNA synthesis, while TLS activity may be more restricted directly by the DNA lesion. This provides a plausible mechanism for limiting TLS, however, the mechanism for releasing a TLS Pol from DNA after translesion synthesis is not well understood.
Pols have several interaction sites with the clamp, but in eukaryotes and archaea, the primary interaction site is through a hydrophobic PCNA-Interacting Protein (PIP) patch, adjacent to the interdomain connecting loop within a single PCNA monomer (Figure 2). This PIP binding site is located on the front (or leading face) of PCNA and interacts with proteins through a specific motif in a partner protein containing the general consensus sequence, Qxxhxxaa, where h is hydrophobic and a is aromatic (Warbrick, 1998). Glutamine binds to a Q-pocket through specific hydrogen bonding contacts, while the aromatic residues lay within the hydrophobic pocket. Although more than 75 proteins, including several DNA repair proteins and cell cycle regulators, contain this consensus PIP motif, TLS Pols generally encode a nonconsensus PIP motif, (K/G)xx(I/L)xx(FY/L)(FY/L) that bypasses the Q-pocket (Slade, 2018; Dash and Hadden, 2021). One to three PIP motifs are located in the unstructured C-terminal extensions of Y-family Pols (Powers and Washington, 2018). The variability within the consensus and nonconsensus PIP motifs can alter the Kd of interaction widely from 10 nM to >30 µM (Slade, 2018; Prestel et al., 2019).
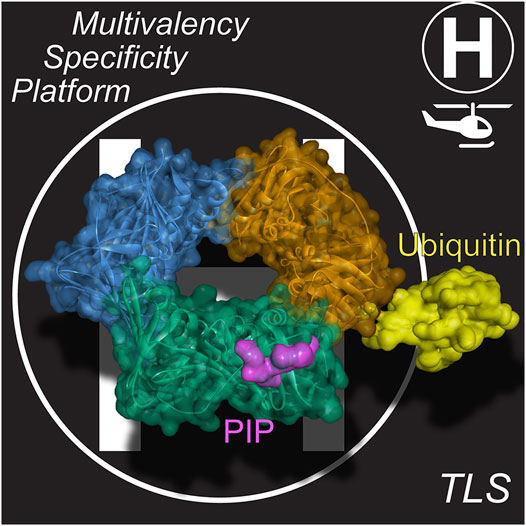
FIGURE 2. PCNA is a dynamic “landing-pad” (or platform). PIP binding sites at the interdomain connecting loop (IDL) and ubiquitination at K164 are available on each of the three PCNA subunits, providing for multivalency. Structural model derived from PCNA/p21 peptide structure (PDBID: 1AXC) (Gulbis et al., 1996) and monoubiquitinated PCNA structure (PDBID: 3TBL) (Zhang et al., 2012).
It is well known that upon extreme stalling, PCNA becomes monoubiquitinated (mUB) at K164 (Figure 2) (Stelter and Ulrich, 2003; Kannouche et al., 2004; Chang et al., 2006). This additional ubiquitin facet on PCNA can further increase the binding affinity of TLS Pols though a ubiquitin-binding zinc finger (UBZ) motif (Bomar et al., 2007; Pillaire et al., 2014) or a ubiquitin-binding motif (UBM) (McIntyre et al., 2015; Vanarotti et al., 2018) also within the unstructured C-terminal tails of Y-family TLS Pols (Yang and Gao, 2018). mUb-PCNA acts to not only recruit TLS Pols at stalled replication forks but also to increase their residence times to carry out TLS (Sabbioneda et al., 2008; Sabbioneda et al., 2009). The covalent addition of Ub to the back side of PCNAK164 and the flexibility inherent within the unstructured C-terminal tails containing these motifs allow these TLS Pols to adopt several conformations on PCNA in inactive carrier or active polymerizing states (Shen et al., 2021). Therefore, based on this multivalency for trimeric mUb-PCNA, there are now at least six specific contact points for recruiting and interacting with Pols, providing a landing platform for assemblies (Figure 2) in what has been described previously as a “tool-belt” model (Kath et al., 2014; Boehm et al., 2016b; Cranford et al., 2017). A holoenzyme complex, consisting of the synthesizing Pol bound to PCNA, or a supraholoenzyme complex, comprised of multiple Pols bound to PCNA, contain many protein-protein interactions (PPIs) for added stability during DNA synthesis.
Mechanisms for the Translesion Synthesis Switch or Exchange
Substitution of a HiFi Pol for a TLS Pol to hand off the primer opposite a damaged template is required to insert opposite a variety of template lesions, however, the exact mechanism for the polymerase exchange or switch is uncharacterized. Moreover, the diversity of lesion types and abundance of Pols within the cell would not suggest that a single mechanism is utilized. Instead, it is likely that several mechanisms are employed dependent on the specific lesion, the DNA context, and the signaling pathways. Conceptually, substitution of one Pol for another that relies primarily on kinetic modulation (i.e., koff) can be considered equilibrium exchange events, while those that are mediated through PPIs directly between Pols or through PCNA in a “tool-belt” or “landing-pad” fashion are switches (Figure 3). It is probable that aspects of both of these basic mechanisms are employed. For example, recruitment of a TLS Pol to a stalled HiFi holoenzyme may be brought in by specific contacts with PCNA (Figure 2) or combined with Pol-Pol interactions creating a transient supraholoenzyme to initiate a switch only to then have the HiFi Pol and/or PCNA dissociate in a subsequent exchange for insertion opposite a lesion.
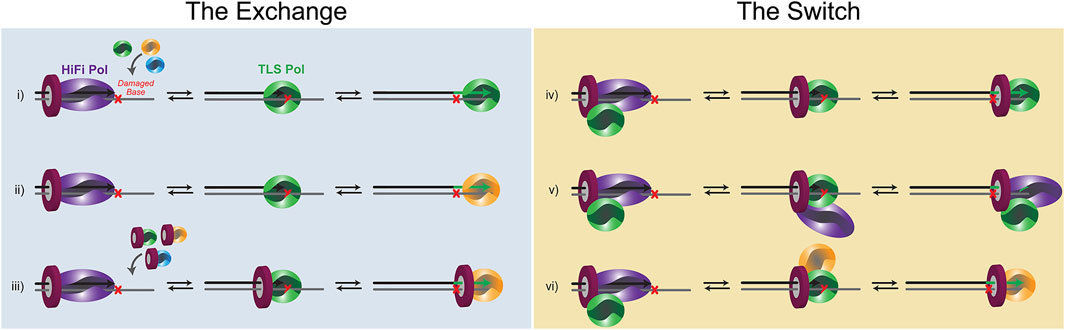
FIGURE 3. Schematic of the potential equilibrium exchanges and PCNA-directed switches that occur for TLS. (i) a distributive Pol exchange before two-substitution TLS. (ii) A distributive Pol exchange before three-substitution TLSs.(iii) a distributive PCNA/Pol exchange before two (or three)-substitution TLS. (iv) A concerted Pol switch with a pre-associated TLS Pol. (v) A concerted Pol switch with the HiFi Pol remaining bound to PCNA throughout TLS. (vi) Concerted Pol switches during three-substitution TLS.
A Distributive Mechanism of Polymerase “Exchange”
Stalling of a DNA Pol at a lesion or template-blocking event will render it catalytically inactive. In that case, kpol becomes extremely slow, and the koff event will predominate, dissociating the Pol from DNA. koff can be increased further by minor DNA distortions caused by the lesion itself (Broyde et al., 2008) or from shuttling between polymerase and exonuclease active sites within the HiFi Pol (Khare and Eckert, 2002). In this situation, multiequilibrium processes will predominate, and depending on the affinity of an exchanged Pol for the lesion and the concentration of Pols within the cell, a new TLS Pol would be selected (Figure 3). In the exchange model, very little, if any, contacts are retained between an incoming TLS Pol and either the HiFi Pol or PCNA. Instead, the template becomes accessible after a koff event, and a new TLS Pol can bind the damaged template for an insertion event. After insertion, the same TLS Pol can extend for several bases past the lesion, dependent on its own processivity and synthesis properties. Alternatively, another Pol substitution event can be utilized to synthesize past the lesion functioning as an “extender” Pol. Thus, the “hand-off” is indirect and is more consistent with an external equilibrium Pol active site exchange of the primer/template.
A Concerted Polymerase ‘Switching’ Mechanism
There is also evidence for several PPIs or contacts within a Pol holoenzyme. This occurs primarily through multivalency of binding to the trimeric PCNA clamp but can also be through direct Pol-Pol interactions. Several Pol interactions have been discovered between B- and Y-family Pols (designated YB sites) (Baldeck et al., 2015; Cranford et al., 2017). The key to a successful switch is that enzyme dissociation-association events from DNA are restricted in favor of pre-bound Pols releasing and swapping active-site binding to the damaged template (Figure 3). The affinity of TLS Pols for PCNA or the HiFi Pol itself effectively increases the local concentrations of TLS Pols at the replication fork, allowing for a more coordinated switch. To be clear, there can be several koff events involved in a switch. The first, and more traditional event, would be koff-DNA of the HiFi Pol from DNA, but in that case, localized binding can be retained by PCNA. Should the HiFi Pol dissociate from PCNA, a second koff-PCNA event can occur. Of course, further contacts such as koff-Pol between Pols (i.e., YB site) can also help mediate a switching event. In support, mutation of the YB site in the archaeal Saccharalobus solfataricus (Sso)Dpo4 enzyme results in decreased processivity of DNA synthesis suggesting that the supraholoenzyme complex is a more stable synthesizer (Cranford et al., 2017). Upon successful switching, the fate of the HiFi Pol is not always clear. It can dissociate from PCNA and the DNA template and be lost to solution, or it can be held in check within the supraholoenzyme complex. In a TLS switch, the “hand-off” is direct with contacts between Pols or coordinated through multivalent PCNA contacts facilitating primer/template binding to a preassociated Pol.
The kinetics of the entire TLS process from stall, substitution, insertion, and extension will be influential in determining whether an exchange or switch is more likely. For example, weaker PPI contacts and slower rates of insertion opposite more difficult lesions may favor a more distributive exchange of Pols from solution. However, stronger interactions and faster insertion kinetics may allow a concerted switch to be more likely, through the confines of a supraholoenzyme complex. Therefore, the PPI affinities, the equilibria concentrations of Pols, and the catalytic efficiency of an insertion (dependent on the lesion type) will be linked, making this highly dynamic process difficult to measure and accurately characterize.
Limitations for Biochemical Assays Designed to Examine Polymerase Substitutions
There are several inherent complexities for a biochemical experiment to fully examine Pol substitutions preceding, during, or after a TLS event. Typically, an in vitro dynamic biochemical kinetic characterization can be performed with only a limited and feasible number of biological components in a single experiment. Countless kinetic fidelity measurements have been made for many DNA Pols for even more DNA lesions in a single enzyme—single substrate experiment (Figure 4A), providing valuable quantitative understanding of lesion bypass and fidelity (Berdis, 2009; Liu et al., 2016; Powers and Washington, 2017; Raper et al., 2018). As the overall TLS process can include several proteins, numerous PPIs, differing kinetics, and variable equilibria, this experimental setup can be complicated. It is common to mutate the exonuclease proofreading site of the HiFi Pol to eliminate the degradation of the primer strand that complicates the extension kinetics, but the shuttling between Pol and Exo sites may be influential in directing the TLS process (Reha-Krantz, 2010). Moreover, it is also common to truncate TLS Pols to their core polymerase domain, removing important PIP/RIR/UBM/UBZ motifs in the unstructured C-terminal tails and making the examination of switching impossible.
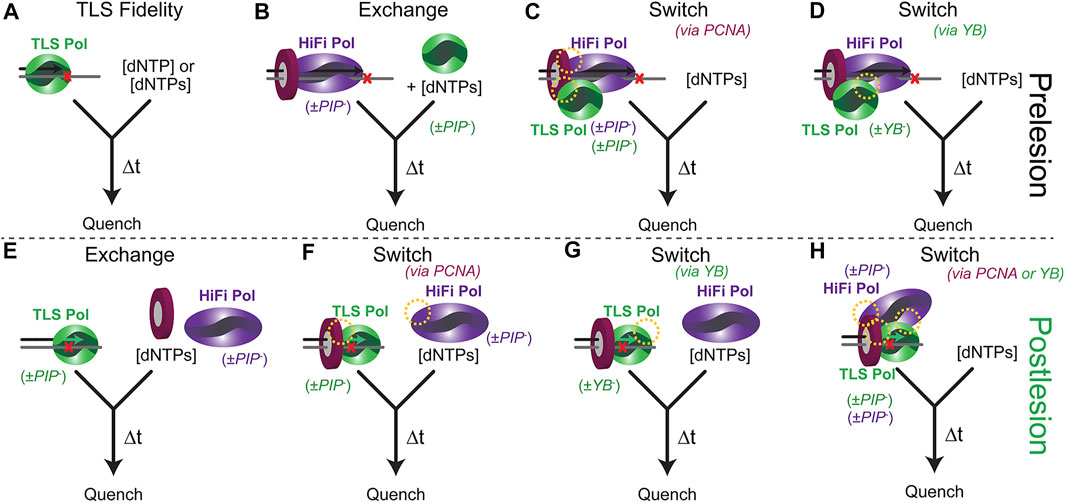
FIGURE 4. Experimental presteady-state schemes to examine the TLS process both pre and post lesion. (A) TLS Pol and various DNA template lesions to examine inherent fidelity in isolation. (B) Prebound HiFi Pol with PCNA stalled to examine a kinetic exchange process. Preassembled supraholoenzyme complex with HiFi and TLS Pols bound to DNA to examine kinetics of switching directed (C) by interactions with PCNA (PIP sites) or (D) through Pol-Pol interactions (YB site) (yellow circles). (E) A TLS Pol with a primer that is a defined number of bases past a lesion is used to examine a distributive exchange, (F) a PCNA directed switch, or (G) a YB directed switch. (H) The kinetics of a preassembled PCNA/TLS/HiFi Pol directed switch back to high fidelity synthesis can be validated by utilizing PIP− or YB− mutants of all Pols.
The presteady-state kinetics of TLS insertions and extensions can be monitored to better understand the impact of various exchange or switch processes using a rapid quench setup (Figures 4B–D). The experimental design is important as it impacts the desired measured outcome. For example, the kon for a TLS pol is usually diffusion-limited and rapid. However, if that is not the case then the method used to examine an exchange (Figure 4B) may be affected. Addition of a TLS Pol after a stalled HiFi Pol will simulate an incoming (or recruited) TLS Pol. The TLS Pol concentration can be varied to examine critical aspects of competitive binding exchange. Preincubation of HiFi and TLS Pols with PCNA and DNA can simulate and test a “tool-belt” model of concerted Pol switching. The kinetics of insertion and extension may vary depending on whether the TLS Pol is included initially or subsequently to help validate one particular model. The DNA primer length can also be altered to examine any kinetic differences of an approaching DNA Pol holoenzyme with a pre-stalled one in a “running start” assay. Catalytically deficient mutations (cat−) in either the HiFi or TLS Pol active sites can be used to distinguish extension products when multiple Pols are included. PPIs can be disrupted through site-specific mutations of the PIP (PIP−) or YB (YB−) sites for each Pol to test the impact of these contacts in facilitating a TLS switch. To fully understand the implications of Pol interactions with PCNA in a switch, full length, and not truncated core forms, of TLS Pols should be utilized.
After insertion, the stability and processivity of the TLS Pol can be examined. The design of these experiments will again test whether there is a second distributive exchange or facilitated switch to resume processive synthesis (Figures 4E–H). The DNA primer is systematically lengthened to determine whether there is a position of inherent destabilization of the TLS Pol that limits further extension. PCNA can be excluded or included with the TLS Pol (+/− Rev1) to initiate extension with mixing the HiFi Pol. The resulting product distributions and maximal kinetics of extension will inform on the preferred scheme. For eukaryotic systems, Ub can be covalently added to PCNA through several chemical biology approaches (Freudenthal et al., 2010; Hibbert and Sixma, 2012; Yang et al., 2016; Gong et al., 2018). Combined with UBM/UBZ mutations in the C-termini of TLS Pols, the role for mUb-PCNA in the TLS process can also be explored through the kinetics of extension.
Prelesion: Stalling of the High Fidelity Polymerase
Upon encountering a template lesion or a difficult to replicate region from secondary structure or physical impediments, the HiFi Pol will stall (Marians, 2018; Maiorano et al., 2021). The HiFi Pol will attempt insertion of a nucleotide opposite the lesion, however it is almost always unsuccessful (McCulloch and Kunkel, 2008). If insertion opposite a lesion is successful, then further extension is severely inhibited because of distortion in the duplex that is sensed, and the terminal nucleotide is removed at the exonuclease proofreading active site of the HiFi Pol. This stalling (or shuttling between active sites) will set in motion a series of events that act to either stabilize the replication fork for downstream repair or initiate TLS to rapidly continue (Aguilera and Gomez-Gonzalez, 2008; Marechal and Zou, 2013).
After stalling, the HiFi Pol will decouple from the replication helicase causing a buildup of single-strand DNA (ssDNA) (Byun et al., 2005; Chang et al., 2006; Guilliam and Yeeles, 2020). ssDNA alone is fragile and must be protected from breakage by RPA or Rad51 to prevent loss of genetic material and can be a checkpoint signal for repair through the ATR kinase signaling pathway (Cimprich and Cortez, 2008; Blackford and Jackson, 2017; Lanz et al., 2019). Transient stalls, where no direct DNA damage is present, may be relieved by a host of fork stabilizers, accessory factors, and restart proteins that can remove a physical block and allow synthesis and coupling to resume (Lopes et al., 2006; Rickman and Smogorzewska, 2019; Qiu et al., 2021). It has even been shown that the eukaryotic lagging strand Pol δ can efficiently substitute for a decoupled leading strand Pol ε when smaller lesions such as 8-oxoG or thymine glycol are present (Guilliam and Yeeles, 2021). However, if a template lesion is more severe and unsurpassable by the HiFi Pol, then other TLS Pols will be recruited to attempt bypass (Ma et al., 2020).
Prelesion: The First Translesion Synthesis Polymerase Substitution
After stalling of the HiFi Pol at a lesion, it must dissociate from the primer/template DNA through one of the above substitution mechanisms (Figure 3). The replicative Pol will either fully dissociate from the primer-template junction into solution, following a distributive exchange mechanism, or it will remain bound to PCNA in accordance with a concerted switching mechanism. For both the archaeal Pol B1 and eukaryotic Pol δ HiFi Pol holoenzyme systems, it was found that the HiFi Pol readily dissociates in a distributive exchange mechanism even during normal DNA synthesis (Bauer et al., 2013; Hedglin et al., 2016b). The lagging strand HiFi Pol in humans, Pol δ, has PIP-sites on three of its four subunits, however only mutation of the PIP-site in the large catalytic subunit significantly reduces Pol activity (Lancey et al., 2020). This potentiates multiple Pols being bound to other PCNA subunits simultaneously in a supraholoenzyme, or at least that multiple PCNA subunits can facilitate exchange of Pols (as a landing pad) during TLS. Aside from specific contacts with PCNA, there are several other factors regulating TLS Pol recruitment and selection prior to lesion bypass.
Translesion Synthesis Pol Recruitment and Selection
In mammalian cells, Pol δ is known to convert from a four-subunit to a three-subunit enzyme complex both upon S-phase entry (Zhang et al., 2013) and specifically in response to DNA damage (Zhang et al., 2007; Lee et al., 2014a). Degradation of the smallest subunit, p12, serves as a regulatory mechanism to activate Pol δ for synthesis normally and is further directed by ATR signaling (Zhang et al., 2007; Lee et al., 2019). From here, TLS Pol recruitment and selection can depend on a multitude of factors, including the aforementioned PIP or UBZ/UBM interactions with (Ub-)PCNA (Figure 2), Pol affinity for the damaged DNA substrate, and Pol-Pol interactions. Various types of DNA-damaging conditions have been shown to localize TLS Pols in replication foci in nuclei, indicating that Pol concentration is an early contributor of selection for TLS (Maiorano et al., 2021). Once TLS Pols are in the vicinity of the damage site, Pol affinity for the DNA substrate becomes a driver of Pol selection. Accommodation of lesions by TLS Pols can be influenced by template-strand sequence and steric hindrance between bulkier lesions and Pol active sites (Zhao and Washington, 2017; Thomforde et al., 2021). Sequence context has also been shown to affect Pol efficiency and accuracy during TLS opposite various lesions, however the molecular basis for this effect is not well understood (Shriber et al., 2015; Bacurio et al., 2021).
Despite these qualifications for TLS by certain Pols to occur, it should be noted that backup TLS pathways exist in eukaryotes. Certain lesions deter recruitment of some TLS Pols in favor of more suitable inserters, however in the absence of the favored Pol, a less-favored TLS Pol(s) can bypass these lesions. Secondary TLS mechanisms have been demonstrated both in vivo, through Pol knockdowns, and in vitro, by examining multiple TLS Pol bypass capabilities opposite an assortment of lesions (Yoon et al., 2009; Livneh et al., 2010; Jha and Ling, 2018; Inomata et al., 2021). The presence of backup TLS pathways indicates that steric effects may not fully regulate Pol selection, but likely contribute significantly.
Postlesion: Extension Past the Lesion, a Possible Second Substitution
After insertion of a base opposite a lesion, the TLS Pol can continue synthesizing and extending downstream until inherent enzymatic properties, PPIs, or multiequilibria processes allow for substitution of other Pols. TLS Pols have no exonuclease proofreading domains and lower fidelities compared to HiFi Pols (Figure 1C), and so, their lower processivities serve a vital function to limit further DNA synthesis past a lesion to maintain genome integrity. However, the mechanisms for extension and reestablishing the HiFi holoenzyme past a TLS event are not well studied.
Translesion Synthesis Polymerases: From Inserters to Extenders
Beyond the lesion, “inserter” TLS Pols extend the nascent DNA strand to a position where either an “extender” TLS Pol, such as eukaryotic Pol ζ, can continue TLS or where the HiFi Pol can resume synthesis. A TLS mechanism in which an “inserter” Pol is the only Pol required for TLS can be termed “two-substitution” TLS. In the event both an “inserter” and “extender” Pol is required for TLS, the term “three-substitution” TLS can be used. The vocabulary of “substitution” is preferred over other vague describers including “one-“ or “two-polymerase” TLS (Johnson et al., 2000a) to focus more on the mechanistic process. With Rev1 potentially being involved as a scaffold in both two- and three-substitution mechanisms, defining the TLS by the number of Pols may be inaccurate and confusing. Therefore, nomenclature referring to the number of “substitutions” is preferred. Pol ζ has been shown to function as the primary “extender” Pol after the “inserter” in a “three-substitution” TLS process for certain lesions (Shachar et al., 2009; Lee Y.-S. et al., 2014). A-family Pol θ has also displayed some “extender” activity, although its exact role is still not fully defined (Seki et al., 2004; Bacurio et al., 2021). Recently, Sulfolobus islandicus B-family polymerase Dpo2 has been shown to act as the “extender” Pol in combination with “inserter” Pol Dpo4 during bypass of abasic sites (Feng et al., 2021).
Pol ζ Recruitment and Assembly
Assembly of the five-subunit extender Pol ζ is facilitated by HiFi Pol δ and the Y-family Rev1. Although the precise timing of this mechanism is not clear, Pol δ can rearrange and share subunits with Pol ζ to form the active Pol ζ complex. Pol δ shares subunits Pol31 and Pol32 in yeast and subunits p50 (Polδ2) and p66 (Polδ3) in humans with Pol ζ (Baranovskiy et al., 2012; Malik et al., 2020). Aside from the shared subunits, Pol ζ is comprised of Rev3, the catalytic subunit, simultaneously bound to two Rev7 subunits (Rizzo et al., 2018). The entire Pol ζ complex interacts in a 1:1 ratio with the scaffold TLS Pol Rev1. Rev1 is presumably bound to the replisome during an initial response to DNA damage and is soon thereafter able to recruit Pol ζ to the primer/template junction, coordinating the inserter-to-extender three-substitution mechanism (Makarova and Burgers, 2015; Liu et al., 2018). In a two-substitution mechanism, such as Pol η bypass of a UV-induced CPD, there is no requirement for an extender Pol. Under such circumstances, Pol ζ recruitment and assembly would likely not occur, even though the initial DNA damage response may generate mUb-PCNA and recruit Rev1. Interestingly, when Pol η is absent, Pol ζ functions in subsequence to Pol ι or Pol κ in TLS opposite this exact lesion (Ziv et al., 2009). The ability of the extender Pol ζ to function in a backup TLS mechanism when it is not required in the primary bypass mechanism for a certain lesion indicates that Pol ζ recruitment and assembly may always occur in response to DNA damage. Another possibility is that Pol ζ assembly does not occur until after the “inserter” Pol has performed the first step of TLS, at which time synthesis is stalled until a suitable “extender” can be recruited to the DNA substrate. Further experimentation needs to be performed to ascertain the exact timing of Pol ζ recruitment and assembly.
Postlesion: Resuming High Fidelity Synthesis, a Final Substitution
While most TLS studies have focused on the capability of TLS Pols to insert nucleotides opposite a template-strand DNA lesion, less focus has been placed on resumption of high-fidelity synthesis beyond the lesion. As TLS Pols are often inaccurate and less processive for synthesis opposite an undamaged DNA template, they are limited in their extension capabilities after lesion bypass (Vaisman and Woodgate, 2017). Limiting extension by TLS Pols requires a mechanism for substitution back to a HiFi Pol. The length of extension beyond the lesion by TLS Pols can vary, and likely has to do with the native processivity of the TLS Pol on DNA templates (Figure 5A), additional Pols in the vicinity of the primer/template junction (Figure 5B), the contacts that the TLS Pol active site residues make with the DNA substrate (Figure 5C), and possibly even structural impediments to extension (Figure 5D). After TLS has been completed, the TLS Pols extend to a position beyond the lesion, which is now outside of the Pol active site. At this point, structural characteristics of the TLS Pol can cause dissociation from the DNA substrate and initiate a substitution back to the HiFi Pol.
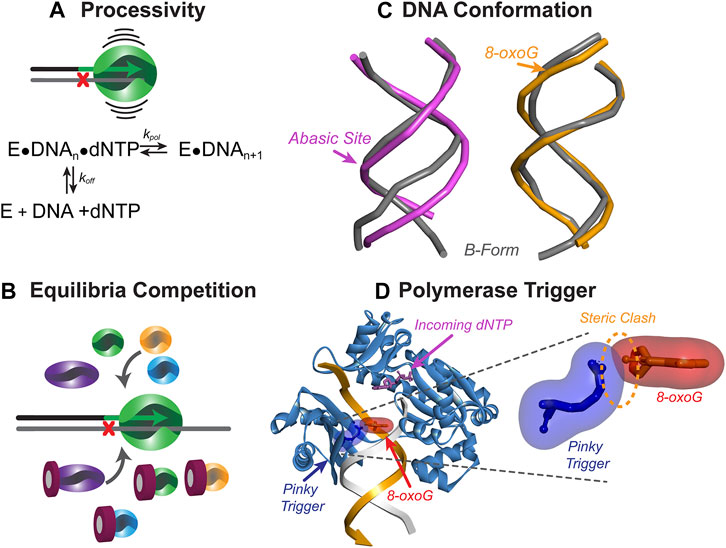
FIGURE 5. Mechanisms to initiate resumption of high-fidelity synthesis. (A) The processivity of a Pol represents the number of nucleotides inserted before dissociating from DNA and calculated as a ratio of kpol to koff. TLS Pols have lower processivity than HiFi Pols and can readily dissociate from DNA when the bypassed lesion is no longer in the active site. (B) DNA-damaging events can cause upregulation of TLS Pols, leading to concentration-dependent competition between Pols in solution for access to the DNA substrate. (C) DNA lesions can alter the conformation of the DNA, making extension of the nascent strand more difficult for the synthesizing Pol. Abasic site duplex (pink, PDB: 2HSS (Chen et al., 2007)) show more distortion than 8-oxo-G duplex (orange, PDB: 5IZP (Hoppins et al., 2016)) compared to B-form DNA (grey). Arrow shows the position of the lesion (D) Template-strand lesions are bypassed by TLS Pols, however as the lesion is exiting the active site, a steric clash may occur between a specific amino acid residue (i.e., Pinky Trigger) and the damaged template base to destabilize binding and further extension. Shown is the ternary SsoDpo4 complex (PDB:ID 1JX4) with oxygen modelled in the 8-position of the guanine in the −3 position on the primer strand.
Inherent Translesion Synthesis Pol Processivity
A primary reason for why TLS Pol synthesis is limited is due to low processivity on undamaged substrates (Figure 5A). TLS Pols have the structural features to allow bulky lesions into the active site (Figure 1C), but they dissociate readily from undamaged DNA (Raper et al., 2016). The large, tolerant active sites of TLS Pols are optimal for TLS but make the TLS Pol inherently less stable, less processive, and more mutagenic on undamaged substrates. “Extender” Pols have slightly increased processivity compared to “inserter” Pols but remain limited in synthesis capability once far enough downstream from the lesion. Recent structural studies have identified the position of the N-terminal domain-palm domain linker in the catalytic subunit of human Pol ζ as paramount in allowing the extender to tolerate distorted templates (Malik et al., 2020). Beyond active-site recognition of the DNA damage, Pol ζ processivity is greatly reduced. This finding is supported in vivo by examination of human Pol ζ mutational signatures in extension past damaged DNA. Recently, human Pol ζ was found to extend the nascent strand of DNA roughly 30 nucleotides from BPDE-induced damage (Table 1) (Suzuki et al., 2021). The mutational frequency data also indicated that Pol ζ may be recruited multiple times during extension, even after Pol δ has performed some synthesis. Therefore, further research needs to be performed to confirm the exact mechanism of limiting “extender” Pols in TLS. Adding to this complexity are the shared subunits (PolD2 and D3) between replicative Pol δ and extender Pol ζ. It also remains unclear the exact position of the exchange or switch back following Pol ζ extension, and whether or not this position is the same for extension past all types of DNA lesions.

TABLE 1. The position of substitution back to high fidelity synthesis.
Direct contacts between the TLS Pol active site and the damaged DNA template also contribute to the position of the hand-off to a replicative Pol. Likely contributing to processivity, the ability of a TLS Pol to interact with the lesion as it leaves the active site can dictate how many additional nucleotides the Pol will synthesize before dissociating from DNA. For certain Pols, such as human Pol η and its bypass of UV-induced TT-dimers, the TLS mechanism has been thoroughly examined. Crystal structures of Pol η in complex with CPD-containing DNA have shown that the Pol η active site can easily accommodate two template-strand nucleotides (Biertumpfel et al., 2010). Having the dimer within the Pol η active site is crucial to maintaining interaction with the DNA and allows for efficient TLS across dimerized lesions in eukaryotes (Prakash and Prakash, 2002). During bypass of a CPD, it has been shown that Pol η inserts nucleotides opposite the dimer, extends two positions beyond the lesion, and then destabilizes from DNA (Kusumoto et al., 2004; McCulloch et al., 2004). When Pol η is three nucleotides from the template-strand dimer it becomes deprived of these stabilizing contacts (Biertumpfel et al., 2010). This correlates with the position at which the HiFi Pol is able to resume synthesis after TLS. For bypass of this particular DNA lesion in humans, an “extender” Pol is not necessary, confirming +2 nucleotides past the lesion as the position for the HiFi substitution (Table 1).
Equilibrium Competition for DNA Binding
The concentrations of TLS Pols are generally kept lower than HiFi Pols to prevent their equilibrium association and low fidelity synthesis. In fact, altered expression of the TLS Pols have been linked with increased mutation rates (Pavlov et al., 2001; Qi et al., 2012; Sasatani et al., 2017), correlated with various cancers (O-Wang et al., 2001; Albertella et al., 2005b; Flanagan et al., 2007; Wang et al., 2010), and give rise to chemotherapeutic resistance (Saha et al., 2021). There is evidence that TLS Pols are cell cycle regulated under normal conditions, peaking in G2 phase to facilitate any required TLS prior to cell division (Waters and Walker, 2006; Plachta et al., 2015; Sobolewska et al., 2020). Even reorganization of Pol δ in eukaryotes from a three to four subunit enzyme (Zhang et al., 2007; Lee et al., 2019) or exchanging with Rev3/7 to form Pol ζ (Rizzo et al., 2018) can impact activity. The return of the p12 subunit to reestablish a four subunit Pol δ is influential in providing greater processivity and strand displacement activity possibly reserved for other DNA repair pathways (Meng et al., 2010; Lin et al., 2013).
Several eukaryotic TLS Pols, including Pol η (Tomicic et al., 2014), Rev1 (Uchiyama et al., 2015), and Pol κ (Velasco-Miguel et al., 2003) are upregulated after specific DNA damage, however the prototypical upregulation of TLS Pols occurs during the SOS response in bacteria to overcome substantial DNA damage and induce DNA mutagenesis for survival (Kenyon and Walker, 1980; Napolitano et al., 2000; Yeiser et al., 2002). Deubiquitination of PCNA may also serve to recruit HiFi Pols back after insertion/extension events, although this may be more influential in the yeast system (Zhuang et al., 2008) compared to mammalian systems (Niimi et al., 2008; Brown et al., 2009). Therefore, with all of these potential expression changes occurring normally during the cell cycle and more specifically in response to DNA damage and in cells with multiple TLS Pols, it is likely that even small changes in the cellular multiequilibrium will affect Pol binding and selection (Figure 5B).
Altered DNA Conformation
Some TLS Pols have active sites evolved for specific lesions (Sale et al., 2012; Sale, 2013) either to recognize the lesion itself or the alternative conformation of the primer-template DNA duplex induced by the lesion. Of the most common lesions, 8-oxoG does not induce significant structural duplex distortion (Plum et al., 1995), unlike that for an abasic site (Bignon et al., 2016) (Figure 5C). Other lesions such as benzopyrene dG adducts (Menzies et al., 2015), CPDs (Mcateer et al., 1998), or cis-Pt (Hartog et al., 1983) also induce major structural distortions (Pages and Fuchs, 2002; Lukin and De los Santos, 2006; Zhao et al., 2012). After insertion of a base across these lesions, the “inserter” Pol will move to the +1 position. From there, the “inserter” or “extender” will continue to synthesize downstream of the lesion. The structural perturbations that these Pols encounter past the lesions has not been studied in great detail and provide a plausible model for binding destabilization that limits further extension. Interestingly, the extender Pol ζ includes structural features to tolerate lesion-distorted DNA upstream (Malik et al., 2020).
Steric Impediments to Extension
While the aforementioned contacts with DNA stabilize the Pol during TLS, some interactions between the Pol active site and the lesion can promote dissociation from the substrate. Recent studies on the bypass of 8-oxoG lesions by SsoDpo4 have indicated that this Pol is able to insert a nucleotide opposite the lesion and extend three bases beyond the lesion before becoming catalytically inefficient (Table 1) (Cranford et al., 2020). This coincided with the exact position (+3) that the replicative Pol B1 is capable of reengaging and efficiently extending the primer to resume high fidelity synthesis. Upon structural examination of Dpo4 in complex with DNA, specific residues of the little finger domain may be directly clashing with the 8-oxoG lesion as it exits the active site. This hypothetical “pinky trigger” may dictate the position where HiFi synthesis resumes for the archaeal system (Figure 5D). The ability of TLS Pols to sense the template lesion outside of the active site may destabilize binding of the TLS Pol downstream and provide an opportunity for a HiFi Pol to resume synthesis.
Therapeutic Strategies Targeting TLS Steps
Polymerase inhibitors have become part of the ever-growing list of chemotherapeutics. As accurate and complete replication of DNA is vital to cellular growth and viability, inhibition of this process can block growth and promote apoptosis of aberrant cells. By specifically targeting cancer cells with molecules designed to impede DNA replication, one could limit tumor growth. Chain-terminating nucleoside analogs are among the most popular in this class of therapeutics (Berdis, 2017). These nucleotide mimics are inserted by Pols to a nascent strand of DNA during replication and promptly conclude extension of that strand. The potential impact of these chain terminators can be escalated when combined with DNA-damaging agents. TLS Pols are known to bypass lesions generated by platinum chemotherapeutics (Albertella et al., 2005a) and are upregulated in multiple cancer cell lines (Tomicic et al., 2014). The overzealous activity of these TLS Pols desensitizes cancer cells to chemotherapeutics by enabling cell survival amid DNA-damaging conditions. By inhibiting these TLS Pols and preventing insertions opposite chemotherapy-induced lesions or other steps in the TLS process, the efficacy of the treatment is amplified overall.
In accordance with this principle, alternate methods of TLS Pol inhibition would suffice for ameliorated chemotherapy. Pol-Pol and PCNA-Pol contacts that are involved in TLS Pol recruitment, stabilization, and substitutions make ideal targets for cancer drug research (Altieri and Kelman, 2018). TLS in humans across cisplatin-induced intrastrand crosslinks is a three-substitution mechanism requiring Pol η as the inserter, Pol ζ as the extender, and Rev1 as a scaffold. Recently, JH-RE-06, a small molecule inhibitor of this bypass, has been shown to dimerize Rev1 and prevent Pol ζ from binding (Wojtaszek et al., 2019). Without an efficient extender, TLS opposite Pt-GG lesions is reduced, and cisplatin becomes more effective at counteracting tumors. JH-RE-06, along with other small molecule inhibitors of protein-protein interactions, serve to block a specific element of TLS mechanisms in order to minimize obstructions to DNA-damaging agents (Wojtaszek et al., 2019; Dash and Hadden, 2021).
Discussion
All of the examples described in the Postlesion: Resuming High Fidelity Synthesis, a Final Substitution section are mechanisms used to limit synthesis by a TLS Pol after insertion to maintain downstream genome fidelity. The same structural features of TLS Pols that allow bulky lesions to enter the Pol active site are the features that make TLS Pols inaccurate and unstable on undamaged DNA substrates. TLS Pols are more mutagenic on undamaged templates than on suitable damaged substrates; so strict regulation outside of a specific TLS insertion is necessary. Thus, TLS Pols must insert nucleotides opposite the lesion, extend beyond the lesion, and then hand the DNA back to a HiFi Pol at a position that is both favorable to TLS Pol dissociation and subsequent HiFi Pol extension. Integral to the positioning of the resumption of HiFi synthesis are direct Pol-DNA, Pol-PCNA, and Pol-Pol interactions. Pol-Pol binding has been shown to stabilize the replicating Pol throughout TLS (Cranford et al., 2017). Pol-PCNA contacts through PIP motifs, UBZ/UBM contacts, and Rev1 bridges are also known to improve Pol stability and processivity (Boehm et al., 2016b; Acharya et al., 2020). These precise interactions that provide stability during TLS may also be involved in regulation of steps after TLS insertion. Further experimentation needs to be performed to identify the exact substitution mechanism(s) and steps that occur during lesion bypass.
Many in vitro experiments examine the TLS lesion bypass capability of truncated core TLS Pols in isolation. However, these absent regions are precisely the ones that facilitate Pol-Pol and PCNA-Pol binding and are needed to examine the entire TLS process. Full-length TLS and HiFi Pols, in the presence of PCNA and a suitable “extender” Pol will allow for interactions that may influence extension beyond the lesion, thus providing confidence in the kinetic characterizations required to determine a position where high fidelity synthesis resumes. Ultimately, translesion synthesis needs to be rapid, but distributive, to limit successive nucleotide incorporations so that replicative Pols can resume high fidelity synthesis and recouple with the replisome. The entire TLS mechanism, from the initial HiFi Pol stalling, “inserter” Pol recruitment, and TLS Pol(s) selection to the second (and possibly third) Pol substitutions to an “extender” Pol or HiFi Pol, respectively, remains to be fully understood. Location of the substitution positions and identification of the kinetically favored TLS mechanisms for bypass of DNA damage has direct applications in drug discovery. Future experiments should seek to expand our understanding of the complete TLS process to identify additional therapeutic targets within TLS.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
Work in the Trakselis laboratory is generously supported by the NSF (MCB 2105167 and CHE 2104242 to MT), the NIH R15 (GM13791 to MT), and Baylor University.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Acharya, N., Patel, S. K., Sahu, S. R., and Kumari, P. (2020). 'PIPs' in DNA Polymerase: PCNA Interaction Affairs. Biochem. Soc. Trans. 48, 2811–2822. doi:10.1042/bst20200678
Aguilera, A., and Gómez-González, B. (2008). Genome Instability: A Mechanistic View of its Causes and Consequences. Nat. Rev. Genet. 9, 204–217. doi:10.1038/nrg2268
Albertella, M. R., Green, C. M., Lehmann, A. R., and O'connor, M. J. (2005a). A Role for Polymerase η in the Cellular Tolerance to Cisplatin-Induced Damage. Cancer Res. 65, 9799–9806. doi:10.1158/0008-5472.can-05-1095
Albertella, M. R., Lau, A., and O’Connor, M. J. (2005b). The Overexpression of Specialized DNA Polymerases in Cancer. DNA Repair 4, 583–593. doi:10.1016/j.dnarep.2005.01.005
Alt, A., Lammens, K., Chiocchini, C., Lammens, A., Pieck, J. C., Kuch, D., et al. (2007). Bypass of DNA Lesions Generated during Anticancer Treatment with Cisplatin by DNA Polymerase η. Science 318, 967–970. doi:10.1126/science.1148242
Altieri, A. S., and Kelman, Z. (2018). DNA Sliding Clamps as Therapeutic Targets. Front. Mol. Biosci. 5, 87. doi:10.3389/fmolb.2018.00087
Bacurio, J. H. T., Yang, H., Naldiga, S., Powell, B. V., Ryan, B. J., Freudenthal, B. D., et al. (2021). Sequence Context Effects of Replication of FapydG in Three Mutational Hot Spot Sequences of the P53 Gene in Human Cells. DNA Repair 108, 103213. doi:10.1016/j.dnarep.2021.103213
Baldeck, N., Janel-Bintz, R., Wagner, J., Tissier, A., Fuchs, R. P., Burkovics, P., et al. (2015). FF483-484 Motif of Human Polη Mediates its Interaction with the POLD2 Subunit of Polδ and Contributes to DNA Damage Tolerance. Nucleic Acids Res. 43, 2116–2125. doi:10.1093/nar/gkv076
Baranovskiy, A. G., Lada, A. G., Siebler, H. M., Zhang, Y., Pavlov, Y. I., and Tahirov, T. H. (2012). DNA Polymerase δ and ζ Switch by Sharing Accessory Subunits of DNA Polymerase δ. J. Biol. Chem. 287, 17281–17287. doi:10.1074/jbc.m112.351122
Bauer, R. J., Wolff, I. D., Zuo, X., Lin, H.-K., and Trakselis, M. A. (2013). Assembly and Distributive Action of an Archaeal DNA Polymerase Holoenzyme. J. Mol. Biol. 425, 4820–4836. doi:10.1016/j.jmb.2013.09.003
Berdis, A. J. (2017). Inhibiting DNA Polymerases as a Therapeutic Intervention against Cancer. Front. Mol. Biosci. 4, 78. doi:10.3389/fmolb.2017.00078
Berdis, A. J. (2009). Mechanisms of DNA Polymerases. Chem. Rev. 109, 2862–2879. doi:10.1021/cr800530b
Biertümpfel, C., Zhao, Y., Kondo, Y., Ramón-Maiques, S., Gregory, M., Lee, J. Y., et al. (2010). Structure and Mechanism of Human DNA Polymerase η. Nature 465, 1044–1048. doi:10.1038/nature09196
Bignon, E., Gattuso, H., Morell, C., Dehez, F., Georgakilas, A. G., Monari, A., et al. (2016). Correlation of Bistranded Clustered Abasic DNA Lesion Processing with Structural and Dynamic DNA helix Distortion. Nucleic Acids Res. 44, 8588–8599. doi:10.1093/nar/gkw773
Blackford, A. N., and Jackson, S. P. (2017). ATM, ATR, and DNA-PK: The trinity at the Heart of the DNA Damage Response. Mol. Cel 66, 801–817. doi:10.1016/j.molcel.2017.05.015
Boehm, E. M., Gildenberg, M. S., and Washington, M. T. (2016a). The many Roles of PCNA in Eukaryotic DNA Replication. Enzymes 39, 231–254. doi:10.1016/bs.enz.2016.03.003
Boehm, E. M., Spies, M., and Washington, M. T. (2016b). PCNA Tool Belts and Polymerase Bridges Form during Translesion Synthesis. Nucleic Acids Res. 44, 8250–8260. doi:10.1093/nar/gkw563
Bomar, M. G., Pai, M. T., Tzeng, S. R., Li, S. S. C., and Zhou, P. (2007). Structure of the Ubiquitin‐Binding Zinc finger Domain of Human DNA Y‐Polymerase η. EMBO Rep. 8, 247–251. doi:10.1038/sj.embor.7400901
Boudsocq, F., Kokoska, R. J., Plosky, B. S., Vaisman, A., Ling, H., Kunkel, T. A., et al. (2004). Investigating the Role of the Little Finger Domain of Y-Family DNA Polymerases in Low Fidelity Synthesis and Translesion Replication. J. Biol. Chem. 279, 32932–32940. doi:10.1074/jbc.m405249200
Brown, S., Niimi, A., and Lehmann, A. R. (2009). Ubiquitination and Deubiquitination of PCNA in Response to Stalling of the Replication Fork. Cell Cycle 8, 689–692. doi:10.4161/cc.8.5.7707
Broyde, S., Wang, L., Rechkoblit, O., Geacintov, N. E., and Patel, D. J. (2008). Lesion Processing: High-Fidelity Versus Lesion-Bypass DNA Polymerases. Trends Biochem. Sci. 33, 209–219. doi:10.1016/j.tibs.2008.02.004
Byun, T. S., Pacek, M., Yee, M.-c., Walter, J. C., and Cimprich, K. A. (2005). Functional Uncoupling of MCM Helicase and DNA Polymerase Activities Activates the ATR-Dependent Checkpoint. Genes Dev. 19, 1040–1052. doi:10.1101/gad.1301205
Chang, D. J., Lupardus, P. J., and Cimprich, K. A. (2006). Monoubiquitination of Proliferating Cell Nuclear Antigen Induced by Stalled Replication Requires Uncoupling of DNA Polymerase and Mini-Chromosome Maintenance Helicase Activities. J. Biol. Chem. 281, 32081–32088. doi:10.1074/jbc.m606799200
Chatterjee, N., and Walker, G. C. (2017). Mechanisms of DNA Damage, Repair, and Mutagenesis. Environ. Mol. Mutagen. 58, 235–263. doi:10.1002/em.22087
Chen, J., Dupradeau, F.-Y., Case, D. A., Turner, C. J., and Stubbe, J. (2007). Nuclear Magnetic Resonance Structural Studies and Molecular Modeling of Duplex DNA Containing Normal and 4'-Oxidized Abasic Sites,. Biochemistry 46, 3096–3107. doi:10.1021/bi6024269
Cimprich, K. A., and Cortez, D. (2008). ATR: An Essential Regulator of Genome Integrity. Nat. Rev. Mol. Cel Biol. 9, 616–627. doi:10.1038/nrm2450
Cranford, M. T., Chu, A. M., Baguley, J. K., Bauer, R. J., and Trakselis, M. A. (2017). Characterization of a Coupled DNA Replication and Translesion Synthesis Polymerase Supraholoenzyme from Archaea. Nucleic Acids Res. 45, 8329–8340. doi:10.1093/nar/gkx539
Cranford, M. T., Kaszubowski, J. D., and Trakselis, M. A. (2020). A Hand-Off of DNA between Archaeal Polymerases Allows High-Fidelity Replication to Resume at a Discrete Intermediate Three Bases Past 8-Oxoguanine. Nucleic Acids Res. 48, 10986–10997. doi:10.1093/nar/gkaa803
Dash, R. C., and Hadden, K. (2021). Protein-Protein Interactions in Translesion Synthesis. Molecules 26, 5544. doi:10.3390/molecules26185544
Du, H., Wang, P., Wu, J., He, X., and Wang, Y. (2020). The Roles of Polymerases ν and θ in Replicative Bypass of O6- and N2-Alkyl-2′-Deoxyguanosine Lesions in Human Cells. J. Biol. Chem. 295, 4556–4562. doi:10.1074/jbc.ra120.012830
Feng, X., Zhang, B., Xu, R., Gao, Z., Liu, X., Yuan, G., et al. (2021). Enzymatic Switching Between Archaeal DNA Polymerases Facilitates Abasic Site Bypass. Front. Microbiol. 12, 802670. doi:10.3389/fmicb.2021.802670
Flanagan, A., Rafferty, G., O'neill, A., Rynne, L., Kelly, J., Mccann, J., et al. (2007). The Human POLH Gene Is Not Mutated, and Is Expressed in a Cohort of Patients with Basal or Squamous Cell Carcinoma of the Skin. Int. J. Mol. Med. 19, 589–596. doi:10.3892/ijmm.19.4.589
Freudenthal, B. D., Gakhar, L., Ramaswamy, S., and Washington, M. T. (2010). Structure of Monoubiquitinated PCNA and Implications for Translesion Synthesis and DNA Polymerase Exchange. Nat. Struct. Mol. Biol. 17, 479–484. doi:10.1038/nsmb.1776
Fujii, S., and Fuchs, R. P. (2020). A Comprehensive View of Translesion Synthesis in Escherichia C. Microbiol. Mol. Biol. Rev. 84, e00002–20. doi:10.1128/MMBR.00002-20
Ghodke, P. P., Mali, J. R., Patra, A., Rizzo, C. J., Guengerich, F. P., and Egli, M. (2021). Enzymatic Bypass and the Structural Basis of Miscoding Opposite the DNA Adduct 1,N2-Ethenodeoxyguanosine by Human DNA Translesion Polymerase η. J. Biol. Chem. 296, 100642. doi:10.1016/j.jbc.2021.100642
Gong, P., Davidson, G. A., Gui, W., Yang, K., Bozza, W. P., and Zhuang, Z. (2018). Activity-Based Ubiquitin-Protein Probes Reveal Target Protein Specificity of Deubiquitinating Enzymes. Chem. Sci. 9, 7859–7865. doi:10.1039/c8sc01573b
Goodman, M. F., and Woodgate, R. (2013). Translesion DNA Polymerases. Cold Spring Harb. Perspect. Biol. 5, a010363. doi:10.1101/cshperspect.a010363
Gowda, A. S. P., and Spratt, T. E. (2016). DNA Polymerase ν Rapidly Bypasses O6-Methyl-dG but Not O6-[4-(3-Pyridyl)-4-Oxobutyl-dG and O2-Alkyl-dTs. Chem. Res. Toxicol. 29, 1894–1900. doi:10.1021/acs.chemrestox.6b00318
Guilliam, T. A., and Yeeles, J. T. (2021). The Eukaryotic Replisome Tolerates Leading-Strand Base Damage by Replicase Switching. EMBO J. 40, e107037. doi:10.15252/embj.2020107037
Guilliam, T. A., and Yeeles, J. T. P. (2020). Reconstitution of Translesion Synthesis Reveals a Mechanism of Eukaryotic DNA Replication Restart. Nat. Struct. Mol. Biol. 27, 450–460. doi:10.1038/s41594-020-0418-4
Gulbis, J. M., Kelman, Z., Hurwitz, J., O'donnell, M., and Kuriyan, J. (1996). Structure of the C-Terminal Region of p21WAF1/CIP1 Complexed with Human PCNA. Cell 87, 297–306. doi:10.1016/s0092-8674(00)81347-1
Hakem, R. (2008). DNA-Damage Repair; the Good, the Bad, and the Ugly. EMBO J. 27, 589–605. doi:10.1038/emboj.2008.15
Hartog, J. H. J., Altona, C., Boom, J. H., Marcelis, A. T. M., Marel, G. A., Rinkel, L. J., et al. (1983). Cis-Platinum Induced Distortions in DNA. Conformational Analysis of d(GpCpG) and cis-Pt(NH3)2{d(GpCpG)}, Studied by 500-MHz NMR. Eur. J. Biochem. 134, 485–495. doi:10.1111/j.1432-1033.1983.tb07593.x
Hedglin, M., Pandey, B., and Benkovic, S. J. (2016a). Characterization of Human Translesion DNA Synthesis across a UV-Induced DNA Lesion. Elife 5, e19788. doi:10.7554/elife.19788
Hedglin, M., Pandey, B., and Benkovic, S. J. (2016b). Stability of the Human Polymerase δ Holoenzyme and its Implications in Lagging Strand DNA Synthesis. Proc. Natl. Acad. Sci. USA 113, E1777–E1786. doi:10.1073/pnas.1523653113
Hibbert, R. G., and Sixma, T. K. (2012). Intrinsic Flexibility of Ubiquitin on Proliferating Cell Nuclear Antigen (PCNA) in Translesion Synthesis. J. Biol. Chem. 287, 39216–39223. doi:10.1074/jbc.m112.389890
Hippel, P. H. V., Fairfield, F. R., and Dolejsi, M. K. (1994). On the Processivity of Polymerases. Ann. N.Y. Acad. Sci. 726, 118–131. doi:10.1111/j.1749-6632.1994.tb52803.x
Hoeijmakers, J. H. J. (2009). DNA Damage, Aging, and Cancer. N. Engl. J. Med. 361, 1475–1485. doi:10.1056/nejmra0804615
Hoppins, J. J., Gruber, D. R., Miears, H. L., Kiryutin, A. S., Kasymov, R. D., Petrova, D. V., et al. (2016). 8-Oxoguanine Affects DNA Backbone Conformation in the EcoRI Recognition Site and Inhibits its Cleavage by the Enzyme. Plos One 11, e0164424. doi:10.1371/journal.pone.0164424
Inomata, Y., Abe, T., Tsuda, M., Takeda, S., and Hirota, K. (2021). Division of Labor of Y-Family Polymerases in Translesion-DNA Synthesis for Distinct Types of DNA Damage. PLoS One 16, e0252587. doi:10.1371/journal.pone.0252587
Jha, V., and Ling, H. (2018). Structural Basis for Human DNA Polymerase Kappa to Bypass Cisplatin Intrastrand Cross-Link (Pt-GG) Lesion as an Efficient and Accurate Extender. J. Mol. Biol. 430, 1577–1589. doi:10.1016/j.jmb.2018.04.023
Johnson, R. E., Washington, M. T., Haracska, L., Prakash, S., and Prakash, L. (2000a). Eukaryotic Polymerases ι and ζ Act Sequentially to Bypass DNA Lesions. Nature 406, 1015–1019. doi:10.1038/35023030
Johnson, R. E., Washington, M. T., Prakash, S., and Prakash, L. (2000b). Fidelity of Human DNA Polymerase η. J. Biol. Chem. 275, 7447–7450. doi:10.1074/jbc.275.11.7447
Jung, H., and Lee, S. (2020). Promutagenic Bypass of 7,8-Dihydro-8-Oxoadenine by Translesion Synthesis DNA Polymerase Dpo4. Biochem. J. 477, 2859–2871. doi:10.1042/bcj20200449
Jung, H., Rayala, N. K., and Lee, S. (2020). Translesion Synthesis of the Major Nitrogen Mustard-Induced DNA Lesion by Human DNA Polymerase η. Biochem. J. 477, 4543–4558. doi:10.1042/bcj20200767
Kannouche, P. L., Wing, J., and Lehmann, A. R. (2004). Interaction of Human DNA Polymerase η with Monoubiquitinated PCNA. Mol. Cel 14, 491–500. doi:10.1016/s1097-2765(04)00259-x
Kath, J. E., Jergic, S., Heltzel, J. M. H., Jacob, D. T., Dixon, N. E., Sutton, M. D., et al. (2014). Polymerase Exchange on Single DNA Molecules Reveals Processivity Clamp Control of Translesion Synthesis. Proc. Natl. Acad. Sci. 111, 7647–7652. doi:10.1073/pnas.1321076111
Kenyon, C. J., and Walker, G. C. (1980). DNA-damaging Agents Stimulate Gene Expression at Specific Loci in Escherichia C. Proc. Natl. Acad. Sci. 77, 2819–2823. doi:10.1073/pnas.77.5.2819
Khare, V., and Eckert, K. A. (2002). The Proofreading 3′→5′ Exonuclease Activity of DNA Polymerases: A Kinetic Barrier to Translesion DNA Synthesis. Mutat. Research/Fundamental Mol. Mech. Mutagenesis 510, 45–54. doi:10.1016/s0027-5107(02)00251-8
Kropp, H. M., Betz, K., Wirth, J., Diederichs, K., and Marx, A. (2017). Crystal Structures of Ternary Complexes of Archaeal B-Family DNA Polymerases. PLoS One 12, e0188005. doi:10.1371/journal.pone.0188005
Kusumoto, R., Masutani, C., Iwai, S., and Hanaoka, F. (2002). Translesion Synthesis by Human DNA Polymerase η across Thymine Glycol Lesions. Biochemistry 41, 6090–6099. doi:10.1021/bi025549k
Kusumoto, R., Masutani, C., Shimmyo, S., Iwai, S., and Hanaoka, F. (2004). DNA Binding Properties of Human DNA Polymerase η: Implications for Fidelity and Polymerase Switching of Translesion Synthesis. Genes Cells 9, 1139–1150. doi:10.1111/j.1365-2443.2004.00797.x
Lancey, C., Tehseen, M., Raducanu, V.-S., Rashid, F., Merino, N., Ragan, T. J., et al. (2020). Structure of the Processive Human Pol δ Holoenzyme. Nat. Commun. 11, 1109. doi:10.1038/s41467-020-14898-6
Lanz, M. C., Dibitetto, D., and Smolka, M. B. (2019). DNA Damage Kinase Signaling: Checkpoint and Repair at 30 Years. EMBO J. 38, e101801. doi:10.15252/embj.2019101801
Lee, M. Y. W. T., Zhang, S., Hua Lin, S., Wang, X., Darzynkiewicz, Z., Zhang, Z., et al. (2014a). The Tail that Wags the Dog: P12, the Smallest Subunit of DNA Polymerase δ, Is Degraded by Ubiquitin Ligases in Response to DNA Damage and during Cell Cycle Progression. Cell Cycle 13, 23–31. doi:10.4161/cc.27407
Lee, M. Y. W. T., Zhang, S., Wang, X., Chao, H. H., Zhao, H., Darzynkiewicz, Z., et al. (2019). Two Forms of Human DNA Polymerase δ: Who Does what and Why? DNA Repair 81, 102656. doi:10.1016/j.dnarep.2019.102656
Lee, Y.-S., Gregory, M. T., and Yang, W. (2014b). Human Pol Purified with Accessory Subunits Is Active in Translesion DNA Synthesis and Complements Pol in Cisplatin Bypass. Proc. Natl. Acad. Sci. 111, 2954–2959. doi:10.1073/pnas.1324001111
Lin, H.-K., Chase, S. F., Laue, T. M., Jen-Jacobson, L., and Trakselis, M. A. (2012). Differential Temperature-Dependent Multimeric Assemblies of Replication and Repair Polymerases on DNA Increase Processivity. Biochemistry 51, 7367–7382. doi:10.1021/bi300956t
Lin, S. H. S., Wang, X., Zhang, S., Zhang, Z., Lee, E. Y. C., and Lee, M. Y. W. T. (2013). Dynamics of Enzymatic Interactions during Short Flap Human Okazaki Fragment Processing by Two Forms of Human DNA Polymerase δ. DNA Repair 12, 922–935. doi:10.1016/j.dnarep.2013.08.008
Liu, B., Xue, Q., Tang, Y., Cao, J., Guengerich, F. P., and Zhang, H. (2016). Mechanisms of Mutagenesis: DNA Replication in the Presence of DNA Damage. Mutat. Research/Reviews Mutat. Res. 768, 53–67. doi:10.1016/j.mrrev.2016.03.006
Liu, F., Yang, Y., and Zhou, Y. (2018). Polymerase delta in Eukaryotes: How Is it Transiently Exchanged with Specialized DNA Polymerases during Translesion DNA Synthesis? Curr. Protein Pept. Sci. 19, 790–804. doi:10.2174/1389203719666180430155625
Livneh, Z., Z, O., and Shachar, S. (2010). Multiple Two-Polymerase Mechanisms in Mammalian Translesion DNA Synthesis. Cell Cycle 9, 729–735. doi:10.4161/cc.9.4.10727
Lopes, M., Foiani, M., and Sogo, J. M. (2006). Multiple Mechanisms Control Chromosome Integrity after Replication fork Uncoupling and Restart at Irreparable UV Lesions. Mol. Cel 21, 15–27. doi:10.1016/j.molcel.2005.11.015
Lukin, M., and De Los Santos, C. (2006). NMR Structures of Damaged DNA. Chem. Rev. 106, 607–686. doi:10.1021/cr0404646
Ma, X., Tang, T. S., and Guo, C. (2020). Regulation of Translesion DNA Synthesis in Mammalian Cells. Environ. Mol. Mutagen. 61, 680–692. doi:10.1002/em.22359
Maiorano, D., El Etri, J., Franchet, C., and Hoffmann, J. S. (2021). Translesion Synthesis or Repair by Specialized DNA Polymerases Limits Excessive Genomic Instability upon Replication Stress. Int. J. Mol. Sci. 22, 3924. doi:10.3390/ijms22083924
Makarova, A. V., and Burgers, P. M. (2015). Eukaryotic DNA Polymerase ζ. DNA Repair 29, 47–55. doi:10.1016/j.dnarep.2015.02.012
Malik, R., Kopylov, M., Gomez-Llorente, Y., Jain, R., Johnson, R. E., Prakash, L., et al. (2020). Structure and Mechanism of B-Family DNA Polymerase ζ Specialized for Translesion DNA Synthesis. Nat. Struct. Mol. Biol. 27, 913–924. doi:10.1038/s41594-020-0476-7
Marechal, A., and Zou, L. (2013). DNA Damage Sensing by the ATM and ATR Kinases. Cold Spring Harb. Perspect. Biol. 5, a012716. doi:10.1101/cshperspect.a012716
Marians, K. J. (2018). Lesion Bypass and the Reactivation of Stalled Replication Forks. Annu. Rev. Biochem. 87, 217–238. doi:10.1146/annurev-biochem-062917-011921
Mcateer, K., Jing, Y., Kao, J., Taylor, J.-S., and Kennedy, M. A. (1998). Solution-State Structure of a DNA Dodecamer Duplex Containing a Cis-Syn Thymine Cyclobutane Dimer, the Major UV Photoproduct of DNA. J. Mol. Biol. 282, 1013–1032. doi:10.1006/jmbi.1998.2062
McCulloch, S. D., Kokoska, R. J., Masutani, C., Iwai, S., Hanaoka, F., and Kunkel, T. A. (2004). Preferential Cis-Syn Thymine Dimer Bypass by DNA Polymerase η Occurs with Biased Fidelity. Nature 428, 97–100. doi:10.1038/nature02352
McCulloch, S. D., and Kunkel, T. A. (2008). The Fidelity of DNA Synthesis by Eukaryotic Replicative and Translesion Synthesis Polymerases. Cell Res. 18, 148–161. doi:10.1038/cr.2008.4
McIntyre, J., Mclenigan, M. P., Frank, E. G., Dai, X., Yang, W., Wang, Y., et al. (2015). Posttranslational Regulation of Human DNA Polymerase ι. J. Biol. Chem. 290, 27332–27344. doi:10.1074/jbc.m115.675769
Meng, X., Zhou, Y., Lee, E. Y. C., Lee, M. Y. W. T., and Frick, D. N. (2010). The P12 Subunit of Human Polymerase δ Modulates the Rate and Fidelity of DNA Synthesis. Biochemistry 49, 3545–3554. doi:10.1021/bi100042b
Menzies, G. E., Reed, S. H., Brancale, A., and Lewis, P. D. (2015). Base Damage, Local Sequence Context andTP53mutation Hotspots: a Molecular Dynamics Study of Benzo[a]pyrene Induced DNA Distortion and Mutability. Nucleic Acids Res. 43, 9133–9146. doi:10.1093/nar/gkv910
Minko, I. G., Harbut, M. B., Kozekov, I. D., Kozekova, A., Jakobs, P. M., Olson, S. B., et al. (2008). Role for DNA Polymerase κ in the Processing of N2-N2-Guanine Interstrand Cross-Links. J. Biol. Chem. 283, 17075–17082. doi:10.1074/jbc.m801238200
Napolitano, R., Janel-Bintz, R., Wagner, J., and Fuchs, R. P. (2000). All Three SOS-Inducible DNA Polymerases (Pol II, Pol IV and Pol V) Are Involved in Induced Mutagenesis. EMBO J. 19, 6259–6265. doi:10.1093/emboj/19.22.6259
Niimi, A., Brown, S., Sabbioneda, S., Kannouche, P. L., Scott, A., Yasui, A., et al. (2008). Regulation of Proliferating Cell Nuclear Antigen Ubiquitination in Mammalian Cells. Proc. Natl. Acad. Sci. 105, 16125–16130. doi:10.1073/pnas.0802727105
O-Wang, J., Kawamura, K., Tada, Y., Ohmori, H., Kimura, H., Sakiyama, S., et al. (2001). DNA Polymerase Kappa, Implicated in Spontaneous and DNA Damage-Induced Mutagenesis, Is Overexpressed in Lung Cancer. Cancer Res. 61, 5366–5369.
Pagès, V., and Fuchs, R. P. (2002). How DNA Lesions Are Turned into Mutations within Cells? Oncogene 21, 8957–8966. doi:10.1038/sj.onc.1206006
Pata, J. D. (2010). Structural Diversity of the Y-Family DNA Polymerases. Biochim. Biophys. Acta (Bba) - Proteins Proteomics 1804, 1124–1135. doi:10.1016/j.bbapap.2010.01.020
Pavlov, Y. I., Nguyen, D., and Kunkel, T. A. (2001). Mutator Effects of Overproducing DNA Polymerase η (Rad30) and its Catalytically Inactive Variant in Yeast. Mutat. Research/Fundamental Mol. Mech. Mutagenesis 478, 129–139. doi:10.1016/s0027-5107(01)00131-2
Phi, K. K., Smith, M. C., Tokarsky, E. J., and Suo, Z. (2019). Kinetic Investigation of Translesion Synthesis across a 3-Nitrobenzanthrone-Derived DNA Lesion Catalyzed by Human DNA Polymerase Kappa. Chem. Res. Toxicol. 32, 1699–1706. doi:10.1021/acs.chemrestox.9b00219
Pillaire, M.-J., Bétous, R., and Hoffmann, J.-S. (2014). Role of DNA Polymerase κ in the Maintenance of Genomic Stability. Mol. Cell Oncol. 1, e29902. doi:10.4161/mco.29902
Plachta, M., Halas, A., Mcintyre, J., and Sledziewska-Gojska, E. (2015). The Steady-State Level and Stability of TLS Polymerase Eta Are Cell Cycle Dependent in the Yeast S. C. DNA Repair 29, 147–153. doi:10.1016/j.dnarep.2015.02.015
Plum, G. E., Grollman, A. P., Johnson, F., and Breslauer, K. J. (1995). Influence of the Oxidatively Damaged Adduct 8-oxodeoxyguanosine on the Conformation, Energetics, and Thermodynamic Stability of a DNA Duplex. Biochemistry 34, 16148–16160. doi:10.1021/bi00049a030
Powers, K. T., and Washington, M. T. (2017). Analyzing the Catalytic Activities and Interactions of Eukaryotic Translesion Synthesis Polymerases. Methods Enzymol. 592, 329–356. doi:10.1016/bs.mie.2017.04.002
Powers, K. T., and Washington, M. T. (2018). Eukaryotic Translesion Synthesis: Choosing the Right Tool for the Job. DNA Repair 71, 127–134. doi:10.1016/j.dnarep.2018.08.016
Prakash, S., and Prakash, L. (2002). Translesion DNA Synthesis in Eukaryotes: A One- or Two-Polymerase Affair. Genes Dev. 16, 1872–1883. doi:10.1101/gad.1009802
Prestel, A., Wichmann, N., Martins, J. M., Marabini, R., Kassem, N., Broendum, S. S., et al. (2019). The PCNA Interaction Motifs Revisited: Thinking outside the PIP-Box. Cell. Mol. Life Sci. 76, 4923–4943. doi:10.1007/s00018-019-03150-0
Pustovalova, Y., Magalhães, M. T. Q., D’Souza, S., Rizzo, A. A., Korza, G., Walker, G. C., et al. (2016). Interaction between the Rev1 C-Terminal Domain and the PolD3 Subunit of Polζ Suggests a Mechanism of Polymerase Exchange upon Rev1/Polζ-Dependent Translesion Synthesis. Biochemistry 55, 2043–2053. doi:10.1021/acs.biochem.5b01282
Qi, H., Zhu, H., Lou, M., Fan, Y., Liu, H., Shen, J., et al. (2012). Interferon Regulatory Factor 1 Transactivates Expression of Human DNA Polymerase η in Response to Carcinogen N-Methyl-N′-nitro-N-Nitrosoguanidine. J. Biol. Chem. 287, 12622–12633. doi:10.1074/jbc.m111.313429
Qin, Z., Lu, M., Xu, X., Hanna, M., Shiomi, N., and Xiao, W. (2013). DNA-Damage Tolerance Mediated by PCNAUb Fusions in Human Cells Is Dependent on Rev1 but Not Polη. Nucleic Acids Res. 41, 7356–7369. doi:10.1093/nar/gkt542
Qiu, S., Jiang, G. X., Cao, L. P., and Huang, J. (2021). Replication fork Reversal and Protection. Front. Cel Dev. Biol. 9, 670392. doi:10.3389/fcell.2021.670392
Raper, A. T., Gadkari, V. V., Maxwell, B. A., and Suo, Z. (2016). Single-Molecule Investigation of Response to Oxidative DNA Damage by a Y-Family DNA Polymerase. Biochemistry 55, 2187–2196. doi:10.1021/acs.biochem.6b00166
Raper, A. T., Reed, A. J., and Suo, Z. (2018). Kinetic Mechanism of DNA Polymerases: Contributions of Conformational Dynamics and a Third Divalent Metal Ion. Chem. Rev. 118, 6000–6025. doi:10.1021/acs.chemrev.7b00685
Reha-Krantz, L. J. (2010). DNA Polymerase Proofreading: Multiple Roles Maintain Genome Stability. Biochim. Biophys. Acta (Bba) - Proteins Proteomics 1804, 1049–1063. doi:10.1016/j.bbapap.2009.06.012
Rickman, K., and Smogorzewska, A. (2019). Advances in Understanding DNA Processing and protection at Stalled Replication forks. J. Cel Biol. 218, 1096–1107. doi:10.1083/jcb.201809012
Rizzo, A. A., Vassel, F.-M., Chatterjee, N., D’Souza, S., Li, Y., Hao, B., et al. (2018). Rev7 Dimerization Is Important for Assembly and Function of the Rev1/Polζ Translesion Synthesis Complex. Proc. Natl. Acad. Sci. USA 115, E8191–E8200. doi:10.1073/pnas.1801149115
Sabbioneda, S., Gourdin, A. M., Green, C. M., Zotter, A., Giglia-Mari, G., Houtsmuller, A., et al. (2008). Effect of Proliferating Cell Nuclear Antigen Ubiquitination and Chromatin Structure on the Dynamic Properties of the Y-Family DNA Polymerases. Mol. Biol. Cel. 19, 5193–5202. doi:10.1091/mbc.e08-07-0724
Sabbioneda, S., Green, C. M., Bienko, M., Kannouche, P., Dikic, I., and Lehmann, A. R. (2009). Ubiquitin-Binding Motif of Human DNA Polymerase η Is Required for Correct Localization. Proc. Natl. Acad. Sci. U. S. A. 106, E20. doi:10.1073/pnas.0812744106
Saha, P., Mandal, T., Talukdar, A. D., Kumar, D., Kumar, S., Tripathi, P. P., et al. (2021). DNA Polymerase Eta: A Potential Pharmacological Target for Cancer Therapy. J. Cel. Physiol. 236, 4106–4120. doi:10.1002/jcp.30155
Sale, J. E., Lehmann, A. R., and Woodgate, R. (2012). Y-family DNA Polymerases and Their Role in Tolerance of Cellular DNA Damage. Nat. Rev. Mol. Cel Biol. 13, 141–152. doi:10.1038/nrm3289
Sale, J. E. (2013). Translesion DNA Synthesis and Mutagenesis in Eukaryotes. Cold Spring Harbor Perspect. Biol. 5, a012708. doi:10.1101/cshperspect.a012708
Sasatani, M., Xi, Y., Kajimura, J., Kawamura, T., Piao, J., Masuda, Y., et al. (2017). Overexpression of Rev1 Promotes the Development of Carcinogen-Induced Intestinal Adenomas via Accumulation of point Mutation and Suppression of Apoptosis Proportionally to the Rev1 Expression Level. Carcinogenesis 38, 570–578. doi:10.1093/carcin/bgw208
Seki, M., Masutani, C., Yang, L. W., Schuffert, A., Iwai, S., Bahar, I., et al. (2004). High-Efficiency Bypass of DNA Damage by Human DNA Polymerase Q. EMBO J. 23, 4484–4494. doi:10.1038/sj.emboj.7600424
Shachar, S., Ziv, O., Avkin, S., Adar, S., Wittschieben, J., Reissner, T., et al. (2009). Two-Polymerase Mechanisms Dictate Error-Free and Error-Prone Translesion DNA Synthesis in Mammals. EMBO J. 28, 383–393. doi:10.1038/emboj.2008.281
Shen, S., Davidson, G. A., Yang, K., and Zhuang, Z. (2021). Photo-Activatable Ub-PCNA Probes Reveal New Structural Features of the Saccharomyces C Polη/PCNA Complex. Nucleic Acids Res. 49, 9374–9388. doi:10.1093/nar/gkab646
Shilkin, E. S., Boldinova, E. O., Stolyarenko, A. D., Goncharova, R. I., Chuprov-Netochin, R. N., Khairullin, R. F., et al. (2020). Translesion DNA Synthesis and Carcinogenesis. Biochem. Mosc. 85, 425–435. doi:10.1134/s0006297920040033
Shriber, P., Leitner-Dagan, Y., Geacintov, N., Paz-Elizur, T., and Livneh, Z. (2015). DNA Sequence Context Greatly Affects the Accuracy of Bypass across an Ultraviolet Light 6-4 Photoproduct in Mammalian Cells. Mutat. Res. 780, 71–76. doi:10.1016/j.mrfmmm.2015.08.002
Slade, D. (2018). Maneuvers on PCNA Rings during DNA Replication and Repair. Genes (Basel) 9, 416. doi:10.3390/genes9080416
Sobolewska, A., Halas, A., Plachta, M., Mcintyre, J., and Sledziewska-Gojska, E. (2020). Regulation of the Abundance of Y-Family Polymerases in the Cell Cycle of Budding Yeast in Response to DNA Damage. Curr. Genet. 66, 749–763. doi:10.1007/s00294-020-01061-3
Stelter, P., and Ulrich, H. D. (2003). Control of Spontaneous and Damage-Induced Mutagenesis by SUMO and Ubiquitin Conjugation. Nature 425, 188–191. doi:10.1038/nature01965
Suzuki, T., Sassa, A., Grúz, P., Gupta, R. C., Johnson, F., Adachi, N., et al. (2021). Error-prone Bypass Patch by a Low-Fidelity Variant of DNA Polymerase Zeta in Human Cells. DNA Repair 100, 103052. doi:10.1016/j.dnarep.2021.103052
Swan, M. K., Johnson, R. E., Prakash, L., Prakash, S., and Aggarwal, A. K. (2009). Structural Basis of High-Fidelity DNA Synthesis by Yeast DNA Polymerase δ. Nat. Struct. Mol. Biol. 16, 979–986. doi:10.1038/nsmb.1663
Takata, K.-I., Shimizu, T., Iwai, S., and Wood, R. D. (2006). Human DNA Polymerase N (POLN) Is a Low Fidelity Enzyme Capable of Error-free Bypass of 5S-Thymine Glycol. J. Biol. Chem. 281, 23445–23455. doi:10.1074/jbc.m604317200
Thomforde, J., Fu, I., Rodriguez, F., Pujari, S. S., Broyde, S., and Tretyakova, N. (2021). Translesion Synthesis Past 5-Formylcytosine-Mediated DNA-Peptide Cross-Links by hPolη Is Dependent on the Local DNA Sequence. Biochemistry 60, 1797–1807. doi:10.1021/acs.biochem.1c00130
Tomicic, M. T., Aasland, D., Naumann, S. C., Meise, R., Barckhausen, C., Kaina, B., et al. (2014). Translesion Polymerase η Is Upregulated by Cancer Therapeutics and Confers Anticancer Drug Resistance. Cancer Res. 74, 5585–5596. doi:10.1158/0008-5472.can-14-0953
Trakselis, M. A., and Benkovic, S. J. (2001). Intricacies in ATP-Dependent Clamp Loading: Variations across Replication Systems. Structure 9, 999–1004. doi:10.1016/s0969-2126(01)00676-1
Trakselis, M. A., Cranford, M. T., and Chu, A. M. (2017). Coordination and Substitution of DNA Polymerases in Response to Genomic Obstacles. Chem. Res. Toxicol. 30, 1956–1971. doi:10.1021/acs.chemrestox.7b00190
Uchiyama, M., Terunuma, J., and Hanaoka, F. (2015). The Protein Level of Rev1, a TLS Polymerase in Fission Yeast, Is Strictly Regulated during the Cell Cycle and after DNA Damage. PLoS One 10, e0130000. doi:10.1371/journal.pone.0130000
Vaisman, A., Tissier, A., Frank, E. G., Goodman, M. F., and Woodgate, R. (2001). Human DNA Polymerase ι Promiscuous Mismatch Extension. J. Biol. Chem. 276, 30615–30622. doi:10.1074/jbc.m102694200
Vaisman, A., and Woodgate, R. (2020). Mysterious and Fascinating: DNA Polymerase Ɩ Remains Enigmatic 20 Years after its Discovery. DNA Repair 93, 102914. doi:10.1016/j.dnarep.2020.102914
Vaisman, A., and Woodgate, R. (2017). Translesion DNA Polymerases in Eukaryotes: What Makes Them Tick? Crit. Rev. Biochem. Mol. Biol. 52, 274–303. doi:10.1080/10409238.2017.1291576
Vanarotti, M., Grace, C. R., Miller, D. J., Actis, M. L., Inoue, A., Evison, B. J., et al. (2018). Structures of REV1 UBM2 Domain Complex with Ubiquitin and with a Small-Molecule that Inhibits the REV1 UBM2-Ubiquitin Interaction. J. Mol. Biol. 430, 2857–2872. doi:10.1016/j.jmb.2018.05.042
Velasco-Miguel, S., Richardson, J. A., Gerlach, V. L., Lai, W. C., Gao, T., Russell, L. D., et al. (2003). Constitutive and Regulated Expression of the Mouse Dinb (Polκ) Gene Encoding DNA Polymerase Kappa. DNA Repair 2, 91–106. doi:10.1016/s1568-7864(02)00189-1
Volkova, N. V., Meier, B., González-Huici, V., Bertolini, S., Gonzalez, S., Vöhringer, H., et al. (2020). Mutational Signatures Are Jointly Shaped by DNA Damage and Repair. Nat. Commun. 11, 2169. doi:10.1038/s41467-020-15912-7
Wang, H., Wu, W., Wang, H.-W., Wang, S., Chen, Y., Zhang, X., et al. (2010). Analysis of Specialized DNA Polymerases Expression in Human Gliomas: Association with Prognostic Significance. Neuro. Oncol. 12, 679–686. doi:10.1093/neuonc/nop074
Warbrick, E. (1998). PCNA Binding through a Conserved Motif. Bioessays 20, 195–199. doi:10.1002/(sici)1521-1878(199803)20:3<195:aid-bies2>3.0.co;2-r
Waters, L. S., Minesinger, B. K., Wiltrout, M. E., D'souza, S., Woodruff, R. V., and Walker, G. C. (2009). Eukaryotic Translesion Polymerases and Their Roles and Regulation in DNA Damage Tolerance. Microbiol. Mol. Biol. Rev. 73, 134–154. doi:10.1128/mmbr.00034-08
Waters, L. S., and Walker, G. C. (2006). The Critical Mutagenic Translesion DNA Polymerase Rev1 Is Highly Expressed during G2/M Phase rather Than S Phase. Proc. Natl. Acad. Sci. 103, 8971–8976. doi:10.1073/pnas.0510167103
Wojtaszek, J. L., Chatterjee, N., Najeeb, J., Ramos, A., Lee, M., Bian, K., et al. (2019). A Small Molecule Targeting Mutagenic Translesion Synthesis Improves Chemotherapy. Cell 178, 152–159. doi:10.1016/j.cell.2019.05.028
Wong, J. H., Brown, J. A., Suo, Z., Blum, P., Nohmi, T., and Ling, H. (2010). Structural Insight into Dynamic Bypass of the Major Cisplatin-DNA Adduct by Y-Family Polymerase Dpo4. EMBO J. 29, 2059–2069. doi:10.1038/emboj.2010.101
Yang, K., Li, G., Gong, P., Gui, W., Yuan, L., and Zhuang, Z. (2016). Chemical Protein Ubiquitylation with Preservation of the Native Cysteine Residues. ChemBioChem 17, 995–998. doi:10.1002/cbic.201600042
Yang, W. (2014). An Overview of Y-Family DNA Polymerases and a Case Study of Human DNA Polymerase η. Biochemistry 53, 2793–2803. doi:10.1021/bi500019s
Yang, W., and Gao, Y. (2018). Translesion and Repair DNA Polymerases: Diverse Structure and Mechanism. Annu. Rev. Biochem. 87, 239–261. doi:10.1146/annurev-biochem-062917-012405
Yeiser, B., Pepper, E. D., Goodman, M. F., and Finkel, S. E. (2002). SOS-induced DNA Polymerases Enhance Long-Term Survival and Evolutionary Fitness. Proc. Natl. Acad. Sci. 99, 8737–8741. doi:10.1073/pnas.092269199
Yoon, J.-H., Prakash, L., and Prakash, S. (2009). Highly Error-fFee Role of DNA Polymerase in the Replicative Bypass of UV-Induced Pyrimidine Dimers in Mouse and Human Cells. Proc. Natl. Acad. Sci. 106, 18219–18224. doi:10.1073/pnas.0910121106
Yoon, J. H., Basu, D., Sellamuthu, K., Johnson, R. E., Prakash, S., and Prakash, L. (2021). A Novel Role of DNA Polymerase Lambda in Translesion Synthesis in Conjunction with DNA Polymerase Zeta. Life Sci. Alliance 4, e202000900. doi:10.26508/lsa.202000900
Zhang, S., Zhao, H., Darzynkiewicz, Z., Zhou, P., Zhang, Z., Lee, E. Y. C., et al. (2013). A Novel Function of CRL4Cdt2. J. Biol. Chem. 288, 29550–29561. doi:10.1074/jbc.m113.490466
Zhang, S., Zhou, Y., Trusa, S., Meng, X., Lee, E. Y. C., and Lee, M. Y. W. T. (2007). A Novel DNA Damage Response. J. Biol. Chem. 282, 15330–15340. doi:10.1074/jbc.m610356200
Zhang, Z., Zhang, S., Lin, S. H. S., Wang, X., Wu, L., Lee, E. Y. C., et al. (2012). Structure of Monoubiquitinated PCNA. Cell Cycle 11, 2128–2136. doi:10.4161/cc.20595
Zhao, L., and Washington, M. T. (2017). Translesion Synthesis: Insights into the Selection and Switching of DNA Polymerases. Genes (Basel) 8, 24. doi:10.3390/genes8010024
Zhao, Y., Biertumpfel, C., Gregory, M. T., Hua, Y.-J., Hanaoka, F., and Yang, W. (2012). Structural Basis of Human DNA Polymerase -Mediated Chemoresistance to Cisplatin. Proc. Natl. Acad. Sci. 109, 7269–7274. doi:10.1073/pnas.1202681109
Zhuang, Z., Johnson, R. E., Haracska, L., Prakash, L., Prakash, S., and Benkovic, S. J. (2008). Regulation of Polymerase Exchange between Pol and Pol by Monoubiquitination of PCNA and the Movement of DNA Polymerase Holoenzyme. Proc. Natl. Acad. Sci. 105, 5361–5366. doi:10.1073/pnas.0801310105
Keywords: DNA replication, polymerase, switching, translesion synthesis, PCNA, DNA damage, DNA lesion, substitution
Citation: Kaszubowski JD and Trakselis MA (2022) Beyond the Lesion: Back to High Fidelity DNA Synthesis. Front. Mol. Biosci. 8:811540. doi: 10.3389/fmolb.2021.811540
Received: 09 November 2021; Accepted: 16 December 2021;
Published: 05 January 2022.
Edited by:
Whitney Yin, University of Texas Medical Branch at Galveston, United StatesReviewed by:
Linlin Zhao, University of California, Riverside, United StatesChris Putnam, University of California, San Diego, United States
Copyright © 2022 Kaszubowski and Trakselis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael A. Trakselis, bWljaGFlbF90cmFrc2VsaXNAYmF5bG9yLmVkdQ==
†ORCID: Joseph D. Kaszubowski, 0000-0003-0153-9043; Michael A. Trakselis, 0000-0001-7054-8475