 Sandra Torres1†
Sandra Torres1† Maximilian J Brol1†
Maximilian J Brol1† Fernando Magdaleno2†
Fernando Magdaleno2† Robert Schierwagen1
Robert Schierwagen1 Frank E. Uschner1
Frank E. Uschner1 Sabine Klein1
Sabine Klein1 Cristina Ortiz1
Cristina Ortiz1 Olaf Tyc1
Olaf Tyc1 Nadine Bachtler1James Stunden3
Nadine Bachtler1James Stunden3 Damien Bertheloot3,4Ana Kitanovic3Brian Sanchez5Jacob Schrum3William R. Roush3Luigi Franchi5Kate Byth3Eicke Latz3,4
Damien Bertheloot3,4Ana Kitanovic3Brian Sanchez5Jacob Schrum3William R. Roush3Luigi Franchi5Kate Byth3Eicke Latz3,4 Jonel Trebicka1,6*
Jonel Trebicka1,6*- 1Translational Hepatology, Department of Internal Medicine I, Universitätsklinikum/ Goethe-Universität, Frankfurt, Germany
- 2Department of Internal Medicine I, University Clinic Bonn, Bonn, Germany
- 3IFM Therapeutics, Boston, MA, United States
- 4Institute of Innate Immunity, University Clinic Bonn, Bonn, Germany
- 5IFM Therapeutics, Ann Arbor, MI, United States
- 6European Foundation for the Study of Chronic Liver Failure – EF Clif, Barcelona, Spain
Background and Aims: Activation of the inflammasome NLRP3 (NOD-, LRR- and pyrin domain containing 3) contributes to the development of non-alcoholic fatty liver disease (NAFLD) and progression to non-alcoholic steatohepatitis (NASH). Therefore, this study explored the therapeutic effects of a novel and selective NLRP3 antagonist in a murine dietary model of NASH.
Methods: Groups of 12-week-old ApoE-/- mice were fed ad lib for 7 weeks with a methionine/choline deficient (MCD) and western diet (WD). After 3 weeks of diet-induced injury, mice were injected i. p. with the NLRP3 antagonist IFM-514 (100 mg/kg body weight) or vehicle (0.5% carmellose) every day, 5 days/week for a further 4 weeks. Several markers of inflammation, fibrosis and steatosis were evaluated. Whole transcriptome sequencing and panel RNA expression analysis (NanoString) were performed.
Results: IFM-514 inhibited IL-1β production in mice challenged with 20 mg/kg lipopolysaccharide, and in mouse and human inflammatory cells in vitro. IFM-514 inhibited hepatic inflammation in the in vivo non-alcoholic steatohepatitis model assessed by H&E staining and in the hepatic gene expression of inflammasome-related proinflammatory cytokines. This effect was associated with significant reduction in caspase-1 activation. Similarly, IFM-514 was efficacious in vivo in MDC-fed ApoE-/- mice, markedly reducing portal pressure, Sirius red staining and 4-hydroxyproline content compared to vehicle-treated mice. Moreover, IFM-514 significantly reduced hepatic steatosis in MCD-fed ApoE-/- mice, as evidenced by NAFLD scores, oil red O staining, hepatic triglycerides and gene expression. In WD treated animals, similar trends in inflammation and fibrosis were observed, although not sufficient IFM-514 levels were reached.
Conclusion: Overall, IFM-514 reduced liver inflammation and fibrosis, with mild effects on liver steatosis in experimental murine NASH. Blocking of NLRP3 may be an attractive therapeutic approach for NASH patients.
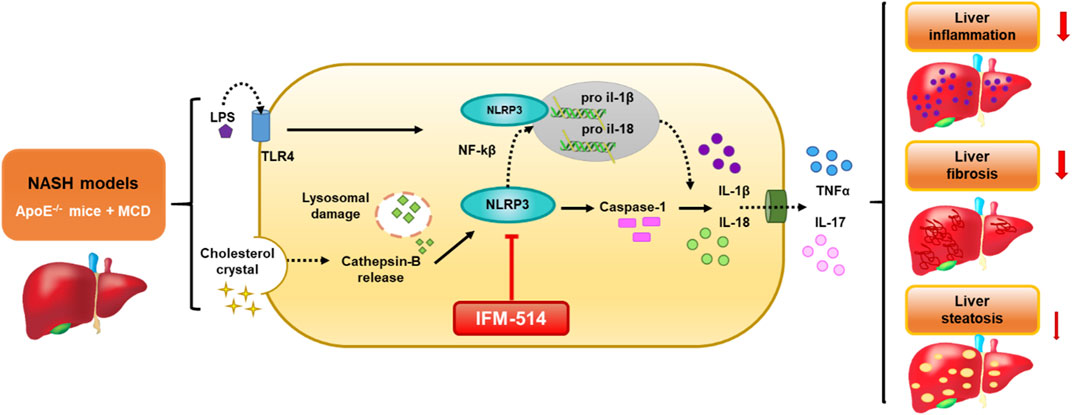
GRAPHICAL ABSTRACT. Schematic representation of the IFM-514 treatment effect in experimental murine NASH. The activation of NLRP3 inflammasome in response to NASH models is due to the gut-derived PAMPs, such as LPS, and cholesterol crystals. LPS activates nuclear factor kappa B (Nf-kB) via triggering of the TLR4 receptor and consequently promotes the expression of pro-IL-1β and pro-IL-18. Cholesterol crystals produce lysosomal damage with cathepsin-B release that also activates NLRP3, followed by caspase-1 activation that allows the release of IL-1β and IL-18 into the extracellular space and increasing TNF-α and IL-17. All these cytokines promote an increase in liver inflammation, fibrosis and steatosis. In our study, IFM-514, an NRLP3 antagonist, could protect prior to the pathogenesis produced by NASH murine model, with mild effect in liver steatosis.
Highlights
⁃ IFM-514 reduced liver inflammation and fibrosis in MCD-fed ApoE-/- mice.
⁃ IFM-514 produced a mild reduction in liver steatosis in MCD-fed ApoE-/- mice.
⁃ Blocking NLRP3 might be an attractive therapeutic approach for NASH patients.
Lay Summary
The activation of NLRP3 inflammasome in response to NASH models is due to the gut-derived PAMPs, such as LPS, and cholesterol crystals. LPS activates nuclear factor kappa B (Nf-kB) via triggering of the TLR4 receptor and consequently promotes the expression of pro-IL-1β and pro-IL-18. Cholesterol crystals produce lysosomal damage with cathepsin-B release that also activates NLRP3, followed by caspase-1 activation that allows the release of IL-1β and IL-18 into the extracellular space and increasing TNF-α and IL-17. All these cytokines promote an increase in liver inflammation, fibrosis and steatosis. In our study, IFM-514, an NRLP3 antagonist, could protect prior to the pathogenesis produced by NASH murine model, with mild effect in liver steatosis.
Introduction
Non-alcoholic fatty liver disease (NAFLD), characterized by fat accumulation in the liver (steatosis) in the absence of chronic alcohol use, is a common and emerging cause of chronic liver disease (Williams et al., 2011; Rinella, 2015). Interestingly, NAFLD is present in obese as well as lean patients. However, little is known about the differences between these two NAFLD types and thus, no tailored therapeutic approaches are available. Moreover, an estimated 10–15% of NAFLD patients develop non-alcoholic steatohepatitis (NASH), while a quarter of those will develop liver cirrhosis and potentially hepatocellular carcinoma (Macaluso et al., 2015). Therefore, NASH represents the severe and dangerous form of NAFLD characterized by hepatocyte injury, inflammation and fibrosis - the most critical outcome in NASH - , that may result from inflammasome activation (Ganz et al., 2015).
Inflammasomes are cytoplasmic multiprotein complexes that can sense danger signals from damaged cells and pathogens. They assemble to mediate caspase-1 activity, secretion of cytokines and other pro-inflammatory mediators, including IL-1β and IL-18 (Martinon et al., 2002; Schroder and Tschopp, 2010; Szabo and Petrasek, 2015), as a result of tissue damage or cellular stress. Several members of the NLR family (nucleotide-binding and oligomerization domain and leucine-rich-repeat-containing proteins), including the NOD-like receptor protein 3 (NLRP3), have been linked to the pathophysiology of NASH (Abstract diagram). This has inspired strategies to block inflammasome activation by pharmacological targeting of NLRP3 (Próchnicki et al., 2016). In fact, inhibition of NLRP3 can be achieved by limiting Toll-like receptor- and tumor necrosis factor-mediated increases in NLRP3 expression. However, since this approach lacks specificity and is likely to produce many off-target effects (Bahia et al., 2015), this study tested a novel selective NLRP3 antagonist for therapeutic effects in murine models of NASH. Since both NASH and NLRP3 have been linked to metabolic syndrome we have chosen murine models for which we have previously demonstrated that it is suitable for investigation of liver of metabolic syndrome (Schierwagen et al., 2015, 2016).
Results
IFM-514 Inhibits the NLRP3 Inflammasome In Vivo and In Vitro
In human cells, IFM-514 inhibited gramicidin-induced IL-1β release in PMA-primed human THP-1 cells (half maximal inhibitory concentration, IC50 0.275 ± 0.115 µM, n = 9) and was a potent inhibitor of gramicidin-induced IL-1β release in LPS-primed monocyte-derived macrophages (IC50 0.156 ± 0.042 µM, n = 8). Similarly, IFM-514 was a potent inhibitor of gramicidin-induced IL-1β release in LPS-primed immortalized macrophages from WT C57BL/6 mice (IC50 0.146 ± 0.042 µM, n = 6), displayed similar potency in primary bone marrow-derived macrophages (BMDM) from C57BL/6 mice (data not shown) and in BMDM from Balb/c mice (IC50 0.313 ± 0.013 µM, n = 6; free IC50 10.9 ± 0.5 nM. To determine the recommended dose in vivo, WT C57BL/6 mice were used in a pharmacodynamic (PD) model of lipopolysaccharide (LPS) mediated cytokine release. IFM-514 was dosed orally in mice at 0.1, 1, 10 and 100 mg/kg 1 h before LPS challenge (20 mg/kg i. p.) and plasma cytokines were measured 5 h later by ELISA (Figure 1A). IFM-514 strongly and dose-dependently abrogated the LPS-induced production of NLRP3-dependent plasma cytokines IL-1β and IL-18, and had a mild but significant effect on TNFα and IL-6 at the highest dose tested (Figures 1B–E). Moreover, the 100 mg/kg dose of IFM-514 inhibited IL-1β production in vivo by more than 90%, even when dosed orally. The plasma concentration of IFM-514 was measured at the time of cytokine measurement and plotted against the percentage inhibition of IL-1β production, resulting in an IC50 of 947 ng/ml (95% CI 499.5–1,683 ng/ml) (Figure 1F). IFM-514 was highly protein bound in plasma (99.59% bound), to an extent that, when adjusted for plasma protein binding, the free IC50in vivo was 3.9 ng/ml or 9.2 nM, which corresponded with the free in vitro IC50 in mouse BMDM. Consistently, single-dose pharmacokinetic (PK) studies in WT C57BL/6 mice suggested that the recommended dose for in vivo use was 100 mg/kg free base (corresponding to 105 mg/kg Na + salt) when delivered i. p. or orally once-per-day (Supplementary Table 1). Using modeled PK data, this dose gave an average free liver concentration above the free in vivo IC90 in the LPS challenge model (83 nM) and the free in vitro IC90 in the BMDM assay (40.0 ± 3.5 nM) for approximately 24 h. Therefore, this dose was used for the in vivo experiments in NASH-induced ApoE-/- mice (Table 1).
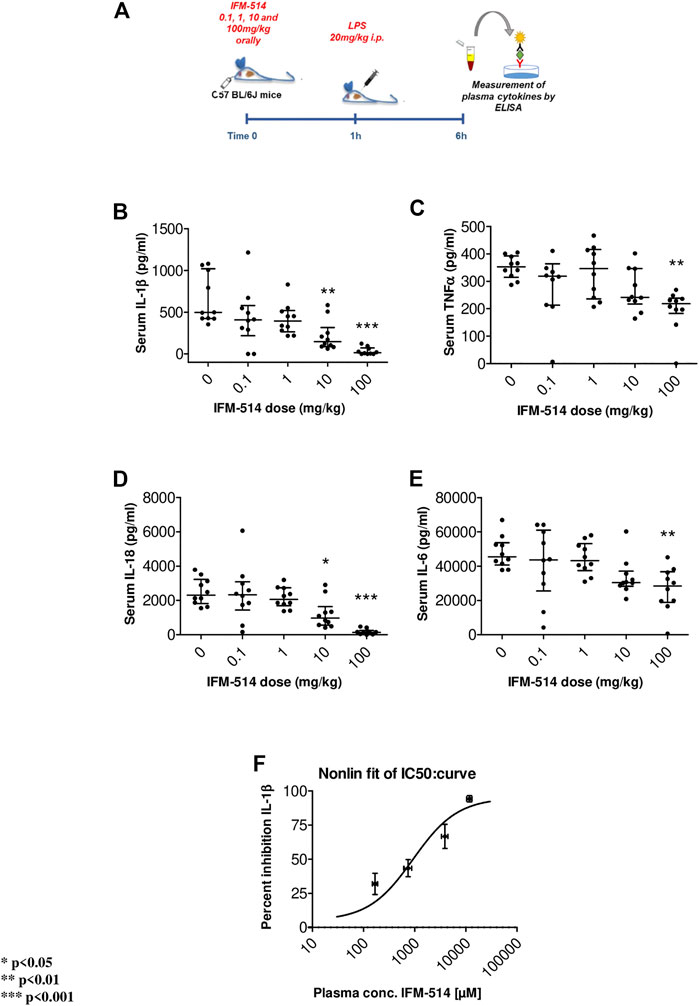
FIGURE 1. Experimental design for mouse in vivo pharmacodynamics model: LPS stimulation of plasma cytokines. IFM-514 was dosed orally in WT C57BL/6 mice at 0.1, 1, 10 and 100 mg/kg 1 h before LPS (20 mg/kg i. p.) (A). LPS-induced production of plasma cytokines IL-1β(B), TNFα(C), IL-18 (D) and IL-6 (E) measured by ELISA 5 h after LPS challenge. IFM-514 significantly inhibited IL-1β production in vivo when dosed orally with an IC50 = 947 ng/ml; 95% CI 499.5–1,683 ng/ml (F). Results are expressed as mean ± standard error of the mean (SEM); n = 10/group, *p < 0.05, **p < 0.01 and ***p < 0.001. Abbreviations: ELISA, enzyme-linked immunosorbent assay; IC50, half maximal inhibitory concentration; IL1β, interleukin 1β; IL-16, interleukin 16; IL18, interleukin 18; LPS, lipopolysaccharide; TNFα, tumor necrosis factor α.
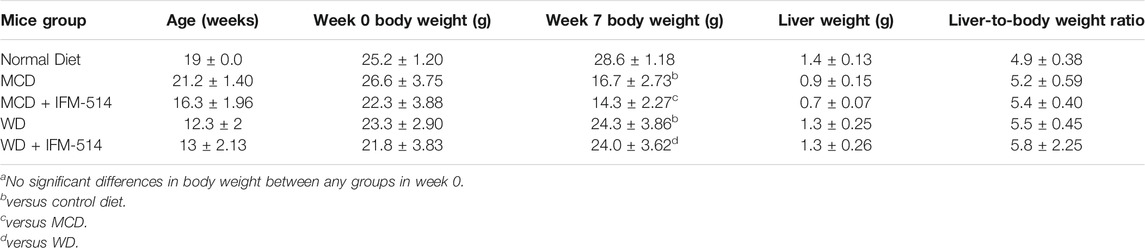
TABLE 1. Age, body weight (week 0 and 7), liver weight and liver-to-body weight ratio of ApoE-/- mice receiving normal diet, MCD, WD and IFM-514-treated MCD-fed and WD-fed mice.
Here, 12-week-old ApoE-/- mice were fed for a total of 7 weeks with MCD diet. After 3 weeks of diet-induced injury, mice received i. p. injections of IFM-514 (100 mg/kg) or vehicle for 5 days/week for a further 4 weeks (Figure 2A). To determine whether the liver was adequately exposed to IFM-514, the concentration of IFM-514 was measured by HPLC-MS in liver and serum 8 hours after the last dose of IFM-514 in MCD-fed ApoE-/- mice (Figures 2B,C). The free IFM-514 concentration in the liver was above the free plasma concentration and sufficient to inhibit 90% of the IL-1β synthesis in the LPS-PK/PD model. IFM-514-treated NASH ApoE-/- mice had similar circulating IL-1β and IL-1α as vehicle-treated NASH ApoE-/- mice (Figures 2D,E).
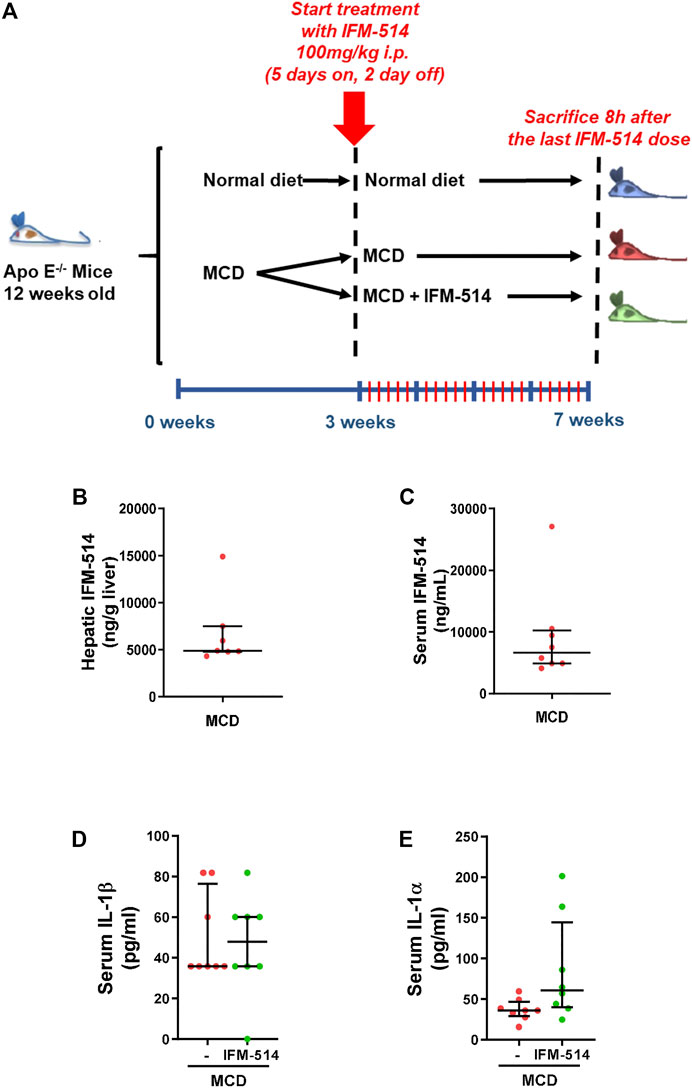
FIGURE 2. Study design and interleukin levels. Groups (n = 10) of 12-week-old ApoE-/- mice were fed ad lib for 7 weeks with methionine/choline deficient (MCD) diet. After 3 weeks of diet-induced injury, mice were injected i. p. with the NLRP3 antagonist IFM-514 (100 mg/kg body weight) or vehicle (0.5% carmellose) every day, 5 days/week for a further 4 weeks (A). Concentration of IFM-514 in liver (ng/g of tissue) and serum (ng/ml) following i. p. administration of IFM-514 in NASH-induced mice for 20 consecutive days. The concentration was measured by HPLC-MS (B–C). Circulating IL-1β and IL-1α in IFM-514-treated MCD-fed ApoE-/- mice (D–E). Results are expressed as mean ± standard error of the mean (SEM); n = 10/group, *p < 0.05, **p < 0.01 and ***p < 0.001. Abbreviations: IL1α, interleukin 1α.
IFM-514 Reduces Hepatic Inflammation in MCD-Fed ApoE-/- Mice
Since NLRP3-mediated inflammation is linked to NASH (Mridha et al., 2017), we sought to test whether IFM-514 has an effect in MCD-fed ApoE-/- mice. Hematoxylin and eosin (H&E) staining (Figure 3A), inflammation score (Figure 3B) and NAS scores (Figure 3C) showed that IFM-514 reduces hepatic inflammation in MCD-fed ApoE-/- mice, being significant the NAS score. Liver samples were further processed to analyze the activation of caspase-1 and IL-1β protein expression, a pathway that is extremely important in NASH (Szabo and Petrasek, 2015). Protein analysis showed that the activation of p20 and p33-caspase-1 subunits and pro-IL-1β were significantly reduced in IFM-514-treated vs vehicle-treated MCD-fed ApoE-/- mice, but mature IL-1β and pro-caspase-1 showed a tendency to decrease with IFM-514-treatment (Figure 3D). To further pinpoint the mechanism of IFM-514 in hepatic inflammation, liver RNA was extracted for NanoString mouse myeloid innate immunity panel of 770 genes. MCD diet induced upregulation of genes at least 1.5-fold, and some of these genes were clearly reduced in IFM-514-treated vs vehicle-treated NASH ApoE-/- mice. This set of genes included several pro-inflammatory genes, such as chemokine ligands (Ccl) 1, 4 and 28, Il10 and Tnfα (Figure 3E), also shown in the heatmap (Figure 3F). In agreement with these data, IFM-514 had a pronounced effect on cytokine and inflammasome-related genes in the MCD-fed mice. Moreover, IFM-514 clearly reduced the hepatic expression of many genes related to the cytokine-cytokine receptor interaction (C-C) pathway, which is not limited to inflammasome function, but inflammation in general. These results suggest that IFM-514 inhibits the NLRP3 inflammasome and the subsequent activation of caspase-1 in MCD-fed ApoE-/- mice. The effect of IFM-514 decreasing liver inflammation was supported by immunohistochemistry of hepatic macrophages and activated kuffer cells (F4/80-positive cells) (Supplementary Figure 1), such as the reduction of Adgre1 expression indicated in the heatmap (Figure 3F).
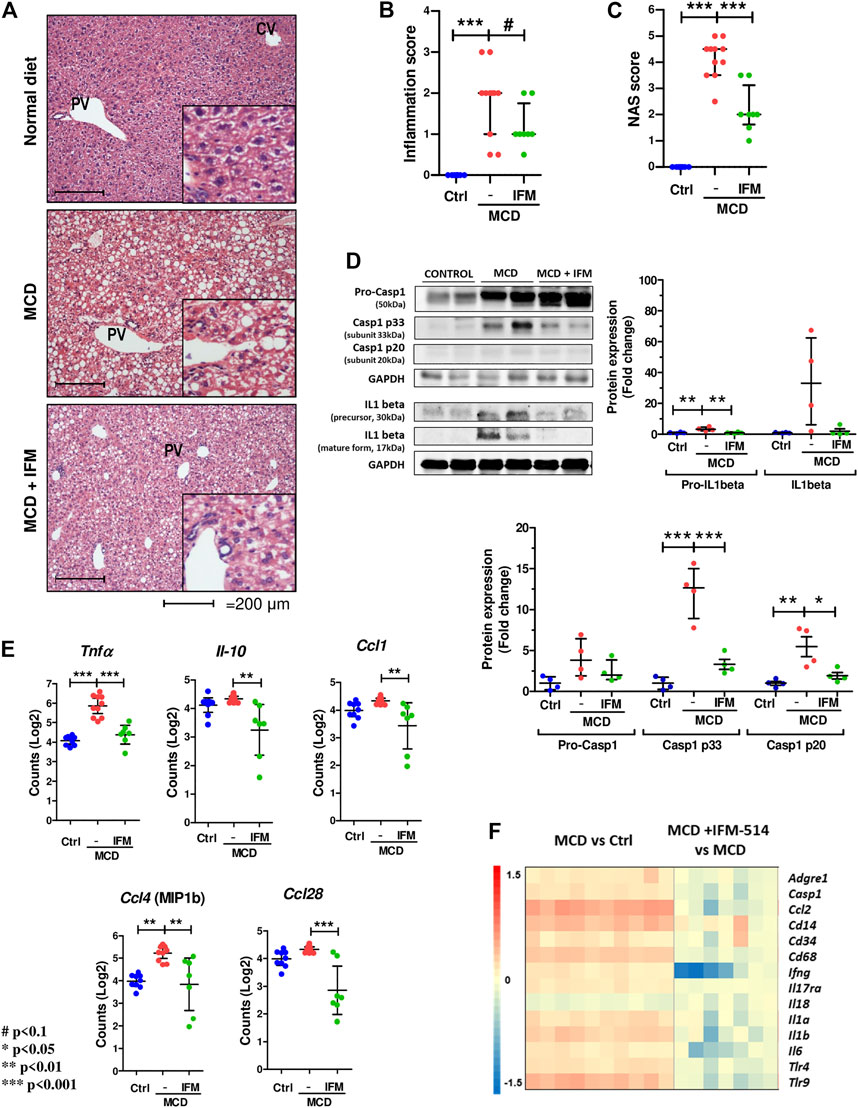
FIGURE 3. IFM-514 reduces hepatic inflammation in MCD-fed ApoE-/- mice. H&E staining (A) inflammation score (B) and NAS score (C) in IFM-514-treated MCD-fed ApoE-/- mice. Caspase-1 p33, caspase-1 p20, pro-caspase-1, mature IL-1β and pro-IL-1β protein expression (D). Inflammasome-related gene expression (E). Heatmap for hepatic inflammation set of genes (F). Scale and genes are provided on heatmap. Results are expressed as the mean ± standard error of the mean (SEM); n = 10/group, #p < 0.1. *p < 0.05, **p < 0.01 and ***p < 0.001. Representative photomicrographs were captured at ×100 (scale bars = 200 μm) and ×200 magnification (scale bars = 100 μm). Abbreviations: Ccl1, chemokine (C-C motif) ligand 1; Ccl4, chemokine (C-C motif) ligand 4; Ccl28, chemokine (C-C motif) ligand 28; CV, central vein; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; H&E, hematoxylin and eosin; Il-10, interleukin 10; PV, portal vein. See Supplementary Table 3 for Heatmap genes.
IFM-514 Reduces Hepatic Fibrosis in MCD-Fed ApoE-/- Mice
Given that MCD feeding induced hepatic fibrosis in ApoE-/- mice, we tested whether the inhibition of NLRP3 with IFM-514 had an effect on markers of fibrosis. IFM-514 treatment significantly reduced liver fibrosis as shown by quantitative Sirius red (SR) staining (Figures 4A,B), and the 4-hydroxyproline content (Figure 4C). Collagen type 1 α1 chain (Col1a1) protein expression (Figure 4D) had a significant reduction and hepatic mRNA expression levels (Figure 4E) also had a tendency towards reduction in MCD-fed ApoE-/- mice treated with IFM-514. In addition, NanoString gene expression analyses indicated that several fibrotic genes that were increased by MCD feeding were decreased by treatment with IFM-514, as shown in the heatmap (Figure 4F). Taken together, these data indicate that IFM-514 treatment markedly reduces key players in liver fibrosis.
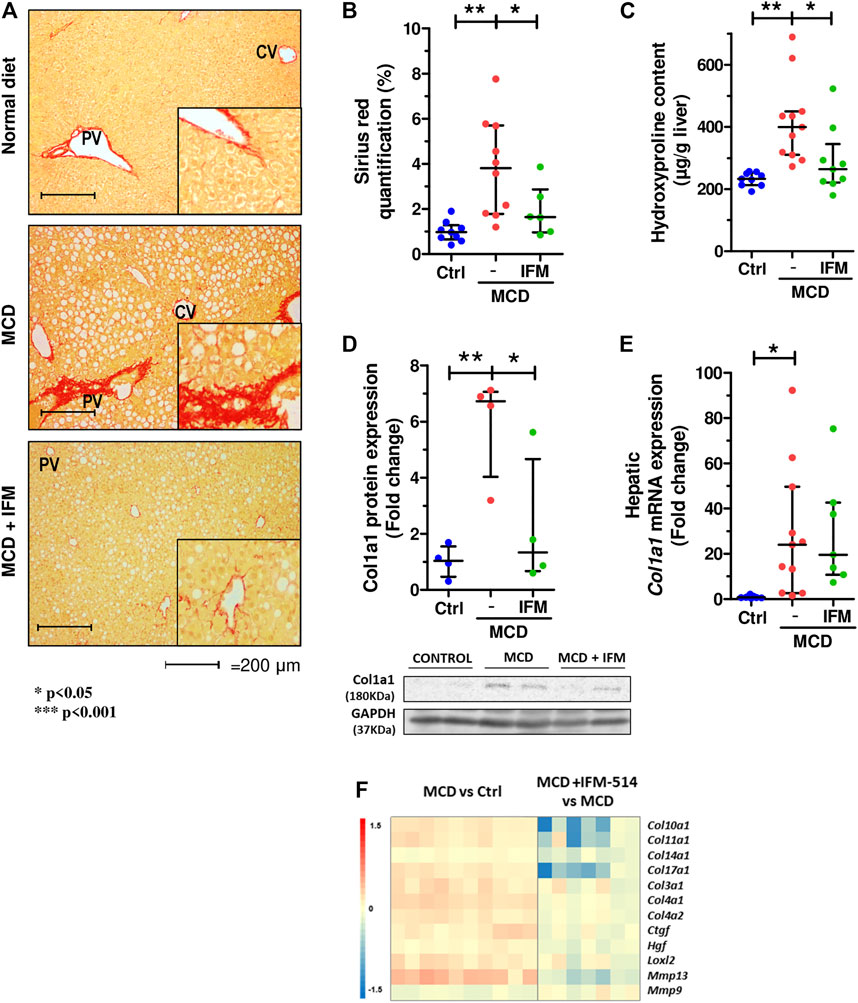
FIGURE 4. IFM-514 reduces liver fibrosis in MCD-fed ApoE-/- mice. Sirius red (SR) staining (A) with SR quantification (B) and hepatic 4-hydroxyproline content (C). Hepatic Col1a1 protein (D) and mRNA expression (E) from IFM-514-treated MCD-fed ApoE-/- mice. Heatmap for hepatic fibrosis set of genes (F). Results are expressed as mean ± standard error of the mean (SEM); n = 10/group, #p < 0.1, *p < 0.05, **p < 0.01 and ***p < 0.001. $p < 0.05, if outliers are excluded. Scale bars = 200 μm. Abbreviations: col1a1, collagen type 1 α1. See Supplementary Table 3 for Heatmap genes.
Hepatic Stellate Cell Activation and Portal Hypertension in MCD-Fed ApoE-/- Mice
We next examined hepatic stellate cell activation (HSC) as an important mechanism for development of portal hypertension in addition to fibrosis (Abdulla et al., 2014), using quantitative immunostaining for α-smooth muscle actin (Acta2) (Figures 5A,B). Activation of HSC in MCD-fed mice was increased compared to controls, while IFM-514 treatment showed a tendency towards reduced αSMA expression. Similarly, the mRNA expression level of Acta2 had a decreasing trend in IFM-treated MCD-fed ApoE-/- mice (Figure 5C). Portal pressure-measured in the spleen pulp-was significantly reduced in IFM-514-treated MCD-fed ApoE-/- mice (Figure 5D). Genes of key players in portal hypertension and activation of HSC were analyzed by NanoString gene expression (see heatmap, Figure 5E), showing a reduction of these genes with IFM-514 treatment.
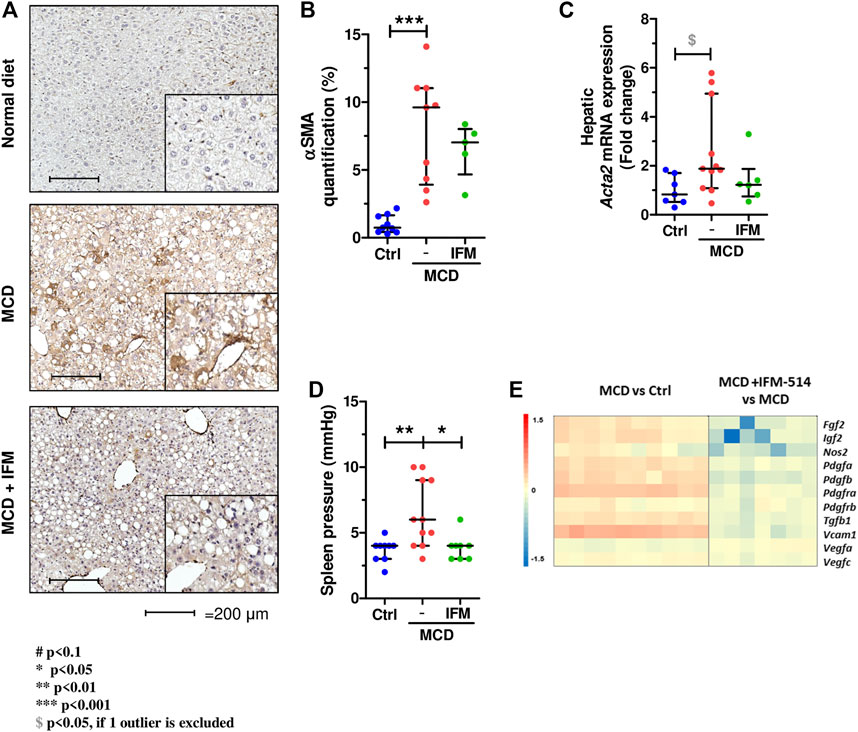
FIGURE 5. Hepatic stellate cell activation in MCD-fed ApoE-/- mice. α-smooth muscle actin immunostaining (A) with quantification (B) from IFM-514-treated MCD-fed ApoE-/- mice. Acta2 mRNA expression (C). Spleen pressure analyses (D). Heatmap for portal hypertension set of genes (E). All mRNA data were normalized to the expression of 18s. Results are expressed as mean ± standard error of the mean (SEM); n = 10/group, #p < 0.1, *p < 0.05, **p < 0.01 and ***p < 0.001. $p < 0.05, if outliers are excluded. Scale bars = 200 μm. Abbreviations: CV, central vein; PV, portal vein; acta2, α-smooth muscle actin gene; αSMA, α-smooth muscle actin. See Supplementary Table 3 for Heatmap genes.
IFM-514 Reduces Hepatic Steatosis in MCD-Fed ApoE-/- Mice
The steatosis score assessed by a liver pathologist demonstrated a significant reduction in mice treated with IFM-514 compared to vehicle treated mice (Figure 6A). Similarly, analysis of hepatic steatosis using quantitative hepatic triglycerides and oil red O staining revealed a trend towards a reduction in IFM-treated mice, although this was not significant, likely due to the persistence of numerous oil red O-positive small lipid droplets (Figures 6B–D). Similar trends were seen on protein and mRNA expression levels of central regulators of lipid homeostasis, sterol regulatory element binding factor 1 (Srebf1) and fatty acid synthase (Fasn), being significant for Fasn gene expression (Dinarello et al., 2012) (Figures 6E–G). The heatmap of the steatosis selected genes confirms the effect of IFM-514 treatment in MCD-fed mice (Figure 6H).
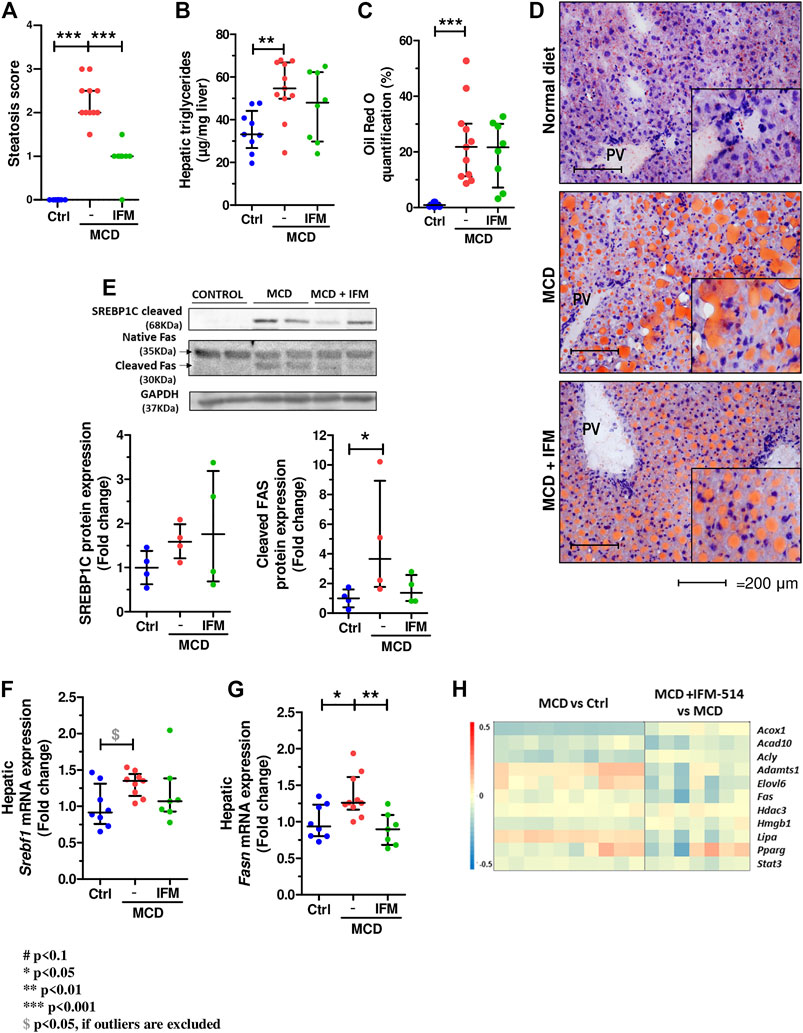
FIGURE 6. IFM-514 reduces hepatic steatosis in MCD-fed ApoE-/- mice. Steatosis score (A). Hepatic triglycerides levels (B). Oil red O staining and quantification as shown as percentage of the positive area (C–D). Srebp1c, native Fas and cleaved Fas protein (E) and mRNA expression (F–G) from IFM-514-treated MCD-fed ApoE-/- mice. Heatmap for hepatic steatosis set of genes (H). All mRNA data were normalized to the expression of 18s. Results are expressed as the mean ± standard error of the mean (SEM); n = 10/group, *p < 0.05, **p < 0.01 and ***p < 0.001. Abbreviations: srebp1c; sterol regulatory element-binding protein 1c; srebf1, sterol regulatory element-binding transcription factor 1; fas, fatty acid synthase. See Supplementary Table 3 for Heatmap genes.
IFM-514 Reduces Hepatic Gene Expression of Inflammasome-Related and Proinflammatory Cytokines in MCD-Fed ApoE-/- Mice
To assess the extent of the genes differentially expressed following IFM-514 treatment in MCD-fed ApoE-/- mice, we analyzed the sets of genes upregulated in MCD-fed ApoE-/- mice (Figures 7A–C). To further characterize the upregulated pathways, we performed gene ontology (GO) analysis of all significantly upregulated genes in MCD-fed ApoE-/- mice. In biological processes, we found a one- to eight-fold enrichment in inflammation-related GO terms, such as regulation of tumor necrosis factor superfamily cytokine production and inflammatory response (Figure 7A). Through Venn analyses, a total of 236 genes were significantly upregulated in MCD-fed ApoE-/- mice (Figure 7B). Venn analyses revealed that a total of 126 genes specifically upregulated by MCD diet were found to be downregulated by IFM-514 (Figure 7B; Table 2). To examine the transcriptomic inflammatory signature in response to the IFM-514 treatment, we compared the sets of genes which were downregulated following IFM-514 treatment in MCD-fed ApoE-/- mice. GO analysis revealed enrichment for several genes related to calcium-mediated signaling, G protein-coupled receptor signaling pathway, inflammatory and immune responses in IFM-514-treated MCD-fed ApoE-/- mice (Figure 7C).
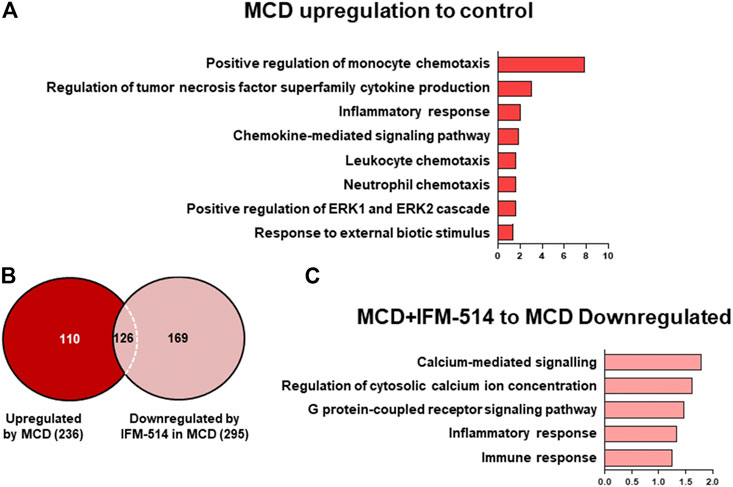
FIGURE 7. Transcriptomic analysis of IFM-514-treated MCD-fed ApoE-/- mice. Gene ontology (GO) analysis and fold enrichment of genes of biological process upregulated in MCD-fed (A). Venn diagram of downregulated genes in IFM-514-treated MCD-fed (B). Enriched GO terms for genes downregulated in IFM-514-treated MCD-fed (C). The signaling pathways are based on statistical significance. Genes were considered significantly down expressed (p < 0.05) in IFM-treated mice by a minimum of 1.5 fold above that of vehicle-treated MCD-fed ApoE-/- mice.
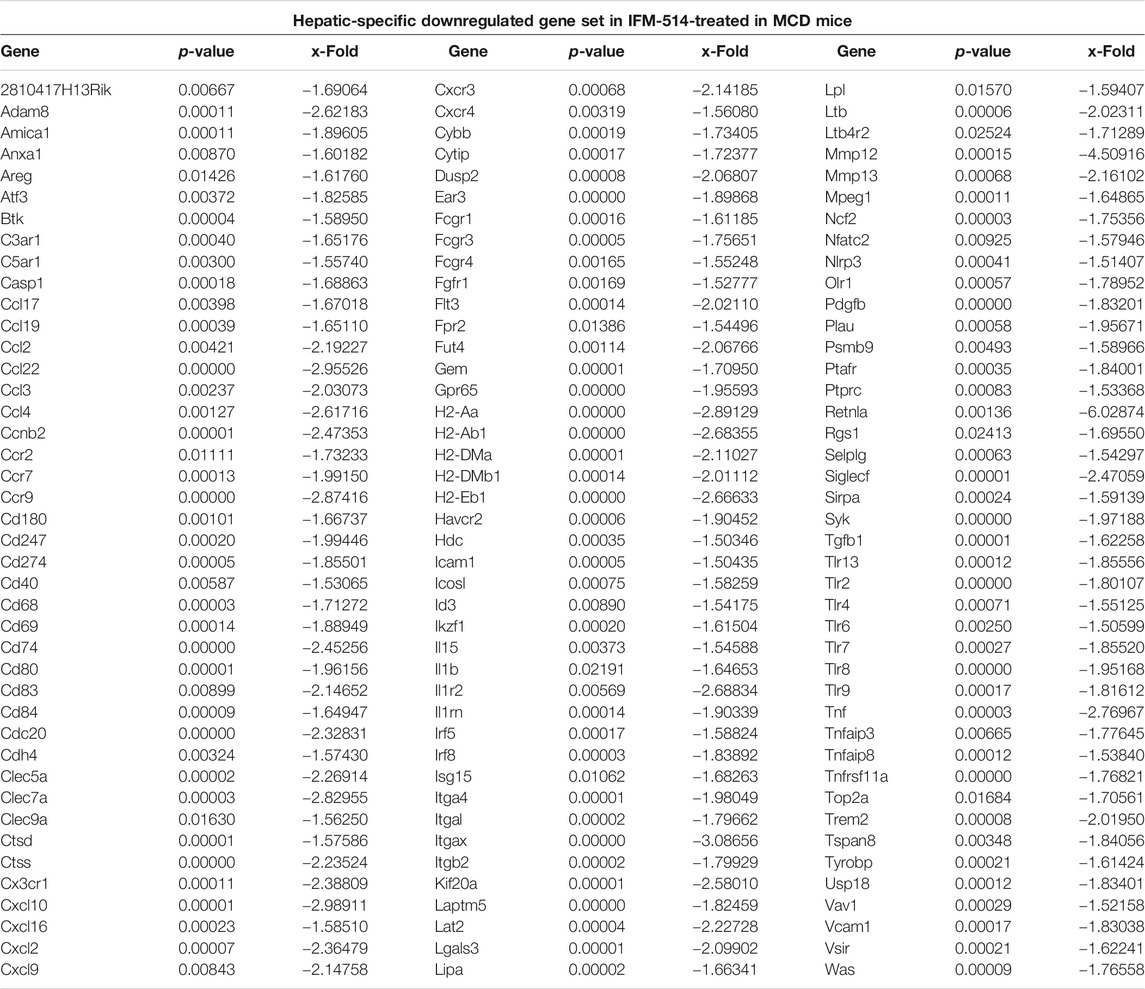
TABLE 2. IFM-514 changes the hepatic gene expression of inflammasome-related and proinflammatory cytokines in MCD-fed ApoE-/- mice. Table shows all genes significantly downregulated in IFM-514-treated MCD-fed mice.
Thus, IFM-514 clearly reduced the expression of several genes specifically upregulated in the MCD model, not limited to inflammasome function, but related to inflammation in general. All in all, these results strongly support that the primary targets of the NLRP3 blockade are liver inflammation and fibrosis, with a mild effect on hepatic steatosis in MCD-fed ApoE-/- mice.
IFM-514 Effect on Hepatic Inflammation and Fibrosis in WD-Fed ApoE-/- Mice
Since MCD-fed mice recapitulate the histology of human NAFLD (hepatic steatosis, and fibrosis) but lack the metabolic component (obesity, high cholesterol and diabetes) to assess the impact of metabolic imbalances in NLRP3 activation in NASH, ApoE-/- mice were fed with WD. 12-week-old ApoE-/- mice were fed for a total of 7 weeks with WD diet. After 3 weeks of diet-induced injury, mice received i. p. injections of IFM-514 (100 mg/kg) or vehicle for 5 days/week for a further 4 weeks (Supplementary Figure 2A). IFM-514 was measured by HPLC-MS in liver and serum 8 h after the last dose of IFM-514 in WD-fed ApoE-/- mice which was rather low compared to MCD fed ApoE-/- (Supplementary Figures 2B–C). IFM-514-treated NASH ApoE-/- mice had similar circulating IL-1β and IL-1α as vehicle-treated NASH ApoE-/- mice (Supplementary Figures 2D–E).
We further investigated the effect of IFM-514 treatment on inflammation and fibrosis in WD-fed ApoE-/- mice. No difference in inflammation score between ApoE-/- and WD fed ApoE-/- mice was found (Supplementary Figure 3B), but WD induced an increase in NAS score (Supplementary Figure 3C). In addition, WD significantly increased Tnf-α gene expression (Supplementary Figure 3E) while IFM-514 showed a tendency towards reduction in some inflammatory genes, being significant for Ccl1 and Ccl28 (Supplementary Figures 3F–G). Liver fibrosis was assessed by sirius red staining and hepatic hydroxyproline content, which also tend to decrease by the IFM-514 treatment (Supplementary Figures 3A,C,E). Activation of hepatic stellate cells assessed by α-sma staining (Supplementary Figures 4B,D), was significantly increased in WD fed ApoE-/- mice, and showed a tendency to decrease after the IFM-514. Similar effects showed spleen pressure (Supplementary Figure 4F) and the heatmaps of fibrotic and portal hypertension related genes (Supplementary Figure 4G–H).
Discussion
Currently, therapies targeting NLRP3-dependent cytokines, i.e. canakinumab, an IL-1β-neutralizing antibody, and rilonacept, a soluble receptor that binds IL-1β and IL-1α, are applied in humans. However, they have notable immunosuppressive disadvantages compared to selective NLRP3 antagonism (Dinarello et al., 2012; Ridker et al., 2017). This is of particular relevance when considering that in a targeted anti-NLRP3 therapy, other pathogen-recognizing inflammasomes can be engaged to produce IL-1β, thus reducing the risk of immune suppression and opportunistic infections. Supporting this idea, similar concentrations of circulating IL-1β and IL-1α detected in IFM-514-treated vs untreated NASH mice might lower the risk of immunosuppressive effects of this anti-inflammasome therapy, rendering the IFM-514 treatment very suitable in NASH.
A recent study investigated the effect of the NLRP3 antagonist CP-456,773 (Jiang et al., 2017) (also called MCC950 or CRID3) on the development of NASH in dietary models (Mridha et al., 2017). Similar to our data, CRID3 caused a significant reduction in hepatic infiltration of macrophages and neutrophils and modulated fibrotic progression of steatohepatitis in MCD-fed as well as foz/foz mice fed with atherogenic diet. These effects correlated with a significant reduction of NLRP3 activation in the liver, suggesting an important role for NLRP3 in the progression of NASH. Nevertheless, further work is still required regarding the precise molecular target and safety of CRID3. Indeed, liver toxicity has been observed in humans, probably due to either the high dose of CRID3 (1,200 mg per day) or a reactive metabolite of its furan moiety or both (Mangan et al., 2018). The latter two are well-known causes of drug-induced liver toxicity. By contrast, in our study, the daily administration of 100 mg/kg of IFM-514 reached adequate concentrations in liver and serum. These concentrations are low enough not to induce liver toxicity, but still sufficient to inhibit NLRP3 in vitro, as shown by the observed effects of IFM-514 in the in vivo LPS-PD/PK studies.
NAFLD is generally considered the hepatic manifestation of the metabolic syndrome and its predominant underlying risk factors appear to be increased body weight and obesity. However, NAFLD can occur in lean subjects with a body mass index <25 kg/m2 (Voss et al., 2011), suggesting that other predisposing factors and inherited disorders play a critical role in lean NAFLD patients, which is an important consideration in Asian populations (Wong et al., 2018). In the MCD diet, the absence of methionine leads to increased hepatic inflammation, fibrosis, liver damage and macrovesicular steatosis (Caballero et al., 2010). Therefore, MCD-fed mice show severe steatohepatitis but is not caused by metabolic syndrome or insulin resistance and show weight loss and increased mortality (Larter and Yeh, 2008). For that reason, this study was conducted in ApoE-/- mice, which have a genetic predisposition to develop chronic inflammation (Harja et al., 2008), including metabolic syndrome (Schierwagen et al., 2015) as a useful tool to explore the therapeutic effects of a novel selective NLRP3 antagonist in lean NAFLD.
There is compelling evidence that NLRP3 inflammasome acts as a mediator of inflammation, lipotoxicity, and fibrosis (Duewell et al., 2010; Bracey et al., 2014; Mehta et al., 2014). Also, NLRP3 has been identified as a central insult sensor that triggers and sustains disease driven by metabolic dysfunction and fibrosis following either acute tissue injury or chronic inflammation (Christ et al., 2018). Similarly, NLRP3 activation occurs when oxidation of LDL, cholesterol, and fats are increased (Shi et al., 2006; Saberi et al., 2009). Thus, it is likely that blockage of the NLRP3 signaling pathway will decelerate the progression from NAFLD to NASH. Therefore, we tested a novel selective NLRP3 antagonist in a dietary in vivo study to reduce fibrosis and inflammation in NASH-induced ApoE-/- mice.
The essential findings of the present study are that an anti-NLRP3 approach in NASH can arrest established liver fibrosis and chronic inflammation in ApoE-/- mice. As for liver fibrosis, this was confirmed by robust readouts. Importantly, the ability of IFM-514 to reduce liver fibrosis is highly relevant as liver fibrosis is associated with adverse liver outcomes in NASH (Angulo et al., 2015). Concerning hepatic chronic inflammation, the NanoString panel and the analysis of the specific gene set enrichment accompanied by reduced Tnfα mRNA expression clearly demonstrate a reduction in cytokine- and inflammasome-related genes by IFM-514. These results indicate that IFM-514 exerts both anti-fibrotic and anti-inflammatory effects in NASH ApoE-/- mice.
Several lines of evidence suggest that a specific NLRP3 antagonist could be an effective novel therapy in NASH for several reasons. First, the transition from NAFLD to NASH correlates with the accumulation of hepatic cholesterol crystals, a known NLRP3 trigger (Duewell et al., 2010; Ioannou et al., 2013). Second, genetic NLRP3 deficiency (Stienstra et al., 2011; Vandanmagsar et al., 2011; Wree et al., 2014a) as well as a specific NLRP3 antagonist (Mridha et al., 2017) halt the progression of NAFLD into NASH. Third, the gain-of-function NLRP3 knock-in mice exhibited enhanced NASH-induced fibrosis when fed with MCD (Wree et al., 2014a). Collectively, these data suggest that NLRP3 inhibitors are potential targets in obesity-induced inflammation and insulin resistance (Vandanmagsar et al., 2011), NAFLD (Henao-Mejia et al., 2012; Wree et al., 2014b) and NASH (Mridha et al., 2017). IFM-514 inhibited NLRP3 inflammasome and the subsequent caspase-1 proteolytic activation, thus preventing the development of NASH in ApoE-/- mice.
Our study has several limitations. The treatment regimen, although based on previous data using NLRP3 inhibitor (Mridha et al., 2017), may have been more efficient if the treatment would be administrated daily. In addition, the lower exposure from IP dosing in those mice may be the reason for reduced efficacy. Moreover, MCD diet in ApoE-/- mice is not the standard model of NASH, but still a model with high liver inflammation and fibrosis, and without substantial weight loss (Schierwagen et al., 2015). Finally, although NLRP3 inhibition with IFM-514 reduces lipogenic genes, it fails to decrease oil red staining in the livers of treated animals. One explanation may be that the NLRP3 inhibition decreases the inflammatory response and thereby the lipogenic stress in a second step.
Our data indicate that the beneficial effects of IFM-514 against steatohepatitis in the MCD ApoE-/- model were due to reduction of hepatic inflammation and fibrosis with marginal changes in lipid accumulation. Thus, these findings demonstrate a link between NLRP3 inflammasome activation and the progression to NASH.
Conclusion
IFM-514 reduced liver inflammation and fibrosis, with mild effect in liver steatosis in MCD-fed ApoE-/- mice. These data suggest that blocking NLRP3 might be an attractive therapeutic approach for lean NASH patients.
Methods
Formulation of IFM-514
NLRP3 inhibitor, IFM-514, was kindly provided by the manufacturer (IFM Therapeutics, Boston, MA, United States) and it was supplied in the form of a sodium salt that is soluble in water at concentrations up to 2 mM. To prepare a suspension for i. p. or oral (100 mg/kg, 10 ml/kg) dosing, the appropriate amount of dry powder was weighed out and added to an appropriate volume of 0.5% carboxymethylcellulose (CMC) in PBS to obtain a 10 mg/ml suspension. The physicochemical properties of IFM-514 are shown in Supplementary Table 1.
In Vitro Experiments
To analyze the in vitro potency and selectivity of IFM-514, mouse and human inflammatory cells were used. To determine the activity of IFM-514 on gramicidin-induced release of IL-1β in cell lines, PMA-differentiated human THP-1 cells were treated with IFM-514 for 1 h and then stimulated with gramicidin (5 μM) for 2 h; an immortalized murine macrophage cell line was primed with LPS (200 ng/ml) for 2 h, treated with IFM-514 for 1 h and then stimulated with gramicidin (2 μM) for 2 h. Primary human CD14 + monocytes were treated with GM-CSF and IL-4 for 6 days to induce differentiation to macrophages. The cells were pre-treated with IFM-514 for 1 h, primed with LPS for 2 h and then triggered with gramicidin for 2 h. In addition, BALB/c bone marrow-derived cells were treated with M-CSF for 6 days to induce differentiation to macrophages. After an overnight rest, cells were pre-treated with compounds for 1 h, primed with LPS (10 ng/ml) for 3 h and stimulated with gramicidin (5 μM final) for 1 h. In all cases, the activity of NLRP3 was evaluated by measuring the production of IL-1β in the supernatant. In all experiments, the IL-1β production was analyzed by homogeneous time resolved fluorescence (HTRF) in cell-free supernatant.
Mice
Female C57BL/6 mice used for the acute lipopolysaccaride (LPS)-induced peritoneal inflammation model (pK/PD) were obtained from Jackson Laboratory. Mice were kept under specific pathogen-free conditions and provided with food and water ad libitum. The animal studies were conducted under protocols approved by the TSRL Institutional Animal Care and Use Committee (IACUC) (application number EFF-21001-02).
A total number of 30 male Apolipoprotein E-deficient mice (ApoE-/- mice) were used for all studies. ApoE-/- mice are predisposed to hypercholesterolemia, atherosclerosis, and obesity. All experiments were performed in accordance with the German animal protection law and statutory guidelines of the animal care facility (Haus für experimentelle Therapie, University Clinics Bonn, Germany), and approved by the North Rhine-Westphalia State Agency for Nature, Environment, and Consumer Protection (LANUV, file reference LANUV NRW, 84-02.04.2014.A137).
LPS-Induced Cytokine Release in C57BL/6 Mice
C57BL/6 mice (8–10 weeks of age, Jackson Laboratories) in groups of 10 were pre-dosed orally 1 h before study start with IFM-514 or vehicle (0.5% CMC in water) at various concentrations in a total volume of 10 ml/kg. The mice were then returned to their cage and allowed food and water ad libitum. After 1 h, mice were then injected IP with 20 mg/kg of LPS (0.1 ml in saline) and again returned to their cages. After an five additional hours (t = 6 h post drug), blood was collected for cytokine analysis by ELISA and quantification of IFM-514 was performed by HPLC using standard reverse-phase conditions.
Induction of NASH With Liver Fibrosis in Mice
10–12-week-old ApoE-/- male and female C57BL/6 mice were fed ad lib for 7 weeks with a methionine/choline deficient (MCD) diet (Diet#E15653-94, Ssniff Spezialdiäten GmbH, Soest, Germany) or with a high-fat cholesterol-rich Western diet (WD) (Diet#S0279-S011, 1.25% cholesterol, Ssniff Spezialdiäten GmbH, Soest, Germany). In MCD-fed mice, hepatic steatosis became histologically evident after 10 days and fibrosis after 8–10 weeks (Schierwagen et al., 2015, 2016). The MCD model shows severe steatohepatitis but is not caused by overnutrition or associated with insulin resistance (Larter and Yeh, 2008). After 3 weeks of diet-induced injury, mice were injected i. p. with 100 mg/kg/day of an NLRP3 antagonist IFM-514 (IFM Therapeutics Inc., Boston, United States) (100 mg/kg body weight) or vehicle (0.5% carmellose) every day, 5 days/week for a further 4 weeks. Serum and liver were collected for histological and molecular readouts of fibrosis, inflammation and steatosis. Portal pressure measured invasively in the spleen pulp, a cannula made from a 25-gauge needle connected to a saline-filled manometer was inserted into the spleen pulp, as previously described (Ackerman et al., 2008). The catheters were connected to a pressure transducer (Hugo Sachs Elektronik, March-Hugstetten, Germany) for blood pressure measurement. Splenic pulp pressure was measured as an index of portal venous pressure (Ppv). For assessment of liver injury, inflammation, fibrosis and steatosis, liver samples were fixed in 10% neutral-buffered formalin and paraffin embedded. Liver sections were stained with H&E and Sirius red (SR). Fibrosis severity was determined by SR morphometry and hepatic 4-hydroxyproline content as previously described (Schierwagen et al., 2015, 2016). To demonstrate steatosis, hepatic triglycerides were measured using an enzyme-linked colorimetric assay TG liquicolor mono (Human Diagnostics, Wiesbaden, Germany) and fresh-frozen hepatic sections were stained with oil red O for neutral fat.
Immunohistochemical Staining
Immunohistochemical staining for αSMA was performed in paraffin-embedded sections (5 μm). The sections were incubated with a mouse-anti-SMA antibody (Actin clone 1A4; Dako, Hamburg, Germany). Thereafter, biotinylated goat-anti-mouse (Dako, Hamburg, Germany) secondary antibody was used. Finally, sections were counterstained with hematoxylin.
Pathology
Blind analysis by a liver pathologist was carried out to determine the activity scores according to Kleiner et al. (Kleiner et al., 2005). The NAFLD Activity Score (NAS) was calculated as the sum of scores for steatosis, lobular inflammation, pigmented macrophages, and fibrosis.
Mouse Serum Chemokines
Blood was collected at the time of euthanasia, maintained at RT for 30 min to retract the clot and centrifuged at 2,000 rpm for 5 min for serum collection. Cytokines in mouse sera were measured with the BioPlex mouse cytokine assay on a Bio-Plex 200 system powered by Luminex xMAP Technology. The analysis was performed according to the manufacturer’s protocol.
Gene Expression Analysis by NanoString Technology
We determined hepatic expression of specific mRNAs by reverse-transcriptase quantitative PCR (RT-qPCR), following extraction of total RNA from liver tissue. Primer sequences are shown in Supplementary Table 2. Expression of mRNA was relative to 18 s.
The prevalence of hepatic innate inflammation was evaluated by determining expression of a panel of mouse myeloid innate immunity (770 immune-related genes) using the NanoString technology (NanoString Technologies, Seattle, United States) according to manufacturer’s instructions. Briefly, 200 ng of total RNA was hybridized, quantified, and loaded into Partek Genomics Suite (Partek Inc. St. Louis, MO, United States). The gene enrichment analysis was performed using Gene Ontology and Consensus Pathway DB. Heatmaps were generated using the gplots library in R. Genes were considered significantly down expressed (p < 0.05) in IFM-treated mice by a minimum of 1.5 fold above that of vehicle-treated MCD-fed ApoE-/- mice, using a hypergeometric test with FDR <0.05. Venn diagram was used to intersect the predicted target genes and the distribution of the 1.5-fold differentially expressed genes, showing the overlap in genes significantly upregulated and downregulated.
Western Blotting
Protein levels were analyzed by Western blot as described previously (Granzow et al., 2014; Brol et al., 2019). Briefly, snap-frozen livers were homogenized and diluted. Protein quantification was performed using a colorimetric BCA protein assay kit (Cat 23225, Thermo Fisher Scientific Inc., IL, United States). Forty micrograms of protein samples was subjected to SDS-PAGE under reducing conditions (10% gels), and proteins were blotted on nitrocellulose membranes. The membranes were blocked and incubated with primary antibody against collagen 1α1 (SC-12895; Santa Cruz Biotechnology, Santa Cruz, CA), srebp1c (ab28481, Abcam), fas (ab82419, Abcam), caspase-1 (sc-56036, Santa Cruz Biotechnology, Santa Cruz, CA), IL-1β (NB600-633). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as an endogenous control (sc-47724; Santa Cruz Biotechnology, Santa Cruz, CA). Membranes were incubated with the corresponding secondary antibody, and blots were developed using enhanced chemiluminescence. Protein quantification was performed by ImageJ (version 1.51q, NIH, United States) and results were corrected for GAPDH levels.
Statistical Analysis
Statistical analyses among groups were performed using Prism V.5.0 (GraphPad, San Diego, CA). Comparisons between two groups were done by non-parametric Mann-Whitney U t-tests and one-way ANOVA followed by Tukey’s Multiple Comparison test were used for statistical comparisons between the three groups, with p < 0.05 considered as significant (#< 0.1. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). $ p < 0.05, if the point is excluded as an outlier. Data were expressed as mean ± SEM. All experiments were performed in triplicate at least four times and a representative image or blots are shown on the manuscript. For transcriptome analysis, statistical parameters were computed between groups, and results are shown as log2-fold change and visualized by heatmaps. p-values were calculated using paired t-test and corrected according to the adaptive Benjamini-Hochberg procedure. A FDR-adjusted p-value below 0.05 was considered statistically significant.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by the TSRL Institutional Animal Care and Use Committee (IACUC) (application number EFF-21001-02). All experiments were performed in accordance with the German animal protection law and statutory guidelines of the animal care facility (Haus für experimentelle Therapie, University Clinics Bonn, Germany), and approved by the North Rhine-Westphalia State Agency for Nature, Environment, and Consumer Protection (LANUV, file reference LANUV NRW, 84-02.04.2014.A137).
Author Contributions
ST, FM, JT drafted the manuscript. FM, ST, MB, JSc and JSt acquired, analysed and interpreted the data. DB, AK, FU, CO, OT, BS and RS acquired and analysed the data. WR designed the compound. RSI performed pathology analysis. KB and LF provided substantial material and methods and analysed and interpreted data. EL, JT designed the original study, interpreted the data, supervised the study and obtained financial support for the study. All authors reviewed the draft for important intellectual content and approved the final article for submission.
Funding
This study received funding from the Deutsche Forschungsgemeinschaft (SFB TRR57 and CRC1382), European Union's Horizon 2020 Research and Innovation Programme (Galaxy, No. 668031, MICROB-PREDICT, No. 825694, and DECISION No. 84794), Societal Challenges-Health, Demographic Change, and Well-Being (No. 731875), Cellex Foundation (PREDICT), LIVERHOPE (No. 731875), Enable (funded by Hessian Ministry of Higher Education, Research, Science and the Arts) and deep-HCC (German Federal Ministry of Education and Research). These funders had no involvement in the design of the study.
Conflict of Interest
JSt, DB, AK, BS, JSc, WR, LF, KB and EL are employed by IFM Therapeutics LLC. IFM Therapeutics LLC has provided the drug and financed parts of the results.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
All the authors are very grateful to Gudrun Hack, Silke Bellinghausen, Christiane Esch and Dikra Zouiten for their excellent technical assistance, and to Robert Sigler, Unit for Laboratory Animal Medicine, University of Michigan Medical School, Ann Arbor for performing the NAFLD scoring.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2021.715765/full#supplementary-material
References
Abdulla, A., Zhang, Y., Hsu, F.-N., Xiaoli, A. M., Zhao, X., Yang, E. S. T., et al. (2014). Regulation of Lipogenic Gene Expression by Lysine-specific Histone Demethylase-1 (LSD1). J. Biol. Chem. 289, 29937–29947. doi:10.1074/jbc.M114.573659
Ackerman, Z., Karmeli, F., Amir, G., and Rachmilewitz, D. (2008). Gastric and Colonic Inflammatory and Vasoactive Mediators in Experimental portal Hypertension. Liver 16, 12–18. doi:10.1111/j.1600-0676.1996.tb00697.x
Angulo, P., Kleiner, D. E., Dam-Larsen, S., Adams, L. A., Bjornsson, E. S., Charatcharoenwitthaya, P., et al. (2015). Liver Fibrosis, but No Other Histologic Features, Is Associated with Long-Term Outcomes of Patients with Nonalcoholic Fatty Liver Disease. Gastroenterology 149, 389–397.e10. doi:10.1053/j.gastro.2015.04.043
Bahia, M. S., Kaur, M., Silakari, P., and Silakari, O. (2015). Interleukin-1 Receptor Associated Kinase Inhibitors: Potential Therapeutic Agents for Inflammatory- and Immune-Related Disorders. Cell Signal. 27, 1039–1055. doi:10.1016/j.cellsig.2015.02.025
Bracey, N. A., Gershkovich, B., Chun, J., Vilaysane, A., Meijndert, H. C., Wright, J. R., et al. (2014). Mitochondrial NLRP3 Protein Induces Reactive Oxygen Species to Promote Smad Protein Signaling and Fibrosis Independent from the Inflammasome. J. Biol. Chem. 289, 19571–19584. doi:10.1074/jbc.M114.550624
Brol, M. J., Rösch, F., Schierwagen, R., Magdaleno, F., Uschner, F. E., Manekeller, S., et al. (2019). Combination of CCl4 with Alcoholic and Metabolic Injuries Mimics Human Liver Fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 317, G182–G194. doi:10.1152/ajpgi.00361.2018
Caballero, F., Fernández, A., Matías, N., Martínez, L., Fucho, R., Elena, M., et al. (2010). Specific Contribution of Methionine and Choline in Nutritional Nonalcoholic Steatohepatitis. J. Biol. Chem. 285, 18528–18536. doi:10.1074/jbc.M109.099333
Christ, A., Günther, P., Lauterbach, M. A. R., Duewell, P., Biswas, D., Pelka, K., et al. (2018). Western Diet Triggers NLRP3-dependent Innate Immune Reprogramming. Cell 172, 162–175.e14. doi:10.1016/j.cell.2017.12.013
Dinarello, C. A., Simon, A., and Van Der Meer, J. W. M. (2012). Treating Inflammation by Blocking Interleukin-1 in a Broad Spectrum of Diseases. Nat. Rev. Drug Discov. 11, 633–652. doi:10.1038/nrd3800
Duewell, P., Kono, H., Rayner, K. J., Sirois, C. M., Vladimer, G., Bauernfeind, F. G., et al. (2010). NLRP3 Inflammasomes Are Required for Atherogenesis and Activated by Cholesterol Crystals. Nature 464, 1357–1361. doi:10.1038/nature08938
Ganz, M., Bukong, T. N., Csak, T., Saha, B., Park, J.-K., Ambade, A., et al. (2015). Progression of Non-alcoholic Steatosis to Steatohepatitis and Fibrosis Parallels Cumulative Accumulation of Danger Signals that Promote Inflammation and Liver Tumors in a High Fat-Cholesterol-Sugar Diet Model in Mice. J. Transl. Med. 13, 193. doi:10.1186/s12967-015-0552-7
Granzow, M., Schierwagen, R., Klein, S., Kowallick, B., Huss, S., Linhart, M., et al. (2014). Angiotensin-II Type 1 Receptor-Mediated Janus Kinase 2 Activation Induces Liver Fibrosis. Hepatology 60, 334–348. doi:10.1002/hep.27117
Harja, E., Bu, D.-x., Hudson, B. I., Chang, J. S., Shen, X., Hallam, K., et al. (2008). Vascular and Inflammatory Stresses Mediate Atherosclerosis via RAGE and its Ligands in apoE-/- Mice. J. Clin. Invest. 118, 183–194. doi:10.1172/JCI32703
Henao-Mejia, J., Elinav, E., Jin, C., Hao, L., Mehal, W. Z., Strowig, T., et al. (2012). Inflammasome-mediated Dysbiosis Regulates Progression of NAFLD and Obesity. Nature 482, 179–185. doi:10.1038/nature10809
Ioannou, G. N., Haigh, W. G., Thorning, D., and Savard, C. (2013). Hepatic Cholesterol Crystals and crown-like Structures Distinguish NASH from Simple Steatosis. J. Lipid Res. 54, 1326–1334. doi:10.1194/jlr.M034876
Jiang, H., He, H., Chen, Y., Huang, W., Cheng, J., Ye, J., et al. (2017). Identification of a Selective and Direct NLRP3 Inhibitor to Treat Inflammatory Disorders. J. Exp. Med. 214, 3219–3238. doi:10.1084/jem.20171419
Kleiner, D. E., Brunt, E. M., Van Natta, M., Behling, C., Contos, M. J., Cummings, O. W., et al. (2005). Design and Validation of a Histological Scoring System for Nonalcoholic Fatty Liver Disease. Hepatology 41, 1313–1321. doi:10.1002/hep.20701
Larter, C. Z., and Yeh, M. M. (2008). Animal Models of NASH: Getting Both Pathology and Metabolic Context Right. J. Gastroenterol. Hepatol. 23, 1635–1648. doi:10.1111/j.1440-1746.2008.05543.x
Macaluso, F. S., Maida, M., and Petta, S. (2015). Genetic Background in Nonalcoholic Fatty Liver Disease: A Comprehensive Review. Wjg 21, 11088–11111. doi:10.3748/wjg.v21.i39.11088
Mangan, M. S. J., Olhava, E. J., Roush, W. R., Seidel, H. M., Glick, G. D., and Latz, E. (2018). Targeting the NLRP3 Inflammasome in Inflammatory Diseases. Nat. Rev. Drug Discov. 17, 588–606. doi:10.1038/nrd.2018.97
Martinon, F., Burns, K., and Tschopp, J. (2002). The Inflammasome. Mol. Cel 10, 417–426. doi:10.1016/S1097-2765(02)00599-3
Mehta, R., Neupane, A., Wang, L., Goodman, Z., Baranova, A., and Younossi, Z. M. (2014). Expression of NALPs in Adipose and the Fibrotic Progression of Non-alcoholic Fatty Liver Disease in Obese Subjects. BMC Gastroenterol. 14, 208. doi:10.1186/s12876-014-0208-8
Mridha, A. R., Wree, A., Robertson, A. A. B., Yeh, M. M., Johnson, C. D., Van Rooyen, D. M., et al. (2017). NLRP3 Inflammasome Blockade Reduces Liver Inflammation and Fibrosis in Experimental NASH in Mice. J. Hepatol. 66, 1037–1046. doi:10.1016/j.jhep.2017.01.022
Próchnicki, T., Mangan, M. S., and Latz, E. (2016). Recent Insights into the Molecular Mechanisms of the NLRP3 Inflammasome Activation [version 1; Referees: 2 Approved, F1000Res 5, 1469. doi:10.12688/F1000RESEARCH.8614.1
Ridker, P. M., Everett, B. M., Thuren, T., MacFadyen, J. G., Chang, W. H., Ballantyne, C., et al. (2017). Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 377, 1119–1131. doi:10.1056/NEJMoa1707914
Rinella, M. E. (2015). Nonalcoholic Fatty Liver Disease. Jama 313, 2263–2273. doi:10.1001/jama.2015.5370
Saberi, M., Woods, N.-B., de Luca, C., Schenk, S., Lu, J. C., Bandyopadhyay, G., et al. (2009). Hematopoietic Cell-specific Deletion of Toll-like Receptor 4 Ameliorates Hepatic and Adipose Tissue Insulin Resistance in High-Fat-Fed Mice. Cel Metab. 10, 419–429. doi:10.1016/j.cmet.2009.09.006
Schierwagen, R., Maybüchen, L., Hittatiya, K., Klein, S., Uschner, F. E., Braga, T. T., et al. (2016). Statins Improve NASH via Inhibition of RhoA and Ras. Am. J. Physiol. Gastrointest. Liver Physiol. 311, G724–G733. doi:10.1152/ajpgi.00063.2016
Schierwagen, R., Maybüchen, L., Zimmer, S., Hittatiya, K., Bäck, C., Klein, S., et al. (2015). Seven Weeks of Western Diet in Apolipoprotein-E-Deficient Mice Induce Metabolic Syndrome and Non-alcoholic Steatohepatitis with Liver Fibrosis. Sci. Rep. 5, 12931. doi:10.1038/srep12931
Schroder, K., and Tschopp, J. (2010). The Inflammasomes. Cell 140, 821–832. doi:10.1016/j.cell.2010.01.040
Shi, H., Kokoeva, M. V., Inouye, K., Tzameli, I., Yin, H., and Flier, J. S. (2006). TLR4 Links Innate Immunity and Fatty Acid-Induced Insulin Resistance. J. Clin. Invest. 116, 3015–3025. doi:10.1172/JCI28898
Stienstra, R., Van Diepen, J. A., Tack, C. J., Zaki, M. H., Van De Veerdonk, F. L., Perera, D., et al. (2011). Inflammasome Is a central Player in the Induction of Obesity and Insulin Resistance. Proc. Natl. Acad. Sci. 108, 15324–15329. doi:10.1073/pnas.1100255108
Szabo, G., and Petrasek, J. (2015). Inflammasome Activation and Function in Liver Disease. Nat. Rev. Gastroenterol. Hepatol. 12, 387–400. doi:10.1038/nrgastro.2015.94
Vandanmagsar, B., Youm, Y.-H., Ravussin, A., Galgani, J. E., Stadler, K., Mynatt, R. L., et al. (2011). The NLRP3 Inflammasome Instigates Obesity-Induced Inflammation and Insulin Resistance. Nat. Med. 17, 179–188. doi:10.1038/nm.2279
Vos, B., Moreno, C., Nagy, N., Féry, F., Cnop, M., Vereerstraeten, P., et al. (2011). Lean Non-alcoholic Fatty Liver Disease (Lean-NAFLD): a Major Cause of Cryptogenic Liver Disease. Acta Gastroenterol. Belg. 74, 389–394.
Williams, C. D., Stengel, J., Asike, M. I., Torres, D. M., Shaw, J., Contreras, M., et al. (2011). Prevalence of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis Among a Largely Middle-Aged Population Utilizing Ultrasound and Liver Biopsy: A Prospective Study. Gastroenterology 140, 124–131. doi:10.1053/j.gastro.2010.09.038
Wong, V. W.-S., Wong, G. L.-H., Chan, R. S.-M., Shu, S. S.-T., Cheung, B. H.-K., Li, L. S., et al. (2018). Beneficial Effects of Lifestyle Intervention in Non-obese Patients with Non-alcoholic Fatty Liver Disease. J. Hepatol. 69, 1349–1356. doi:10.1016/j.jhep.2018.08.011
Wree, A., Eguchi, A., Mcgeough, M. D., Pena, C. A., Johnson, C. D., Canbay, A., et al. (2014a). NLRP3 Inflammasome Activation Results in Hepatocyte Pyroptosis, Liver Inflammation, and Fibrosis in Mice. Hepatology 59, 898–910. doi:10.1002/hep.26592
Keywords: inflammasome, liver fibrosis, steatosis, NASH, caspase-1
Citation: Torres S, Brol MJ, Magdaleno F, Schierwagen R, Uschner FE, Klein S, Ortiz C, Tyc O, Bachtler N, Stunden J, Bertheloot D, Kitanovic A, Sanchez B, Schrum J, Roush WR, Franchi L, Byth K, Latz E and Trebicka J (2021) The Specific NLRP3 Antagonist IFM-514 Decreases Fibrosis and Inflammation in Experimental Murine Non-Alcoholic Steatohepatitis. Front. Mol. Biosci. 8:715765. doi: 10.3389/fmolb.2021.715765
Received: 27 May 2021; Accepted: 27 July 2021;
Published: 13 August 2021.
Edited by:
Enis Kostallari, Mayo Clinic, United StatesReviewed by:
Hannah Kristin Drescher, Massachusetts General Hospital and Harvard Medical School, United StatesEkihiro Seki, Cedars Sinai Medical Center, United States
Copyright © 2021 Torres, Brol, Magdaleno, Schierwagen, Uschner, Klein, Ortiz, Tyc, Bachtler, Stunden, Bertheloot, Kitanovic, Sanchez, Schrum, Roush, Franchi, Byth, Latz and Trebicka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonel Trebicka, Sm9uZWwuVHJlYmlja2FAa2d1LmRl
†These authors share first co-authorship