 Giuseppe Deganutti1*
Giuseppe Deganutti1* Silvia Atanasio2
Silvia Atanasio2 Roxana-Maria Rujan1Patrick M. Sexton3,4
Roxana-Maria Rujan1Patrick M. Sexton3,4 Denise Wootten3,4Christopher A. Reynolds1*
Denise Wootten3,4Christopher A. Reynolds1*- 1Centre for Sport, Exercise and Life Sciences, Coventry University, Coventry, United Kingdom
- 2School of Life Sciences, University of Essex, Colchester, United Kingdom
- 3Drug Discovery Biology Theme, Monash Institute of Pharmaceutical Sciences, Monash University, Parkville, VIC, Australia
- 4ARC Centre for Cryo-Electron Microscopy of Membrane Proteins, Monash Institute of Pharmaceutical Sciences, Monash University, Parkville, VIC, Australia
Class B1 G protein-coupled receptors (GPCRs) are important targets for many diseases, including cancer, diabetes, and heart disease. All the approved drugs for this receptor family are peptides that mimic the endogenous activating hormones. An understanding of how agonists bind and activate class B1 GPCRs is fundamental for the development of therapeutic small molecules. We combined supervised molecular dynamics (SuMD) and classic molecular dynamics (cMD) simulations to study the binding of the calcitonin gene-related peptide (CGRP) to the CGRP receptor (CGRPR). We also evaluated the association and dissociation of the antagonist telcagepant from the extracellular domain (ECD) of CGRPR and the water network perturbation upon binding. This study, which represents the first example of dynamic docking of a class B1 GPCR peptide, delivers insights on several aspects of ligand binding to CGRPR, expanding understanding of the role of the ECD and the receptor-activity modifying protein 1 (RAMP1) on agonist selectivity.
Introduction
G protein-coupled receptors, the largest family of cell surface receptors (Rosenbaum et al., 2009), are targeted by approximatively 34% of all approved drugs (Hauser et al., 2017). Among the four major classes of vertebrate GPCRs (A, B, C, and F), class B1 are endogenously activated by peptide hormones involved in homeostatic control, e.g., of bone and energy metabolism, and cardiovascular and immune responses (Hollenstein et al., 2014). Drugs acting on class B1 GPCRs could be tremendously useful for the treatment of a range of disorders such as cancer, diabetes, heart disease, hypercalcemia, obesity, and osteoporosis (Hollenstein et al., 2014).
Since the first active structure, published in 2017 (Liang et al., 2017), the structures of most class B1 GPCRs in the active, G protein complexed, state have been determined through cryo-electron microscopy (cryo-EM), advancing the knowledge of their activation mechanism (García-Nafría and Tate, 2019; Liang et al., 2020b; Ma et al., 2020; Qiao et al., 2020; Liang et al., 2020a; Dong et al., 2020; Duan et al., 2020; Zhou et al., 2020). GPCRs are characterized by a 7-transmembrane helix domain (TMD) and, in class B1, by a N-terminal extracellular domain that contributes to agonist binding, receptor activation, and signaling (de Graaf et al., 2017) (Figure 1A). Heterodimerization with accessory receptor activity-modifying proteins (RAMPs, Figure 1A) can modulate hormone binding and signaling (Hay and Pioszak, 2016) through an allosteric mechanism (Liang et al., 2020a; Pham et al., 2019). The transmembrane spanning RAMPs interact with the calcitonin receptor (CTR) and CTR-like (CLR) receptors at the transmembrane level, making contacts with TM3, TM4 and TM5, and at the extracellular level, where they interface with the ECD (Figures 1A,D). The heterodimer of CLR and RAMP1 displays selectivity for calcitonin gene-related peptide and therefore constitutes the prototypical CGRP receptor (CGRPR, Figures 1A,D) (Liang et al., 2018a). The metastable binding of the neuropeptide CGRP to CGRPR facilitates the intracellular G protein engagement, which in turn allows the peptide to reach the final bound state (Josephs et al., 2021). In this complex, CGRP inserts into the receptor TMD core via a disulfide-bridged N-terminal loop and short amphipathic α-helix, with an unstructured C-terminal tail extended towards the ECD, where it forms an additional anchoring point (Figures 1A,D). The aforementioned stable binding mode can be achieved only after the engagement of the G protein (De Lean et al., 1980; Chung et al., 2011; DeVree et al., 2016), which allosterically facilitates an outward movement of ECL3 and an upward shift of ECL2 (Josephs et al., 2021). The CGRP-bound receptor, prior to G protein coupling, more closely resembles the apo structure with stable binding of the peptide C-terminus to the ECD, and where the peptide N-terminus only transiently and dynamically engaged with the receptor core (Josephs et al., 2021).
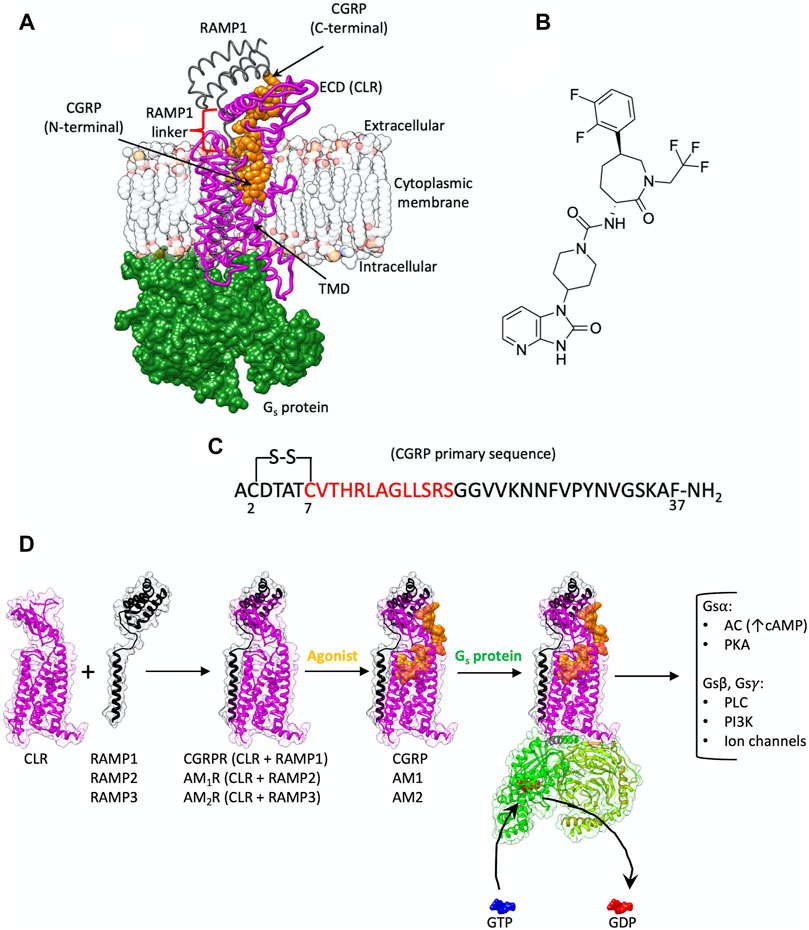
FIGURE 1. (A) CGRPR is a heterodimer membrane protein composed of CLR (purple ribbon) and RAMP1 (gray ribbon); CGRP (orange van der Waals spheres) binds to the TMD and extends towards the ECD. Agonist-bound CGRPR couples to Gs protein (green surface). (B) Chemical structure of the CGRPR antagonist telcagepant. (C) CGRP primary sequence; the residues in the N-terminal helix are indicated in red. (D) Schematic representation of the events from the heterodimerization between CLR and RAMPs to G protein signaling. The formation of complexes between CLR and different RAMPs generate receptors with selectivity towards CGRP, AM1 or AM2; upon agonist binding, the trimeric Gs protein is recruited and activated, favoring the exchange between GDP and GTP at the level of the Gsα subunit. After Gs subunits dissociation multiple intracellular signaling pathways are triggered (AC, adenylyl cyclase; PKA, protein kinase A; PLC, phospholipase C; PI3K, phosphoinositide 3-kinases).
CGRP, which is involved in nociceptive transmission and modulation of vasodilatation, is widely expressed in both peripheral and central regions of the nervous systems, including the trigeminal vascular system (Rujan and Reynolds, 2019). Multiple studies have shown that CGRP is associated with migraine pathophysiology (Durham, 2006), making CGRP and its receptor an important target for the treatment of migraine (Rujan and Reynolds, 2019). Despite its involvement in the underlying mechanisms associated with migraine, CGRP also plays a protective role in the cardiovascular system (Kee et al., 2018). Indeed, a novel peptide analogue of CGRP produces a decrease in angiotensin II-induced hypertension as well as a protective effect against heart failure (Aubdool et al., 2017). The heterodimers of CLR and RAMP2 or RAMP3, instead, are more selective for adrenomedullin (AM) and adrenomedullin 2 (AM2) peptides and constitute the AM1 and AM2 receptors, respectively (AM1R and AM2R, Figure 1D) (Hay and Pioszak, 2016; Hay et al., 2018; Liang et al., 2020a).
The role played by CGRP in migraine has stimulated the pursuit of agents able to block CGRPR activation (Rujan and Reynolds, 2019). Clinical approval of agents targeting this system was first achieved for monoclonal antibodies targeting CGRP or CGRPR. To date, there are three approved monoclonal antibodies against CGRP (galcanezumab, fremanezumab and eptinezumab), and one against CGRPR [erenumab, the first FDA-approved monoclonal antibody targeting a GPCR (Garces et al., 2020)]. Erenumab binds to the ECD of CGRPR, interacting with both CLR and RAMP1 (Garces et al., 2020). The pursuit of orally bioavailable CGRPR antagonists led to the discovery of phenylcaprolactam derivatives. Ubrogepant became the first-in-class oral CGRP antagonist approved for the acute treatment of migraine (December 2019) (Scott, 2020b). This was followed by the approval of rimegepant in February 2020 making it the second approved acute anti-migraine small-molecule drug and the first fast-acting orally disintegrating tablet (Scott, 2020a).
Telcagepant (Figure 1B), a phenylcaprolactam prototype, binds to the ECD of CGRPR with a Ki of 0.8 nM, inhibiting the binding of the CGRP C-terminal domain (Paone et al., 2007; Salvatore et al., 2008; Aksoydan and Durdagi, 2021). CGRP displays biphasic competition with telcagepant (Moore et al., 2009), consistent with the two-step binding mechanism proposed for class B1 GPCR peptides (Pal et al., 2012). According to this model, preliminary interactions between the receptor ECD and the ligand C-terminal domain facilitates the binding of the N-terminal domain within the receptor transmembrane domain (TMD) (Castro et al., 2005; Pal et al., 2012). This increases the local concentration of the N-terminal activation domain of the peptide in the proximity of the target, productively orienting the peptide and favoring the transition of the TMD core from an inactive to an active conformation. The distinct timescales characterizing the two different steps (microsecond and several millisecond, respectively (Castro et al., 2005) suggest that the productive engagement of the peptide N-terminal domain with the receptor TMD core is the rate-limiting step for agonist binding and receptor activation. However, at high concentrations of CGRP, in the presence of telcagepant, monophasic saturation curves are observed (Moore et al., 2009). This is putatively due to an ability of the CGRP N-terminal domain to bind to the TMD of the receptor without the prior formation of the metastable complex between the ECD and the peptide C-terminal domain.
GPCR activation mechanisms also involve water molecules within the TMD (Venkatakrishnan et al., 2019), including the orthosteric (endogenous) ligand binding site (Higgs et al., 2010), and the G protein binding site. From a drug design perspective, discerning whether a region of the binding site is solvated by stable or unstable water molecules can drive the development of tighter or more selective binders. Chemical modifications can be designed to displace unstable water molecules or further stabilize stable ones (Ladbury, 1996; Bortolato et al., 2013).
In the present study, we employed supervised molecular dynamics (Sabbadin and Moro, 2014; Cuzzolin et al., 2016; Salmaso et al., 2017; Deganutti et al., 2020) and classic molecular dynamics simulations to sample the binding of CGRP (Figures 1A,C) and the inhibitor telcagepant (Figure 1B) to CGRPR. The dynamic docking of CGRP to the TMD highlighted residues located within loop 4 of the ECD as a possible key player in the association mechanism. Simulated telcagepant binding and unbinding paths identified residues involved in metastable binding states as well as interactions that could constitute putative kinetic bottlenecks in formation of the high-affinity binding pose. We also simulated the active state CGRPR after removal of any ligand to compare the hydrated regions of the receptor before and after CGRP or telcagepant binding to assess potential changes to CGRPR hydration that occur upon ligand binding.
Methods
Telcagepant Force Field Parameters
All the systems (Table 1) were prepared for MD using the CHARMM36 (Huang and MacKerell, 2013; Huang et al., 2017)/CGenFF 3.0.1 (Vanommeslaeghe et al., 2012; Vanommeslaeghe and MacKerell, 2012; Yu et al., 2012) force field combination. The initial telcagepant force field, topology and parameter files were obtained from the ParamChem webserver (Vanommeslaeghe and MacKerell, 2012).

TABLE 1. Summary of the MD simulations performed and analyzed.
Protein Preparation
In order to speed up simulations involving CGRP, only the Gs C-terminal helix (residues N371-L394) of the G protein from the PDB entry 6E3Y was retained, after modelling of the missing segments in the ICL3 (residues 324–328), ECL3 (residues 356–363), ECD (residues 55–63), and CGRP (residues 24–26) as reported in our previous work (Liang et al., 2018a). The resulting active CGRP:CLR:RAMP1 complex was then prepared as follows. Hydrogen atoms were added by means of the pdb2pqr (Dolinsky et al., 2004) and propka (Olsson et al., 2011) software (considering a simulated pH of 7.0); the protonation of titratable side chains was checked by visual inspection. The resulting receptor was inserted in a square 100 Å × 100 Å 1-palmitoyl-2-oleyl-sn-glycerol-3-phosphocholine (POPC) bilayer (previously built by using the VMD Membrane Builder plugin 1.1, Membrane Plugin, Version 1.1. at http://www.ks.uiuc.edu/Research/vmd/plugins/membrane/), through an insertion method (Sommer, 2013). The receptor orientation was obtained by superposing the coordinates on the corresponding structure retrieved from the OPM database (Lomize et al., 2006). Lipids overlapping the receptor transmembrane helical bundle were removed and TIP3P water molecules (Jorgensen et al., 1983) were added to the simulation box by means of the VMD Solvate plugin 1.5 (Solvate Plugin, Version 1.5. at http://www.ks.uiuc.edu/Research/vmd/plugins/solvate/). Finally, overall charge neutrality was reached by adding Na+/Cl− counter ions up to the final concentration of 0.150 M), using the VMD Autoionize plugin 1.3 (Autoionize Plugin, Version 1.3. at http://www.ks.uiuc.edu/Research/vmd/plugins/autoionize/).
Simulations involving telcagepant were carried out considering only the CGRPR extracellular domain (CLR residues Q33ECD-T1311.29, RAMP1 residues E29-P114). Telcagepant was placed about 30 Å away from the ECD and the system was solvated and neutralized as described above.
System Equilibration and General MD Settings
The MD engine ACEMD (Harvey et al., 2009) was employed for both the equilibration and productive simulations. The equilibration of the CGRP:CLR:RAMP1 complex was achieved in isothermal-isobaric conditions (NPT) using the Berendsen barostat (Berendsen et al., 1984) (target pressure 1 atm) and the Langevin thermostat (Loncharich et al., 1992) (target temperature 300 K) with low damping of 1 ps−1. A four-stage procedure was performed (integration time step of 2 fs): first, clashes between protein and lipid atoms were reduced through 2000 conjugate-gradient minimization steps, then a 2 ns long MD simulation was run with a positional constraint of 1 kcal mol−1 Å−2 on protein and lipid phosphorus atoms. During the second stage, 20 ns of MD simulation was performed constraining only the protein atoms, while in the last equilibration stage, positional constraints were applied only to the protein backbone alpha carbons, for a further 20 ns.
ECD equilibration was achieved in two steps: after 500 cycles of conjugate-gradient minimization, the system was simulated for 5 ns, employing an integration time step of 2 fs, in the isothermal-isobaric conditions (NPT). The system comprising telcagepant and ECD was equilibrated in the NPT ensemble for 2 ns restraining the protein backbone alpha carbons.
Productive trajectories (Table 1) were computed with an integration time step of 4 fs, using the hydrogen mass repartition (Hopkins et al., 2015), in the canonical ensemble (NVT). The target temperature was set at 300 K, using a thermostat damping of 0.1 ps−1; the M-SHAKE algorithm (Forester and Smith, 1998; Kräutler et al., 2001) was employed to constrain the bond lengths involving hydrogen atoms. The cut-off distance for electrostatic interactions was set at 9 Å, with a switching function applied beyond 7.5 Å. Long range Coulomb interactions were handled using the particle mesh Ewald summation method (PME) (Essmann et al., 1995) by setting the mesh spacing to 1.0 Å.
Metadynamics Simulation of CGRP:CLR:RAMP1 Complex
The CGRP:CLR:RAMP1 complex was subjected to a metadynamics simulation to release the agonist from the bound state and relax the ECD in the absence of bound ligand. The well-tempered version of metadynamics (Barducci et al., 2008) was performed employing PLUMED 2.3 (Tribello et al., 2014), biasing the distance between the residues C7-L16 (CGRP) and K1341.32-V3917.64 (CLR) centroids. Gaussian energy functions were seeded every 1 ps (height = 0.1 kcal/mol, width = 0.1 Å, with a bias factor = 20), at a simulated temperature of 300 K, until the biased distance reached 50 Å. This final frame was used as the starting point for SuMD binding simulations of CGRP.
The Supervised MD Protocol
The supervised molecular dynamics (SuMD) is an adaptive sampling method (Deganutti and Moro, 2017a) for speeding up simulation of the binding (Cuzzolin et al., 2016; Deganutti et al., 2015; Deganutti and Moro, 2017b; Salmaso et al., 2017; Sabbadin and Moro, 2014; Bower et al., 2018; Bissaro et al., 2019; Bissaro et al., 2020) and unbinding processes (Deganutti et al., 2020). In the first SuMD implementation (Sabbadin and Moro, 2014; Cuzzolin et al., 2016), sampling is gained without the introduction of any energetic bias, by applying a tabu–like algorithm to monitor the distance between the centers of mass (or the geometrical centers) of the ligand and the predicted binding site or the receptor. However, the supervision of a second metric of the system can be considered (Atanasio et al., 2020). A series of short unbiased MD simulations are performed, and, after each simulation, the distances (collected at regular time intervals) are fitted to a linear function. If the resulting slope is negative (for binding), the next simulation step starts from the last set of coordinates and velocities produced, otherwise if the slope is positive, the simulation is restarted by randomly assigning the atomic velocities.
Settings for SuMD Binding
The binding of telcagepant to the CGRPR ECD was obtained by supervising the distance between the ligand and CGRPR residue W72ECD (which is roughly in the center of the telcagepant binding site). A series of 500 ps-long time windows were simulated until the distance reached a value of less than 4 Å. Frames were saved every 50 ps and used to interpolate the linear function of the distance during the simulated 500 ps.
To simulate the CGRP binding to CGRPR, the distance between the centroids of residues C7-L16 (CGRP) and K1341.32-V3917.64 (corresponding to the CLR TMD) was supervised. Four replicas were started from the last frame extracted from the metadynamics, by moving CGRP further away from the receptor and randomly reorienting it. After 250 ns of productive SuMD simulations time, the replica with lower RMSD values to the cryo-EM structure (CGRP residues C7-L16) was used to seed four 1 µs-long classical MD (cMD) simulations. This unsupervised step was performed to facilitate the reorganization of the CGRP:CGRPR metastable states minimizing external influence due to the supervision. The cMD replica characterized by the lowest RMSD value to the cryo-EM structure (CGRP residues C7-L16) was used as a starting point for four additional SuMD binding simulations until visual inspection confirmed appropriate binding of the CGRP N-terminus.
Settings for SuMD Unbinding of Telcagepant
The frame with the lowest RMSD value to the X-ray complex 3N7R (ter Haar et al., 2010), representing the ECD in complex with telcagepant, was extracted from SuMD binding trajectories and used as a starting point for CGRPR ECD-telcagepant unbinding simulations (ECD residues Q33ECD-T1311.29, RAMP1 residues E29-P114). A double supervision was performed: both of the ligand-W72ECD distance and the number of water oxygen atoms within 4 Å of protein atoms and that are hydrogen bonding with telcagepant (if the slope of the linear function plotted on each of the two data series was positive then the time window was productive). The protocol for unbinding differs from the original SuMD binding algorithm, in that the length (Δt) of the short simulations performed increased along the unbinding pathway, according to the formula:
Δt0 is the duration of the very first MD time window and Nti represents a factor that is picked from three user-defined values (Nt1, Nt2, and Nt3), according to the last ligand-protein distance detected (Deganutti et al., 2020). Three distance threshold values (D1, D2, and D3) were set and the ligand-protein distance (rL) at the end of each MD run was compared to these threshold values, allowing a decision on the value of the Nti factor according to the following conditions:
The goal of increasing the simulation time window (Δt in Eq. 1) along the unbinding pathway is to facilitate the sampling of metastable states, which could otherwise be poorly visited. Frames were saved every 50 ps. The initial time window length was 300 ps, with Nt1, Nt2, and Nt3 set to 3, 6, and 10. Values of 5, 8, and 10 Å were used as D1, D2, and D3 distances.
The unbinding was iterated until no ligand-protein van der Waals contact was detected by means of the GetContacts scripts tools (https://getcontacts.github.io). The ligand-protein distance and the number of water oxygen atoms within 4 Å of protein donor/acceptor atoms were computed using PLUMED 2.3. After each productive MD time window, GetContacts was employed to detect and update the protein atoms involved in hydrogen bonds with the ligand, considering a distance of 3.5 Å and an angle value of 120° as geometrical cut-offs. Notably, if no hydrogen bond between the ligand and the protein was present at the end of a productive MD time window, then protein atoms involved in water-mediated or van der Waals interactions were considered.
SuMD Path Sampling Protocol
SuMD path sampling (Deganutti et al., 2021) (Table 1) was performed considering the output from each SuMD replica, for binding of both CGRP and telcagepant and the unbinding of telcagepant. Each trajectory was aligned on the protein alpha carbon atoms and the frames were clustered according to the ligand RMSD to the starting positions (bin of 1 Å). A frame from each group was randomly extracted and used as a starting point for 20 ns (for telcagepant unbinding) or 30 ns (for CGRPR binding) cMD simulations.
Analysis of the MD Trajectories
Only the MD trajectories from the SuMD path sampling were analyzed in the case of telcagepant unbinding and CGRPR binding. Interatomic contacts and root mean square deviations (RMSD) were computed using VMD (Humphrey et al., 1996). A contact was considered productive if the distance between two atoms was less than 3.5 Å. Ligand-protein hydrogen bonds were detected using the GetContacts scripts tool (https://getcontacts.github.io), setting a hydrogen bond donor-acceptor distance of 3.5 Å and an angle value of 120° as geometrical cut-offs. Contacts and hydrogen bond persistency are quantified as the percentage of frames (over all the frames obtained by merging the different replicas) in which protein residues formed contacts or hydrogen bonds with the ligand.
Distances between atoms were computed using PLUMED 2.3 (Tribello et al., 2014). The molecular mechanics energy combined with the generalized Born surface area (MM/GBSA) was computed with the MMPBSA.py (Miller et al., 2012) script (AmberTools17 suite at http://ambermd.org/) after transforming the CHARMM psf topology files to an Amber prmtop format using ParmEd (documentation at http://parmed.github.io/ParmEd/html/index.html).
Detection of hydrated spots within CGRPR in the peptide-bound active state (PDB: 6E3Y) was performed on 100 ns long cMD simulations by means of AquaMMapS (Cuzzolin et al., 2018), in the holo state or following removal of CGRP from the complex. This allowed taking into account the flexibility of the systems and thermal fluctuation within the orthosteric binding site.
Numbering System
Throughout the manuscript, the class B1 GPCR Wootten residue numbering system (Wootten et al., 2013) is displayed as superscripts to the CLR residues numbers.
Results
Binding Path of CGRP
The current study focuses on the mechanism of binding for the N-terminal domain of CGRP that is critical to activation of CGRPR. Recent work elucidated the structures of apo and peptide-bound CGRPR in the absence of transducer protein (Josephs et al., 2021) and revealed that, as expected, the peptide C-terminus is stably engaged with the ECD of the receptor, albeit that the ECD is highly dynamic. However, the N-terminus of the peptide only transiently engages with the core of the receptor. As such, there is a gap in understanding of how the CGRP N-terminus (that includes the disulfide-bridged N-terminal loop and short α-helix that extends from this loop) engages with CGRPR to reach the fully-active, G protein-coupled complex that has been experimentally characterized by cryo-EM (Liang et al., 2018a).
During the dynamic docking of the N-terminal helix of CGRP (Supplementary Video S1), the unstructured C-terminal domain spontaneously approached the ECD, forming transitory interactions between F37CGRP and W72ECD or W84RAMP1 (Supplementary Table S1). High flexibility of the CGRP C-terminal segment bound to the receptor was also suggested by MD simulations of the active, CGRP:CGRPR:G protein complex (Liang et al., 2018a). These contacts, along with the hydrogen bond between T122ECD backbone and the terminal NH2 at F37CGRP, are present in the cryo-EM structure and are likely important for the first step of CGRP binding. Very few interactions were formed with RAMP1 (Figures 2C,D; Supplementary Figures S1A,B; Supplementary Tables S1, S2).
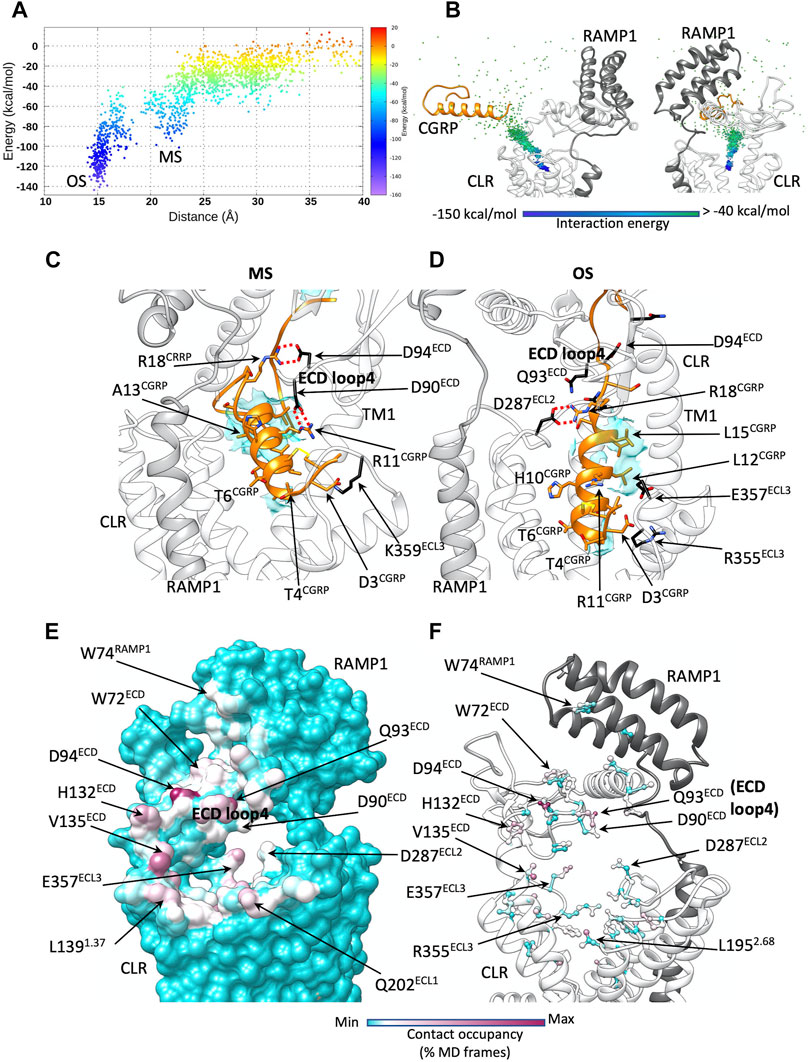
FIGURE 2. CGRP dynamic docking. (A) CGRP binding energy landscape; x axes report the distance between the TMD centroid and the CGRP N-terminal helix (the metastable macrostate, MS, is indicated, alongside the deep bound states OS); (B) binding path of the centroid of the CGRP N-terminal helix, colored according to the interaction energy with CGRPR; (C) representative configuration from the macrostate MS. CGRPR ECD loop 4 forms salt bridges with CGRP R11 and R18; (D) representative configuration from the deep bound states OS. Electrostatic interactions are depicted as red dashed lines, while hydrophobic contacts are depicted as cyan transparent surfaces. (E) CGRP-CGRPR contacts plotted on CGRPR surface and colored according to the occupancy (% MD frames) of the interactions; (F) CGRP-CGRPR contacts plotted on CGRPR atoms and colored according to the occupancy (% MD frames) of the interactions.
CGRP transition from the bulk solvent to the orthosteric site within the TMD was progressively stabilized by interactions of the peptide and receptor (Figures 2A,B). The first contacts between CGRP and CLR were formed at the level of the tunnel entrance shaped by the ECD, TM1, TM2, and ECL1 (Figures 2C–E, Supplementary Figures S1A,B). The peptide disulfide-bridged N-terminal loop (residues 1–7) formed transitory polar interactions with Q33ECD, Q93ECD, D90ECD, N200ECL1, N201ECL1, and Q202ECL1, before V8CGRP, L12CGRP, and L15CGRP on the hydrophobic side of the amphipathic α-helix of CGRP engaged the side chains of V1351.33, L1391.37, F1421.40, L1952.68 in hydrophobic contacts. While the initial polar interactions are not present in the CGRP:CGRPR:Gs cryo-EM structure, the latter hydrophobic contacts are reflected in the experimental structure, and throughout the binding simulation acted as a glidant, favoring the peptide insertion into the TMD. While entering the TMD orthosteric site, CGRP formed further interactions with D94ECD, F92ECD, W3546.58, D3667.39, and H3707.43 (Figures 2C,D; Supplementary Figures S1A,B; Supplementary Tables S1, S2). In the intermediate macrostate, MS (Figure 2A), the agonists sampled several metastable configurations before reaching the bound orthosteric microstate, OS (Figure 2A). This latter state was in remarkable agreement with the experimental coordinates of the fully active complex (Liang et al., 2018a) (Supplementary Video S1). The RMSD of the CGRP Cα carbons to the cryo-EM conformation of CGRP, indeed, reached values lower than 1 Å (3 Å considering the side chain heavy atoms of the peptide helix, Supplementary Figure S2). The agonist residues forming the most frequent interactions during the binding (considering the whole binding trajectory) were D3CGRP, T4CGRP, T6CGRP, T9CGRP, H10CGRP, R11CGRP, L15CGRP, R18CGRP, and F37CGRP (Supplementary Figures S1C,D; Supplementary Tables S1, S2).
The gradual engagement of the top of TM6, TM7 and ECL3 by the peptide N-terminal domain was accompanied by an inward movement of ECL3 (Supplementary Video S1); ECL3 is important for CGRPR signaling (Barwell et al., 2011) but its dynamics during receptor activation are still unclear. A comparison between the consensus maps of CGRP:CGRPR and CGRP:CGRPR:Gs complexes (Josephs et al., 2021) shows that the location of the top of TM6/7/ECL3 in the former structure partially overlaps with the location of the peptide binding pocket in the active CGRPR. Analysis of the conformational dynamics of the CGRP:CGRPR complex revealed that the top of TM6/7/ECL3 dynamically opens and closes in association with transient interaction of the peptide N-terminus and the TMD core (Josephs et al., 2021). This reinforces the concept that the presence of the G protein at the intracellular side of the receptor is needed to allosterically stabilize peptide N-terminal domain binding to the receptor core. Our simulations started from the fully active CGRPR in complex with both CGRP and the Gα helix 5, which could favor the productive engagement of ECL3 with the peptide N-terminus.
The binding energy landscape of CGRP shows low prevalence of states between MS and OS (Figure 2A), indicative of the presence of a transition state. In MS, the positively charged agonist residues R11CGRP, R18CGRP formed electrostatic interactions with the side chains of loop 4 residues D90ECD and D94ECD (Figure 2E; Supplementary Figure S1D). In OS, the same CGRP residues interacted with D287ECL2, E357ECL3 and R355ECL3 (Figure 2F; Supplementary Table S2). It follows that during the transition from MS to OS, the ionic network between CGRP and D90ECD, D94ECD becomes disrupted to allow new interactions with the TMD.
In the TMD of the active CGRPR, after removal of any ligand (Figure 3A), two hydration clusters were detected in close proximity to ECL2 (namely ECL2 water clusters “up” and “down,” according to the position respective to the backbone of the loop). A further hydrated region was detected near ECL3 (ECL3 water cluster). Two structural water molecules are positioned close to the conserved polar core of the receptor (N1872.60 and TM2-TM4 water molecules in Figure 3A); the polar core is important for class B1 receptor activation and biased agonism (Wootten et al., 2013; Wootten et al., 2016; Yin et al., 2017). Upon binding and insertion of the N-terminal helix within the TMD, CGRP stabilizes several water molecules located in proximity to ECL2 and ECL3 (Figure 3B), without overlapping any hydrated region present in the active CGRPR in the absence of ligand. Interactions between CGRPR and the TMD changes the hydration of the TMD by destabilizing the N1872.60 and TM2-TM4 water molecules, which are not detected in the peptide-occupied complex (Figure 3B). Overall, these data suggest an efficient solvation profile and formation of a favorable water network during the binding of CGRP.
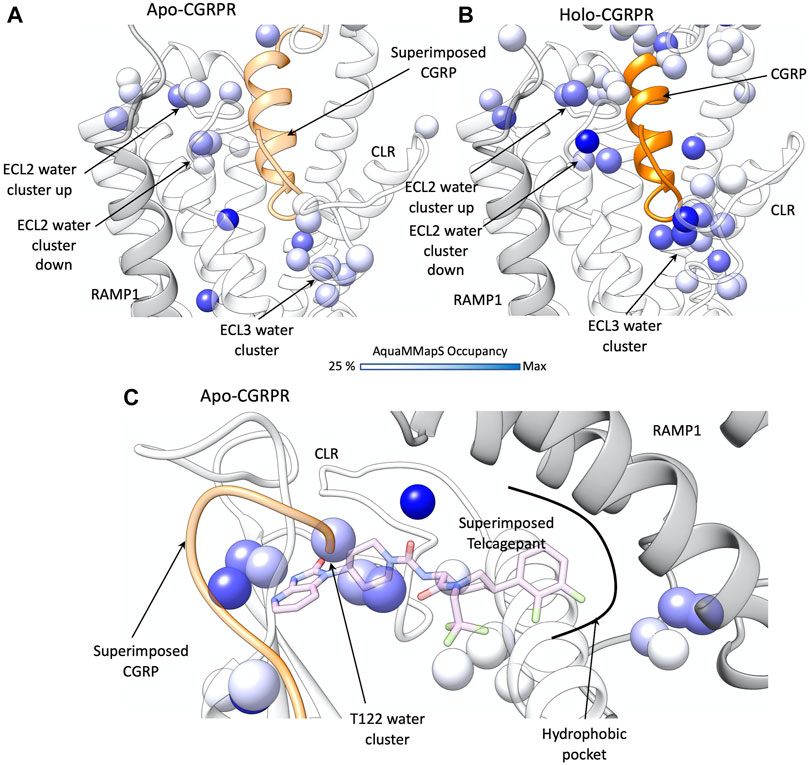
FIGURE 3. Water network perturbation upon CGRP and telcagepant binding. (A) hydration positions in the TMD of the active CGRPR (white ribbon, representative conformation from the first frame of MD simulation), after removal of ligand; CLR and RAMP1 are shown as transparent white and grey ribbons, CGRP from PDB 6E3Y is superimposed (by superimposing TMD scaffolds) and shown as a transparent orange ribbon; (B) hydration positions in the holo CGRPR TMD (representative conformation from the first frame of MD simulation). CLR and RAMP1 are shown as transparent white and grey ribbons, CGRP as an orange ribbon; (C) hydration positions in the ECD of the active CGRPR, after removal of any ligand; CLR and RAMP1 are shown as transparent white and grey ribbons, CGRP from PDB 6E3Y and telcagepant from PDB 3N7R are superimposed and shown as transparent orange ribbon and pink sticks, respectively. In (A–C) the color scale of the hydrated spots corresponds to the occupancy of water molecules during the MD simulation (in dark blue the maximum occupancy).
Telcagepant Binding and Unbinding Paths
The antagonist telcagepant (Figure 1B) binds into a pocket delimited by the ECD and RAMP1 (ter Haar et al., 2010). During dynamic docking simulations (Figure 4; Supplementary Video S2), the ligand rapidly reached the crystallographic conformation following two possible paths (Paths A and B in Figure 4B) and forming intermediate metastable states in correspondence of macrostates, denoted MS1 and MS2 (Figures 4A,B). The most persistent contacts with the receptor were formed around the binding site (Figures 4C,D; Supplementary Tables S3, S4) and comprised interactions with side chains of W72ECD, I41ECD, M42ECD, R38ECD, Q45ECD, W74RAMP1, and W84RAMP1, which are also involved in the final bound state. Besides the hydrogen bonds with W72ECD and the backbone of T122ECD, the antagonist formed transitory polar interactions with N128ECD, R119ECD, and W121ECD (Supplementary Figure S3; Supplementary Table S4). Binding Path A was facilitated by interactions with the ECD helix 1 residues Q33ECD, V36ECD, T37ECD, K40ECD (Figure 4E). Binding Path B involved interactions with the RAMP1 residues N31RAMP1 and F83RAMP1, located over the binding site (Figure 4F). Consistent with mutagenesis studies (Moore et al., 2010), no significant hydrogen bonds were formed between telcagepant and R67RAMP1, D71RAMP1, and E78RAMP1 (Supplementary Table S4).
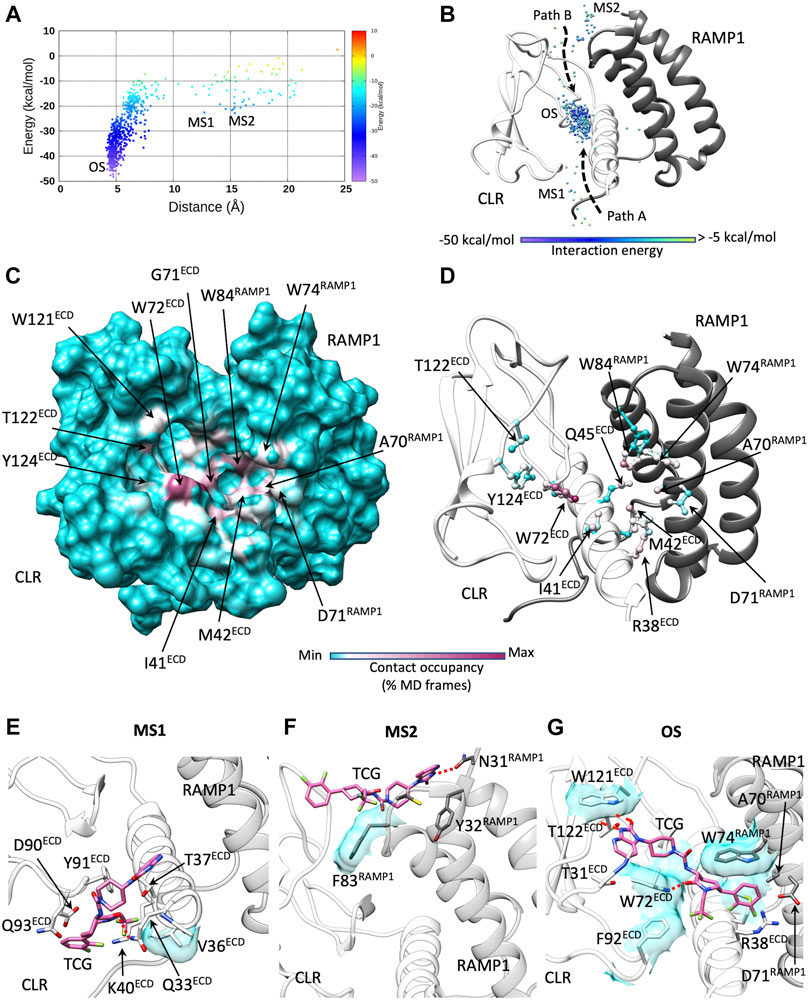
FIGURE 4. Telcagepant dynamic docking. (A) Telcagepant binding energy landscape (the metastable macrostates, MS1 and MS2, are indicated, alongside the bound states, OS, corresponding to the pose in solved structure); (B) binding paths of the centroid of telcagepant, colored according to the interaction energy with CGRPR (the metastable macrostates, MS1 and MS2, are indicated, alongside the stable bound states OS); (C) Telcagepant-CGRPR contacts plotted on CGRPR surface and colored according to the occupancy (% MD frames) of the interactions; (D) Telcagepant-CGRPR contacts plotted on CGRPR atoms and colored according to the occupancy (% MD frames) of the interactions; (E) representative configuration from the macrostate, MS1; (F) representative configuration from the macrostate, MS2; (G) representative configuration from the bound states OS. Electrostatic interactions are depicted as red dashed lines, while hydrophobic contacts are shown as cyan transparent surfaces.
Unbinding simulations (Figure 5; Supplementary Figure S4; Supplementary Tables S5, S6) sampled one notable metastable macrostate (MS in Figures 5A,B) close to the bound configuration observed in the static crystallographic structure (ter Haar et al., 2010) (OS in Figures 5A,B). The transition between OS and MS took place in the early stages of ligand dissociation and involved the rupture of the hydrogen bonds with the backbone of T122ECD in favor of new transient interactions with D94ECD and N128ECD (Figures 5E,F). After release from the hydrophobic interactions between the 1,2-dichlorobenzene group and W74RAMP1, W84RAMP1, and M42ECD, telcagepant followed two possible unbinding routes from MS (Figure 5B): unbinding Path A roughly represented the inverse of binding Path A, while unbinding Path B followed an alternative direction, away from RAMP1, which involved interactions with W72ECD.
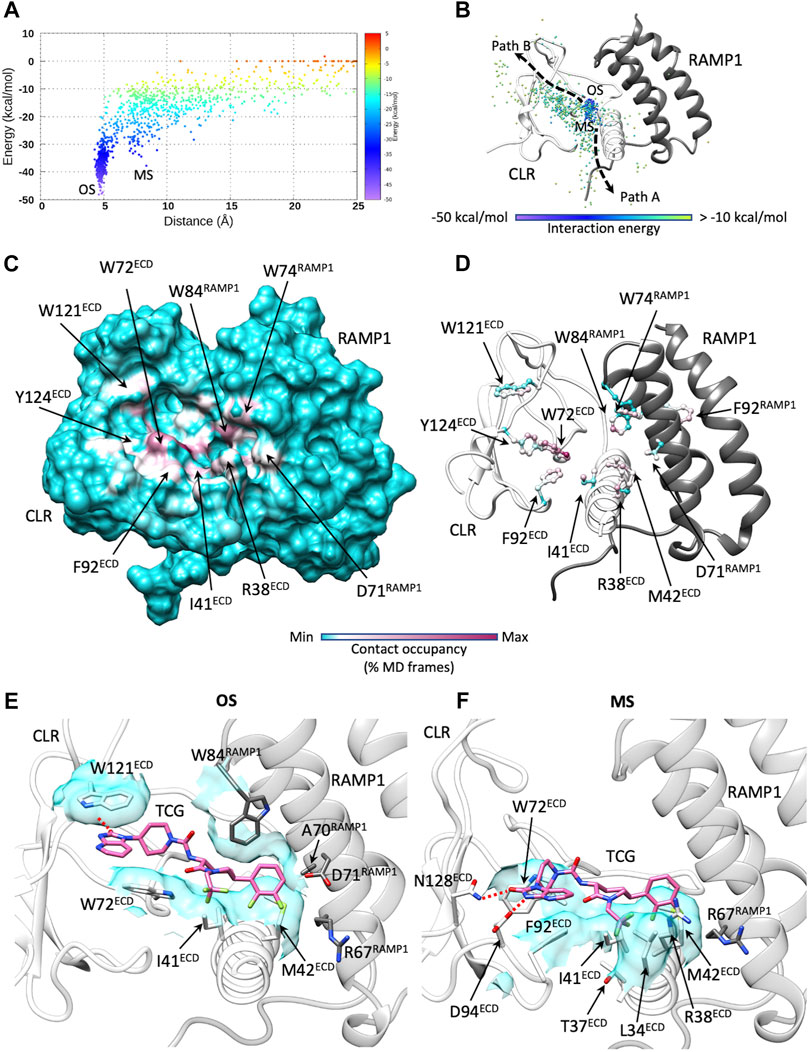
FIGURE 5. Telcagepant dynamic unbinding. (A) Telcagepant unbinding energy landscape (the metastable macrostates, MS, are indicated, alongside the stable bound states OS); (B) unbinding paths of the centroid of telcagepant, colored according to the interaction energy with CGRPR (the metastable macrostates, MS, are indicated, alongside the stable bound states, OS); (C) Telcagepant-CGRPR contacts plotted on CGRPR surface and colored according to the occupancy (% MD frames) of the interactions; (D) Telcagepant-CGRPR contacts plotted on CGRPR atoms and colored according to the occupancy (% MD frames) of the interactions; (E) representative configuration from the stable bound state, OS; (F) representative configuration from the metastable macrostate, MS. Electrostatic interactions are depicted as red dashed lines, while hydrophobic contacts are shown as cyan transparent surfaces.
The ECD is the first site of stable contacts for class B1 peptides during the proposed two-stage peptide agonist binding (Hoare, 2005), as well as the target of the antagonist telcagepant (Figure 3C). In the active CGRPR, following removal of the ligand, AquaMMapS analysis highlighted two clusters of stable water molecules in the proximity of T122ECD (Figure 3C), within the binding site of telcagepant and the distal residues of the CGRP C-terminal domain. The backbone of T122ECD forms hydrogen bonds with either the primary amide of the CGRP C-terminal residue F37CGRP or with the phthalimide moiety of telcagepant. It follows that both ligands likely displace stable water molecules to form hydrogen bonds with the T122ECD backbone. Upon binding, telcagepant extends the difluorophenyl moiety into a hydrophobic pocket and displaces unstable water molecules, as suggested by the absence of hydrated spots in this region of ECD (Figure 3C).
Discussion
Recently resolved class B1 GPCR structures reveal a heterogenous conformational landscape that involves the extracellular elements of the receptor (Liang et al., 2020b; Zhang et al., 2020). For example, different glucagon-like peptide receptor (GLP-1R) agonists imprint divergent orientations of the ECD and alternative conformations of the top of TM1, TM6, TM7, ECL2, and ECL3. Some of these structural features, such as an outward conformation of ECL3, appear linked to biased agonism of different ligands (Liang et al., 2018b; Zhang et al., 2020).
The structural mechanism underlining the agonist two-step binding and class B1 receptor activation is still unclear. The recent structures of CGRPR in apo and CGRP-bound (before G protein coupling) forms have delivered new insights into the transition from the resting to the fully active state of the receptor (Josephs et al., 2021). Interestingly, in the consensus cryo-EM maps only minor conformational changes in the backbone of the receptor were observed before and after the binding of CGRP. Given the intrinsic flexibility that characterizes GPCRs (Latorraca et al., 2017), orthogonal methods such as hydrogen-deuterium exchange mass spectrometry (Yang et al., 2015; Josephs et al., 2021), 3D variance analysis of cryo-EM conformational continuums (Liang et al., 2020a; Dong et al., 2020; Zhang et al., 2020; Josephs et al., 2021) and molecular dynamics simulations (Deganutti et al., 2019) are required to characterize CGRPR molecular motions and advance the understanding of its pharmacology.
The adaptive sampling protocol employed in the current study focused on the peptide N-terminal approach to the CGRPR TMD, and therefore did not interrogate the preliminary interactions between the ECD and the peptide C-terminal domain. The simulation of the complete two-stage binding mechanism requires a more thorough computational sampling and should take into account the secondary structure characterizing the class B1 peptides in solution before any interaction with the ECD occurs. GLP-1, secretin (Sec), glucagon (GCG), pituitary adenylate cyclase activating polypeptide (PACAP), vasoactive intestinal polypeptide (VIP), parathyroid hormone 1 (PTH1), and corticotrophin-releasing factor (CRF) bind with an extended α-helix conformation spanning from the TMD to the ECD binding sites. However, in solution, many of them present a disordered N-terminal segment (Gronenborn et al., 1987; Neidigh et al., 2001). This suggests that the formation of the α-helix is a hallmark of the binding. On the other hand, agonists from the calcitonin sub-family, such as CGRP, in solution show only partial folding of the N-terminal in α-helix motif (Breeze et al., 1991) and engage their target by retaining this structural organization.
This structural conservation between solution and receptor-bound CGRP structures rules out any major conformational changes of the CGRP during the binding, reinforcing our findings. RAMPs allosterically alter the dynamics of the ECD, ECL2 and ECL3 and the signaling profile of CLR, in a peptide-dependent manner (Liang et al., 2020a). However, the structural reasons for the binding selectivity exerted on CLR is still unclear. We speculate that different RAMPs may modulate the affinity of CLR agonists by driving divergent metastable states of ECD loop 4 during binding to the TMD. The membrane-proximal linker region of the RAMP1 (residues 102–118, Figures 1A, 2D,E) is an important contributor to the allosteric modulation of CLR (Liang et al., 2020a). The different rotameric states of D113RAMP1 in the apo and CGRP-bound states of CGRPR (Josephs et al., 2021) along with dynamic differences in this region reported for the RAMPs (Supplementary Figure S5) suggests that the flexibility of the linker could play a more complex role in selectivity of agonist binding. The position of ECD loop 4 in the CGRP:CGRPR:Gs, AM:AM1R:Gs, AM:AM2R:Gs and AM2:AM2R:Gs complexes (Liang et al., 2020a) is different, implying a possible correlation with the linker region. The exchange of the RAMP1 linker with the RAMP2 linker produces a loss of CGRP potency ascribable to a correlated dynamic between the ECD and the G protein (Liang et al., 2020a).
Our simulations showed that ECD loop 4 may act as a selectivity filter for the incoming agonist. The shape and dynamics of the gate formed by ECD, TM1, TM2 and ECL1 appear to be linked to ECD mobility and, in turn, to the interactions between the RAMP linker and CLR. We speculate that different intramolecular interactions with CLR differently modulate ECD flexibility. These differences in the dynamics of loop 4 residues D90ECD, Q93ECD, and D94ECD could contribute to the selectivity displayed by CLR in complex with the RAMPs, thus influencing the overall kinetics of binding.
We cannot rule out an involvement of the extracellular surface of the membrane during the first steps of the association between the N-terminal domain of CGRPR and TMD extracellular vestibule. However, the preliminary binding of the peptide C-terminus to the ECD should restrain the path of the N-terminal domain, reducing the possibility of diffusion from the membrane.
Simulations of telcagepant highlighted key features and a partial overlap of association and dissociation paths from the ECD. The telcagepant binding site is easily accessible from the bulk solvent without hindrances and presents stable water molecules only in the proximity of T122ECD, which the phthalimide moiety is likely to occupy without unfavorable energy contributions thanks to the hydrogen bonds formed by the amide group. It follows that desolvation of the pocket should be ruled out as a bottleneck to binding (Sykes et al., 2019), consistent with the “fast-on” nature of telcagepant. Our simulations also suggest that the low-nanomolar/high picomolar affinity of telcagepant for CGRPR is due to the fast association rather than a long residence time, which is reported to be close to 2 min (Moore et al., 2009).
In summary, we propose an updated scenario for the binding to CGRPR by the endogenous agonist and a prototypic antagonist. Exploiting the recent structural information on different states of the CGRPR (Josephs et al., 2021) and GLP-1R in the absence of endogenous agonist bound (Wu et al., 2020), future work will be directed to establishing MD protocols able to reconstruct the different phases of the proposed two-step binding mechanism of all class B peptides, taking into account the conformational transition occurring upon binding.
Data Availability Statement
SuMD simulations dataset can be download from the Zenodo repository at https://zenodo.org/record/5109537.
Author Contributions
GD and CAR designed the study. GD carried out the simulations, GD, SA, and R-MR analyzed the data. GD wrote the manuscript with significant input from CAR, SA, R-MR, DW, and PMS.
Funding
The project was funded by the BBSCR grant BB/M006883/1 (P.I. CAR).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2021.720561/full#supplementary-material
References
Aksoydan, B., and Durdagi, S. (2021). Molecular Simulations Reveal the Impact of RAMP1 on Ligand Binding and Dynamics of CGRPR Heterodimer. BioRxiv. doi:10.1101/2021.05.10.443383
Atanasio, S., Deganutti, G., and Reynolds, C. A. (2020). Addressing Free Fatty Acid Receptor 1 (FFAR1) Activation Using Supervised Molecular Dynamics. J. Comput. Aided Mol. Des. 34, 1181–1193. doi:10.1007/s10822-020-00338-6
Aubdool, A. A., Thakore, P., Argunhan, F., Smillie, S.-J., Schnelle, M., Srivastava, S., et al. (2017). A Novel α-Calcitonin Gene-Related Peptide Analogue Protects against End-Organ Damage in Experimental Hypertension, Cardiac Hypertrophy, and Heart Failure. Circulation 136, 367–383. doi:10.1161/CIRCULATIONAHA.117.028388
Barducci, A., Bussi, G., and Parrinello, M. (2008). Well-tempered Metadynamics: a Smoothly Converging and Tunable Free-Energy Method. Phys. Rev. Lett. 100, 020603. doi:10.1103/PhysRevLett.100.020603
Barwell, J., Conner, A., and Poyner, D. R. (2011). Extracellular Loops 1 and 3 and Their Associated Transmembrane Regions of the Calcitonin Receptor-like Receptor Are Needed for CGRP Receptor Function. Biochim. Biophys. Acta (Bba) - Mol. Cel Res. 1813, 1906–1916. doi:10.1016/j.bbamcr.2011.06.005
Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., DiNola, A., and Haak, J. R. (1984). Molecular Dynamics with Coupling to an External bath. J. Chem. Phys. 81, 3684–3690. doi:10.1063/1.448118
Bissaro, M., Bolcato, G., Deganutti, G., Sturlese, M., and Moro, S. (2019). Revisiting the Allosteric Regulation of Sodium Cation on the Binding of Adenosine at the Human A2A Adenosine Receptor: Insights from Supervised Molecular Dynamics (SuMD) Simulations. Molecules 24, 2752. doi:10.3390/molecules24152752
Bissaro, M., Sturlese, M., and Moro, S. (2020). Exploring the RNA-Recognition Mechanism Using Supervised Molecular Dynamics (SuMD) Simulations: Toward a Rational Design for Ribonucleic-Targeting Molecules?. Front. Chem. 8, 107. doi:10.3389/fchem.2020.00107
Bortolato, A., Tehan, B. G., Bodnarchuk, M. S., Essex, J. W., and Mason, J. S. (2013). Water Network Perturbation in Ligand Binding: Adenosine A2A Antagonists as a Case Study. J. Chem. Inf. Model. 53, 1700–1713. doi:10.1021/ci4001458
Bower, R. L., Yule, L., Rees, T. A., Deganutti, G., Hendrikse, E. R., Harris, P. W. R., et al. (2018). Molecular Signature for Receptor Engagement in the Metabolic Peptide Hormone Amylin. ACS Pharmacol. Transl. Sci. 1, 32–49. doi:10.1021/acsptsci.8b00002
Breeze, A. L., Harvey, T. S., Bazzo, R., and Campbell, I. D. (1991). Solution Structure of Human Calcitonin Gene-Related Peptide by Proton NMR and Distance Geometry with Restrained Molecular Dynamics. Biochemistry 30, 575–582. doi:10.1021/bi00216a036
Castro, M., Nikolaev, V. O., Palm, D., Lohse, M. J., and Vilardaga, J.-P. (2005). Turn-on Switch in Parathyroid Hormone Receptor by a Two-step Parathyroid Hormone Binding Mechanism. Proc. Natl. Acad. Sci. 102, 16084–16089. doi:10.1073/pnas.0503942102
Chung, K. Y., Rasmussen, S. G. F., Liu, T., Li, S., DeVree, B. T., Chae, P. S., et al. (2011). Conformational Changes in the G Protein Gs Induced by the β2 Adrenergic Receptor. Nature 477, 611–615. doi:10.1038/nature10488
Cuzzolin, A., Deganutti, G., Salmaso, V., Sturlese, M., and Moro, S. (2018). AquaMMapS: An Alternative Tool to Monitor the Role of Water Molecules during Protein-Ligand Association. ChemMedChem 13, 522–531. doi:10.1002/cmdc.201700564
Cuzzolin, A., Sturlese, M., Deganutti, G., Salmaso, V., Sabbadin, D., Ciancetta, A., et al. (2016). Deciphering the Complexity of Ligand-Protein Recognition Pathways Using Supervised Molecular Dynamics (SuMD) Simulations. J. Chem. Inf. Model. 56, 687–705. doi:10.1021/acs.jcim.5b00702
de Graaf, C., Song, G., Cao, C., Zhao, Q., Wang, M.-W., Wu, B., et al. (2017). Extending the Structural View of Class B Gpcrs. Trends Biochem. Sci. 42, 946–960. doi:10.1016/j.tibs.2017.10.003
De Lean, A., Stadel, J. M., and Lefkowitz, R. J. (1980). A Ternary Complex Model Explains the Agonist-specific Binding Properties of the Adenylate Cyclase-Coupled Beta-Adrenergic Receptor. J. Biol. Chem. 255, 7108–7117. doi:10.1016/S0021-9258(20)79672-9
Deganutti, G., Barkan, K., Preti, B., Leuenberger, M., Wall, M., Frenguelli, B. G., et al. (2021). Deciphering the Agonist Binding Mechanism to the Adenosine A1 Receptor. ACS Pharmacol. Transl. Sci. 4, 314–326. doi:10.1021/acsptsci.0c00195
Deganutti, G., Cuzzolin, A., Ciancetta, A., and Moro, S. (2015). Understanding Allosteric Interactions in G Protein-Coupled Receptors Using Supervised Molecular Dynamics: A Prototype Study Analysing the Human A3 Adenosine Receptor Positive Allosteric Modulator LUF6000. Bioorg. Med. Chem. 23, 4065–4071. doi:10.1016/j.bmc.2015.03.039
Deganutti, G., and Moro, S. (2017a). Estimation of Kinetic and Thermodynamic Ligand-Binding Parameters Using Computational Strategies. Future Med. Chem. 9, 507–523. doi:10.4155/fmc-2016-0224
Deganutti, G., Moro, S., and Reynolds, C. A. (2020). A Supervised Molecular Dynamics Approach to Unbiased Ligand-Protein Unbinding. J. Chem. Inf. Model. 60, 1804–1817. doi:10.1021/acs.jcim.9b01094
Deganutti, G., Moro, S., and Reynolds, C. A. (2019). Peeking at G-Protein-Coupled Receptors through the Molecular Dynamics Keyhole. Future Med. Chem. 11, 599–615. doi:10.4155/fmc-2018-0393
Deganutti, G., and Moro, S. (2017b). Supporting the Identification of Novel Fragment-Based Positive Allosteric Modulators Using a Supervised Molecular Dynamics Approach: A Retrospective Analysis Considering the Human A2A Adenosine Receptor as a Key Example. Molecules 22, 818. doi:10.3390/molecules22050818
DeVree, B. T., Mahoney, J. P., Vélez-Ruiz, G. A., Rasmussen, S. G. F., Kuszak, A. J., Edwald, E., et al. (2016). Allosteric Coupling from G Protein to the Agonist-Binding Pocket in GPCRs. Nature 535, 182–186. doi:10.1038/nature18324
Dolinsky, T. J., Nielsen, J. E., McCammon, J. A., and Baker, N. A. (2004). PDB2PQR: an Automated Pipeline for the Setup of Poisson-Boltzmann Electrostatics Calculations. Nucleic Acids Res. 32, W665–W667. doi:10.1093/nar/gkh381
Dong, M., Deganutti, G., Piper, S. J., Liang, Y.-L., Khoshouei, M., Belousoff, M. J., et al. (2020). Structure and Dynamics of the Active Gs-Coupled Human Secretin Receptor. Nat. Commun. 11, 4137. doi:10.1038/s41467-020-17791-4
Duan, J., Shen, D.-D., Zhou, X. E., Bi, P., Liu, Q.-F., Tan, Y.-X., et al. (2020). Cryo-EM Structure of an Activated VIP1 Receptor-G Protein Complex Revealed by a NanoBiT Tethering Strategy. Nat. Commun. 11, 4121. doi:10.1038/s41467-020-17933-8
Durham, P. L. (2006). Calcitonin Gene-Related Peptide (CGRP) and Migraine. Headache 46 (Suppl. 1), S3–S8. doi:10.1111/j.1526-4610.2006.00483.x
Essmann, U., Perera, L., Berkowitz, M. L., Darden, T., Lee, H., and Pedersen, L. G. (1995). A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 103, 8577–8593. doi:10.1063/1.470117
Forester, T. R., and Smith, W. (1998). SHAKE, Rattle, and Roll: Efficient Constraint Algorithms for Linked Rigid Bodies. J. Comput. Chem. 19, 102–111. doi:10.1002/(sici)1096-987x(19980115)19:1<102::aid-jcc9>3.0.co;2-t
Garces, F., Mohr, C., Zhang, L., Huang, C.-S., Chen, Q., King, C., et al. (2020). Molecular Insight into Recognition of the CGRPR Complex by Migraine Prevention Therapy Aimovig (Erenumab). Cel Rep. 30, 1714–1723. e6. doi:10.1016/j.celrep.2020.01.029
García-Nafría, J., and Tate, C. G. (2019). Cryo-EM Structures of GPCRs Coupled to Gs, Gi and Go. Mol. Cell Endocrinol. 488, 1–13. doi:10.1016/j.mce.2019.02.006
Gronenborn, A. M., Bovermann, G., and Clore, G. M. (1987). A 1 H-NMR Study of the Solution Conformation of Secretin Resonance Assignment and Secondary Structure. FEBS Lett. 215, 88–94. doi:10.1016/0014-5793(87)80119-9
Harvey, M. J., Giupponi, G., and Fabritiis, G. D. (2009). ACEMD: Accelerating Biomolecular Dynamics in the Microsecond Time Scale. J. Chem. Theor. Comput. 5, 1632–1639. doi:10.1021/ct9000685
Hauser, A. S., Attwood, M. M., Rask-Andersen, M., Schiöth, H. B., and Gloriam, D. E. (2017). Trends in GPCR Drug Discovery: New Agents, Targets and Indications. Nat. Rev. Drug Discov. 16, 829–842. doi:10.1038/nrd.2017.178
Hay, D. L., Garelja, M. L., Poyner, D. R., and Walker, C. S. (2018). Update on the Pharmacology of Calcitonin/CGRP Family of Peptides: IUPHAR Review 25. Br. J. Pharmacol. 175, 3–17. doi:10.1111/bph.14075
Hay, D. L., and Pioszak, A. A. (2016). Receptor Activity-Modifying Proteins (RAMPs): New Insights and Roles. Annu. Rev. Pharmacol. Toxicol. 56, 469–487. doi:10.1146/annurev-pharmtox-010715-103120
Higgs, C., Beuming, T., and Sherman, W. (2010). Hydration Site Thermodynamics Explain SARs for Triazolylpurines Analogues Binding to the A2A Receptor. ACS Med. Chem. Lett. 1, 160–164. doi:10.1021/ml100008s
Hoare, S. (2005). Mechanisms of Peptide and Nonpeptide Ligand Binding to Class B G-Protein-Coupled Receptors. Drug Discov. Today 10, 417–427. doi:10.1016/S1359-6446(05)03370-2
Hollenstein, K., de Graaf, C., Bortolato, A., Wang, M.-W., Marshall, F. H., and Stevens, R. C. (2014). Insights into the Structure of Class B GPCRs. Trends Pharmacol. Sci. 35, 12–22. doi:10.1016/j.tips.2013.11.001
Hopkins, C. W., Le Grand, S., Walker, R. C., and Roitberg, A. E. (2015). Long-Time-Step Molecular Dynamics through Hydrogen Mass Repartitioning. J. Chem. Theor. Comput. 11, 1864–1874. doi:10.1021/ct5010406
Huang, J., and MacKerell, A. D. (2013). CHARMM36 All-Atom Additive Protein Force Field: Validation Based on Comparison to NMR Data. J. Comput. Chem. 34, 2135–2145. doi:10.1002/jcc.23354
Huang, J., Rauscher, S., Nawrocki, G., Ran, T., Feig, M., de Groot, B. L., et al. (2017). CHARMM36m: an Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 14, 71–73. doi:10.1038/nmeth.4067
Humphrey, W., Dalke, A., and Schulten, K. (1996). VMD: Visual Molecular Dynamics. J. Mol. Graph 14 (33–8), 33–38. doi:10.1016/0263-7855(96)00018-5
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W., and Klein, M. L. (1983). Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 79, 926–935. doi:10.1063/1.445869
Josephs, T. M., Belousoff, M. J., Liang, Y.-L., Piper, S. J., Cao, J., Garama, D. J., et al. (2021). Structure and Dynamics of the CGRP Receptor in Apo and Peptide-Bound Forms. Science 372, eabf7258. doi:10.1126/science.abf7258
Kee, Z., Kodji, X., and Brain, S. D. (2018). The Role of Calcitonin Gene Related Peptide (CGRP) in Neurogenic Vasodilation and its Cardioprotective Effects. Front. Physiol. 9, 1249. doi:10.3389/fphys.2018.01249
Kräutler, V., van Gunsteren, W. F., and Hünenberger, P. H. (2001). A Fast SHAKE Algorithm to Solve Distance Constraint Equations for Small Molecules in Molecular Dynamics Simulations. J. Comput. Chem. 22, 501–508. doi:10.1002/1096-987X(20010415)22:5<501::AID-JCC1021>3.0.CO;2-V
Ladbury, J. E. (1996). Just Add Water! the Effect of Water on the Specificity of Protein-Ligand Binding Sites and its Potential Application to Drug Design. Chem. Biol. 3, 973–980. doi:10.1016/S1074-5521(96)90164-7
Latorraca, N. R., Venkatakrishnan, A. J., and Dror, R. O. (2017). GPCR Dynamics: Structures in Motion. Chem. Rev. 117, 139–155. doi:10.1021/acs.chemrev.6b00177
Liang, Y.-L., Belousoff, M. J., Fletcher, M. M., Zhang, X., Khoshouei, M., Deganutti, G., et al. (2020a). Structure and Dynamics of Adrenomedullin Receptors AM1 and AM2 Reveal Key Mechanisms in the Control of Receptor Phenotype by Receptor Activity-Modifying Proteins. ACS Pharmacol. Transl. Sci. 3, 263–284. doi:10.1021/acsptsci.9b00080
Liang, Y.-L., Belousoff, M. J., Zhao, P., Koole, C., Fletcher, M. M., Truong, T. T., et al. (2020b). Toward a Structural Understanding of Class B GPCR Peptide Binding and Activation. Mol. Cel 77, 656–668. e5. doi:10.1016/j.molcel.2020.01.012
Liang, Y.-L., Khoshouei, M., Deganutti, G., Glukhova, A., Koole, C., Peat, T. S., et al. (2018a). Cryo-EM Structure of the Active, Gs-Protein Complexed, Human CGRP Receptor. Nature 561, 492–497. doi:10.1038/s41586-018-0535-y
Liang, Y.-L., Khoshouei, M., Glukhova, A., Furness, S. G. B., Zhao, P., Clydesdale, L., et al. (2018b). Phase-plate Cryo-EM Structure of a Biased Agonist-Bound Human GLP-1 Receptor-Gs Complex. Nature 555, 121–125. doi:10.1038/nature25773
Liang, Y.-L., Khoshouei, M., Radjainia, M., Zhang, Y., Glukhova, A., Tarrasch, J., et al. (2017). Phase-plate Cryo-EM Structure of a Class B GPCR-G-Protein Complex. Nature 546, 118–123. doi:10.1038/nature22327
Lomize, M. A., Lomize, A. L., Pogozheva, I. D., and Mosberg, H. I. (2006). OPM: Orientations of Proteins in Membranes Database. Bioinformatics 22, 623–625. doi:10.1093/bioinformatics/btk023
Loncharich, R. J., Brooks, B. R., and Pastor, R. W. (1992). Langevin Dynamics of Peptides: The Frictional Dependence of Isomerization Rates ofN-Acetylalanyl-N?-Methylamide. Biopolymers 32, 523–535. doi:10.1002/bip.360320508
Ma, S., Shen, Q., Zhao, L.-H., Mao, C., Zhou, X. E., Shen, D.-D., et al. (2020). Molecular Basis for Hormone Recognition and Activation of Corticotropin-Releasing Factor Receptors. Mol. Cel 77, 669–680. e4. doi:10.1016/j.molcel.2020.01.013
Miller, B. R., McGee, T. D., Swails, J. M., Homeyer, N., Gohlke, H., and Roitberg, A. E. (2012). MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theor. Comput. 8, 3314–3321. doi:10.1021/ct300418h
Moore, E. L., Burgey, C. S., Paone, D. V., Shaw, A. W., Tang, Y. S., Kane, S. A., et al. (2009). Examining the Binding Properties of MK-0974: a CGRP Receptor Antagonist for the Acute Treatment of Migraine. Eur. J. Pharmacol. 602, 250–254. doi:10.1016/j.ejphar.2008.11.050
Moore, E. L., Gingell, J. J., Kane, S. A., Hay, D. L., and Salvatore, C. A. (2010). Mapping the CGRP Receptor Ligand Binding Domain: Tryptophan-84 of RAMP1 Is Critical for Agonist and Antagonist Binding. Biochem. Biophysical Res. Commun. 394, 141–145. doi:10.1016/j.bbrc.2010.02.131
Neidigh, J. W., Fesinmeyer, R. M., Prickett, K. S., and Andersen, N. H. (2001). Exendin-4 and Glucagon-Like-Peptide-1: NMR Structural Comparisons in the Solution and Micelle-Associated States†. Biochemistry 40, 13188–13200. doi:10.1021/bi010902s
Olsson, M. H. M., Søndergaard, C. R., Rostkowski, M., and Jensen, J. H. (2011). PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theor. Comput. 7, 525–537. doi:10.1021/ct100578z
Pal, K., Melcher, K., and Xu, H. E. (2012). Structure and Mechanism for Recognition of Peptide Hormones by Class B G-Protein-Coupled Receptors. Acta Pharmacol. Sin 33, 300–311. doi:10.1038/aps.2011.170
Paone, D. V., Shaw, A. W., Nguyen, D. N., Burgey, C. S., Deng, J. Z., Kane, S. A., et al. (2007). Potent, Orally Bioavailable Calcitonin Gene-Related Peptide Receptor Antagonists for the Treatment of Migraine: Discovery of N-[(3R,6S)-6-(2,3-difluorophenyl)-2-oxo-1- (2,2,2-Trifluoroethyl)azepan-3-Yl]-4- (2-Oxo-2,3-Dihydro-1h-Imidazo[4,5-B]pyridin- 1-Yl)piperidine-1-Carboxamide (MK-0974). J. Med. Chem. 50, 5564–5567. doi:10.1021/jm070668p
Pham, V., Zhu, Y., Dal Maso, E., Reynolds, C. A., Deganutti, G., Atanasio, S., et al. (2019). Deconvoluting the Molecular Control of Binding and Signaling at the Amylin 3 Receptor: RAMP3 Alters Signal Propagation through Extracellular Loops of the Calcitonin Receptor. ACS Pharmacol. Transl. Sci. 2, 183–197. doi:10.1021/acsptsci.9b00010
Qiao, A., Han, S., Li, X., Li, Z., Zhao, P., Dai, A., et al. (2020). Structural Basis of Gsand Girecognition by the Human Glucagon Receptor. Science 367, 1346–1352. doi:10.1126/science.aaz5346
Rosenbaum, D. M., Rasmussen, S. G. F., and Kobilka, B. K. (2009). The Structure and Function of G-Protein-Coupled Receptors. Nature 459, 356–363. doi:10.1038/nature08144
Rujan, R.-M., and Reynolds, C. A. (2019). Calcitonin Gene-Related Peptide Antagonists and Therapeutic Antibodies. Handb Exp. Pharmacol. 255, 169–192. doi:10.1007/164_2018_173
Sabbadin, D., and Moro, S. (2014). Supervised Molecular Dynamics (SuMD) as a Helpful Tool to Depict GPCR-Ligand Recognition Pathway in a Nanosecond Time Scale. J. Chem. Inf. Model. 54, 372–376. doi:10.1021/ci400766b
Salmaso, V., Sturlese, M., Cuzzolin, A., and Moro, S. (2017). Exploring Protein-Peptide Recognition Pathways Using a Supervised Molecular Dynamics Approach. Structure 25, 655–662. doi:10.1016/j.str.2017.02.009
Salvatore, C. A., Hershey, J. C., Corcoran, H. A., Fay, J. F., Johnston, V. K., Moore, E. L., et al. (2008). Pharmacological Characterization of MK-0974 [N-[(3R,6S)-6-(2,3-difluorophenyl)-2-oxo-1-(2,2,2-trifluoroethyl)azepan-3-yl]-4-(2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-1-yl)piperidine-1-carboxamide], a Potent and Orally Active Calcitonin Gene-Related Peptide Receptor Antagonist for the Treatment of Migraine. J. Pharmacol. Exp. Ther. 324, 416–421. doi:10.1124/jpet.107.130344
Sommer, B. (2013). Membrane Packing Problems: A Short Review on Computational Membrane Modeling Methods and Tools. Comput. Struct. Biotechnol. J. 5, e201302014. doi:10.5936/csbj.201302014
Sykes, D. A., Stoddart, L. A., Kilpatrick, L. E., and Hill, S. J. (2019). Binding Kinetics of Ligands Acting at GPCRs. Mol. Cell Endocrinol. 485, 9–19. doi:10.1016/j.mce.2019.01.018
ter Haar, E., Koth, C. M., Abdul-Manan, N., Swenson, L., Coll, J. T., Lippke, J. A., et al. (2010). Crystal Structure of the Ectodomain Complex of the CGRP Receptor, a Class-B GPCR, Reveals the Site of Drug Antagonism. Structure 18, 1083–1093. doi:10.1016/j.str.2010.05.014
Tribello, G. A., Bonomi, M., Branduardi, D., Camilloni, C., and Bussi, G. (2014). PLUMED 2: New Feathers for an Old Bird. Comp. Phys. Commun. 185, 604–613. doi:10.1016/j.cpc.2013.09.018
Vanommeslaeghe, K., and MacKerell, A. D. (2012). Automation of the CHARMM General Force Field (CGenFF) I: Bond Perception and Atom Typing. J. Chem. Inf. Model. 52, 3144–3154. doi:10.1021/ci300363c
Vanommeslaeghe, K., Raman, E. P., and MacKerell, A. D. (2012). Automation of the CHARMM General Force Field (CGenFF) II: Assignment of Bonded Parameters and Partial Atomic Charges. J. Chem. Inf. Model. 52, 3155–3168. doi:10.1021/ci3003649
Venkatakrishnan, A. J., Ma, A. K., Fonseca, R., Latorraca, N. R., Kelly, B., Betz, R. M., et al. (2019). Diverse GPCRs Exhibit Conserved Water Networks for Stabilization and Activation. Proc. Natl. Acad. Sci. USA 116, 3288–3293. doi:10.1073/pnas.1809251116
Wootten, D., Reynolds, C. A., Smith, K. J., Mobarec, J. C., Furness, S. G. B., Miller, L. J., et al. (2016). Key Interactions by Conserved Polar Amino Acids Located at the Transmembrane Helical Boundaries in Class B GPCRs Modulate Activation, Effector Specificity and Biased Signalling in the Glucagon-like Peptide-1 Receptor. Biochem. Pharmacol. 118, 68–87. doi:10.1016/j.bcp.2016.08.015
Wootten, D., Simms, J., Miller, L. J., Christopoulos, A., and Sexton, P. M. (2013). Polar Transmembrane Interactions Drive Formation of Ligand-specific and Signal Pathway-Biased Family B G Protein-Coupled Receptor Conformations. Proc. Natl. Acad. Sci. 110, 5211–5216. doi:10.1073/pnas.1221585110
Wu, F., Yang, L., Hang, K., Laursen, M., Wu, L., Han, G. W., et al. (2020). Full-length Human GLP-1 Receptor Structure without Orthosteric Ligands. Nat. Commun. 11, 1272. doi:10.1038/s41467-020-14934-5
Yang, L., Yang, D., de Graaf, C., Moeller, A., West, G. M., Dharmarajan, V., et al. (2015). Conformational States of the Full-Length Glucagon Receptor. Nat. Commun. 6, 7859. doi:10.1038/ncomms8859
Yin, Y., de Waal, P. W., He, Y., Zhao, L.-H., Yang, D., Cai, X., et al. (2017). Rearrangement of a Polar Core Provides a Conserved Mechanism for Constitutive Activation of Class B G Protein-Coupled Receptors. J. Biol. Chem. 292, 9865–9881. doi:10.1074/jbc.M117.782987
Yu, W., He, X., Vanommeslaeghe, K., and MacKerell, A. D. (2012). Extension of the CHARMM General Force Field to Sulfonyl-Containing Compounds and its Utility in Biomolecular Simulations. J. Comput. Chem. 33, 2451–2468. doi:10.1002/jcc.23067
Zhang, X., Belousoff, M. J., Zhao, P., Kooistra, A. J., Truong, T. T., Ang, S. Y., et al. (2020). Differential GLP-1R Binding and Activation by Peptide and Non-peptide Agonists. Mol. Cel 80, 485–500. doi:10.1016/j.molcel.2020.09.020
Keywords: GPCR, CGRPR, CGRP, telcagepant, binding, molecular dynamics, supervised molecular dynamics
Citation: Deganutti G, Atanasio S, Rujan R-M, Sexton PM, Wootten D and Reynolds CA (2021) Exploring Ligand Binding to Calcitonin Gene-Related Peptide Receptors. Front. Mol. Biosci. 8:720561. doi: 10.3389/fmolb.2021.720561
Received: 04 June 2021; Accepted: 13 August 2021;
Published: 26 August 2021.
Edited by:
Antonella Di Pizio, Technical University of Munich, GermanyReviewed by:
Hugo Gutiérrez De Teran, Uppsala University, SwedenTomasz Stepniewski, Institute of Metallurgy and Materials Science (PAN), Poland
Antonios Kolocouris, National and Kapodistrian University of Athens, Greece
Copyright © 2021 Deganutti, Atanasio, Rujan, Sexton, Wootten and Reynolds. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giuseppe Deganutti, YWQ1Mjg4QGNvdmVudHJ5LmFjLnVr; Christopher A. Reynolds, YWQ1MjkxQGNvdmVudHJ5LmFjLnVr