 Alexander A. Malär1†
Alexander A. Malär1† Morgane Callon1†
Morgane Callon1† Albert A. Smith2
Albert A. Smith2 Shishan Wang3
Shishan Wang3 Lauriane Lecoq3Carolina Pérez-Segura4Jodi A. Hadden-Perilla4
Lauriane Lecoq3Carolina Pérez-Segura4Jodi A. Hadden-Perilla4 Anja Böckmann3*
Anja Böckmann3* Beat H. Meier1*
Beat H. Meier1*- 1Physical Chemistry, ETH Zürich, Zürich, Switzerland
- 2Institute of Medical Physics and Biophysics, Universität Leipzig, Leipzig, Germany
- 3Molecular Microbiology and Structural Biochemistry (MMSB), UMR 5086 CNRS-Université de Lyon, Labex Ecofect, Lyon, France
- 4Department of Chemistry and Biochemistry, University of Delaware, Newark, DE, United States
Protein plasticity and dynamics are important aspects of their function. Here we use solid-state NMR to experimentally characterize the dynamics of the 3.5 MDa hepatitis B virus (HBV) capsid, assembled from 240 copies of the Cp149 core protein. We measure both T1 and T1ρ relaxation times, which we use to establish detectors on the nanosecond and microsecond timescale. We compare our results to those from a 1 microsecond all-atom Molecular Dynamics (MD) simulation trajectory for the capsid. We show that, for the constituent residues, nanosecond dynamics are faithfully captured by the MD simulation. The calculated values can be used in good approximation for the NMR-non-detected residues, as well as to extrapolate into the range between the nanosecond and microsecond dynamics, where NMR has a blind spot at the current state of technology. Slower motions on the microsecond timescale are difficult to characterize by all-atom MD simulations owing to computational expense, but are readily accessed by NMR. The two methods are, thus, complementary, and a combination thereof can reliably characterize motions covering correlation times up to a few microseconds.
Introduction
Characterizing the dynamics of proteins and their assemblies is often key to understanding their function, and NMR has proven to be particularly suitable to study dynamical behavior at the atomic level. For large systems with a limited monomer size, such as oligomeric proteins and multimeric assemblies like fibrils and sub-viral particles, solid-state NMR is the method of choice, since it allows evaluation of dynamical properties on a per-residue basis. The latter is achieved by measuring NMR relaxation parameters, in particular the 15N spin-lattice relaxation T1 that addresses fast motions (correlation times around 1 ns), as well as the rotating frame relaxation, T1ρ, which probes hundreds-of-nanosecond to millisecond motions (Krushelnitsky et al., 2010; Schanda et al., 2010; Quinn and McDermott, 2012; Tollinger et al., 2012; Lamley et al., 2015; Schanda and Ernst, 2016; Rovó et al., 2019; Shi et al., 2019). The advent of proton detection under fast magic-angle spinning (Agarwal et al., 2014; Andreas et al., 2015, 2016; Penzel et al., 2019; Schledorn et al., 2020) allowed for the sensitive determination of the 15N relaxation parameters in a series of two-dimensional (hNH) or, for larger proteins, three-dimensional (e.g. hCANH) experiments (Vasa et al., 2018; Schubeis et al., 2020) to characterize the protein dynamics for each spectrally resolved residue. Important timescales of motion in the context of protein function are slow components, spanning the hundreds-of-nanosecond to low-millisecond time range. While they can be easily accessed in NMR by rotating-frame relaxation measurements, they are difficult to measure by most other techniques.
In the following study, we use solid-state NMR spectroscopy for which, when compared to solution-state NMR, the interpretation of relaxation data is simplified by the absence of overall molecular tumbling. However, the internal protein motions can be rather complex, and the description in terms of an extended model-free approach (Clore et al., 1990; Chevelkov et al., 2009) with several correlation times τc can be ambiguous (Smith et al., 2017). To mitigate this, the detector analysis approach has been introduced to analyze such data with minimal bias (Smith et al., 2018), as well as to facilitate comparison with molecular dynamics (MD) simulations (Smith et al., 2019; Zumpfe and Smith, 2021).
In contrast to experimental observations of dynamical effects, which are only sensitive to certain windows of correlation times τc, and motions of certain atoms (often the amide proton/nitrogen pairs), MD simulations do not have these restrictions. All-atom MD simulations can provide dynamical details at fully atomic resolution, however, owing to computational expense, sampling timescales are often limited, especially for large protein oligomers or assemblies. With increasing computational power, longer all-atom MD simulations of large systems, including complete virus capsids, have become accessible, revealing insights into functional motions observable over 1 microsecond of sampling (Perilla et al., 2015; Pérez-Segura et al., 2021). However, as simulations must extend over a time significantly longer than the longest correlation time of interest to accurately characterize protein behavior, examination of the most interesting and biologically-relevant motions that occur over multi-microsecond and millisecond timescales remains a challenge.
An all-atom MD simulation describing the Cp149 HBV capsid (with a number of mutations, vide infra) for 1 microsecond was recently reported (Hadden et al., 2018). While the timescale of the simulation characterizes, with good statistics, motions with correlation times of a few hundred nanoseconds, increasing the sampling time by an order of magnitude to study slower motions represents a significant computational expense. We herein examine HBV capsid dynamics by NMR in order to experimentally test the simulation predictions for the NMR-observable spins and time windows. We show that results of the two methods coincide, demonstrating the validity of using MD simulation data to extrapolate correlation functions for atoms and dynamic timescales which are not observable by NMR.
To shortly describe the objects investigated: HBV particles enclose their genetic material inside mainly T = 4-icosahedral capsids formed by 240 copies of the 183-amino-acid residues core protein (Cp) (Böttcher et al., 1997; Wynne et al., 1999; Yu et al., 2013). The first 149 residues constitute the assembly domain, Cp149, and they are sufficient for in-vitro capsid formation (Gallina et al., 1989; Birnbaum and Nassal, 1990). Cp149 forms dimers in solution, and self-assembles, at sufficiently high concentration, into capsids. The asymmetric unit of the icosahedral capsid contains four sequentially but not conformationally identical protein monomers denoted as A, B, C, and D, which are organized as AB and CD dimers (Wynne et al., 1999). The C-terminal 34 residues in Cp183 are disordered and not observed in NMR or cryo-EM (Zlotnick et al., 1997; Wang et al., 2019). The capsid geometry of Cp149 is virtually indistinguishable from the ones formed by the full-length core protein Cp183 (Wang et al., 2019). High-resolution solid-state NMR spectra of Cp149 have previously been described and the 1H, 13C and 15N NMR resonances and have been sequentially assigned (Lecoq et al., 2018, 2019). Indeed, some resonances split into four, representing the four conformationally different core protein monomers, whose structural flexibility is essential to form the capsid.
While the structure of the HBV capsid is well characterized, this is less true for its dynamic behavior; still, it becomes more and more clear that viral capsids are not rigid “tin cans” (Sherman et al., 2020) but plastic and dynamic entities. While plasticity and dynamics are sometimes used interchangeably, we here use plasticity to indicate the deformability of the protein under external forces, as well as its structural flexibility, while we use dynamics to indicate reversible motional processes like backbone- and side-chain motions and conformational exchange between structures (Henzler-Wildman and Kern, 2007). For HBV, the plasticity of the capsid was demonstrated e.g. by (Böttcher et al., 2006) by showing that induced stress (by the biochemical introduction of foreign amino-acid sequences at the spike tips) onto the capsid structure could be accommodated by conformational changes still preserving the capsid form. MD simulations of the capsid by Hadden et al. revealed its ability to distort asymmetrically (Hadden et al., 2018) and alter its morphology to further accommodate the binding of small molecules (Hadden et al., 2018). Another indication for structural plasticity comes from the four different conformations of the monomers in the asymmetric unit discussed above and from the recent observation of a conformational switch of the capsid conformation triggered by a pocket factor (Lecoq et al., 2021).
The importance of dynamics for the functional characterization of viruses has recently been demonstrated for the HIV-1 capsid by measuring the dynamical averaging of chemical-shift tensors (Zhang et al., 2016). Plasticity and dynamics are intimately linked to function (Sherman et al., 2020) e.g. in HBV for the transmission of the “maturation signal” (Summers and Mason, 1982; Böttcher et al., 2006) which is believed to trigger interaction with the envelope, and thus virion production once the internal genome is mature. This process thus transmits information on the capsid’s internal genome status to the capsid surface. The mechanism is yet unknown, but must harness conformational plasticity to modulate the envelopment-proficiency of the capsid surface.
In the following, we present an experimental approach to characterize the HBV Cp149 capsid dynamics using the amide nitrogen-proton vector as the probe of dynamics and using amide proton-detected solid-state NMR spectroscopy, allowing the use of fast MAS experiments for characterizing slow motions (Lakomek et al., 2017). We measure data which offer three different observation windows into capsid dynamics: one in the nanosecond range (measuring 15N T1), and two in the upper nanoseconds-to-microsecond range (measuring 15N T1ρ). We find, using the detectors approach, overall good agreement between MD simulations and NMR for the localized fast motions, yet still with some significant differences. Importantly, we then obtain for the first time a detailed picture also on the microsecond motions of the HBV capsid, which cannot be faithfully predicted from currently available simulation data.
Materials and Methods
Sample Preparation
Uniformly 2H-13C-15N labeled Cp149 empty capsids (not containing nucleic acids) were produced as described in (Lecoq et al., 2019) and purified as described in (Lecoq et al., 2018). In brief, Cp149 protein was expressed in E. coli BL21 (DE3)*CP strain, grown at 37°C in M9 minimal medium containing 1 g/L of 15NH4Cl and 2 g/L of deuterated 13C-glucose in D2O and supplemented with 100 μg/ml ampicillin and 34 μg/ml chloramphenicol. When the OD600 reached 0.7, the expression was induced with 1 mM IPTG and the culture was further incubated for 6 h at 25°C.
Harvested cells were resuspended in 15 ml of lysis buffer (50 mM Tris pH 7.5, 300 mM NaCl, 5 mM DTT) and incubated on ice for 45 min with 1 mg/ml of chicken lysozyme, 1x of protease inhibitor cocktail solution and 0.5% of Triton-X-100, then mixed with 4 µL of benzonase nuclease for 30 min at room temperature. Cells were broken by sonication and centrifuged for 1 h at 8,000×g to remove cell debris. Cp149 capsids in the supernatant were purified on a stepwise sucrose gradient from 10 to 60% (m/v) sucrose (buffered in 50 mM Tris pH 7.5, 300 mM NaCl) which was centrifuged at 28,800×g for 3 h at 4°C. The fractions containing Cp149 were further purified by (NH4)2SO4 precipitation (up to 35% saturation) and finally dialyzed in the solid-state-NMR buffer (50 mM TRIS pH 7.5, 5 mM DTT) overnight at 4°C. 20 µL of saturated 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) solution were added to the protein for chemical-shift referencing prior to the sedimentation step. Sub-milligram amounts of capsids were filled into 0.5 and 0.7 mm rotors using home-made filling tools (Böckmann et al., 2009) by centrifugation (200,000 g, 17 h, 4°C).
NMR Spectroscopy, Data Processing and Analysis
Solid-state NMR experiments were recorded using a static magnetic-field strength of 20 T (wide-bore 850 MHz Bruker Avance III Spectrometer) on uniformly (2H, 13C, 15N)-labeled Cp149 capsids sedimented (Bertini et al., 2011, 2012; Gardiennet et al., 2012) in H2O to re-protonate amide protons. The 2D hNH spectra for the R1ρ(15N) determination were measured in a 0.5 mm triple-resonance probe head constructed by Ago Samoson and coworkers (Darklands OÜ, Tallinn, Estonia) (Schledorn et al., 2020), at 80 kHz and 160 kHz MAS frequency, respectively, and at a sample temperature of ∼28°C as determined from the supernatant water resonance frequency (Gottlieb et al., 1997; Böckmann et al., 2009). The 3D hCANH spectra for the R1ρ(15N) determination were recorded at 80 and 110 kHz MAS in a 0.7 mm triple-resonance Bruker probe, at ∼21°C sample temperature. Both 2D and 3D R1ρ relaxation sequences contain a 15N spin lock with an rf-field strength of 13 kHz and a temperature-compensation block before the actual experiment to ensure that the temperature change caused by rf heating is the same for all spin-lock lengths (Lakomek et al., 2017). The variable spin-lock times range from 1 μs to 371 ms for the 2D experiments, and from 1 μs to 251 ms for the 3D experiments. Further details on the hCANH pulse sequence can be found in Supplementary Figure S1. In the case of both 2D- and 3D-based relaxation experiments, one series of measurements was composed of eight experiments with different spin-lock times. The inter-scan delay for the 3D measurements has been set to the optimal value of 2.19 s, based on the bulk protein T1(1HN) time of 1.73 s. All 2D R1ρ measurements were performed at 28°C, and all 3D measurements at 21°C.
The site-specific R1(15N) relaxation-rate constants were recorded at 108 kHz MAS (0.7 mm Bruker probe head) at ∼28°C using the 2D hNH-based sequence described in ref. (Lakomek et al., 2017). Detailed information about all acquisition parameters can be found in Table S1.
Spectroscopic data has been processed using Topspin 3.5pl6 (Bruker Biospin) with zero filling to the double amount of data points and a shifted sine-bell apodization function in direct and indirect dimensions with SSB = 2.5. Resonance assignments were transferred from earlier work (Lecoq et al., 2019), BMRB accession number 27845, using CcpNmr Analysis 2.4.2 (Vranken et al., 2005; Stevens et al., 2011). Peak positions and peak intensities were then exported to MATLAB (MATLAB R2019a, 9.6.0.) for the relaxation analysis.
Relaxation Analysis
Site-specific relaxation-rate constants were obtained within MATLAB using the spectral fitting package INFOS(Smith, 2017), error bars were obtained using bootstrapping methods and are reported throughout the work as twice the standard deviation,
Detector Approach for Comparison of Amplitudes and Correlation Times of Motion From NMR Measurements and MD Trajectories
The detector approach (Smith et al., 2017, 2018, 2019) was used to extract, from the experimental NMR data as well as from the MD trajectory, site-specific amplitudes of motion over ranges of timescales quantified by detector responses
An all-atom MD simulation provides the coordinates of each atom as function of time, and allows the calculation of residue-specific relaxation-rate constants from the autocorrelation function
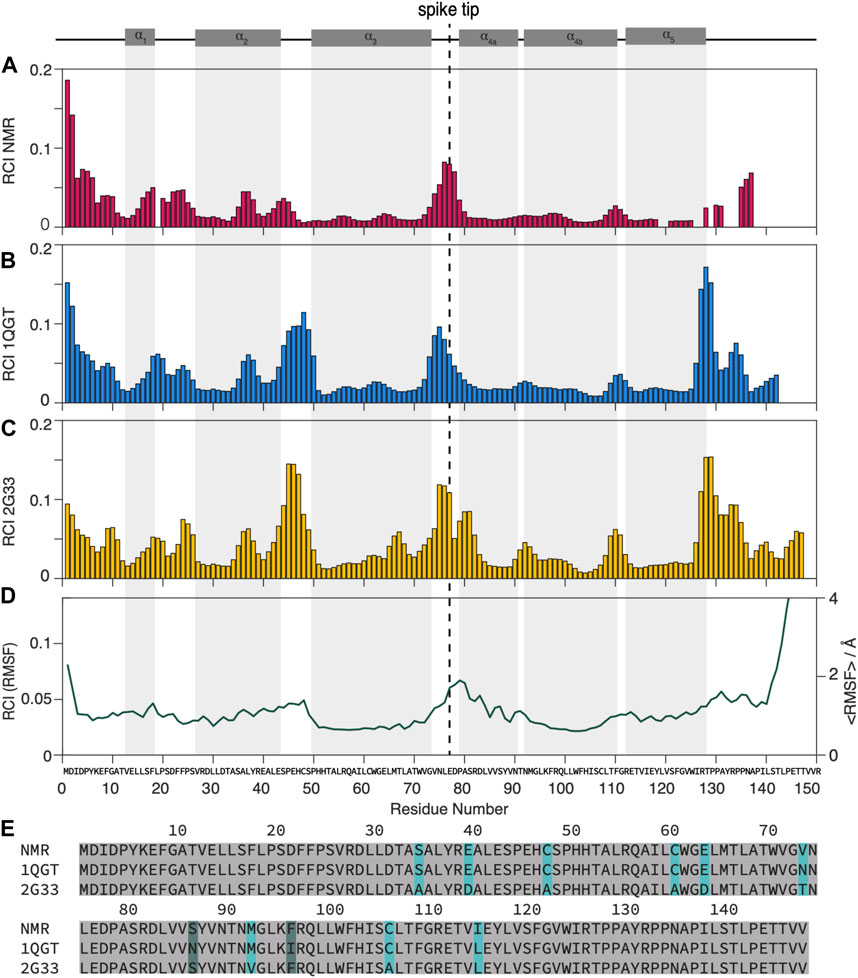
FIGURE 1. Dynamic and static regions as distinguished by the random coil index.(A) RCI from experimental chemical-shift data (Lecoq et al., 2018, 2019), (B) from the X-ray structures used for NMR (PDB 1QGT) and (C) for MD (PDB 2G33). (D) RMSF from the MD trajectory (Hadden et al., 2018) (left scale) and RCI calculated from the RMSF (right scale) by multiplying it by an empirical factor of 28.3 A according to (Berjanskii and Wishart, 2008). (E) Alignment of the different strains used: strain D for NMR, strain CW for the 1QGT crystal structure, strain ADYW for the 2G33 crystal structure and the MD calculations. The mutations between strains D and ADYW (NMR and MD) are highlighted in cyan.
Results
A Rough Overview: Random Coil Index
A first overview over dynamic and rigid regions of the capsids is provided by the random coil index (RCI) (Berjanskii and Wishart, 2005, 2008; Fowler et al., 2020), an empirical quantity calculated from the NMR chemical shifts. While the RCI does not provide detailed information about the timescale of the dynamics, it specifies the amplitude of the motion and distinguishes dynamic and rigid regions, allowing for an overview of the molecule’s dynamic behavior. The RCI values from the experimental NMR data were calculated using HN, 15N, 13Cα, 13Cβ, and C′ chemical shifts from (Lecoq et al., 2018, 2019) and are shown in Figure 1A. The RCIs from the X-ray structures are calculated by converting the X-ray data to chemical shifts using the software SHIFTX2 (Han et al., 2011) RCI values from the X-ray structures of the genotype used for NMR (PDB 1GQT) and MD (PDB 2G33) are shown in Figures 1B, C respectively and compared to the root mean square fluctuation (RMSF) from MD (from (Hadden et al., 2018) in Figure 1D.
Although some significant differences are observed (mainly in the loop from residues 44 to 49 and towards the C-terminal around 130), overall the RCIs calculated from the experimental NMR data match well those calculated from the X-ray structures and they highlight similar dynamic regions than the MD RMSF. The RCI method is an experimentally rather straightforward approach and requires only resonance assignment. For a detailed dynamic characterization, particularly disentangling the timescales of the underlying motions, NMR relaxation-rate constants need to be evaluated.
NMR Relaxation Measurements
The solid-state NMR relaxation measurements were performed on assembled HBV capsids formed from the N-terminal assembly domain (Cp149). Figure 2A shows, in green, a 2D hNH spectrum recorded at 110 kHz MAS in a 0.7 mm rotor at 20.0 T magnetic field. The spectrum shows the expected good spectral resolution, with proton linewidths in the order of 110 ± 50 Hz as reported previously (Lecoq et al., 2019). To characterize a higher number of residues by reducing spectral overlap, 3D hCANH-type relaxation experiments were recorded. We obtained two sets of eight R1ρ(15N) 3D spectra, at 80 kHz and 110 kHz MAS (and 21°C), using optimized acquisition parameters to fit each spectrum into one single day of measurement time (for details on the optimization see Supplementary Figure S8). A series of 2D slices (δ(13C) from 59 to 65 ppm) of the first 3D hCANH spectrum in the series (with essentially zero relaxation delay) is shown in steel blue in Figure 2A (for all slices see Supplementary Figure S9). In the 3D hCANH spectra, 73 peaks could be resolved (Figure 2D).
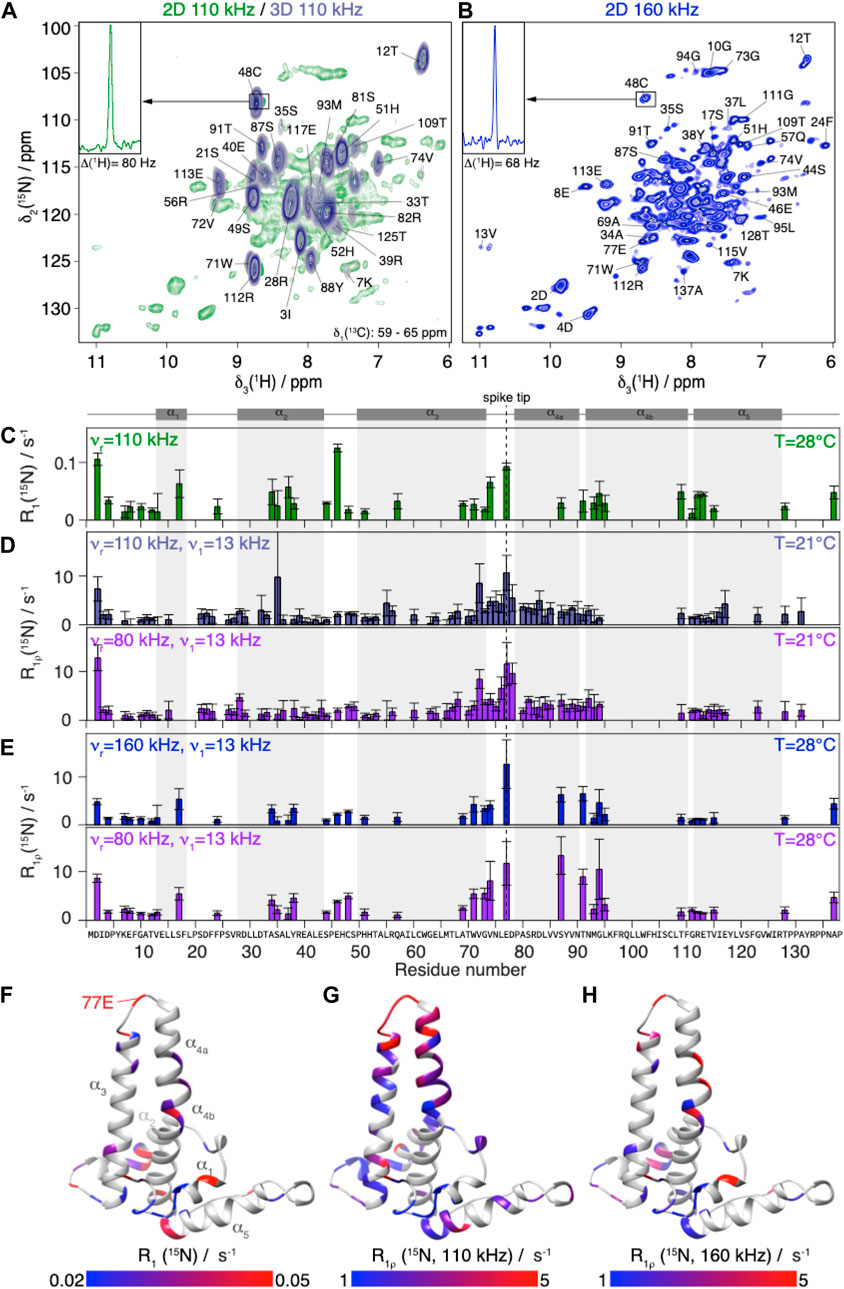
FIGURE 2. NMR 15N relaxation-rate constants of Cp149 capsid.(A) Overlay of Cp149 2D hNH spectrum (green) and slices of 3D hCANH (δ(13C) from 58.95 to 65.51 ppm, steel blue) recorded at 110 kHz MAS. The proton line of 48C in the 2D spectrum is shown in the insert.(B) 2D hNH spectrum of Cp149 capsids recorded at 160 kHz MAS. The proton line of 48C is shown in the insert. The assignment is transferred from Lecoq et al. (Lecoq et al., 2019). Experimental site-specific values of (C) R1(15N, 86 MHz) measured at 110 kHz,(D) R1ρ(15N) measured at 110 kHz (steel blue) and 80 kHz (purple) MAS using ν1 = 13 kHz spin-lock with the 3D hCANH in the 0.7 mm rotor and (E) R1ρ(15N) measured with 2D hNH at 160 kHz (blue) and 80 kHz (purple) MAS in the 0.5 mm rotor using ν 1 = 13 kHz spin-lock. The error bars depicted are calculated using a bootstrapping method and are given by
In order to extend the time windows of the relaxation analysis, we also recorded hNH-type relaxation experiments in an 0.5 mm rotor at currently fastest MAS frequencies (160 kHz) at 28°C, shown in Figure 2B. A representative trace (48C in the inset in Figure 1A, B) reports the concomitant linewidth improvement from 80 to 68 Hz achieved thanks to the higher spinning frequency (Schledorn et al., 2020). In these 2D spectra, 35 well-resolved backbone amide proton/nitrogen peaks, labelled in Figure 2B, were used for the relaxation analysis of the data, roughly half as many site-specific R1ρ(15N) rate constants as from the three-dimensional experiments due to peak overlap.
R1 (15N) relaxation was investigated using hNH-based experiments at 110 kHz MAS and 28°C (being to a large degree MAS-frequency independent). The relaxation-rate constants resulting from the analysis of the R1(15N) measurements are shown in Figure 2C. The rate constants contain information for each individual residue about motions of the NH vector on a timescale on the order of the inverse Larmor angular frequency for 15N, 1.8 ns. The secondary structure of the protein shown in grey above the graph indicates that the fastest relaxing residues are located in loops; for a more detailed discussion see below where the resulting detector response is interpreted. In general, relaxation-rate constants of roughly 0.1 s−1 and smaller are observed, which are very similar to values reported for rigid proteins studied under similar conditions, as for example microcrystalline ubiquitin, where however the dynamic β1-β2 turn (residues 10–12) relaxes faster (Ma et al., 2015; Lakomek et al., 2017). Other examples from the literature include (Knight et al., 2012; Lamley et al., 2015; Smith et al., 2016). This observation indicates that the capsid is quite rigid on the nanosecond timescale.
The R1ρ (15N) relaxation-rate constants resulting from the analysis of the 3D hCANH spectra are shown in Figure 2D. They are mostly below 10 s−1 at 110 kHz, and slightly higher at the lower MAS frequency, 80 kHz. R1ρ(15N) rotating-frame relaxation-rate constants inform about motions at frequencies of the spin-lock frequency plus or minus once or twice the MAS frequency:
The relaxation-rate constants resulting from the analysis of the 2D hNH spectra at 160 and 80 kHz MAS at slightly higher temperature (28°C instead of 21°C) are shown in Figure 2E. Despite the overall rather rigid behavior on the nano as well as microsecond scale, we indeed find significant differences between the backbone-relaxation properties of the different residues for all five relaxation-rate constants determined. The fastest R1ρ (15N) relaxation is observed for residues around 77E (the spike tip) and for some residues at the spike base, as illustrated in the color-coded representations of the rate constants on the capsid monomer given in Figures 2F–H.
MD simulations have been performed at 37°C, while NMR relaxation measurements could not be conducted at these temperatures, due to limited protein stability over time. In order to assess the temperature-dependent R1ρ(15N) rate constants in a site-resolved manner, we measured the faster-to-record two-dimensional spectra at different temperatures between 11°C and 34°C at 80 kHz MAS (Figure 3A).
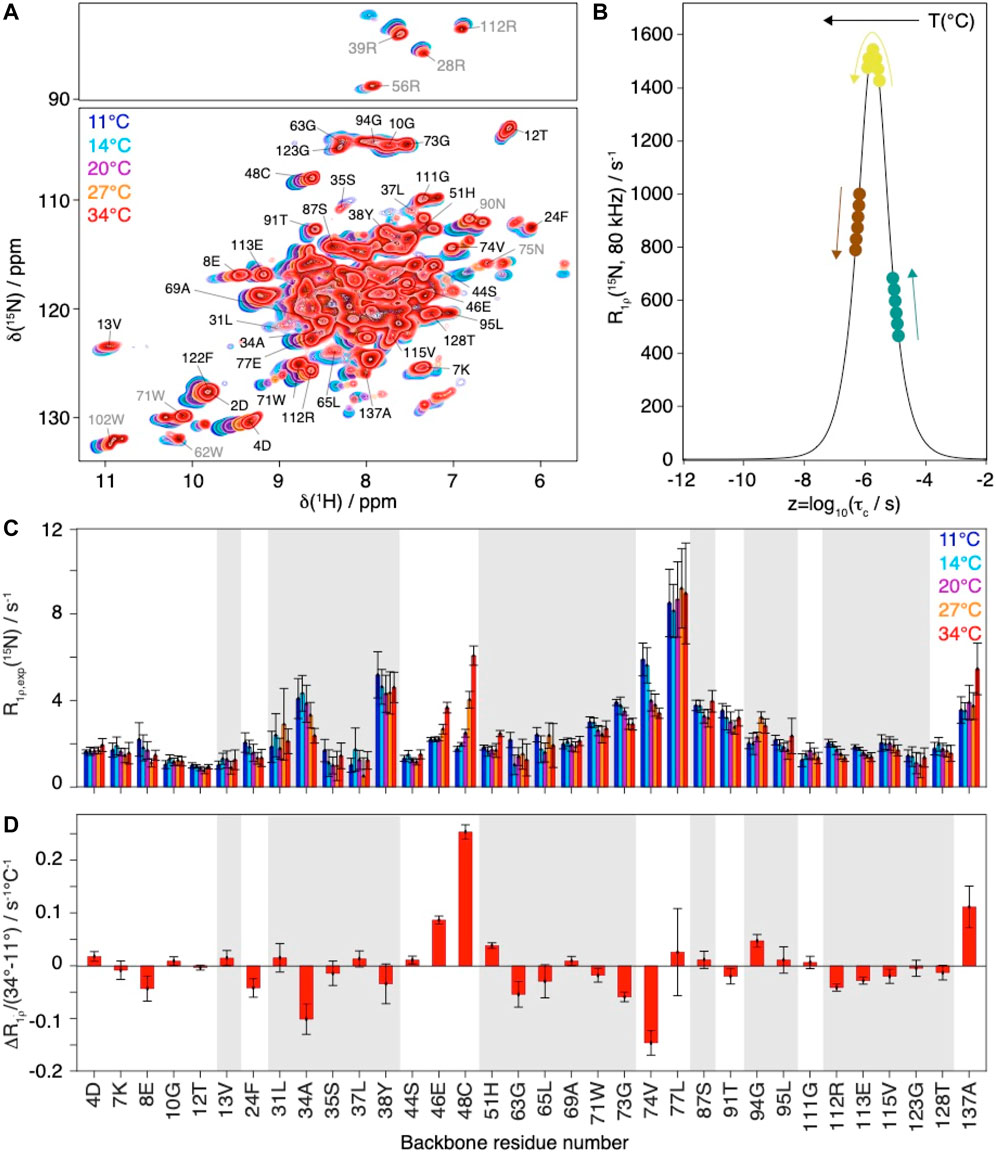
FIGURE 3. Evolution of Cp149 relaxation-rate constants as function of the temperature. (A) Overlay of 2D hNH spectra of Cp149 capsids recorded at 80 kHz MAS, 28 scans and 15.8 ms acquisition in the 15N dimension at 11°C (blue), 14°C (cyan), 20°C (purple), 27°C (orange) and 34°C (red). The assignment is transferred from Lecoq et al. (Lecoq et al., 2019). (B) Normalized analytical R1ρ (15N, νr = 80 kHz, ν1 = 13 kHz) rate constant as function of the correlation time. The colored dots and arrows indicate the evolution of the rate constant with temperature depending on its original position on the curve. (C) Selected experimental site-specific values of R1ρ (15N, νr = 80 kHz, ν1 = 13 kHz) measured between 11°C (blue) and 34°C (red) for the backbone residues and (D) Site-specific gradients for the change in relaxation rate constant between 34°C and 11°C, ΔR1ρ/ΔT = (R1ρ(34°C) - R1ρ(11°C))/(34°C-11°C). The error bars have been calculated with Gaussian error propagation. In both graphs the bars for residues 2D and 122F have been marked by an (*), due to the relaxation data being biased by the overlap of the two resonances in the hNH at higher temperatures. Grey shaded areas indicate
The relaxation-rate constants for resolved backbone amide HN spins are shown in Figure 3C. In a schematic picture, Figure 3B illustrates that, while motion becomes faster with increasing temperature, the corresponding relaxation-rate constants can increase, decrease or go through a maximum. As discussed above NMR rotating-frame rate constants under MAS become particularly large for motions on timescales with corresponding frequency approaching the order of magnitude of the inverse spinning, typically corresponding to values in the low microsecond timescale. For residues with characteristic motions slower than microseconds, the increase of temperature shifts them closer to the timescale regime which leads to a larger rate constant response under the experimental conditions and would result in a monotonic increase in measured rate constant. Experimentally, we observe all three scenarios (Figure 3): some rate constants increase with increasing temperature (e.g. residues 46E, 48C, 51H, 137A), some show signs of increase followed by a decrease (e.g. residues 37L, 94G and 111G) and some decrease with increasing temperature (e.g. residues 8E, 24F, 34A, 74V, 112R, 113E, 115V). The temperature dependence is further illustrated by comparing the difference in the relaxation-rate constants between 11°C and 34°C in Figure 3D. One can see that most gradients are small, below 0.05°C−1 s−1. A strong increase in relaxation-rate constants with temperature is observed for residues 46E, 48C, and 137A, all located in loop regions. These residues thus show motions with correlation times above 1.9 µs (maximum of the curve in Figure 3B. The
Detector Analysis of NMR Relaxation Data and MD Simulations
In order to compare the NMR relaxation-rate constants with the MD simulation, a detector analysis was performed on both data sets.(Smith et al., 2019). The general procedure is summarized in Figure 4A, distinguishing NMR experiments and MD simulation with red and blue boxes respectively.
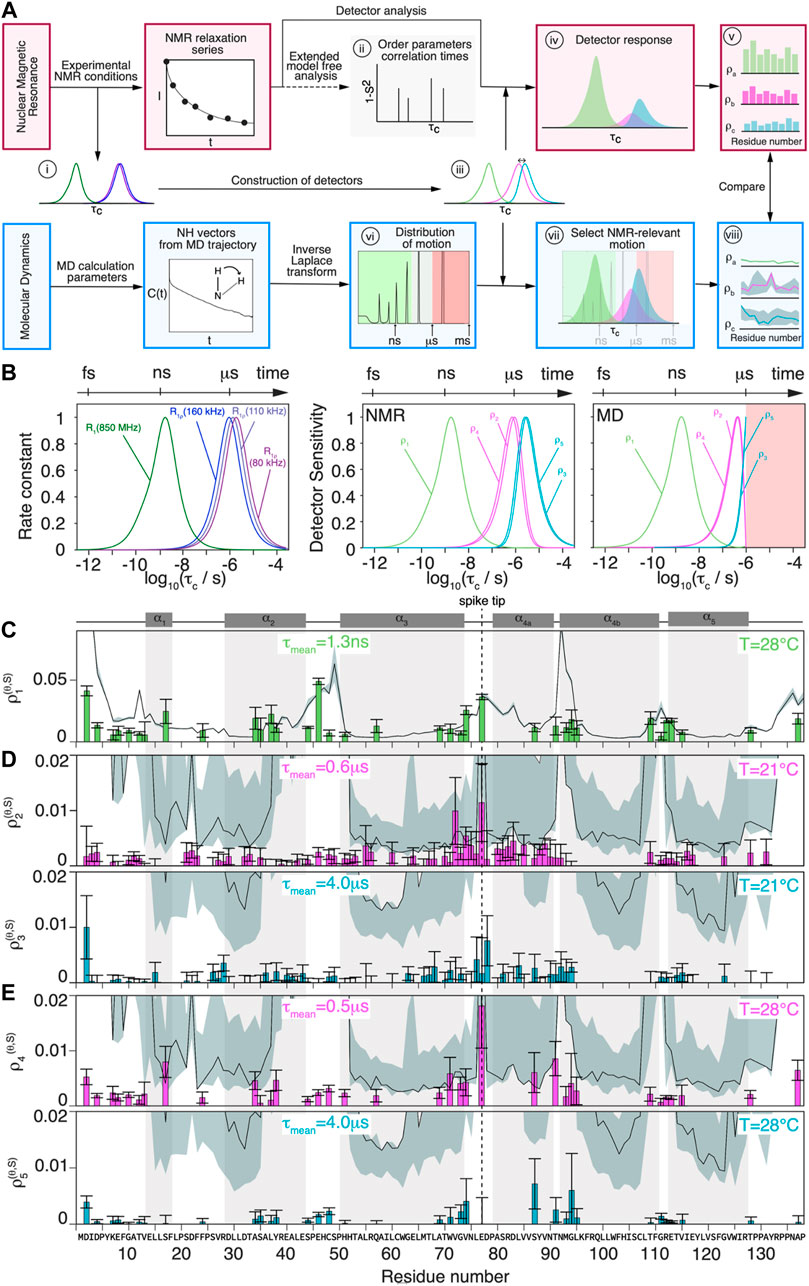
FIGURE 4. Comparison of MD simulation and NMR dynamics detector responses. (A) Schematic flowchart of the method used to obtain detector responses from the NMR data (orange boxes) and MD simulation trajectory (blue boxes). (B) Normalized analytical R1(15N, 86 MHz) and R1ρ (15N, νr = 160/110/80 kHz, ν1 = 13 kHz) rate constants as function of the correlation time (left) and detector sensitivities constructed from linear combination of analytical rate constant functions depicted beside, where
The NMR relaxation-rate constants are sensitive to the motion of an N-H vector with a correlation time τc at a certain amplitude as shown in panel (i) of Figure 4A (note the logarithmic scale in τc). If the order parameter S, which characterizes the amplitude of the motion, is known, τc can in principle be determined from the measured 15N relaxation-rate constants
For a motion with an arbitrary distribution
Figure 4B (left) shows the calculated relaxation-rate constant as function of
The MD detector sensitivities have been obtained allowing an optimization only for a correlation timescale range up to
The detector responses are plotted in Figures 4C–E for all residues that could be evaluated. The detectors used are most sensitive to motions on the nanosecond timescale for R1 and on the microsecond timescale for R1ρ. While the former can be directly compared with the detector responses from the MD simulation, the latter lies, for MD, right at the edge of the sampled timescale. It is clear that the simulation predictions lose their statistical relevance once the timescale approaches the length of the trajectory. We estimate the reliability of the data by an error analysis presented in the Supplementary Information section S1. This information is represented, together with the detector responses from MD simulation, as grey area (error) and solid black line (response) in Figures 4C–E.
Comparison of NMR Relaxation Rates and NMR Detector Response
The detector response of
For the slower motions, the corresponding detector responses
Many mobile residues are located in the tip of the spike (residues 73G to 79P, see Figure 2F). The residues forming the lumen of the capsid, in contrast, stay rather rigid (e.g. 7K-10G, 111G-115V), with the exception of 46E. Other areas of elevated amplitudes of motion on the microsecond timescales are found at the N-terminal part of the protein, in the loop regions between helices
The detector responses from the 2D measurements at 80/160 kHz MAS at 28°C are shown in Figure 4E. Due to the larger separation of spinning frequencies (80/160 kHz), the resulting two detectors are further apart:
Comparison of NMR and MD Detector Responses
One can compare the detector responses obtained by NMR and MD simulation in Figures 4C–E, where simulation detector responses are given as a solid black line, with error estimates shown as grey shaded areas. As expected, the error is substantially larger for the microsecond than for the nanosecond MD detector responses.
For the
The comparison for the MD simulation and NMR predictions at the microsecond timescales (
Assessing the Impact of Sequence Substitutions on NMR Versus MD Detector Responses
Differences in the Cp149 amino-acid sequence could account for part of the disparities in detector responses used to compare NMR and MD simulations for sub-microsecond time: nine residues are different in the MD (strain ADYW) and NMR (strain D) samples (see Figure 1E and Supplementary Figure S15) representing a 6% change in sequence identity. In Figure 1, the difference in RCI between the two X-ray structures which are different in nine residues show only small differences. Still such substitutions may slightly reduce the thermodynamic stability of the simulation model, based on a decrease of up to 5% in the predicted melting temperature index (Ku et al., 2009). The latter suggests the sequence studied with MD simulation may be more responsive to increases in temperature, compared to the one used in NMR, raising the possibility of increased native flexibility at 37°C. Notably, root-mean-square fluctuations calculated from the simulation agree reasonably with crystallographic B-factors extracted for HBV capsid crystal structures of the same sequence. (Hadden et al., 2018). as well as with extracted detector responses (Supplementary Figure S14).
The presence of alanine at S35, C48, C61, and C107 in the simulation model incurs the loss of hydrogen-bonding capability, thus eliminating stabilizing interactions; the shortening of the carboxylate side chains at E40 and E64 likewise alters the potential for salt bridges. A detailed examination of the simulation trajectory suggests that C48, which showed a notable disparity in the detector response, may be in proximity to interact with S44, E46, H47, S49, H52, T53, or R56 in the sequence studied by NMR, and the absence of such contacts may account for enhanced motion of the residue and its parent loop (43E-51H) connecting the
Discussion
The measured solid-state NMR observables (
Detector responses on different timescales of motion were compared between NMR and MD simulation for the Cp149 capsid. The structures shown in Figure 5 graphically summarize and compare the dynamics found by NMR experiments and MD simulation for the intact capsid structure and a single Cp149 dimer, evaluated for the timescale windows accessible by the NMR experiment. While the dimer structures in the first row show the NMR results, the second row displays the MD simulation from (Hadden et al., 2018) for the NMR-visible residues only, and the third row shows the full information available from simulation.
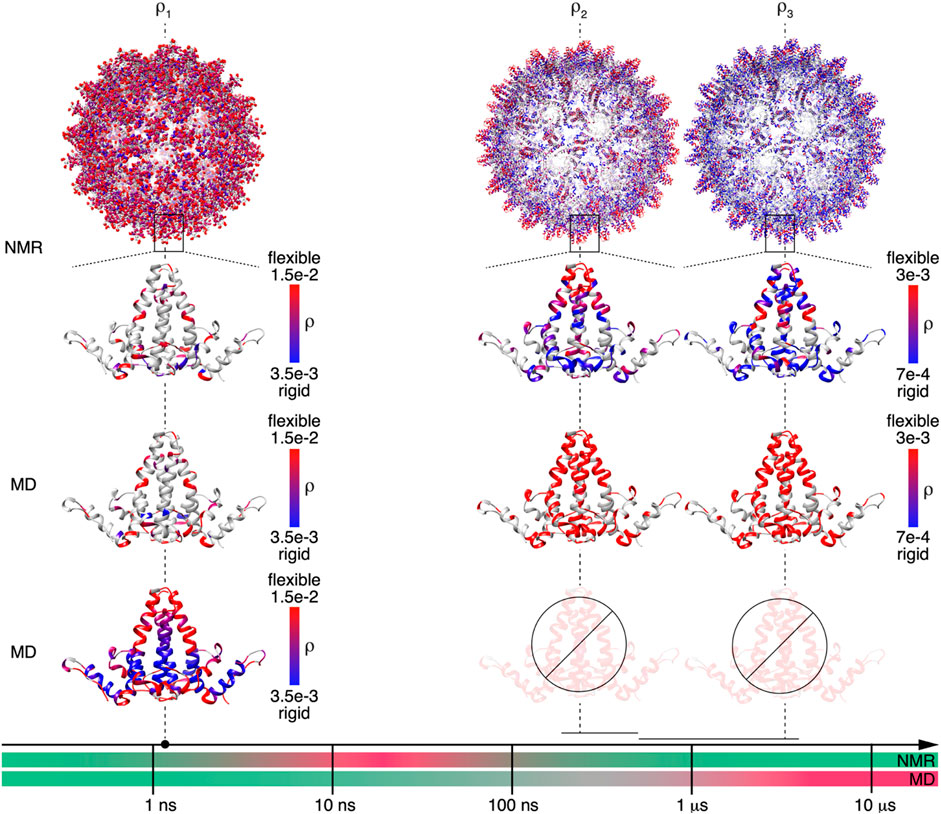
FIGURE 5. NMR and MD are complementary methods for characterizing Cp149 capsid dynamics up to the microsecond timescale. Comparison of the MD and NMR amplitude of motions
On the nanosecond timescale (amplitudes of motion centered at
The faster of the 2 microsecond detectors
Finally, the comparison between MD simulation and NMR for the slower microsecond amplitudes of motion (
In summary, for short timescales, the two methods ideally provide identical results in the time-windows accessible to NMR. In this range, the NMR measurements validate the MD simulation results, which can then be used to characterize additional residues not resolved in the spectra, as well as additional timescales by interpolation/extrapolation. For slower timescales with correlation times approaching the length of the trajectory, the MD simulation becomes less reliable; the error increases significantly already around a timescale of 100 ns, approximately an order of magnitude smaller than the trajectory length (SI section S1). In the present study, the MD simulation data could not be used for extrapolations in timescale windows above 100 ns. In these windows, the predictions lose statistical viability, and in the absence of extended conformational sampling, access to experimental data becomes crucial.
The timescale axis shown in Figure 5 accounts for these observations by subdividing the timescales for the MD simulation predictions in three portions. The green one indicates motions that are well-described by the trajectory (
Conclusions
As predicted by a previous MD simulation study, the HBV capsid is not a rigid “tin can” but indeed shows considerable motion. These motions were detected by NMR on different timescales, from nanoseconds to microseconds. MD simulations and NMR both come to similar conclusions on the dynamics of HBV capsids for short timescales (nanoseconds), which experimentally validates the MD trajectory. Some loops were found to be significantly more rigid than predicted by MD simulation, however, this discrepancy may be, at least in part, attributed to differences in experimental conditions (e.g. temperature) and a 6% difference in sequence identity. The motional parameters for those residues not observed in the NMR spectra, e.g. because of signal overlap, can therefore be accurately estimated from the MD results. Predicting microsecond motion from the MD simulation trajectory is difficult because of insufficient statistics, and a rough error estimation indicates large uncertainties. For the microsecond detectors, the length of the MD trajectory is clearly too short for a good prediction, and for the slower two detectors, the experimental detector responses are more reliable. The accuracy and utility of the combined NMR/MD detector analysis is expected to increase as longer timescale MD simulations of large biomolecular assemblies become more accessible and common.
In summary, we found that for obtaining a full dynamic picture of HBV capsids, a combination of NMR and MD simulations approaches is a promising tool. Since on the faster timescales both methods come to similar results, MD simulations can be used to predict motions in the time window from 10 to 100 ns, where NMR is at the current state of technology has a blind spot. On the timescales from 100 ns-
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author Contributions
AM, MC, AB and BM designed research; AM and MC performed research; SW and LL produced the protein sample; AM, AC, AS, LL, BM and AB analyzed data; CP-S and JAH-P compared to the MD data; AM, MC, AB and BM wrote the paper with contributions of all authors.
Funding
Financial support by an ERC Advanced Grant (BM, grant number 741863, Faster), by the Swiss National Science Foundation (BM, grant number 200020_159707 and 200020_188711), the French Agence Nationale de Recherche (AB, ANR-14-CE09-0024B) and the LABEX ECOFECT (AB, ANR-11-LABX-0048) within the Université de Lyon program Investissements d’Avenir (AB, ANR-11-IDEX-0007) and the DFG (AS-P, SM 576/1-1) are acknowledged. Open access publication fees were covered by the library of ETH Zurich.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2021.807577/full#supplementary-material
References
Agarwal, V., Penzel, S., Székely, K., Cadalbert, R., Testori, E., Oss, A., et al. (2014). De Novo 3D Structure Determination from Sub-Milligram Protein Samples by Solid-State 100 kHz MAS NMR Spectroscopy. Angew. Chem. Int. Ed. 53, 12253–12256. doi:10.1002/anie.201405730
Andreas, L. B., Jaudzems, K., Stanek, J., Lalli, D., Bertarello, A., Le Marchand, T., et al. (2016). Structure of Fully Protonated Proteins by Proton-Detected Magic-Angle Spinning NMR. Proc. Natl. Acad. Sci. USA. 113, 9187–9192. doi:10.1073/pnas.1602248113
Andreas, L. B., Le Marchand, T., Jaudzems, K., and Pintacuda, G. (2015). High-Resolution Proton-Detected NMR of Proteins at Very Fast MAS. J. Magn. Reson. 253, 36–49. doi:10.1016/j.jmr.2015.01.003
Berjanskii, M. V., and Wishart, D. S. (2005). A Simple Method to Predict Protein Flexibility Using Secondary Chemical Shifts. J. Am. Chem. Soc. 127, 14970–14971. doi:10.1021/ja054842f
Berjanskii, M. V., and Wishart, D. S. (2008). Application of the Random Coil Index to Studying Protein Flexibility. J. Biomol. NMR. 40, 31–48. doi:10.1007/s10858-007-9208-0
Bertini, I., Engelke, F., Gonnelli, L., Knott, B., Luchinat, C., Osen, D., et al. (2012). On the Use of Ultracentrifugal Devices for Sedimented Solute NMR. J. Biomol. NMR. 54, 123–127. doi:10.1007/s10858-012-9657-y
Bertini, I., Luchinat, C., Parigi, G., Ravera, E., Reif, B., and Turano, P. (2011). Solid-State NMR of Proteins Sedimented by Ultracentrifugation. Proc. Natl. Acad. Sci. 108, 10396–10399. doi:10.1073/pnas.1103854108
Birnbaum, F., and Nassal, M. (1990). Hepatitis B Virus Nucleocapsid Assembly: Primary Structure Requirements in the Core Protein. J. Virol. 64, 3319–3330. doi:10.1128/JVI.64.7.3319-3330.1990
Böckmann, A., Gardiennet, C., Verel, R., Hunkeler, A., Loquet, A., Pintacuda, G., et al. (2009). Characterization of Different Water Pools in Solid-State NMR Protein Samples. J. Biomol. NMR. 45, 319–327. doi:10.1007/s10858-009-9374-3
Böttcher, B., Vogel, M., Ploss, M., and Nassal, M. (2006). High Plasticity of the Hepatitis B Virus Capsid Revealed by Conformational Stress. J. Mol. Biol. 356, 812–822. doi:10.1016/j.jmb.2005.11.053
Böttcher, B., Wynne, S. A., and Crowther, R. A. (1997). Determination of the Fold of the Core Protein of Hepatitis B Virus by Electron Cryomicroscopy. Nature 386, 88–91. doi:10.1038/386088a0
Bourne, C. R., Finn, M. G., and Zlotnick, A. (2006). Global Structural Changes in Hepatitis B Virus Capsids Induced by the Assembly Effector HAP1. J. Virol. 80, 11055–11061. doi:10.1128/JVI.00933-06
Chevelkov, V., Fink, U., and Reif, B. (2009). Quantitative Analysis of Backbone Motion in Proteins Using MAS Solid-State NMR Spectroscopy. J. Biomol. NMR. 45, 197–206. doi:10.1007/s10858-009-9348-5
Clore, G. M., Szabo, A., Bax, A., Kay, L. E., Driscoll, P. C., and Gronenborn, A. M. (1990). Deviations from the Simple Two-Parameter Model-Free Approach to the Interpretation of Nitrogen-15 Nuclear Magnetic Relaxation of Proteins. J. Am. Chem. Soc. 112, 4989–4991. doi:10.1021/ja00168a070
Fowler, N. J., Sljoka, A., and Williamson, M. P. (2020). A Method for Validating the Accuracy of NMR Protein Structures. Nat. Commun. 11, 6321. doi:10.1038/s41467-020-20177-1
Gallina, A., Bonelli, F., Zentilin, L., Rindi, G., Muttini, M., and Milanesi, G. (1989). A Recombinant Hepatitis B Core Antigen Polypeptide with the Protamine-like Domain Deleted Self-Assembles into Capsid Particles but Fails to Bind Nucleic Acids. J. Virol. 63, 4645–4652. doi:10.1128/JVI.63.11.4645-4652.1989
Gardiennet, C., Schütz, A. K., Hunkeler, A., Kunert, B., Terradot, L., Böckmann, A., et al. (2012). A Sedimented Sample of a 59 kDa Dodecameric Helicase Yields High-Resolution Solid-State NMR Spectra. Angew. Chem. Int. Ed. 51, 7855–7858. doi:10.1002/anie.201200779
Gottlieb, H. E., Kotlyar, V., and Nudelman, A. (1997). NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. J. Org. Chem. 62, 7512–7515. doi:10.1021/jo971176v
Hadden, J. A., Perilla, J. R., Schlicksup, C. J., Venkatakrishnan, B., Zlotnick, A., and Schulten, K. (2018). All-Atom Molecular Dynamics of the HBV Capsid Reveals Insights into Biological Function and Cryo-EM Resolution Limits. eLife. 7, 165101. doi:10.7554/eLife.32478
Han, B., Liu, Y., Ginzinger, S. W., and Wishart, D. S. (2011). SHIFTX2: Significantly Improved Protein Chemical Shift Prediction. J. Biomol. NMR. 50, 43–57. doi:10.1007/s10858-011-9478-4
Henzler-Wildman, K., and Kern, D. (2007). Dynamic Personalities of Proteins. Nature 450, 964–972. doi:10.1038/nature06522
Knight, M. J., Pell, A. J., Bertini, I., Felli, I. C., Gonnelli, L., Pierattelli, R., et al. (2012). Structure and Backbone Dynamics of a Microcrystalline Metalloprotein by Solid-State NMR. Proc. Natl. Acad. Sci. 109, 11095–11100. doi:10.1073/pnas.1204515109
Krushelnitsky, A., Zinkevich, T., Reichert, D., Chevelkov, V., and Reif, B. (2010). Microsecond Time Scale Mobility in a Solid Protein as Studied by the 15N R1ρ Site-Specific NMR Relaxation Rates. J. Am. Chem. Soc. 132, 11850–11853. doi:10.1021/ja103582n
Ku, T., Lu, P., Chan, C., Wang, T., Lai, S., Lyu, P., et al. (2009). Predicting Melting Temperature Directly from Protein Sequences. Comput. Biol. Chem. 33, 445–450. doi:10.1016/j.compbiolchem.2009.10.002
Lakomek, N.-A., Penzel, S., Lends, A., Cadalbert, R., Ernst, M., and Meier, B. H. (2017). Microsecond Dynamics in Ubiquitin Probed by Solid-State 15 N NMR Spectroscopy R 1ρ Relaxation Experiments under Fast MAS (60-110 kHz). Chem. Eur. J. 23, 9425–9433. doi:10.1002/chem.201701738
Lamley, J. M., Öster, C., Stevens, R. A., and Lewandowski, J. R. (2015). Intermolecular Interactions and Protein Dynamics by Solid-State NMR Spectroscopy. Angew. Chem. Int. Ed. 54, 15374–15378. doi:10.1002/anie.201509168
Lecoq, L., Schledorn, M., Wang, S., Smith-Penzel, S., Malär, A. A., Callon, M., et al. (2019). 100 kHz MAS Proton-Detected NMR Spectroscopy of Hepatitis B Virus Capsids. Front. Mol. Biosci. 6, 58. doi:10.3389/fmolb.2019.00058
Lecoq, L., Wang, S., Dujardin, M., Zimmermann, P., Schuster, L., Fogeron, M.-L., et al. (2021). A Pocket-Factor-Triggered Conformational Switch in the Hepatitis B Virus Capsid. Proc. Natl. Acad. Sci. USA. 118, e2022464118. doi:10.1073/pnas.2022464118
Lecoq, L., Wang, S., Wiegand, T., Bressanelli, S., Nassal, M., Meier, B. H., et al. (2018). Solid-State [13C-15N] NMR Resonance Assignment of Hepatitis B Virus Core Protein. Biomol. NMR Assign. 12, 205–214. doi:10.1007/s12104-018-9810-y
Ma, P., Xue, Y., Coquelle, N., Haller, J. D., Yuwen, T., Ayala, I., et al. (2015). Observing the Overall Rocking Motion of a Protein in a Crystal. Nat. Commun. 6, 8361. doi:10.1038/ncomms9361
Penzel, S., Oss, A., Org, M.-L., Samoson, A., Böckmann, A., Ernst, M., et al. (2019). Spinning Faster: Protein NMR at MAS Frequencies up to 126 kHz. J. Biomol. NMR. 73, 19–29. doi:10.1007/s10858-018-0219-9
Pérez-Segura, C., Goh, B. C., and Hadden-Perilla, J. A. (2021). All-Atom MD Simulations of the HBV Capsid Complexed with AT130 Reveal Secondary and Tertiary Structural Changes and Mechanisms of Allostery. Viruses 13, 564. doi:10.3390/v13040564
Perilla, J. R., Goh, B. C., Cassidy, C. K., Liu, B., Bernardi, R. C., Rudack, T., et al. (2015). Molecular Dynamics Simulations of Large Macromolecular Complexes. Curr. Opin. Struct. Biol. 31, 64–74. doi:10.1016/j.sbi.2015.03.007
Quinn, C. M., and McDermott, A. E. (2012). Quantifying Conformational Dynamics Using Solid-State R1ρ Experiments. J. Magn. Reson. 222, 1–7. doi:10.1016/j.jmr.2012.05.014
Rovó, P., Smith, C. A., Gauto, D., de Groot, B. L., and Linser, R. (2019). Mechanistic Insights into Microsecond Time-Scale Motion of Solid Proteins Using Complementary 15N and 1H Relaxation Dispersion Techniques. J. Am. Chem. Soc. 141, 858–869. doi:10.1021/jacs.8b09258
Schanda, P., and Ernst, M. (2016). Studying Dynamics by Magic-Angle Spinning Solid-State NMR Spectroscopy: Principles and Applications to Biomolecules. Prog. Nucl. Magn. Reson. Spectrosc. 96, 1–46. doi:10.1016/j.pnmrs.2016.02.001
Schanda, P., Meier, B. H., and Ernst, M. (2010). Quantitative Analysis of Protein Backbone Dynamics in Microcrystalline Ubiquitin by Solid-State NMR Spectroscopy. J. Am. Chem. Soc. 132, 15957–15967. doi:10.1021/ja100726a
Schledorn, M., Malär, A. A., Torosyan, A., Penzel, S., Klose, D., Oss, A., et al. (2020). Protein NMR Spectroscopy at 150 kHz Magic-Angle Spinning Continues to Improve Resolution and Mass Sensitivity. ChemBioChem. 21, 2540–2548. doi:10.1002/cbic.202000341
Schubeis, T., Le Marchand, T., Daday, C., Kopec, W., Tekwani Movellan, K., Stanek, J., et al. (2020). A β-barrel for Oil Transport through Lipid Membranes: Dynamic NMR Structures of AlkL. Proc. Natl. Acad. Sci. USA. 117, 21014–21021. doi:10.1073/pnas.2002598117
Sherman, M. B., Smith, H. Q., and Smith, T. J. (2020). The Dynamic Life of Virus Capsids. Viruses 12, 618–621. doi:10.3390/v12060618
Shi, C., Öster, C., Bohg, C., Li, L., Lange, S., Chevelkov, V., et al. (2019). Structure and Dynamics of the Rhomboid Protease GlpG in Liposomes Studied by Solid-State NMR. J. Am. Chem. Soc. 141, 17314–17321. doi:10.1021/jacs.9b08952
Smith, A. A., Ernst, M., and Meier, B. H. (2017). Because the Light Is Better Here: Correlation-Time Analysis by NMR Spectroscopy. Angew. Chem. Int. Ed. 56, 13590–13595. doi:10.1002/anie.201707316
Smith, A. A., Ernst, M., and Meier, B. H. (2018). Optimized "detectors" for Dynamics Analysis in Solid-State NMR. J. Chem. Phys. 148, 045104–045118. doi:10.1063/1.5013316
Smith, A. A., Ernst, M., Riniker, S., and Meier, B. H. (2019). Localized and Collective Motions in HET-S(218-289) Fibrils from Combined NMR Relaxation and MD Simulation. Angew. Chem. Int. Ed. 58, 9383–9388. doi:10.1002/anie.201901929
Smith, A. A. (2017). INFOS: Spectrum Fitting Software for NMR Analysis. J. Biomol. NMR. 67, 77–94. doi:10.1007/s10858-016-0085-2
Smith, A. A., Testori, E., Cadalbert, R., Meier, B. H., and Ernst, M. (2016). Characterization of Fibril Dynamics on Three Timescales by Solid-State NMR. J. Biomol. NMR. 65, 171–191. doi:10.1007/s10858-016-0047-8
Stevens, T. J., Fogh, R. H., Boucher, W., Higman, V. A., Eisenmenger, F., Bardiaux, B., et al. (2011). A Software Framework for Analysing Solid-State MAS NMR Data. J. Biomol. NMR. 51, 437–447. doi:10.1007/s10858-011-9569-2
Summers, J., and Mason, W. S. (1982). Replication of the Genome of a Hepatitis B-Like Virus by Reverse Transcription of an RNA Intermediate. Cell. 29, 403–415. doi:10.1016/0092-8674(82)90157-x
Tollinger, M., Sivertsen, A. C., Meier, B. H., Ernst, M., and Schanda, P. (2012). Site-Resolved Measurement of Microsecond-To-Millisecond Conformational-Exchange Processes in Proteins by Solid-State NMR Spectroscopy. J. Am. Chem. Soc. 134, 14800–14807. doi:10.1021/ja303591y
Vasa, S. K., Rovó, P., and Linser, R. (2018). Protons as Versatile Reporters in Solid-State NMR Spectroscopy. Acc. Chem. Res. 51, 1386–1395. doi:10.1021/acs.accounts.8b00055
Vranken, W. F., Boucher, W., Stevens, T. J., Fogh, R. H., Pajon, A., Llinas, M., et al. (2005). The CCPN Data Model for NMR Spectroscopy: Development of a Software Pipeline. Proteins 59, 687–696. doi:10.1002/prot.20449
Wang, S., Fogeron, M.-L., Schledorn, M., Dujardin, M., Penzel, S., Burdette, D., et al. (2019). Combining Cell-Free Protein Synthesis and NMR into a Tool to Study Capsid Assembly Modulation. Front. Mol. Biosci. 6, 67–11. doi:10.3389/fmolb.2019.00067
Wynne, S. A., Crowther, R. A., and Leslie, A. G. W. (1999). The Crystal Structure of the Human Hepatitis B Virus Capsid. Mol. Cel. 3, 771–780. doi:10.1016/s1097-2765(01)80009-5
Yu, X., Jin, L., Jih, J., Shih, C., and Hong Zhou, Z. (2013). 3.5Å cryoEM Structure of Hepatitis B Virus Core Assembled from Full-Length Core Protein. PLoS ONE 8, e69729–11. doi:10.1371/journal.pone.0069729
Zhang, H., Hou, G., Lu, M., Ahn, J., Byeon, I. -J. L., Langmead, C. J., et al. (2016). HIV-1 Capsid Function Is Regulated by Dynamics: Quantitative Atomic-Resolution Insights by Integrating Magic-Angle-Spinning NMR, QM/MM, and MD. J. Am. Chem. Soc. 138, 14066–14075. doi:10.1021/jacs.6b08744
Zhao, Z., Wang, J. C.-Y., Segura, C. P., Hadden-Perilla, J. A., and Zlotnick, A. (2020). The Integrity of the Intradimer Interface of the Hepatitis B Virus Capsid Protein Dimer Regulates Capsid Self-Assembly. ACS Chem. Biol. 15, 3124–3132. doi:10.1021/acschembio.0c00277
Zlotnick, A., Cheng, N., Stahl, S. J., Conway, J. F., Steven, A. C., and Wingfield, P. T. (1997). Localization of the C Terminus of the Assembly Domain of Hepatitis B Virus Capsid Protein: Implications for Morphogenesis and Organization of Encapsidated RNA. Proc. Natl. Acad. Sci. 94, 9556–9561. doi:10.1073/pnas.94.18.9556
Keywords: solid-state NMR, virus, dynamics, relaxation, molecular dynamics
Citation: Malär AA, Callon M, Smith AA, Wang S, Lecoq L, Pérez-Segura C, Hadden-Perilla JA, Böckmann A and Meier BH (2022) Experimental Characterization of the Hepatitis B Virus Capsid Dynamics by Solid-State NMR. Front. Mol. Biosci. 8:807577. doi: 10.3389/fmolb.2021.807577
Received: 02 November 2021; Accepted: 06 December 2021;
Published: 03 January 2022.
Edited by:
Steffen P. Graether, University of Guelph, CanadaReviewed by:
Eric R. May, University of Connecticut, United StatesRachel Wagner Martin, University of California, Irvine, United States
Copyright © 2022 Malär, Callon, Smith, Wang, Lecoq, Pérez-Segura, Hadden-Perilla, Böckmann and Meier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anja Böckmann, YS5ib2NrbWFubkBpYmNwLmZy; Beat H. Meier, YmVtZUBldGh6LmNo
†These authors have contributed equally to this work