
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Biosci. , 31 August 2021
Sec. Cellular Biochemistry
Volume 8 - 2021 | https://doi.org/10.3389/fmolb.2021.723003
 Ireti Eni-Aganga1,2,3
Ireti Eni-Aganga1,2,3 Zeljka Miletic Lanaghan1,4
Zeljka Miletic Lanaghan1,4 Muthukumar Balasubramaniam1,5
Muthukumar Balasubramaniam1,5 Chandravanu Dash1,2,5
Chandravanu Dash1,2,5 Jui Pandhare1,2,3*
Jui Pandhare1,2,3*Prolidase (peptidase D), encoded by the PEPD gene, is a ubiquitously expressed cytosolic metalloproteinase, the only enzyme capable of cleaving imidodipeptides containing C-terminal proline or hydroxyproline. Prolidase catalyzes the rate-limiting step during collagen recycling and is essential in protein metabolism, collagen turnover, and matrix remodeling. Prolidase, therefore plays a crucial role in several physiological processes such as wound healing, inflammation, angiogenesis, cell proliferation, and carcinogenesis. Accordingly, mutations leading to loss of prolidase catalytic activity result in prolidase deficiency a rare autosomal recessive metabolic disorder characterized by defective wound healing. In addition, alterations in prolidase enzyme activity have been documented in numerous pathological conditions, making prolidase a useful biochemical marker to measure disease severity. Furthermore, recent studies underscore the importance of a non-enzymatic role of prolidase in cell regulation and infectious disease. This review aims to provide comprehensive information on prolidase, from its discovery to its role in health and disease, while addressing the current knowledge gaps.
Prolidase is the only dipeptidase known to catalyze the hydrolysis of a dipeptide containing a C-terminal proline or hydroxyproline into constituent amino acids (Endo et al., 1989; Kitchener and Grunden 2012) (Figure 1). It has many aliases, including peptidase D (PEPD), Xaa-Pro dipeptidase, X-Pro dipeptidase, proline dipeptidase, or imidodipeptidase. Prolidase is present in all three domains of life - archaea, bacteria, and eukaryotes. The enzymatic activity of prolidase releases proline or hydroxyproline in the final stages of the catabolism of endogenous and dietary proteins. The most studied substrate of prolidase are imidodipeptides that are generated during breakdown of collagen - the predominant component of the extracellular matrix (ECM) (Kitchener and Grunden 2012). Collagen is the most abundant structural protein in the human body, attributing to approximately 33% of total protein mass. Proline and hydroxyproline constitute over 20% of collagen (Ramshaw et al., 1998); therefore, collagen degradation generates a considerable amount of dipeptides containing proline or hydroxyproline (Laurent, 1987; McAnulty and Laurent 1987). Prolidase plays an essential role in ECM remodeling by facilitating the intracellular digestion of these dipeptides and promoting the recycling of proline for collagen synthesis (Duong et al., 2006; Surazynski et al., 2008b). Thus, prolidase is required in several physiological and pathological processes such as wound healing, inflammation, angiogenesis, cell proliferation, and carcinogenesis. In this review, we start with a historical snapshot of the discovery of prolidase, then provide an overview of the current body of research, followed by a discussion of the existing knowledge gaps in prolidase research, and conclude with future prospective.
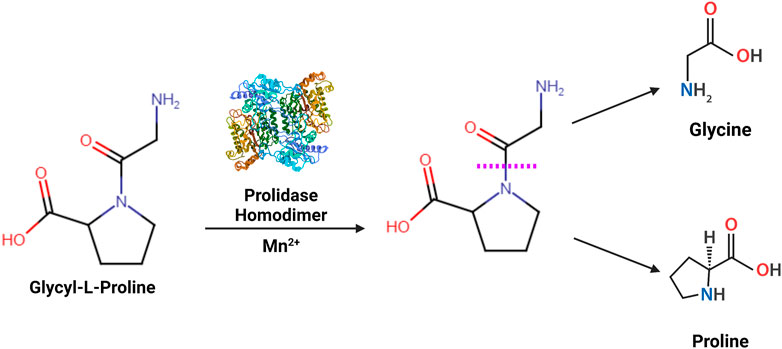
FIGURE 1. Enzymatic reaction catalyzed by prolidase- Prolidase is a metallopeptidase that functions as a homodimer catalyzing the hydrolysis of imidodipeptides containing a C-terminal proline or hydroxyproline residue (Xaa-Pro). The scissile amide linkage hydrolyzed by prolidase is shown with a purple dashed line.
The term “Prolidase” was first coined in 1937 (Grassmann et al., 1932; Bergmann and Fruton 1937; Smith and Bergmann 1979). Researchers, while studying digestion in the intestinal tract, found that crude preparations of erepsin (protein fractions found in the intestinal fluid) had the ability to cleave dipeptides containing a C-terminus proline (glycyl-proline (gly-pro) and alanyl-proline (ala-pro)). It was unclear whether it was a new activity of aminopeptidase or an unknown enzyme. Aminopeptidases can act only on peptide linkages with a hydrogen, while proline with its α-imino group forms special peptide linkages that lack a peptide hydrogen. Thus, the possibility of the enzyme being an aminopeptidase was ruled out. Moreover, upon further study, the enzyme was found to lack the ability to cleave carbobenzoxyglycyl-L-proline (Cbz-pro) and glycylsarcosine. Simultaneously, phenylhydrazine did not inhibit the activity, confirming that this enzyme did not require a peptide hydrogen for action but adapted to cleave linkages containing a proline nitrogen (Bergmann and Fruton 1937). Therefore, the enzyme was given the name “Prolidase,” specifically to classify it as the enzyme that cleaves proline on the C-terminus of a proline containing dipeptide (Grassmann et al., 1932) (Figure 1) and to differentiate it from another enzyme involved in proline metabolism known as prolinase, which cleaves proline on the N-terminus of a proline containing dipeptide (prolyl-glycine). Prolidase was first discovered to cleave gly-pro, but later found to also cleave glycyl-hydroxyproline (Bergmann and Fruton, 1937). C-terminal prolines are not exclusive to dipeptides and may also occur in tripeptides during intestinal digestion. Prolidase activity was narrowed down to dipeptides since the catalytic activity was undetectable with tripeptides and was classified as an imidodipeptidase (Adams and Smith, 1952). Prolidase activation requires manganese and is thus identified as a metal-activated peptidase (Smith and Bergmann, 1979). It requires four manganese ions, but is also functional with other divalent cations like zinc and cobalt, albeit to a lesser degree (Besio et al., 2010; Besio et al., 2013), on the other hand prolidase activity is inhibited by nickel (Miltyk et al., 2008). Prolidase enzymatic function and cofactor requirements are further explored later in this review.
The discovery of prolidase in the intestinal mucosa is significant because prolidase is required for the complete breakdown of proteins such as collagen and gelatin that contain imino-group peptide linkages. Although prolidase’s discovery in intestinal mucosa and its imidodipeptidase activity contributed to increasing knowledge of intestinal fluid composition and protein metabolism, questions arose about prolidase expression in other tissues or cell types. Prolidase was found in human erythrocytes, human kidneys, and swine kidneys (Davis and Smith, 1957). Among the cell types, erythrocytes of both human and horse use manganese as a cofactor and do not differ in substrate specificity (Adams, McFadden et al., 1952; Adams and Smith, 1952). In horse erythrocytes, manganese increases prolidase activity by 26-fold while pyrophosphate, ethylenediaminetetraacetate, fluoride, and citrate were found to inhibit prolidase activity (Adams and Smith, 1952).
Human prolidase is encoded by the PEPD gene, located on the long arm of chromosome 19 at position 13.11 (Gene ID: 5184) (Figure 2). PEPD has no TATA box, but S1 nuclease mapping revealed a “CAAT”-like box upstream of the transcription start site, as well as sequences that resemble the Sp1 transcription factor binding sites in this region. (Tanoue et al., 1990a). The 130 kilobases long gene contains 15 exons encoding three mRNA transcript variants (Figure 2). Translation of transcript variant 1 — the longest isoform—yields the canonical 493 amino acid protein. In contrast, the transcript variants 2 and 3 lack two alternate in-frame exons in the central coding region and are translated into 452 and 429 amino acid proteins, respectively (https://www.ncbi.nlm.nih.gov/gene/5184). It is currently unclear whether the three isoforms vary in terms of subcellular localization or functional characteristics. PEPD is found in mice, chimps, rhesus monkeys, dogs, cows, rats, chickens, frogs, zebrafish, fruit flies, mosquitos, Caenorhabditis elegans, and Arabidopsis thaliana (https://www.ncbi.nlm.nih.gov/gene/5184/). The mouse PEPD located on chromosome 7 containing 15 exons encodes a 493 amino acid protein. Mouse prolidase is 83.2% homologous at the nucleotide level and 87.2% homologous at the amino acid level to human prolidase (Ishii et al., 1996).
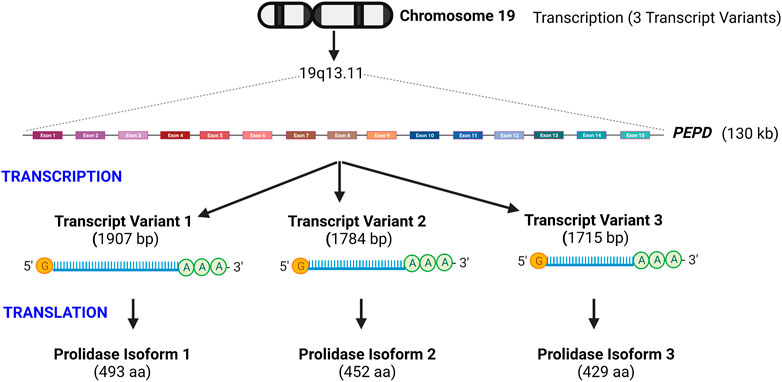
FIGURE 2. Schematic illustration of prolidase gene organization- Prolidase encoded by the PEPD gene is located on chromosome 19q13.11. PEPD gene is 130 kb long and encodes three transcript variants that yield three protein isoforms. Isoform 1 is the longest and predominantly studied. Transcript variants 2 and 3 lack two alternate in-frame exons in the central coding region and are translated into proteins of shorter length.
Human prolidase protein has been shown to exists in two isoforms; I and II (Uramatsu et al., 2009). Prolidase I (56 kDa), the most common, has a higher affinity for gly-pro and ala-pro dipeptides. In comparison, prolidase II (95 kDa) has a higher affinity for met-pro and a very low affinity for gly-pro (Ohhashi et al., 1988a) (Oono et al., 1990) (Uramatsu et al., 2009). In addition to manganese, calcium can act as a cofactor for prolidase I and cobalt for prolidase II (Ohhashi et al., 1988b). Though the two isoforms are reported, prolidase I is most abundant in human plasma with little to no prolidase II (Cosson et al., 1992). The function of prolidase II is unclear; however, research indicates that patients who are deficient in prolidase have prolidase I deficiency rather than a prolidase II deficiency since in these patients the ability to degrade gly-pro dipeptides is compromised while other C-terminus proline containing dipeptides are degraded (Uramatsu et al., 2009).
Prolidase is a homodimeric enzyme. Hydrophobic and hydrophilic residues are distributed evenly throughout the protein’s amino acid composition (Endo et al., 1989; Tanoue et al., 1990b). Sequence and structure analysis of prolidase shows that it shares similar binding motifs with methionine aminopeptidase, aminopeptidase P (APPro), and creatinase (Bazan et al., 1994). Phenylglyoxal or 1-cyclohexyl-3-(2-morpholinoethyl) carbodiimide metho-p-toluenesulfonate inactivate prolidase while protection is conferred by competitive inhibitor N-acetylcholine, suggesting aspartic and glutamic acid are in the active site (Mock and Zhuang, 1991). Magnesium renders prolidase inactive while occupying the same position as manganese in crystal structures (Wilk et al., 2017). The first structure of prolidase was deduced from the hyperthermophilic archaeon Pyrococcus furiosus. The enzyme’s folding pattern was similar to that of APPro and creatinase, confirming previous similarities between these enzymes (Maher et al., 2004). The crystal structure of human prolidase has been determined (Mueller et al., 2006) and is available in the protein data bank (PDB).
The homodimer of human prolidase is oriented in a C2 symmetric pattern. The N-terminal domain of each subunit comprises of amino acid residues 1–184, while the C-terminal domain, which houses the enzyme’s active site, comprises of amino acid residues 185–493. The C-terminal domain contains the “pita-bread fold” found in methionine aminopeptidase, APPro, and creatinase (Maher et al., 2004). Enzymes possessing a pita-bread fold carry a canonical bimetallic center at the bottom of their active site cleft formed by two structurally similar domains of two α-helices and an antiparallel β-sheet. An intermolecular disulfide bond was observed between residues Cys58A and Cys158B, linking two monomers within one dimer. Manganese is the preferred co-factor for prolidase, and the crystal structure suggests that one Mn2+ ion is more tightly bound adjacent to His370, while another is weakly bound adjacent to Asp276 in each subunit. The bridging moiety between the two manganese is essential for catalysis due to the distance, and it is most likely a hydroxide ion instead of a water molecule (Mueller et al., 2006).
Prolidase activity begins with the abstraction of a proton from the bridge water molecule by Glu412 (Wilk et al., 2017). The substrate (gly-pro) diffuses into the active site, leading to a conformational change from an open to a closed state. The Gly-N atom of the substrate interacts with one of the manganese ions, while the Gly-O of the scissile peptide bond interacts with the second manganese ion. His255 is essential for substrate binding, and His377 stabilizes the transitional state. A network of hydrogen bonds from His377, Arg398, His255 holds Gly-Pro to the protein’s active site. His255 binds to the carboxylate group of the substrate proline. The hydroxide ion is held by the manganese ions and the nitrogen on the proline. Manganese ions are stabilized by Asp287, Asp276, Glu452, Glu412, His370, along with the amino group of glycine. The polarization builds a partial positive charge on the carbonyl C-atom of the scissile bond. A nucleophilic attack occurs on the hydroxide ion onto the carbonyl C-atom forming a tetrahedral intermediate. The proton’s shift from the hydroxide to the Pro-N atom coincides with the breaking of the peptide bond. The first product, glycine, leaves the active site while the protein is still in a closed conformation. Another conformation change returns the protein to the open state, and His255 and Trp107 release the proline (Wilk et al., 2017). Other pita-bread enzymes have similar activity and homologs, solidifying the evolutionary relationship (Bazan et al., 1994; Maher et al., 2004).
Collagen is the most common structural protein in the human body, supporting connective tissues such as skin, bones, cartilage, and ligaments (Laurent, 1987). It accounts for approximately 33% of total protein mass and the predominant component of the extracellular matrix (ECM). Roughly 22% of amino acid residues in collagen strands are either proline or hydroxyproline (Ramshaw et al., 1998). Matrix metalloproteinases (MMPs) are endopeptidases involved in collagen metabolism (Laronha and Caldeira, 2020). The sequential cleavage by multiple MMPs degrades collagens into dipeptides containing proline/hydroxyproline at the C-terminal end that serve as substrates for prolidase (Verma and Hansch, 2007; Yu et al., 2012). Accordingly, the completion of ECM-associated collagen degradation is dependent on the catalytic activity of prolidase. Prolidase catalyzes the final and rate-limiting step in the degradation of collagen, releasing free proline/hydroxyproline, which can recycle into collagen synthesis during matrix remodeling (Surazynski et al., 2008a). Synthesis of collagen is dependent on the availability of proline, which is primarily provided by prolidase, via hydrolysis of imidodipeptides generated during collagen breakdown (Albaugh et al., 2017) (Karna et al., 2020). Alternately, proline can be synthesized from glutamine and glutamate or from arginine via its conversion to ornithine (Phang et al., 2010). Interestingly, studies have shown that, dietary supplementation of proline does not increase collagen synthesis nor do supplementation of glutamine or glutamate. However, dietary supplementation of arginine and ornithine have been shown to be most effective in increasing collagen deposition (Barbul, 2008). Thus, the homeostasis of collagen synthesis is dependent on the availability of proline via cellular metabolism and collagen breakdown generating imidodipeptides. Due to its involvement in collagen remodeling, fluctuations in prolidase activity may therefore, indicate dysfunction in collagen metabolism, altered disease states, or homeostatic imbalance.
Wound healing is a complex and highly regulated process in which different cellular types participate to rapidly and efficiently reconstitute tissue integrity (Gonzalez et al., 2016). It involves intricate and interconnected processes, which occur in four phases: hemostasis, inflammation, proliferation, and remodeling. During hemostasis, damaged tissue triggers the formation of a blood clot. Platelets release growth factors to stimulate the propagation of neutrophils and macrophages, which remove necrotic tissue, debris, and bacteria from the wound. Macrophages release various growth factors and cytokines required for recruiting fibroblasts during the inflammatory phase, such as transforming growth factor-beta (TGFβ). Fibroblasts, the most common cell type of connective tissues, are the primary cells of the proliferative phase that then initiate angiogenesis and collagen formation (Gonzalez et al., 2016; DesJardins-Park et al., 2018). Fibroblasts play a crucial part in synthesizing ECM components, mainly collagen, which provides structure to the wound and replaces the fibronectin–fibrin matrix from the blood clot formed during hemostasis. Collagen turnover provides tensile strength to wounds during the acute and late stages of wound healing. During the early stages of wound healing, fibroblasts actively produce type III collagen. During the remodeling phase, type III collagen is replaced by type I collagen to restore the normal dermal collagen composition (Xue and Jackson, 2015). Collagen turnover drives the remodeling of the ECM and is critical for effective wound healing. Wound repair depends upon a supply of proline via collagen recycling for the healing process. The recycling and remodeling of collagen is therefore dependent on prolidase activity to make free proline available from collagen degradation for resynthesis. This was demonstrated in rats where during wound healing, prolidase activity increased in rats with burned skin compared to rats without burns (Umemura et al., 1980). Prolidase activity has been reported to be higher in wound fluid from patients with chronic pressure wounds, and prolidase mRNA levels were found to be higher in scar tissue fibroblasts, keratinocytes, and endothelial cells (Senboshi et al., 1996; Oono et al., 1997). Also, prolidase gene expression is upregulated in old scar tissue compared to uninjured skin (Senboshi et al., 1996). On the other hand, research on aged fibroblasts shows a decrease in collagen, along with a decrease in prolidase activity (Pałka et al., 1996). Over time, fibroblasts in the dermis become senescent, and the number of collagen metabolic enzymes, including prolidase, decreases. Because manganese is a co-factor for prolidase, drugs that chelate metal ions, such as daunorubicin, doxycycline, and anthracycline, inhibit prolidase activity and lead to poor wound healing as a side effect (Muszyńska et al., 2000; Karna et al., 2001; Muszyńska et al., 2001). A common symptom and identifier of Prolidase deficiency (PD) is defective wound healing. Proline availability is critical for proper collagen synthesis, and the body maintains a tight balance over proline availability (Albaugh et al., 2017). PD patients suffer from unhealing ulcerative wounds and bone deformities due to the excretion of proline-containing dipeptides due to inactive prolidase.
Diabetes mellitus (DM) may impair wound healing, though the pathophysiology of impaired wound healing in DM is not fully understood (Huang and Kyriakides, 2020). High blood glucose decreases collagen production and fibroblast migration and inhibits inflammatory cell proliferation, leading to chronically non-healing ulcers (Okonkwo and DiPietro, 2017). Serum prolidase levels are elevated in patients with Type 2 diabetes and diabetic foot ulcers (Eren et al., 2013). Lower or defective prolidase is associated with impaired wound healing, suggesting that in the case of DM, other factors may also affect wound healing.
Keloids are an abnormal proliferation of scar tissue that form at sites of a cutaneous wound (Hinz, 2016). Keloids result from inflammation in the dermis, overactivation of myofibroblasts and other connective tissue cells, and excess ECM proteins. This leads to an atypical overgrowth of the scar that extends past the area of the wound. Collagen synthesis, mainly accumulation of type I collagen, is about 20-fold higher in keloid than in healthy skin. Collagen accumulates due to an imbalance in the homeostasis of collagen turnover by fibroblasts, and collagenase activities are also significantly higher in keloid tissues (Hinz, 2016; Karppinen et al., 2019). Prolidase mRNA and protein expression are increased in keloids in comparison to healthy skin. This increase in activity may enhance collagen biosynthesis due to the increase in the proline pool, providing a constant resource that enhances collagen deposition (Duong et al., 2006).
A plethora of reports on the clinical importance of prolidase, especially in the context of prolidase deficiency, are available. However, studies focused on the regulation of prolidase are limited. As aforementioned, prolidase plays a pivotal role in collagen metabolism, mainly in recycling collagen via its degradation and resynthesis. Studies suggest that prolidase activity is regulated by interactions between the ECM and surface receptors because prolidase activity increases with cell growth and density. Thus, it was logical to presume that factors involved in the regulation of collagen may also regulate prolidase activity. The first reference on the regulation of prolidase activity was through the study of collagen and β1 integrin receptor (Palka and Phang, 1997) (Figure 3). Integrin receptors are essential for integrating ECM signals by transmitting signals bidirectionally between the ECM and cytoplasm. They are heterodimeric transmembrane receptors composed of eighteen α subunits and eight β subunits that are non-covalently assembled into at least 24 different functional receptors (Mezu-Ndubuisi and Maheshwari, 2021). The most prominent integrin receptor, β1 integrin, is mainly responsible for attachment to the ECM. Upon stimulation, β1 integrin has been shown to mediate cell motility, survival, proliferation and differentiation. Type I collagen is a ligand for the β1 integrin receptor, and it was shown that the addition of collagen led to an increase in prolidase activity (Palka and Phang, 1997; Zeltz et al., 2014). Subsequent studies demonstrated that β1 integrin triggers the Ras-Raf-MEK-ERK pathway to stimulate prolidase catalytic activity independent of prolidase protein expression. They further demonstrated that inhibitors of β1 integrin inhibit the Ras-Raf-MEK-ERK pathway with a concomitant decrease in prolidase activity, strongly suggesting that prolidase activity is regulated by this pathway (Surazyński et al., 2004). Another example of the factor that stimulates collagen biosynthesis and regulates prolidase activity is the insulin-like growth factor-1 (IGF1) (Figure 3). When IGF1 interacts with its receptor IGF1R, it stimulates collagen biosynthesis, and this interaction results in an increase in prolidase activity in human dermal fibroblasts (Miltyk et al., 1998; Blackstock et al., 2014). Prolidase activity is also influenced by the induction of oxidative stress that affects IGF1 receptor signaling (Sienkiewicz et al., 2004).
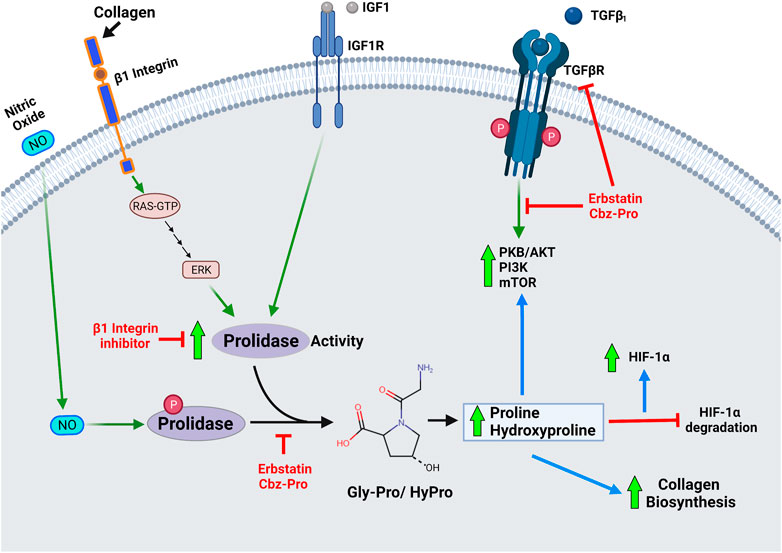
FIGURE 3. Diagram depicting role of prolidase enzymatic activity in cellular regulation- Prolidase plays a pivotal role in collagen metabolism, matrix remodeling and cell growth. Therefore, prolidase, is essential in several physiological processes such as wound healing, inflammation, angiogenesis, cell proliferation, and carcinogenesis. Accordingly, primary mechanisms that are known to regulate prolidase activity include-activation of the β1 integrin receptor, insulin-like growth factor 1 receptor (IGF-1R), and transforming growth factor (TGF) receptor. Stimulation of these receptors by their ligands-collagen, IGF1 and TGFβ1 respectively affect prolidase enzyme activity and downstream processes associated with cell proliferation and growth. Nitric oxide (NO) mediated phosphorylation of prolidase has been shown to increase enzyme activity, while inhibition of phosphorylation renders a less active prolidase enzyme. The products of prolidase activity-proline and hydroxyproline stimulate collagen biosynthesis, stabilize HIF-1α by inhibiting its degradation and induce TGF receptor mediated pathways.
Prolidase requires activation for its enzymatic activity. Phosphorylation of prolidase has been identified as a potential mechanism for regulating its enzymatic activity (Figure 3). Prolidase activity was reduced in fibroblasts treated with erbstatin, a naturally occurring inhibitor of tyrosine-specific protein kinase. The same study showed that an anti-phosphotyrosine antibody also reduced prolidase activity in a dose-dependent manner while phosphotyrosine phosphatase 1B increased prolidase activity (Surazyński et al., 2001). Serine/threonine phosphorylation of prolidase was also reported in fibroblasts treated with nitric oxide, which mediates inflammatory responses, wound repair, and angiogenesis (Surazynski et al., 2005). In another study, prolidase activity was shown to be decreased in fibroblasts from fasting rats. Fasting results in ATP depletion, leading to accumulation of inorganic phosphate and pyruvate kinase activity inhibition, affecting phosphoenolpyruvate (PEP) conversion into pyruvate. The authors postulated that the accumulating PEP may form a complex with prolidase and prevent its phosphorylation or stimulate its dephosphorylation; however, the exact mechanism remains unknown (Cechowska-Pasko et al., 2004). The same research group demonstrated that thrombin, the enzyme that converts fibrinogen to fibrin in the blood clotting cascade, inhibits prolidase expression while inducing its phosphorylation in colorectal adenocarcinoma cells (Lundblad et al., 2004; Karna et al., 2012). However, thrombin binds to the β1 integrin receptor and increases prolidase activity in human dermal fibroblasts (Surazyński et al., 2005).
Proline and hydroxyproline, the products of prolidase enzyme activity, have been shown to increase nuclear hypoxia-inducible factor levels (HIF-1α) levels by inhibiting HIF-1α degradation (Surazynski et al., 2008a) (Figure 3). HIF-1α is associated with wound repair as it enhances genes associated with inflammation, angiogenesis, and fibrosis such as vascular endothelial growth factor (VEGF), glucose transporter-1 (GLUT-1), and transforming growth factor-β1 and 2 (TGF-β1 and 2) (Lokmic et al., 2012). HIF-1α is hydroxylated at proline residues by prolyl hydroxylase under normoxic conditions, allowing for HIF-1α ubiquitination by the von Hippel-Lindau (VHL) E3 ubiquitin ligase, leading to proteasomal degradation. In the absence of proteasomal degradation, HIF1α accumulates and translocates into the nucleus to transcribe associated genes. Overexpression of prolidase results in increased HIF-1α levels - leading to elevated HIF-1α-dependent gene products such as VEGF and GLUT-1. This increase in HIF-1α-dependent gene products was attributed to products of prolidase activity. To determine if prolidase was directly involved in the increased production or decreased degradation of HIF-1α, the oxygen-dependent degradation domain (ODD) of HIF-1α was fused to a luciferase gene (Luc-ODD). Overexpression of prolidase increased Luc-ODD levels, suggesting that HIF1 degradation was disrupted. The same effect was also observed with the addition of proline or hydroxyproline in a dose-dependent manner. The authors concluded that prolidase activity, which produces proline and hydroxyproline, inhibits the degradation of HIF-1α via the VHL dependent proteasomal pathway (Surazynski et al., 2008a).
TGF-β1 is a cytokine that plays a key role in wound healing, vascular injury, cell proliferation, and differentiation. It has been linked to fibrosis development due to its ability to regulate and stimulate collagen biosynthesis (Kubiczkova et al., 2012). TGF- β1 can cause fibrosis by inducing protease inhibitor production, which prevents the enzymatic breakdown of the ECM, particularly collagen (Kim et al., 2018). Inhibitors of prolidase activity decreased TGF-β1 and its receptor expression in fibroblasts (Surazynski et al., 2010). This results in the downregulation of TGFβ receptor-stimulated kinases such as Protein Kinase B (PKB or AKT), phosphatidylinositide 3-kinases (PI3K), and mammalian target of rapamycin (mTOR). Furthermore, the addition of proline and hydroxyproline reversed the effect, thus leading to the conclusion that the products of prolidase activity, rather than the prolidase protein, affect TGFβ downstream signaling (Figure 3).
Estrogen, the primary female sex hormone, is frequently linked to breast cancer and the female reproductive system. Estrogen affects the balance of collagen biosynthesis and acts as a regulator in collagen metabolism. Estrogen has been linked to an overall increase in collagen deposition in wound healing, as estrogen levels have been shown to decrease with age, resulting in decreased collagen and impaired wound healing (Horng et al., 2017). Estrogen was shown to upregulate prolidase activity with a concomitant increase in collagen biosynthesis in MCF7 breast cancer cells (Miltyk et al., 1999). In the presence of estradiol, prolactin inhibited prolidase activity while estrogen receptor α (ERα) upregulated prolidase activity (Surazynski et al., 2013a; Surazynski et al., 2013b).
Prolidase has been recently shown to play a novel role in exacerbation of HIV-1 infection by cocaine treatment. HIV-1 neuropathogenesis has been reported to worsen in HIV-1 infected patients who abuse drugs like cocaine. HIV-1 is usually unable to cross the blood-brain barrier (BBB). The BBB is a semipermeable and selective barrier that separates the circulating blood from the brain to protect the central nervous system. The BBB consists of the basement membrane, which is primarily composed of type IV collagen. Cocaine has been shown to increase prolidase activity in brain microvascular endothelial cells, causing a weakened basement membrane and increased permeability in HIV-1-infected monocytes. Cocaine-dependent nitric oxide induction was also shown to increase prolidase phosphorylation, resulting in increased activity (Ysrayl et al., 2019).
The majority of research on prolidase focuses on its catalytic activity in collagen metabolism. However, recently enzyme activity independent functions of prolidase have emerged (Figure 4). In a novel finding, prolidase was shown to function as a ligand of EGFR (ErbB1), and its binding stimulated downstream signaling proteins in the EGFR pathway (Yang et al., 2013) (Figure 4). This activation of signaling was not dependent on the catalytic activity of prolidase. Interestingly, prolidase does not have any structural homologies to other EGFR ligands. The binding of prolidase resulted in the phosphorylation of EGFR and the activation of key signaling molecules downstream of EGFR, such as Akt (Protein kinase B), STAT3 (Signal transducer and activator of transcription 3), and ERK (Extracellular signal-regulated kinase). This activation occurred only when prolidase was present extracellularly. This is intriguing because prolidase is generally considered a cytosolic protein and is not known to be secreted in the extracellular milieu even during collagen or ECM remodeling. It is plausible that prolidase is released from damaged cells and tissue, which then binds to EGFR. The binding affinity of prolidase to EGFR was much lower than EGF, the known ligand of EGFR; however, mutated prolidase was shown to silence EGFR signaling (Yang et al., 2016). Follow up studies by the same group also demonstrated that prolidase and an enzymatically inactive mutant function as ligands for the ErbB2 (HER2) receptor (Figure 4). ErbB2 is an oncogenic receptor tyrosine kinase overexpressed in a subset of human breast cancer and other cancers (Joshi and Press, 2018). Recombinant human prolidase binds as a dimer with a high affinity to ErbB2 monomers resulting in their dimerization. Prolidase binding was shown to strongly inhibit ErbB2-overexpressing tumors in mice, whereas it did not impact tumors without ErbB2 overexpression (Yang et al., 2014). In cells overexpressing ErbB2, recombinant prolidase was shown to bind to preformed ErbB2 dimers. The inhibition in tumors due to prolidase binding was attributed to ErbB2 depletion and disruption in oncogenic Src signaling. The binding of prolidase and its enzymatic mutant to the receptor led to potent inhibition of ErbB2 (dimeric form)-overexpressing tumors in mice, and the phenomena was observed only when ErbB2 is overexpression is not present (Yang et al., 2015; Yang et al., 2016).
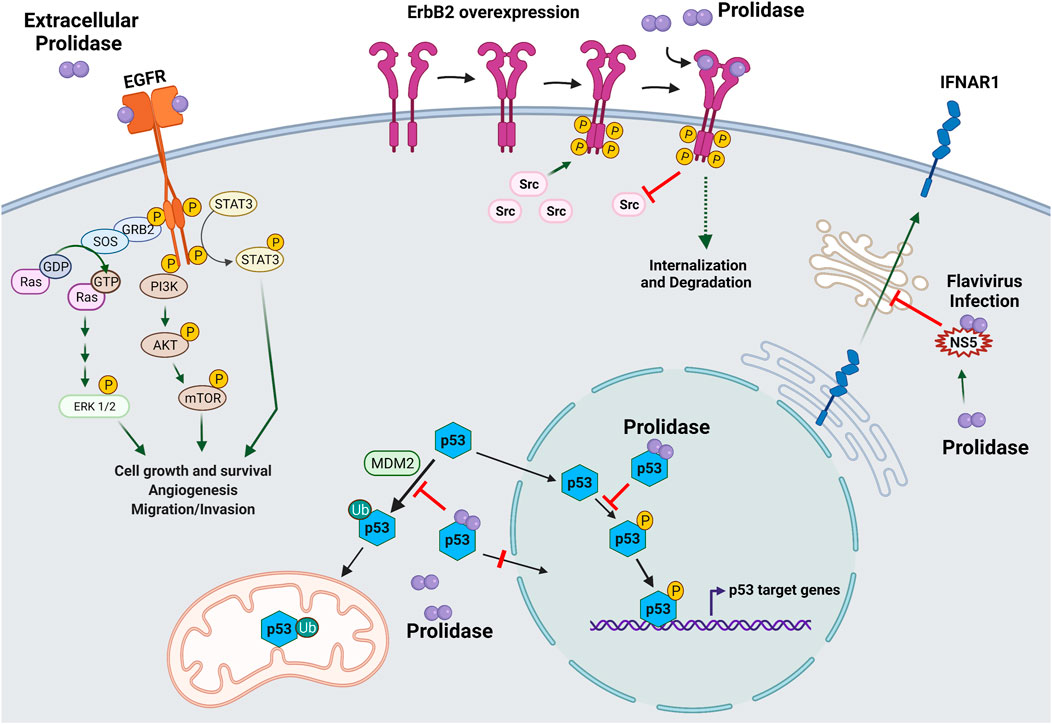
FIGURE 4. Enzyme activity independent role of prolidase in cellular regulation. A novel role of prolidase as a biological regulatory protein independent of its catalytic activity has recently emerged. Prolidase was demonstrated to function as a ligand for epidermal growth factor receptor (EGFR/ErbB2) and HER2/ErbB2 receptor. Binding of prolidase extracellularly to EGFR receptor stimulated downstream pathways important in cell proliferation and growth such as extracellular signal-regulated kinase (ERK)1/2, phosphoinositide 3-kinase (PI3K), protein kinase B (Akt), mammalian target of rapamycin (mTOR), and signal transducer and activator of transcription 3 (STAT3). Likewise, prolidase was shown to bind overexpressed HER2/ErbB2 receptor and inhibit downstream signaling via disruption of its association with Src causing internalization and degradation. Prolidase, independent of its catalytic activity was demonstrated to regulate p53 tumor suppressor protein via direct binding. A dual mechanism involving inhibition of p53 phosphorylation and disruption of its interaction with MDM2 resulted in sequestration of p53 in the cytosol thereby preventing both transcription-dependent and transcription-independent activities of p53. Prolidase plays a novel role as a regulator of type I interferon (IFN-I) immune response during flavivirus infection. It is essential for maturation and trafficking of IFN-I receptor (IFNAR1). During infection, the nonstructural protein 5 (NS5) produced by flaviviruses was shown to bind prolidase compromising IFN-1 response.
Another novel non-enzymatic activity of prolidase (and its mutants) is as a regulator of p53 tumor suppressor (Yang et al., 2017). Prolidase was shown to bind to p53 in the nucleus and in the cytoplasm and suppress both transcription-dependent and transcription-independent activities of p53 (Figure 4). Prolidase suppresses p53 from functioning as a transcription factor by sequestering nuclear p53 and inhibiting its phosphorylation. In addition, prolidase acts as a mouse double minute 2 homolog (MDM2) competitor preventing its association with p53, and consequent ubiquitination and translocation to mitochondria. This inhibitory effect is abrogated upon disruption of the association of prolidase with p53 and the ensuing p53 transcription-dependent and independent functions, result in cell death and tumor regression. Interestingly, this is the first report demonstrating the presence of prolidase in the nucleus (Yang et al., 2017).
In addition to the above mentioned non-enzymatic roles of prolidase, a unique role of prolidase in flavivirus infection has been reported (Lubick et al., 2015). During viral infections, a type I interferon (IFN-I) immune response is induced that signals through IFN-I receptor (IFNAR1). To counter this immune response, viruses are known to produce proteins that antagonize the response. For instance, the nonstructural protein 5 (NS5) produced by flaviviruses, acts as an antagonist of IFN-I. Investigation of flavivirus antagonism of IFN-1 response revealed that prolidase is required for IFNAR1 receptor maturation and that flavivirus NS5 downregulates IFNAR1 by sequestering prolidase and thus inhibiting the maturation of IFNAR1 (Lubick et al., 2015) (Figure 4). Improper maturation of IFNAR1 in prolidase deficient patients may contribute to immune issues observed in these patients (Lubick et al., 2015).
Prolidase Deficiency (PD) is a rare autosomal recessive disorder marked by elevated levels of imidodipeptides (proline/hydroxyproline-containing dipeptides excreted in urine) (Cottin et al., 2020). One of the first reports of PD described a patient with an unusual bone disorder characterized by excessive excretion of dipeptides containing proline, implying an increase in bone collagen turnover. Though the patient’s bones were deformed, the skin appeared normal; while it was concluded that there was a defect in collagen synthesis, the causative agent was however, unknown (Seakins, 1963). Another subsequent study, described a patient with skeletal, facial, and cardiovascular abnormalities, as well as high imidodipeptide excretion. The patient excreted increased quantities of bound hydroxyproline, indicating increased collagen turnover or defective catabolism by hydroxyproline oxidase (Goodman et al., 1968). Although the patient’s condition resembled lathyrism, the decreased collagen stability did not fully match the symptoms, implying other unidentified mechanisms. The dipeptides found in urine were extracted and tested in vitro as prolidase substrates. The excreted dipeptides were hydrolyzed by exogenous prolidase, leading to the conclusion that the patient had a prolidase function defect due to a deficiency. Additional studies by another research group defined the lathyrism-like symptoms as PD (Buist et al., 1972).
PD was categorized as an autosomal recessive disorder by studying the immediate family tree in a patient with imidopeptiduria (Powell et al., 1974). Prolidase activity was reduced in the patient’s red and white blood cells, in addition to the clinical indication of defective collagen metabolism. White blood cells from the patient’s mother and maternal grandfather also had lower prolidase activity compared to healthy controls. Prolidase enzymatic activity was normal in the maternal grandmother, signifying that prolidase is an autosomal recessive disorder (Powell et al., 1974). Both hydroxyproline and proline were found on the C-terminus of excreted dipeptides, and excreted hydroxyproline level was ten times higher than that of healthy people. Prolinase levels remained normal, bolstering the theory that the dipeptides were excreted due to a prolidase deficiency. This was the first time that the symptoms of prolidase deficiency were linked to genetics.
The efficiency of recycling proline is approximately 90–95% in healthy individuals (Jackson and Heininger, 1975). Prolidase is essential in collagen turnover, and synthesis, and defects in prolidase lead to blockage in regular recycling of collagen because of a decreased pool of available proline (Spodenkiewicz et al., 2020). Fibroblasts are primarily known for leading collagen metabolism and have high levels of prolidase activity. Disturbances in prolidase activity in fibroblasts are associated with connective tissue abnormalities like hypermobile joints. Proline deprivation due to reduced prolidase activity may cause improper wound healing (Dolenga and Hechtman, 1992). Prolidase is necessary for cell survival, and cultured fibroblasts from PD patients exhibited necrosis-like cell death due to increase in undigested prolidase substrates (Forlino et al., 2002). Collagen metabolism is disrupted in all PD patients, affecting bone structure and wound healing. Improperly formed bone structure causes dysmorphic facial features such as a low anterior and posterior hairline, widely spaced/bulging eyes, a depressed nasal bridge, thin upper lip vermilion, and prognathism (Falik-Zaccai et al., 2010). Due to the importance of collagen turnover during wound healing events, most PD patients have large ulcerative wounds that are unable to heal correctly (Goodman et al., 1968; Dunn et al., 2011; Süßmuth et al., 2020). Collagen constituents are excreted rather than recycled, which may impede wound healing in PD patients. Various other symptoms are associated with PD including dermatological lesions, developmental delay, splenomegaly, repetitive infections, and autoimmune manifestations (Nir et al., 2016; Spodenkiewicz et al., 2020; Kenig et al., 2021). In addition to chronic and recurrent ulcers, other skin manifestations including dermatitis, erythematous papular eruptions, impetigo-like eruptions, pseudo-psoriasis skin lesions, and pruritic eczematous lesions have been reported (Falik-Zaccai et al., 2010; Spodenkiewicz et al., 2020).
Another common symptom is breathing problems stemming from a cystic fibrosis (CF)-like phenotype (Luder et al., 2007; Cottin et al., 2020). Immunoglobulin levels are higher in PD patients due to the binding of gamma globulins to prolidase substrates in blood serum, and PD patients suffer from recurrent infections due to open sores on the skin and infections in the respiratory tract (Jackson and Heininger, 1975; Sheffield et al., 1977; Cleary et al., 1994). Other than collagen, these imidodipeptide substrates are common in immunoglobulins and Clq in complement (Reid, 2018). Another immune related symptom includes a systemic lupus erythematosus (SLE)-like phenotype (Butbul Aviel et al., 2012; Kurien et al., 2013; Chidambaram et al., 2021). The mechanism by which SLE is associated with PD is not clear although, as in the case of CF, a role for a high proline containing defective C1q complement has been proposed (Chidambaram et al., 2021).
Numerous clinical studies on PD have observed intellectual disabilities and developmental delays in patients. The deficiency of prolidase in mice also shows intellectual delays and disabilities (Besio et al., 2015) which have now become associated with PD, implicating the importance of prolidase in brain health and function. However, the role of prolidase in brain health is understudied. A study suggested that the presence of iminodipeptides leads to weakened cerebral vessels inducing superoxide production (Yasuda et al., 1999). Prolidase and other mental disorders are discussed later in this review. Though there are multiple phenotypes associated with PD, not all exhibit the same severity amongst all patients and even patients let alone in patients belonging to the same family tree.
Studies of prolidase activity in healthy patients’ sera revealed that a dysfunctional enzyme more likely causes PD rather than a reduction in the amount of enzyme available. Manganese is required for prolidase activity, and the addition of manganese did not activate prolidase in PD patients suggesting that an inactive enzyme may be due to mutations in the PEPD gene (Ohhashi et al., 1988a). Amplifying the coding region of prolidase from PD patients revealed a G to A substitution in position 826 in exon 12, resulting in an aspartic acid instead of asparagine and consequently producing a defective enzyme. Gene expression studies in NIH3T3 cells confirmed that, unlike the enzymatically active wild-type prolidase, the mutant prolidase lacked activity thus establishing the importance of G to A substitution in PD. This discovery, at the time, meant gene replacement therapy might be possible (Tanoue et al., 1990a). However, in addition to the G to A substitution, the absence of the 14th exon of PEPD gene was reported to result in prolidase deficiency in another PD patient, (Tanoue et al., 1990b).
Based on previous studies utilizing a monoclonal antibody specific to wild-type human prolidase, it was suggested that PD was due to a missing portion of the enzyme since the antibody was unable to detect the enzyme in patients (Endo et al., 1987). A single nucleotide base substitution of C to T at position 551 in exon 8 of PEPD gene was identified in a PD patient, that yielded a stop codon instead of arginine, resulting in a 20 kDa defective prolidase (Kikuchi et al., 2000). Recently, a single nucleotide deletion, causing a frameshift mutation and encoding a premature stop codon in the PEPD gene was reported to disrupt the enzymatic function of prolidase (Kiratli Nalbant et al., 2019). Likewise, another study reported a homozygous nonsense variation in exon 5 of the PEPD gene that resulted in a stop codon and premature truncation of the protein at codon 140 resulting in PD (Chidambaram et al., 2021). Overall, 35 PEPD variants have been identified. There are 16 missense/nonsense variants, nine splice variants, nine insertions/deletions, and one large deletion. The majority of missense mutations are found in the catalytic domain, located in exon 12 (six mutations). A large number of mutations are also found in exons 8 (six mutations) and 14 (four mutations), all of which are in the C-terminus. Due to the wide range of phenotypes, the extent of prolidase inactivation varies. Disruption of the catalytic center, displacement of important active site residues, and the rigidity of the open-closed conformation required for substrate uptake and release are likely mechanisms for inactivation, that are based on the location of the mutations (Spodenkiewicz et al., 2020).
To date, different methods have been developed for diagnosing prolidase deficiency. They are mainly based 1) on the determination of enzyme activity in erythrocytes, leukocytes, and/or skin fibroblast cultures of PD patients or 2) on the screening of X-Pro imidodipeptides massively excreted in their urine. The oldest method for detecting prolidase activity utilizes Chinard’s reagent, which relies on a colorimetric reaction of proline upon hydrolysis of the X-Pro peptide substrate (Myara et al., 1982). Other methods to determine prolidase activity and imidodipeptides include amino acid analysis, HPLC-API/MS, and MALDI-TOF (Viglio et al., 2006). The preferred method of establishing PD diagnosis is sequence analysis of PEPD, followed by gene-targeted deletion/duplication analysis if only one or no pathogenic variant is found (Lupi et al., 2008). Alternately, in specific populations, targeted analysis for pathogenic variants such as Arg265Ter in exon 11 and Ser202Phe in exon 8 is performed first followed by sequence analysis (Ferreira and Wang, 1993; Spodenkiewicz et al., 2020).
Prolidase deficiency being a rare disorder, not many therapeutics are currently available. Gene therapy is a viable option but has not been accomplished so far. The clinical features of PD are characterized by chronic recurrent ulcers, especially on the legs; therefore, treatment with topical ointments has been suggested as a possible therapy. Ointments containing glycine and proline as well as growth hormone (GH) have been used since prolidase deficiency is associated with GH deficiency (Arata et al., 1986; Monafo et al., 2000). A few therapies have tried to alleviate symptoms by replacing deficient enzyme in PD patients (Viglio et al., 2006). This included the use of manganese-activated red blood cells from healthy patients to overcome prolidase deficiency; however, they have been largely unsuccessful (Hechtman et al., 1988). Another approach was adenovirus-mediated gene therapy using human prolidase cDNA transferred into PD fibroblasts from patients. Prolidase activity increased up to approximately 7.5 times in PD fibroblasts compared to normal fibroblasts (Ikeda et al., 1997). Other approaches include targeted delivery of liposome-encapsulated prolidase to PD patients’ fibroblasts (Perugini et al., 2005). Unfortunately, these proposed therapies for PD are understudied, short-lived, inaccessible, and have a low success rate.
The majority of research on prolidase focuses on PD and defective wound healing, however, perturbations in prolidase activity have been reported in various other disorders and diseases including cancer, diabetes, cardiovascular disease and others. These alterations in prolidase activity are mainly associated in pathological conditions with enhanced collagen breakdown or turnover, while in some conditions the cause of the observed variations in activity remains to be elucidated. Prolidase activity has been shown to be directly associated with oxidative stress and suggested to be a useful biochemical marker of oxidative stress in various disorders (Yildiz et al., 2008; Gecit et al., 2012; Kokacya et al., 2014; Ozbay et al., 2020). Oxidative stress is described as an imbalance between reactive oxygen species (ROS) and antioxidants due to overproduction of ROS (Pizzino et al., 2017). Multiple studies provide evidence that oxidative stress stimulates collagen breakdown via inductions of MMPs and inducing an inflammatory microenvironment (Kim et al., 2016). These ROS-induced alterations of the ECM microenvironment have been suggested to be responsible for modulating prolidase activity by making available the substrate via breakdown of collagen.
Excessive accumulation of collagen and other matrix components causes liver fibrosis, which can progress to liver cirrhosis in most chronic liver diseases. Chronic hepatitis C virus (HCV) infection, alcohol abuse, and non-alcoholic steatohepatitis (NASH) are some of the most common causes of liver fibrosis (Bataller and Brenner, 2005). Myara et al. discovered a link between serum prolidase activity (SPA) and chronic liver diseases, suggesting that plasma prolidase activity monitoring could help assess fibrotic processes in chronic liver disease (Myara et al., 1984). Patients with chronic liver diseases had higher SPA than healthy individuals; however, only a few biopsy-positive cirrhosis patients had increased SPA. A study using a rat model of cirrhotic liver fibrosis suggested that SPA levels may be higher in the early stages of fibrosis and then drop as the disease progresses. This increase in circulating prolidase was not due to leakage as there was no correlation with alanine aminotransferase, an enzyme that leaks into the bloodstream following liver damage (Abraham et al., 2000). Prolidase may increase in the first stages of fibrosis due to the sudden accumulation of collagen and falls at later stages since the liver may not sustain prolidase production.
Brosset et al. investigated prolidase enzyme activity in patients with alcoholic liver disease (Brosset et al., 1988). Patients with alcoholic hepatitis have been shown to have significantly higher prolidase activity in their plasma than patients with stable cirrhosis. NASH is a form of advanced non-alcoholic fatty liver disease (NAFLD) due to buildup of fat in the liver, causing inflammation and liver damage. NASH also leads to fibrosis before cirrhosis. The serum prolidase levels have been used as a non-invasive indicator of the disease’s progression, distinguishing between simple steatosis and non-alcoholic steatohepatitis (Kayadibi et al., 2009; Arora and Sharma, 2012). Another study revealed that liver biopsy specimens from patients with NASH had significantly higher serum prolidase enzyme activity than healthy controls, (Horoz et al., 2010).
HCV is a single-stranded enveloped positive-sense RNA virus belonging to the Flaviviridae family. It infects the liver, leading to inflammation and fibrosis. It also causes cirrhosis, steatosis, and hepatocellular carcinoma (Ray and Thomas, 2014). Studies suggest that structural and non-structural HCV proteins may directly trigger stellate cell (liver fibroblasts) activation, initiating fibrogenesis (Mazzocca et al., 2005). TGFβ-1, which can initiate as a pro-fibrotic pathway, is upregulated in response to HCV replication and may contribute to collagen metabolism during HCV infection (Lin et al., 2010). Prolidase activity increases in HCV infection, and the combination of prolidase activity and oxidative damage leads to chronic viral hepatitis (Duygu et al., 2012). Liver fibrosis is due to the constant destruction of collagen in hepatic tissue and collagen accumulation in the ECM. Similar to HCV, patients with hepatitis B virus (HBV) infection have also been reported to display increased prolidase activity. HBV has a partially double-stranded DNA genome and belongs to the Hepadnaviridae family. However, in contrast to HCV, the authors found that HBV infection did not correlate with liver fibrosis (Duygu et al., 2013). A similar increase in prolidase activity was seen in pediatric HBV infection (Şen et al., 2014).
The bone comprises of approximately 90% collagen (mostly type I collagen) (Clarke, 2008) PD patients exhibit symptoms such as a shortened stature, hypertelorism, and saddle nose. In addition, bone deformations were used to identify the first patient with PD (Seakins, 1963). Accordingly, PD mice are small with a short femur snout compared to healthy mice. This phenotype was prominent in mice at an early age (post-natal and pubertal), when collagen synthesis and degradation are the highest, demonstrating that prolidase is required for normal skeletogenesis (Besio et al., 2015). Serum prolidase activity is significantly lower in patients with benign joint hypermobility syndrome than in healthy individuals, similar to PD patients (Em et al., 2014).
Osteogenesis imperfecta (OI) is a genetic disorder characterized by defects in COL1A1 and COL1A2, affecting bone, skin, and other tissue with high collagen content. The mutated gene results in improper collagen metabolism (Nijhuis et al., 2019). OI and PD have similar phenotypes, and both have decreased prolidase activity. In OI fibroblasts, prolidase activity is diminished despite the amount of prolidase protein remaining the same. This, suggests a defect at the post-transcriptional and/or post-translation level, in contrast with PD patients wherein the effect is mainly genetic. Expression of beta-1 integrin and IGFR, which have been previously shown to regulate prolidase activity are decreased in OI (Galicka et al., 2001; Galicka et al., 2003). Decreased collagen synthesis is accompanied by decreased prolidase activity, β1 integrin, and IGFI receptors in OI fibroblasts (Galicka et al., 2005). Pharmacological therapy using type-1 bioactive compounds in OI treatment stimulate collagen biosynthesis by increasing prolidase activity and is accompanied by increased IGF-I receptor expression (Galicka and Nazaruk, 2007). Ankylosing spondylitis and Rheumatoid Arthritis have lower serum prolidase levels associated with decreased collagen turnover (Uçar et al., 2013). On the other hand, prolidase activity is increased in thalassemia major, which is associated with osteoporosis (Cakmak et al., 2010). There are no mechanisms reported so far for prolidase regulation in bone-associated diseases.
Prolidase activity has been found to be altered in a variety of cancers. An increase or decrease in prolidase activity can indicate the presence of a cancer and its progression. As a result, prolidase activity measurement can be a useful diagnostic marker for cancer detection and progression. Prolidase activity is elevated in several cancers, such as breast cancer, endometrial cancer, lung cancer, melanoma, and others (Karna et al., 2000; Cechowska-Pasko et al., 2006; Mittal et al., 2007; Arioz et al., 2009). The increase in prolidase activity in these cancers is due to increased collagen biosynthesis allowing for tumor metastasis. In most cancers, the benefit of early detection dramatically enhances the chance of post-diagnosis survival. Importantly, monitoring prolidase activity could be a way to evaluate the success of a treatment or cancer progression.
Collagen has been shown to regulate cellular growth and differentiation through interaction with integrin receptors and plays a vital role in tumorigenesis and invasiveness. The main factor contributing to collagen metabolism disturbances in cancer tissue is the deregulation of the balance between collagen biosynthesis and degradation. A decrease in tissue collagen content and β1-integrin receptor accompanied by an increase in prolidase activity was observed in breast cancer (infiltrating ductal carcinoma) tissue compared to normal tissue. The significantly higher prolidase activity levels indicated an increase in collagen turnover rate in tumor tissue than controls (Cechowska-Pasko et al., 2006). Another study found a correlation between elevated serum prolidase activity to the stage of the disease, tumor size, and prognosis of breast carcinoma (Bayhan et al., 2016). Enhancement of collagen turnover, accompanied by an increase in prolidase activity, was reported in stomach cancer (Guszczyn and Sobolewski, 2004). Prolidase activity was also significantly higher in patients with inoperable gastric cancer than in operable cases and the control group. There is also a strong correlation between tumor volume and prolidase activity (Celik et al., 2017). It has been reported that serum prolidase activity was significantly higher in patients with epithelial ovarian carcinomas (EOC) than healthy controls (Camuzcuoglu et al., 2009). The study observed that more advanced stages, higher tumor grades, and higher cancer antigen 125 (CA-125) levels among patients with EOC were associated with higher serum prolidase activity. Similarly, higher serum prolidase activity was observed in esophageal squamous cell carcinoma (ESCC) patients compared to healthy controls with a positive correlation between the stage of ESCC and prolidase activity (Sayır, 2019). In most cancers studied, aberrations in collagen metabolism were accompanied by significant differences in prolidase activity. Cumulatively, these studies suggest that enhanced prolidase activity in cancer tissues may contribute towards matrix degradation and remodeling of the tumor microenvironment towards a more invasive phenotype. As a result, it has been speculated that compounds that inhibit prolidase activity may have therapeutic potential as anticancer drugs.
Idiopathic pulmonary fibrosis (IPF) is a chronic lung disease that results in fibrosis of the lungs (Barratt et al., 2018). Although the reasons are unknown, the pathobiology of IPF begins with fibroblastic foci (local accumulation of fibroblasts and myofibroblasts), and excessive deposition of collagen and ECM components, which result in the deformation of the lungs. Intraluminal fibrosis is a characteristic of IPF due to the recruitment of inflammatory cells, resulting in the synthesis and degradation of extracellular-associated components in the lung microenvironment (Oikonomidi et al., 2009; Barratt et al., 2018). Animal models of IPF show elevated prolidase activity that correlates with observed histopathology of lung fibrosis (Türkbeyler et al., 2012). IPF is associated with an increased risk of lung cancer in which collagen and prolidase activity also increase (Karna et al., 2000; Ballester et al., 2019). Unlike other cancers wherein beta-1 integrin levels were shown to be associated with prolidase regulation, there was no difference in β1 integrin levels in healthy lung cells versus cancerous cells, suggesting a different mechanism for prolidase regulation in lung cancer (Karna et al., 2000).
Asthma is characterized by inflamed and narrowed airways that produce excessive mucus, triggering breathing problems. Due to inflammation, increased collagen deposition narrows the airways (Oikonomidi et al., 2009). PD patients experience asthma-like manifestations, highlighting the crucial role of prolidase activity in lung health and pathologies. In blood sera collected from adults with bronchial asthma, prolidase activity was increased (Kaleli et al., 2006). However, another study found that serum prolidase activity decreased in children with bronchial asthma (Cakmak et al., 2009). These conflicting studies suggests the possibility of differential impact of age on prolidase activity. Some other collagenolytic enzymes are also associated with an age-related increase (Yu et al., 2013). There is no explanation why a decrease in prolidase activity, as in PD, causes the same pathology as an increase in prolidase activity in adults with bronchial asthma.
Pulmonary tuberculosis (PTB) is a severe bacterial infection of the lungs and one of the most common infectious diseases worldwide (Gumus et al., 2011). Serum prolidase activity was increased in moderate to severe stages of PTB. The increase in serum prolidase activity may be due to tissue destruction, increased immunoglobulin, increased complement levels, or increased fibroblastic activity, which occurs in PTB (Gumus et al., 2011; Wang et al., 2014). The same is seen in Tuberculous Pleurisy (Ipcioglu et al., 2008). Chronic obstructive pulmonary disease (COPD), another inflammatory lung disease that obstructs airflow in the lungs caused by smaller air passageways and irreversibly damaged alveoli, is marked by significantly lower plasma prolidase activity than healthy controls (Gencer et al., 2011).
Influenza is a highly infectious respiratory illness caused by either influenza A and influenza B viruses. Symptoms include sore throat, runny nose, cough, fever, headache, muscle pain, and fatigue (Krammer et al., 2018). Prolidase has been reported to function as a novel entry factor for influenza A virus. Prolidase knockdown and inhibition negatively affected influenza A virus fusion events and its colocalization with endosomal markers. Prolidase was shown to be essential for early endosomal routing upon viral entry (Pohl et al., 2014).
Persistent hyperglycemia due to DM causes pronounced effects on the expression, organization, and modification of ECM components in many organs (Brownlee, 2001; Kolset et al., 2012). These complications lead to the development of diabetes-specific microvascular pathologies. Diabetic nephropathy (DN) is one of the major complication of long term DM and is the most common cause of end stage renal disease (ESRD). DN is associated with structural alterations in the glomerular basement membrane, a specialized ECM composed of type IV collagen, laminin, nidogen, and heparan sulfate proteoglycan, that connects podocytes and endothelial cells in the kidneys (Miner, 2012). In type II DN patients, the substantial damage to kidneys is associated with increased collagen accumulation due to increased thickening of the basement membrane. The development of kidney disease in these patients is marked by excretion of albumin into the urine and renal impairment. Serum prolidase activity is reported to be higher in type 2 DM patients with microalbuminuria than patients without microalbuminuria (Sabuncu et al., 2016). A previous study also reported that, DN patients with ESRD had higher serum prolidase activity, which positively correlated with glucose and creatinine levels (Verma et al., 2014). On the contrary, Erbagci et al. have reported decreased prolidase activity in patients with type II DM than in healthy volunteers with or without diabetic nephropathy (Erbaǧci et al., 2002). These studies thus suggest that the different stages of DN may have a variable effect on prolidase enzyme activity.
The amino acid proline has been suggested to act as a neurotransmitter in brain, and high levels of proline have been associated with neurological symptoms and brain abnormalities (Wyse and Netto, 2011). Since proline is a product of prolidase catalytic activity, higher prolidase activity will increase proline levels. Proline that is not incorporated into collagen or other proteins is further sequentially catabolized to glutamate by proline oxidase/proline dehydrogenase and pyrroline-5-carboxylate dehydrogenase (Pandhare et al., 2009; Phang et al., 2010). A dysfunction or disorder in these enzymes can cause either type I or II hyperprolinemia (Wang et al., 2013; Plecko and Steinfeld, 2017). Hyperprolinemia is associated with cognitive deficits and intellectual disabilities and is associated with schizophrenia, Parkinson’s disease, Alzheimer’s disease, and cerebral ischemia. In addition, proline levels can affect glutamate levels in the brain, and an increase in prolidase may affect glutamate-proline dynamics. High activity of prolidase in the hippocampus and cerebellum regions has been reported in adult male rat brains (Chi et al., 2009). Rat brain prolidase also has a high affinity for Ser-Pro dipeptides. Serine activates a type of neurotransmitter receptor called the N-methyl-D-aspartate (NMDA) receptor (Van-Horn et al., 2013). The hippocampus is vital for connecting short-term memory to long-term memory and spatial memory to enable navigation (Squire et al., 2015). Damage or improper function of the hippocampus is associated with Alzheimer’s disease, stress, and post-traumatic disorder. The potentiation of glutamate excitotoxicity via NMDA activation has been suggested to be a possible mechanism for neurological dysfunction in hyperprolinemia (Cohen and Nadler, 1997; Delwing et al., 2003; Arikanoglu et al., 2013). Hyperstimulation by glutamate of NMDA receptors can lead to neuronal death. Elevated prolidase activity is thought to induce NMDA dysregulation by increasing plasma proline levels in schizophrenia (Bahceci et al., 2015; Güneş et al., 2016). T-Cell proteome of Parkinson’s disease patients treated with dopamine receptor antagonist showed increased levels of prolidase (Alberio et al., 2012). Similarly, in patients of Alzheimer’s disease and major depressive disorder, prolidase activity has been reported to be higher (Arikanoglu et al., 2013) (Kokacya et al., 2014). Recently, a study reported elevated prolidase activity in epileptic patients taking some antiepileptic drug treatment, suggesting an increased risk of subclinical vascular damage (Karacan et al., 2020). Patients with post-traumatic stress disorder have defects in the hippocampus and show decreased prolidase activity (Kitayama et al., 2005; Demir et al., 2016). Despite the lack of research demonstrating the direct impact of prolidase in these disorders, prolidase activity may serve as a useful marker in brain health and mental disorders, especially at different stages of the disease.
PD phenotype has been shown to include a moderate to severe degree of intellectual disabilities and developmental delays. High amounts of accumulating proline-containing peptides have been suggested as a possible explanation (Hui and Lajtha, 1980). Post-natal prolidase deficient model mice exhibit vascularization defects in the cerebral and cerebellar cortices linked to the role of prolidase in angiogenic signaling processes. Improper ECM remodeling causes meningeal defects affecting brain development resulting in impairments, loss of basement membrane integrity, and disorganization of the cytoarchitecture of the cerebral cortex (Insolia and Piccolini, 2014).
Helicobacter pylori (H. pylori) is a non-invasive spiral-shaped microaerophilic microorganism associated with severe gastric pathologies, such as peptic ulcers, chronic active gastritis, mucosal atrophy, and gastric adenocarcinoma. Upon infection, gastric atrophy occurs due to replacement of the gastric mucosal glands with collagen fibers. Persistent infection results in the architectural disruption of the ECM and fibrosis of the stomach’s mucosal layer. A study observed that H. pylori-positive subjects had higher serum prolidase activity than H. pylori-negative subjects (Aslan et al., 2007). Since this was an observational study, no mechanism was offered to explain why the serum prolidase activity may be higher. Though patients were selected based on health, other fibrotic events may contribute to the increased serum prolidase levels.
Organophosphorus acid anhydrolases (OPAA) are enzymes that hydrolyze highly toxic compounds such as acetylcholinesterase-inhibiting compounds, chemical warfare G-type nerve agents, and pesticides. OPAA-2 from Alteromonas sp. strain JD6.5 EcoRI-lZAPII chromosomal library showed sequence homology with E. coli aminopeptidase P and human prolidase. OPAA-2 also demonstrates prolidase-like activity by cleaving leu-pro and ala-pro, suggesting an evolutionary relationship. Though OPAA performs this activity, it hydrolyzes P-F and P-O bonds while prolidase hydrolyzes C-N (Cheng et al., 1996). Further study suggested that Alteromonas haloplanktis OPAA is a type of prolidase. Alteromonas prolidase can hydrolyze G-type nerve agents and DFP, soman, sarin, and cyanide containing tabun, therefore is used as an antidote for poisoning (Cheng et al., 1997; Cheng et al., 1999; Petrikovics et al., 2000). Native and recombinant human prolidase also catalyzes these nerve agents with A252R or P365R substitution mutation of the enzyme, improving the catalytic activity against organophosphorus compounds (Chandrasekaran et al., 2013; Yun et al., 2017).
Prolidase is an enzyme that catalyzes dipeptides containing a C-terminal proline or hydroxyproline. These dipeptides are produced primarily during collagen turnover, thus making prolidase mediated catalysis as the rate-limiting step in collagen biosynthesis. The generated pool of proline is recycled during collagen synthesis and turnover. This process is important in several physiological and pathological processes. Therefore, alterations in prolidase activity are associated with not only prolidase deficiency, a rare genetic disorder, but also with several pathological conditions. Prolidase activity is vital for wound healing, and prolidase deficiency is associated with poor wound healing in both mice and humans. Although prolidase is essential in various processes, fundamental research on the enzyme is limited. Thus, a better understanding of the regulation of prolidase both during normal and diseased conditions, will help identify novel pathways that can be exploited for possible therapeutic application, especially in patients suffering from chronic unhealing wounds.
IE, ZL, and JP wrote the manuscript. IE, MB, CD, and JP edited and finalized the manuscript.
This work was supported by the Research Centers in Minority Institutions (RCMI) grant U54MD007586 to JP and CD, National Institutes of Health Grants R01 AI136740, R56 AI122960. IE was in part supported by the NIH 5R25GM059994 (RISE). This work is also in part supported by the Meharry Translational Research Center (MeTRC) grant 5U54MD007593-10 and Tennessee CFAR Grant P30 AI110527 from the National Institutes of Health to CD. All figures for the review article were created with BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abraham, P., Wilfred, G., and Ramakrishna, B. (2000). Plasma Prolidase May Be an Index of Liver Fibrosis in the Rat. Clinica Chim. Acta 295, 199–202. doi:10.1016/s0009-8981(00)00183-2
Adams, E., McFadden, M., and Smith, E. L. (1952). Peptidases of Erythrocytes. I. Distribution in Man and Other Species. J. Biol. Chem. 198 (2), 663–670. doi:10.1016/s0021-9258(18)55523-x
Adams, E., and Smith, E. L. (1952). Peptidases of Erythrocytes. II. Isolation and Properties of Prolidase. J. Biol. Chem. 198 (2), 671–682. doi:10.1016/s0021-9258(18)55524-1
Albaugh, V. L., Mukherjee, K., and Barbul, A. (2017). Proline Precursors and Collagen Synthesis: Biochemical Challenges of Nutrient Supplementation and Wound Healing. J. Nutr. 147 (11), 2011–2017. doi:10.3945/jn.117.256404
Alberio, T., Pippione, A. C., Comi, C., Olgiati, S., Cecconi, D., Zibetti, M., et al. (2012). Dopaminergic Therapies Modulate the T-CELL Proteome of Patients with Parkinson's Disease. IUBMB Life 64, 846–852. doi:10.1002/iub.1073
Arata, J., Hatakenaka, K., and Oono, T. (1986). Effect of Topical Application of Glycine and Proline on Recalcitrant Leg Ulcers of Prolidase Deficiency. Arch. Dermatol. 122 (6), 626–627. doi:10.1001/archderm.122.6.626
Arikanoglu, A., Akil, E., Varol, S., Yucel, Y., Yuksel, H., Cevik, M. U., et al. (2013). Relationship of Cognitive Performance with Prolidase and Oxidative Stress in Alzheimer Disease. Neurol. Sci. 34, 2117–2121. doi:10.1007/s10072-013-1346-4
Arioz, D. T., Camuzcuoglu, H., Toy, H., Kurt, S., Celik, H., and Aksoy, N. (2009). Serum Prolidase Activity and Oxidative Status in Patients with Stage I Endometrial Cancer. Int. J. Gynecol. Cancer 19 (7), 1244–1247. doi:10.1111/igc.0b013e3181af711e
Arora, A., and Sharma, P. (2012). Non-invasive Diagnosis of Fibrosis in Non-alcoholic Fatty Liver Disease. J. Clin. Exp. Hepatol. 2, 145–155. doi:10.1016/s0973-6883(12)60103-0
Aslan, M., Nazligul, Y., Horoz, M., Bolukbas, C., Bolukbas, F. F., Aksoy, N., et al. (2007). Serum Prolidase Activity and Oxidative Status in Helicobacter pylori Infection. Clin. Biochem. 40, 37–40. doi:10.1016/j.clinbiochem.2006.08.006
Bahceci, B., Bagcioglu, E., Kokacya, M. H., Dilek, A. R., Bahceci, I., and Selek, S. (2015). Prolidase Activity and Oxidative Stress in Patients with Schizophrenia: A Preliminary Study. JPMA. J. Pakistan Med. Assoc. 65, 131–135.
Ballester, B., Milara, J., and Cortijo, J. (2019). Idiopathic Pulmonary Fibrosis and Lung Cancer: Mechanisms and Molecular Targets. Int. J. Mol. Sci. MDPI AG 20, 593. doi:10.3390/ijms20030593
Barbul, A. (2008). Proline Precursors to Sustain Mammalian Collagen Synthesis. J. Nutr. 138 (10), 2021S–2024S. doi:10.1093/jn/138.10.2021s
Barratt, S., Creamer, A., Hayton, C., and Chaudhuri, N. (2018). Idiopathic Pulmonary Fibrosis (IPF): An Overview. J. Clin. Med. 7, 201. doi:10.3390/jcm7080201
Bataller, R., and Brenner, D. A. (2005). Liver Fibrosis. J. Clin. Invest. 115, 209–218. doi:10.1172/jci24282
Bayhan, Z., Zeren, S., Kocak, C., Kocak, F. E., Duzgun, S. A., Algin, M. C., et al. (2016). Prolidase Activity and Oxidative Stress in Patients with Breast Carcinoma A Prospective Randomized Case-Controlled Study. Ann. Ital. Chir 87 (6), 517–524.
Bazan, J. F., Weaver, L. H., Roderick, S. L., Huber, R., and Matthews, B. W. (1994). Sequence and Structure Comparison Suggest that Methionine Aminopeptidase, Prolidase, Aminopeptidase P, and Creatinase Share a Common Fold. Proc. Natl. Acad. Sci. United States America 91, 2473–2477. doi:10.1073/pnas.91.7.2473
Bergmann, M., and Fruton, J. S. (1937). On Proteolytic Enzymes Xii. Regarding the Specificity of Aminopeptidase and Carboxypeptidase. A New Type of Enzyme in the Intestinal Tract. J. Biol. Chem. 117, 189–202. doi:10.1016/s0021-9258(18)74600-0
Besio, R., Alleva, S., Forlino, A., Lupi, A., Meneghini, C., Minicozzi, V., et al. (2010). Identifying the Structure of the Active Sites of Human Recombinant Prolidase. Eur. Biophys. J. 39, 935–945. doi:10.1007/s00249-009-0459-4
Besio, R., Baratto, M. C., Gioia, R., Monzani, E., Nicolis, S., Cucca, L., et al. (2013). A Mn(II)–Mn(II) Center in Human Prolidase. Biochim. Biophys. Acta (Bba) - Proteins Proteomics 1834, 197–204. doi:10.1016/j.bbapap.2012.09.008
Besio, R., Maruelli, S., Gioia, R., Villa, I., Grabowski, P., Gallagher, O., et al. (2015). Lack of Prolidase Causes a Bone Phenotype Both in Human and in Mouse. Bone 72, 53–64. doi:10.1016/j.bone.2014.11.009
Blackstock, C. D., Higashi, Y., Sukhanov, S., Shai, S. Y., Stefanovic, B., Tabony, A. M., et al. (2014). Insulin-like Growth Factor-1 Increases Synthesis of Collagen Type I via Induction of the mRNA-Binding Protein LARP6 Expression and Binding to the 5′ Stem-Loop of COL1a1 and COL1a2 mRNA. J. Biol. Chem. 289, 7264–7274. doi:10.1074/jbc.m113.518951
Brosset, B., Myara, I., Fabre, M., and Lemonnier, A. (1988). Plasma Prolidase and Prolinase Activity in Alcoholic Liver Disease. Clinica Chim. Acta 175, 291–295. doi:10.1016/0009-8981(88)90105-2
Brownlee, M. (2001). Biochemistry and Molecular Cell Biology of Diabetic Complications. Nature 414 (6865), 813–820. doi:10.1038/414813a
Buist, N. R. M., Strandholm, J. J., Bellinger, J. F., and Kennaway, N. G. (1972). Further Studies on a Patient with Iminodipeptiduria: A Probable Case of Prolidase Deficiency. Metabolism 21, 1113–1123. doi:10.1016/0026-0495(72)90106-0
Butbul Aviel, Y., Mandel, H., Avitan Hersh, E., Bergman, R., Adiv, O. E., Luder, A., et al. (2012). Prolidase Deficiency Associated with Systemic Lupus Erythematosus (SLE): Single Site Experience and Literature Review. Pediatr. Rheumatol. 10, 18. doi:10.1186/1546-0096-10-18
Cakmak, A., Soker, M., Koc, A., and Aksoy, N. (2010). Prolidase Activity and Oxidative Status in Patients with Thalassemia Major. J. Clin. Lab. Anal. 24, 6–11. doi:10.1002/jcla.20361
Cakmak, A., Zeyrek, D., Atas, A., Celik, H., Aksoy, N., and Erel, O. (2009). Serum Prolidase Activity and Oxidative Status in Patients with Bronchial Asthma. J. Clin. Lab. Anal. 23, 132–138. doi:10.1002/jcla.20303
Camuzcuoglu, H., Arioz, D. T., Toy, H., Kurt, S., Celik, H., and Aksoy, N. (2009). Assessment of Preoperative Serum Prolidase Activity in Epithelial Ovarian Cancer. Eur. J. Obstet. Gynecol. Reprod. Biol. 147, 97–100. doi:10.1016/j.ejogrb.2009.07.012
Cechowska-Pasko, M., Palka, J., and Bańkowski, E. (2004). Fasting-induced Inhibition of Collagen Biosynthesis in Rat Skin. A Possible Role for Phosphoenolpyruvate in This Process. Mol. Cell. Biochem. 265, 203–208. doi:10.1023/b:mcbi.0000044397.32748.23
Cechowska-Pasko, M., Pałka, J., and Wojtukiewicz, M. Z. (2006). Enhanced Prolidase Activity and Decreased Collagen Content in Breast Cancer Tissue. Int. J. Exp. Pathol. 87, 289–296. doi:10.1111/j.1365-2613.2006.00486.x
Celik, S., Kiziltan, R., Yilmaz, E. M., Yilmaz, Ö., and Demir, H. (2017). Potential Diagnostic and Prognostic Significance of Plasma Prolidase Activity in Gastric Cancer. Biomarkers Med. 11, 319–327. doi:10.2217/bmm-2016-0367
Chandrasekaran, L., Belinskaya, T., and Saxena, A. (2013). In Vitro Characterization of Organophosphorus Compound Hydrolysis by Native and Recombinant Human Prolidase. Toxicol. Vitro 27, 499–506. doi:10.1016/j.tiv.2012.05.012
Cheng, T.-c., DeFrank, J. J., and Rastogi, V. K. (1999). Alteromonas Prolidase for Organophosphorus G-Agent Decontamination. Chemico-Biological Interactions 119-120, 455–462. doi:10.1016/s0009-2797(99)00058-7
Cheng, T. C., Harvey, S. P., and Chen, G. L. (1996). Cloning and Expression of a Gene Encoding a Bacterial Enzyme for Decontamination of Organophosphorus Nerve Agents and Nucleotide Sequence of the Enzyme. Appl. Environ. Microbiol. 62, 1636–1641. doi:10.1128/aem.62.5.1636-1641.1996
Cheng, T., Liu, L., Wang, B., Wu, J., DeFrank, J. J., Anderson, D. M., et al. (1997). Nucleotide Sequence of a Gene Encoding an Organophosphorus Nerve Agent Degrading Enzyme from Alteromonas Haloplanktis. J. Ind. Microbiol. Biotechnol. 18, 49–55. doi:10.1038/sj.jim.2900358
Chi, H., Lu, J., Liu, G., Tong, J., Nakayama, K., Yamashita, K., et al. (2009). Activity of Prolidase Isoenzymes in the Rat Brain: Subcellular and Regional Distribution during Development. Brain Res. 1303, 8–14. doi:10.1016/j.brainres.2009.09.081
Chidambaram, A. C., Thangaraju, S. K., Sarangarajan, S., Maulik, K., Ramamoorthy, J. G., and Gunasekaran, D. (2021). Macrophage Activation Syndrome in a Patient with Prolidase Deficiency: A Rare Genetic Disorder Associated with Elevated IgE and Lupus-like Syndrome. J. Clin. Immunol. doi:10.1007/s10875-021-01096-2
Clarke, B. (2008). Normal Bone Anatomy and Physiology. Clin. J. Am. Soc. Nephrol. 3, S131–S139. doi:10.2215/cjn.04151206
Cleary, M. A., Heaney, M., Couriel, J. M., and Walter, J. H. (1994). Immune Function in Prolidase Deficiency. J. Inherit. Metab. Dis. 17, 345–348. doi:10.1007/bf00711826
Cohen, S. M., and Nadler, J. V. (1997). Sodium-Dependent Proline and Glutamate Uptake by Hippocampal Synaptosomes During Postnatal Development. Develop. Brain Res. 100, 230–233. doi:10.1016/s0165-3806(97)00045-x
Cosson, C., Myara, I., Miech, G., Moatti, N., and Lemonnier, A. (1992). Only Prolidase I Activity Is Present in Human Plasma. Int. J. Biochem. 24, 427–432. doi:10.1016/0020-711x(92)90035-y
Cottin, V., Nasser, M., Traclet, J., Chalabreysse, L., Lèbre, A. S., Si-Mohamed, S., et al. (2020). Prolidase Deficiency: A New Genetic Cause of Combined Pulmonary Fibrosis and Emphysema Syndrome in the Adult. Eur. Respir. J. 55, 1901952. doi:10.1183/13993003.01952-2019
Davis, N. C., and Smith, E. L. (1957). Purification and Some Properties of Prolidase of Swine Kidney. J. Biol. Chem. 224 (1), 261–275. doi:10.1016/s0021-9258(18)65027-6
Delwing, D., Tagliari, B., Streck, E. L., Wannamacher, C. M. D., Wajner, M., and Wyse, A. T. D. S. (2003). Reduction of Energy Metabolism in Rat hippocampus by Arginine Administration. Brain Res. 983, 58–63. doi:10.1016/s0006-8993(03)03029-4
Demir, S., Bulut, M., Atli, A., Kaplan, İ., Kaya, M. C., Bez, Y., et al. (2016). Decreased Prolidase Activity in Patients with Posttraumatic Stress Disorder. Psychiatry Invest. 13, 420. doi:10.4306/pi.2016.13.4.420
DesJardins-Park, H. E., Foster, D. S., and Longaker, M. T. (2018). Fibroblasts and Wound Healing: An Update. Regenerative Med. 13, 491–495. doi:10.2217/rme-2018-0073
Dolenga, M., and Hechtman, P. (1992). Prolidase Deficiency in Cultured Human Fibroblasts: Biochemical Pathology and Iminodipeptide-Enhanced Growth. Pediatr. Res. 32, 479–482. doi:10.1203/00006450-199210000-00020
Dunn, R., Varigos, G., and Winship, I. (2011). A Photographic Essay of Prolidase Deficiency. Clin. Dysmorphol. Clin Dysmorphol. 20, 194–199. doi:10.1097/mcd.0b013e3283486cbd
Duong, H. S., Zhang, Q.-Z., Le, A. D., Kelly, A. P., Kamdar, R., and Messadi, D. V. (2006). Elevated Prolidase Activity in Keloids: Correlation with Type I Collagen Turnover. Br. J. Dermatol. 154, 820–828. doi:10.1111/j.1365-2133.2006.07167.x
Duygu, F., Aksoy, N., Cicek, A. C., Butun, I., and Unlu, S. (2013). Does Prolidase Indicate Worsening of Hepatitis B Infection?. J. Clin. Lab. Anal. 27, 398–401. doi:10.1002/jcla.21617
Duygu, F., Koruk, S. T., Karsen, H., Aksoy, N., Taskin, A., and Hamidanoglu, M. (2012). Prolidase and Oxidative Stress in Chronic Hepatitis C. J. Clin. Lab. Anal. 26, 232–237. doi:10.1002/jcla.21510
Em, S., Ucar, D., Oktayoglu, P., Bozkurt, M., Caglayan, M., Yıldız, I., et al. (2014). Serum Prolidase Activity in Benign Joint Hypermobility Syndrome. BMC Musculoskelet. Disord. 15, 75. doi:10.1186/1471-2474-15-75
Endo, F., Motohara, K., Indo, Y., and Matsuda, I. (1987). Immunochemical Studies of Human Prolidase with Monoclonal and Polyclonal Antibodies:Absence of the Subunit of Prolidase in Erythrocytes from a Patient with Prolidase Deficiency. Pediatr. Res. 22, 627–633. doi:10.1203/00006450-198712000-00002
Endo, F., Tanoue, A., Nakai, H., Hata, A., Indo, Y., Titani, K., et al. (1989). Primary Structure and Gene Localization of Human Prolidase. J. Biol. Chem. 264, 4476–4481. doi:10.1016/s0021-9258(18)83768-1
Erbaǧci, A. B., Araz, M., Erbaǧci, A., Tarakçioǧlu, M., and Namiduru, E. S. (2002). Serum Prolidase Activity as a Marker of Osteoporosis in Type 2 Diabetes Mellitus. Clin. Biochem. 35, 263–268. doi:10.1016/s0009-9120(02)00305-3
Eren, M. A., Torun, A. N., Tabur, S., Ulas, T., Demir, M., Sabuncu, T., et al. (2013). Serum Prolidase Activity in Diabetic Foot Ulcers. Acta Diabetol. 50, 423–427. doi:10.1007/s00592-012-0448-4
Falik-Zaccai, T. C., Khayat, M., Luder, A., Frenkel, P., Magen, D., Brik, R., et al. (2010). A Broad Spectrum of Developmental Delay in a Large Cohort of Prolidase Deficiency Patients Demonstrates Marked Interfamilial and Intrafamilial Phenotypic Variability. Am. J. Med. Genet. B Neuropsychiatr. Genet. 153B (1), 46–56. doi:10.1002/ajmg.b.30945
Ferreira, C., and Wang, H. (1993). “Prolidase Deficiency,” in GeneReviews((R)). Editors M. P. Adam, H. H. Ardinger, and R. A. Pagon (Seattle, WA: University of Washington).
Forlino, A., Lupi, A., Vaghi, P., Cornaglia, A. I., Calligaro, A., Campari, E., et al. (2002). Mutation Analysis of Five New Patients Affected by Prolidase Deficiency: The Lack of Enzyme Activity Causes Necrosis-Like Cell Death in Cultured Fibroblasts. Hum. Genet. 111, 314–322. doi:10.1007/s00439-002-0792-5
Galicka, A., and Nazaruk, J. (2007). Stimulation of Collagen Biosynthesis by Flavonoid Glycosides in Skin Fibroblasts of Osteogenesis Imperfecta Type I and the Potential Mechanism of Their Action. Int. J. Mol. Med. 20, 889–895. doi:10.3892/ijmm.20.6.889
Galicka, A., Surazyński, A., Wołczyński, S., Pałka, J., Popko, J., and Gindzieński, A. (2005). Phenotype Variability in a Daughter and Father with Mild Osteogenesis Imperfecta Correlated with Collagen and Prolidase Levels in Cultured Skin Fibroblasts. Ann. Clin. Biochem. 42, 80–84. doi:10.1258/0004563053026871
Galicka, A., Wolczyñski, S., Anchim, T., Surazyñski, A., Lesniewicz, R., and Palka, J. (2001). Defects of Type I Procollagen Metabolism Correlated with Decrease of Prolidase Activity in a Case of Lethal Osteogenesis Imperfecta. Eur. J. Biochem. 268, 2172–2178. doi:10.1046/j.1432-1327.2001.02099.x
Galicka, A., Wołczyński, S., Gindzieński, A., Surazyński, A., and Pałka, J. (2003). Gly511 to Ser Substitution in the COL1A1 Gene in Osteogenesis Imperfecta Type III Patient with Increased Turnover of Collagen. Mol. Cell. Biochem. 248, 49–56. doi:10.1023/a:1024197213525
Gecit, I., Aslan, M., Gunes, M., Pirincci, N., Esen, R., Demir, H., et al. (2012). Serum Prolidase Activity, Oxidative Stress, and Nitric Oxide Levels in Patients with Bladder Cancer. J. Cancer Res. Clin. Oncol. 138 (5), 739–743. doi:10.1007/s00432-011-1136-4
Gencer, M., Aksoy, N., Dagli, E. C., Uzer, E., Aksoy, S., Selek, S., et al. (2011). Prolidase Activity Dysregulation and its Correlation with Oxidative-Antioxidative Status in Chronic Obstructive Pulmonary Disease. J. Clin. Lab. Anal. 25, 8–13. doi:10.1002/jcla.20347
Gonzalez, A. C., Costa, T. F., Andrade, Z. A., and Medrado, A. R. (2016). Wound Healing - A Literature Review. Bras Dermatol. 91 (5), 614–620. doi:10.1590/abd1806-4841.20164741
Goodman, S. I., Solomons, C. C., Muschenheim, F., McIntyre, C. A., Miles, B., and O'Brien, D. (1968). A Syndrome Resembling Lathyrism Associated with Iminodipeptiduria. Am. J. Med. 45, 152–159. doi:10.1016/0002-9343(68)90016-8
Grassmann, W., Schoenebeck, O. V., and Auerbach, G. (1932). Uber die enzymatische Spaltbarkeit der Prolinpeptide. II. Physiol. Chem. 210, 1–14. doi:10.1515/bchm2.1932.210.1-2.1
Gumus, S., Yaman, H., Ozcan, O., Deniz, O., Karaman, B., Cakir, E., et al. (2011). Serum Prolidase Activity in Patients with Pulmonary Tuberculosis. Scand. J. Clin. Lab. Invest. 71, 467–472. doi:10.3109/00365513.2011.587021
Güneş, M., Bulut, M., Demir, S., İbiloğlu, A. O., Kaya, M. C., Atlı, A., et al. (2016). Diagnostic Performance of Increased Prolidase Activity in Schizophrenia. Neurosci. Lett. 613, 36–40. doi:10.1016/j.neulet.2015.12.036
Guszczyn, T., and Sobolewski, K. (2004). Deregulation of Collagen Metabolism in Human Stomach Cancer. Pathobiology 71, 308–313. doi:10.1159/000081726
Hechtman, P., Richter, A., Corman, N., and Leong, Y. M. (1988). In Situ Activation of Human Erythrocyte Prolidase: Potential for Enzyme Replacement Therapy in Prolidase Deficiency. Pediatr. Res. 24, 709–712. doi:10.1203/00006450-198812000-00012
Hinz, B. (2016). The Role of Myofibroblasts in Wound Healing. Curr. Res. Translational Med. 64, 171–177. doi:10.1016/j.retram.2016.09.003
Horng, H. C., Chang, W. H., Yeh, C. C., Huang, B. S., Chang, C. P., Chen, Y. J., et al. (2017). Estrogen Effects on Wound Healing. Int. J. Mol. Sci. MDPI AG 18, 2325. doi:10.3390/ijms18112325
Horoz, M., Aslan, M., Bolukbas, F. F., Bolukbas, C., Nazligul, Y., Celik, H., et al. (2010). Serum Prolidase Enzyme Activity and its Relation to Histopathological Findings in Patients with Non-alcoholic Steatohepatitis. J. Clin. Lab. Anal. 24, 207–211. doi:10.1002/jcla.20334
Huang, Y., and Kyriakides, T. R. (2020). The Role of Extracellular Matrix in the Pathophysiology of Diabetic Wounds. Matrix Biol. Plus 6-7, 100037. doi:10.1016/j.mbplus.2020.100037
Hui, K.-S., and Lajtha, A. (1980). Activation and Inhibition of Cerebral Prolidase. J. Neurochem. 35, 489–494. doi:10.1111/j.1471-4159.1980.tb06292.x
Ikeda, K., Tohyama, J., Tsujlno, S., Sato, K., Oono, T., Arata, J., et al. (1997). Amelioration of Prolidase Deficiency in Fibroblasts Using Adenovirus Mediated Gene Transfer. Jpn. J. Hum. Genet. 42, 401–408. doi:10.1007/bf02766940
Insolia, V., and Piccolini, V. M. (2014). Brain Morphological Defects in Prolidase Deficient Mice: First Report. Eur. J. Histochem. 58, 2417. doi:10.4081/ejh.2014.2417
Ipcioglu, O. M., Ozcan, O., Gultepe, M., Deniz, O., and Akgul, E. O. (2008). Prolidase Activity in Serum and Pleural Fluids in Patients with Tuberculous Pleural Effussion. Clin. Biochem. 41, 670–675. doi:10.1016/j.clinbiochem.2008.03.006
Ishii, T., Tsujino, S., Matsunobu, S., Endo, F., Sato, K., and Sakuragawa, N. (1996). Cloning of Mouse Prolidase cDNA: Predominant Expression of Prolidase mRNA in Kidney. Biochim. Biophys. Acta 1308 (1), 15–16. doi:10.1016/0167-4781(96)00084-x
Jackson, S. H., and Heininger, J. A. (1975). Proline Recycling During Collagen Metabolism as Determined by Concurrent 18O2- and 3H-Labeling. BBA - Gen. Subjects 381, 359–367. doi:10.1016/0304-4165(75)90241-x
Joshi, H., and Press, M. F. (2018). “Molecular Oncology of Breast Cancer,” in The Breast: Comprehensive Management of Benign and Malignant Diseases (Amsterdam: Elsevier), 282–307. doi:10.1016/b978-0-323-35955-9.00022-2
Kaleli, S., Akkaya, A., Akdogan, M., and Gültekin, F. (2006). The Effects of Different Treatments on Prolidase and Antioxidant Enzyme Activities in Patients with Bronchial Asthma. Environ. Toxicol. Pharmacol. 22, 35–39. doi:10.1016/j.etap.2005.11.001
Karacan, N., Calik, M., Kazanasmaz, H., Ethemoglu, O., Guzelcicek, A., Yasin, S., et al. (2020). The Serum Prolidase Enzyme Activity as a Biomarker for Evaluation of the Subclinical Vascular Damage in Children with Epilepsy. Ann. Indian Acad. Neurol. 23 (6), 787–791. doi:10.4103/aian.AIAN_640_19
Karna, E., Pałka, J., and Wołczyński, S. (2001). Doxycycline-Induced Inhibition of Prolidase Activity in Human Skin Fibroblasts and its Involvement in Impaired Collagen Biosynthesis. Eur. J. Pharmacol. 430, 25–31. doi:10.1016/s0014-2999(01)01372-3
Karna, E., Surazynski, A., and Palka, J. (2000). Collagen Metabolism Disturbances Are Accompanied by an Increase in Prolidase Activity in Lung Carcinoma Planoepitheliale. Int. J. Exp. Pathol. 81, 341–347. doi:10.1046/j.1365-2613.2000.00168.x
Karna, E., Szoka, L., Huynh, T. Y. L., and Palka, J. A. (2020). Proline-Dependent Regulation of Collagen Metabolism. Cell Mol Life Sci 77 (10), 1911–1918. doi:10.1007/s00018-019-03363-3
Karna, E., Szoka, L., and Palka, J. (2012). Thrombin-Dependent Modulation of β1-Integrin-Mediated Signaling Up-Regulates Prolidase and HIF-1α through P-FAK in Colorectal Cancer Cells. Mol. Cell Biochem. 361, 235–241. doi:10.1007/s11010-011-1108-7
Karppinen, S.-M., Heljasvaara, R., Gullberg, D., Tasanen, K., and Pihlajaniemi, T. (2019). Open Peer Review Toward Understanding Scarless Skin Wound Healing and Pathological Scarring [version 1; Peer Review: 2 Approved]. F1000Research 2019 8, 787. doi:10.12688/f1000research.18293.1
Kayadibi, H., Gültepe, M., Yasar, B., Ince, A. T., Ozcan, O., Ipcioglu, O. M., et al. (2009). Diagnostic Value of Serum Prolidase Enzyme Activity to Predict the Liver Histological Lesions in Non-Alcoholic Fatty Liver Disease: A Surrogate Marker to Distinguish Steatohepatitis from Simple Steatosis. Dig. Dis. Sci. 54, 1764–1771. doi:10.1007/s10620-008-0535-0
Kenig, A., Perzon, O., Tal, Y., Sviri, S., Abutbul, A., Romain, M., et al. (2021). An Adult with Recurrent Severe Pneumococcal Pneumonia Secondary to Prolidase Deficiency. Isr. Med. Assoc. J. 23 (3), 193–195.
Kikuchi, S., Tanoue, A., Endo, F., Wakasugi, S., Matsuo, N., and Tsujimoto, G. (2000). A Novel Nonsense Mutation of the PEPD Gene in a Japanese Patient with Prolidase Deficiency. J. Hum. Genet. 45, 102–104. doi:10.1007/s100380050023
Kim, K. E., Cho, D., and Park, H. J. (2016). Air Pollution and Skin Diseases: Adverse Effects of Airborne Particulate Matter on Various Skin Diseases. Life Sci. 152, 126–134. doi:10.1016/j.lfs.2016.03.039
Kim, K. K., Sheppard, D., and Chapman, H. A. (2018). TGF-β1 Signaling and Tissue Fibrosis. Cold Spring Harbor Perspect. Biol. 10, a022293. doi:10.1101/cshperspect.a022293
Kiratli Nalbant, E., Karaosmanoglu, N., Kutlu, O., Ceylaner, S., and Eksioglu, H. M. (2019). A Rare Case of Prolidase Deficiency with Situs Inversus Totalis, Identified by a Novel Mutation in the PEPD Gene. JAAD Case Rep. 5 (5), 436–438. doi:10.1016/j.jdcr.2019.03.008
Kitayama, N., Vaccarino, V., Kutner, M., Weiss, P., and Bremner, J. D. (2005). Magnetic Resonance Imaging (MRI) Measurement of Hippocampal Volume in Posttraumatic Stress Disorder: A Meta-Analysis. J. Affective Disord. 88, 79–86. doi:10.1016/j.jad.2005.05.014
Kitchener, R. L., and Grunden, A. M. (2012). Prolidase Function in Proline Metabolism and its Medical and Biotechnological Applications. J. Appl. Microbiol. 113 (2), 233–247. doi:10.1111/j.1365-2672.2012.05310.x
Kokacya, M. H., Bahceci, B., Bahceci, I., Dilek, A. R., and Dokuyucu, R. (2014). Prolidase Activity and Oxidative Stress in Patients with Major Depressive Disorder. Psychiatria Danubina 26, 314–318.
Kolset, S. O., Reinholt, F. P., and Jenssen, T. (2012). Diabetic Nephropathy and Extracellular Matrix. J. Histochem. Cytochem. 60 (12), 976–986. doi:10.1369/0022155412465073
Krammer, F., Smith, G. J. D., Fouchier, R. A. M., Peiris, M., Kedzierska, K., Doherty, P. C., et al. (2018). Influenza. Nat. Rev. Dis. Primers 4 (1), 3. doi:10.1038/s41572-018-0002-y
Kubiczkova, L., Sedlarikova, L., Hajek, R., and Sevcikova, S. (2012). TGF-β - An Excellent Servant but a Bad Master. J. Translational Med. BioMed Cent. 10, 1–24. doi:10.1186/1479-5876-10-183
Kurien, B. T., D'Sousa, A., Bruner, B. F., Gross, T., James, J. A., Targoff, I. N., et al. (2013). Prolidase Deficiency Breaks Tolerance to Lupus-Associated Antigens. Int. J. Rheum. Dis. 16, 674–680. doi:10.1111/1756-185x.12254
Laronha, H., and Caldeira, J. (2020). Structure and Function of Human Matrix Metalloproteinases. Cells, NLM (Medline). 9, 1076. doi:10.3390/cells9051076
Laurent, G. J. (1987). Dynamic State of Collagen: Pathways of Collagen Degradation In Vivo and Their Possible Role in Regulation of Collagen Mass. Am. J. Physiol. 252 (1 Pt 1), C1–C9. doi:10.1152/ajpcell.1987.252.1.C1
Lin, W., Tsai, W. L., Shao, R. X., Wu, G., Peng, L. F., Barlow, L. L., et al. (2010). Hepatitis C Virus Regulates Transforming Growth Factor β1 Production Through the Generation of Reactive Oxygen Species in a Nuclear Factor κB-Dependent Manner. Gastroenterology 138, 250918. doi:10.1053/j.gastro.2010.03.008
Lokmic, Z., Musyoka, J., Hewitson, T. D., and Darby, I. A. (2012). Hypoxia and Hypoxia Signaling in Tissue Repair and Fibrosis. Int. Rev. Cel Mol. Biol. 296, 139–185. doi:10.1016/b978-0-12-394307-1.00003-5
Lubick, K. J., Robertson, S. J., McNally, K. L., Freedman, B. A., Rasmussen, A. L., Taylor, R. T., et al. (2015). Flavivirus Antagonism of Type I Interferon Signaling Reveals Prolidase as a Regulator of IFNAR1 Surface Expression. Cell Host & Microbe 18, 61–74. doi:10.1016/j.chom.2015.06.007
Luder, A. S., Mandel, H., Khayat, M., Gurevich, I., Frankel, P., Rivlin, J., et al. (2007). Chronic Lung Disease and Cystic Fibrosis Phenotype in Prolidase Deficiency: A Newly Recognized Association. J. Pediatr. 150, 656–658. doi:10.1016/j.jpeds.2007.03.025
Lundblad, R. L., Bradshaw, R. A., Gabriel, D., Ortel, T. L., Lawson, J., and Mann, K. G. (2004). A Review of the Therapeutic Uses of Thrombin. Thrombosis and Haemostasis. Schattauer GmbH 91, 851–860. doi:10.1160/TH03-12-0792
Lupi, A., Tenni, R., Rossi, A., Cetta, G., and Forlino, A. (2008). Human Prolidase and Prolidase Deficiency: An Overview on the Characterization of the Enzyme Involved in Proline Recycling and on the Effects of its Mutations. Amino Acids 35, 739–752. doi:10.1007/s00726-008-0055-4
Maher, M. J., Ghosh, M., Grunden, A. M., Menon, A. L., Adams, M. W., Freeman, H. C., et al. (2004). Structure of the Prolidase from Pyrococcus Furiosus. Biochemistry 43 (10), 2771–2783. doi:10.1021/bi0356451
Mazzocca, A., Sciammetta, S. C., Carloni, V., Cosmi, L., Annunziato, F., Harada, T., et al. (2005). Binding of Hepatitis C Virus Envelope Protein E2 to CD81 Up-Regulates Matrix Metalloproteinase-2 in Human Hepatic Stellate Cells. J. Biol. Chem. 280, 11329–11339. doi:10.1074/jbc.m410161200
McAnulty, R. J., and Laurent, G. J. (1987). Collagen Synthesis and Degradation In Vivo. Evidence for Rapid Rates of Collagen Turnover with Extensive Degradation of Newly Synthesized Collagen in Tissues of the Adult Rat. Coll. Relat. Res. 7 (2), 93–104. doi:10.1016/s0174-173x(87)80001-8
Mezu-Ndubuisi, O. J., and Maheshwari, A. (2021). The Role of Integrins in Inflammation and Angiogenesis. Pediatr. Res. 89 (7), 1619–1626. doi:10.1038/s41390-020-01177-9
Miltyk, W., Anchim, T., Wolczynski, S., and Palka, J. (1999). Estrogen-Dependent Regulation of Prolidase Activity in Breast Cancer MCF-7 Cells. Gynecol. Endocrinol. 13, 166–174. doi:10.3109/09513599909167551
Miltyk, W., Karna, E., Wołczyński, S., and Pałka, J. (1998). Insulin-Like Growth Factor I-Dependent Regulation of Prolidase Activity in Cultured Human Skin Fibroblasts. Mol. Cell. Biochem. 189, 177–183. doi:10.1023/a:1006958116586
Miltyk, W., Surazynski, A., Grabowska, J., and Palka, J. A. (2008). Prolidase Dependent Inhibition of Collagen Biosynthesis in Chinese Hamster Ovary Cells. J. Biochem. 144, 409–414. doi:10.1093/jb/mvn083
Miner, J. H. (2012). The Glomerular Basement Membrane. Exp. Cel Res 318 (9), 973–978. doi:10.1016/j.yexcr.2012.02.031
Mittal, S., Song, X., Vig, B. S., and Amidon, G. L. (2007). Proline Prodrug of Melphalan Targeted to Prolidase, A Prodrug Activating Enzyme Overexpressed in Melanoma. Pharm. Res. 24, 1290–1298. doi:10.1007/s11095-007-9249-9
Mock, W. L., and Zhuang, H. (1991). Chemical Modification Locates Guanidinyl and Carboxylate Groups within the Active Site of Prolidase. Biochem. Biophysical Res. Commun. 180, 401–406. doi:10.1016/s0006-291x(05)81307-5
Monafo, V., Marseglia, G. L., Maghnie, M., Dyne, K. M., and Cetta, G. (2000). Transient Beneficial Effect of GH Replacement Therapy and Topical GH Application on Skin Ulcers in a Boy with Prolidase Deficiency. Pediatr. Dermatol. 17 (3), 227–230. doi:10.1046/j.1525-1470.2000.01760.x
Mueller, U., Niesen, F. H., Roske, Y., Goetz, F., Behlke, J., Buessow, K., et al. (2006). RCSB PDB - 2IW2: Crystal Structure of Human Prolidase. Homo sapiens. doi:10.2210/pdb2iw2/pdb
Muszyńska, A., Pałka, J., and Gorodkiewicz, E. (2000). The Mechanism of Daunorubicin-Induced Inhibition of Prolidase Activity in Human Skin Fibroblasts and its Implication to Impaired Collagen Biosynthesis. Exp. Toxicologic Pathol. 52, 149–155. doi:10.1016/S0940-2993(00)80108-6
Muszyńska, A., Wołczyński, S., and Pałka, J. (2001). The Mechanism for Anthracycline-Induced Inhibition of Collagen Biosynthesis. Eur. J. Pharmacol. 411, 17–25. doi:10.1016/s0014-2999(00)00847-5
Myara, I., Charpentier, C., and Lemonnier, A. (1982). Optimal Conditions for Prolidase Assay by Proline Colorimetric Determination: Application to Iminodipeptiduria. Clinica Chim. Acta 125, 193–205. doi:10.1016/0009-8981(82)90196-6
Myara, I., Myara, A., Mangeot, M., Fabre, M., Charpentier, C., and Lemonnier, A. (1984). Plasma Prolidase Activity: A Possible index of Collagen Catabolism in Chronic Liver Disease. Clin. Chem. 30, 211–215. doi:10.1093/clinchem/30.2.211
Nijhuis, W. H., Eastwood, D. M., Allgrove, J., Hvid, I., Weinans, H. H., Bank, R. A., et al. (2019). Current Concepts Review Current Concepts in Osteogenesis Imperfecta: Bone Structure, Biomechanics and Medical Management. J. Child. Orthop. 13, 1–11. doi:10.1302/1863-2548.13.180190
Nir, V., Ilivitky, A., Hakim, F., Yoseph, R. B., Gur, M., Mandel, H., et al. (2016). Pulmonary Manifestations of Prolidase Deficiency. Pediatr. Pulmonol 51 (11), 1229–1233. doi:10.1002/ppul.23435
Ohhashi, T., Ohno, T., Arata, J., and Kodama, H. (1988a). Biochemical Studies on Prolidase in Sera from Control, Patients with Prolidase Deficiency and Their Mother. J. Inherit. Metab. Dis. 11, 166–173. doi:10.1007/bf01799867
Ohhashi, T., Ohno, T., Arata, J., and Kodama, H. (1988b). Characteristics of Partially Purified Prolidase from Erythrocytes of Normal Individuals, of Two Patients with Prolidase Deficiency, and of the Patients' Mother. J. Clin. Biochem. Nutr. 5, 183–192. doi:10.3164/jcbn.5.183
Oikonomidi, S., Kostikas, K., Tsilioni, I., Tanou, K., Gourgoulianis, K., and Kiropoulos, T. (2009). Matrix Metalloproteinases in Respiratory Diseases: From Pathogenesis to Potential Clinical Implications. Curr. Med. Chem. 16, 1214–1228. doi:10.2174/092986709787846587
Okonkwo, U., and DiPietro, L. (2017). Diabetes and Wound Angiogenesis. Int. J. Mol. Sci. 18, 1419. doi:10.3390/ijms18071419
Oono, T., Fujiwara, Y., Yoshioka, T., and Arata, J. (1997). Prolidase Activity in Chronic Wound and Blister Fluids. J. Dermatol. 24 (10), 626–629. doi:10.1111/j.1346-8138.1997.tb02306.x
Oono, T., Yasutomi, H., Ohhashi, T., Kodama, H., and Arata, J. (1990). Characterization of Fibroblast-Derived Prolidase. The Presence of Two Forms of Prolidase. J. Dermatol. Sci. 1, 319–323. doi:10.1016/0923-1811(90)90588-5
Ozbay, I., Topuz, M. F., Oghan, F., Kocak, H., and Kucur, C. (2020). Serum Prolidase, Malondialdehyde and Catalase Levels for the Evaluation of Oxidative Stress in Patients with Peripheral Vertigo. Eur. Arch. Otorhinolaryngol. 278, 3773–3776. doi:10.1007/s00405-020-06466-x
Palka, J. A., and Phang, J. M. (1997). Prolidase Activity in Fibroblasts Is Regulated by Interaction of Extracellular Matrix with Cell Surface Integrin Receptors. J. Cell. Biochem. 67, 166–175. doi:10.1002/(sici)1097-4644(19971101)67:2<166::aid-jcb2>3.0.co;2-v
Pandhare, J., Donald, S. P., Cooper, S. K., and Phang, J. M. (2009). Regulation and Function of Proline Oxidase under Nutrient Stress. J. Cel Biochem 107 (4), 759–768. doi:10.1002/jcb.22174
Pałka, J. A., Miltyk, W., Karna, E., and Wołczyński, S. (1996). Modulation of Prolidase Activity during In Vitro Aging of Human Skin Fibroblasts the Role of Extracellular Matrix Collagen. Tokai J. Exp. Clin. Med. 21, 207–213.
Perugini, P., Hassan, K., Genta, I., Modena, T., Pavanetto, F., Cetta, G., et al. (2005). Intracellular Delivery of Liposome-Encapsulated Prolidase in Cultured Fibroblasts from Prolidase-Deficient Patients. J. Controlled Release 102, 181–190. doi:10.1016/j.jconrel.2004.09.013
Petrikovics, I., Cheng, T. C., Papahadjopoulos, D., Hong, K., Yin, R., DeFrank, J. J., et al. (2000). Long Circulating Liposomes Encapsulating Organophosphorus Acid Anhydrolase in Diisopropylfluorophosphate Antagonism. Toxicol. Sci. 57, 16–21. doi:10.1093/toxsci/57.1.16
Phang, J. M., Liu, W., and Zabirnyk, O. (2010). Proline Metabolism and Microenvironmental Stress. Annu. Rev. Nutr. Annu. Rev. Inc. 30, 441–463. doi:10.1146/annurev.nutr.012809.104638
Pizzino, G., Irrera, N., Cucinotta, M., Pallio, G., Mannino, F., Arcoraci, V., et al. (2017). Oxidative Stress: Harms and Benefits for Human Health. Oxid Med. Cel Longev 2017, 8416763. doi:10.1155/2017/8416763
Plecko, B., and Steinfeld, R. (2017). Disorders of Vitamin Metabolism. Swaiman's Pediatric Neurology: Principles and Practice. 6th Edn. Elsevier, 373–382. doi:10.1016/b978-0-323-37101-8.00046-1
Pohl, M. O., Edinger, T. O., and Stertz, S. (2014). Prolidase Is Required for Early Trafficking Events during Influenza A Virus Entry. J. Virol. 88 (19), 11271–11283. doi:10.1128/jvi.00800-14
Powell, G. F., Rasco, M. A., and Maniscalco, R. M. (1974). A Prolidase Deficiency in Man with Iminopeptiduria. Metabolism 23, 505–513. doi:10.1016/0026-0495(74)90078-x
Ramshaw, J. A. M., Shah, N. K., and Brodsky, B. (1998). Gly-X-Y Tripeptide Frequencies in Collagen: A Context for Host-Guest Triple-Helical Peptides. J. Struct. Biol. 122, 86–91. doi:10.1006/jsbi.1998.3977
Ray, S. C., and Thomas, D. L. (2014). Hepatitis C. Mandell, Douglas, and Bennett's Principles and Practice of Infectious Diseases, 2. Philadelphia, Pennsylvania: Elsevier, 1904–1927.
Reid, K. B. M. (2018). Complement Component C1q: Historical Perspective of a Functionally Versatile, and Structurally Unusual, Serum Protein. Front. Immunol. 9, 1. doi:10.3389/fimmu.2018.00764
Sabuncu, T., Boduroglu, O., Eren, M. A., Torun, A. N., and Aksoy, N. (2016). The Value of Serum Prolidase Activity in Progression of Microalbuminuria in Patients with Type 2 Diabetes Mellitus. J. Clin. Lab. Anal. 30 (5), 557–562. doi:10.1002/jcla.21902
Sayır, F. (2019). Serum Prolidase Activity, Total Oxidant/antioxidant, and Nitric Oxide Levels in Patients with Esophageal Squamous Cell Carcinoma. Turkish J. Thorac. Cardiovasc. Surg. 27, 206–211. doi:10.5606/tgkdc.dergisi.2019.16888
Seakins, J. W. (1963). Peptiduria in an Unusual Bone Disorder. Isolation of Two Peptides. Arch. Dis. Child. 38, 215–219. doi:10.1136/adc.38.199.215
Şen, V., Uluca, Ü., Ece, A., Kaplan, İ., Bozkurt, F., Aktar, F., et al. (2014). Serum Prolidase Activity and Oxidant–Antioxidant Status in Children with Chronic Hepatitis B Virus Infection. Ital. J. Pediatr. 40, 95. doi:10.1186/s13052-014-0095-1
Senboshi, Y., Oono, T., and Arata, J. (1996). Localization of Prolidase Gene Expression in Scar Tissue Using In Situ Hybridization. J. Dermatol. Sci. 12, 163–171. doi:10.1016/0923-1811(95)00505-6
Sheffield, L. J., Schlesinger, P., Faull, K., Halpern, B. J., Schier, G. M., Cotton, R. G. H., et al. (1977). Iminopeptiduria, Skin Ulcerations, and Edema in a Boy with Prolidase Deficiency. J. Pediatr. 91, 578–583. doi:10.1016/s0022-3476(77)80506-4
Sienkiewicz, P., Pałka, M., and Pałka, J. (2004). Oxidative Stress Induces IGF-I Receptor Signaling Disturbances in Cultured Human Dermal Fibroblasts. A Possible Mechanism for Collagen Biosynthesis Inhibition. Cell Mol. Biol. Lett. 9, 643–650.
Smith, E., and Bergmann, M. (1979). The Peptidases of Intestinal Mucosa. UCLA Forum Med. Sci. 21, 2–19.
Spodenkiewicz, M., Spodenkiewicz, M., Cleary, M., Massier, M., Fitsialos, G., Cottin, V., et al. (2020). Clinical Genetics of Prolidase Deficiency: An Updated Review. Biology 9, 108. doi:10.3390/biology9050108
Squire, L. R., Genzel, L., Wixted, J. T., and Morris, R. G. (2015). Memory Consolidation. Cold Spring Harbor Perspect. Biol. 7, a021766. doi:10.1101/cshperspect.a021766
Surazynski, A., Donald, S. P., Cooper, S. K., Whiteside, M. A., Salnikow, K., Liu, Y., et al. (2008a). Extracellular Matrix and HIF-1 Signaling: The Role of Prolidase. Int. J. Cancer 122, 1435–1440. doi:10.1002/ijc.23263
Surazynski, A., Liu, Y., Miltyk, W., and Phang, J. M. (2005). Nitric Oxide Regulates Prolidase Activity by Serine/threonine Phosphorylation. J. Cel Biochem 96 (5), 1086–1094. doi:10.1002/jcb.20631
Surazynski, A., Miltyk, W., Palka, J., and Phang, J. M. (2008b). Prolidase-dependent Regulation of Collagen Biosynthesis. Amino Acids 35, 731–738. doi:10.1007/s00726-008-0051-8
Surazynski, A., Miltyk, W., Prokop, I., and Palka, J. (2010). Prolidase-Dependent Regulation of TGF Beta (Corrected) and TGF Beta Receptor Expressions in Human Skin Fibroblasts. Eur. J. Pharmacol. 649 (1-3), 115–119. doi:10.1016/j.ejphar.2010.09.034
Surazynski, A., Miltyk, W., Prokop, I., and Palka, J. (2013a). The Effect of Estrogen on Prolidase-Dependent Regulation of HIF-1α Expression in Breast Cancer Cells. Mol. Cell Biochem. 379, 29–36. doi:10.1007/s11010-013-1623-9
Surazynski, A., Miltyk, W., Wolczynski, S., and Palka, J. (2013b). The Effect of Prolactin and Estrogen Cross-Talk on Prolidase– Dependent Signaling in MCF-7 Cells. Neoplasma 60, 355–363. doi:10.4149/neo_2013_047
Surazyński, A., Pałka, J., and Wołczyński, S. (2004). Acetylsalicylic Acid-Dependent Inhibition of Collagen Biosynthesis and Beta1-Integrin Signaling in Cultured Fibroblasts. Med. Sci. monitor : Int. Med. J. Exp. Clin. Res. 10, BR175–179.
Surazyński, A., Pałka, J., and Wołczyński, S. (2001). Phosphorylation of Prolidase Increases the Enzyme Activity. Mol. Cell. Biochem. 220, 95–101. doi:10.1023/a:1010849100540
Surazyński, A., Sienkiewicz, P., Wołczyński, S., and Pałka, J. (2005). Differential Effects of Echistatin and Thrombin on Collagen Production and Prolidase Activity in Human Dermal Fibroblasts and Their Possible Implication in β1-Integrin-Mediated Signaling. Pharmacol. Res. 51, 217–221. doi:10.1016/j.phrs.2004.08.004
Süßmuth, K., Metze, D., Muresan, A. M., Lehmberg, K., Zur Stadt, U., Speckmann, C., et al. (2020). Ulceration in Prolidase Deficiency: Successful Treatment with Anticoagulants. Acta Dermato-Venereologica 100, adv00002. doi:10.2340/00015555-3324
Tanoue, A., Endo, F., Kitano, A., and Matsuda, I. (1990a). A Single Nucleotide Change in the Prolidase Gene in Fibroblasts from Two Patients with Polypeptide Positive Prolidase Deficiency. Expression of the Mutant Enzyme in NIH 3T3 Cells. J. Clin. Invest. 86, 351–355. doi:10.1172/jci114708
Tanoue, A., Endo, F., and Matsuda, I. (1990b). Structural Organization of the Gene for Human Prolidase (Peptidase D) and Demonstration of a Partial Gene Deletion in a Patient with Prolidase Deficiency. J. Biol. Chem. 265, 11306–11311. doi:10.1016/s0021-9258(19)38592-8
Türkbeyler, I., Demir, T., Pehlivan, Y., Kaplan, D. S., Ceribasi, A. O., Orkmez, M., et al. (2012). Prolidase Could Act as a Diagnosis and Treatment Mediator in Lung Fibrosis. Inflammation 35, 1747–1752. doi:10.1007/s10753-012-9493-y
Umemura, S., Arata, J., and Nohara, N. (1980). Prolidase Activity During Healing of Skin Burns in Rats. J. Dermatol. 7 (3), 217–219. doi:10.1111/j.1346-8138.1980.tb03536.x
Uramatsu, S., Liu, G., Yang, Q., Uramatsu, M., Chi, H., Lu, J., et al. (2009). Characterization of Prolidase I and II Purified from Normal Human Erythrocytes: Comparison with Prolidase in Erythrocytes from a Patient with Prolidase Deficiency. Amino Acids 37, 543–551. doi:10.1007/s00726-009-0262-7
Uçar, D., Em, S., Bozkurt, M., Oktayoglu, P., Yüksel, H. K., Caglayan, M., et al. (2013). Serum Prolidase Activity in Ankylosing Spondylitis and Rheumatoid Arthritis. Clin. Med. insights. Arthritis Musculoskelet. Disord. 6, 29–33. doi:10.4137/cmamd.s12602
Van-Horn, M. R., Sild, M., and Ruthazer, E. S. (2013). D-serine as a Gliotransmitter and its Roles in Brain Development and Disease. Front. Cell Neurosci. 7, 39. doi:10.3389/fncel.2013.00039
Verma, A. K., Chandra, S., Singh, R. G., Singh, T. B., Srivastava, S., and Srivastava, R. (2014). Serum Prolidase Activity and Oxidative Stress in Diabetic Nephropathy and End Stage Renal Disease: A Correlative Study with Glucose and Creatinine. Biochem. Res. Int. 2014, 291458. doi:10.1155/2014/291458
Verma, R. P., and Hansch, C. (2007). Matrix Metalloproteinases (MMPs): Chemical-Biological Functions and (Q)SARs. Bioorg. Med. Chem. 15 (6), 2223–2268. doi:10.1016/j.bmc.2007.01.011
Viglio, S., Annovazzi, L., Conti, B., Genta, I., Perugini, P., Zanone, C., et al. (2006). The Role of Emerging Techniques in the Investigation of Prolidase Deficiency: From Diagnosis to the Development of a Possible Therapeutical Approach. J. Chromatogr. B 832, 1–8. doi:10.1016/j.jchromb.2005.12.049
Wang, C., Li, Y.-Y., Li, X., Wei, L.-L., Yang, X.-Y., Xu, D.-D., et al. (2014). Serum Complement C4b, Fibronectin, and Prolidase Are Associated with the Pathological Changes of Pulmonary Tuberculosis. BMC Infect. Dis. 14, 52. doi:10.1186/1471-2334-14-52
Wang, R. Y., Wilcox, W. R., and Cederbaum, S. D. (2013). Amino Acid metabolism, Emery and Rimoin's Principles and Practice of Medical Genetics. Amsterdam: Elsevier, Chap. 92, 2564–2605. doi:10.1016/B978-0-12-801238-3.05564-1
Wilk, P., Uehlein, M., Kalms, J., Dobbek, H., Mueller, U., and Weiss, M. S. (2017). Substrate Specificity and Reaction Mechanism of Human Prolidase. FEBS J. 284, 2870–2885. doi:10.1111/febs.14158
Wyse, A. T. S., and Netto, C. A. (2011). Behavioral and Neurochemical Effects of Proline. Metab. Brain Dis. 26, 159–172. doi:10.1007/s11011-011-9246-x
Xue, M., and Jackson, C. J. (2015). Extracellular Matrix Reorganization During Wound Healing and its Impact on Abnormal Scarring. Adv. Wound Care (New Rochelle) 4 (3), 119–136. doi:10.1089/wound.2013.0485
Yang, L., Li, Y., Bhattacharya, A., and Zhang, Y. (2016). Dual Inhibition of ErbB1 and ErbB2 in Cancer by Recombinant Human Prolidase Mutant hPEPD-G278D. Oncotarget 7, 42340–42352. doi:10.18632/oncotarget.9851
Yang, L., Li, Y., Bhattacharya, A., and Zhang, Y. (2015). Inhibition of ERBB2-Overexpressing Tumors by Recombinant Human Prolidase and its Enzymatically Inactive Mutant. EBioMedicine 2, 396–405. doi:10.1016/j.ebiom.2015.03.016
Yang, L., Li, Y., Bhattacharya, A., and Zhang, Y. (2017). PEPD Is a Pivotal Regulator of P53 Tumor Suppressor. Nat. Commun. 8, 2052. doi:10.1038/s41467-017-02097-9
Yang, L., Li, Y., Ding, Y., Choi, K.-S., Kazim, A. L., and Zhang, Y. (2013). Prolidase Directly Binds and Activates Epidermal Growth Factor Receptor and Stimulates Downstream Signaling. J. Biol. Chem. 288, 2365–2375. doi:10.1074/jbc.m112.429159
Yang, L., Li, Y., and Zhang, Y. (2014). Identification of Prolidase as a High Affinity Ligand of the ErbB2 Receptor and its Regulation of ErbB2 Signaling and Cell Growth. Cel Death Dis. 5, e1211. doi:10.1038/cddis.2014.187
Yasuda, K., Ogata, K., Kariya, K., Kodama, H., Zhang, J., Sugahara, K., et al. (1999). Corticosteroid Treatment of Prolidase Deficiency Skin Lesions by Inhibiting Iminodipeptide-Primed Neutrophil Superoxide Generation. Br. J. Dermatol. 141, 846–851. doi:10.1046/j.1365-2133.1999.03157.x
Yildiz, A., Demirbag, R., Yilmaz, R., Gur, M., Altiparmak, I. H., Akyol, S., et al. (2008). The Association of Serum Prolidase Activity with the Presence and Severity of Coronary Artery Disease. Coron. Artery Dis. 19 (5), 319–325. doi:10.1097/mca.0b013e32830042ba
Ysrayl, B. b., Balasubramaniam, M., Albert, I., Villalta, F., Pandhare, J., and Dash, C. (2019). A Novel Role of Prolidase in Cocaine-Mediated Breach in the Barrier of Brain Microvascular Endothelial Cells. Scientific Rep. 9, 2567. doi:10.1038/s41598-018-37495-6
Yu, T. Y., Pang, J. H. S., Wu, K. P. H., Chen, M. J. L., Chen, C. H., and Tsai, W. C. (2013). Aging Is Associated with Increased Activities of Matrix Metalloproteinase-2 and -9 in Tenocytes. BMC Musculoskelet. Disord. 14, 2. doi:10.1186/1471-2474-14-2
Yu, Z., Visse, R., Inouye, M., Nagase, H., and Brodsky, B. (2012). Defining Requirements for Collagenase Cleavage in Collagen Type III Using a Bacterial Collagen System. J. Biol. Chem. 287, 22988–22997. doi:10.1074/jbc.m112.348979
Yun, H., Lee, S., Kim, S., Yu, J., Lee, N., Lee, J., et al. (2017). Improved Hydrolysis of Organophosphorus Compounds by Engineered Human Prolidases. Protein Pept. Lett. 24, 617–625. doi:10.2174/0929866524666170428143759
Keywords: prolidase, collagen, wound healing, prolidase deficiency, regulation
Citation: Eni-Aganga I, Lanaghan ZM, Balasubramaniam M, Dash C and Pandhare J (2021) PROLIDASE: A Review from Discovery to its Role in Health and Disease. Front. Mol. Biosci. 8:723003. doi: 10.3389/fmolb.2021.723003
Received: 09 June 2021; Accepted: 18 August 2021;
Published: 31 August 2021.
Edited by:
Prasun K. Datta, Tulane University, United StatesReviewed by:
Miriam Zacchia, University of Campania Luigi Vanvitelli, ItalyCopyright © 2021 Eni-Aganga, Lanaghan, Balasubramaniam, Dash and Pandhare. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jui Pandhare, anBhbmRoYXJlQG1tYy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.