 Lei Chen1
Lei Chen1 Guohua Gong
Guohua Gong
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Biosci. , 07 May 2021
Sec. Cellular Biochemistry
Volume 8 - 2021 | https://doi.org/10.3389/fmolb.2021.681237
This article is part of the Research Topic Organellar Dynamics in Cell Fate View all 6 articles
Mitofusin 2 (Mfn2) is a transmembrane GTPase located on the mitochondrial outer membrane that contributes to mitochondrial network regulation. It is an essential multifunctional protein that participates in various biological processes under physical and pathological conditions, including mitochondrial fusion, reticulum–mitochondria contacts, mitochondrial quality control, and apoptosis. Mfn2 dysfunctions have been found to contribute to cardiovascular diseases, such as ischemia-reperfusion injury, heart failure, and dilated cardiomyopathy. Here, this review mainly focuses on what is known about the structure and function of Mfn2 and its crucial role in heart failure.
In mammals, cardiomyocytes are highly metabolically active cells. Mitochondria, as a cellular energy power plant, produce ∼90% cellular ATP and occupy ∼40% of the adult cardiomyocyte volume (Marin-Garcia and Akhmedov, 2016). The dysfunctional mitochondria have been linked with various heart diseases (Ong et al., 2012; Dorn, 2013). In most cells, mitochondria are highly dynamic organelles organized in a tubular, dynamic network that constantly undergo movement, fusion, and fission events. Adult cardiomyocytes exhibit a fragmented mitochondrial network. Spheroid mitochondria are densely confined among myofibrils—limiting the movement of mitochondria (Gong et al., 2015), which leads to the low frequency of mitochondrial dynamics events, especially mitochondrial fission and fusion (Eisner et al., 2017; Qin et al., 2018).
Mitochondria fusion and fission, termed as major mitochondrial dynamics, are mediated by mitofusin1/2 (Mfn1/2), optic atrophy 1 (Opa1), and dynamin-related protein 1 (Drp1). Although mitochondrial dynamics events are rare in adult cardiomyocytes (Qin et al., 2018), these dynamism proteins are highly expressed in the adult cardiac.
Mitofusin 2 (Mfn2) is abundantly expressed in the heart, in which it is located at the outer mitochondrial membrane (OMM) to modulate its fusion (Eura et al., 2003). It has been found that Mfn2 participates in multiple cellular biological processes under physical and pathological conditions. Apart from mitochondria fusion, Mfn2 is also involved in the endoplasmic reticulum (ER)–mitochondria tether, mitophagy, cellular apoptosis, and proliferation. Mfn2 as the central player of the Charcot Marie tooth disease, also participated in the onset and development of various heart diseases, including transverse aortic constriction (TAC), induced heart failure, and myocardial infarction (MI) (Givvimani et al., 2014).
In this review, we focus on the current understanding of Mfn2 structure, function, and its role in heart failure.
The first mitochondrial fusion protein was Drosophila fuzzy onion (Fzo) found by Margaret Fuller and colleagues in 1997 (Hales and Fuller, 1997). Mfn2, one of the human Fzo, is dynamin-like GTPases encoded by the mitofusin 2 (MFN2) gene, which is located on the outer mitochondrial membrane. Mfn2 has 757 amino acids that consist of a GTPase domain, a first coiled-coil domain (heptad repeat HR1), two C-terminal transmembrane domains (TM), and a second coiled-coil domain (heptad repeat HR2) (Chandhok et al., 2018; Filadi et al., 2018) (Figure 1A). Mfn2 shares ∼82% similar and 66% identical with mitofusin 1 (Mfn1), and their most extensive homology exists in the same relevant functional domains (Filadi et al., 2018; Santel and Fuller, 2001). The major difference between Mfn1 and Mfn2 is that Mfn2 has a unique proline-rich domain between HR1 and the TM domains, which are tightly associated with protein–protein interactions (Filadi et al., 2018). In the canonical model, the GTPase and coiled-coil domains (HR1 and HR2) are exposed to the cytosol and transmembrane domains crosse the OMM that constitutes a hairpin curve structure mediating the mitochondrial fusion process (Figure 1B). However, Mattie et al. revised the topology of Mfn2. In the revised model, the GTPase and a coiled-coil (HR1) are still in the cytosol, a single transmembrane domain spans the OMM and a second coiled-coil (HR), and the C-terminal domain resides within intermembrane space (IMS) (Giacomello and Scorrano, 2018; Mattie et al., 2018) (Figure 1C).
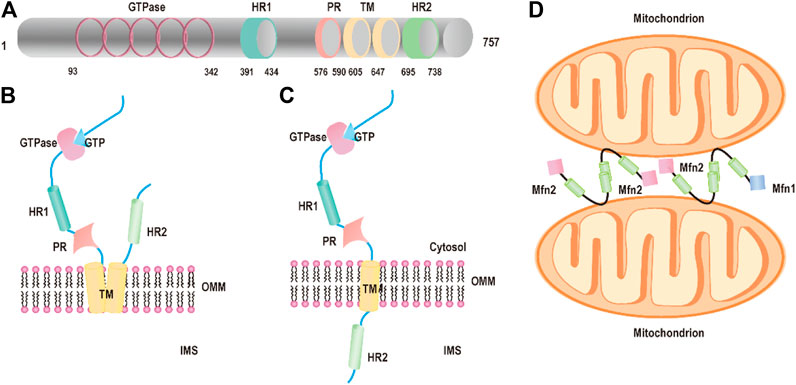
FIGURE 1. Structure of Mfn2. (A) The functional domains of mitofusin 2: the GTPase domains, heptad repeat (HR) coiled-coil regions 1 and 2, the PR domain, and the transmembrane (TM) domain. (B) Schematic diagram of Mfn2 topology: P21RAS domain, a proline-rich (PR) domain, the GTPase, and coiled-coil domains (HR1 and HR2) are exposed to the cytosol and transmembrane domains crosse the OMM. (C) Schematic diagram of revised Mfn2 topology: P21RAS domain, a GTPase, a coiled-coil (HR1), and a proline-rich (PR) domain are exposed into the cytosol, a transmembrane domain spans the OMM, and a second coiled-coil (HR) C-terminal domain protrudes into the IMS. (D) Mitochondrial tethering is mediated by Mfn2 or Mfn1.
Mitochondrial fusion has been studied for around two decades, and numerous excellent articles are focused on the molecules and the mechanism of mitochondrial fusion. Yet, the molecular details of the processes of fusion are still unclear. First, the HR2 domains initiate tethering between adjacent mitochondria through a dimeric antiparallel coiled-coil structure forming either homodimers (Mfn1–Mfn1 or Mfn2–Mfn2) or heterodimers (Mfn1–Mfn2) (Chen et al., 2003; Koshiba et al., 2004) (Figure 1D). The dimeric antiparallel coiled-coil is 95 Å long, constituting a large interaction interface located at opposite ends of the 95 Å coiled-coil, which links two mitochondria together (Koshiba et al., 2004). After the formation of mitochondria–mitochondria tethers, the GTPase domain further promotes fusion events accomplishment. The GTPase domain induced GTP hydrolysis to self-assembly and conformational changes, which lead to membrane remodeling and the two OMMs mixings (Sloat et al., 2019). A new study shows that Mfn2 maintains dimer’s form even after GTP hydrolysis through the GTPase domain (G) interface compared with other dynamin superfamily members including Mfn1, which ensures its higher efficient membrane tethering.(Li et al., 2019).
Recent studies provide novel insights into the mechanism of mitochondrial fusion. Qi et al. analyzed the crystal structures of human Mfn1 and observed a helix bundle (HB) containing three helices extending from the GTPase and one extending from the C-terminal tail (CT) closely attach to the GTPase domain (Qi et al., 2016). Based on the structure, the fusion model is proposed that HB1 (helix bundle1) rotates upon GTP hydrolysis, which allows HB2 (helix bundle2) to bend over and further attach to the GTPase contributing to apposing membranes merge (Qi et al., 2016). In other models, Franco et al. unveiled that in the resting or inactive state, HR2 binds with HR1 of intramolecular by an antiparallel way. In an active state, HR1–HR2 binding unfolds and extends to bind with an HR2 from a different Mfn2 molecule mediating tethering of mitochondria in trans (Franco et al., 2016). HR1 and HR2 domains exposed in the cytoplasm of OMM are necessary for all the models mentioned above to complete fusion. However, Mattie et al. have found the revised model of the C-terminal HR2 domain located in IMS. In the mammalian system, levels of oxidized glutathione disulfide (GSSG) increase, where redox mediates the formation of disulfide bridges between adjacent Mfn molecules, resulting in mitochondrial fusion (Shutt et al., 2012; Mattie et al., 2018). This result provides a new conceptual understanding of the mechanisms that activate mitochondrial fusion, but a few questions still exist. For example, what are mediators of mitochondria tethering in trans for fusion? Which domain mediates mitochondrial fusion? Besides its sensitivity to redox, does the C-terminal have other special functions? Thus, further work will aim to research on how the revised model of the Mfn2 structure drives mitochondrial fusion.
Mfn2 was first identified by Santel and Fuller, and its crucial function is to mediate mitochondria fusion and alter mitochondrial morphology (Santel and Fuller, 2001). After 20 years of intense research, increasing studies reveal that beyond its core function, mitochondrial fusion, Mfn2 is implicated in an expanding array of mitochondrial and cellular processes, including ER–mitochondria contacts (de Brito and Scorrano, 2008), cell apoptosis (Zhang et al., 2013), and autophagy (Chen and Dorn, 2013).
Mitochondria are dynamic in morphology that undergo continuous remodeling by fusion and fission events. Mitochondrial fusion implicates that two mitochondria merge into an elongated one. This process permits the exchange of contents between different mitochondria and maintains genetic and biochemical uniformity via elimination of superoxide species and mutated DNA and the repolarization of membranes, which avoids accumulation of the mutation mitochondrial genome and loss of mitochondria (Yang et al., 2015; Bertholet et al., 2016; El-Hattab et al., 2018). Accumulating studies have reported that it has a critical role in tethering and fusing the OMM while maintaining normal mitochondria morphology. Appropriate overexpression of Mfn2 leads to mitochondrial fusion and elongation (Qin et al., 2020). However, highly overexpressed Mfn2 leads to mitochondrial aggregation around the nucleus (Huang et al., 2007). Deleting both Mfn1 and Mfn2 in mouse embryonic fibroblasts (MEFs) leads to a complete lack of mitochondrial fusion, which causes the mitochondria to become globular (Chen et al., 2003). MEFs deficient in either Mfn1 or Mfn2 genes show fragmentation of mitochondrial tubules, resulting in an aberrant mitochondrial network (Chen et al., 2003).
Surprisingly, the loss of either Mfn1 or Mfn2 displays different forms of fragmented mitochondria. In comparison, cells lacking Mfn1 exhibit short mitochondrial tubules or tiny mitochondrial spheres with uniform size, and cells lacking Mfn2 show some swollen spherical or short-rod mitochondria with largely different sizes and a diameter several times larger than those in wild-type (wt) cells (Figure 2) (Chen et al., 2003), which indicates a functional difference in the mitochondrial fusion. Interestingly, overexpression of Mfn1 in Mfn1 or Mfn2-deletion MEFs could restore 75–80% of cells' mitochondrial morphology (Chen et al., 2003). In contrast, overexpression of Mfn2 in Mfn2-deletion MEFs could rescue 91% cell fusion activity (Figure 2), but only 25% in Mfn1-deficient cells (Chen et al., 2003). These suggest different but partial overlapping functions between the Mfn1 and Mfn2 in the OMM fusion. Mfn1 is predominant for OMM fusion because Mfn1-harboring mitochondria show higher efficient mitochondrial membrane tethering dependent on GTPase than Mfn2-harboring mitochondria (Ishihara et al., 2004). The abundant expression of Mfn2 in the heart is non-predominant for mitochondrial fusion function, suggesting that Mfn2 integrates other biological functions.
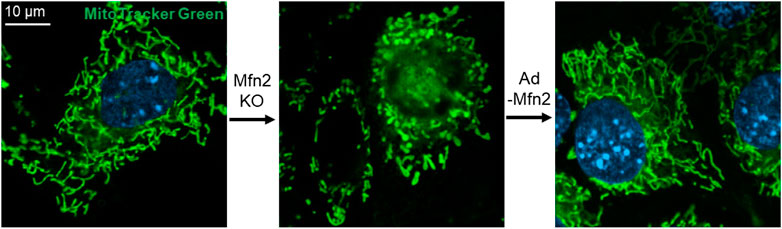
FIGURE 2. Mfn2 shapes the morphology of mitochondria in MEFs. Mfn2 deletion decreases the fusion activity that leads to mitochondrial fragmentation. The fragmented mitochondria network caused by Mfn2 deletion can be restored by adenovirus-mediated Mfn2 overexpression.
The physical contacts of ER and mitochondria have been observed initially by electron microscopy (EM) in the 50 s (Bernhard et al., 1952). Yet, the character has been regarded as artifact of fixation for a long time. By 1998, Rosario and colleagues found numerous close contacts and dynamic networks between ER and mitochondria in living HeLa cells by using high resolution in three dimensions (Rizzuto et al., 1998). The mitochondria and ER are closely connected but never touch with each other. There is a gap between them, the thickness ranging from ∼10 nm up to 80–100 nm (Giacomello and Pellegrini, 2016). Therefore, mitochondria or ER-bound proteins are required for maintaining the spatial relationship between ER and mitochondria. Recently, de Brito and Scorrano firstly revealed that Mfn2 acts as a critical regulator of directly mediating mitochondrial tethering in mammals (de Brito and Scorrano, 2008). This study showed that Mfn2 proteins are enriched at resides in ER membranes, especially in contact sites between ER and mitochondria called mitochondria-associated membranes (MAMs). Mfn2 on the ER interacted with Mfn1 or 2 located in the OMM to engage in homotypic and heterotypic complexes, which mediates the two organelles tethering (de Brito and Scorrano, 2008) (Figure 3). Mfn2-deficient cells disrupted ER morphology, decreased ER–mitochondria contacts, and thus reduced mitochondrial Ca2+ uptake upon activation of ER Ca2+ release (de Brito and Scorrano, 2008). However, Pierre and his coworker observed that the percentage of the OMM engaged in the formation of close contacts (distance <20 nm) with the ER in Mfn2−/− MEFs (the same cell line used in Scorrano’s group) was more than doubled (∼4.9%) compared to its wt MEFs (∼2.2%) using the quantitative electron microscopy (EM) approach (Cosson et al., 2012). Overexpression of Mfn2 in Mfn2 KO cells significantly reduced the number of ER–mitochondria contacts. However, this study also found the reduced ER–mitochondria colocalization index (Manders’ B colocalization coefficient) in Mfn2−/− MEFs by confocal microscopy, as Scorrano’s studied. Similarly, other studies also demonstrated the increase of the structural and functional ER–mitochondria coupling in Mfn2-deficient cells or Mfn2-knockdown (KD) cells obtained by EM and confocal microscopy (Filadi et al., 2015; Leal et al., 2016) and the reduction of ER–mitochondria colocalization by fluorescence microscopy (Filadi et al., 2015). The apparent paradox is because the loss of Mfn2 alters the morphology of both the ER and mitochondria, obtained an artifact result by fluorescence microscopy. From a functional point of view, uptake of Ca2+ by mitochondria upon Ca2+ release from the ER depends on close contacts of the two organelles (Csordas et al., 2010). Therefore, the increase of their interconnection promotes this process. Riccardo et al. observed the reduced mitochondrial Ca2+ uptake upon ER Ca2+ release in Mfn2−/− MEFs. Besides, they also found that expression of the mitochondrial Ca2+ uniporter (MCU) was reduced by almost 50% in Mfn2−/− MEFs compared with wt MEFs(Csordas et al., 2010). Therefore, it is decreased MCU not reduction of ER–mitochondria tethering that results in the reduced mitochondrial Ca2+ uptake. One independent experiment could support this conclusion. Acute downregulation of Mfn2 expression in wt MEFs does not affect the MCU level but induces mitochondrial Ca2+ rise in response to an IP3-induced ER Ca2+ release.
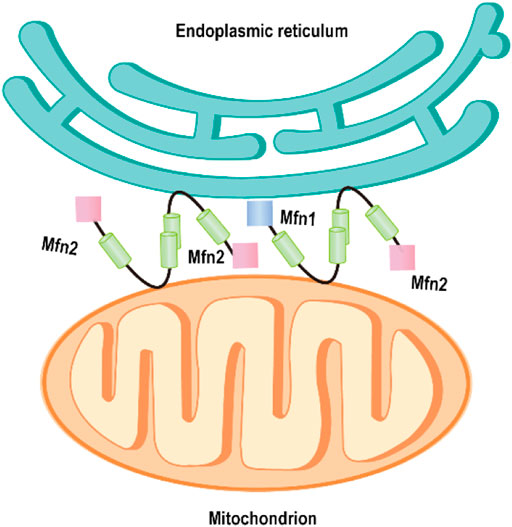
FIGURE 3. Mfn2 tethers endoplasmic reticulum to mitochondria. Mfn2 on the ER membrane interacts with Mfn2 or Mfn1 on the outer mitochondrial membrane, mediating the ER–mitochondria tethering.
Recently, a study from Scorrano’s group further confirms the ER–mitochondria tether role for Mfn2 by employing distinct techniques as they originally reported (Naon et al., 2016). However, some conclusions are still being challenged. By using the new method: split-GFP–based contact site sensor (SPLICS), Tito’s group unraveled increased ER–mitochondria short-range (8–10 nm) contacts in acute Mfn2-KD-HeLa cells but the decreased long-range (40–50 nm) contacts (Cieri et al., 2018). Although some evidence supports the negative role of Mfn2 in the formation of close contacts (∼10 nm) between the two organelles, more evidence confirms the positive role of MFN2 in ER–mitochondria tethering. Whether Mfn2 tethers (Schneeberger et al., 2013; Li et al., 2015; Naon et al., 2016) or divides (Cosson et al., 2012; Filadi et al., 2016; Leal et al., 2016; Naon et al., 2016), ER and mitochondria remain controversial. Therefore, more useful tools, more efficient methods, and further investigations will be necessary to explore the accurate function of Mfn2 in ER and mitochondrial tethering.
Mitochondria are critical for the initiation of intrinsic apoptosis. Upon apoptotic stimuli, BAX and BAK are activated and furtherly form higher-order oligomers to increase mitochondrial outer membrane permeabilization (MOMP), which is independent of the mitochondrial permeability transition pore (mPTP) opening (Bock and Tait, 2020). The increased MOMP leads to cytochrome c release into the cytosol, and subsequently, caspases are activated to initiate apoptosis. Compelling evidence exhibit that Mfn2 is integrated with proteins of the B-cell lymphoma-2 (BCL-2) family to modulate apoptosis. On apoptotic stimuli, BAX is recruited to the Mfn2-containing puncta contributing to cell apoptosis (Neuspiel et al., 2005). However, activation of BAX and release of cytochrome c are repressed in the presence of the dominant active mutants of Mfn2. Overexpression of Mfn2 promotes BAX translocation from the cytoplasm to the mitochondrial membrane, which induces apoptosis in many tumor cells, including hepatocellular carcinoma cells, breast carcinoma cells, urinary bladder carcinoma cells, and cervical carcinoma cells. These results indicate the antitumor efficacy of Mfn2 (Wu et al., 2008; Jin et al., 2011; Wang et al., 2012; Wang et al., 2018).
Overexpression of Mfn2 in hepatocellular carcinoma cells leads to calcium ion (Ca2+) influx from the ER. The higher mitochondrial Ca2+ triggers mitochondrial reactive oxygen species (ROS) generation that stimulates mPTP opening, cytochrome c release, and apoptosis (Wang et al., 2015). Mfn2 is downregulated in clinical colorectal cancer (CRC) tissues. Overexpression of Mfn2 results in a cell cycle arrest at the G2/M phase in CRC cells, and it increases the levels of activated caspase-3 and cleaved PARP (Cheng et al., 2013).
On the contrary, results from studies show the anti-apoptotic role of Mfn2. An increase of ubiquitylation and proteasomal degradation of Mfn2 in response to oxidative stress and other apoptotic stimuli leads to mitochondrial fragmentation and induced apoptotic cell death (Leboucher et al., 2012). In agreement with this result, Mfn2 overexpression could restore mitochondrial fragmentation and prevents neuronal apoptosis (Jahani-Asl et al., 2007). Similarly, Mfn2 complexed with BCL-XL maintains the anti-apoptotic function (Du et al., 2020). In brief, what is the role of Mfn2 in cell apoptosis might be context-dependent (types of cells, different apoptosis stimuli, and the interaction of distinct molecules).
In order to maintain a healthy mitochondrial network and function in response to all sorts of stress, the mitochondrial quality control mechanism needs to be activated in cells, termed mitophagy (Green and Van Houten, 2011; Gong et al., 2015). Mitophagy is a cellular mechanism that selectively eliminates damaged, dysfunctional mitochondria. In this process, autophagosomes engulf mitochondria, transfer to lysosomes, and subsequently fuse with the lysosome, where degradation takes place. Studies show Mfn2 has associated with the increase in autophagosome formation and fusion of autophagosome–lysosome, suggesting that Mfn2 is a mediator of mitophagy (Sebastian et al., 2016; Peng et al., 2018). Consistently, Mfn2-KO in specific tissue such as the heart and brain directly impaired mitophagy and caused the accumulation of damaged mitochondria (Lee et al., 2012; Chen et al., 2014). The molecular mechanism of Mfn2-mediated mitophagy is uncovered by Dr. Dorn’s lab in 2013 (Chen and Dorn, 2013). The degradation of PINK1 in the depolarized mitochondria matrix will lead to PINK1 accumulates at the OMM. The phosphorylated Mfn2 at T111 and S442 by PINK1 as the Parkin receptor recruits Parkin to translocate to OMM. Subsequently, Parkin is activated via PINK1-mediated Ser65 phosphorylation in the Parkin ubiquitin domain. Activation of Parkin ubiquitinates MFN2 and other OMM proteins (Gegg et al., 2010; Poole et al., 2010; Tanaka et al., 2010; Ziviani et al., 2010). Furthermore, the ubiquitinated proteins bind to LC3-II via specific autophagy-related receptors, such as optineurin (optin), NBR1, and p62, to form autophagosomes. The formation of autophagosomes abrogates mitochondrial fusion events, which leads to mitochondrial fragmentation (Chen and Dorn, 2013; Lazarou et al., 2015; Khaminets et al., 2016; McLelland et al., 2018) (Figure 4).
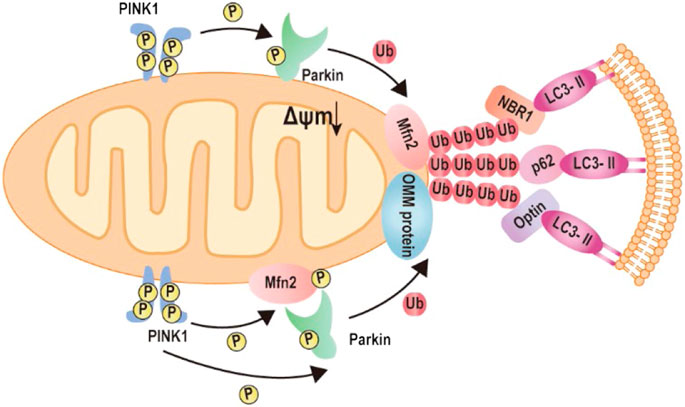
FIGURE 4. Mechanism of Mfn2-mediated mitophagy. 1) Upon mitochondrial depolarization, the kinase PINK1 accumulates at the mitochondrial outer membrane and initiates phosphorylation and recruitment of Parkin. Activated Parkin ubiquitylates Mfn2. The formation of ubiquitin chains on mitochondrial surface proteins promotes its binding to LC3-II via the mitochondrial receptors such as optineurin, NBR, or p62. 2) Upon mitochondrial depolarization, accumulation of kinase PINK1 phosphorylates Mfn2, which recruits Parkin to the mitochondrial outer membrane. Parkin phosphorylated by PKIN1 ubiquitylates several outer mitochondrial membrane proteins. The formation of ubiquitin chains on mitochondrial surface proteins promotes its binding to LC3-II via the mitochondrial receptors such as optineurin, NBR, or p62.
Cardiac hypertrophy (CH) occurs in a number of disease states in response to increased cardiac workload and can readily progress to ventricular dilatation, contractile dysfunction, and heart failure. Although there is limited literature that decodes the function of Mfn2 in heart failure, it has been reported that Mfn2 has associated with the onset and progression of cardiac hypertrophy and heart failure. Mfn2 expression is decreased in various rat models of cardiac hypertrophy, including MI and TAC (Fang et al., 2007). Similarly, Mfn2 expression is downregulated in Ang II-induced or PE-induced cardiomyocyte hypertrophy. Upregulation of Mfn2 could inhibit the hypertrophic progression (Yu et al., 2011; Guan et al., 2016; Sun et al., 2019).
Mfn2 deletion interrupts multiple molecular signal pathways that gradually lead to cardiac vulnerability and dysfunction. Recently, genetic ablation mice are used to explore the function of Mfn2 in cardiac hypertrophy and heart failure. A report from Dorn’s group exhibits that hearts deficient in Mfn1 and Mfn2 contribute to progressive dilated cardiomyopathy at five weeks and heart failure from seven to eight weeks (Chen et al., 2011). Furthermore, dilated cardiomyopathy is observed in mice lacking Mfn2 at 16 weeks, and the severity increases with age (Chen and Dorn, 2013). However, another study shows that cardiac deletion of Mfn1 and Mfn2 in mice is no sign of cardiac hypertrophy at 8–10 weeks, with normal left ventricular volume and normal contractile performance (Hall et al., 2016). Conditional deletion of Mfn2 only leads to mild myocyte hypertrophy accompanied by mild deterioration of left ventricular function and does not develop heart failure (Papanicolaou et al., 2011). Mfn2 deletion or Mfn1 and Mfn2 deletion protect the heart against ischemia and reperfusion injury (Papanicolaou et al., 2011; Hall et al., 2016). Genetic deletion mice from different study groups display distinct phenotypes, which might attribute to deletion methods, the location and timing of Mfn2 deletion, the genetic background, and the ages of the mice.
Mfn2 plays a critical mediator in the development of cardiac hypertrophy and heart failure via regulation of various biological processes, including mitochondrial fusion, mitophagy, and apoptosis. Vivo studies demonstrated that specific deletion of Mfn2 in cardiomyocytes impaired mitochondrial fusion and modest cardiac hypertrophy (Papanicolaou et al., 2011). Similarly, in vitro studies observed that Mfn2 was decreased in Ang II-induced cardiac myocytes hypertrophy, accompanied by the change of mitochondria morphology from the filament to short and small. The aspect ratio of the mitochondria is also reduced (Xiong et al., 2019; Yu et al., 2011). Mfn2 overexpression on Ang II-induced cardiomyocyte rescue mitochondrial fusion, which resulted in two short and small mitochondria to form large mitochondria and attenuated myocyte hypertrophy (Xiong et al., 2019). Cardiac deletion of Mfn2 in mice increases the proportion of enlarged mitochondria and impairs mitophagy that leads to cardiomyopathy. Upon mitochondria depolarization, PINK1-phosphorylated Mfn2 recruits Parkin to the OMM and facilitates mitophagy (Figure 5). Mfn2 ablation defects Parkin signaling, initiating aberrant mitophagy (Chen and Dorn, 2013). Interestingly, the effects of Mfn2 overexpression ameliorated Ang II-induced cardiac hypertrophy via facilitating Parkin translocation and phosphorylation, triggering mitophagy (Xiong et al., 2019). Besides, autophagosome–lysosome fusion is impaired in Mfn2-deficient mice, leading to accumulation of autophagosomes and progressive heart failure (Zhao et al., 2012). Mfn2 mediates autophagosome–lysosome fusion in the heart via attracting and binding RAB7 to the autophagosomal membrane (Zhao et al., 2012).
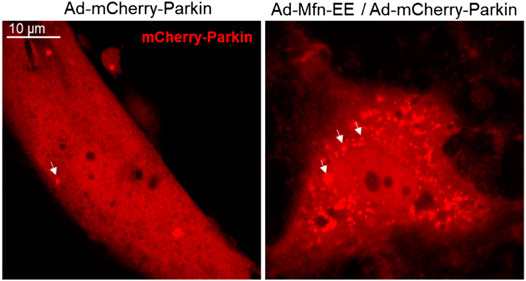
FIGURE 5. Phosphorylated Mfn2 recruits Parkin to OMM. Mfn2 Thr111 (T111) and Ser442 (S442) were replaced by Glu (E) that mimics PINK1 phosphorylation of Mfn2 (T111E/S442E) conferred it PINK1-independent binding activity to Parkin.
Besides, Mfn2 expression is associated with proliferation and apoptosis of VSMCs and myocytes in cardiac hypertrophy (Chen et al., 2004). Neonatal rat cardiomyocytes treated with H2O2 inducing oxidative stress lead to concurrent increases in Mfn2 expression and apoptosis (Guo et al., 2007; Shen et al., 2007). Further study shows that upregulation of Mfn2 significantly increases caspase-9, caspase-3, and BAX/BCL-2 ratio, initiates cytochrome c release, and decreases the level of phosphorylated Akt, thereby triggering apoptosis (Guo et al., 2007). Additionally, Papanicolaou et al. observed that cardiomyocytes by genetically deleting Mfn2 can be resistant to cell death, owing to more tolerance to Ca2+-induced MPTP opening (Papanicolaou et al., 2011). Mfn2 could regulate ER–mitochondria tethering, which is related to Ca2+ influx. ER and mitochondria contacts are more close, and Ca2+ influx in the mitochondrial matrix is highly increased. High-level mitochondrial Ca2+ facilitates the opening of MPTP which leads to apoptosis (Filadi et al., 2015). Cumulatively, these results indicate that Mfn2 is a necessary component for cardiac hypertrophy and heart failure.
Although the functions of Mfn2 in cardiac hypertrophy and heart failure are far more complex than what we have yet known, enhancing Mfn2 function or upregulating expression may be useful as a therapeutic strategy for cardiac hypertrophy and heart failure. Recently, reporters have observed that 6-phenylhexanamide derivative Mfn2 activators or Mfn2 agonists could enhance Mfn2 function for the treatment of diseases (Rocha et al., 2018; Dang et al., 2020). Besides, sex hormones (estrogen and testosterone) also upregulate Mfn1 and Mfn2 via the regulation of PGC1α in cardiac tissue (Lynch et al., 2020). Although, direct modulation of Mfn1 and Mfn2 by the sex hormones is still unclear in heart failure, several studies have indicated estrogen and testosterone are cardioprotective factors (Volterrani et al., 2012; Lynch et al., 2020). It is also convenient for the patient to monitor and analyze the concentration of hormones by modern medical techniques.
The role of Mfn2 is essential for heart diseases, owing to its ability to regulate mitochondria fusion, ER–mitochondria contacts, cell metabolism, apoptosis, and autophagy. Alteration of Mfn2 expression or dysfunctions of Mfn2 has been observed in heart diseases. Some studies indicate that overexpression of Mfn2 in heart diseases, including heart failure and myocardial ischemia, could attenuate cardiac hypertrophy and dysfunction in response to various stresses. Other studies show that deletion of Mfn2 in cardiac myocytes is able to protect against ischemia and reperfusion injury. Therefore, further studies are needed to explore detailed molecular mechanisms of Mfn2 in cardiovascular diseases, which might provide a novel therapeutic target for patients.
LC wrote the first draft of the manuscript. GG revised the manuscript. BL, MG, AL, and YQ helped in manuscript writing and editing. LC and GG prepared the images. All authors contributed to the article and approved the submitted version.
This work was partially supported by the National Key Research and Development Program of China (Grant Nos. 2017YFA0105601 and 2018YFA0107102), the National Natural Science Foundation of China (Grant Nos. 31771524, 81970333, and 31901044), and the Program for Professor of Special Appointment at Shanghai Institutions of Higher Learning (Grant No. TP2017036). Major Program of Development Fund for Shanghai Zhangjiang National Innovation Demonstration Zone, Stem Cell Strategic Biobank and Stem Cell Clinical Technology Transformation Platform, (Grant No. ZJ2018-ZD-004).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ang II angiotensin II
BCL-2 B-cell lymphoma-2
CDK cyclin-dependent kinases
CT C-terminal tail
Drp1 dynamin-related protein 1
EM electron microscopy
ER endoplasmic reticulum
Fzo Drosophila fuzzy onion
HB helix bundle
GSSG glutathione disulfide
KD knockdown
MAM mitochondria-associated membranes
MCU mitochondrial Ca2+ uniporter
MEFs embryonic fibroblasts
MFN1 mitofusin 1
MFN2 mitofusin 2
MFNs mitofusins
MI myocardial infarction
MOMP mitochondrial outer membrane permeabilization
OMM outer mitochondrial membrane
Opa1 optic atrophy 1
Optin optineurin
PE phenylephedrine
SHR spontaneously hypertensive rats
SPLICS split-GFP–based contact site sensor
TAC transverse aortic constriction
TM transmembrane domains
VSMCs vascular smooth muscle cells
WKY Wistar Kyoto rat
WT wild type
Bernhard, W., Haguenau, F., Gautier, A., and Oberling, C. (1952). La structure submicroscopique des elements basophiles cytoplasmiques dans le foie, le pancreas et les glandes salivaires. Z. Zellforschung 37, 281–300. doi:10.1007/bf00343816
Bertholet, A. M., Delerue, T., Millet, A. M., Moulis, M. F., David, C., Daloyau, M., et al. (2016). Mitochondrial Fusion/fission Dynamics in Neurodegeneration and Neuronal Plasticity. Neurobiol. Dis. 90, 3–19. doi:10.1016/j.nbd.2015.10.011
Bock, F. J., and Tait, S. W. G. (2020). Mitochondria as Multifaceted Regulators of Cell Death. Nat. Rev. Mol. Cel Biol 21, 85–100. doi:10.1038/s41580-019-0173-8
Chandhok, G., Lazarou, M., and Neumann, B. (2018). Structure, Function, and Regulation of Mitofusin-2 in Health and Disease. Biol. Rev. 93, 933–949. doi:10.1111/brv.12378
Chen, H., Detmer, S. A., Ewald, A. J., Griffin, E. E., Fraser, S. E., and Chan, D. C. (2003). Mitofusins Mfn1 and Mfn2 Coordinately Regulate Mitochondrial Fusion and Are Essential for Embryonic Development. J. Cel Biol 160, 189–200. doi:10.1083/jcb.200211046
Chen, K.-H., Guo, X., Ma, D., Guo, Y., Li, Q., Yang, D., et al. (2004). Dysregulation of HSG Triggers Vascular Proliferative Disorders. Nat. Cel Biol 6, 872–883. doi:10.1038/ncb1161
Chen, Y., and Dorn, G. W. (2013). PINK1-phosphorylated Mitofusin 2 Is a Parkin Receptor for Culling Damaged Mitochondria. Science 340, 471–475. doi:10.1126/science.1231031
Chen, Y., Liu, Y., and Dorn, G. W. (2011). Mitochondrial Fusion Is Essential for Organelle Function and Cardiac Homeostasis. Circ. Res. 109, 1327–1331. doi:10.1161/circresaha.111.258723
Chen, Y., Sparks, M., Bhandari, P., Matkovich, S. J., Dorn, G. W., and 2nd, (2014). Mitochondrial Genome Linearization Is a Causative Factor for Cardiomyopathy in Mice and Drosophila. Antioxid. Redox Signaling 21, 1949–1959. doi:10.1089/ars.2013.5432
Cheng, X., Zhou, D., Wei, J., and Lin, J. (2013). Cell-cycle Arrest at G2/M and Proliferation Inhibition by Adenovirus-Expressed Mitofusin-2 Gene in Human Colorectal Cancer Cell Lines. Neoplasma 60, 620–626. doi:10.4149/neo_2013_080
Cieri, D., Vicario, M., Giacomello, M., Vallese, F., Filadi, R., Wagner, T., et al. (2018). SPLICS: a Split Green Fluorescent Protein-Based Contact Site Sensor for Narrow and Wide Heterotypic Organelle Juxtaposition. Cell Death Differ 25, 1131–1145. doi:10.1038/s41418-017-0033-z
Cosson, P., Marchetti, A., Ravazzola, M., and Orci, L. (2012). Mitofusin-2 Independent Juxtaposition of Endoplasmic Reticulum and Mitochondria: an Ultrastructural Study. PLoS One 7, e46293. doi:10.1371/journal.pone.0046293
Csordás, G., Várnai, P., Golenár, T., Roy, S., Purkins, G., Schneider, T. G., et al. (2010). Imaging Interorganelle Contacts and Local Calcium Dynamics at the ER-Mitochondrial Interface. Mol. Cel 39, 121–132. doi:10.1016/j.molcel.2010.06.029
Dang, X., Zhang, L., Franco, A., Li, J., Rocha, A. G., Devanathan, S., et al. (2020). Discovery of 6-Phenylhexanamide Derivatives as Potent Stereoselective Mitofusin Activators for the Treatment of Mitochondrial Diseases. J. Med. Chem. 63, 7033–7051. doi:10.1021/acs.jmedchem.0c00366
de Brito, O. M., and Scorrano, L. (2008). Mitofusin 2 Tethers Endoplasmic Reticulum to Mitochondria. Nature 456, 605–610. doi:10.1038/nature07534
Dorn, G. W. (2013). Mitochondrial Dynamics in Heart Disease. Biochim. Biophys. Acta (Bba) - Mol. Cel Res. 1833, 233–241. doi:10.1016/j.bbamcr.2012.03.008
Du, M., Yu, S., Su, W., Zhao, M., Yang, F., Liu, Y., et al. (2020). Mitofusin 2 but Not Mitofusin 1 Mediates Bcl-XL-Induced Mitochondrial Aggregation. J. Cel Sci 133. doi:10.1242/jcs.245001
Eisner, V., Cupo, R. R., Gao, E., Csordás, G., Slovinsky, W. S., Paillard, M., et al. (2017). Mitochondrial Fusion Dynamics Is Robust in the Heart and Depends on Calcium Oscillations and Contractile Activity. Proc. Natl. Acad. Sci. USA 114, E859–E868. doi:10.1073/pnas.1617288114
El-Hattab, A. W., Suleiman, J., Almannai, M., and Scaglia, F. (2018). Mitochondrial Dynamics: Biological Roles, Molecular Machinery, and Related Diseases. Mol. Genet. Metab. 125, 315–321. doi:10.1016/j.ymgme.2018.10.003
Eura, Y., Ishihara, N., Yokota, S., and Mihara, K. (2003). Two Mitofusin Proteins, Mammalian Homologues of FZO, with Distinct Functions Are Both Required for Mitochondrial Fusion. J. Biochem. 134, 333–344. doi:10.1093/jb/mvg150
Fang, L., Moore, X.-L., Gao, X.-M., Dart, A. M., Lim, Y. L., and Du, X.-J. (2007). Down-regulation of Mitofusin-2 Expression in Cardiac Hypertrophy In Vitro and In Vivo. Life Sci. 80, 2154–2160. doi:10.1016/j.lfs.2007.04.003
Filadi, R., Greotti, E., Turacchio, G., Luini, A., Pozzan, T., and Pizzo, P. (2015). Mitofusin 2 Ablation Increases Endoplasmic Reticulum-Mitochondria Coupling. Proc. Natl. Acad. Sci. USA 112, E2174–E2181. doi:10.1073/pnas.1504880112
Filadi, R., Greotti, E., Turacchio, G., Luini, A., Pozzan, T., and Pizzo, P. (2016). Presenilin 2 Modulates Endoplasmic Reticulum-Mitochondria Coupling by Tuning the Antagonistic Effect of Mitofusin 2. Cel Rep. 15, 2226–2238. doi:10.1016/j.celrep.2016.05.013
Filadi, R., Pendin, D., and Pizzo, P. (2018). Mitofusin 2: from Functions to Disease. Cell Death Dis 9, 330. doi:10.1038/s41419-017-0023-6
Franco, A., Kitsis, R. N., Fleischer, J. A., Gavathiotis, E., Kornfeld, O. S., Gong, G., et al. (2016). Correcting Mitochondrial Fusion by Manipulating Mitofusin Conformations. Nature 540, 74–79. doi:10.1038/nature20156
Gegg, M. E., Cooper, J. M., Chau, K.-Y., Rojo, M., Schapira, A. H. V., and Taanman, J.-W. (2010). Mitofusin 1 and Mitofusin 2 Are Ubiquitinated in a PINK1/parkin-dependent Manner upon Induction of Mitophagy. Hum. Mol. Genet. 19, 4861–4870. doi:10.1093/hmg/ddq419
Giacomello, M., and Pellegrini, L. (2016). The Coming of Age of the Mitochondria-ER Contact: a Matter of Thickness. Cel Death Differ 23, 1417–1427. doi:10.1038/cdd.2016.52
Giacomello, M., and Scorrano, L. (2018). The INs and OUTs of Mitofusins. J. Cel Biol 217, 439–440. doi:10.1083/jcb.201801042
Givvimani, S., Pushpakumar, S., Veeranki, S., and Tyagi, S. C. (2014). Dysregulation of Mfn2 and Drp-1 Proteins in Heart Failure. Can. J. Physiol. Pharmacol. 92, 583–591. doi:10.1139/cjpp-2014-0060
Gong, G., Song, M., Csordas, G., Kelly, D. P., Matkovich, S. J., and Dorn, G. W. (2015). Parkin-mediated Mitophagy Directs Perinatal Cardiac Metabolic Maturation in Mice. Science 350, aad2459. doi:10.1126/science.aad2459
Green, D. R., and Van Houten, B. (2011). SnapShot: Mitochondrial Quality Control. Cell 147, 950–950.e951. doi:10.1016/j.cell.2011.10.036
Guan, X., Wang, L., Liu, Z., Guo, X., Jiang, Y., Lu, Y., et al. (2016). miR-106a Promotes Cardiac Hypertrophy by Targeting Mitofusin 2. J. Mol. Cell Cardiol. 99, 207–217. doi:10.1016/j.yjmcc.2016.08.016
Guo, X., Chen, K.-H., Guo, Y., Liao, H., Tang, J., and Xiao, R.-P. (2007). Mitofusin 2 Triggers Vascular Smooth Muscle Cell Apoptosis via Mitochondrial Death Pathway. Circ. Res. 101, 1113–1122. doi:10.1161/circresaha.107.157644
Hales, K. G., and Fuller, M. T. (1997). Developmentally Regulated Mitochondrial Fusion Mediated by a Conserved, Novel, Predicted GTPase. Cell 90, 121–129. doi:10.1016/s0092-8674(00)80319-0
Hall, A. R., Burke, N., Dongworth, R. K., Kalkhoran, S. B., Dyson, A., Vicencio, J. M., et al. (2016). Hearts Deficient in Both Mfn1 and Mfn2 Are Protected against Acute Myocardial Infarction. Cel Death Dis 7, e2238. doi:10.1038/cddis.2016.139
Huang, P., Yu, T., and Yoon, Y. (2007). Mitochondrial Clustering Induced by Overexpression of the Mitochondrial Fusion Protein Mfn2 Causes Mitochondrial Dysfunction and Cell Death. Eur. J. Cel Biol. 86, 289–302. doi:10.1016/j.ejcb.2007.04.002
Ishihara, N., Eura, Y., and Mihara, K. (2004). Mitofusin 1 and 2 Play Distinct Roles in Mitochondrial Fusion Reactions via GTPase Activity. J. Cel Sci. 117, 6535–6546. doi:10.1242/jcs.01565
Jahani-Asl, A., Cheung, E. C. C., Neuspiel, M., MacLaurin, J. G., Fortin, A., Park, D. S., et al. (2007). Mitofusin 2 Protects Cerebellar Granule Neurons against Injury-Induced Cell Death*. J. Biol. Chem. 282, 23788–23798. doi:10.1074/jbc.m703812200
Jin, B., Fu, G., Pan, H., Cheng, X., Zhou, L., Lv, J., et al. (2011). Anti-tumour Efficacy of Mitofusin-2 in Urinary Bladder Carcinoma. Med. Oncol. 28 (Suppl. 1), S373–S380. doi:10.1007/s12032-010-9662-5
Khaminets, A., Behl, C., and Dikic, I. (2016). Ubiquitin-Dependent and Independent Signals in Selective Autophagy. Trends Cell Biology 26, 6–16. doi:10.1016/j.tcb.2015.08.010
Koshiba, T., Detmer, S. A., Kaiser, J. T., Chen, H., McCaffery, J. M., and Chan, D. C. (2004). Structural Basis of Mitochondrial Tethering by Mitofusin Complexes. Science 305, 858–862. doi:10.1126/science.1099793
Lazarou, M., Sliter, D. A., Kane, L. A., Sarraf, S. A., Wang, C., Burman, J. L., et al. (2015). The Ubiquitin Kinase PINK1 Recruits Autophagy Receptors to Induce Mitophagy. Nature 524, 309–314. doi:10.1038/nature14893
Leal, N. S., Schreiner, B., Pinho, C. M., Filadi, R., Wiehager, B., Karlström, H., et al. (2016). Mitofusin-2 Knockdown Increases ER-Mitochondria Contact and Decreases Amyloid β-peptide Production. J. Cel. Mol. Med. 20, 1686–1695. doi:10.1111/jcmm.12863
Leboucher, G. P., Tsai, Y. C., Yang, M., Shaw, K. C., Zhou, M., Veenstra, T. D., et al. (2012). Stress-induced Phosphorylation and Proteasomal Degradation of Mitofusin 2 Facilitates Mitochondrial Fragmentation and Apoptosis. Mol. Cel 47, 547–557. doi:10.1016/j.molcel.2012.05.041
Lee, S., Sterky, F. H., Mourier, A., Terzioglu, M., Cullheim, S., Olson, L., et al. (2012). Mitofusin 2 Is Necessary for Striatal Axonal Projections of Midbrain Dopamine Neurons. Hum. Mol. Genet. 21, 4827–4835. doi:10.1093/hmg/dds352
Li, D., Li, X., Guan, Y., and Guo, X. (2015). Mitofusin-2-mediated Tethering of Mitochondria and Endoplasmic Reticulum Promotes Cell Cycle Arrest of Vascular Smooth Muscle Cells in G0/G1 Phase. Acta Biochim. Biophys. Sinica 47, 441–450. doi:10.1093/abbs/gmv035
Li, Y. J., Cao, Y. L., Feng, J. X., Qi, Y., Meng, S., Yang, J. F., et al. (2019). Structural Insights of Human Mitofusin-2 into Mitochondrial Fusion and CMT2A Onset. Nat. Commun. 10, 4914. doi:10.1038/s41467-019-12912-0
Lynch, S., Boyett, J. E., Smith, M. R., and Giordano-Mooga, S. (2020). Sex Hormone Regulation of Proteins Modulating Mitochondrial Metabolism, Dynamics and Inter-organellar Cross Talk in Cardiovascular Disease. Front Cel Dev Biol. 8, 610516. doi:10.3389/fcell.2020.610516
Marín-García, J., and Akhmedov, A. T. (2016). Mitochondrial Dynamics and Cell Death in Heart Failure. Heart Fail. Rev. 21, 123–136. doi:10.1007/s10741-016-9530-2
Mattie, S., Riemer, J., Wideman, J. G., and McBride, H. M. (2018). A New Mitofusin Topology Places the Redox-Regulated C Terminus in the Mitochondrial Intermembrane Space. J. Cel Biol 217, 507–515. doi:10.1083/jcb.201611194
McLelland, G. L., Goiran, T., Yi, W., Dorval, G., Chen, C. X., Lauinger, N. D., et al. (2018). Mfn2 Ubiquitination by PINK1/parkin Gates the P97-dependent Release of ER from Mitochondria to Drive Mitophagy. Elife 7. doi:10.7554/elife.32866
Naon, D., Zaninello, M., Giacomello, M., Varanita, T., Grespi, F., Lakshminaranayan, S., et al. (2016). Critical Reappraisal Confirms that Mitofusin 2 Is an Endoplasmic Reticulum-Mitochondria Tether. Proc. Natl. Acad. Sci. USA 113, 11249–11254. doi:10.1073/pnas.1606786113
Neuspiel, M., Zunino, R., Gangaraju, S., Rippstein, P., and McBride, H. (2005). Activated Mitofusin 2 Signals Mitochondrial Fusion, Interferes with Bax Activation, and Reduces Susceptibility to Radical Induced Depolarization*. J. Biol. Chem. 280, 25060–25070. doi:10.1074/jbc.m501599200
Ong, S.-B., Hall, A. R., and Hausenloy, D. J. (2012). Mitochondrial Dynamics in Cardiovascular Health and Disease. Antioxid. Redox Signal. 19, 400–414. doi:10.1089/ars.2012.4777
Papanicolaou, K. N., Khairallah, R. J., Ngoh, G. A., Chikando, A., Luptak, I., O'Shea, K. M., et al. (2011). Mitofusin-2 Maintains Mitochondrial Structure and Contributes to Stress-Induced Permeability Transition in Cardiac Myocytes. Mol. Cell Biol. 31, 1309–1328. doi:10.1128/mcb.00911-10
Peng, C., Rao, W., Zhang, L., Gao, F., Hui, H., Wang, K., et al. (2018). Mitofusin 2 Exerts a Protective Role in Ischemia Reperfusion Injury through Increasing Autophagy. Cell Physiol Biochem 46, 2311–2324. doi:10.1159/000489621
Poole, A. C., Thomas, R. E., Yu, S., Vincow, E. S., and Pallanck, L. (2010). The Mitochondrial Fusion-Promoting Factor Mitofusin Is a Substrate of the PINK1/parkin Pathway. PLoS One 5, e10054. doi:10.1371/journal.pone.0010054
Qi, Y., Yan, L., Yu, C., Guo, X., Zhou, X., Hu, X., et al. (2016). Structures of Human Mitofusin 1 Provide Insight into Mitochondrial Tethering. J. Cel Biol 215, 621–629. doi:10.1083/jcb.201609019
Qin, Y., Gao, M., Li, A., Sun, J., Liu, B., and Gong, G. (2018). Mitoflash Lights Single Mitochondrial Dynamics Events in Mature Cardiomyocytes. Biochem. Biophysical Res. Commun. 503, 729–736. doi:10.1016/j.bbrc.2018.06.068
Qin, Y., Li, A., Liu, B., Jiang, W., Gao, M., Tian, X., et al. (2020). Mitochondrial Fusion Mediated by Fusion Promotion and Fission Inhibition Directs Adult Mouse Heart Function toward a Different Direction. FASEB j. 34, 663–675. doi:10.1096/fj.201901671r
Rizzuto, R., Pinton, P., Carrington, W., Fay, F. S., Fogarty, K. E., Lifshitz, L. M., et al. (1998). Close Contacts with the Endoplasmic Reticulum as Determinants of Mitochondrial Ca2+ Responses. Science 280, 1763–1766. doi:10.1126/science.280.5370.1763
Rocha, A. G., Franco, A., Krezel, A. M., Rumsey, J. M., Alberti, J. M., Knight, W. C., et al. (2018). MFN2 Agonists Reverse Mitochondrial Defects in Preclinical Models of Charcot-Marie-Tooth Disease Type 2A. Science 360, 336–341. doi:10.1126/science.aao1785
Santel, A., and Fuller, M. T. (2001). Control of Mitochondrial Morphology by a Human Mitofusin. J. Cel Sci 114, 867–874. doi:10.1080/152165401753544269
Schneeberger, M., Dietrich, M. O., Sebastián, D., Imbernón, M., Castaño, C., Garcia, A., et al. (2013). Mitofusin 2 in POMC Neurons Connects ER Stress with Leptin Resistance and Energy Imbalance. Cell 155, 172–187. doi:10.1016/j.cell.2013.09.003
Sebastián, D., Sorianello, E., Segalés, J., Irazoki, A., Ruiz‐Bonilla, V., Sala, D., et al. (2016). Mfn2 Deficiency Links Age‐related Sarcopenia and Impaired Autophagy to Activation of an Adaptive Mitophagy Pathway. EMBO J. 35, 1677–1693. doi:10.15252/embj.201593084
Shen, T., Zheng, M., Cao, C., Chen, C., Tang, J., Zhang, W., et al. (2007). Mitofusin-2 Is a Major Determinant of Oxidative Stress-Mediated Heart Muscle Cell Apoptosis. J. Biol. Chem. 282, 23354–23361. doi:10.1074/jbc.m702657200
Shutt, T., Geoffrion, M., Milne, R., and McBride, H. M. (2012). The Intracellular Redox State Is a Core Determinant of Mitochondrial Fusion. EMBO Rep. 13, 909–915. doi:10.1038/embor.2012.128
Sloat, S. R., Whitley, B. N., Engelhart, E. A., and Hoppins, S. (2019). Identification of a Mitofusin Specificity Region that Confers Unique Activities to Mfn1 and Mfn2. MBoC 30, 2309–2319. doi:10.1091/mbc.e19-05-0291
Sun, D., Li, C., Liu, J., Wang, Z., Liu, Y., Luo, C., et al. (2019). Expression Profile of microRNAs in Hypertrophic Cardiomyopathy and Effects of microRNA-20 in Inducing Cardiomyocyte Hypertrophy through Regulating Gene MFN2. DNA Cel Biol. 38, 796–807. doi:10.1089/dna.2019.4731
Tanaka, A., Cleland, M. M., Xu, S., Narendra, D. P., Suen, D.-F., Karbowski, M., et al. (2010). Proteasome and P97 Mediate Mitophagy and Degradation of Mitofusins Induced by Parkin. J. Cel Biol 191, 1367–1380. doi:10.1083/jcb.201007013
Volterrani, M., Rosano, G., and Iellamo, F. (2012). Testosterone and Heart Failure. Endocrine 42, 272–277. doi:10.1007/s12020-012-9725-9
Wang, W., Liu, X., Guo, X., and Quan, H. (2018). Mitofusin-2 Triggers Cervical Carcinoma Cell Hela Apoptosis via Mitochondrial Pathway in Mouse Model. Cel Physiol Biochem 46, 69–81. doi:10.1159/000488410
Wang, W., Lu, J., Zhu, F., Wei, J., Jia, C., Zhang, Y., et al. (2012). Pro-apoptotic and Anti-proliferative Effects of Mitofusin-2 via Bax Signaling in Hepatocellular Carcinoma Cells. Med. Oncol. 29, 70–76. doi:10.1007/s12032-010-9779-6
Wang, W., Xie, Q., Zhou, X., Yao, J., Zhu, X., Huang, P., et al. (2015). Mitofusin-2 Triggers Mitochondria Ca2+ Influx from the Endoplasmic Reticulum to Induce Apoptosis in Hepatocellular Carcinoma Cells. Cancer Lett. 358, 47–58. doi:10.1016/j.canlet.2014.12.025
Wu, L., Li, Z., Zhang, Y., Zhang, P., Zhu, X., Huang, J., et al. (2008). Adenovirus-expressed Human Hyperplasia Suppressor Gene Induces Apoptosis in Cancer Cells. Mol. Cancer Ther. 7, 222–232. doi:10.1158/1535-7163.mct-07-0382
Xiong, W., Ma, Z., An, D., Liu, Z., Cai, W., Bai, Y., et al. (2019). Mitofusin 2 Participates in Mitophagy and Mitochondrial Fusion against Angiotensin II-Induced Cardiomyocyte Injury. Front. Physiol. 10, 411. doi:10.3389/fphys.2019.00411
Yang, L., Long, Q., Liu, J., Tang, H., Li, Y., Bao, F., et al. (2015). Mitochondrial Fusion Provides an 'initial Metabolic Complementation' Controlled by mtDNA. Cell. Mol. Life Sci. 72, 2585–2598. doi:10.1007/s00018-015-1863-9
Yu, H., Guo, Y., Mi, L., Wang, X., Li, L., and Gao, W. (2011). Mitofusin 2 Inhibits Angiotensin II-Induced Myocardial Hypertrophy. J. Cardiovasc. Pharmacol. Ther. 16, 205–211. doi:10.1177/1074248410385683
Zhang, G.-E., Jin, H.-L., Lin, X.-K., Chen, C., Liu, X.-S., Zhang, Q., et al. (2013). Anti-Tumor Effects of Mfn2 in Gastric Cancer. Ijms 14, 13005–13021. doi:10.3390/ijms140713005
Zhao, T., Huang, X., Han, L., Wang, X., Cheng, H., Zhao, Y., et al. (2012). Central Role of Mitofusin 2 in Autophagosome-Lysosome Fusion in Cardiomyocytes. J. Biol. Chem. 287, 23615–23625. doi:10.1074/jbc.m112.379164
Keywords: Mfn2, mitochondria fusion, endoplasmic reticulum–mitochondria contacts, mitophagy, heart failure
Citation: Chen L, Liu B, Qin Y, Li A, Gao M, Liu H and Gong G (2021) Mitochondrial Fusion Protein Mfn2 and Its Role in Heart Failure. Front. Mol. Biosci. 8:681237. doi: 10.3389/fmolb.2021.681237
Received: 16 March 2021; Accepted: 21 April 2021;
Published: 07 May 2021.
Edited by:
Sang-Bing Ong, The Chinese University of Hong Kong, ChinaReviewed by:
Claudia Penna, University of Turin, ItalyCopyright © 2021 Chen, Liu, Qin, Li, Gao, Liu and Gong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guohua Gong, Z3VvaGdvbmdAdG9uZ2ppLmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.