 Jill L. Johnson
Jill L. Johnson- Department of Biological Sciences and Center for Reproductive Biology, University of Idaho, Moscow, ID, United States
The Hsp90 molecular chaperone, along with a set of approximately 50 cochaperones, mediates the folding and activation of hundreds of cellular proteins in an ATP-dependent cycle. Cochaperones differ in how they interact with Hsp90 and their ability to modulate ATPase activity of Hsp90. Cochaperones often compete for the same binding site on Hsp90, and changes in levels of cochaperone expression that occur during neurodegeneration, cancer, or aging may result in altered Hsp90-cochaperone complexes and client activity. This review summarizes information about loss-of-function mutations of individual cochaperones and discusses the overall association of cochaperone alterations with a broad range of diseases. Cochaperone mutations result in ciliary or muscle defects, neurological development or degeneration disorders, and other disorders. In many cases, diseases were linked to defects in established cochaperone-client interactions. A better understanding of the functional consequences of defective cochaperones will provide new insights into their functions and may lead to specialized approaches to modulate Hsp90 functions and treat some of these human disorders.
Introduction
Cytosolic Hsp90 is an abundant and essential molecular chaperone that mediates the folding of hundreds of cellular proteins, called clients. Hsp90 inhibition directly or indirectly impacts the function of up to 10–15% of all human proteins (Wu et al., 2012). Humans have two isoforms of cytosolic Hsp90: Hsp90α (encoded by HSP90AA1), and Hsp90β (encoded by HSP90AB1) (Chen et al., 2006). While both isoforms are abundantly expressed, there are some tissue-specific differences in expression (Dean and Johnson, 2021). In addition to Hsp90, client proteins interact with the molecular chaperone Hsp70 in an ordered cycle characterized by the presence of differing cochaperones. These cochaperones have a wide variety of functions, such as modulating Hsp90 ATPase activity or conformational changes or targeting clients to Hsp90. The most complete model of Hsp90 function is based on an ATP-dependent cycle of glucocorticoid receptor (GR) folding. Unfolded GR bound to Hsp70 progresses through a loading complex consisting of Hsp70, Hsp90 and Hop (encoded by STI1P) to a mature complex consisting of folded GR, Hsp90, and p23 (encoded by PTGES3). Additional cochaperones also interact with GR and influence its function (Panaretou et al., 2002; Pratt and Toft, 2003; Kirschke et al., 2014; Schopf et al., 2017). Altered cochaperone levels and/or complexes have dramatic effects on GR activity (Reynolds et al., 1999; Echeverria et al., 2016).
As the understanding of the range of Hsp90 functions has expanded (Gvozdenov et al., 2019), the number of identified Hsp90 cochaperones has also increased. There are roughly 50 human cochaperones that vary in client specificity, abundance, and tissue distribution (Taipale et al., 2014; Dean and Johnson, 2021). These cochaperones are roughly grouped by how they interact with Hsp90. One large group of contains a p23-like domain that shares homology with crystallin, termed a CS domain (Garcia-Ranea et al., 2002), which interacts with the amino terminus of Hsp90. Two of the main proteins in this group are p23 and Sgt1 (encoded by SUGT1). Although each of these cochaperones share a similar domain, they differ with regard for preference for the ATP-bound or nucleotide-free conformation of Hsp90 and client preference (Ali et al., 2006; Zhang et al., 2008).
Another large group, which contains Hop, contains tetratricopeptide repeat (TPR) or similar domains, and interacts with the carboxy-terminus of Hsp90 (Schopf et al., 2017). Although they share a common binding site, cochaperones in this group bind different Hsp90 conformations and have differing impacts on client function and Hsp90 ATPase activity (Prodromou et al., 1999; Riggs et al., 2004). A third set, which interacts primarily with the middle domain, does not contain either TPR or CS domains. This set includes Aha1, which strongly activates ATPase activity of Hsp90 (Panaretou et al., 2002). Some of the other cochaperones in this group regulate Hsp90, in part, by competing with Aha1 for Hsp90 interaction (Woodford et al., 2016). Studies in yeast suggest that a small set of cochaperones are part of the core Hsp90 machinery required for the folding of a wide range of clients, while others may have functions restricted to a few clients (Sahasrabudhe et al., 2017; Biebl et al., 2020). In humans, some cochaperones preferentially interact with one Hsp90 isoform or the other, which further contributes to the specificity of their effects (Chadli et al., 2008; Echeverria et al., 2016).
The functions of Hsp90 and cochaperones in cancer cells is well established and has been the subject of recent reviews (Calderwood and Gong, 2016; Vartholomaiou et al., 2016). Mutations in many cochaperones have been identified in tumor cells (https://www.cbioportal.org/). However, a detailed analysis of those changes is beyond the scope of this review. Changes in the expression of some of the cochaperones during aging or neurodegenerative disease has been noted (Brehme et al., 2014; Herold et al., 2016; Calderwood and Murshid, 2017; Lackie et al., 2017; Shelton et al., 2017; Bohush et al., 2019). In addition to those disorders listed above, altered cochaperone levels have been linked to inflammatory and heart diseases, asthma susceptibility, and dementia (Metcalfe et al., 2005; Sinclair et al., 2013; Patel et al., 2016; Bonham et al., 2018; Lee et al., 2019; Huang et al., 2020; Salhi et al., 2021). While most of the observed changes are at the transcriptional level, copy number variations in HSP90AB1 and some cochaperones have been identified (Metcalfe et al., 2005; Tomita-Mitchell et al., 2012; Zhang et al., 2019; Xu et al., 2021).
Mutations in genes encoding chaperones, most notably mitochondrial chaperonins Hsp60 and Hsp10, which are homologous to bacterial GroEL/GroES, and the small Hsp, Hsp27, have been previously found to cause a variety of pathologic conditions such as abnormalities in the nervous, muscular, or other tissues. These disorders have been termed chaperonopathies, and since most chaperones assist the folding of multiple proteins, the specific phenotypes of a chaperonopathy will depend on what function is impaired or abolished. (Macario et al., 2005; Macario et al., 2010; Cappello et al., 2013; Cappello et al., 2014; Lupo et al., 2016; Bulone et al., 2019; Comert et al., 2019). The goal of this review is to take advantage of recent advances and dissemination of genome sequencing technology to examine defects associated with mutation or deletion of specific human Hsp90 cochaperones. The results link chaperonopathies and altered Hsp90 cochaperone function. The variety of phenotypes demonstrates the unique functions of individual cochaperones and helps identify Hsp90-client-cochaperone interactions critical for human health.
Disorders Linked to Mutations in Cochaperone Proteins
The reasons that cytosolic Hsp90 requires such large group of cochaperones is largely unknown. There is evidence that cochaperones exhibit client specificity, which suggests that the large number of cochaperones increases the Hsp90 clientele (Taipale et al., 2014). However, the folding pathway has been elucidated for only a small number of clients and it is largely unknown whether there are truly distinct folding pathways for diverse clients. As an alternate approach to identify unique functions of cochaperones, a literature review was used to identify whether disease-linked mutations have been found in any of the roughly 50 human cochaperones. Mutations in about 20 different cochaperones were found. Further analysis of the role of individual cochaperones in disease could help elucidate functions of both Hsp90 and cochaperones. In some cases, specific disorders were linked to mutations in single cochaperones, highlighting unique cochaperone functions. Since many cochaperones share common Hsp90 interacting domains, analysis of these interactions may be useful to understand the basis of selective cochaperone effects. As summarized in Table 1, the identified mutations result in a variety of phenotypes that affect heart, muscle, eye, and brain functions. For simplicity, cochaperones are identified by the gene name unless there is specific mention of the encoded protein.
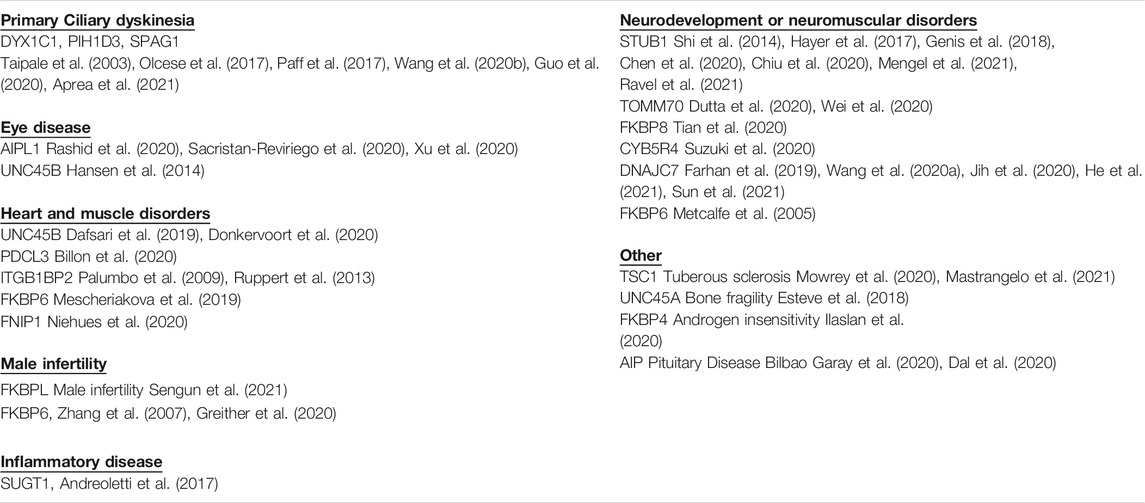
TABLE 1. Hsp90 cochaperone-encoding genes with mutations linked to disease.
Cilia Defects
A recent review of discussed the role of Hsp90 cochaperones in the assembly of dynein arm complexes (Fabczak and Osinka, 2019). Primary ciliary dyskinesia results from defective cilia and flagella beating and is characterized by airway disease, infertility, and laterality defects. Mutations in DYX1C1, PIH1D3 and SPAG1 result in primary ciliary dyskinesia. Most are deletion or truncation mutants that disrupt protein production (Taipale et al., 2003; Olcese et al., 2017; Paff et al., 2017; Guo et al., 2020; Wang et al., 2020b; Aprea et al., 2021). However, an inactivating missense mutation in PIH1D3 alters a conserved residue within the Hsp90-interacting CS domain (Olcese et al., 2017). Additional cochaperones with suspected links to cilia-related diseases are encoded by PIH1D1, PIH1D2, SUGT1, NUDC, NUDCD3, and RPAP3 ((Fabczak and Osinka, 2019) and (https://diseases.jensenlab.org/Search)) (Table 2). Studies with model organisms containing mutations or deletions of some of these cochaperones confirm the presence of ciliary defects (Chandrasekar et al., 2013; Tarkar et al., 2013). This indicates that a significant part of the Hsp90 machinery cooperates in the assembly of dynein complexes.
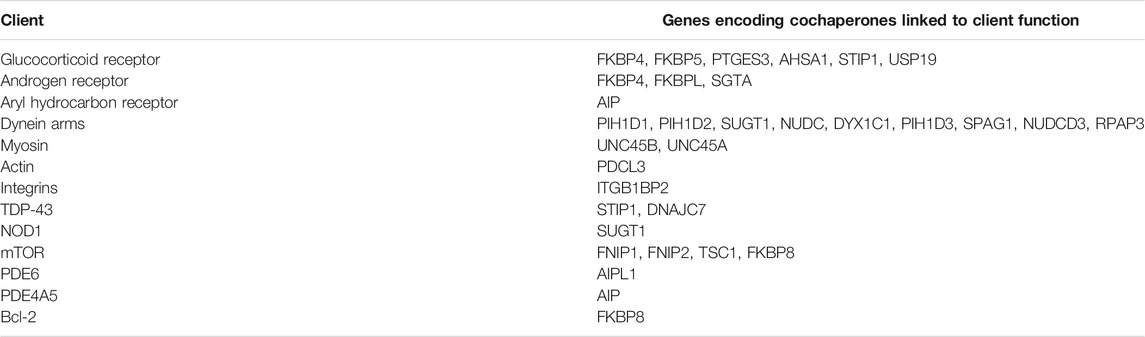
TABLE 2. Cochaperone-client associations linked to human disorders.
Leber Congenital Amaurosis
AIPL1 (encoded by AIPL1) cooperates with Hsp70 and Hsp90 to facilitate folding and assembly of retinal cGMP phosphodiesterase (PDE6) (Hidalgo-de-Quintana et al., 2008; Sacristan-Reviriego and van der Spuy, 2018). PDE6 is a known Hsp90 client, and prolonged treatment with Hsp90 inhibitors results in reduced PDE6 levels (Aguila et al., 2014). Mutations in AIPL1 are associated with Leber congenital amaurosis, a severe form of inherited retinal degeneration (Yadav et al., 2017; Rashid et al., 2020; Sacristan-Reviriego et al., 2020; Xu et al., 2020). Mice lacking AIPL1 serve as a disease model of Leber congenital amaurosis. Retinas from AIPL1 knockout mice exhibit rapid degeneration of both rods and cones and destabilization of cGMP phosphodiesterase (Ramamurthy et al., 2004; Singh et al., 2014). Multiple missense mutations in AIPL1 have been analyzed to determine their effect on Hsp90 interaction and/or PDE6 functions (Sacristan-Reviriego et al., 2020). Mutations that altered either the FKBP domain or the TPR domain of AIPL1 resulted in reduced Hsp90 interaction and reduced PDE6 activity in cells coexpressing AIPL1 and PDE6. Thus, both the FKBP and TPR domains are critical for AIPL1 function.
Neurodevelopmental and Neurodegeneration Disorders
CHIP (encoded by STUB1) is a ubiquitin ligase that plays a key role in regulating targeting misfolded Hsp90 clients for degradation (Connell et al., 2001). Mutations in STUB1 have been found in patients with both recessive and dominant forms of ataxia, which is characterized by a lack of muscle control or coordination of voluntary movements (Ravel et al., 2021). Some other mutations cause ataxia plus additional severe phenotypes (Shi et al., 2014; Hayer et al., 2017). Mice lacking the gene exhibit defects in motor and sensory function, plus additional defects (Shi et al., 2014). The disease correlates with overall loss of CHIP function, and missense mutations in the TPR domain or the ubiquitin-ligase domain are known to affect localization and/or activity (Shi et al., 2014; Hayer et al., 2017; Genis et al., 2018; Chen et al., 2020; Chiu et al., 2020; Mengel et al., 2021; Pakdaman et al., 2021).
TOMM70 encodes a protein that is part of the mitochondrial preprotein import machinery (Young et al., 2003). Missense mutations in TOMM70 cause ataxia combined with white matter abnormalities and other defects. Both of those mutations exhibited loss of function in an animal model (Dutta et al., 2020). Additional TOMM70 variants have been linked to reduced overall mitochondrial and OXPHOS deficiencies, resulting in severe anemia, lactic acidosis, and developmental delay (Wei et al., 2020). Some of the mutations alter residues in the TPR domains, although direct analysis of their impact on Hsp90 interaction is unknown.
Mutations in a different TPR-containing cochaperone, FKBP38 (encoded by FKBP8), have been identified as risk factors for spina bifida, a neural tube defect. FKBP38 localizes to the mitochondria and helps regulate apoptosis through Bcl-2 interaction (Shirane and Nakayama, 2003). Mice deficient in FKBP8 exhibited neural tube defects due to unrestrained apoptosis (Shirane-Kitsuji and Nakayama, 2014). Each of the mutants identified resulted in an increase in cellular apoptosis and/or altered FKBP38 levels. The structure of FKBP38 bound to the TPR acceptor site is known (Blundell et al., 2017), and two of the mutants identified altered conserved residues in the TPR domain (Tian et al., 2020).
DNAJC7 encodes TPR2, a protein that contains TPR domains in addition to a J domain, a domain involved in Hsp70 interaction (Brychzy et al., 2003). Mutations in DNAJC7 have been linked to amyotrophic lateral sclerosis, a progressive nervous system disease that affects nerve cells in the brain and spinal cord. Most of observed mutations were protein-truncating variants, and in some cases protein loss was confirmed (Farhan et al., 2019; Jih et al., 2020; He et al., 2021). However, one missense mutation that alters a residue in one of the TPR domains was found (Wang et al., 2020a). A specific role for TPR2 in pathogenic events has not yet been identified, but yeast models suggest a role for Hsp90 and cochaperones in TDP-43 misfolding, a hallmark of the disease (Lin et al., 2021).
NADH cytochrome b5 oxidoreductase (Ncb5or) protects beta-cells against oxidative stress and lipotoxicity (Kalman et al., 2013). In mice, loss of Ncb5or has been linked to insulin-deficient diabetes and other deficiencies (Xie et al., 2004; Stroh et al., 2016; Stroh et al., 2018) Ncb5or contains a CS domain that is shared with other Hsp90 cochaperones, but its functional interaction with Hsp90 has not been characterized (Benson et al., 2019). A mutation in CYB5R4 that leads to a truncated version of Ncb5or has been implicated in neurodevelopmental disorders (Suzuki et al., 2020), but a potential mechanistic defect is unknown.
Heart and Muscle Disorders
The basic mechanism in muscle involves the interaction of the protein filaments myosin and actin, both of which are affected by mutations in Hsp90 cochaperones. The TPR-containing cochaperones encoded by UNC45A and UNC45B promote myosin folding (Lee et al., 2014; Lehtimaki et al., 2017; Bujalowski et al., 2018). Mutations that alter the myosin-binding domain of UNC45B were found in a patient with congenital muscle disorder (Dafsari et al., 2019). Additional mutations were associated with progressive myopathy (Donkervoort et al., 2020). Yet another mutation in UNC45B resulted in cataracts, a result supported by studies in an animal model (Hansen et al., 2014). Mutations in UNC45A, which encodes GCUNC45, a related cochaperone, were associated with a range of symptoms, including impaired hearing and bone fragility (Esteve et al., 2018).
PhLP2A (encoded by PDCL3) is an Hsp90 interacting protein that does not contain either TPR or CS domain that is involved in the folding of actin (Krzemien-Ojak et al., 2017). Loss of function alleles of PDCL3 were identified in patients with megacystis-microcolon-intestinal-hypoperistalsis syndrome, which affects muscles in the bladder and intestines (Billon et al., 2020). Integrins are transmembrane links between extracellular contacts and the actin microfilaments. Melusin (encoded by ITGB1BP2) is a muscle-specific integrin-interacting protein that contain a CS domain and has important cardioprotective functions (Brancaccio et al., 2003; Sbroggio et al., 2008; Tarone and Brancaccio, 2015). Mutations in ITGB1BP2 have been detected in families of patients affected by hypertension or cardiomyopathy (Palumbo et al., 2009; Ruppert et al., 2013).
Loss of folliculin interacting protein 1 (encoded by FNIP1) due to inactivating mutations results in immunodeficiency and heart defects due to disruption of essential metabolic regulators AMPK and mTOR (Saettini et al., 2021). Hsp90 inhibition is known to target multiple components of mTOR signaling (Giulino-Roth et al., 2017). One of the FNIP1 mutations associated with disease is a predicted missense mutation in a domain known to be involved in regulation of Hsp90 interaction and FNIP stability (Sager et al., 2019; Niehues et al., 2020), suggesting that this alteration affects Hsp90-related functions. Two additional cochaperones, encoded by FNIP2 and TSC1, are also involved in mTOR signaling, and the three all cochaperones function to inhibit Hsp90 (Woodford et al., 2016; Woodford et al., 2017; Sager et al., 2018; Sager et al., 2019). Mutation of TSC1 is associated with tuberous sclerosis complex, an autosomal dominant multisystem disorder that affects the brain, skin, heart, kidneys, and lung (Mallela and Kumar, 2021). Most of the mutations disrupt protein production, but a few examples of missense mutations in the region implicated in Hsp90 interaction have been identified (Bykhovskaya et al., 2017; Woodford et al., 2017).
Mutations in FK506-Binding Proteins have Been Linked to Various Disorders
One set of cochaperones, including the ones encoded by FKBP8 and AIPL1, which were described above, contains a FK506-binding domain in addition to TPR domains (Annett et al., 2020). A mutation that alters a residue in the TPR domain of FKBP6 was identified in patients with multiple sclerosis, but it has not been conclusively linked to disease (Mescheriakova et al., 2019). Other genetic variations in FKBP6 have been linked to the neurodevelopmental disorder Williams-Beuren syndrome (Metcalfe et al., 2005), congenital heart malformations (Tomita-Mitchell et al., 2012) and male infertility (Zhang et al., 2007; Greither et al., 2020). Loss of function alleles of FKBPL have also been associated with male infertility (Sengun et al., 2021), and a mutation in FKBP4 has been associated with androgen insensitivity (Ilaslan et al., 2020).
Aryl hydrocarbon receptor interacting protein AIP is similar to AIPL1. Mutations in the AIP gene are the most frequent genetic cause for familial isolated pituitary adenomas, which predispose individuals to acromegaly and gigantism (Bilbao Garay et al., 2020). Mutations that disrupted the ability of AIP to interact with clients, such as the aryl hydrocarbon receptor and phosphodiesterase 4A5 were linked to disease (Morgan et al., 2012).
Link Between Cochaperones and Inflammatory Disease
Inflammatory bowel disease is an umbrella term for a group of illnesses that includes Crohn’s disease, ulcerative colitis, and other diseases. Mutations that affect the NOD signaling pathway are a major contributor to the disease. The Sgt1 cochaperone (encoded by SUGT1) is essential for NOD1 activation (da Silva Correia et al., 2007). SUGT1 mutations have been identified but not yet definitely linked to disease. One of those is located in a TPR domain (Andreoletti et al., 2017). Variation in the cochaperones encoded by PTGES3L and FKBPL has also been potentially linked to various inflammatory disease (Hocevar et al., 2018; Bianchi et al., 2021), but more analysis is required. Single-nucleotide variants in the HSP90AB1 and HSP90B1 genes have been proposed to modulate chronic obstructive pulmonary disease (Ambrocio-Ortiz et al., 2021), and Hsp90 is known to regulate inflammatory processes (Nizami et al., 2021). More research is needed to understand the role of Hsp90 and cochaperones in inflammatory disease.
Mutations in Cochaperones for the Hsp90 Isoform in the Endoplasmic Reticulum have Also Been Associated With Disease
While the main focus of this review is on cytosolic Hsp90, higher eukaryotes express Hsp90 isoforms in both the mitochondria (TRAP1) and the ER (GRP94) (Chen et al., 2006; Yoshida et al., 2013). Both TRAP1 and GRP94 have important functions and there is interest in developing isoform specific inhibitors (Seo, 2015; Sanchez-Martin et al., 2020). These isoforms of Hsp90 do not require a large number of cochaperones. However, there are a few examples of cochaperones that play critical roles in client maturation. GRP94 is required for folding of immunoglobulin light chains, Toll-like receptors and other proteins. A few different cochaperones for GRP94 have been identified, including OS-9 (Christianson et al., 2008), MZB1 (Rosenbaum et al., 2014), and CNPY3 (Liu et al., 2010). Mutations in CNPY3 are associated with epileptic encephalopathy (Mutoh et al., 2018), demonstrating that cochaperones for other Hsp90 isoforms also have critical functions relevant to human disease.
Hsp90-Cochaperone-Client Interactions Relevant to Human Disorders
Hsp90 cochaperones are a diverse group of proteins that, in addition to the Hsp90 interacting domain, usually contain other domains with known functions. Figure 1 shows the domain structure of cochaperones with missense mutations implicated in disease. In many cases, missense mutations are located within the Hsp90-interacting domain, implying that the defective Hsp90-cochaperone interaction results in disease. However, it should be noted that many of these mutations are newly identified, and more studies are needed to determine whether the mutations alter Hsp90 interaction. It is also possible that the mutations affect Hsp90-independent functions of the cochaperones. Further, some of these cochaperones, including but not limited to TOM70, DNAJC7 and HOP (Scheufler et al., 2000; Young et al., 2003; Timsit and Negishi, 2014; Assimon et al., 2015), interact with both Hsp90 and Hsp70 and thus it is not possible to assign the defects to Hsp90 interaction without further analysis. Finally, it is also possible that the mutations affect the ability of cochaperones to be post-translationally modified. As summarized in recent reviews, there is growing evidence that post-translational modifications on chaperones and cochaperones regulate their interactions and function (Backe et al., 2020; Nitika et al., 2020).
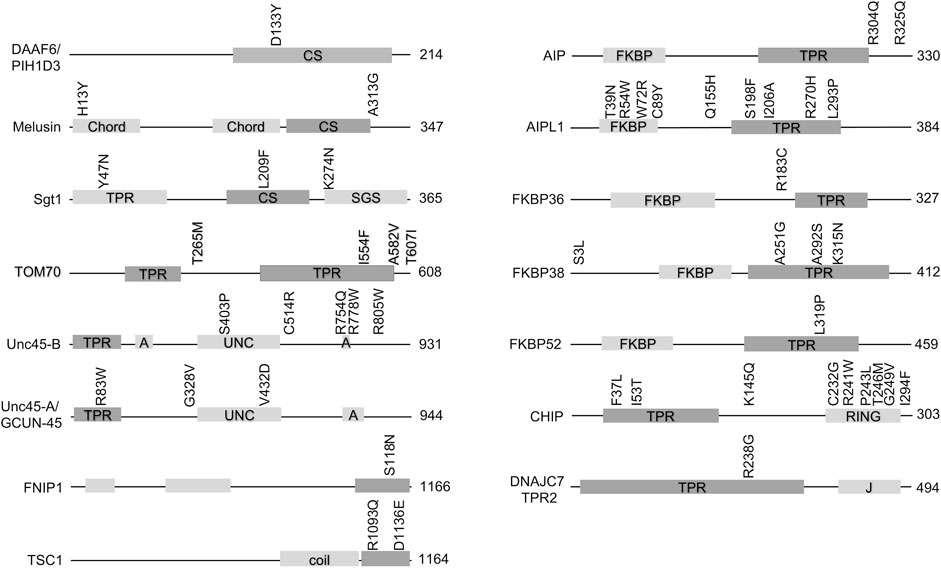
FIGURE 1. Missense mutations in cochaperones linked to human disease. Schematic of cochaperone protein domains and the location of disease-associated mutations. The size of each protein (in amino acids is shown). The domain known or predicted to mediate Hsp90 interaction is shaded in the darker color. Abbreviations: CS= (Chord and Sgt1 like), Chord (Cysteine- and histidine-rich domains), TPR (tetratricopeptide repeat), SGS (Sgt1-specific), A (armadillo), UNC (uncoordinated, myosin-interacting), coil (coiled-coil), FKBP (FK506-binding protein), RING (RING-finger U-box). J = DnaJ-like domain).
Identification of Hsp90-client-cochaperone interactions important for human health may be helpful to develop assays to test for Hsp90 inhibitor selectivity or potential negative side effects. A list of Hsp90 clients known or suspected to be affected by mutation of the cochaperones listed above are summarized in Table 2. In some cases, additional cochaperones are also known to interact with some of those listed. For example, the GR is one of the most widely studied Hsp90 clients, and changes in activity of GR and/or other clients due to altered abundance of associated cochaperones has been linked to mood disorders, autism, anxiety, psychotic illness, depression, and altered pain susceptibility (Sinclair et al., 2013; Patel et al., 2016; Baker et al., 2018; Lee et al., 2019; Lou et al., 2021; Mokha et al., 2021; Salhi et al., 2021; Szczepankiewicz et al., 2021). Similarly, androgen receptor activity is highly dependent on cochaperones encoded by FKBP4, FKBPL, and SGTA (Paul et al., 2014; Ilaslan et al., 2020; Sengun et al., 2021).
Cochaperones Without Identified Human Mutations
Deletion of some mammalian cochaperones results in inviability, while loss of others has mild effects. Mice lacking HSP90AA1 survive to adulthood, but deletion of HSP90AB1, PTGES3 or STIP1 caused embryonic lethality (Grad et al., 2006; Grad et al., 2010; Beraldo et al., 2013). Deletion of SGTA resulted in reduced viability and growth defects (Philp et al., 2016), while deletion of PPP5 or FNIP2 in mice causes mild effects (Amable et al., 2011; Hasumi et al., 2015). In contrast, deletion of USP19 protected mice against muscle wasting in response to glucocorticoids (Bedard et al., 2015). The Genome Aggregation Database (https://gnomad.broadinstitute.org/) is a publicly available database on human genetic variation. Based on the number of protein truncating variants observed in the dataset, each gene has a pLI score that reflects the likely tolerance of gene loss. A score of 0 suggests that loss of the gene may be tolerated, while a score of 1 suggests that it is not tolerated (Karczewski et al., 2020). Although there are known pitfalls of relying too heavily on pLI scores (Ziegler et al., 2019), this provides a useful comparison of the likely effect of cochaperone mutation. As shown in Table 3, available data suggests that deletion of some cochaperones is not tolerated. This includes genes encoding cochaperones like Hop, p23 and Cdc37 that are required for activity of a wide range of Hsp90 clients and likely have essential functions. The yeast homologs of those cochaperones are known to be required for activity of multiple clients (Sahasrabudhe et al., 2017; Schopf et al., 2017; Verba and Agard, 2017). In other cases, there may be functional redundancy with other cochaperones or other cellular pathways that masks the effect of cochaperone mutation. Alternatively, the encoded cochaperone may interact with either a limited set of clients or only nonessential clients.
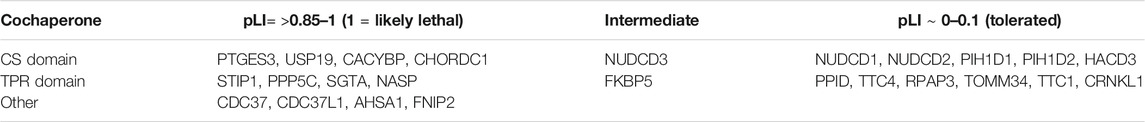
TABLE 3. Hsp90 cochaperones without known genomic mutations linked to disease. pLI (probably of loss interference) numbers are from https://gnomad.broadinstitute.org/.
Conclusion
This review is an attempt to consolidate available information about known genetic variation in genes encoding human Hsp90 cochaperones. One conclusion is that there are many similarities between previously characterized chaperonopathies and disorders linked to Hsp90 cochaperone mutations. This includes ciliary defects, cataracts and other eye diseases, neuropathies or neurodegenerative diseases, and various myopathies (Macario et al., 2005). More research is needed to understand these complex disease associations and determine the extent of functional overlap between different types of chaperones. Another conclusion is that loss of different cochaperones causes distinct effects. This is consistent with prior studies that indicate that even cochaperones that share homologous Hsp90-interacting domains exhibit client specificity (Riggs et al., 2004; Taipale et al., 2014). Another conclusion is that while some processes, such as maturation of the glucocorticoid receptor or the assembly of dynein arms, require groups of cochaperones that may be able to compensate for each other, in other cases, such as with AIPL1, cochaperone function may be limited to a very small number of critical clients. A better understanding of the specific role of AIPL1 in PDE6 function may provide important information about why closely related cochaperones, such as AIP, are apparently unable to compensate for the loss of AIPL1.
Due to the importance of Hsp90 in supporting cancerous growth, development of Hsp90 inhibitors remains a priority as a tool to treat cancer. The general Hsp90 inhibitors that bind the ATP-binding site are associated with unwanted side effects. Alternative strategies include development of Hsp90 isoform specific inhibitors (Khandelwal et al., 2018; Mak et al., 2019; Mishra et al., 2021) or compounds that block functions of specific cochaperones (Guy et al., 2015; Stiegler et al., 2017; Wang et al., 2017; Singh et al., 2020; Serwetnyk and Blagg, 2021). A greater understanding of the possible consequences of cochaperone inhibition is necessary to assess the potential negative side effects of some of these more selective inhibitors of Hsp90 function.
Author Contributions
JLJ conceived the idea for the publication and wrote the manuscript.
Funding
Research in the Johnson lab is supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R01GM127675. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
I wish to thank Ella Dahl, Peyton Hamel, Damian Placencia, and Marissa Dean for assistance in gathering data about cochaperones, and Paul LaPointe and Melody Fulton for helpful comments on the manuscript.
References
Aguilà, M., Bevilacqua, D., McCulley, C., Schwarz, N., Athanasiou, D., Kanuga, N., et al. (2014). Hsp90 Inhibition Protects against Inherited Retinal Degeneration. Hum. Mol. Genet. 23 (8), 2164–2175. doi:10.1093/hmg/ddt613
Ali, M. M. U., Roe, S. M., Vaughan, C. K., Meyer, P., Panaretou, B., Piper, P. W., et al. (2006). Crystal Structure of an Hsp90-Nucleotide-p23/Sba1 Closed Chaperone Complex. Nature 440 (7087), 1013–1017. doi:10.1038/nature04716
Amable, L., Grankvist, N., Largen, J. W., Ortsäter, H., Sjöholm, Å., Honkanen, R. E., et al. (2011). Disruption of Serine/threonine Protein Phosphatase 5 (PP5:PPP5c) in Mice Reveals a Novel Role for PP5 in the Regulation of Ultraviolet Light-Induced Phosphorylation of Serine/threonine Protein Kinase Chk1 (CHEK1). J. Biol. Chem. 286 (47), 40413–40422. doi:10.1074/jbc.M111.244053
Ambrocio-Ortiz, E., Pérez-Rubio, G., Ramírez-Venegas, A., Hernández-Zenteno, R. d. J., Paredes-López, A., Sansores, R. H., et al. (2021). Protective Role of Genetic Variants in HSP90 Genes-Complex in COPD Secondary to Biomass-Burning Smoke Exposure and Non-severe COPD Forms in Tobacco Smoking Subjects. Cimb 43 (2), 887–899. doi:10.3390/cimb43020063
Andreoletti, G., Shakhnovich, V., Christenson, K., Coelho, T., Haggarty, R., Afzal, N. A., et al. (2017). Exome Analysis of Rare and Common Variants within the NOD Signaling Pathway. Sci. Rep. 7, 46454. doi:10.1038/srep46454
Annett, S., Moore, G., and Robson, T. (2020). FK506 Binding Proteins and Inflammation Related Signalling Pathways; Basic Biology, Current Status and Future Prospects for Pharmacological Intervention. Pharmacol. Ther. 215, 107623. doi:10.1016/j.pharmthera.2020.107623
Aprea, I., Raidt, J., Höben, I. M., Loges, N. T., Nöthe-Menchen, T., Pennekamp, P., et al. (2021). Defects in the Cytoplasmic Assembly of Axonemal Dynein Arms Cause Morphological Abnormalities and Dysmotility in Sperm Cells Leading to Male Infertility. Plos Genet. 17 (2), e1009306. doi:10.1371/journal.pgen.1009306
Assimon, V. A., Southworth, D. R., and Gestwicki, J. E. (2015). Specific Binding of Tetratricopeptide Repeat Proteins to Heat Shock Protein 70 (Hsp70) and Heat Shock Protein 90 (Hsp90) Is Regulated by Affinity and Phosphorylation. Biochemistry 54 (48), 7120–7131. doi:10.1021/acs.biochem.5b00801
Backe, S. J., Sager, R. A., Woodford, M. R., Makedon, A. M., and Mollapour, M. (2020). Post-translational Modifications of Hsp90 and Translating the Chaperone Code. J. Biol. Chem. 295 (32), 11099–11117. doi:10.1074/jbc.REV120.011833
Baker, J., Ozsan, I., Rodriguez Ospina, S., Gulick, D., and Blair, L. (2018). Hsp90 Heterocomplexes Regulate Steroid Hormone Receptors: From Stress Response to Psychiatric Disease. Ijms 20 (1), 79. doi:10.3390/ijms20010079
Bédard, N., Jammoul, S., Moore, T., Wykes, L., Hallauer, P. L., Hastings, K. E. M., et al. (2015). Inactivation of the Ubiquitin-specific Protease 19 Deubiquitinating Enzyme Protects against Muscle Wasting. FASEB J. 29 (9), 3889–3898. doi:10.1096/fj.15-270579
Benson, D. R., Lovell, S., Mehzabeen, N., Galeva, N., Cooper, A., Gao, P., et al. (2019). Crystal Structures of the Naturally Fused CS and Cytochrome B5 Reductase (b5R) Domains of Ncb5or Reveal an Expanded CS Fold, Extensive CS-b5R Interactions and Productive Binding of the NAD(P)+ Nicotinamide Ring. Acta Crystallogr. D Struct. Biol. 75 (Pt 7), 628–638. doi:10.1107/S205979831900754X
Beraldo, F. H., Soares, I. N., Goncalves, D. F., Fan, J., Thomas, A. A., Santos, T. G., et al. (2013). Stress‐inducible Phosphoprotein 1 Has Unique Cochaperone Activity during Development and Regulates Cellular Response to Ischemia via the Prion Protein. FASEB j. 27 (9), 3594–3607. doi:10.1096/fj.13-232280
Bianchi, M., Kozyrev, S. V., Notarnicola, A., Hultin Rosenberg, L., Karlsson, Å., Pucholt, P., et al. (2021). Contribution of Rare Genetic Variation to Disease Susceptibility in a Large Scandinavian Myositis Cohort. Arthritis Rheumatol. Online ahead of print. doi:10.1002/art.41929 Available at: https://onlinelibrary.wiley.com/doi/10.1002/art.41929
Biebl, M. M., Riedl, M., and Buchner, J. (2020). Hsp90 Co-chaperones Form Plastic Genetic Networks Adapted to Client Maturation. Cel Rep. 32 (8), 108063. doi:10.1016/j.celrep.2020.108063
Bilbao Garay, I., Daly, A. F., Egaña Zunzunegi, N., and Beckers, A. (2020). Pituitary Disease in AIP Mutation-Positive Familial Isolated Pituitary Adenoma (FIPA): A Kindred-Based Overview. Jcm 9 (6), 2003. doi:10.3390/jcm9062003
Billon, C., Molin, A., Poirsier, C., Clemenson, A., Dauge, C., Grelet, M., et al. (2020). Fetal Megacystis‐microcolon: Genetic Mutational Spectrum and Identification of PDCL3 as a Novel Candidate Gene. Clin. Genet. 98 (3), 261–273. doi:10.1111/cge.13801
Blundell, K. L. I. M., Pal, M., Roe, S. M., Pearl, L. H., and Prodromou, C. (2017). The Structure of FKBP38 in Complex with the MEEVD Tetratricopeptide Binding-Motif of Hsp90. PLoS One 12 (3), e0173543. doi:10.1371/journal.pone.0173543
Bohush, A., Bieganowski, P., and Filipek, A. (2019). Hsp90 and its Co-chaperones in Neurodegenerative Diseases. Ijms 20 (20), 4976. doi:10.3390/ijms20204976
Bonham, L. W., Steele, N. Z. R., Karch, C. M., Manzoni, C., Geier, E. G., Wen, N., et al. (2018). Protein Network Analysis Reveals Selectively Vulnerable Regions and Biological Processes in FTD. Neurol. Genet. 4 (5), e266. doi:10.1212/NXG.0000000000000266
Brancaccio, M., Fratta, L., Notte, A., Hirsch, E., Poulet, R., Guazzone, S., et al. (2003). Melusin, a Muscle-specific Integrin β1-interacting Protein, Is Required to Prevent Cardiac Failure in Response to Chronic Pressure Overload. Nat. Med. 9 (1), 68–75. doi:10.1038/nm805
Brehme, M., Voisine, C., Rolland, T., Wachi, S., Soper, J. H., Zhu, Y., et al. (2014). A Chaperome Subnetwork Safeguards Proteostasis in Aging and Neurodegenerative Disease. Cel Rep. 9 (3), 1135–1150. doi:10.1016/j.celrep.2014.09.042
Brychzy, A., Rein, T., Winklhofer, K. F., Hartl, F. U., Young, J. C., and Obermann, W. M. (2003). Cofactor Tpr2 Combines Two TPR Domains and a J Domain to Regulate the Hsp70/Hsp90 Chaperone System. EMBO J. 22 (14), 3613–3623. doi:10.1093/emboj/cdg362
Bujalowski, P. J., Nicholls, P., Garza, E., and Oberhauser, A. F. (2018). The central Domain of UNC-45 Chaperone Inhibits the Myosin Power Stroke. FEBS Open Bio 8 (1), 41–48. doi:10.1002/2211-5463.12346
Bulone, D., San Biagio, P. L., Quiñones-Ruiz, T., Rosario-Alomar, M., Lednev, I. K., Robb, F. T., et al. (2019). A Multipronged Method for Unveiling Subtle Structural-Functional Defects of Mutant Chaperone Molecules Causing Human Chaperonopathies. Methods Mol. Biol. 1873, 69–92. doi:10.1007/978-1-4939-8820-4_5
Bykhovskaya, Y., Fardaei, M., Khaled, M. L., Nejabat, M., Salouti, R., Dastsooz, H., et al. (2017). TSC1 Mutations in Keratoconus Patients with or without Tuberous Sclerosis. Invest. Ophthalmol. Vis. Sci. 58 (14), 6462–6469. doi:10.1167/iovs.17-22819
Calderwood, S. K., and Gong, J. (2016). Heat Shock Proteins Promote Cancer: It's a Protection Racket. Trends Biochem. Sci. 41 (4), 311–323. doi:10.1016/j.tibs.2016.01.003
Calderwood, S. K., and Murshid, A. (2017). Molecular Chaperone Accumulation in Cancer and Decrease in Alzheimer's Disease: The Potential Roles of HSF1. Front. Neurosci. 11, 192. doi:10.3389/fnins.2017.00192
Cappello, F., Angileri, F., Conway de Macario, E., and J. L. Macario, A. (2013). Chaperonopathies and Chaperonotherapy. Hsp60 as Therapeutic Target in Cancer: Potential Benefits and Risks. Curr. Pharm. Des. 19 (3), 452–457. doi:10.2174/138161213804143653
Cappello, F., Marino Gammazza, A., Palumbo Piccionello, A., Campanella, C., Pace, A., Conway de Macario, E., et al. (2014). Hsp60 Chaperonopathies and Chaperonotherapy: Targets and Agents. Expert Opin. Ther. Targets 18 (2), 185–208. doi:10.1517/14728222.2014.856417
Chadli, A., Felts, S. J., and Toft, D. O. (2008). GCUNC45 Is the First Hsp90 Co-chaperone to Show α/β Isoform Specificity. J. Biol. Chem. 283 (15), 9509–9512. doi:10.1074/jbc.c800017200
Chandrasekar, G., Vesterlund, L., Hultenby, K., Tapia-Páez, I., and Kere, J. (2013). The Zebrafish Orthologue of the Dyslexia Candidate Gene DYX1C1 Is Essential for Cilia Growth and Function. PLoS One 8 (5), e63123. doi:10.1371/journal.pone.0063123
Chen, B., Zhong, D., and Monteiro, A. (2006). Comparative Genomics and Evolution of the HSP90 Family of Genes across All Kingdoms of Organisms. BMC Genomics 7, 156. doi:10.1186/1471-2164-7-156
Chen, D.-H., Latimer, C., Yagi, M., Ndugga-Kabuye, M. K., Heigham, E., Jayadev, S., et al. (2020). Heterozygous STUB1 Missense Variants Cause Ataxia, Cognitive Decline, and STUB1 Mislocalization. Neurol. Genet. 6 (2), e397–13. doi:10.1212/NXG.0000000000000397
Chiu, H.-H., Hsaio, C.-T., Tsai, Y.-S., Liao, Y.-C., Lee, Y.-C., and Soong, B.-W. (2020). Clinical and Genetic Characterization of Autosomal Recessive Spinocerebellar Ataxia Type 16 (SCAR16) in Taiwan. Cerebellum 19 (4), 544–549. doi:10.1007/s12311-020-01136-4
Christianson, J. C., Shaler, T. A., Tyler, R. E., and Kopito, R. R. (2008). OS-9 and GRP94 Deliver Mutant α1-antitrypsin to the Hrd1-Sel1l Ubiquitin Ligase Complex for ERAD. Nat. Cel Biol 10 (3), 272–282. doi:10.1038/ncb1689
Cömert, C., Fernandez-Guerra, P., and Bross, P. (2019). A Cell Model for HSP60 Deficiencies: Modeling Different Levels of Chaperonopathies Leading to Oxidative Stress and Mitochondrial Dysfunction. Methods Mol. Biol. 1873, 225–239. doi:10.1007/978-1-4939-8820-4_14
Connell, P., Ballinger, C. A., Jiang, J., Wu, Y., Thompson, L. J., Höhfeld, J., et al. (2001). The Co-chaperone CHIP Regulates Protein Triage Decisions Mediated by Heat-Shock Proteins. Nat. Cel Biol 3 (1), 93–96. doi:10.1038/35050618
da Silva Correia, J., Miranda, Y., Leonard, N., and Ulevitch, R. (2007). SGT1 Is Essential for Nod1 Activation. Proc. Natl. Acad. Sci. 104 (16), 6764–6769. doi:10.1073/pnas.0610926104
Dafsari, H. S., Kocaturk, N. M., Daimagüler, H.-S., Brunn, A., Dötsch, J., Weis, J., et al. (2019). Bi-allelic Mutations in Uncoordinated Mutant Number-45 Myosin Chaperone B Are a Cause for Congenital Myopathy. Acta Neuropathol. Commun. 7 (1), 211. doi:10.1186/s40478-019-0869-1
Dal, J., Nielsen, E. H., Klose, M., Feldt‐Rasmussen, U., Andersen, M., Vang, S., et al. (2020). Phenotypic and Genotypic Features of a Large kindred with a Germline AIP Variant. Clin. Endocrinol. 93 (2), 146–153. doi:10.1111/cen.14207
Dean, M. E., and Johnson, J. L. (2021). Human Hsp90 Cochaperones: Perspectives on Tissue-specific Expression and Identification of Cochaperones with Similar In Vivo Functions. Cell Stress and Chaperones 26 (1), 3–13. doi:10.1007/s12192-020-01167-0
Donkervoort, S., Kutzner, C. E., Hu, Y., Lornage, X., Rendu, J., Stojkovic, T., et al. (2020). Pathogenic Variants in the Myosin Chaperone UNC-45B Cause Progressive Myopathy with Eccentric Cores. Am. J. Hum. Genet. 107 (6), 1078–1095. doi:10.1016/j.ajhg.2020.11.002
Dutta, D., Briere, L. C., Kanca, O., Marcogliese, P. C., Walker, M. A., High, F. A., et al. (2020). De Novo mutations in TOMM70, a Receptor of the Mitochondrial Import Translocase, Cause Neurological Impairment. Hum. Mol. Genet. 29 (9), 1568–1579. doi:10.1093/hmg/ddaa081
Echeverría, P. C., Briand, P.-A., and Picard, D. (2016). A Remodeled Hsp90 Molecular Chaperone Ensemble with the Novel Cochaperone Aarsd1 Is Required for Muscle Differentiation. Mol. Cel Biol 36 (8), 1310–1321. doi:10.1128/MCB.01099-15
Esteve, C., Francescatto, L., Tan, P. L., Bourchany, A., De Leusse, C., Marinier, E., et al. (2018). Loss-of-Function Mutations in UNC45A Cause a Syndrome Associating Cholestasis, Diarrhea, Impaired Hearing, and Bone Fragility. Am. J. Hum. Genet. 102 (3), 364–374. doi:10.1016/j.ajhg.2018.01.009
Fabczak, H., and Osinka, A. (2019). Role of the Novel Hsp90 Co-chaperones in Dynein Arms' Preassembly. Ijms 20 (24), 6174. doi:10.3390/ijms20246174
Farhan, S. M. K., Howrigan, D. P., Howrigan, D. P., Abbott, L. E., Klim, J. R., Topp, S. D., et al. (2019). Exome Sequencing in Amyotrophic Lateral Sclerosis Implicates a Novel Gene, DNAJC7, Encoding a Heat-Shock Protein. Nat. Neurosci. 22 (12), 1966–1974. doi:10.1038/s41593-019-0530-0
Garcia-Ranea, J. A., Mirey, G., Camonis, J., and Valencia, A. (2002). p23 and HSP20/alpha-Crystallin Proteins Define a Conserved Sequence Domain Present in Other Eukaryotic Protein Families. FEBS Lett. 529 (2-3), 162–167. doi:10.1016/s0014-5793(02)03321-5
Genis, D., Ortega-Cubero, S., San Nicolás, H., Corral, J., Gardenyes, J., de Jorge, L., et al. (2018). Heterozygous STUB1 Mutation Causes Familial Ataxia with Cognitive Affective Syndrome (SCA48). Neurology 91 (21), e1988–e1998. doi:10.1212/WNL.0000000000006550
Giulino-Roth, L., van Besien, H. J., Dalton, T., Totonchy, J. E., Rodina, A., Taldone, T., et al. (2017). Inhibition of Hsp90 Suppresses PI3K/AKT/mTOR Signaling and Has Antitumor Activity in Burkitt Lymphoma. Mol. Cancer Ther. 16 (9), 1779–1790. doi:10.1158/1535-7163.MCT-16-0848
Grad, I., Cederroth, C. R., Walicki, J., Grey, C., Barluenga, S., Winssinger, N., et al. (2010). The Molecular Chaperone Hsp90α Is Required for Meiotic Progression of Spermatocytes beyond Pachytene in the Mouse. PLoS One 5 (12), e15770. doi:10.1371/journal.pone.0015770
Grad, I., McKee, T. A., Ludwig, S. M., Hoyle, G. W., Ruiz, P., Wurst, W., et al. (2006). The Hsp90 Cochaperone P23 Is Essential for Perinatal Survival. Mol. Cel Biol 26 (23), 8976–8983. doi:10.1128/MCB.00734-06
Greither, T., Schumacher, J., Dejung, M., Behre, H. M., Zischler, H., Butter, F., et al. (2020). Fertility Relevance Probability Analysis Shortlists Genetic Markers for Male Fertility Impairment. Cytogenet. Genome Res. 160 (9), 506–522. doi:10.1159/000511117
Guo, Z., Chen, W., Wang, L., and Qian, L. (2020). Clinical and Genetic Spectrum of Children with Primary Ciliary Dyskinesia in China. J. Pediatr. 225, 157–165. doi:10.1016/j.jpeds.2020.05.052
Guy, C. N., A. Garcia, Y., and B. Cox, M. (2015). Therapeutic Targeting of the FKBP52 Co-chaperone in Steroid Hormone Receptor-Regulated Physiology and Disease. Cmp 9 (2), 109–125. doi:10.2174/1874467208666150519114115
Gvozdenov, Z., Kolhe, J., and Freeman, B. C. (2019). The Nuclear and DNA-Associated Molecular Chaperone Network. Cold Spring Harb Perspect. Biol. 11 (10), a034009. doi:10.1101/cshperspect.a034009
Hansen, L., Comyn, S., Mang, Y., Lind-Thomsen, A., Myhre, L., Jean, F., et al. (2014). The Myosin Chaperone UNC45B Is Involved in Lens Development and Autosomal Dominant Juvenile Cataract. Eur. J. Hum. Genet. 22 (11), 1290–1297. doi:10.1038/ejhg.2014.21
Hasumi, H., Baba, M., Hasumi, Y., Lang, M., Huang, Y., Oh, H. F., et al. (2015). Folliculin-interacting Proteins Fnip1 and Fnip2 Play Critical Roles in Kidney Tumor Suppression in Cooperation with Flcn. Proc. Natl. Acad. Sci. USA 112 (13), E1624–E1631. doi:10.1073/pnas.1419502112
Hayer, S. N., Deconinck, T., Bender, B., Smets, K., Züchner, S., Reich, S., et al. (2017). STUB1/CHIP Mutations Cause Gordon Holmes Syndrome as Part of a Widespread Multisystemic Neurodegeneration: Evidence from Four Novel Mutations. Orphanet J. Rare Dis. 12 (1), 31. doi:10.1186/s13023-017-0580-x
He, J., Ma, X., Yu, W., Tang, L., Fu, J., Liu, X., et al. (2021). Validation of the Pathogenic Role of Rare DNAJC7 Variants in Chinese Patients with Amyotrophic Lateral Sclerosis. Neurobiol. Aging 106, e1–314. doi:10.1016/j.neurobiolaging.2021.04.026
Herold, C., Hooli, B. V., Mullin, K., Liu, T., Roehr, J. T., Mattheisen, M., et al. (2016). Family-based Association Analyses of Imputed Genotypes Reveal Genome-wide Significant Association of Alzheimer's Disease with OSBPL6, PTPRG, and PDCL3. Mol. Psychiatry 21 (11), 1608–1612. doi:10.1038/mp.2015.218
Hidalgo-de-Quintana, J., Evans, R. J., Cheetham, M. E., and van der Spuy, J. (2008). The Leber Congenital Amaurosis Protein AIPL1 Functions as Part of a Chaperone Heterocomplex. Invest. Ophthalmol. Vis. Sci. 49 (7), 2878–2887. doi:10.1167/iovs.07-1576
Hočevar, K., Maver, A., Kunej, T., and Peterlin, B. (2018). Sarcoidosis Related Novel Candidate Genes Identified by Multi-Omics Integrative Analyses. OMICS: A J. Integr. Biol. 22 (5), 322–331. doi:10.1089/omi.2018.0027
Huang, Q., Deng, G., Wei, R., Wang, Q., Zou, D., and Wei, J. (2020). Comprehensive Identification of Key Genes Involved in Development of Diabetes Mellitus-Related Atherogenesis Using Weighted Gene Correlation Network Analysis. Front. Cardiovasc. Med. 7, 580573. doi:10.3389/fcvm.2020.580573
Ilaslan, E., Markosyan, R., Sproll, P., Stevenson, B. J., Sajek, M., Sajek, M. P., et al. (2020). The FKBP4 Gene, Encoding a Regulator of the Androgen Receptor Signaling Pathway, Is a Novel Candidate Gene for Androgen Insensitivity Syndrome. Ijms 21 (21), 8403. doi:10.3390/ijms21218403
Jih, K.-Y., Tsai, P.-C., Tsai, Y.-S., Liao, Y.-C., and Lee, Y.-C. (2020). Rapid Progressive ALS in a Patient with a DNAJC7 Loss-Of-Function Mutation. Neurol. Genet. 6 (5), e503. doi:10.1212/NXG.0000000000000503
Kálmán, F. S., Lizák, B., Nagy, S. K., Mészáros, T., Zámbó, V., Mandl, J., et al. (2013). Natural Mutations lead to Enhanced Proteasomal Degradation of Human Ncb5or, a Novel Flavoheme Reductase. Biochimie 95 (7), 1403–1410. doi:10.1016/j.biochi.2013.03.004
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., et al. (2020). The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 581 (7809), 434–443. doi:10.1038/s41586-020-2308-7
Khandelwal, A., Kent, C. N., Balch, M., Peng, S., Mishra, S. J., Deng, J., et al. (2018). Structure-guided Design of an Hsp90β N-Terminal Isoform-Selective Inhibitor. Nat. Commun. 9 (1), 425. doi:10.1038/s41467-017-02013-1
Kirschke, E., Goswami, D., Southworth, D., Griffin, P. R., and Agard, D. A. (2014). Glucocorticoid Receptor Function Regulated by Coordinated Action of the Hsp90 and Hsp70 Chaperone Cycles. Cell 157 (7), 1685–1697. doi:10.1016/j.cell.2014.04.038
Krzemień-Ojak, Ł., Góral, A., Joachimiak, E., Filipek, A., and Fabczak, H. (2017). Interaction of a Novel Chaperone PhLP2A with the Heat Shock Protein Hsp90. J. Cel. Biochem. 118 (2), 420–429. doi:10.1002/jcb.25669
Lackie, R. E., Maciejewski, A., Ostapchenko, V. G., Marques-Lopes, J., Choy, W.-Y., Duennwald, M. L., et al. (2017). The Hsp70/Hsp90 Chaperone Machinery in Neurodegenerative Diseases. Front. Neurosci. 11, 254. doi:10.3389/fnins.2017.00254
Lee, C. F., Melkani, G. C., and Bernstein, S. I. (2014). The UNC-45 Myosin Chaperone. Int. Rev. Cel Mol Biol 313, 103–144. doi:10.1016/B978-0-12-800177-6.00004-9
Lee, C. H., Sinclair, D., O'Donnell, M., Galletly, C., Liu, D., Weickert, C. S., et al. (2019). Transcriptional Changes in the Stress Pathway Are Related to Symptoms in Schizophrenia and to Mood in Schizoaffective Disorder. Schizophrenia Res. 213, 87–95. doi:10.1016/j.schres.2019.06.026
Lehtimäki, J. I., Fenix, A. M., Kotila, T. M., Balistreri, G., Paavolainen, L., Varjosalo, M., et al. (2017). UNC-45a Promotes Myosin Folding and Stress Fiber Assembly. J. Cel Biol 216 (12), 4053–4072. doi:10.1083/jcb.201703107
Lin, L. T. W., Razzaq, A., Di Gregorio, S. E., Hong, S., Charles, B., Lopes, M. H., et al. (2021). Hsp90 and its Co‐chaperone Sti1 Control TDP‐43 Misfolding and Toxicity. FASEB j. 35 (5), e21594. doi:10.1096/fj.202002645R
Liu, B., Yang, Y., Qiu, Z., Staron, M., Hong, F., Li, Y., et al. (2010). Folding of Toll-like Receptors by the HSP90 Paralogue Gp96 Requires a Substrate-specific Cochaperone. Nat. Commun. 1, 79. doi:10.1038/ncomms1070
Lou, Q.-Y., Li, Z., Teng, Y., Xie, Q.-M., Zhang, M., Huang, S.-W., et al. (2021). Associations of FKBP4 and FKBP5 Gene Polymorphisms with Disease Susceptibility, Glucocorticoid Efficacy, Anxiety, Depression, and Health-Related Quality of Life in Systemic Lupus Erythematosus Patients. Clin. Rheumatol. 40 (1), 167–179. doi:10.1007/s10067-020-05195-0
Lupo, V., Aguado, C., Knecht, E., and Espinós, C. (2016). Chaperonopathies: Spotlight on Hereditary Motor Neuropathies. Front. Mol. Biosci. 3, 81. doi:10.3389/fmolb.2016.00081
Macario, A. J. L., Cappello, F., Zummo, G., and Conway de Macario, E. (2010). Chaperonopathies of Senescence and the Scrambling of Interactions between the Chaperoning and the Immune Systems. Ann. N. Y Acad. Sci. 1197, 85–93. doi:10.1111/j.1749-6632.2010.05187.x
Macario, A. J. L., Grippo, T. M., and de Macario, E. C. (2005). Genetic Disorders Involving Molecular-Chaperone Genes: a Perspective. Genet. Med. 7 (1), 3–12. doi:10.1097/01.gim.0000151351.11876.c3
Mak, O. W., Chand, R., Reynisson, J., and Leung, I. K. H. (2019). Identification of Isoform-Selective Ligands for the Middle Domain of Heat Shock Protein 90 (Hsp90). Ijms 20 (21), 5333. doi:10.3390/ijms20215333
Mallela, K., and Kumar, A. (2021). Role of TSC1 in Physiology and Diseases. Mol. Cel Biochem 476 (6), 2269–2282. doi:10.1007/s11010-021-04088-3
Mastrangelo, M., Commone, C., Greco, C., and Leuzzi, V. (2021). TSC1 as a Novel Gene for Sleep-Related Hypermotor Epilepsy: A Child with a Mild Phenotype of Tuberous Sclerosis. Neuropediatrics 52 (2), 146–149. doi:10.1055/s-0041-1722881
Mengel, D., Traschütz, A., Reich, S., Leyva-Gutiérrez, A., Bender, F., Hauser, S., et al. (2021). A De Novo STUB1 Variant Associated with an Early Adult-Onset Multisystemic Ataxia Phenotype. J. Neurol. 268, 3845–3851. doi:10.1007/s00415-021-10524-7
Mescheriakova, J. Y., Verkerk, A. J., Amin, N., Uitterlinden, A. G., van Duijn, C. M., and Hintzen, R. Q. (2019). Linkage Analysis and Whole Exome Sequencing Identify a Novel Candidate Gene in a Dutch Multiple Sclerosis Family. Mult. Scler. 25 (7), 909–917. doi:10.1177/1352458518777202
Metcalfe, K., Simeonov, E., Beckett, W., Donnai, D., and Tassabehji, M. (2005). Autosomal Dominant Inheritance of Williams-Beuren Syndrome in a Father and Son with Haploinsufficiency for FKBP6. Clin. Dysmorphol. 14 (2), 61–65. doi:10.1097/00019605-200504000-00002
Mishra, S. J., Khandelwal, A., Banerjee, M., Balch, M., Peng, S., Davis, R. E., et al. (2021). Selective Inhibition of the Hsp90α Isoform. Angew. Chem. Int. Ed. 60 (19), 10547–10551. doi:10.1002/anie.202015422
Mokha, J. S., Hyams, J. S., Glidden, N. C., Balarezo, F., and Young, E. (2021). Characterizing Clinical Features and Location‐specific Gene Expression Profiles Associated with Pain burden in Children with Functional Dyspepsia. Neurogastroenterology Motil. 33, e14185. doi:10.1111/nmo.14185
Morgan, R. M. L., Hernández-Ramírez, L. C., Trivellin, G., Zhou, L., Roe, S. M., Korbonits, M., et al. (2012). Structure of the TPR Domain of AIP: Lack of Client Protein Interaction with the C-Terminal α-7 Helix of the TPR Domain of AIP Is Sufficient for Pituitary Adenoma Predisposition. PLoS One 7 (12), e53339. doi:10.1371/journal.pone.0053339
Mowrey, K., Koenig, M. K., Szabo, C. A., Samuels, J., Mulligan, S., Pearson, D. A., et al. (2020). Two Different Genetic Etiologies for Tuberous Sclerosis Complex (TSC) in a Single Family. Mol. Genet. Genomic Med. 8 (7), e1296. doi:10.1002/mgg3.1296
Mutoh, H., Kato, M., Akita, T., Shibata, T., Wakamoto, H., Ikeda, H., et al. (2018). Biallelic Variants in CNPY3, Encoding an Endoplasmic Reticulum Chaperone, Cause Early-Onset Epileptic Encephalopathy. Am. J. Hum. Genet. 102 (2), 321–329. doi:10.1016/j.ajhg.2018.01.004
Niehues, T., Özgür, T. T., Bickes, M., Waldmann, R., Schöning, J., Bräsen, J., et al. (2020). Mutations of the Gene FNIP1 Associated with a Syndromic Autosomal Recessive Immunodeficiency with Cardiomyopathy and Pre‐excitation Syndrome. Eur. J. Immunol. 50 (7), 1078–1080. doi:10.1002/eji.201948504
Nitika, , , Porter, C. M., Truman, A. W., and Truttmann, M. C. (2020). Post-translational Modifications of Hsp70 Family Proteins: Expanding the Chaperone Code. J. Biol. Chem. 295 (31), 10689–10708. doi:10.1074/jbc.REV120.011666
Nizami, S., Arunasalam, K., Green, J., Cook, J., Lawrence, C. B., Zarganes‐Tzitzikas, T., et al. (2021). Inhibition of the NLRP3 Inflammasome by HSP90 Inhibitors. Immunology 162 (1), 84–91. doi:10.1111/imm.13267
Olcese, C., Patel, M. P., Patel, M. P., Shoemark, A., Kiviluoto, S., Legendre, M., et al. (2017). X-linked Primary Ciliary Dyskinesia Due to Mutations in the Cytoplasmic Axonemal Dynein Assembly Factor PIH1D3. Nat. Commun. 8, 14279. doi:10.1038/ncomms14279
Paff, T., Loges, N. T., Aprea, I., Wu, K., Bakey, Z., Haarman, E. G., et al. (2017). Mutations in PIH1D3 Cause X-Linked Primary Ciliary Dyskinesia with Outer and Inner Dynein Arm Defects. Am. J. Hum. Genet. 100 (1), 160–168. doi:10.1016/j.ajhg.2016.11.019
Pakdaman, Y., Berland, S., Bustad, H. J., Erdal, S., Thompson, B. A., James, P. A., et al. (2021). Genetic Dominant Variants in STUB1, Segregating in Families with SCA48, Display In Vitro Functional Impairments Indistinctive from Recessive Variants Associated with SCAR16. Ijms 22 (11), 5870. doi:10.3390/ijms22115870
Palumbo, V., Segat, L., Padovan, L., Amoroso, A., Trimarco, B., Izzo, R., et al. (2009). Melusin Gene (ITGB1BP2) Nucleotide Variations Study in Hypertensive and Cardiopathic Patients. BMC Med. Genet. 10, 140. doi:10.1186/1471-2350-10-140
Panaretou, B., Siligardi, G., Meyer, P., Maloney, A., Sullivan, J. K., Singh, S., et al. (2002). Activation of the ATPase Activity of Hsp90 by the Stress-Regulated Cochaperone Aha1. Mol. Cel 10 (6), 1307–1318. doi:10.1016/s1097-2765(02)00785-2
Patel, N., Crider, A., Pandya, C. D., Ahmed, A. O., and Pillai, A. (2016). Altered mRNA Levels of Glucocorticoid Receptor, Mineralocorticoid Receptor, and Co-chaperones (FKBP5 and PTGES3) in the Middle Frontal Gyrus of Autism Spectrum Disorder Subjects. Mol. Neurobiol. 53 (4), 2090–2099. doi:10.1007/s12035-015-9178-2
Paul, A., Garcia, Y. A., Zierer, B., Patwardhan, C., Gutierrez, O., Hildenbrand, Z., et al. (2014). The Cochaperone SGTA (Small Glutamine-Rich Tetratricopeptide Repeat-Containing Protein Alpha) Demonstrates Regulatory Specificity for the Androgen, Glucocorticoid, and Progesterone Receptors. J. Biol. Chem. 289 (22), 15297–15308. doi:10.1074/jbc.M113.535229
Philp, L. K., Day, T. K., Butler, M. S., Laven-Law, G., Jindal, S., Hickey, T. E., et al. (2016). Small Glutamine-Rich Tetratricopeptide Repeat-Containing Protein Alpha (SGTA) Ablation Limits Offspring Viability and Growth in Mice. Sci. Rep. 6, 28950. doi:10.1038/srep28950
Pratt, W. B., and Toft, D. O. (2003). Regulation of Signaling Protein Function and Trafficking by the Hsp90/hsp70-Based Chaperone Machinery. Exp. Biol. Med. (Maywood) 228 (2), 111–133. doi:10.1177/153537020322800201
Prodromou, C., Siligardi, G., O'Brien, R., Woolfson, D. N., Regan, L., Panaretou, B., et al. (1999). Regulation of Hsp90 ATPase Activity by Tetratricopeptide Repeat (TPR)-domain Co-chaperones. Embo J. 18 (3), 754–762. doi:10.1093/emboj/18.3.754
Ramamurthy, V., Niemi, G. A., Reh, T. A., and Hurley, J. B. (2004). Leber Congenital Amaurosis Linked to AIPL1: a Mouse Model Reveals Destabilization of cGMP Phosphodiesterase. Proc. Natl. Acad. Sci. 101 (38), 13897–13902. doi:10.1073/pnas.0404197101
Rashid, M., Qasim, M., Ishaq, R., Bukhari, S. A., Sajid, Z., Ashfaq, U. A., et al. (2020). Pathogenic Variants of AIPL1, MERTK, GUCY2D, and FOXE3 in Pakistani Families with Clinically Heterogeneous Eye Diseases. PLoS One 15 (9), e0239748. doi:10.1371/journal.pone.0239748
Ravel, J.-M., Benkirane, M., Calmels, N., Marelli, C., Ory-Magne, F., Ewenczyk, C., et al. (2021). Expanding the Clinical Spectrum of STIP1 Homology and U-Box Containing Protein 1-associated Ataxia. J. Neurol. 268 (5), 1927–1937. doi:10.1007/s00415-020-10348-x
Reynolds, P. D., Ruan, Y., Smith, D. F., and Scammell, J. G. (1999). Glucocorticoid Resistance in the Squirrel Monkey Is Associated with Overexpression of the Immunophilin FKBP51. J. Clin. Endocrinol. Metab. 84 (2), 663–669. doi:10.1210/jc.84.2.663
Riggs, D. L., Cox, M. B., Cheung-Flynn, J., Prapapanich, V., Carrigan, P. E., and Smith, D. F. (2004). Functional Specificity of Co-chaperone Interactions with Hsp90 Client Proteins. Crit. Rev. Biochem. Mol. Biol. 39 (5-6), 279–295. doi:10.1080/10409230490892513
Rosenbaum, M., Andreani, V., Kapoor, T., Herp, S., Flach, H., Duchniewicz, M., et al. (2014). MZB1 Is a GRP94 Cochaperone that Enables Proper Immunoglobulin Heavy Chain Biosynthesis upon ER Stress. Genes Develop. 28 (11), 1165–1178. doi:10.1101/gad.240762.114
Ruppert, V., Meyer, T., Richter, A., Maisch, B., Pankuweit, S., and German Competence Network of Heart, F. (2013). Identification of a Missense Mutation in the Melusin-Encoding ITGB1BP2 Gene in a Patient with Dilated Cardiomyopathy. Gene 512 (2), 206–210. doi:10.1016/j.gene.2012.10.055
Sacristan-Reviriego, A., Le, H. M., Georgiou, M., Meunier, I., Bocquet, B., Roux, A.-F., et al. (2020). Clinical and Functional Analyses of AIPL1 Variants Reveal Mechanisms of Pathogenicity Linked to Different Forms of Retinal Degeneration. Sci. Rep. 10 (1), 17520. doi:10.1038/s41598-020-74516-9
Sacristan-Reviriego, A., and van der Spuy, J. (2018). The Leber Congenital Amaurosis-Linked Protein AIPL1 and its Critical Role in Photoreceptors. Adv. Exp. Med. Biol. 1074, 381–386. doi:10.1007/978-3-319-75402-4_47
Saettini, F., Poli, C., Vengoechea, J., Bonanomi, S., Orellana, J. C., Fazio, G., et al. (2021). Absent B Cells, Agammaglobulinemia, and Hypertrophic Cardiomyopathy in Folliculin-Interacting Protein 1 Deficiency. Blood 137 (4), 493–499. doi:10.1182/blood.2020006441
Sager, R. A., Woodford, M. R., Backe, S. J., Makedon, A. M., Baker-Williams, A. J., DiGregorio, B. T., et al. (2019). Post-translational Regulation of FNIP1 Creates a Rheostat for the Molecular Chaperone Hsp90. Cel Rep. 26 (5), 1344e1345–1356. doi:10.1016/j.celrep.2019.01.018
Sager, R. A., Woodford, M. R., and Mollapour, M. (2018). The mTOR Independent Function of Tsc1 and FNIPs. Trends Biochem. Sci. 43 (12), 935–937. doi:10.1016/j.tibs.2018.09.018
Sahasrabudhe, P., Rohrberg, J., Biebl, M. M., Rutz, D. A., and Buchner, J. (2017). The Plasticity of the Hsp90 Co-chaperone System. Mol. Cel 67 (6), 947–961. doi:10.1016/j.molcel.2017.08.004
Salhi, M., Lahmar, O., Salah, M. O., Banić, I., Binghao, B., Malik, W., et al. (2021). GLCCI1 and STIP1 Variants Are Associated with Asthma Susceptibility and Inhaled Corticosteroid Response in a Tunisian Population. J. Asthma 58 (2), 197–206. doi:10.1080/02770903.2019.1666867
Sanchez-Martin, C., Moroni, E., Ferraro, M., Laquatra, C., Cannino, G., Masgras, I., et al. (2020). Rational Design of Allosteric and Selective Inhibitors of the Molecular Chaperone TRAP1. Cel Rep. 31 (3), 107531. doi:10.1016/j.celrep.2020.107531
Sbroggiò, M., Ferretti, R., Percivalle, E., Gutkowska, M., Zylicz, A., Michowski, W., et al. (2008). The Mammalian CHORD-Containing Protein Melusin Is a Stress Response Protein Interacting with Hsp90 and Sgt1. FEBS Lett. 582 (13), 1788–1794. doi:10.1016/j.febslet.2008.04.058
Scheufler, C., Brinker, A., Bourenkov, G., Pegoraro, S., Moroder, L., Bartunik, H., et al. (2000). Structure of TPR Domain-Peptide Complexes. Cell 101 (2), 199–210. doi:10.1016/s0092-8674(00)80830-2
Schopf, F. H., Biebl, M. M., and Buchner, J. (2017). The HSP90 Chaperone Machinery. Nat. Rev. Mol. Cel Biol 18 (6), 345–360. doi:10.1038/nrm.2017.20
Sengun, A. D., Guzel Tanoglu, E., and Ulucan, H. (2021). A Novel Mutation in FK506 Binding Protein-like (FKBPL) Causes Male Infertility. Croat. Med. J. 62 (3), 227–232. doi:10.3325/cmj.2021.62.227
Seo, Y. H. (2015). Organelle-specific Hsp90 Inhibitors. Arch. Pharm. Res. 38 (9), 1582–1590. doi:10.1007/s12272-015-0636-1
Serwetnyk, M. A., and Blagg, B. S. J. (2021). The Disruption of Protein−protein Interactions with Co-chaperones and Client Substrates as a Strategy towards Hsp90 Inhibition. Acta Pharmaceutica Sinica B 11 (6), 1446–1468. doi:10.1016/j.apsb.2020.11.015
Shelton, L. B., Koren, J., and Blair, L. J. (2017). Imbalances in the Hsp90 Chaperone Machinery: Implications for Tauopathies. Front. Neurosci. 11, 724. doi:10.3389/fnins.2017.00724
Shi, C.-H., Schisler, J. C., Rubel, C. E., Tan, S., Song, B., McDonough, H., et al. (2014). Ataxia and Hypogonadism Caused by the Loss of Ubiquitin Ligase Activity of the U Box Protein CHIP. Hum. Mol. Genet. 23 (4), 1013–1024. doi:10.1093/hmg/ddt497
Shirane, M., and Nakayama, K. I. (2003). Inherent Calcineurin Inhibitor FKBP38 Targets Bcl-2 to Mitochondria and Inhibits Apoptosis. Nat. Cel Biol 5 (1), 28–37. doi:10.1038/ncb894
Shirane-Kitsuji, M., and Nakayama, K. I. (2014). Mitochondria: FKBP38 and Mitochondrial Degradation. Int. J. Biochem. Cel Biol. 51, 19–22. doi:10.1016/j.biocel.2014.03.007
Sinclair, D., Fillman, S. G., Webster, M. J., and Weickert, C. S. (2013). Dysregulation of Glucocorticoid Receptor Co-factors FKBP5, BAG1 and PTGES3 in Prefrontal Cortex in Psychotic Illness. Sci. Rep. 3, 3539. doi:10.1038/srep03539
Singh, J. K., Hutt, D. M., Tait, B., Guy, N. C., Sivils, J. C., Ortiz, N. R., et al. (2020). Management of Hsp90-dependent Protein Folding by Small Molecules Targeting the Aha1 Co-chaperone. Cel Chem. Biol. 27 (3), 292–305. e296. doi:10.1016/j.chembiol.2020.01.008
Singh, R. K., Kolandaivelu, S., and Ramamurthy, V. (2014). Early Alteration of Retinal Neurons inAipl1−/−Animals. Invest. Ophthalmol. Vis. Sci. 55 (5), 3081–3092. doi:10.1167/iovs.13-13728
Stiegler, S. C., Rübbelke, M., Korotkov, V. S., Weiwad, M., John, C., Fischer, G., et al. (2017). A Chemical Compound Inhibiting the Aha1-Hsp90 Chaperone Complex. J. Biol. Chem. 292 (41), 17073–17083. doi:10.1074/jbc.M117.797829
Stroh, M. A., Winter, M. K., McCarson, K. E., Thyfault, J. P., and Zhu, H. (2018). NCB5OR Deficiency in the Cerebellum and Midbrain Leads to Dehydration and Alterations in Thirst Response, Fasted Feeding Behavior, and Voluntary Exercise in Mice. Cerebellum 17 (2), 152–164. doi:10.1007/s12311-017-0880-7
Stroh, M. A., Winter, M. K., Swerdlow, R. H., McCarson, K. E., and Zhu, H. (2016). Loss of NCB5OR in the Cerebellum Disturbs Iron Pathways, Potentiates Behavioral Abnormalities, and Exacerbates Harmaline-Induced Tremor in Mice. Metab. Brain Dis. 31 (4), 951–964. doi:10.1007/s11011-016-9834-x
Sun, X., Zhao, X., Liu, Q., Zhang, K., Liu, S., Wang, Z., et al. (2021). Mutations of DNAJC7 Are Rare in Chinese Amyotrophic Lateral Sclerosis Patients. Amyotroph. Lateral Scler. Frontotemporal Degeneration 22 (3-4), 312–315. doi:10.1080/21678421.2020.1813314
Suzuki, T., Suzuki, T., Raveau, M., Miyake, N., Sudo, G., Tsurusaki, Y., et al. (2020). A Recurrent PJA1 Variant in Trigonocephaly and Neurodevelopmental Disorders. Ann. Clin. Transl Neurol. 7 (7), 1117–1131. doi:10.1002/acn3.51093
Szczepankiewicz, D., Narożna, B., Celichowski, P., Sakrajda, K., Kołodziejski, P., Banach, E., et al. (2021). Genes Involved in Glucocorticoid Receptor Signalling Affect Susceptibility to Mood Disorders. World J. Biol. Psychiatry 22 (2), 149–160. doi:10.1080/15622975.2020.1766109
Taipale, M., Kaminen, N., Nopola-Hemmi, J., Haltia, T., Myllyluoma, B., Lyytinen, H., et al. (2003). A Candidate Gene for Developmental Dyslexia Encodes a Nuclear Tetratricopeptide Repeat Domain Protein Dynamically Regulated in Brain. Proc. Natl. Acad. Sci. 100 (20), 11553–11558. doi:10.1073/pnas.1833911100
Taipale, M., Tucker, G., Peng, J., Krykbaeva, I., Lin, Z.-Y., Larsen, B., et al. (2014). A Quantitative Chaperone Interaction Network Reveals the Architecture of Cellular Protein Homeostasis Pathways. Cell 158 (2), 434–448. doi:10.1016/j.cell.2014.05.039
Tarkar, A., Loges, N. T., Loges, N. T., Slagle, C. E., Francis, R., Dougherty, G. W., et al. (2013). DYX1C1 Is Required for Axonemal Dynein Assembly and Ciliary Motility. Nat. Genet. 45 (9), 995–1003. doi:10.1038/ng.2707
Tarone, G., and Brancaccio, M. (2015). The Muscle-specific Chaperone Protein Melusin Is a Potent Cardioprotective Agent. Basic Res. Cardiol. 110 (2), 10. doi:10.1007/s00395-015-0466-9
Tian, T., Cao, X., Kim, S.-E., Lin, Y. L., Steele, J. W., Cabrera, R. M., et al. (2020). FKBP8 Variants Are Risk Factors for Spina Bifida. Hum. Mol. Genet. 29 (18), 3132–3144. doi:10.1093/hmg/ddaa211
Timsit, Y. E., and Negishi, M. (2014). Coordinated Regulation of Nuclear Receptor CAR by CCRP/DNAJC7, HSP70 and the Ubiquitin-Proteasome System. PLoS One 9 (5), e96092. doi:10.1371/journal.pone.0096092
Tomita-Mitchell, A., Mahnke, D. K., Struble, C. A., Tuffnell, M. E., Stamm, K. D., Hidestrand, M., et al. (2012). Human Gene Copy Number Spectra Analysis in Congenital Heart Malformations. Physiol. Genomics 44 (9), 518–541. doi:10.1152/physiolgenomics.00013.2012
Vartholomaiou, E., Echeverría, P. C., and Picard, D. (2016). Unusual Suspects in the Twilight Zone between the Hsp90 Interactome and Carcinogenesis. Adv. Cancer Res. 129, 1–30. doi:10.1016/bs.acr.2015.08.001
Verba, K. A., and Agard, D. A. (2017). How Hsp90 and Cdc37 Lubricate Kinase Molecular Switches. Trends Biochem. Sci. 42 (10), 799–811. doi:10.1016/j.tibs.2017.07.002
Wang, L., Li, L., Gu, K., Xu, X.-L., Sun, Y., and You, Q.-D. (2017). Targeting Hsp90-Cdc37: A Promising Therapeutic Strategy by Inhibiting Hsp90 Chaperone Function. Cdt 18 (13), 1572–1585. doi:10.2174/1389450117666160527125522
Wang, M., Liu, Z., Yuan, Y., Ni, J., Li, W., Hu, Y., et al. (2020a). A Novel Potentially Pathogenic Rare Variant in the DNAJC7 Gene Identified in Amyotrophic Lateral Sclerosis Patients from Mainland China. Front. Genet. 11, 821. doi:10.3389/fgene.2020.00821
Wang, Y., Tu, C., Nie, H., Meng, L., Li, D., Wang, W., et al. (2020b). Novel DNAAF6 Variants Identified by Whole-Exome Sequencing Cause Male Infertility and Primary Ciliary Dyskinesia. J. Assist. Reprod. Genet. 37 (4), 811–820. doi:10.1007/s10815-020-01735-4
Wei, X., Du, M., Xie, J., Luo, T., Zhou, Y., Zhang, K., et al. (2020). Mutations in TOMM70 lead to Multi-OXPHOS Deficiencies and Cause Severe Anemia, Lactic Acidosis, and Developmental Delay. J. Hum. Genet. 65 (3), 231–240. doi:10.1038/s10038-019-0714-1
Woodford, M. R., Dunn, D. M., Blanden, A. R., Capriotti, D., Loiselle, D., Prodromou, C., et al. (2016). The FNIP Co-chaperones Decelerate the Hsp90 Chaperone Cycle and Enhance Drug Binding. Nat. Commun. 7, 12037. doi:10.1038/ncomms12037
Woodford, M. R., Sager, R. A., Marris, E., Dunn, D. M., Blanden, A. R., Murphy, R. L., et al. (2017). Tumor Suppressor Tsc1 Is a New Hsp90 Co‐chaperone that Facilitates Folding of Kinase and Non‐kinase Clients. EMBO J. 36 (24), 3650–3665. doi:10.15252/embj.201796700
Wu, Z., Gholami, A. M., and Kuster, B. (2012). Systematic Identification of the HSP90 Candidate Regulated Proteome. Mol. Cel Proteomics 11 (6), M111–M016675. doi:10.1074/mcp.M111.016675
Xie, J., Zhu, H., Larade, K., Ladoux, A., Seguritan, A., Chu, M., et al. (2004). Absence of a Reductase, NCB5OR, Causes Insulin-Deficient Diabetes. Proc. Natl. Acad. Sci. 101 (29), 10750–10755. doi:10.1073/pnas.0404044101
Xu, K., Xie, Y., Sun, T., Zhang, X., Chen, C., and Li, Y. (2020). Genetic and Clinical Findings in a Chinese Cohort with Leber Congenital Amaurosis and Early Onset Severe Retinal Dystrophy. Br. J. Ophthalmol. 104 (7), 932–937. doi:10.1136/bjophthalmol-2019-314281
Xu, Y., Ogawa, S., Adachi, Y., Sone, N., Gotoh, S., Ikejiri, M., et al. (2021). A Pediatric Case of Primary Ciliary Dyskinesia Caused by Novel Copy Number Variation in PIH1D3. Auris Nasus Larynx. S0385-8146, 00087. doi:10.1016/j.anl.2021.03.012
Yadav, R. P., Gakhar, L., Yu, L., and Artemyev, N. O. (2017). Unique Structural Features of the AIPL1-FKBP Domain that Support Prenyl Lipid Binding and Underlie Protein Malfunction in Blindness. Proc. Natl. Acad. Sci. USA 114 (32), E6536–E6545. doi:10.1073/pnas.1704782114
Yoshida, S., Tsutsumi, S., Muhlebach, G., Sourbier, C., Lee, M.-J., Lee, S., et al. (2013). Molecular Chaperone TRAP1 Regulates a Metabolic Switch between Mitochondrial Respiration and Aerobic Glycolysis. Proc. Natl. Acad. Sci. 110 (17), E1604–E1612. doi:10.1073/pnas.1220659110
Young, J. C., Hoogenraad, N. J., and Hartl, F. U. (2003). Molecular Chaperones Hsp90 and Hsp70 Deliver Preproteins to the Mitochondrial Import Receptor Tom70. Cell 112 (1), 41–50. doi:10.1016/s0092-8674(02)01250-3
Zhang, M., Botër, M., Li, K., Kadota, Y., Panaretou, B., Prodromou, C., et al. (2008). Structural and Functional Coupling of Hsp90- and Sgt1-Centred Multi-Protein Complexes. Embo J. 27 (20), 2789–2798. doi:10.1038/emboj.2008.190
Zhang, M., Gu, Y., Huang, S., Lou, Q., Xie, Q., Xu, Z., et al. (2019). Copy Number Variations and Polymorphisms in HSP90AB1 and Risk of Systemic Lupus Erythematosus and Efficacy of Glucocorticoids. J. Cel Mol Med 23 (8), 5340–5348. doi:10.1111/jcmm.14410
Zhang, W., Zhang, S., Xiao, C., Yang, Y., and Zhoucun, A. (2007). Mutation Screening of the FKBP6 Gene and its Association Study with Spermatogenic Impairment in Idiopathic Infertile Men. Reproduction 133 (2), 511–516. doi:10.1530/REP-06-0125
Keywords: CS domain, Aha1, tetratricopeptide repeat, FKBP, FNIP1, chaperonopathy
Citation: Johnson JL (2021) Mutations in Hsp90 Cochaperones Result in a Wide Variety of Human Disorders. Front. Mol. Biosci. 8:787260. doi: 10.3389/fmolb.2021.787260
Received: 30 September 2021; Accepted: 08 November 2021;
Published: 08 December 2021.
Edited by:
Ilda D'Annessa, Study and Scientific Solutions, ItalyReviewed by:
Andrew Truman, University of North Carolina at Charlotte, United StatesGeorgios Karras, University of Texas MD Anderson Cancer Center, United States
Copyright © 2021 Johnson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jill L. Johnson, amlsbGpvaG5AdWlkYWhvLmVkdQ==