 Thomas Paß
Thomas Paß Rudolf J. Wiesner1,2
Rudolf J. Wiesner1,2 David Pla-Martín
David Pla-Martín- 1Center for Physiology and Pathophysiology, Institute of Vegetative Physiology, University of Cologne, Cologne, Germany
- 2Cologne Excellence Cluster on Cellular Stress Responses in Aging Associated Diseases (CECAD), University of Cologne, Cologne, Germany
Mitochondrial dysfunction is a central feature of neurodegeneration within the central and peripheral nervous system, highlighting a strong dependence on proper mitochondrial function of neurons with especially high energy consumptions. The fitness of mitochondria critically depends on preservation of distinct processes, including the maintenance of their own genome, mitochondrial dynamics, quality control, and Ca2+ handling. These processes appear to be differently affected in common neurodegenerative diseases, such as Alzheimer’s and Parkinson’s disease, as well as in rare neurological disorders, including Huntington’s disease, Amyotrophic Lateral Sclerosis and peripheral neuropathies. Strikingly, particular neuron populations of different morphology and function perish in these diseases, suggesting that cell-type specific factors contribute to the vulnerability to distinct mitochondrial defects. Here we review the disruption of mitochondrial processes in common as well as in rare neurological disorders and its impact on selective neurodegeneration. Understanding discrepancies and commonalities regarding mitochondrial dysfunction as well as individual neuronal demands will help to design new targets and to make use of already established treatments in order to improve treatment of these diseases.
Introduction
Mitochondria are dynamic, double-membrane-surrounded organelles executing a wide range of essential functions within the cell, including ATP production, metabolism of amino acids, lipids and nucleotides, iron-sulfur cluster synthesis, ion homeostasis, and programmed cell death. The pivotal role of mitochondria for cellular survival is highlighted by the variety of diseases that are associated with mitochondrial dysfunction in diverse tissues. Neurons especially depend on proper mitochondrial function due to their extremely high energetic demands, with maintaining resting membrane potentials and firing of action potentials being the largest energy guzzlers (Howarth et al., 2012). Whereas accounting for only 2% of the whole body mass, the brain consumes 20% of the body’s total oxygen amounts during ATP generation (Attwell and Laughlin 2001). 75–80% of the brains’ energy is thereby used up by neurons (Harris et al., 2012; Hyder et al., 2013), and in contrast to other neural cells, mitochondrial OXPHOS carries most of the burden (Belanger et al., 2011; Rangaraju et al., 2014; Zhang et al., 2014). Therefore, neurons are locally supported by astrocytes, providing additional lactate (Weber and Barros 2015). Moreover, neuronal mitochondria serve as dynamic key regulators of intracellular Ca2+. In collaboration with the endoplasmic reticulum (ER), mitochondria control somato-dendritic Ca2+ levels (Rizzuto et al., 1998; Kornmann et al., 2009; Hirabayashi et al., 2017) and buffer Ca2+ at presynaptic terminals as well as axonal boutons, in order to regulate neurotransmission (Billups and Forsythe 2002; Kwon et al., 2016; Marland et al., 2016; de Juan-Sanz et al., 2017; Vaccaro et al., 2017). Maintenance of mitochondrial fitness is therefore of great importance and requires efficient quality control mechanisms (Rugarli and Langer 2012), which are challenging regarding the extended and complex neuron morphology (Misgeld and Schwarz 2017).
Perturbations of mitochondrial functions, whether they are of primary cause or not, are accordingly associated with neuronal death in common as well as rare neurological disorders. Mitochondrial dysfunction becomes apparent by impaired activity of respiratory chain complexes, which eventually impacts oxidative phosphorylation (OXPHOS), and thereby ATP generation. Respiratory chain deficiency is found in patients suffering from Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), Amyotrophic Lateral Sclerosis (ALS) (Golpich et al., 2017) spinocerebellar ataxia (Lax et al., 2012), and other peripheral neuropathies, such as Charcot-Marie-Tooth disease (Rizzo et al., 2016). However, defects of the respiratory chain are only the tip of the iceberg. In general, they are preceded by disruption of distinct other mitochondrial processes, including mitochondrial DNA (mtDNA) maintenance, mitochondrial dynamics, quality control, and Ca2+ handling.
Remarkably, neurodegenerative diseases usually show selective vulnerability of diverse neuron populations, even in familial cases with monogenic mutations ubiquitously present in the body: 1) In AD, pyramidal neurons in the entorhinal cortex layer II (ECII) and the hippocampal CA1 (cornu ammonis 1) region degenerate first, causing cognitive decline and memory loss (Hyman et al., 1984; Arnold et al., 1991; Gomez-Isla et al., 1996; Fukutani et al., 2000; Bussiere et al., 2003). 2) In PD patients, characteristic motor symptoms are induced by the loss of midbrain dopaminergic neurons in the substantia nigra pars compacta (SNc) (Michel et al., 2016; Obeso et al., 2017), whereas 3) in HD, motor impairment is based on the decline of GABAergic medium spiny neurons located in the striatum (Halliday et al., 1998; Vonsattel 2008). 4) Spinal motor neurons innervating fast-twitch muscles selectively perish in ALS and result in muscle atrophy and spasticity (Nijssen et al., 2017). 5) Spinocerebellar ataxia is mainly caused by the loss of cerebellar Purkinje cells (Durr 2010), and lastly, 6) Charcot-Marie-Tooth disease is characterized by the decline of peripheral nerves (Reilly et al., 2010; Saporta et al., 2011).
The selective neurodegeneration associated with disruption of distinct mitochondrial processes, raises the question whether cell-type specific properties contribute to an enhanced susceptibility to mitochondrial defects. Here, we review distinct sources for mitochondrial dysfunction in selected common and rare neurological disorders, discuss their primary role in degeneration of specific neuron types and summarize current and potential approaches against neurodegeneration. Understanding the vulnerability of different neuronal populations to specific mitochondrial impairment will prove crucial to disease-specific development of therapies as well as to usage of established drugs for disease-spanning treatment.
Mitochondrial Pathways Affected in Neurological Disorders
For a long time, impaired mitochondrial OXPHOS has been the most obvious and exclusive explanation for neuronal death associated with mitochondrial dysfunction. With growing knowledge, however, mitochondria have no longer been solely restricted to energy supply. Today we know that mitochondria form a mobile and interactive network. Disturbances in mobility, fusion and fission are observed in common as well as rare neurological disorders, many of them being even caused by mitochondrial-related gene mutations, and led to a better understanding of pathological mechanisms underlying those diseases.
Mitochondrial DNA Maintenance and Disorders
Mitochondria contain their own double-stranded DNA, which is densely packed, containing 37 genes in 16.6 kb of its sequence, and present in thousands of copies in neurons. All of the 13 mRNAs encode subunits of the respiratory chain complexes, while 22 tRNAs and two rRNAs are necessary for the mitochondrial translation machinery. Although presenting only the minority of OXPHOS proteins, the 13 mtDNA-encoded subunits are essential since OXPHOS collapses in the absence of mtDNA expression (Larsson et al., 1998; Gustafsson et al., 2016). Regarding that the mitochondrial proteome consists of ∼1.200 different proteins (Sickmann et al., 2003; Foster et al., 2006), the vast majority of mitochondrial proteins are nuclear-encoded and imported into mitochondria while being translated. Consequently, transcription, translation, maintenance, and replication of mtDNA are regulated by hundreds of nuclear-encoded and imported proteins (Gustafsson et al., 2016). Therefore, inherited mutations of mtDNA but also nuclear genes encoding respiratory chain subunits and proteins regulating mtDNA maintenance, respectively, are causing mtDNA diseases with a wide range of clinical manifestations (Schapira 2012). Symptoms of mtDNA diseases that are related to mutations in the mtDNA only arise when the copy number of mutated molecules surpasses a certain threshold (Picard et al., 2016). The presence of both wild type and mutant mtDNA molecules is known as mtDNA heteroplasmy.
Transcription of mtDNA is initiated by TFAM (mitochondrial transcription factor A), which binds to mitochondrial promoters (Ngo et al., 2011; Shi et al., 2012) and enables recruitment of the mitochondrial RNA polymerase (Yakubovskaya et al., 2014). Simultaneously, TFAM is also responsible for mtDNA maintenance since it entirely coats mtDNA molecules and thus mediates the formation of nucleo-protein structures called nucleoids (Figure 1) (Kaufman et al., 2007; Kukat et al., 2015). In humans, mutation of TFAM causes severe mitochondrial depletion syndrome, whereas in mice, the whole body knockout (KO) displayed embryonic lethality due to severe mtDNA depletion (Table 1; Stiles et al., 2016). Interestingly, cell type-specific KO of Tfam in dopaminergic neurons mimics the key features of PD pathology in mice. These so called “MitoPark” animals showed rapid and selective loss of dopaminergic neurons in the SNc due to mtDNA depletion, which was accompanied by progressive motor impairment (Ekstrand et al., 2007). Accordingly, low protein levels of TFAM together with reduced mtDNA copy number and complex I deficiency have been detected in SNc dopamine neurons from idiopathic PD patients (Grunewald et al., 2016), suggesting mtDNA depletion to be a critical factor for selective neurodegeneration following mitochondrial dysfunction in PD. Reduced TFAM levels and/or mtDNA copy number have been measured in affected regions of the nervous system from patients of other neurodegenerative diseases as well, including AD (Coskun et al., 2004; Coskun et al., 2012; Rice et al., 2014; Wei et al., 2017), ALS (Keeney and Bennett 2010; Thau et al., 2012; Ladd et al., 2017) and HD (Kim et al., 2010). The vital importance of reduced mtDNA copy number in these disorders is however still disputed. Recently, increased mtDNA levels within the cytosol have been associated with neurodegeneration (Jauhari et al., 2021). Nevertheless, the release of mtDNA, which has been linked to inflammation, is likely to be a consequence of mitochondrial dysfunction in the first place (Sprenger et al., 2021), questioning its potential as a therapeutic target in common and rare neurological disorders.
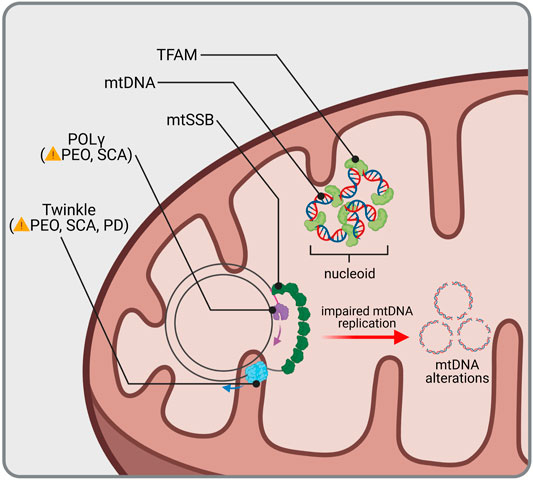
FIGURE 1. mtDNA homeostasis and related diseases. mtDNA is protected and condensed by TFAM, building an organized DNA-protein complex known as nucleoid. Replication of mtDNA requires coordination of the mitochondrial helicase Twinkle and DNA polymerase γ (POLγ). Mutations in genes encoding for Twinkle and POLγ lead to Progressive External Ophthalmoplegia (PEO), Spinocerebellar Ataxia (SCA) and Parkinsonism (PD). Impaired mtDNA replication is accompanied with mtDNA alterations, such as loss of wild type mtDNA or accumulation of mtDNA deletions, which have not only been found in rare or common diseases but also during aging.

TABLE 1. Proteins related to disturbed mtDNA maintenance.
Besides mtDNA depletion, mitochondrial dysfunction in PD is further linked to the presence of deleted mtDNA molecules. In healthy aged individuals (Kraytsberg et al., 2006) but especially in patients with idiopathic PD (Bender et al., 2006), SNc dopaminergic neurons accumulate high loads of mtDNA deletions which are accompanied with respiratory chain deficiency. Accumulation of deletions is driven by catecholamine metabolism (Neuhaus et al., 2014; Neuhaus et al., 2017), explaining why especially dopamine-expressing neurons are hotspots for such alterations in the mtDNA. Importantly, the high deletion load replaces populations of wild-type mtDNA in PD patients, whereas during normal aging, a sufficient pool of wild-type mtDNA can be maintained by upregulation of the total mtDNA copy number (Dolle et al., 2016). The mechanism for the formation of deleted mtDNA molecules is still not fully understood, however, it is suggested that deletions are generated by misrepaired double strand breaks upon mtDNA damage (Krishnan et al., 2008), followed by nuclease activity preceding re-ligation and during incorrect mtDNA replication (Reeve et al., 2008).
Replication of mtDNA is mediated by DNA polymerase-γ (POLγ). Together with the replicative mtDNA helicase TWINKLE and the mitochondrial single-stranded DNA-binding protein (mtSSB), it forms the mitochondrial replisome, which is able to replicate both mtDNA strands in a continuous manner (Figure 1; Gustafsson et al., 2016). Multiple mutations in POLG as well as TWNK have been identified and associated with a wide range of diseases usually showing neuromuscular defects, such as progressive external ophthalmoplegia, as a consequence of mtDNA depletion or accumulation of mtDNA deletions (Table 1; Copeland 2014). Ataxia is a prominent clinical feature among mitochondrial diseases related to mutated POLG (Van Goethem et al., 2004; Hakonen et al., 2005). Whereas it was first reported to be mostly of sensory nature with no or only mild cerebellar atrophy (Synofzik et al., 2012), Turnbull and colleagues showed that degeneration of Purkinje cells following mitochondrial dysfunction was most pronounced in patients with POLG mutations (Lax et al., 2012). In line with this, mtDNA heteroplasmy associated with complex I deficiency was found in cerebellar Purkinje cells of mitochondrial disease patients suffering from ataxia, including patients with POLG mutations (Chrysostomou et al., 2016), supporting a cerebellar involvement in ataxias. Patients harboring TWNK mutations are presenting with spinocerebellar ataxias (Hudson et al., 2005; Hakonen et al., 2007; Lonnqvist et al., 2009). Correspondingly, respiratory chain-deficient Purkinje cells were detected in mice expressing mutant Twinkle (Tyynismaa et al., 2005). Insights of case studies thereby point to special vulnerability of cerebellar Purkinje cells to mtDNA alterations due to impaired replication.
In line with the high load of mtDNA deletions in SNc dopaminergic neurons of idiopathic PD patients, parkinsonian features were additionally seen in some patients harboring mutations of POLG (Luoma et al., 2004; Hudson et al., 2007) and TWNK (Suomalainen et al., 1997; Baloh et al., 2007; Kiferle et al., 2013). Moreover, mtDNA deletions of idiopathic PD patients were very similar to those found in POLG patients (Reeve et al., 2008). Interestingly, extensive study of POLG cases indeed revealed severe nigrostriatal degeneration, but lacking characteristic motor symptoms for PD (Tzoulis et al., 2013). The same was true for patients with TWNK mutations (Palin et al., 2013), whereas a case of familial parkinsonism with heterozygous mutation in TWNK was recently reported (Breen et al., 2020). Taken together, these data indicate that mtDNA maintenance plays a decisive role for survival of SNc dopaminergic neurons and PD pathogenesis.
Mitochondrial Morphology, Fission and Fusion
From isolated organelles being fixed at a certain place within the cell, the image of mitochondria has changed to an interconnected reticulum, which is continuously separating and fusing. Mitochondrial fission and fusion are dynamic processes which are influenced by the cell’s metabolic needs. Whereas fission generates new mitochondrial particles and can contribute to removal of defective compartments at the same time, fusion is crucial to distribute the load upon increased metabolic demands and mitochondrial damage, respectively (Youle and van der Bliek 2012), leading to constant rejuvenation of the mitochondrial pool. In contrast to their hosting neurons, proteins only last for weeks or even days (Goldberg 2003). In addition, the majority of mitochondrial proteins are encoded in the nucleus (Calvo et al., 2016). Together with the fact that intact mitochondria are substantially transported anterogradely along the axon (Lin et al., 2017), mitochondrial biogenesis is thought to mainly occur in the Soma. Considering mitochondrial transport velocity along the axon (∼0.5 μm/s) and the lifetime of mitochondrial proteins, rejuvenation of those mitochondrial populations, which reside in large distance from their point of origin, is of great importance for neuronal functionality (Misgeld and Schwarz, 2017). In support of this, a recent study suggests the fitness of mitochondria to also depend on its distance from the cell body (Baranov et al., 2019).
Mitochondrial fission is initiated by tubules of the ER constricting a mitochondrial segment, followed by recruitment of the GTPase DRP1 (dynamin-related protein 1), which completes separation (Figure 2) (Friedman et al., 2011). Mutations in DNML1, which encodes DRP1, have been associated with rare cases of neonatal encephalopathy and epilepsy (Table 2; Bitoun et al., 2007; Fahrner et al., 2016; Vanstone et al., 2016). Remarkably, other mutations in genes encoding for regulators of DRP1 function have been linked to neurodegenerative disorders: Mutations in SACSIN lead to the development of Spastic Ataxia of Charlevoix Saguenay (ARSACS) due to decreased recruitment of DRP1 to mitochondria (Bradshaw et al., 2016). Furthermore, mutations in REEP1 causing Hereditary Spastic Paraplegia 31 (SPG31) induce hyperphosphorylation of DRP1, hence inhibiting its activity (Lavie et al., 2017).
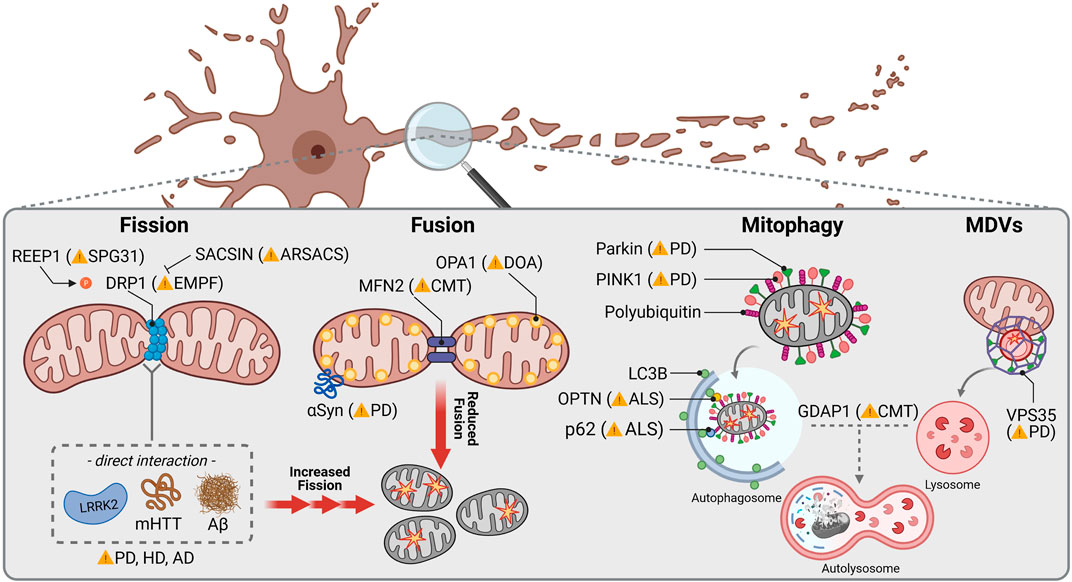
FIGURE 2. Mitochondrial dynamics in common and rare neurodegenerative diseases. Mitochondrial fission is orchestrated by DRP1, which mutations have been linked to autosomal dominant encephalopathy and neonatal lethality caused by defective mitochondrial and peroxisomal fission. Mutations in REEP1 are associated with autosomal dominant Spastic Paraplegia Type 31 (SPG31) due to hyperphosphorylation of DRP1 causing increased mitochondrial fission. In contrast, SACSIN mutations lead to autosomal recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS) underlying reduced DRP1 recruitment to mitochondrial membranes and decreased fission. PD, HD and AD associated proteins LRKK2, mHTT and Aβ, respectively, have been reported to directly interact with DRP1 stimulating mitochondrial fission. Mitochondrial fusion is regulated by the OMM protein MFN2 and the inner mitochondrial membrane protein OPA1, whose mutations cause autosomal dominant Charcot-Marie-Tooth disease (CMT) and Optic atrophy (DOA), respectively. αSyn interacts with TOM20 which is accompanied with a lower mitochondrial fusion rate. Specific removal of mitochondria through mitophagy in neurons is controlled by PINK1 and Parkin, mutations of both being related to early juvenile recessive PD. PINK1 signalizes depolarized mitochondria by inducing recruitment of Parkin and starting a signaling cascade to remove defective mitochondria through autophagosome-lysosome degradation. Other receptors linked to recessive forms of ALS are the mitophagy specific adaptor protein OPTN and the general autophagy adaptor p62. CMT-related GDAP1 mediates contacts between mitochondria and lysosomes and is hence thought to be involved in mitochondrial removal as well. A higher specialized pathway to eliminate mitochondrial fragments is known to pursue the mitochondrial-derived vesicles (MDVs) trail, where VPS35, linked to dominant forms of PD, triggers the force to generate vesicles which eventually fuse with lysosomes.
TABLE 2. Proteins related to impaired mitochondrial dynamics.
Mitochondrial fusion requires merging of the outer mitochondrial membrane (OMM), mediated by mitofusins (MFN1/2), as well as coupling of the inner mitochondrial membrane through OPA1 (optic atrophy 1) (Chen et al., 2003; Eura et al., 2003). While OPA1 mutations are associated with optic atrophy (Amati-Bonneau et al., 2005; Zanna et al., 2008), mutations in MFN2 are found in patients with Charcot-Marie-Tooth disease axonal form type 2A (CMT2A), an autosomal dominant form of Charcot-Marie-Tooth (Table 2; Zuchner et al., 2006). This subtype is characterized by peripheral neuropathy primarily affecting motor neurons (Verhoeven et al., 2006; Feely et al., 2011). According to the absence of OPA1, loss of MFN2 has been shown to cause fragmentation of the mitochondrial network in cultured embryonic fibroblasts and membrane potential breakdown in a subpopulation of the arising fragmented mitochondria (Chen et al., 2007; Chen et al., 2010). Besides fusion, MFN2 is necessary for transport of axonal mitochondria through interaction with MIRO1 (mitochondrial Rho GTPase 1) (Misko et al., 2010). MIRO1 is an OMM protein anchoring the microtubule motors kinesin and dynein to mitochondria (Melkov and Abdu 2018). Accordingly, mitochondria are inadequately distributed along axons in a transgenic mouse model of CMT2A carrying a pathogenic Mfn2 mutation (Detmer et al., 2008).
In line with the strong dependence of SNc dopaminergic neurons on proper mitochondrial functionality, KO of Mfn2 in dopaminergic neurons leads to severe motor symptoms in mice due to early loss of axonal projections (Lee et al., 2012; Pham et al., 2012). PINK1 and Parkin, whose encoding genes are associated with recessive PD when mutated, have been shown to regulate MFN1 and MFN2 levels (Yang et al., 2008; Tanaka et al., 2010; Glauser et al., 2011). Impaired mitochondrial fusion is suggested to contribute to progressive degeneration of SNc dopaminergic neurons in PD. Indeed, investigation of post mortem brain samples of idiopathic PD patients revealed decreased levels of the short form of OPA1 (Zilocchi et al., 2018).
Furthermore, mutations of LRRK2 causing autosomal dominant PD (Zimprich et al., 2004), are linked to altered mitochondrial morphology (Yue et al., 2015), including elongated mitochondria (Mortiboys et al., 2010). However, most studies reported mitochondrial fragmentation, probably induced by decreased levels of the short form of OPA1 (Stafa et al., 2014) as well as direct interaction of mutant LRRK2 with DRP1, promoting mitochondrial fission (Figure 2; Wang et al., 2012; Su and Qi 2013; Smith et al., 2016; Perez Carrion et al., 2018). Mutant LRRK2 further affects mitochondrial trafficking. Whereas under physiological conditions LRRK2 is thought to promote MIRO1 removal, mutant LRRK2 disrupts this function, resulting in accumulation of the anchoring protein and hence in delay of proper mitochondrial removal. According to similarly enhanced protein levels of MIRO1 in fibroblasts from idiopathic PD patients, mitochondrial trafficking is thought to be generally altered in PD (Hsieh et al., 2016). Noteworthy, mitochondrial dynamics in PD can be affected by α-Syn. Aggregates of α-Syn are the main component of intracellular Lewy bodies, the cellular hallmark for many idiopathic PD cases (Spillantini et al., 1997) as well as familial forms showing mutations in the α-Syn encoding gene SNCA (Polymeropoulos et al., 1997). α-Syn, and especially its aggregated form (Wang et al., 2019), is able to bind to the OMM (Di Maio et al., 2016; Hu et al., 2019). Interaction with α-Syn has been shown to decrease mitochondrial fusion rate (Figure 2; Devoto et al., 2017). Moreover, transgenic mice overexpressing α-Syn revealed reduced levels of MFN1 and MFN2, which was accompanied by shortened mitochondria (Xie and Chung 2012), whereas knockdown of α-Syn resulted in mitochondrial elongation (Kamp et al., 2010).
Morphological alterations of mitochondria were one of the first observations made in affected neurons of ALS patients (Atsumi 1981; Sasaki and Iwata 2007). Like in CMT2A, long motor neurons primarily perish. In contrast, only 10% of ALS cases have a familial background, including the most frequently mutated genes SOD1 (superoxide dismutase 1), TDP-43 (TAR DNA binding protein) and FUS (fused in sarcoma). The majority of cases, however, remain idiopathic (Zou et al., 2017). In vitro as well as in vivo models of ALS, which have been established in order to investigate the consequences upon expression of mutant SOD1, TDP-43 and FUS, respectively, revealed both aggregation and fragmentation of mitochondria (Dal Canto and Gurney 1994; Higgins et al., 2003; De Vos et al., 2007; Vande Velde et al., 2011; Hong et al., 2012; Wang W et al., 2013; Magrane et al., 2014), suggesting impaired mitochondrial dynamics to be a disease-contributing factor. According to this, many groups reported an imbalance between levels of fission and fusion protein: transgenic mouse as well as cell culture models expressing SOD1 G93A and mutant TDP-43, respectively, showed decreased levels of fusion-mediating proteins, such as MFN1 and OPA1, with simultaneously increased fission-mediating proteins, like DRP1 and FIS1 (Ferri et al., 2010; Xu et al., 2010; Liu et al., 2013; Onesto et al., 2016). Protein levels thereby point to a condition promoting fission, explaining shortened and fragmented mitochondria.
In HD, which is known to be caused by a CAG repeat expansion in the huntingtin gene (HTT) generating an expanded polyglutamine stretch in the HTT protein (MacDonald et al., 1993), analysis of striatal lysates from patients indicates a shift to mitochondrial fission, too. Similar to observations made in ALS, increased DRP1 and simultaneously reduced MFN1 levels were measured (Kim et al., 2010), which was further supported by mRNA expression levels showing upregulation of DRP1 and FIS1 towards downregulation of MFN1, MFN2 and OPA1 (Shirendeb et al., 2011). Correspondingly, fragmented mitochondria have been detected in striatal cells from mutant HTT (mHTT) transgenic mice (Napoli et al., 2013). In particular, mHTT abnormally binds DRP1 (Figure 2), which led to mitochondrial fragmentation in transgenic rodent HD models as well as in fibroblasts from HD patients (Song et al., 2011). Simultaneously, mHTT blocks mitochondrial transport (Orr et al., 2008; Tian et al., 2014), especially when it contains polyglutamine repeats (Chang et al., 2006).
Due to their clinical manifestation in mitochondrial DNA diseases (Lax et al., 2012), cerebellar ataxias have been investigated for abnormalities in mitochondrial morphology as well. Interestingly, both fragmentation and elongation of mitochondria have been observed in related model organisms. Mutant ATXN3, which is linked to spinocerebellar ataxia type 3 (Kawaguchi et al., 1994), caused mitochondrial fission in neuroblastoma cells and transgenic mice, with decreased protein levels of MFN1 and MFN2 (Hsu et al., 2017). Based on a study expressing mutant ppp2r2b in Drosophila melanogaster, mitochondrial fragmentation induced by fission is also thought to occur in spinocerebellar ataxia type 12 (Wang et al., 2011). Another type of spinocerebellar ataxia is referred to mutations in AFG3L2. AFG3L2 is a subunit of m-AAA proteases, whose loss affects mitochondrial protein synthesis and respiration (Patron et al., 2018). The absence of AFG3L2 in Purkinje cells triggers mitochondrial fragmentation as well as altered distribution of mitochondria in the dendritic tree (Almajan et al., 2012). Fragmented mitochondria and defective mitochondrial trafficking were further observed in murine cortical neurons following AFG3L2 depletion (Kondadi et al., 2014). In particular, it is suggested that the inner membrane peptidase OMA1 is activated by absence of the m-AAA protease (Ehses et al., 2009), triggering OPA1 cleavage (Head et al., 2009) and thereby facilitating mitochondrial fission (Anand et al., 2014). Mice expressing mutant ATXN7, which is linked to spinocerebellar ataxia type 7, revealed mitochondrial network fragmentation in Purkinje cells. Counterintuitively, enlarged mitochondria have been identified by ultrastructural analysis (Ward et al., 2019). In addition, brain-specific loss of DRP1 causes degeneration of Purkinje cells with simultaneously oversized mitochondria failing to spread into neuronal projections (Delettre et al., 2000; Ishihara et al., 2009). These observations are supported by an animal model of spastic ataxia of Charlevoix-Saguenay, in which mice do not express the DRP1-binding protein sacsin: animals showed similar features to mice lacking DRP1, including hyperfused mitochondria and loss of Purkinje cells (Girard et al., 2012), rendering proper mitochondrial fission of critical importance for Purkinje cell survival.
Investigation of patient-derived fibroblasts and in vitro models exposed to high Aβ levels pointed to mitochondrial fragmentation and accumulation of dysfunctional mitochondria in AD (DuBoff et al., 2013; Nixon 2013). Some studies could correspondingly demonstrate increased DRP1 as well as decreased MFN1, MFN2 and OPA1 protein levels in brains of AD patients as well as mice (Cho et al., 2009; Wang et al., 2009), whereas others showed conflicting results (Wang et al., 2008). Aβ aggregates could play a critical role for the observed disturbances in mitochondrial dynamics: impaired transport and fragmentation of mitochondria were observed upon overexpression of the Aβ precursor protein in primary neurons, which was connected to a direct interaction of Aβ and DRP1 (Figure 2; Manczak et al., 2011). Interestingly, inhibition of mitochondrial fission could ameliorate aberrant Aβ accumulation in transgenic mice, which was associated with prevention of cognitive deficits (Wang et al., 2017). This indicates that impaired mitochondrial dynamics is not only a consequence of aberrant Aβ aggregates, but could affect plaque formation and might hence be a critical factor for AD pathogenesis. Besides Aβ, pTau is thought to interfere with mitochondrial trafficking. Usually binding and stabilizing microtubules, Tau detaches from microtubules in its hyperphosphorylated form causing microtubule destabilization (Alonso et al., 1994; LeBoeuf et al., 2008). Although the exact mechanism how pTau affects mitochondrial function still remains unclear, it is most likely, that general impairment of axonal transport due to pTau (Combs et al., 2019) also hits mitochondrial movement. In addition, Aβ also affects the processing of pre-proteins imported from the cytosol and thereby mitochondrial proteostasis (Mossmann et al., 2014), leading to instability of OXPHOS complexes, impaired oxygen consumption and reduced mitochondrial membrane potential.
Mitophagy
Besides constant rejuvenation via fission and fusion, efficient quality control is indispensable in order to maintain a healthy mitochondrial pool in somatic and especially axonal compartments. Autophagy functions as a neuronal safeguard and removes large compartments of the mitochondrial network in a controlled manner, which is replaced by ongoing biogenesis. This so called mitophagy is initiated by the mitochondrial serine/threonine kinase PINK1 (PTEN-induced putative kinase 1). Upon mitochondrial damage, PINK1 is no longer cleaved by mitochondrial proteases, such as PARL (Jin et al., 2010; Deas et al., 2011), which is usually followed by its import via the TOM (translocase of the outer membrane) (Lazarou et al., 2012) and TIM (translocase of the inner membrane) complexes (Sekine and Youle 2018). This leads to the accumulation of PINK1 on the OMM (Lin and Kang 2008; Zhou et al., 2008). PINK1 hence phosphorylates ubiquitin chains and activates the E3 ubiquitin ligase Parkin (Figure 2; Kondapalli et al., 2012; Ordureau et al., 2014). In turn, Parkin further ubiquitinates OMM proteins (Chan et al., 2011; Sarraf et al., 2013) inducing recruitment of autophagosomes and lysosomes (Matsuda et al., 2010). Mutations of both PINK1 and Parkin (PARK2) are the most common cause of autosomal recessive PD with predominantly early onset (Table 2; Kilarski et al., 2012; Truban et al., 2017). Thus, impaired mitochondrial quality control is obviously a key factor contributing to the loss of dopaminergic neurons in PD. However, the extensive functions of PINK1 and Parkin are not fully elucidated. Loss of PINK1 activity has been primarily related to decreased mitochondrial membrane potential as well as complex I and III deficiency (Amo et al., 2014). Furthermore, mitochondrial Ca2+ overload-induced cell death of PINK1-deficient dopaminergic neurons suggests an involvement of PINK1 in Ca2+ handling (Kostic et al., 2015). Despite the conserved role of Parkin in diverse mammalian cell lines, its relevance in neuronal mitophagy has been questioned. Some studies reported an only moderate Parkin contribution to degradation of damaged mitochondria in primary neurons (Cai et al., 2012; Grenier et al., 2013), whereas others confirmed the importance of Parkin for mitophagy in these cells (Grenier et al., 2014) and especially axons (Ashrafi et al., 2014). Interestingly, Parkin-deficient mice (Perez and Palmiter 2005) as well as rats (Dave et al., 2014) did not reveal a PD-related phenotype, and neither did the absence of Parkin influence selective neurodegeneration in MitoPark mice (Sterky et al., 2011). Accordingly, alternative mitophagy key players have been recently identified to regulate mitochondrial turnover in absence of Parkin (Villa et al., 2018). However, Parkin depletion caused degeneration of SNc dopaminergic neurons and motor symptoms upon impaired mtDNA replication (Pickrell et al., 2015), highlighting the importance of Parkin for dopaminergic neuron survival in face of accumulating mtDNA deletions.
Mitophagy depends on specific receptors which interact with the autophagosomal protein LC3 (microtubule-associated protein 1A/1B light chain 3). Among them, p62 was found to act as receptor for degradation of ubiquitinated proteins (Johansen and Lamark 2011). p62 affinity to ubiquitinated cargo is thereby facilitated by the kinases ULK1 (Unc-51 like autophagy activating kinase), CK2 (casein kinase 2) and TBK1 (TANK binding kinase 1) (Deng et al., 2017). Mutations in p62 encoding SQSTM1 have been identified in patients suffering from ALS (Figure 2) associated with frontotemporal dementia (Rubino et al., 2012; Le Ber et al., 2013; Yilmaz et al., 2020). In vitro studies revealed that mutant SQSTM1 is unable to recognize LC3, limiting p62 recruitment to autophagosomes (Goode et al., 2016; Nozaki et al., 2021). According to this, p62-positive aggregates have been detected in the spinal cord of ALS patients (Mizuno et al., 2006; Sasaki 2011) as well as ALS-related animal models (Sasaki and Iwata, 2007; Kato 2008; Hadano et al., 2010). Regarding the large number of distinct proteins that is found to aggregate in ALS, it is likely that motor neuron degeneration underlies impaired selective autophagy. Besides its interaction with LC3, p62 is further involved in the anti-oxidative stress response pathway. SQSTM1 mutation exacerbated TDP-43 dependent stress response due to impaired p62 interaction with KEAP1 (Kelch-like ECH-associated protein 1) (Deng et al., 2020) and gives reason to suggest a pathological dual mode of action of mutated SQSTM1. Recently, homozygous mutations in SQSTM1 have been linked to childhood-onset cerebellar ataxia (Haack et al., 2016; Vedartham et al., 2019). Skin fibroblasts from patients revealed a decreased autophagic flux with concurrently reduced mitochondrial OXPHOS activity. Downregulation of SQSTM1 further led to atrophy in the cerebellum following axonal degeneration in a zebrafish model (Muto et al., 2018), indicating cerebellar susceptibility to disturbed p62 function.
Nevertheless, p62 rather functions as a general autophagic receptor for ubiquitinated proteins and has been further linked to the ubiquitin proteasome system (Liu et al., 2016; Nam et al., 2017). Thus, p62 is thought to play a secondary role in mitophagy, while as primary mitophagic receptors, OPTN and NDP52 have been identified (Wong and Holzbaur 2014; Heo et al., 2015; Lazarou et al., 2015; Richter et al., 2016). It is suggested that those are recruited to mitochondria via their ubiquitin binding domain, which then activates a positive feedback loop in order to recruit autophagosomes (Padman et al., 2019). Mutations of OPTN as well as TBK1, encoding a kinase which contains OPTN as a substrate, are found in patients with ALS (Figure 2; Wong and Holzbaur, 2014; Cirulli et al., 2015; Freischmidt et al., 2015; Feng et al., 2019). In particular, mutant OPTN failed to associate with the mitochondrial surface (Wong and Holzbaur, 2014; Lazarou et al., 2015), while mutant TBK1 reduced recruitment of OPTN and LC3 to damaged mitochondria (Moore and Holzbaur, 2016). Loss of function of either OPTN or TBK1 impaired mitophagy resulting in accumulation of defective mitochondria (Evans and Holzbaur, 2019). Taken together, these data suggest impaired mitochondrial quality control via autophagy to be a critical factor for mitochondrial dysfunction found in ALS. However, the role of disturbed mitophagy in sporadic cases still needs to be elucidated.
Another pathway for mitochondrial quality control has been recently described involving so called mitochondrial-derived vesicles (MDVs) (Figure 2), which shuttle mitochondrial cargo to peroxisomes or lysosomes (Sugiura et al., 2014). Here, selected proteins can be degraded instead of the entire organelle (McLelland et al., 2014), which renders MDV-derived mitochondrial removal more selective than mitophagy. Formation of MDVs is promoted by the retromer complex, which was initially described in endosome-to-Golgi as well as endosome-to-plasma membrane transport (Seaman, 2012). Since VPS35 is part of the retromer complex and mutations in the encoding gene are associated with autosomal dominant PD (Vilarino-Guell et al., 2011; Zimprich et al., 2011), VPS35 is thought to play an important role in mitochondrial quality control. Indeed, VPS35 has been shown to associate with mitochondrial membranes and mediates vesicle transport between mitochondria and peroxisomes (Braschi et al., 2010). VPS35 has also been linked to mitochondrial fusion and fission, since mutant VPS35 caused mitochondrial fragmentation, explained by decreased MFN2 levels in response to mitochondrial E3 ubiquitin ligase 1 (MUL1) degradation (Tang et al., 2015), and increased turnover of Drp1 (Wang et al., 2016). A recent study further showed reduced mitochondrial clearance in dopaminergic neurons derived from patients carrying the VPS35 mutation p.D620N (Hanss et al., 2020), providing first evidence for impaired mitophagy by mutant VPS35 in PD.
In vitro analysis of induced pluripotent stem cell-derived motor neurons from CMT2A patients, revealed upregulation of PINK1 and Parkin together with an increased autophagic flux (Rizzo et al., 2016). The authors therefore suggest that low mitochondrial mass upon MFN2 mutation is rather induced by enhanced mitophagy instead of impaired mitochondrial biogenesis. Indeed, MFN2 has been shown to be further involved in OMM tethering to ER membranes in order to limit mitochondrial ubiquitination and delay mitophagy (McLelland and Fon 2018; McLelland et al., 2018). Mutant MFN2 in CMT2A could hence induce mitophagy by its displacement from contact sites between mitochondria and ER. Simultaneously, decreased protein levels of MFN2 detected in various neurodegenerative diseases could consequently reflect enhanced mitophagy as a consequence of expanding mitochondrial dysfunction. Besides MFN2, Charcot-Marie-Tooth disease can be further caused by mutations in GDAP1 (Zimon et al., 2011). A recent study showed that the OMM protein GDAP1 links mitochondria and lysosomes by binding LAMP1 (lysosomal associated membrane protein 1), while loss of GDPA1 affects basal autophagy (Figure 2) (Cantarero et al., 2020). These data provide new evidence for altered mitochondrial quality control contributing to the pathogenesis of peripheral neuropathies.
Besides gene mutations that are directly linked to mitochondrial removal pathways, accumulation of dysfunctional mitochondria and misfolded or mutant proteins, such as Aβ, Tau, a-Syn, TDP-43 and HTT, point to impaired autophagy in idiopathic cases as well (Monaco and Fraldi 2020). In brains of AD patients, autophagy-related protein Beclin 1 was decreased, which was linked to decreased autophagic flux and higher Aβ accumulation (Pickford et al., 2008). In addition, Parkin-dependent mitophagy was highly induced, while protein levels of Parkin decreased during disease progression (Ye et al., 2015), suggesting disturbed recognition of defective mitochondria at later stages of AD. It is likely, that pTau further interferes with mitophagy by disrupting axonal transport (Combs et al., 2019) and thereby retrograde movement of defective mitochondria. In PD, impaired mitophagy is known to be a shared feature of both idiopathic and familial PD (Hsieh et al., 2016). Interestingly, α-Syn aggregates have been recently shown to cause upregulation of Miro1 leading to delayed mitochondrial removal (Shaltouki et al., 2018). Since HTT physiologically interacts with p62 and the ULK1 complex (Ochaba et al., 2014; Rui et al., 2015), it could be speculated that in HD, mHTT might be unable to provide the proposed scaffold function and thereby affects mitophagy.
Calcium Handling
Mitochondria serve as mobile regulators of local and bulk intracellular Ca2+ concentrations. Upon mitochondrial uptake, Ca2+ boosts mitochondrial metabolism and OXPHOS (McCormack and Denton 1979; Jouaville et al., 1999), and can thereby modulate the cell’s performance (Rizzuto et al., 2012). In neurons, mitochondria further impact on electrophysiological activity by shaping somato-dendritic as well as axonal and presynaptic Ca2+ oscillations (Billups and Forsythe, 2002; Kwon et al., 2016; Marland et al., 2016; de Juan-Sanz et al., 2017; Vaccaro et al., 2017). Under basal conditions, Ca2+ concentrations inside mitochondria resemble those measured in the cytoplasm (100–200 nM), however, mitochondria are able to buffer at 10–20-fold higher concentrations if needed. Pore-forming voltage-dependent anion-selective channel proteins (VDACs) mediate Ca2+ uptake from the cytosol or ER contact sites into the mitochondrial intermembrane space, where Ca2+ is forwarded into the mitochondrial matrix via the mitochondrial Ca2+ uniporter (MCU) complex (Figure 3; Giorgi et al., 2018). Ca2+ thereby follows the negative mitochondrial inner membrane potential (-150 to -180 mV) built up by the respiratory chain (Friel and Tsien 1994; Babcock et al., 1997). Efflux of one Ca2+ from the mitochondrial matrix occurs in exchange of three to four Na+ through the mitochondrial Na+/Ca2+ antiporter (Jung et al., 1995; Dash and Beard 2008). From the intermembrane space, Ca2+ extrudes into the cytosol via VDACs and the Na+/Ca2+ exchanger 3, respectively (Scorziello et al., 2013; Giorgi et al., 2018).
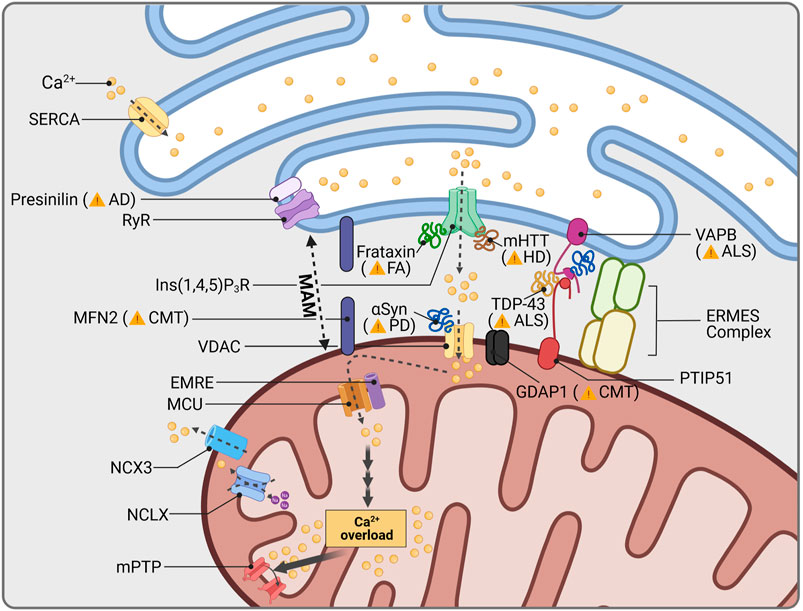
FIGURE 3. Mitochondrial Ca2+ and neurodegeneration. The ER is the main cellular organelle to store Ca2+. ER Ca2+ stores are released into the cytoplasm through Ryanodine Receptor channels (RyR), whose activity is regulated by the AD-related protein Presenilin. Cellular Ca2+ homeostasis is centralized in mitochondria-associated membranes (MAMs), the interphase between mitochondria and the ER. MAM structure is stabilized by a group of proteins, among them CMT2A-related MFN2, as well as ALS-linked VAPB and PTIP51. The OMM protein GDAP1 is further present in MAMs and is linked to impaired Ca2+ signaling, depending on the GDAP1 mutation causing CMT. Other proteins which are associated with human diseases are known to interact with proteins resident in MAMs: Friedreich Ataxia (FA) protein frataxin, stabilizes MAMs through control of ROS levels; aggregates of mHTT increase Ca2+ release through Ins(1,4,5)P3R; mutations in the ALS-related protein TDP-43 disrupt interaction between VAPB and PTIP51; and PD related αSyn has been found to increase mitochondrial Ca2+ permeability through VDAC interaction. Mitochondrial Ca2+ overload induces opening of the mitochondrial permeability transition pore (mPTP), cytochrome c release and apoptosis activation.
Since excessive amounts of cytosolic Ca2+ have been linked to excitotoxicity (Sattler and Tymianski 2000; Hardingham and Bading 2003), and mitochondrial Ca2+ overload has been shown to trigger cell death through opening of the mitochondrial permeability transition pore (Giorgi et al., 2012; Marchi et al., 2018), more and more attention was paid to imbalanced mitochondrial Ca2+ handling as putative cause of neurodegeneration in common and rare disorders. Neuronal Ca2+ oscillations promote pacemaker activity and fuel mitochondrial OXPHOS in SNc dopaminergic neurons (Surmeier et al., 2010). However, Ca2+ oscillations are a disservice to these vulnerable neuron population, as they cause mitochondrial oxidant stress by at that time unknown mechanisms (Guzman et al., 2010). Simultaneously, SNc dopaminergic neurons with low expression of the Ca2+-binding protein calbindin were found to be more affected in post mortem brains of PD patients (Damier et al., 1999) as well as in PD-related animal models (German et al., 1992; Dopeso-Reyes et al., 2014), indicating a potential neuroprotective effect of calbindin. The metabolic challenge following increased Ca2+ levels together with low intrinsic Ca2+ buffer capacities, is thought to render SNc dopaminergic neurons especially vulnerable to mitochondrial dysfunction (Philippart et al., 2016). Correspondingly, various voltage-gated Ca2+ channels have been associated with the selective loss of SNc dopaminergic neurons in PD, including L-type (Chan et al., 2007; Guzman et al., 2010; Surmeier et al., 2012; Duda et al., 2016), T-type (Guzman et al., 2018; Tabata et al., 2018) and R-type channels (Benkert et al., 2019). In line with this, we could recently show that upon respiratory chain deficiency, SNc dopaminergic neurons perish due to high Ca2+ loads, which finally impair the mitochondrial antioxidant defense (Ricke et al., 2020).
The increased levels of cytosolic Ca2+ in SNc dopaminergic neurons have been further linked to the expression of α-Syn, suggesting a converging pathway for these two pathogenic factors in PD (Lieberman et al., 2017). In support of this, a recent study showed that a-Syn dynamically binds to VDACs and modifies their Ca2+ permeability (Figure 3; Rosencrans et al., 2021). It is therefore likely that pathological aggregations of α-Syn might affect VDAC binding and thus mitochondrial Ca2+ buffering. In addition, wild type α-Syn is physiologically localized to mitochondria-ER contact sites called mitochondria-associated ER membranes (MAM) (Poston et al., 2013) which mediates the transfer of Ca2+ (Cali et al., 2012). However, mutant α-Syn dissociates from MAM causing mitochondrial fragmentation and decreased contacts between mitochondria and the ER (Table 3; Guardia-Laguarta et al., 2014). Accordingly, mutant α-Syn disturbs Ca2+ exchange between mitochondria and ER through disruption of the integral ER protein VAPB (vesicle-associated membrane protein-associated protein B) binding (Paillusson et al., 2017), showing mitochondrial Ca2+ homeostasis to be potentially affected by pathogenic α-Syn variations in PD patients. Recently, MAM have been shown to further depend on MIRO1: Heterozygous mutations in the gene encoding MIRO1 (RHOT1) were identified in two PD patients, while fibroblasts from these patients presented a decrease in MAM and impaired Ca2+ homeostasis (Grossmann et al., 2019). These findings were subsequently approved in patient-derived neurons (Berenguer-Escuder et al., 2020) and confirm the importance of mitochondrial Ca2+ handling for SNc dopaminergic neuron survival.
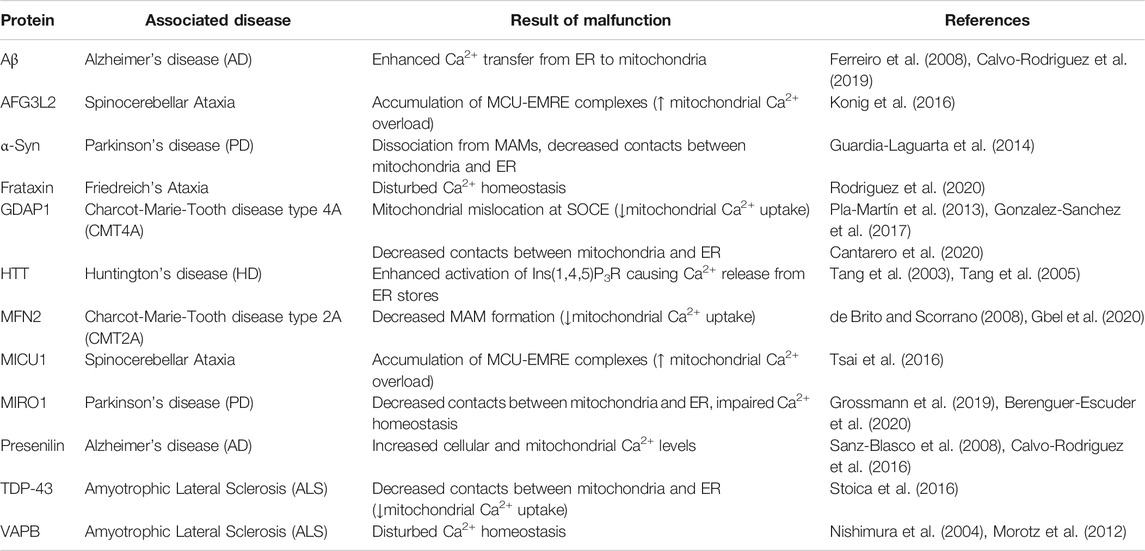
TABLE 3. Proteins related to disturbed mitochondrial Ca2+ handling.
Disturbed Ca2+ homeostasis has been identified early as a characteristic feature in motor neurons of ALS patients (Siklos et al., 1996) and could be mimicked by various in vitro and in vivo models expressing mutant SOD1 (Carri et al., 1997; Siklos et al., 1998; Damiano et al., 2006), TDP-43 (Stoica et al., 2014) and FUS (Stoica et al., 2016), respectively. Interestingly, recovery of physiological Ca2+ concentrations in motor neurons was delayed upon AMPA (α-amino-5-methyl-3-hydroxisoxazolone-4-propionate) receptor activation (Guatteo et al., 2007). Since AMPA receptors are highly expressed at postsynaptic terminals and, similar to SNc dopaminergic neurons, they only possess poor intrinsic Ca2+ buffering capacities, motor neurons in ALS are supposed to perish by Ca2+-induced excitotoxicity (Van Den Bosch et al., 2000; Grosskreutz et al., 2010). Motor neurons therefore especially rely on proper Ca2+ buffering by mitochondria. Accordingly, disruption of MAM has been reported in several ALS-related models (Stoica et al., 2014; Bernard-Marissal et al., 2015; Stoica et al., 2016). VAPB (vesicle associated membrane protein associated protein B and C) interacts with the mitochondrial protein PTPIP51 (protein tyrosine phosphatase-interacting protein 51) in order to regulate Ca2+ exchange (De Vos et al., 2012). Mutations in VAPB have been linked to familial ALS (Figure 3; Nishimura et al., 2004) showing disturbed Ca2+ homeostasis (Morotz et al., 2012). Moreover, TDP-43 was shown to disrupt the VAPB-PTPIP52 pathway (Stoica et al., 2016), suggesting a similar mechanism in idiopathic ALS cases with pathogenic TDP-43 accumulation. In support of this, expression levels of VAPB were found to be reduced in the spinal cord of idiopathic ALS patients (Anagnostou et al., 2010). Recent analysis of motor neurons derived from patients carrying mutations in TDP-43 revealed that Ca2+-permeable AMPA and NMDA receptor upregulation was associated with reduced mitochondrial Ca2+ uptake due to an imbalance between MICU1 and MICU2 (Dafinca et al., 2020). Thus, it is likely that in ALS, glutamate excitotoxicity might generally underlie defective mitochondrial Ca2+ buffering, induced by MCU complex imbalance or MAM disruption. However, a recent study indicated mutation-specific alterations in Ca2+ dynamics of patient-derived motor neurons, which would consequently require differential treatment strategies (Bursch et al., 2019).
In line with its contribution to impaired mitochondrial Ca2+ handling in ALS, disrupted MAM are likely to be involved in motor neuron loss of CMT2A patients (Bernard-Marissal et al., 2019). Besides the scaffolding multisubunit protein complex ERMES (ER-mitochondria encounter structure) (Kornmann et al., 2009), MFN2 is also involved in the connection of mitochondria and ER (McLelland et al., 2018; McLelland et al., 2018). Since silencing or depletion of MFN2 leads to decreased MAM formation associated with impaired mitochondrial Ca2+ uptake (Table 3; de Brito and Scorrano 2008; Gbel et al., 2020), perturbations of mitochondrial Ca2+ handling might play a crucial role for motor neuron survival in Charcot-Marie-Tooth disease as well. In accordance to this, GDAP1 has been linked to the so called store-operated Ca2+ entry (SOCE). In general, SOCE is activated following depletion of ER Ca2+ stores, which is controlled by the ER transmembrane protein STIM1 (stromal interaction protein 1). Low Ca2+ levels within the ER induce STIM1 dimerization and its diffusion to plasma membrane contact sites where it directly binds TRPC channels, in particular ORAI1, activating Ca2+ entry into the cytosol (Liu et al., 2015). These membrane junctions are stabilized by proteins known as junctophilins which also serve as signaling hubs for the process (Sahu et al., 2019). In this context, mitochondria function as local Ca2+ buffer and avoid early inhibition of ORAI1 channels, thereby allowing restoring proper Ca2+ levels in the ER. GDAP1 deficiency has been shown to cause mitochondrial mislocation at SOCE, avoiding proper Ca2+ buffering and inducing the inhibition of ORAI1 (Pla-Martin et al., 2013; Gonzalez-Sanchez et al., 2017). This feature is associated with progressive decline of motor neurons in mice (Barneo-Munoz et al., 2015) and ER stress (Civera-Tregon et al., 2021). It should be noted that missense mutations associated with Charcot-Marie-Tooth disease have different effects in SOCE activity. While dominant inherited missense mutation generate incremented SOCE, recessive inherited mutations lead to a complete loss of function and inhibition of Ca2+ entry (Gonzalez-Sanchez et al., 2017). Noteworthy, mutations in JPH1, encoding Junctophilin-1, were found to increase the severity of a dominant inherited GDAP1 mutation (Pla-Martin et al., 2015).
Multiple types of spinocerebellar ataxia have been found to be associated with alterations of neuronal Ca2+ levels (Patron et al., 2018). The importance of controlled mitochondrial Ca2+ handling, in particular, is evidenced by MICU1-deficient mice, presenting ataxia following degeneration of Purkinje cell projections (Liu et al., 2016). MICU1 depletion leads to mitochondrial Ca2+ overload through accumulation of MCU-EMRE (essential MCU regulator) complexes, allowing unrestricted Ca2+ influx into the mitochondrial matrix (Table 3; Tsai et al., 2016). Moreover, MCU-EMRE complexes accumulate upon depletion of AFG3L2 and again disrupt gatekeeping of the MCU complex (Konig et al., 2016). Lowering Purkinje cell Ca2+ levels in AFG3L2 haploinsufficient mice by decreasing glutamate stimulation, in turn, could rescue Purkinje cell degeneration and the ataxic phenotype (Maltecca et al., 2015). This indicates a beneficial effect of reducing cytosolic Ca2+ concentrations on Purkinje cells of patients carrying AFG3L2 mutations and, simultaneously, points to glutamate-dependent excitotoxicity as cause of neuron death. However, preliminary data by Langer and colleagues showed that MCU deletion in Purkinje cells lacking AFG3L2 did not influence neurodegeneration (Patron et al., 2018), suggesting that modifying mitochondrial Ca2+ levels alone is not sufficient to prevent degeneration of Purkinje cells in AFG3L2-related spinocerebellar ataxia. Nevertheless, it remains to be seen how MCU depletion is truly affecting mitochondrial Ca2+ levels and especially cytosolic Ca2+ concentrations. Accordingly, decreasing cytosolic Ca2+ loads as well as glutamate transmission in Purkinje cells might be the treatment of choice in order to prevent degeneration in spinocerebellar ataxia. Impaired Ca2+ handling might thereby be a general pathological factor underlying ataxias. In support of this, frataxin, whose encoding gene is mutated in Friedreich’s ataxia, has been recently shown to be localized to MAMs and to stabilize them through protection against ROS (Rodriguez et al., 2020). Modulation of Ca2+ levels could further rescue the axonal decline of frataxin-deficient sensory neurons in vitro (Molla et al., 2017).
In HD, abnormalities of mitochondrial Ca2+ handling have been first detected in isolated mitochondria from affected individuals. While having reduced Ca2+ buffering capacities, mitochondria from HD patients further showed mitochondrial membrane depolarization at low Ca2+ concentrations in contrast to those from control subjects (Panov et al., 2002). In accordance to this, investigation of striatal cells from HD-related models revealed that mitochondria were able to buffer low Ca2+ levels, but failed at higher concentrations (Lim et al., 2008; Quintanilla et al., 2013). The failure in mitochondrial buffering of high Ca2+ loads has been subsequently linked to chronic Ca2+ leakage from the ER: in medium spiny neurons, mHTT enhanced activation of the intracellular Ca2+ release channel inositol(1,4,5)-triphosphate receptor (Figure 3; Tang et al., 2003) causing Ca2+ release from ER stores through ryanodine receptors (Tang et al., 2005). Inhibition as well as stabilization of ryanodine receptors correspondingly attenuated Ca2+ leak and cell death (Suzuki et al., 2012), suggesting intracellular Ca2+ store depletion through mHTT to be the causative event for neuronal Ca2+ overload, and impaired mitochondrial Ca2+ handling to be a consequence (Table 3). Furthermore, reduced Ca2+ uptake by mitochondria would exacerbate Ca2+-induced cytotoxicity. However, increased mitochondrial Ca2+ influx has been reported in HD-related models as well (Oliveira et al., 2007; Wang J.-Q et al., 2013; Pellman et al., 2015). Inhibition of the MCU complex thereby prevented mitochondrial permeability transition pore opening and hence degeneration of medium spiny neurons in vitro (Tang et al., 2005). Regarding the effects of mHTT on mitochondrial dynamics, it could be speculated that MAM formation is additionally influenced and would further affect neuronal Ca2+ buffering. It is therefore likely that it needs to target both the ER and mitochondria in order to correct cytosolic Ca2+ levels and prevent Ca2+-induced loss of medium spiny neurons in HD.
Disturbed cytosolic Ca2+ homeostasis had been primarily linked to AD pathogenesis by familial cases associated with mutations of Presenilin 1 (PS1) and 2 (PS2) (Table 3; LaFerla 2002). These mutations cause high accumulation of Aβ aggregates, which have been shown to trigger cell death following increased intracellular (Busche et al., 2008; Kuchibhotla et al., 2008) as well as mitochondrial Ca2+ levels (Sanz-Blasco et al., 2008; Calvo-Rodriguez et al., 2016). Presenilins are directly involved in cytosolic Ca2+ regulation by interaction with ryanodine receptors (Chan et al., 2000) and their modulatory protein sorcin, respectively (Pack-Chung et al., 2000). Moreover, they have been shown to be involved in ER-mitochondria coupling (Filadi et al., 2016), suggesting mutant forms to disrupt neuronal Ca2+ handling by altering ER-mediated Ca2+ storage and MAM formation. In fact, increased contacts between mitochondria and the ER have been found in patient-derived fibroblasts, human brain tissue and AD-related animal models (Hedskog et al., 2013; Area-Gomez and Schon 2017). High MAM number could be detrimental to mitochondria and trigger cell death, especially with regard to the ability of Aβ aggregates to additionally induce Ca2+ transfer from the ER to mitochondria (Ferreiro et al., 2008; Calvo-Rodriguez et al., 2019). Noteworthy, increased ER-mitochondria contacts (Perreault et al., 2009) as well as elevated mitochondrial Ca2+ levels have been linked to AD models showing Tau pathology as well (Quintanilla et al., 2009; Britti et al., 2020). Since Aβ aggregates and pTau are cellular hallmarks for idiopathic AD, limiting mitochondrial Ca2+ uptake could be of general importance in order to preserve mitochondrial functionality and to prevent Ca2+-induced neurodegeneration. In line with this, a recent study revealed that Aβ-induced mitochondrial Ca2+ overload was MCU-dependent and preceded neurodegeneration in mice, indicating putative pharmacological intervention by blocking MCU (Calvo-Rodriguez et al., 2020).
Selective Vulnerability
It is striking that despite the fundamental importance of mitochondria for neurons, specific neuronal populations perish in the disorders presented above. Especially considering familial cases, in which proteins carrying monogenic mutations are widespread in both the central and peripheral nervous system, cell-type specific factors are likely to contribute to the selective vulnerability (Table 4). Concerning the distinct mitochondrial processes being reported to be affected in the diseases mentioned above, cell-intrinsic properties might enhance the risk of neuron death induced by mitochondrial defects.
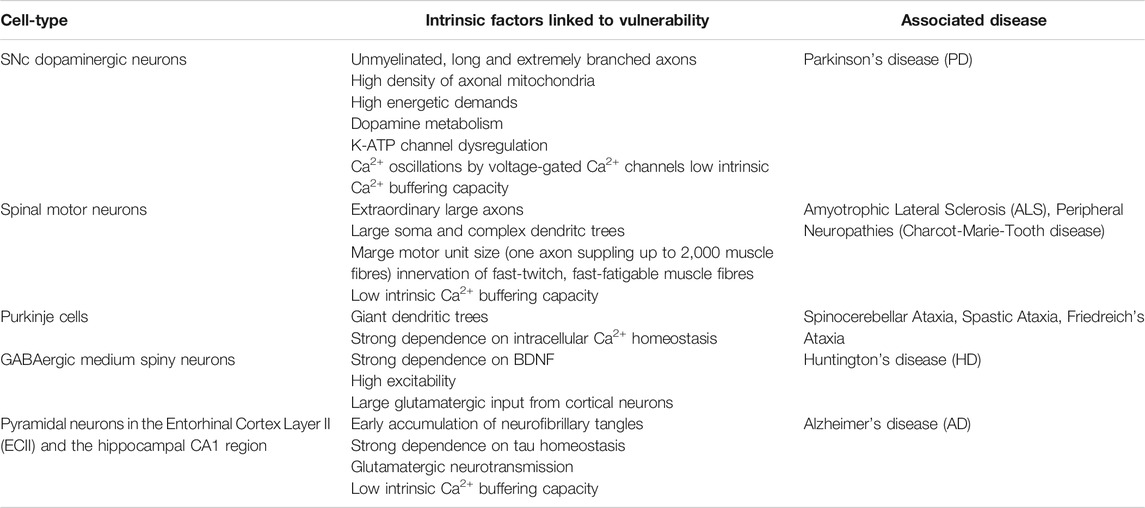
TABLE 4. Most affected neuron populations in selected neurological disorders.
Substantia Nigra Pars Compacta Dopaminergic Neurons
In PD, different neuronal populations are affected in a variety of brain areas, with perished non-dopaminergic neurons being primarily linked to early non-motor symptoms, including anxiety and depression (Postuma et al., 2012). However, characteristic motor symptoms are caused by the loss of dopaminergic neurons in the SNc and the accompanying depletion of dopamine in the dorsal striatum (Michel et al., 2016; Obeso et al., 2017). An important factor for the neurons’ vulnerability is their neurotransmitter. Dopamine metabolism is associated with the production of various intermediate metabolites and reactive oxygen species (ROS), which impede antioxidant processes and can induce oxidative stress (Delcambre et al., 2016), a pathophysiological cellular state which has been early linked to PD pathogenesis (Jenner and Olanow 1996; Alam et al., 1997). Moreover, dopamine metabolism drives the accumulation of mtDNA deletions (Neuhaus et al., 2014; Neuhaus et al., 2017). Nevertheless, dopamine alone cannot be the reason for neurodegeneration in the SNc, since dopaminergic neurons in the neighboring ventral tegmental area (VTA) are much less affected in PD (Alberico et al., 2015). Electrophysiological characterization of these two dopaminergic midbrain populations thereby greatly contributed to a better understanding of potential pathological pathways. In contrast to the VTA (Khaliq and Bean 2010), SNc dopaminergic neurons in the SNc have to face high Ca2+ loads, resulting from activity-dependent Ca2+ oscillations and simultaneously low intrinsic Ca2+ buffering capacities, which have been associated with their selective neurodegeneration, as mentioned above. In addition, activation of mitochondrial OXPHOS by Ca2+ increases generation of ROS as a byproduct (Guzman et al., 2010), which might further promote the formation of mtDNA alterations (Lawless et al., 2020). Furthermore, Ca2+ is involved in the aggregation of αSyn (Dufty et al., 2007; Caraveo et al., 2014; Diepenbroek et al., 2014; Rcom-H'cheo-Gauthier et al., 2014), providing a direct link between enhanced Ca2+ levels and αSyn pathology in SNc dopaminergic neurons. Apart from voltage-gated Ca2+ channels, ATP-sensitive potassium (K-ATP) channels have been linked to SNc dopaminergic neuron death. Whereas protecting the cell from overexcitability and excitotoxicity under physiological conditions by mediating membrane hyperpolarization following metabolic demand (Liss and Roeper 2001), K-ATP channel opening in SNc dopaminergic neurons led to complete loss of electrophysiological activity and promoted cell death in a toxin-based model for PD. Noteworthy, this pathological mechanism was absent in dopaminergic neurons located in the VTA (Liss et al., 2005).
Another critical factor contributing to the vulnerability of SNc dopaminergic neurons is their morphology. Contrary to the VTA or the olfactory bulb, dopaminergic neurons within the SNc possess unusually long and extremely branched axons which are connected to a large number of nerve cells (Matsuda et al., 2009; Pacelli et al., 2015). A single axon of a human SNc dopaminergic neuron is thereby estimated to have a total length of 4.5 m and give rise to up to 2.4 million synapses (Bolam and Pissadaki 2012). This unique architecture of concurrently unmyelinated axon terminals (Braak et al., 2004) is an extreme challenge to mitochondrial bioenergetics and is accompanied by a high density of axonal mitochondria (Pacelli et al., 2015).
Motor Neurons
Motor neuron degeneration is a pathological hallmark of ALS as well as several peripheral neuropathies, including CMT2A. Whether upper (corticospinal) or lower somatic motor neurons, both populations possess extraordinary long axons in order to enable neurotransmission from the primary motor cortex, down the spinal cord to the most distant muscle fibers of our body (Stifani 2014). This renders them critically dependent on proper axonal transport, and especially on mitochondrial trafficking in order to preserve a functional mitochondrial pool in far distance to the cell body. Concerning ALS, however, neurodegeneration seems to be rather complex, since not all motor neurons equally perish. Those neurons within the oculomotor (CNIII), trochlear (CNIV) and abducens (CNVI) nuclei are more or less resistant to neurodegeneration in ALS, allowing patients to maintain communication via eye movements (Kubota et al., 2000; Caligari et al., 2013) when speech problems are too severe. Whereas the majority of skeletal muscles reveal a single contact between each muscle fiber and incoming axon (en plaque), extraocular muscles can be innervated by oculomotor neurons in a grape-like structure (en grappe) (Zimmermann et al., 2013). In addition, oculomotor neurons possess small somas as well as dendritic trees (Ulfhake and Cullheim 1981; Highstein et al., 1982), and only innervate up to five muscle fibers (Enoka 1995). Spinal motor neurons, in contrast, innervate at least 300 muscle fibers, in large muscles such as the medial gastrocnemius supplying up to 2,000 muscle fibers (Burke et al., 1971; Burke and Tsairis 1973). Single fiber innervation and large motor unit size are accompanied with high energetic demands in order to preserve neurotransmission, and hence muscle contraction.
Morphological differences are also invoked concerning the selective vulnerability among spinal motor neurons in ALS: early degenerating motor neurons innervate a large number of fast-twitch, fast-fatigable, glycolytic muscle fibers, which are responsible for high-force movements. Spinal motor neurons which supply slow-twitch, fatigue-resistant muscle fibers, however, are more finely structured and rather robust against cell death (Nijssen et al., 2017). Interestingly, these motor neurons are even temporarily able to compensate the early loss of those which innervate fast-twitch, fast-fatigable muscle fibers (Frey et al., 2000; Schaefer et al., 2005). The robustness of surviving motor neurons in ALS is further related to their high intrinsic Ca2+ buffering capacity (Alexianu et al., 1994), highlighting again the importance of proper neuronal Ca2+ handling especially for susceptible spinal motor neurons innervating fast-twitch muscles.
Purkinje Cells
The cerebellum is essential for the execution of goal-directed movement and coordination of body and limb posture. As they provide the sole output of the cerebellar cortex, Purkinje cells are considered to be the functionally most important cell type of this brain region. These large, pear-shaped neurons receive inputs from excitatory climbing fibers as well as parallel fibers originating from cerebellar granule cells and the inferior olivary nucleus within the brainstem, respectively. With their unbranched axons, Purkinje cells project to cerebellar nuclei located in the white matter, such as the dentate gyrus, and regulate neuronal output from the cerebellum to the brainstem and thalamus by GABAergic transmission (Cerminara et al., 2015). Purkinje cells are primarily characterized by their giant dendritic trees, which enable up to 150,000 synaptic connections per cell (Kandel et al., 2000). In addition, they are able to generate two types of action potential trains: high rate (30–100 Hz) simple spiking upon spontaneous activity or activation by parallel fibers (Armstrong and Rawson 1979), as well as low rate (∼1 Hz) complex spiking following activation by climbing fibers (Eccles et al., 1966). Histological detection of Purkinje cells is enabled by labeling of calbindin, which is abundantly expressed. Loss of calbindin in Purkinje cells caused motor coordination deficits associated with altered time course and amplitude of fast Ca2+ transients (Barski et al., 2003), highlighting the importance of intracellular Ca2+ buffering for Purkinje cell function. Cell-type specific genetic profiling recently revealed a variety of genes to be uniquely present in Purkinje cells. Among them, there were several genes associated with the development of autosomal dominant spinocerebellar ataxia. Interestingly, all of them are involved in intracellular Ca2+ homeostasis by either controlling mitochondrial Ca2+ uptake (AFG3L2), Ca2+ release from ER stores (ITPR1), glutamate receptor-mediated Ca2+ signaling (TRPC3), and fast Ca2+ entry within dendritic spines (CACNA1G) (Huang and Verbeek 2019), highlighting the strong dependence on intracellular Ca2+ homeostasis for Purkinje cell survival as well.
Medium Spiny Neurons
While being located in the striatum, GABAergic medium spiny neurons present the major input region of the basal ganglia. Upon dopaminergic input by the SNc, medium spiny neurons either facilitate (direct pathway) or suppress movement (indirect pathway), depending on whether they express dopamine D1 or D2 receptors. Activation of D1 medium spiny neurons additionally leads to inhibition of the D2 population, and vice versa, according to the current basal ganglia model (McGregor and Nelson 2019). Although neurodegeneration in HD involves diverse brain regions, including the cerebral cortex, thalamus and hypothalamic nuclei, medium spiny neurons within the striatum are most severely affected and responsible for the symptomatic motor defects (Halliday et al., 1998; Vonsattel 2008). Until today, the special vulnerability of these striatal neurons is not fully understood. It is known that HD is caused by the CAG repeat expansion in the HTT gene (MacDonald et al., 1993), however, mHTT is expressed throughout the CNS and expression levels are neither enhanced in the striatum nor in cortical neurons projecting to the striatum (Trottier et al., 1995). One hypothesis for the special vulnerability of the striatum to mHTT involves its strong dependence on brain-derived neurotrophic factor (BDNF). Besides its critical importance during brain development, BDNF is required for long-term survival of adult medium spiny neurons and involved in the regulation of both dendritic morphology and spine number (Baydyuk and Xu 2014). Protein levels of BDNF were found to be reduced in the striatum of HD patients (Ferrer et al., 2000) as well as HD-related animal models expressing mHTT (Apostol et al., 2008; Gharami et al., 2008), which has been linked to interference of mHTT with synthesis and transport of BDNF (Zuccato et al., 2001; Gauthier et al., 2004). Interestingly, striatal neurons depend on BDNF supply through SNc dopaminergic neurons and particularly cortical projection neurons (Altar et al., 1997; Baquet et al., 2004), indicating early loss of cortico-striatal connectivity in HD (Novak et al., 2015) to additionally contribute to lack of striatal BDNF.
Another reason for the striatal vulnerability in HD may be provided by the action of Rhes, a small GTPase which is highly enriched in the striatum. This protein has been shown to interact with mHTT and then promote a toxic effect by causing ubiquitin-like modification (Subramaniam et al., 2009). Correspondingly, Rhes depletion in a HD mouse model improved striatal atrophy and motor symptoms (Swarnkar et al., 2015). A recent study showed that Rhes can form tunneling nanotubes in vitro and thereby enabled transfer of mHTT between striatal cells (Sharma and Subramaniam 2019), persuading the authors to suggest a similar mechanism in vivo. On the other side, the expression of Rhes in medium spiny neurons has not been clearly defined, and other brain regions, which are not affected in HD, have been shown to express it as well.
Interestingly, medium spiny neurons of the indirect pathway are found to degenerate earlier than those involved in the direct pathway. This is in line with the symptomatic course of disease: chorea is part of early HD stages, resulting from missing movement suppression, whereas akinesia and dystonia occur later in disease due to decreased movement initiation (Raymond et al., 2011). One possible explanation for the early death of indirect pathway medium spiny neurons could be the exclusive expression of the BDNF receptor TrkB, which might render those neurons more vulnerable to the lack of BDNF (Baydyuk et al., 2011). Another factor could be the differential innervation. Although both striatal populations are innervated by cortical projection neurons, medium spiny neurons of the indirect pathway receive excitatory inputs from so called pyramidal tract cortical neurons, which is accompanied with larger glutamate release (Ballion et al., 2008). In addition, cortical circuits are thought to be even more active in early HD stages preceding motor symptoms (Burgold et al., 2019) and indirect pathway neurons have been shown to be more excitable than direct pathway neurons (Gertler et al., 2008). Thus, it is likely that medium spiny neurons of the indirect pathway degenerate early due to an enhanced risk for glutamate-mediated excitotoxicity and simultaneously increased energetic demands.
Pyramidal Neurons in the Entorhinal Cortex Layer II and Hippocampal Cornu Ammonis 1 Region
Cognitive decline and memory loss in AD are caused by atrophy of the entorhinal cortex, hippocampus as well as neocortical areas. This atrophy is linked to the accumulation of both Aβ aggregates and neurofibrillary tangles. Whereas Aβ plaques are relatively widespread in the AD brain (Thal et al., 2002), tau pathology reveals the same distribution pattern as neurodegeneration (Braak and Braak 1991) and correlates with clinical symptoms (Brier et al., 2016). In particular, pyramidal neurons of ECII and the hippocampal CA1 region have been shown to selectively perish in early stages of the disease. Other neuron populations, however, such as pyramidal neurons of CA2, CA3, the primary visual cortex, primary somatosensory cortex, and granule cells of the dentate gyrus, are spared from degeneration (Hyman et al., 1984; Arnold et al., 1991; Gomez-Isla et al., 1996; Fukutani et al., 2000; Bussiere et al., 2003). Neurons within the ECII reveal relatively high energetic demands (Hevner and Wong-Riley 1992), indicating an enhanced susceptibility to additional stressors. The same might be true for CA1 pyramidal neurons, which present high sensitivity toward decreased glucose and oxygen delivery (Montagne et al., 2016). In addition, both ECII and CA1 pyramidal neurons are glutamatergic and hence susceptible to NMDA-mediated excitotoxicity, which could be further provoked by Aβ aggregates (Wang and Reddy 2017). Inhibitory interneurons of the neocortex expressing high levels of Ca2+-binding proteins, in contrast, are less affected (Hof et al., 1993; Fu et al., 2017), suggesting intracellular Ca2+ homeostasis to be an important factor for neuron survival in AD.
Interestingly, the spread of Aβ and tau pathology in the brains of AD patients could be recapitulated by clustering of genes whose proteins are likely to co-aggregate with Aβ plaques and neurofibrillary tangles in healthy individuals. Vulnerable brain regions revealed high expression of aggregation-promoting factors, including chaperones and post-translational modifiers (Freer et al., 2016). This suggests that even vulnerable neuron populations have a genetic predisposition to early accumulate Aβ aggregations and neurofibrillary tangles. However, this study did not provide cell-type specific information. Comparison of gene expression profiles between excitatory and inhibitory neurons identified a gene whose protein is involved in the regulation of pathological tau accumulation. Exclusively expressed in inhibitory neurons, BAG3 (BCL2-associated athanogene 3) was consequently linked to the vulnerability of excitatory neurons in AD (Fu et al., 2019). Eventually, a recent study investigating the molecular profile of vulnerable and resistant neurons in AD placed microtubule dynamics at the center of pathogenesis. In particular, a regulator of tau splicing was identified to be dysregulated in vulnerable ECII pyramidal neurons and might explain initial formation of neurofibrillary tangles (Roussarie et al., 2020), which then spread to other brain regions via hippocampal CA1 neurons (Kaufman et al., 2018).
Discussion
It is out of question that mitochondrial defects are involved in neurodegeneration of many common and rare neurodegenerative diseases. However, before considering to target mitochondria, in order to design novel therapeutic strategies against these disorders, it needs to be clarified how much the disrupted mitochondrial pathways eventually contribute to neuron death. Are they crucial factors or do they rather arise secondary to other pathological processes? The development of neurodegenerative diseases is unequivocally linked to aging. During aging, the activity of cellular quality control mechanisms decreases (Rubinsztein et al., 2011) while ROS amounts rise (Stefanatos and Sanz 2018), which should result in a stronger presence of fragmented and defective mitochondria. Interestingly, mitochondrial fragmentation is often observed in both common and rare diseases, with neurons frequently showing altered levels of mitochondrial dynamics proteins. While disrupted mitochondrial fusion or enhanced fission could hence be seen as a disease-spanning feature, mitochondrial fragments could also result from decelerated removal of defective mitochondria in the first place. The presence of fragmented mitochondria observed in neurological disorders must thus be interpreted with caution, since it might reflect a phenomenon related to aging. Impairment of mitochondrial dynamics, mtDNA maintenance or Ca2+ handling, however, could cause additional susceptibility to specific neuron populations, which ultimately tips the balance from survival toward death.
Huntington’s Disease
In HD, mitochondrial defects, such as impaired trafficking and Ca2+ handling, are primarily caused by mHTT. Cell-intrinsic properties of GABAergic medium spiny neurons do not indicate extraordinary vulnerability to mitochondrial dysfunction. In fact, the strong dependence on BDNF signaling rather sustains the special susceptibility of these neurons to mHTT. Instead of facilitating mitochondrial transport or modifying cytosolic Ca2+ levels, which could be beneficial to slow down neurodegeneration for a certain period of time, it is thus wiser to focus on the removal of mutant HTT in order to prevent neuron death in the long term. In this context, a promising new treatment strategy based on antisense oligonucleotide injection via lumbar puncture is currently tested and has already been shown to reduce mHTT levels by 40–60% (Lempriere 2019).
Alzheimer’s Disease
Similar to HD, it is likely that mitochondrial dysfunction plays rather a secondary role for neurodegeneration in AD and arises as a consequence of Aβ and pTau pathology. A probably primary contribution of neurofibrillary tangles and maybe also Aβ aggregates is confirmed by cell-intrinsic properties of ECII and CA1 pyramidal neurons, showing high dependence on tau homeostasis. Correspondingly, reducing Aβ aggregates and pTau levels in the brain is still in the focus of AD research. The antisense oligonucleotide technique has been applied recently and could achieve a reduction of both the amyloid precursor protein (Chang et al., 2018) and pTau (DeVos et al., 2017) in preclinical trials. However, some studies still suggest that mitochondrial defects are upstream of Aβ and pTau pathology, fueling the “mitochondrial cascade hypothesis,” which puts mitochondrial dysfunction at the heart of AD development (Swerdlow et al., 2014). Mitochondrial function has been shown to be impaired even before the accumulation of Aβ deposits (Yao et al., 2009; Leuner et al., 2012; Mao et al., 2012) and mitochondrial impairment via toxins or genetic ablation of ROS suppressing proteins accelerated Aβ pathology (Esposito et al., 2006; Chen et al., 2015). Formation of Aβ aggregates was accordingly facilitated by ROS upon mitochondrial dysfunction (Gwon et al., 2012). Interestingly, complex I inhibition via rotenone treatment in rats leads to increased pTau in neurons, astrocytes and oligodendrocytes (Hoglinger et al., 2005). Furthermore, the loss of mitochondrial proteins, such as the protease AFG3L2 (Kondadi et al., 2014) and the ROS scavenger SOD2 (Melov et al., 2007), is associated with pTau presence, indicating that mitochondrial dysfunction promotes tauopathy in the brain. In line with this, inhibition of mitochondrial fragmentation (Wang et al., 2017) and upregulation of PGC-1α, which induces mitochondrial biogenesis (Katsouri et al., 2016), could ameliorate Aβ pathology and cognitive defects in mice. Recently, by using a series of in vitro and in vivo models, stimulation of mitophagy was shown to reduce Aβ deposits as well as pTau formation, and hence reversed cognitive impairment (Fang et al., 2019). Thus, future approaches to treat AD might be designed in such a way that Aβ and pTau levels in the brain are lowered, and that mitochondrial function is simultaneously improved, e.g., by enhancing mitochondrial quality control. In addition, mitochondrial-targeted reagents suppressing the formation of ROS could be beneficial, as indicated by treatment with tetra-peptide SS31 (Reddy et al., 2017) or Mito-Ubiquinone (MitoQ), which improves cognitive decline in triple transgenic AD mice (McManus et al., 2011) and is currently tested in early onset AD patients (NCT03514875).
Peripheral Neuropathies
In peripheral neuropathies, the mitochondrial impact on selective neurodegeneration might be more extensive. Due to their extraordinary long axons, it is quite comprehensible that motor neurons are especially vulnerable to disruption of mitochondrial dynamics and especially trafficking of mitochondria, as seen in Charcot-Marie-Tooth disease caused by mutations in MFN2, GDAP1 as well as KIF1B, encoding the mitochondrial motor KIF1Bβ (Zhao et al., 2001). Besides mitochondrial fission and fusion, mutations in MFN2 and GDAP1 further affect mitophagy and mitochondrial Ca2+ handling mediated via MAMs. In these cases, disrupted mitochondrial dynamics are clearly the causative event for motor neuron loss. Treatments of peripheral neuropathies might consequently involve genetic approaches to introduce wild type MFNs and GDAP1, as shown recently in a MFN2 transgenic mouse model (Zhou et al., 2019), or pharmacological approaches to target these proteins. In fact, agonists for MFN2 have been identified and shown to rescue mitochondrial fusion as well as trafficking in vitro (Rocha et al., 2018) and could therefore present a promising therapeutic tool for the future, especially for treatment of CMT2A.
Amyotrophic Lateral Sclerosis
For ALS, it is rather difficult to postulate a primary cause of mitochondrial dysfunction for motor neuron death because of the disease’s complex degenerative pattern, the variety of distinct gene mutations, and the presence of aggregate-prone proteins. Whether of primary cause or not, mitochondrial fragmentation at least participates in ALS linked to SOD1 mutation, since inhibition of mitochondrial fission by overexpression of dominant negative Drp1 K38A normalized morphology and transport of mitochondria, leading to increased motor neuron survival (Song et al., 2013). However, in familial cases showing mutations in SQSTM1, OPTN or TBK1, mitochondrial fragmentation might be rather secondary to impaired mitophagy in the first place, resulting in the accumulation of shortened mitochondria. Stimulating mitophagy might hence be a promising target to prevent mitochondrial dysfunction in both familial and sporadic ALS. Simultaneously, for familial cases harboring mutations in SOD1, TDP-43 and FUS, as well as for sporadic cases revealing characteristic protein aggregations, mitochondrial defects likely result from misfolded proteins. In these cases, stimulation of bulk autophagy should address both pathways by promoting mitochondrial turnover and removal of such aggregate-prone proteins. Accordingly, within the last years, different autophagy modulators, including rapamycin, trehalose or rilmenidine, have been tested. However, they provided mixed results depending on the model used, administration route, and off-target effects (Madruga et al., 2021), highlighting both the complexity and heterogeneity of this disease. Finally, targeting cytosolic Ca2+ levels might be an additional promising approach, considering the low Ca2+ buffering capacity and the high risk of glutamate induced excitotoxicity for vulnerable spinal motor neurons innervating fast-twitch muscles. Noteworthy, future therapies would further need to include neuromuscular junctions, since prevention of motor neuron loss per se might not recover the lost axonal connections to muscle fibers, once they have retracted (Gould et al., 2006; Dewil et al., 2007). Focusing only on the maintenance of spinal motor neurons might therefore not improve clinical symptoms at later stages of ALS.
Spinocerebellar Ataxias
In several types of spinocerebellar ataxia fragmented mitochondria are observed in Purkinje cells. However, mitochondrial fragmentation is likely not the primary cause of selective neurodegeneration, but occurs as a side effect of upstream malfunctions, as indicated e.g. in case of AFG3L2 mutations. In addition, shuttling of mitochondria throughout the enormous dendritic tree of Purkinje cells highly depends on prior mitochondrial fission in order to be able to leave the soma (Delettre et al., 2000; Ishihara et al., 2009), with defective fission leading to shortening of the dendritic arbor (Fukumitsu et al., 2016). This renders Purkinje cells rather vulnerable to a pathologically hyperfused mitochondrial network. Consequently, targeting mitochondrial fission could be a potential treatment of patients suffering from spinocerebellar ataxia type 7, which has been linked to mitochondrial elongation (Ward et al., 2019). Furthermore, the enhanced susceptibility to mtDNA alterations following impaired replication highlights a strong dependence of Purkinje cells on mitochondrial OXPHOS, which might underlie the high energetic demand due to the special neuron morphology or an especially high turnover of respiratory chain complexes. Noteworthy, many spinocerebellar ataxias are linked to CAG nucleotide repeat expansions and the subsequent aggregation of toxic polyglutamine protein (Paulson et al., 2017). In these cases, reducing polyglutamine protein levels by using antisense oligonucleotides (Toonen et al., 2016) or small interfering RNA (Ramachandran et al., 2014) is probably of greater priority. After all, concerning the marked susceptibility of Purkinje cells to MCU impairment as well as the variety of monogenic mutations affecting Ca2+ homeostasis that have been linked to spinocerebellar ataxia, disrupted Ca2+ handling is likely to be a crucial factor for Purkinje cell loss. Potentially, future approaches aiming at successfully reducing cytosolic Ca2+ levels, ideally in distinct cell types, could be used to prevent Ca2+-induced cytotoxicity.
Parkinson’s Disease
Mitochondrial dysfunction plays a crucial role for the selective neurodegeneration in PD, as evidenced by complex I inhibiting toxins causing parkinsonism (Langston et al., 1983; Betarbet et al., 2000), an especially high accumulation rate of mtDNA deletions in idiopathic cases, as well as the wide variety of monogenic mutations impairing mitochondrial function in familial forms. Remarkably, all mitochondrial processes mentioned above had been linked to the loss of SNc dopaminergic neurons. It is therefore not surprising that perished SNc dopaminergic neurons or parkinsonian features are further reported in cases of spinocerebellar ataxia (Palin et al., 2013; Tzoulis et al., 2013; Synofzik 2019) and CMT2A (Aerts et al., 2016), emphasizing their special vulnerability to mitochondrial defects. Cell type intrinsic properties are of critical importance for the high susceptibility of SNc dopaminergic neurons, which is highlighted by other dopamine neuron populations being spared upon mitochondrial dysfunction (Pass et al., 2020; Ricke et al., 2020). Thus, a lot of attention is currently paid on voltage-gated Ca2+ channels, since they present promising pharmacological targets to decrease the cytosolic Ca2+ burden and its associated pathology, including mitochondrial dysfunction as well as the formation of αSyn aggregates. The usage of dihydropyridines for instance, which block L-type channels, has been linked to a 20–30% lower risk for PD development in patients treated for high blood pressure (Lang et al., 2015). Despite controversial results regarding dose efficiency as well as specificity in mice, the dihydropyridine isradipine, has been tested in a phase III clinical trial on PD patients (Biglan et al., 2017). This clinical trial failed (Parkinson Study Group, S.-P. D. I. I. I. I., 2020), supporting recent concerns about the possible efficiency of the dosage which could be used (Ortner et al., 2017).
Mitochondrial-targeted reagents suppressing ROS, such as MitoQ, Mito-Apocynin or Szeto-Schiller peptide, reveal neuroprotective effects in vitro as well as in pharmacological-induced PD mouse models (Jin et al., 2014). Moreover, administration of Mito-Apocynin could attenuate motor defects and progressive degeneration of SNc dopaminergic neurons in MitoPark mice (Langley et al., 2017). However, a phase III clinical trial using MitoQ failed (Snow et al., 2010), which led to the oxidative stress hypothesis being fundamentally questioned.
Importantly, PD occurs in a very heterogeneous manner indicating a necessity for adjusted treatments for smaller patient cohorts instead of broadly-based therapy in the future. Considering PD cases presenting αSyn aggregates and mutant LRRK2, which are both likely to be upstream of mitochondrial defects, it appears necessary to decrease their presence in the brain by making use of antisense oligonucleotides or interfering RNA technologies. Noteworthy, not all PD cases reveal αSyn pathology, such as most of the patients harboring Parkin mutations, highlighting the chance for mitochondrial treatment approaches targeting mitochondrial trafficking or quality control.
Conclusion
Despite the complex nature of neurodegenerative diseases, therapies targeting mitochondrial dysfunction represent a promising, potential tool to slow down or even stop neuron death. Depending on the mitochondrial impact on neurodegeneration, which varies from disease to disease, future treatments fueling mitochondrial function might be used either primarily or in a combinatory manner. Pharmacological compounds or therapies stimulating both mitochondrial biogenesis and turnover could therefore be used in addition to already established treatments, which mostly deal with clinical symptoms but not with the progressive loss of neurons. Convergent mitochondrial defects thereby reflect similar pathological pathways and hold the potential for disease-spanning treatment.
Author Contributions
TP: Literature research, Original Draft and Writing, Visualization and Graphics, Review and Editing. RW: Review and Editing. DP-M: Visualization and Graphics, Review and Editing, Supervision.
Funding
This work was supported by the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG)—Project-ID 73111208—SFB 829, PL 895/1-1 and Köln Fortune 341/2019 to DP-M and RW.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
All figures were created with BioRender.com. The table was generated by using Photoshop CS2.
References
Aerts, M. B., Weterman, M. A. J., Quadri, M., Schelhaas, H. J., Bloem, B. R., Esselink, R. A., et al. (2016). A LRSAM1 Mutation Links Charcot-Marie-Tooth Type 2 to P Arkinson’s Disease. Ann. Clin. Transl Neurol. 3 (2), 146–149. doi:10.1002/acn3.281
Alam, Z. I., Jenner, A., Daniel, S. E., Lees, A. J., Cairns, N., Marsden, C. D., et al. (1997). Oxidative DNA Damage in the Parkinsonian Brain: an Apparent Selective Increase in 8-hydroxyguanine Levels in Substantia Nigra. J. Neurochem. 69 (3), 1196–1203. doi:10.1046/j.1471-4159.1997.69031196.x
Alberico, S. L., Cassell, M. D., and Narayanan, N. S. (2015). The Vulnerable Ventral Tegmental Area in Parkinson's Disease. Basal Ganglia 5 (2-3), 51–55. doi:10.1016/j.baga.2015.06.001
Alexianu, M. E., Ho, B.-K., Mohamed, A. H., La Bella, V., Smith, R. G., and Appel, S. H. (1994). The Role of Calcium-Binding Proteins in Selective Motoneuron Vulnerability in Amyotrophic Lateral Sclerosis. Ann. Neurol. 36 (6), 846–858. doi:10.1002/ana.410360608
Almajan, E. R., Richter, R., Paeger, L., Martinelli, P., Barth, E., Decker, T., et al. (2012). AFG3L2 Supports Mitochondrial Protein Synthesis and Purkinje Cell Survival. J. Clin. Invest. 122 (11), 4048–4058. doi:10.1172/jci64604
Alonso, A. C., Zaidi, T., Grundke-Iqbal, I., and Iqbal, K. (1994). Role of Abnormally Phosphorylated Tau in the Breakdown of Microtubules in Alzheimer Disease. Proc. Natl. Acad. Sci. 91 (12), 5562–5566. doi:10.1073/pnas.91.12.5562
Altar, C. A., Cai, N., Bliven, T., Juhasz, M., Conner, J. M., Acheson, A. L., et al. (1997). Anterograde Transport of Brain-Derived Neurotrophic Factor and its Role in the Brain. Nature 389 (6653), 856–860. doi:10.1038/39885
Amati-Bonneau, P., Guichet, A., Olichon, A., Chevrollier, A., Viala, F., Miot, S., et al. (2005). OPA1 R445H Mutation in Optic Atrophy Associated with Sensorineural Deafness. Ann. Neurol. 58 (6), 958–963. doi:10.1002/ana.20681
Amo, T., Saiki, S., Sawayama, T., Sato, S., and Hattori, N. (2014). Detailed Analysis of Mitochondrial Respiratory Chain Defects Caused by Loss of PINK1. Neurosci. Lett. 580, 37–40. doi:10.1016/j.neulet.2014.07.045
Anagnostou, G., Akbar, M. T., Paul, P., Angelinetta, C., Steiner, T. J., and de Belleroche, J. (2010). Vesicle Associated Membrane Protein B (VAPB) Is Decreased in ALS Spinal Cord. Neurobiol. Aging 31 (6), 969–985. doi:10.1016/j.neurobiolaging.2008.07.005
Anand, R., Wai, T., Baker, M. J., Kladt, N., Schauss, A. C., Rugarli, E., et al. (2014). The I-AAA Protease YME1L and OMA1 Cleave OPA1 to Balance Mitochondrial Fusion and Fission. J. Cel Biol 204 (6), 919–929. doi:10.1083/jcb.201308006
Apostol, B. L., Simmons, D. A., Zuccato, C., Illes, K., Pallos, J., Casale, M., et al. (2008). CEP-1347 Reduces Mutant Huntingtin-Associated Neurotoxicity and Restores BDNF Levels in R6/2 Mice. Mol. Cell Neurosci. 39 (1), 8–20. doi:10.1016/j.mcn.2008.04.007
Area-Gomez, E., and Schon, E. A. (2017). On the Pathogenesis of Alzheimer's Disease: The MAM Hypothesis. FASEB j. 31 (3), 864–867. doi:10.1096/fj.201601309
Armstrong, D. M., and Rawson, J. A. (1979). Activity Patterns of Cerebellar Cortical Neurones and Climbing Fibre Afferents in the Awake Cat. J. Physiol. 289, 425–448. doi:10.1113/jphysiol.1979.sp012745
Arnold, S. E., Hyman, B. T., Flory, J., Damasio, A. R., and Van Hoesen, G. W. (1991). The Topographical and Neuroanatomical Distribution of Neurofibrillary Tangles and Neuritic Plaques in the Cerebral Cortex of Patients with Alzheimer's Disease. Cereb. Cortex 1 (1), 103–116. doi:10.1093/cercor/1.1.103
Ashrafi, G., Schlehe, J. S., LaVoie, M. J., and Schwarz, T. L. (2014). Mitophagy of Damaged Mitochondria Occurs Locally in Distal Neuronal Axons and Requires PINK1 and Parkin. J. Cel Biol 206 (5), 655–670. doi:10.1083/jcb.201401070
Atsumi, T. (1981). The Ultrastructure of Intramuscular Nerves in Amyotrophic Lateral Sclerosis. Acta Neuropathol. 55 (3), 193–198. doi:10.1007/bf00691318
Attwell, D., and Laughlin, S. B. (2001). An Energy Budget for Signaling in the Grey Matter of the Brain. J. Cereb. Blood Flow Metab. 21 (10), 1133–1145. doi:10.1097/00004647-200110000-00001
Babcock, D. F., Herrington, J., Goodwin, P. C., Park, Y. B., and Hille, B. (1997). Mitochondrial Participation in the Intracellular Ca2+ Network. J. Cel Biol 136 (4), 833–844. doi:10.1083/jcb.136.4.833
Ballion, B., Mallet, N., Bézard, E., Lanciego, J. L., and Gonon, F. (2008). Intratelencephalic Corticostriatal Neurons Equally Excite Striatonigral and Striatopallidal Neurons and Their Discharge Activity Is Selectively Reduced in Experimental Parkinsonism. Eur. J. Neurosci. 27 (9), 2313–2321. doi:10.1111/j.1460-9568.2008.06192.x
Baloh, R. H., Salavaggione, E., Milbrandt, J., and Pestronk, A. (2007). Familial Parkinsonism and Ophthalmoplegia from a Mutation in the Mitochondrial DNA Helicase Twinkle. Arch. Neurol. 64 (7), 998–1000. doi:10.1001/archneur.64.7.998
Baquet, Z. C., Gorski, J. A., and Jones, K. R. (2004). Early Striatal Dendrite Deficits Followed by Neuron Loss with Advanced Age in the Absence of Anterograde Cortical Brain-Derived Neurotrophic Factor. J. Neurosci. 24 (17), 4250–4258. doi:10.1523/jneurosci.3920-03.2004
Baranov, S. V., Baranova, O. V., Yablonska, S., Suofu, Y., Vazquez, A. L., Kozai, T. D. Y., et al. (2019). Mitochondria Modulate Programmed Neuritic Retraction. Proc. Natl. Acad. Sci. USA 116 (2), 650–659. doi:10.1073/pnas.1811021116
Barneo-Munoz, M., Juarez, P., Civera-Tregon, A., Yndriago, L., Pla-Martin, D., Zenker, J., et al. (2015). Lack of GDAP1 Induces Neuronal Calcium and Mitochondrial Defects in a Knockout Mouse Model of Charcot-Marie-Tooth Neuropathy. Plos Genet. 11 (4), e1005115. doi:10.1371/journal.pgen.1005115
Barski, J. J., Hartmann, J., Rose, C. R., Hoebeek, F., Mörl, K., Noll-Hussong, M., et al. (2003). Calbindin in Cerebellar Purkinje Cells Is a Critical Determinant of the Precision of Motor Coordination. J. Neurosci. 23 (8), 3469–3477. doi:10.1523/jneurosci.23-08-03469.2003
Baydyuk, M., Russell, T., Liao, G.-Y., Zang, K., An, J. J., Reichardt, L. F., et al. (2011). TrkB Receptor Controls Striatal Formation by Regulating the Number of Newborn Striatal Neurons. Proc. Natl. Acad. Sci. 108 (4), 1669–1674. doi:10.1073/pnas.1004744108
Baydyuk, M., and Xu, B. (2014). BDNF Signaling and Survival of Striatal Neurons. Front Cel Neurosci 8, 254. doi:10.3389/fncel.2014.00254
Belanger, M., Yang, J., Petit, J.-M., Laroche, T., Magistretti, P. J., and Allaman, I. (2011). Role of the Glyoxalase System in Astrocyte-Mediated Neuroprotection. J. Neurosci. 31 (50), 18338–18352. doi:10.1523/jneurosci.1249-11.2011
Bender, A., Krishnan, K. J., Morris, C. M., Taylor, G. A., Reeve, A. K., Perry, R. H., et al. (2006). High Levels of Mitochondrial DNA Deletions in Substantia Nigra Neurons in Aging and Parkinson Disease. Nat. Genet. 38 (5), 515–517. doi:10.1038/ng1769
Benkert, J., Hess, S., Roy, S., Beccano-Kelly, D., Wiederspohn, N., Duda, J., et al. (2019). Cav2.3 Channels Contribute to Dopaminergic Neuron Loss in a Model of Parkinson's Disease. Nat. Commun. 10 (1), 5094. doi:10.1038/s41467-019-12834-x
Berenguer-Escuder, C., Grossmann, D., Antony, P., Arena, G., Wasner, K., Massart, F., et al. (2020). Impaired Mitochondrial-Endoplasmic Reticulum Interaction and Mitophagy in Miro1-Mutant Neurons in Parkinson's Disease. Hum. Mol. Genet. 29 (8), 1353–1364. doi:10.1093/hmg/ddaa066
Bernard-Marissal, N., Medard, J. J., Azzedine, H., and Chrast, R. (2015). Dysfunction in Endoplasmic Reticulum-Mitochondria Crosstalk Underlies SIGMAR1 Loss of Function Mediated Motor Neuron Degeneration. Brain 138 (Pt 4), 875–890. doi:10.1093/brain/awv008
Bernard-Marissal, N., van Hameren, G., Juneja, M., Pellegrino, C., Louhivuori, L., Bartesaghi, L., et al. (2019). Altered Interplay between Endoplasmic Reticulum and Mitochondria in Charcot-Marie-Tooth Type 2A Neuropathy. Proc. Natl. Acad. Sci. USA 116 (6), 2328–2337. doi:10.1073/pnas.1810932116
Betarbet, R., Sherer, T. B., MacKenzie, G., Garcia-Osuna, M., Panov, A. V., and Greenamyre, J. T. (2000). Chronic Systemic Pesticide Exposure Reproduces Features of Parkinson's Disease. Nat. Neurosci. 3 (12), 1301–1306. doi:10.1038/81834
Biglan, K. M., Oakes, D., Lang, A. E., Hauser, R. A., Hodgeman, K., Greco, B., et al. (2017). A Novel Design of a Phase III Trial of Isradipine in Early Parkinson Disease (STEADY-PD III). Ann. Clin. Transl Neurol. 4 (6), 360–368. doi:10.1002/acn3.412
Billups, B., and Forsythe, I. D. (2002). Presynaptic Mitochondrial Calcium Sequestration Influences Transmission at Mammalian central Synapses. J. Neurosci. 22 (14), 5840–5847. doi:10.1523/jneurosci.22-14-05840.2002
Bitoun, M., Bevilacqua, J. A., Prudhon, B., Maugenre, S., Taratuto, A. L., Monges, S., et al. (2007). Dynamin 2 Mutations Cause Sporadic Centronuclear Myopathy with Neonatal Onset. Ann. Neurol. 62 (6), 666–670. doi:10.1002/ana.21235
Bolam, J. P., and Pissadaki, E. K. (2012). Living on the edge with too many mouths to feed: why dopamine neurons die. Mov. Disord. 27 (12), 1478–1483. doi:10.1002/mds.25135
Braak, H., and Braak, E. (1991). Neuropathological Stageing of Alzheimer-Related Changes. Acta Neuropathol. 82 (4), 239–259. doi:10.1007/bf00308809
Braak, H., Ghebremedhin, E., Rüb, U., Bratzke, H., and Del Tredici, K. (2004). Stages in the Development of Parkinson's Disease-Related Pathology. Cell Tissue Res 318 (1), 121–134. doi:10.1007/s00441-004-0956-9
Bradshaw, T. Y., Romano, L. E. L., Duncan, E. J., Nethisinghe, S., Abeti, R., Michael, G. J., et al. (2016). A Reduction in Drp1-Mediated Fission Compromises Mitochondrial Health in Autosomal Recessive Spastic Ataxia of Charlevoix Saguenay. Hum. Mol. Genet. 25 (15), 3232–3244. doi:10.1093/hmg/ddw173
Braschi, E., Goyon, V., Zunino, R., Mohanty, A., Xu, L., and McBride, H. M. (2010). Vps35 Mediates Vesicle Transport between the Mitochondria and Peroxisomes. Curr. Biol. 20 (14), 1310–1315. doi:10.1016/j.cub.2010.05.066
Breen, D. P., Munoz, D. G., and Lang, A. E. (2020). Twinkle-associated Familial Parkinsonism with Lewy Pathology. Neurology 95 (14), 644–647. doi:10.1212/wnl.0000000000010674
Brier, M. R., Gordon, B., Friedrichsen, K., McCarthy, J., Stern, A., Christensen, J., et al. (2016). Tau and Abeta Imaging, CSF Measures, and Cognition in Alzheimer's Disease. Sci. Transl Med. 8 (338), 338ra366. doi:10.1126/scitranslmed.aaf2362
Britti, E., Ros, J., Esteras, N., and Abramov, A. Y. (2020). Tau Inhibits Mitochondrial Calcium Efflux and Makes Neurons Vulnerable to Calcium-Induced Cell Death. Cell Calcium 86, 102150. doi:10.1016/j.ceca.2019.102150
Burgold, J., Schulz-Trieglaff, E. K., Voelkl, K., Gutierrez-Angel, S., Bader, J. M., Hosp, F., et al. (2019). Cortical Circuit Alterations Precede Motor Impairments in Huntington's Disease Mice. Sci. Rep. 9 (1), 6634. doi:10.1038/s41598-019-43024-w
Burke, R. E., Levine, D. N., Zajac, F. E., Tsairis, P., and Engel, W. K. (1971). Mammalian Motor Units: Physiological-Histochemical Correlation in Three Types in Cat Gastrocnemius. Science 174 (4010), 709–712. doi:10.1126/science.174.4010.709
Burke, R. E., and Tsairis, P. (1973). Anatomy and Innervation Ratios in Motor Units of Cat Gastrocnemius. J. Physiol. 234 (3), 749–765. doi:10.1113/jphysiol.1973.sp010370
Bursch, F., Kalmbach, N., Naujock, M., Staege, S., Eggenschwiler, R., Abo-Rady, M., et al. (2019). Altered Calcium Dynamics and Glutamate Receptor Properties in iPSC-Derived Motor Neurons from ALS Patients with C9orf72, FUS, SOD1 or TDP43 Mutations. Hum. Mol. Genet. 28 (17), 2835–2850. doi:10.1093/hmg/ddz107
Busche, M. A., Eichhoff, G., Adelsberger, H., Abramowski, D., Wiederhold, K.-H., Haass, C., et al. (2008). Clusters of Hyperactive Neurons Near Amyloid Plaques in a Mouse Model of Alzheimer's Disease. Science 321 (5896), 1686–1689. doi:10.1126/science.1162844
Bussière, T., Giannakopoulos, P., Bouras, C., Perl, D. P., Morrison, J. H., and Hof, P. R. (2003). Progressive Degeneration of Nonphosphorylated Neurofilament Protein-Enriched Pyramidal Neurons Predicts Cognitive Impairment in Alzheimer's Disease: Stereologic Analysis of Prefrontal Cortex Area 9. J. Comp. Neurol. 463 (3), 281–302. doi:10.1002/cne.10760
Cai, Q., Zakaria, H. M., Simone, A., and Sheng, Z.-H. (2012). Spatial Parkin Translocation and Degradation of Damaged Mitochondria via Mitophagy in Live Cortical Neurons. Curr. Biol. 22 (6), 545–552. doi:10.1016/j.cub.2012.02.005
Calì, T., Ottolini, D., Negro, A., and Brini, M. (2012). α-Synuclein Controls Mitochondrial Calcium Homeostasis by Enhancing Endoplasmic Reticulum-Mitochondria Interactions. J. Biol. Chem. 287 (22), 17914–17929. doi:10.1074/jbc.m111.302794
Caligari, M., Godi, M., Guglielmetti, S., Franchignoni, F., and Nardone, A. (2013). Eye Tracking Communication Devices in Amyotrophic Lateral Sclerosis: Impact on Disability and Quality of Life. Amyotroph. Lateral Scler. Frontotemporal Degener 14 (7-8), 546–552. doi:10.3109/21678421.2013.803576
Calvo, S. E., Clauser, K. R., and Mootha, V. K. (2016). MitoCarta2.0: an Updated Inventory of Mammalian Mitochondrial Proteins. Nucleic Acids Res. 44 (D1), D1251–D1257. doi:10.1093/nar/gkv1003
Calvo-Rodríguez, M., García-Durillo, M., Villalobos, C., and Núñez, L. (2016). Aging Enables Ca2+ Overload and Apoptosis Induced by Amyloid-β Oligomers in Rat Hippocampal Neurons: Neuroprotection by Non-steroidal Anti-inflammatory Drugs and R-Flurbiprofen in Aging Neurons. Jad 54 (1), 207–221. doi:10.3233/jad-151189
Calvo-Rodriguez, M., Hernando-Perez, E., Nunez, L., and Villalobos, C. (2019). Amyloid Beta Oligomers Increase ER-Mitochondria Ca(2+) Cross Talk in Young Hippocampal Neurons and Exacerbate Aging-Induced Intracellular Ca(2+) Remodeling. Front. Cel Neurosci 13, 22. doi:10.3389/fncel.2019.00022
Calvo-Rodriguez, M., Hou, S. S., Snyder, A. C., Kharitonova, E. K., Russ, A. N., Das, S., et al. (2020). Increased Mitochondrial Calcium Levels Associated with Neuronal Death in a Mouse Model of Alzheimer's Disease. Nat. Commun. 11 (1), 2146. doi:10.1038/s41467-020-16074-2
Cantarero, L., Juarez-Escoto, E., Civera-Tregon, A., Rodriguez-Sanz, M., Roldan, M., Benitez, R., et al. (2020). Mitochondria-lysosome Membrane Contacts Are Defective in GDAP1-Related Charcot-Marie-Tooth Disease. Hum. Mol. Genet. 29 (22).
Caraveo, G., Auluck, P. K., Whitesell, L., Chung, C. Y., Baru, V., Mosharov, E. V., et al. (2014). Calcineurin Determines Toxic versus Beneficial Responses to -synuclein. Proc. Natl. Acad. Sci. 111 (34), E3544–E3552. doi:10.1073/pnas.1413201111
Carrì, M. T., Ferri, A., Battistoni, A., Famhy, L., Gabbianelli, R., Poccia, F., et al. (1997). Expression of a Cu,Zn Superoxide Dismutase Typical of Familial Amyotrophic Lateral Sclerosis Induces Mitochondrial Alteration and Increase of Cytosolic Ca2+ Concentration in Transfected Neuroblastoma SH-Sy5y Cells. FEBS Lett. 414 (2), 365–368. doi:10.1016/s0014-5793(97)01051-x
Cerminara, N. L., Lang, E. J., Sillitoe, R. V., and Apps, R. (2015). Redefining the Cerebellar Cortex as an Assembly of Non-uniform Purkinje Cell Microcircuits. Nat. Rev. Neurosci. 16 (2), 79–93. doi:10.1038/nrn3886
Chan, C. S., Guzman, J. N., Ilijic, E., Mercer, J. N., Rick, C., Tkatch, T., et al. (2007). ‘Rejuvenation' Protects Neurons in Mouse Models of Parkinson's Disease. Nature 447 (7148), 1081–1086. doi:10.1038/nature05865
Chan, N. C., Salazar, A. M., Pham, A. H., Sweredoski, M. J., Kolawa, N. J., Graham, R. L. J., et al. (2011). Broad Activation of the Ubiquitin-Proteasome System by Parkin Is Critical for Mitophagy. Hum. Mol. Genet. 20 (9), 1726–1737. doi:10.1093/hmg/ddr048
Chan, S. L., Mayne, M., Holden, C. P., Geiger, J. D., and Mattson, M. P. (2000). Presenilin-1 Mutations Increase Levels of Ryanodine Receptors and Calcium Release in PC12 Cells and Cortical Neurons. J. Biol. Chem. 275 (24), 18195–18200. doi:10.1074/jbc.m000040200
Chang, D. T. W., Rintoul, G. L., Pandipati, S., and Reynolds, I. J. (2006). Mutant Huntingtin Aggregates Impair Mitochondrial Movement and Trafficking in Cortical Neurons. Neurobiol. Dis. 22 (2), 388–400. doi:10.1016/j.nbd.2005.12.007
Chang, J. L., Hinrich, A. J., Roman, B., Norrbom, M., Rigo, F., Marr, R. A., et al. (2018). Targeting Amyloid-β Precursor Protein, APP, Splicing with Antisense Oligonucleotides Reduces Toxic Amyloid-β Production. Mol. Ther. 26 (6), 1539–1551. doi:10.1016/j.ymthe.2018.02.029
Chen, H., Detmer, S. A., Ewald, A. J., Griffin, E. E., Fraser, S. E., and Chan, D. C. (2003). Mitofusins Mfn1 and Mfn2 Coordinately Regulate Mitochondrial Fusion and Are Essential for Embryonic Development. J. Cel Biol 160 (2), 189–200. doi:10.1083/jcb.200211046
Chen, H., McCaffery, J. M., and Chan, D. C. (2007). Mitochondrial Fusion Protects against Neurodegeneration in the Cerebellum. Cell 130 (3), 548–562. doi:10.1016/j.cell.2007.06.026
Chen, H., Vermulst, M., Wang, Y. E., Chomyn, A., Prolla, T. A., McCaffery, J. M., et al. (2010). Mitochondrial Fusion Is Required for mtDNA Stability in Skeletal Muscle and Tolerance of mtDNA Mutations. Cell 141 (2), 280–289. doi:10.1016/j.cell.2010.02.026
Chen, L., Na, R., Boldt, E., and Ran, Q. (2015). NLRP3 Inflammasome Activation by Mitochondrial Reactive Oxygen Species Plays a Key Role in Long-Term Cognitive Impairment Induced by Paraquat Exposure. Neurobiol. Aging 36 (9), 2533–2543. doi:10.1016/j.neurobiolaging.2015.05.018
Cho, D.-H., Nakamura, T., Fang, J., Cieplak, P., Godzik, A., Gu, Z., et al. (2009). S-nitrosylation of Drp1 Mediates β-Amyloid-Related Mitochondrial Fission and Neuronal Injury. Science 324 (5923), 102–105. doi:10.1126/science.1171091
Chrysostomou, A., Grady, J. P., Laude, A., Taylor, R. W., Turnbull, D. M., and Lax, N. Z. (2016). Investigating Complex I Deficiency in P Urkinje Cells and Synapses in Patients with Mitochondrial Disease. Neuropathol. Appl. Neurobiol. 42 (5), 477–492. doi:10.1111/nan.12282
Cirulli, E. T., Lasseigne, B. N., Petrovski, S., Sapp, P. C., Dion, P. A., Leblond, C. S., et al. (2015). Exome Sequencing in Amyotrophic Lateral Sclerosis Identifies Risk Genes and Pathways. Science 347 (6229), 1436–1441. doi:10.1126/science.aaa3650
Civera-Tregón, A., Domínguez, L., Martínez-Valero, P., Serrano, C., Vallmitjana, A., Benítez, R., et al. (2021). Mitochondria and Calcium Defects Correlate with Axonal Dysfunction in GDAP1-Related Charcot-Marie-Tooth Mouse Model. Neurobiol. Dis. 152, 105300. doi:10.1016/j.nbd.2021.105300
Combs, B., Mueller, R. L., Morfini, G., Brady, S. T., and Kanaan, N. M. (2019). Tau and Axonal Transport Misregulation in Tauopathies. Adv. Exp. Med. Biol. 1184, 81–95. doi:10.1007/978-981-32-9358-8_7)
Copeland, W. C. (2014). Defects of Mitochondrial DNA Replication. J. Child. Neurol. 29 (9), 1216–1224. doi:10.1177/0883073814537380
Coskun, P. E., Beal, M. F., and Wallace, D. C. (2004). Alzheimer's Brains Harbor Somatic mtDNA Control-Region Mutations that Suppress Mitochondrial Transcription and Replication. Proc. Natl. Acad. Sci. 101 (29), 10726–10731. doi:10.1073/pnas.0403649101
Coskun, P., Wyrembak, J., Schriner, S. E., Chen, H.-W., Marciniack, C., Laferla, F., et al. (2012). A Mitochondrial Etiology of Alzheimer and Parkinson Disease. Biochim. Biophys. Acta (Bba) - Gen. Subjects 1820 (5), 553–564. doi:10.1016/j.bbagen.2011.08.008
Dafinca, R., Barbagallo, P., Farrimond, L., Candalija, A., Scaber, J., Ababneh, N. a. A., et al. (2020). Impairment of Mitochondrial Calcium Buffering Links Mutations in C9ORF72 and TARDBP in iPS-Derived Motor Neurons from Patients with ALS/FTD. Stem Cel Rep. 14 (5), 892–908. doi:10.1016/j.stemcr.2020.03.023
Dal Canto, M. C., and Gurney, M. E. (1994). Development of central Nervous System Pathology in a Murine Transgenic Model of Human Amyotrophic Lateral Sclerosis. Am. J. Pathol. 145 (6), 1271–1279.
Damiano, M., Starkov, A. A., Petri, S., Kipiani, K., Kiaei, M., Mattiazzi, M., et al. (2006). Neural Mitochondrial Ca2+capacity Impairment Precedes the Onset of Motor Symptoms in G93A Cu/Zn-Superoxide Dismutase Mutant Mice. J. Neurochem. 96 (5), 1349–1361. doi:10.1111/j.1471-4159.2006.03619.x
Damier, P., Hirsch, E. C., Agid, Y., and Graybiel, A. M. (1999). The Substantia Nigra of the Human Brain. I. Nigrosomes and the Nigral Matrix, a Compartmental Organization Based on Calbindin D(28K) Immunohistochemistry. Brain 122 (Pt 8), 1421–1436. doi:10.1093/brain/122.8.1421
Dash, R. K., and Beard, D. A. (2008). Analysis of Cardiac Mitochondrial Na+-Ca2+ exchanger Kinetics with a Biophysical Model of Mitochondrial Ca2+handing Suggests a 3: 1 Stoichiometry. J. Physiol. 586 (13), 3267–3285. doi:10.1113/jphysiol.2008.151977
Dave, K. D., De Silva, S., Sheth, N. P., Ramboz, S., Beck, M. J., Quang, C., et al. (2014). Phenotypic Characterization of Recessive Gene Knockout Rat Models of Parkinson's Disease. Neurobiol. Dis. 70, 190–203. doi:10.1016/j.nbd.2014.06.009
de Brito, O. M., and Scorrano, L. (2008). Mitofusin 2 Tethers Endoplasmic Reticulum to Mitochondria. Nature 456 (7222), 605–610. doi:10.1038/nature07534
de Juan-Sanz, J., Holt, G. T., Schreiter, E. R., de Juan, F., Kim, D. S., and Ryan, T. A. (2017). Axonal Endoplasmic Reticulum Ca2+ Content Controls Release Probability in CNS Nerve Terminals. Neuron 93 (4), 867–881. doi:10.1016/j.neuron.2017.01.010
De Vos, K. J., Chapman, A. L., Tennant, M. E., Manser, C., Tudor, E. L., Lau, K.-F., et al. (2007). Familial Amyotrophic Lateral Sclerosis-Linked SOD1 Mutants Perturb Fast Axonal Transport to Reduce Axonal Mitochondria Content. Hum. Mol. Genet. 16 (22), 2720–2728. doi:10.1093/hmg/ddm226
De Vos, K. J., Mórotz, G. M., Stoica, R., Tudor, E. L., Lau, K.-F., Ackerley, S., et al. (2012). VAPB Interacts with the Mitochondrial Protein PTPIP51 to Regulate Calcium Homeostasis. Hum. Mol. Genet. 21 (6), 1299–1311. doi:10.1093/hmg/ddr559
Deas, E., Plun-Favreau, H., Gandhi, S., Desmond, H., Kjaer, S., Loh, S. H. Y., et al. (2011). PINK1 Cleavage at Position A103 by the Mitochondrial Protease PARL. Hum. Mol. Genet. 20 (5), 867–879. doi:10.1093/hmg/ddq526
Delcambre, S., Nonnenmacher, Y., Hiller, K., and Buhlman, L. M. (2016). Dopamine Metabolism and Reactive Oxygen Species Production. Mitochondrial Mechanisms of Degeneration and Repair in Parkinson's Disease. (Cham: Springer International Publishing), 25–47. doi:10.1007/978-3-319-42139-1_2
Delettre, C., Lenaers, G., Griffoin, J.-M., Gigarel, N., Lorenzo, C., Belenguer, P., et al. (2000). Nuclear Gene OPA1, Encoding a Mitochondrial Dynamin-Related Protein, Is Mutated in Dominant Optic Atrophy. Nat. Genet. 26 (2), 207–210. doi:10.1038/79936
Deng, Z., Lim, J., Wang, Q., Purtell, K., Wu, S., Palomo, G. M., et al. (2020). ALS-FTLD-linked Mutations of SQSTM1/p62 Disrupt Selective Autophagy and NFE2L2/NRF2 Anti-oxidative Stress Pathway. Autophagy 16 (5), 917–931. doi:10.1080/15548627.2019.1644076
Deng, Z., Purtell, K., Lachance, V., Wold, M. S., Chen, S., and Yue, Z. (2017). Autophagy Receptors and Neurodegenerative Diseases. Trends Cel Biol. 27 (7), 491–504. doi:10.1016/j.tcb.2017.01.001
Detmer, S. A., Velde, C. V., Cleveland, D. W., and Chan, D. C. (2008). Hindlimb Gait Defects Due to Motor Axon Loss and Reduced Distal Muscles in a Transgenic Mouse Model of Charcot-Marie-Tooth Type 2A. Hum. Mol. Genet. 17 (3), 367–375. doi:10.1093/hmg/ddm314
DeVos, S. L., Miller, R. L., Schoch, K. M., Holmes, B. B., Kebodeaux, C. S., Wegener, A. J., et al. (2017). Tau Reduction Prevents Neuronal Loss and Reverses Pathological Tau Deposition and Seeding in Mice with Tauopathy. Sci. Transl Med. 9 (374). doi:10.1126/scitranslmed.aag0481
Devoto, P. V. M., Dimopoulos, N., Alloatti, M., Pardi, M. B., Saez, T. M., et al. (2017). alphaSynuclein Control of Mitochondrial Homeostasis in Human-Derived Neurons Is Disrupted by Mutations Associated with Parkinson's Disease. Sci. Rep. 7 (1), 5042
Dewil, M., dela Cruz, V. F., Van Den Bosch, L., and Robberecht, W. (2007). Inhibition of P38 Mitogen Activated Protein Kinase Activation and Mutant SOD1G93A-Induced Motor Neuron Death. Neurobiol. Dis. 26 (2), 332–341. doi:10.1016/j.nbd.2006.12.023
Di Maio, R., Barrett, P. J., Hoffman, E. K., Barrett, C. W., Zharikov, A., Borah, A., et al. (2016). α-Synuclein Binds to TOM20 and Inhibits Mitochondrial Protein Import in Parkinson's Disease. Sci. Transl. Med. 8 (342), 342ra78. doi:10.1126/scitranslmed.aaf3634
Diepenbroek, M., Casadei, N., Esmer, H., Saido, T. C., Takano, J., Kahle, P. J., et al. (2014). Overexpression of the Calpain-specific Inhibitor Calpastatin Reduces Human Alpha-Synuclein Processing, Aggregation and Synaptic Impairment in [A30P]αSyn Transgenic Mice. Hum. Mol. Genet. 23 (15), 3975–3989. doi:10.1093/hmg/ddu112
Dolle, C., Flones, I., Nido, G. S., Miletic, H., Osuagwu, N., Kristoffersen, S., et al. (2016). Defective Mitochondrial DNA Homeostasis in the Substantia Nigra in Parkinson Disease. Nat. Commun. 7, 13548. doi:10.1038/ncomms13548
Dopeso-Reyes, I. G., Rico, A. J., Roda, E., Sierra, S., Pignataro, D., Lanz, M., et al. (2014). Calbindin Content and Differential Vulnerability of Midbrain Efferent Dopaminergic Neurons in Macaques. Front. Neuroanat. 8, 146. doi:10.3389/fnana.2014.00146
DuBoff, B., Feany, M., and Götz, J. (2013). Why Size Matters - Balancing Mitochondrial Dynamics in Alzheimer's Disease. Trends Neurosciences 36 (6), 325–335. doi:10.1016/j.tins.2013.03.002
Duda, J., Pötschke, C., and Liss, B. (2016). Converging Roles of Ion Channels, Calcium, Metabolic Stress, and Activity Pattern of Substantia Nigra Dopaminergic Neurons in Health and Parkinson's Disease. J. Neurochem. 139 (Suppl. 1), 156–178. doi:10.1111/jnc.13572
Dufty, B. M., Warner, L. R., Hou, S. T., Jiang, S. X., Gomez-Isla, T., Leenhouts, K. M., et al. (2007). Calpain-Cleavage of α-Synuclein. Am. J. Pathol. 170 (5), 1725–1738. doi:10.2353/ajpath.2007.061232
Durr, A. (2010). Autosomal Dominant Cerebellar Ataxias: Polyglutamine Expansions and beyond. Lancet Neurol. 9 (9), 885–894. doi:10.1016/s1474-4422(10)70183-6
Eccles, J. C., Llinás, R., and Sasaki, K. (1966). The Excitatory Synaptic Action of Climbing Fibres on the Purkinje Cells of the Cerebellum. J. Physiol. 182 (2), 268–296. doi:10.1113/jphysiol.1966.sp007824
Ehses, S., Raschke, I., Mancuso, G., Bernacchia, A., Geimer, S., Tondera, D., et al. (2009). Regulation of OPA1 Processing and Mitochondrial Fusion by M-AAA Protease Isoenzymes and OMA1. J. Cel Biol 187 (7), 1023–1036. doi:10.1083/jcb.200906084
Ekstrand, M. I., Terzioglu, M., Galter, D., Zhu, S., Hofstetter, C., Lindqvist, E., et al. (2007). Progressive Parkinsonism in Mice with Respiratory-Chain-Deficient Dopamine Neurons. Proc. Natl. Acad. Sci. 104 (4), 1325–1330. doi:10.1073/pnas.0605208103
Enoka, R. M. (1995). Morphological Features and Activation Patterns of Motor Units. J. Clin. Neurophysiol. 12 (6), 538–559. doi:10.1097/00004691-199511000-00002
Esposito, L., Raber, J., Kekonius, L., Yan, F., Yu, G. Q., Bien-Ly, N., et al. (2006). Reduction in Mitochondrial Superoxide Dismutase Modulates Alzheimer's Disease-like Pathology and Accelerates the Onset of Behavioral Changes in Human Amyloid Precursor Protein Transgenic Mice. J. Neurosci. 26 (19), 5167–5179. doi:10.1523/jneurosci.0482-06.2006
Eura, Y., Ishihara, N., Yokota, S., and Mihara, K. (2003). Two Mitofusin Proteins, Mammalian Homologues of FZO, with Distinct Functions Are Both Required for Mitochondrial Fusion. J. Biochem. 134 (3), 333–344. doi:10.1093/jb/mvg150
Evans, C. S., and Holzbaur, E. L. F. (2019). Autophagy and Mitophagy in ALS. Neurobiol. Dis. 122, 35–40. doi:10.1016/j.nbd.2018.07.005
Fahrner, J. A., Liu, R., Perry, M. S., Klein, J., and Chan, D. C. (2016). A Novel De Novo Dominant Negative Mutation inDNM1Limpairs Mitochondrial Fission and Presents as Childhood Epileptic Encephalopathy. Am. J. Med. Genet. 170 (8), 2002–2011. doi:10.1002/ajmg.a.37721
Fang, E. F., Hou, Y., Palikaras, K., Adriaanse, B. A., Kerr, J. S., Yang, B., et al. (2019). Mitophagy Inhibits Amyloid-β and Tau Pathology and Reverses Cognitive Deficits in Models of Alzheimer's Disease. Nat. Neurosci. 22 (3), 401–412. doi:10.1038/s41593-018-0332-9
Feely, S. M. E., Laura, M., Siskind, C. E., Sottile, S., Davis, M., Gibbons, V. S., et al. (2011). MFN2 Mutations Cause Severe Phenotypes in Most Patients with CMT2A. Neurology 76 (20), 1690–1696. doi:10.1212/wnl.0b013e31821a441e
Feng, S. M., Che, C. H., Feng, S. Y., Liu, C. Y., Li, L. Y., Li, Y. X., et al. (2019). Novel Mutation in Optineurin Causing Aggressive ALS+/−frontotemporal Dementia. Ann. Clin. Transl Neurol. 6 (12), 2377–2383. doi:10.1002/acn3.50928
Ferreiro, E., Oliveira, C. R., and Pereira, C. M. F. (2008). The Release of Calcium from the Endoplasmic Reticulum Induced by Amyloid-Beta and Prion Peptides Activates the Mitochondrial Apoptotic Pathway. Neurobiol. Dis. 30 (3), 331–342. doi:10.1016/j.nbd.2008.02.003
Ferrer, I., Goutan, E., Marin, C., Rey, M. J., and Ribalta, T. (2000). Brain-derived Neurotrophic Factor in Huntington Disease. Brain Res. 866 (1-2), 257–261. doi:10.1016/s0006-8993(00)02237-x
Ferri, A., Fiorenzo, P., Nencini, M., Cozzolino, M., Pesaresi, M. G., Valle, C., et al. (2010). Glutaredoxin 2 Prevents Aggregation of Mutant SOD1 in Mitochondria and Abolishes its Toxicity. Hum. Mol. Genet. 19 (22), 4529–4542. doi:10.1093/hmg/ddq383
Filadi, R., Greotti, E., Turacchio, G., Luini, A., Pozzan, T., and Pizzo, P. (2016). Presenilin 2 Modulates Endoplasmic Reticulum-Mitochondria Coupling by Tuning the Antagonistic Effect of Mitofusin 2. Cel Rep. 15 (10), 2226–2238. doi:10.1016/j.celrep.2016.05.013
Foster, L. J., de Hoog, C. L., Zhang, Y., Zhang, Y., Xie, X., Mootha, V. K., et al. (2006). A Mammalian Organelle Map by Protein Correlation Profiling. Cell 125 (1), 187–199. doi:10.1016/j.cell.2006.03.022
Freer, R., Sormanni, P., Vecchi, G., Ciryam, P., Dobson, C. M., and Vendruscolo, M. (2016). A Protein Homeostasis Signature in Healthy Brains Recapitulates Tissue Vulnerability to Alzheimer's Disease. Sci. Adv. 2 (8), e1600947. doi:10.1126/sciadv.1600947
Freischmidt, A., Wieland, T., Richter, B., Ruf, W., Schaeffer, V., Müller, K., et al. (2015). Haploinsufficiency of TBK1 Causes Familial ALS and Fronto-Temporal Dementia. Nat. Neurosci. 18 (5), 631–636. doi:10.1038/nn.4000
Frey, D., Schneider, C., Xu, L., Borg, J., Spooren, W., and Caroni, P. (2000). Early and Selective Loss of Neuromuscular Synapse Subtypes with Low Sprouting Competence in Motoneuron Diseases. J. Neurosci. 20 (7), 2534–2542. doi:10.1523/jneurosci.20-07-02534.2000
Friedman, J. R., Lackner, L. L., West, M., DiBenedetto, J. R., Nunnari, J., and Voeltz, G. K. (2011). ER Tubules Mark Sites of Mitochondrial Division. Science 334 (6054), 358–362. doi:10.1126/science.1207385
Friel, D., and Tsien, R. (1994). An FCCP-Sensitive Ca2+ Store in Bullfrog Sympathetic Neurons and its Participation in Stimulus-Evoked Changes in [Ca2+]i. J. Neurosci. 14 (7), 4007–4024. doi:10.1523/jneurosci.14-07-04007.1994
Fu, H., Possenti, A., Freer, R., Nakano, Y., Hernandez Villegas, N. C., Tang, M., et al. (2019). A Tau Homeostasis Signature Is Linked with the Cellular and Regional Vulnerability of Excitatory Neurons to Tau Pathology. Nat. Neurosci. 22 (1), 47–56. doi:10.1038/s41593-018-0298-7
Fu, H., Rodriguez, G. A., Herman, M., Emrani, S., Nahmani, E., Barrett, G., et al. (2017). Tau Pathology Induces Excitatory Neuron Loss, Grid Cell Dysfunction, and Spatial Memory Deficits Reminiscent of Early Alzheimer's Disease. Neuron 93 (3), 533–541. doi:10.1016/j.neuron.2016.12.023
Fukumitsu, K., Hatsukano, T., Yoshimura, A., Heuser, J., Fujishima, K., and Kengaku, M. (2016). Mitochondrial Fission Protein Drp1 Regulates Mitochondrial Transport and Dendritic Arborization in Cerebellar Purkinje Cells. Mol. Cell Neurosci. 71, 56–65. doi:10.1016/j.mcn.2015.12.006
Fukutani, Y., Cairns, N. J., Shiozawa, M., Sasaki, K., Sudo, S., Isaki, K., et al. (2000). Neuronal Loss and Neurofibrillary Degeneration in the Hippocampal Cortex in Late‐onset Sporadic Alzheimer's Disease. Psychiatry Clin. Neurosciences 54 (5), 523–529. doi:10.1046/j.1440-1819.2000.00747.x
Gauthier, L. R., Charrin, B. C., Borrell-Pagès, M., Dompierre, J. P., Rangone, H., Cordelières, F. P., et al. (2004). Huntingtin Controls Neurotrophic Support and Survival of Neurons by Enhancing BDNF Vesicular Transport along Microtubules. Cell 118 (1), 127–138. doi:10.1016/j.cell.2004.06.018
Gbel, J., Engelhardt, E., Pelzer, P., Sakthivelu, V., Jahn, H. M., Jevtic, M., et al. (2020). Mitochondria-Endoplasmic Reticulum Contacts in Reactive Astrocytes Promote Vascular Remodeling. Cell Metab 31 (4), 791–808.
German, D. C., Manaye, K. F., Sonsalla, P. K., and Brooks, B. A. (1992). Midbrain Dopaminergic Cell Loss in Parkinson's Disease and MPTP-Induced Parkinsonism: Sparing of Calbindin-D25k?Containing Cells. Ann. NY Acad. Sci. 648, 42–62. doi:10.1111/j.1749-6632.1992.tb24523.x
Gertler, T. S., Chan, C. S., and Surmeier, D. J. (2008). Dichotomous Anatomical Properties of Adult Striatal Medium Spiny Neurons. J. Neurosci. 28 (43), 10814–10824. doi:10.1523/jneurosci.2660-08.2008
Gharami, K., Xie, Y., An, J. J., Tonegawa, S., and Xu, B. (2008). Brain-derived Neurotrophic Factor Over-expression in the Forebrain Ameliorates Huntington's Disease Phenotypes in Mice. J. Neurochem. 105 (2), 369–379. doi:10.1111/j.1471-4159.2007.05137.x
Giorgi, C., Baldassari, F., Bononi, A., Bonora, M., De Marchi, E., Marchi, S., et al. (2012). Mitochondrial Ca2+ and Apoptosis. Cell Calcium 52 (1), 36–43. doi:10.1016/j.ceca.2012.02.008
Giorgi, C., Marchi, S., and Pinton, P. (2018). The Machineries, Regulation and Cellular Functions of Mitochondrial Calcium. Nat. Rev. Mol. Cel Biol 19 (11), 713–730. doi:10.1038/s41580-018-0052-8
Girard, M., Lariviere, R., Parfitt, D. A., Deane, E. C., Gaudet, R., Nossova, N., et al. (2012). Mitochondrial Dysfunction and Purkinje Cell Loss in Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS). Proc. Natl. Acad. Sci. 109 (5), 1661–1666. doi:10.1073/pnas.1113166109
Glauser, L., Sonnay, S., Stafa, K., and Moore, D. J. (2011). Parkin Promotes the Ubiquitination and Degradation of the Mitochondrial Fusion Factor Mitofusin 1. J. Neurochem. 118 (4), 636–645. doi:10.1111/j.1471-4159.2011.07318.x
Goldberg, A. L. (2003). Protein Degradation and protection against Misfolded or Damaged Proteins. Nature 426 (6968), 895–899. doi:10.1038/nature02263
Golpich, M., Amini, E., Mohamed, Z., Azman Ali, R., Mohamed Ibrahim, N., and Ahmadiani, A. (2017). Mitochondrial Dysfunction and Biogenesis in Neurodegenerative Diseases: Pathogenesis and Treatment. CNS Neurosci. Ther. 23 (1), 5–22. doi:10.1111/cns.12655
Gómez-Isla, T., Price, J. L., McKeel Jr., D. W., Morris, J. C., Growdon, J. H., and Hyman, B. T. (1996). Profound Loss of Layer II Entorhinal Cortex Neurons Occurs in Very Mild Alzheimer's Disease. J. Neurosci. 16 (14), 4491–4500. doi:10.1523/jneurosci.16-14-04491.1996
Gonzalez-Sanchez, P., Pla-Martin, D., Martinez-Valero, P., Rueda, C. B., Calpena, E., Del Arco, A., et al. (2017). CMT-linked Loss-Of-Function Mutations in GDAP1 Impair Store-Operated Ca(2+) Entry-Stimulated Respiration. Sci. Rep. 7, 42993. doi:10.1038/srep42993
Goode, A., Butler, K., Long, J., Cavey, J., Scott, D., Shaw, B., et al. (2016). Defective Recognition of LC3B by Mutant SQSTM1/p62 Implicates Impairment of Autophagy as a Pathogenic Mechanism in ALS-FTLD. Autophagy 12 (7), 1094–1104. doi:10.1080/15548627.2016.1170257
Gould, T. W., Buss, R. R., Vinsant, S., Prevette, D., Sun, W., Knudson, C. M., et al. (2006). Complete Dissociation of Motor Neuron Death from Motor Dysfunction by Bax Deletion in a Mouse Model of ALS. J. Neurosci. 26 (34), 8774–8786. doi:10.1523/jneurosci.2315-06.2006
Grenier, K., Kontogiannea, M., and Fon, E. A. (2014). Short Mitochondrial ARF Triggers Parkin/PINK1-dependent Mitophagy. J. Biol. Chem. 289 (43), 29519–29530. doi:10.1074/jbc.m114.607150
Grenier, K., McLelland, G. L., and Fon, E. A. (2013). Parkin- and PINK1-dependent Mitophagy in Neurons: Will the Real Pathway Please Stand up? Front. Neurol. 4, 100. doi:10.3389/fneur.2013.00100
Grosskreutz, J., Van Den Bosch, L., and Keller, B. U. (2010). Calcium Dysregulation in Amyotrophic Lateral Sclerosis. Cell Calcium 47 (2), 165–174. doi:10.1016/j.ceca.2009.12.002
Grossmann, D., Berenguer-Escuder, C., Bellet, M. E., Scheibner, D., Bohler, J., Massart, F., et al. (2019). Mutations in RHOT1 Disrupt Endoplasmic Reticulum-Mitochondria Contact Sites Interfering with Calcium Homeostasis and Mitochondrial Dynamics in Parkinson's Disease. Antioxid. Redox Signaling 31 (16), 1213–1234. doi:10.1089/ars.2018.7718
Grünewald, A., Rygiel, K. A., Hepplewhite, P. D., Morris, C. M., Picard, M., and Turnbull, D. M. (2016). Mitochondrial DNA Depletion in Respiratory Chain-Deficient P Arkinson Disease Neurons. Ann. Neurol. 79 (3), 366–378. doi:10.1002/ana.24571
Guardia-Laguarta, C., Area-Gomez, E., Rub, C., Liu, Y., Magrane, J., Becker, D., et al. (2014). -Synuclein Is Localized to Mitochondria-Associated ER Membranes. J. Neurosci. 34 (1), 249–259. doi:10.1523/jneurosci.2507-13.2014
Guatteo, E., Carunchio, I., Pieri, M., Albo, F., Canu, N., Mercuri, N. B., et al. (2007). Altered Calcium Homeostasis in Motor Neurons Following AMPA Receptor but Not Voltage-dependent Calcium Channels' Activation in a Genetic Model of Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 28 (1), 90–100. doi:10.1016/j.nbd.2007.07.002
Gustafsson, C. M., Falkenberg, M., and Larsson, N.-G. (2016). Maintenance and Expression of Mammalian Mitochondrial DNA. Annu. Rev. Biochem. 85, 133–160. doi:10.1146/annurev-biochem-060815-014402
Guzman, J. N., Ilijic, E., Yang, B., Sanchez-Padilla, J., Wokosin, D., Galtieri, D., et al. (2018). Systemic Isradipine Treatment Diminishes Calcium-dependent Mitochondrial Oxidant Stress. J. Clin. Invest. 128 (6), 2266–2280. doi:10.1172/jci95898
Guzman, J. N., Sanchez-Padilla, J., Wokosin, D., Kondapalli, J., Ilijic, E., Schumacker, P. T., et al. (2010). Oxidant Stress Evoked by Pacemaking in Dopaminergic Neurons Is Attenuated by DJ-1. Nature 468 (7324), 696–700. doi:10.1038/nature09536
Gwon, A.-R., Park, J.-S., Arumugam, T. V., Kwon, Y.-K., Chan, S. L., Kim, S.-H., et al. (2012). Oxidative Lipid Modification of Nicastrin Enhances Amyloidogenic γ-secretase Activity in Alzheimer's Disease. Aging Cell 11 (4), 559–568. doi:10.1111/j.1474-9726.2012.00817.x
Haack, T. B., Ignatius, E., Calvo-Garrido, J., Iuso, A., Isohanni, P., Maffezzini, C., et al. (2016). Absence of the Autophagy Adaptor SQSTM1/p62 Causes Childhood-Onset Neurodegeneration with Ataxia, Dystonia, and Gaze Palsy. Am. J. Hum. Genet. 99 (3), 735–743. doi:10.1016/j.ajhg.2016.06.026
Hadano, S., Otomo, A., Kunita, R., Suzuki-Utsunomiya, K., Akatsuka, A., Koike, M., et al. (2010). Loss of ALS2/Alsin Exacerbates Motor Dysfunction in a SOD1-Expressing Mouse ALS Model by Disturbing Endolysosomal Trafficking. PLoS One 5 (3), e9805. doi:10.1371/journal.pone.0009805
Hakonen, A. H., Heiskanen, S., Juvonen, V., Lappalainen, I., Luoma, P. T., Rantamäki, M., et al. (2005). Mitochondrial DNA Polymerase W748S Mutation: a Common Cause of Autosomal Recessive Ataxia with Ancient European Origin. Am. J. Hum. Genet. 77 (3), 430–441. doi:10.1086/444548
Hakonen, A. H., Isohanni, P., Paetau, A., Herva, R., Suomalainen, A., and Lonnqvist, T. (2007). Recessive Twinkle Mutations in Early Onset Encephalopathy with mtDNA Depletion. Brain 130 (Pt 11), 3032–3040. doi:10.1093/brain/awm242
Halliday, G. M., McRitchie, D. A., Macdonald, V., Double, K. L., Trent, R. J., and McCusker, E. (1998). Regional Specificity of Brain Atrophy in Huntington's Disease. Exp. Neurol. 154 (2), 663–672. doi:10.1006/exnr.1998.6919
Hanss, Z., Larsen, S. B., Antony, P., Mencke, P., Massart, F., Jarazo, J., et al. (2020). Mitochondrial and Clearance Impairment in p.D620N VPS35 Patient-Derived Neurons. Mov Disord. 36(3):704-715. doi:10.1002/mds.28365
Hardingham, G. E., and Bading, H. (2003). The Yin and Yang of NMDA Receptor Signalling. Trends Neurosciences 26 (2), 81–89. doi:10.1016/s0166-2236(02)00040-1
Harris, J. J., Jolivet, R., and Attwell, D. (2012). Synaptic Energy Use and Supply. Neuron 75 (5), 762–777. doi:10.1016/j.neuron.2012.08.019
Head, B., Griparic, L., Amiri, M., Gandre-Babbe, S., and van der Bliek, A. M. (2009). Inducible Proteolytic Inactivation of OPA1 Mediated by the OMA1 Protease in Mammalian Cells. J. Cel Biol 187 (7), 959–966. doi:10.1083/jcb.200906083
Hedskog, L., Pinho, C. M., Filadi, R., Ronnback, A., Hertwig, L., Wiehager, B., et al. (2013). Modulation of the Endoplasmic Reticulum-Mitochondria Interface in Alzheimer's Disease and Related Models. Proc. Natl. Acad. Sci. 110 (19), 7916–7921. doi:10.1073/pnas.1300677110
Heo, J.-M., Ordureau, A., Paulo, J. A., Rinehart, J., and Harper, J. W. (2015). The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cel 60 (1), 7–20. doi:10.1016/j.molcel.2015.08.016
Hevner, R. F., and Wong-Riley, M. T. (1992). Entorhinal Cortex of the Human, Monkey, and Rat: Metabolic Map as Revealed by Cytochrome Oxidase. J. Comp. Neurol. 326 (3), 451–469. doi:10.1002/cne.903260310
Higgins, C. M., Jung, C., and Xu, Z. (2003). ALS-associated Mutant SOD1G93A Causes Mitochondrial Vacuolation by Expansion of the Intermembrane Space and by Involvement of SOD1 Aggregation and Peroxisomes. BMC Neurosci. 4, 16. doi:10.1186/1471-2202-4-16
Highstein, S. M., Karabelas, A., Baker, R., and McCrea, R. A. (1982). Comparison of the Morphology of Physiologically Identified Abducens Motor and Internuclear Neurons in the Cat: a Light Microscopic Study Employing the Intracellular Injection of Horseradish Peroxidase. J. Comp. Neurol. 208 (4), 369–381. doi:10.1002/cne.902080407
Hirabayashi, Y., Kwon, S.-K., Paek, H., Pernice, W. M., Paul, M. A., Lee, J., et al. (2017). ER-mitochondria Tethering by PDZD8 Regulates Ca2+dynamics in Mammalian Neurons. Science 358 (6363), 623–630. doi:10.1126/science.aan6009
Hof, P. R., Nimchinsky, E. A., Celio, M. R., Bouras, C., and Morrison, J. H. (1993). Calretinin-immunoreactive Neocortical Interneurons Are Unaffected in Alzheimer's Disease. Neurosci. Lett. 152 (1-2), 145–148. doi:10.1016/0304-3940(93)90504-e
Hoglinger, G. U., Lannuzel, A., Khondiker, M. E., Michel, P. P., Duyckaerts, C., Feger, J., et al. (2005). The Mitochondrial Complex I Inhibitor Rotenone Triggers a Cerebral Tauopathy. J. Neurochem. 95 (4), 930–939. doi:10.1111/j.1471-4159.2005.03493.x
Hong, K., Li, Y., Duan, W., Guo, Y., Jiang, H., Li, W., et al. (2012). Full-length TDP-43 and its C-Terminal Fragments Activate Mitophagy in NSC34 Cell Line. Neurosci. Lett. 530 (2), 144–149. doi:10.1016/j.neulet.2012.10.003
Howarth, C., Gleeson, P., and Attwell, D. (2012). Updated Energy Budgets for Neural Computation in the Neocortex and Cerebellum. J. Cereb. Blood Flow Metab. 32 (7), 1222–1232. doi:10.1038/jcbfm.2012.35
Hsieh, C.-H., Shaltouki, A., Gonzalez, A. E., Bettencourt da Cruz, A., Burbulla, L. F., St. Lawrence, E., et al. (2016). Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson's Disease. Cell Stem Cell 19 (6), 709–724. doi:10.1016/j.stem.2016.08.002
Hsu, J. Y., Jhang, Y. L., Cheng, P. H., Chang, Y. F., Mao, S. H., Yang, H. I., et al. (2017). The Truncated C-Terminal Fragment of Mutant ATXN3 Disrupts Mitochondria Dynamics in Spinocerebellar Ataxia Type 3 Models. Front. Mol. Neurosci. 10, 196. doi:10.3389/fnmol.2017.00196
Hu, D., Sun, X., Liao, X., Zhang, X., Zarabi, S., Schimmer, A., et al. (2019). Alpha-synuclein Suppresses Mitochondrial Protease ClpP to Trigger Mitochondrial Oxidative Damage and Neurotoxicity. Acta Neuropathol. 137 (6), 939–960. doi:10.1007/s00401-019-01993-2
Huang, M., and Verbeek, D. S. (2019). Why Do So many Genetic Insults lead to Purkinje Cell Degeneration and Spinocerebellar Ataxia? Neurosci. Lett. 688, 49–57. doi:10.1016/j.neulet.2018.02.004
Hudson, G., Deschauer, M., Busse, K., Zierz, S., and Chinnery, P. F. (2005). Sensory Ataxic Neuropathy Due to a Novel C10Orf2 Mutation with Probable Germline Mosaicism. Neurology 64 (2), 371–373. doi:10.1212/01.wnl.0000149767.51152.83
Hudson, G., Schaefer, A. M., Taylor, R. W., Tiangyou, W., Gibson, A., Venables, G., et al. (2007). Mutation of the Linker Region of the Polymerase γ-1 (POLG1Gene Associated with Progressive External Ophthalmoplegia and Parkinsonism. Arch. Neurol. 64 (4), 553–557. doi:10.1001/archneur.64.4.553
Hyder, F., Rothman, D. L., and Bennett, M. R. (2013). Cortical Energy Demands of Signaling and Nonsignaling Components in Brain Are Conserved across Mammalian Species and Activity Levels. Proc. Natl. Acad. Sci. USA 110 (9), 3549–3554. doi:10.1073/pnas.1214912110
Hyman, B., Van Hoesen, G., Damasio, A., and Barnes, C. (1984). Alzheimer's Disease: Cell-specific Pathology Isolates the Hippocampal Formation. Science 225 (4667), 1168–1170. doi:10.1126/science.6474172
Ishihara, N., Nomura, M., Jofuku, A., Kato, H., Suzuki, S. O., Masuda, K., et al. (2009). Mitochondrial Fission Factor Drp1 Is Essential for Embryonic Development and Synapse Formation in Mice. Nat. Cel Biol 11 (8), 958–966. doi:10.1038/ncb1907
Jauhari, A., Baranov, S. V., Suofu, Y., Kim, J., Singh, T., Yablonska, S., et al. (2021). Melatonin Inhibits Cytosolic Mitochondrial DNA-Induced Neuroinflammatory Signaling in Accelerated Aging and Neurodegeneration. J. Clin. Invest. 131 (9). doi:10.1172/jci150328
Jenner, P., and Olanow, C. W. (1996). Oxidative Stress and the Pathogenesis of Parkinson's Disease. Neurology 47 (6 Suppl. 3), S161–S170. doi:10.1212/wnl.47.6_suppl_3.161s
Jin, H., Kanthasamy, A., Ghosh, A., Anantharam, V., Kalyanaraman, B., and Kanthasamy, A. G. (2014). Mitochondria-targeted Antioxidants for Treatment of Parkinson's Disease: Preclinical and Clinical Outcomes. Biochim. Biophys. Acta (Bba) - Mol. Basis Dis. 1842 (8), 1282–1294. doi:10.1016/j.bbadis.2013.09.007
Jin, S. M., Lazarou, M., Wang, C., Kane, L. A., Narendra, D. P., and Youle, R. J. (2010). Mitochondrial Membrane Potential Regulates PINK1 Import and Proteolytic Destabilization by PARL. J. Cel Biol 191 (5), 933–942. doi:10.1083/jcb.201008084
Johansen, T., and Lamark, T. (2011). Selective Autophagy Mediated by Autophagic Adapter Proteins. Autophagy 7 (3), 279–296. doi:10.4161/auto.7.3.14487
Jouaville, L. S., Pinton, P., Bastianutto, C., Rutter, G. A., and Rizzuto, R. (1999). Regulation of Mitochondrial ATP Synthesis by Calcium: Evidence for a Long-Term Metabolic Priming. Proc. Natl. Acad. Sci. 96 (24), 13807–13812. doi:10.1073/pnas.96.24.13807
Jung, D. W., Baysal, K., and Brierley, G. P. (1995). The Sodium-Calcium Antiport of Heart Mitochondria Is Not Electroneutral. J. Biol. Chem. 270 (2), 672–678. doi:10.1074/jbc.270.2.672
Kamp, F., Exner, N., Lutz, A. K., Wender, N., Hegermann, J., Brunner, B., et al. (2010). Inhibition of Mitochondrial Fusion by α-synuclein Is Rescued by PINK1, Parkin and DJ-1. EMBO J. 29 (20), 3571–3589. doi:10.1038/emboj.2010.223
Kandel, E. R., Schwartz, J. H., and Jessel, T. M. (2000). Principles of Neural Science. New York, USA: McGraw-Hill.
Kato, S. (2008). Amyotrophic Lateral Sclerosis Models and Human Neuropathology: Similarities and Differences. Acta Neuropathol. 115 (1), 97–114. doi:10.1007/s00401-007-0308-4
Katsouri, L., Lim, Y. M., Blondrath, K., Eleftheriadou, I., Lombardero, L., Birch, A. M., et al. (2016). PPARγ-coactivator-1α Gene Transfer Reduces Neuronal Loss and Amyloid-β Generation by Reducing β-secretase in an Alzheimer's Disease Model. Proc. Natl. Acad. Sci. USA 113 (43), 12292–12297. doi:10.1073/pnas.1606171113
Kaufman, B. A., Durisic, N., Mativetsky, J. M., Costantino, S., Hancock, M. A., Grutter, P., et al. (2007). The Mitochondrial Transcription Factor TFAM Coordinates the Assembly of Multiple DNA Molecules into Nucleoid-like Structures. MBoC 18 (9), 3225–3236. doi:10.1091/mbc.e07-05-0404
Kaufman, S. K., Del Tredici, K., Thomas, T. L., Braak, H., and Diamond, M. I. (2018). Tau Seeding Activity Begins in the Transentorhinal/entorhinal Regions and Anticipates Phospho-Tau Pathology in Alzheimer's Disease and PART. Acta Neuropathol. 136 (1), 57–67. doi:10.1007/s00401-018-1855-6
Kawaguchi, Y., Okamoto, T., Taniwaki, M., Aizawa, M., Inoue, M., Katayama, S., et al. (1994). CAG Expansions in a Novel Gene for Machado-Joseph Disease at Chromosome 14q32.1. Nat. Genet. 8 (3), 221–228. doi:10.1038/ng1194-221
Keeney, P. M., and Bennett, J. P. (2010). ALS Spinal Neurons Show Varied and Reduced mtDNA Gene Copy Numbers and Increased mtDNA Gene Deletions. Mol. Neurodegener 5, 21. doi:10.1186/1750-1326-5-21
Khaliq, Z. M., and Bean, B. P. (2010). Pacemaking in Dopaminergic Ventral Tegmental Area Neurons: Depolarizing Drive from Background and Voltage-dependent Sodium Conductances. J. Neurosci. 30 (21), 7401–7413. doi:10.1523/jneurosci.0143-10.2010
Kiferle, L., Orsucci, D., Mancuso, M., Lo Gerfo, A., Petrozzi, L., Siciliano, G., et al. (2013). Twinkle Mutation in an Italian Family with External Progressive Ophthalmoplegia and Parkinsonism: a Case Report and an Update on the State of Art. Neurosci. Lett. 556, 1–4. doi:10.1016/j.neulet.2013.09.034
Kilarski, L. L., Pearson, J. P., Newsway, V., Majounie, E., Knipe, M. D. W., Misbahuddin, A., et al. (2012). Systematic Review and UK-based Study of PARK2 (Parkin), PINK1, PARK7 (DJ-1) and LRRK2 in Early-Onset Parkinson's Disease. Mov. Disord. 27 (12), 1522–1529. doi:10.1002/mds.25132
Kim, J., Moody, J. P., Edgerly, C. K., Bordiuk, O. L., Cormier, K., Smith, K., et al. (2010). Mitochondrial Loss, Dysfunction and Altered Dynamics in Huntington's Disease. Hum. Mol. Genet. 19 (20), 3919–3935. doi:10.1093/hmg/ddq306
Kondadi, A. K., Wang, S., Montagner, S., Kladt, N., Korwitz, A., Martinelli, P., et al. (2014). Loss of the M-AAA Protease Subunit AFG3L2 Causes Mitochondrial Transport Defects and Tau Hyperphosphorylation. EMBO J. 33 (9), 1011–1026. doi:10.1002/embj.201387009
Kondapalli, C., Kazlauskaite, A., Zhang, N., Woodroof, H. I., Campbell, D. G., Gourlay, R., et al. (2012). PINK1 Is Activated by Mitochondrial Membrane Potential Depolarization and Stimulates Parkin E3 Ligase Activity by Phosphorylating Serine 65. Open Biol. 2 (5), 120080. doi:10.1098/rsob.120080
König, T., Tröder, S. E., Bakka, K., Korwitz, A., Richter-Dennerlein, R., Lampe, P. A., et al. (2016). The M -AAA Protease Associated with Neurodegeneration Limits MCU Activity in Mitochondria. Mol. Cel 64 (1), 148–162. doi:10.1016/j.molcel.2016.08.020
Kornmann, B., Currie, E., Collins, S. R., Schuldiner, M., Nunnari, J., Weissman, J. S., et al. (2009). An ER-Mitochondria Tethering Complex Revealed by a Synthetic Biology Screen. Science 325 (5939), 477–481. doi:10.1126/science.1175088
Kostic, M., Ludtmann, M. H. R., Bading, H., Hershfinkel, M., Steer, E., Chu, C. T., et al. (2015). PKA Phosphorylation of NCLX Reverses Mitochondrial Calcium Overload and Depolarization, Promoting Survival of PINK1-Deficient Dopaminergic Neurons. Cel Rep. 13 (2), 376–386. doi:10.1016/j.celrep.2015.08.079
Kraytsberg, Y., Kudryavtseva, E., McKee, A. C., Geula, C., Kowall, N. W., and Khrapko, K. (2006). Mitochondrial DNA Deletions Are Abundant and Cause Functional Impairment in Aged Human Substantia Nigra Neurons. Nat. Genet. 38 (5), 518–520. doi:10.1038/ng1778
Krishnan, K. J., Reeve, A. K., Samuels, D. C., Chinnery, P. F., Blackwood, J. K., Taylor, R. W., et al. (2008). What Causes Mitochondrial DNA Deletions in Human Cells? Nat. Genet. 40 (3), 275–279. doi:10.1038/ng.f.94
Kubota, M., Sakakihara, Y., Uchiyama, Y., Nara, A., Nagata, T., Nitta, H., et al. (2000). New Ocular Movement Detector System as a Communication Tool in Ventilator-Assisted Werdnig-Hoffmann Disease. Dev. Med. Child. Neurol. 42 (1), 61–64. doi:10.1017/s0012162200000116
Kuchibhotla, K. V., Goldman, S. T., Lattarulo, C. R., Wu, H.-Y., Hyman, B. T., and Bacskai, B. J. (2008). Aβ Plaques Lead to Aberrant Regulation of Calcium Homeostasis In Vivo Resulting in Structural and Functional Disruption of Neuronal Networks. Neuron 59 (2), 214–225. doi:10.1016/j.neuron.2008.06.008
Kukat, C., Davies, K. M., Wurm, C. A., Spåhr, H., Bonekamp, N. A., Kühl, I., et al. (2015). Cross-strand Binding of TFAM to a Single mtDNA Molecule Forms the Mitochondrial Nucleoid. Proc. Natl. Acad. Sci. USA 112 (36), 11288–11293. doi:10.1073/pnas.1512131112
Kwon, S. K., Sando, R., Lewis, T. L., Hirabayashi, Y., Maximov, A., and Polleux, F. (2016). LKB1 Regulates Mitochondria-dependent Presynaptic Calcium Clearance and Neurotransmitter Release Properties at Excitatory Synapses along Cortical Axons. Plos Biol. 14 (7), e1002516. doi:10.1371/journal.pbio.1002516
Ladd, A. C., Brohawn, D. G., Thomas, R. R., Keeney, P. M., Berr, S. S., Khan, S. M., et al. (2017). RNA-seq Analyses Reveal that Cervical Spinal Cords and Anterior Motor Neurons from Amyotrophic Lateral Sclerosis Subjects Show Reduced Expression of Mitochondrial DNA-Encoded Respiratory Genes, and rhTFAM May Correct This Respiratory Deficiency. Brain Res. 1667, 74–83. doi:10.1016/j.brainres.2017.05.010
LaFerla, F. M. (2002). Calcium Dyshomeostasis and Intracellular Signalling in Alzheimer's Disease. Nat. Rev. Neurosci. 3 (11), 862–872. doi:10.1038/nrn960
Lang, Y., Gong, D., and Fan, Y. (2015). Calcium Channel Blocker Use and Risk of Parkinson's Disease: a Meta-Analysis. Pharmacoepidemiol. Drug Saf. 24 (6), 559–566. doi:10.1002/pds.3781
Langley, M., Ghosh, A., Charli, A., Sarkar, S., Ay, M., Luo, J., et al. (2017). Mito-Apocynin Prevents Mitochondrial Dysfunction, Microglial Activation, Oxidative Damage, and Progressive Neurodegeneration in MitoPark Transgenic Mice. Antioxid. Redox Signaling 27 (14), 1048–1066. doi:10.1089/ars.2016.6905
Langston, J., Ballard, P., Tetrud, J., and Irwin, I. (1983). Chronic Parkinsonism in Humans Due to a Product of Meperidine-Analog Synthesis. Science 219 (4587), 979–980. doi:10.1126/science.6823561
Larsson, N.-G., Wang, J., Wilhelmsson, H., Oldfors, A., Rustin, P., Lewandoski, M., et al. (1998). Mitochondrial Transcription Factor A Is Necessary for mtDNA Maintance and Embryogenesis in Mice. Nat. Genet. 18 (3), 231–236. doi:10.1038/ng0398-231
Lavie, J., Serrat, R., Bellance, N., Courtand, G., Dupuy, J. W., Tesson, C., et al. (2017). Mitochondrial Morphology and Cellular Distribution Are Altered in SPG31 Patients and Are Linked to DRP1 Hyperphosphorylation. Hum. Mol. Genet. 26 (4), 674–685. doi:10.1093/hmg/ddw425
Lawless, C., Greaves, L., Reeve, A. K., Turnbull, D. M., and Vincent, A. E. (2020). The Rise and Rise of Mitochondrial DNA Mutations. Open Biol. 10 (5), 200061. doi:10.1098/rsob.200061
Lax, N. Z., Hepplewhite, P. D., Reeve, A. K., Nesbitt, V., McFarland, R., Jaros, E., et al. (2012). Cerebellar Ataxia in Patients with Mitochondrial DNA Disease. J. Neuropathol. Exp. Neurol. 71 (2), 148–161. doi:10.1097/nen.0b013e318244477d
Lazarou, M., Jin, S. M., Kane, L. A., and Youle, R. J. (2012). Role of PINK1 Binding to the TOM Complex and Alternate Intracellular Membranes in Recruitment and Activation of the E3 Ligase Parkin. Dev. Cel 22 (2), 320–333. doi:10.1016/j.devcel.2011.12.014
Lazarou, M., Sliter, D. A., Kane, L. A., Sarraf, S. A., Wang, C., Burman, J. L., et al. (2015). The Ubiquitin Kinase PINK1 Recruits Autophagy Receptors to Induce Mitophagy. Nature 524 (7565), 309–314. doi:10.1038/nature14893
Le Ber, I., Camuzat, A., Guerreiro, R., Bouya-Ahmed, K., Bras, J., Nicolas, G., et al. (2013). SQSTM1 Mutations in French Patients with Frontotemporal Dementia or Frontotemporal Dementia with Amyotrophic Lateral Sclerosis. JAMA Neurol. 70 (11), 1403–1410. doi:10.1001/jamaneurol.2013.3849
LeBoeuf, A. C., Levy, S. F., Gaylord, M., Bhattacharya, A., Singh, A. K., Jordan, M. A., et al. (2008). FTDP-17 Mutations in Tau Alter the Regulation of Microtubule Dynamics. J. Biol. Chem. 283 (52), 36406–36415. doi:10.1074/jbc.m803519200
Lee, S., Sterky, F. H., Mourier, A., Terzioglu, M., Cullheim, S., Olson, L., et al. (2012). Mitofusin 2 Is Necessary for Striatal Axonal Projections of Midbrain Dopamine Neurons. Hum. Mol. Genet. 21 (22), 4827–4835. doi:10.1093/hmg/dds352
Lemprière, S. (2019). Antisense Oligonucleotide Reduces Mutant Protein in Patients with HD. Nat. Rev. Neurol. 15 (8), 435. doi:10.1038/s41582-019-0211-3
Leuner, K., Schulz, K., Schütt, T., Pantel, J., Prvulovic, D., Rhein, V., et al. (2012). Peripheral Mitochondrial Dysfunction in Alzheimer's Disease: Focus on Lymphocytes. Mol. Neurobiol. 46 (1), 194–204. doi:10.1007/s12035-012-8300-y
Lieberman, O. J., Choi, S. J., Kanter, E., Saverchenko, A., Frier, M. D., Fiore, G. M., et al. (2017). Alpha-Synuclein-Dependent Calcium Entry Underlies Differential Sensitivity of Cultured SN and VTA Dopaminergic Neurons to a Parkinsonian Neurotoxin. eNeuro 4 (6). doi:10.1523/eneuro.0167-17.2017
Lim, D., Fedrizzi, L., Tartari, M., Zuccato, C., Cattaneo, E., Brini, M., et al. (2008). Calcium Homeostasis and Mitochondrial Dysfunction in Striatal Neurons of Huntington Disease. J. Biol. Chem. 283 (9), 5780–5789. doi:10.1074/jbc.m704704200
Lin, M.-Y., Cheng, X.-T., Tammineni, P., Xie, Y., Zhou, B., Cai, Q., et al. (2017). Releasing Syntaphilin Removes Stressed Mitochondria from Axons Independent of Mitophagy under Pathophysiological Conditions. Neuron 94 (3), 595–610. doi:10.1016/j.neuron.2017.04.004
Lin, W., and Kang, U. J. (2008). Characterization of PINK1 Processing, Stability, and Subcellular Localization. J. Neurochem. 106 (1), 464–474. doi:10.1111/j.1471-4159.2008.05398.x
Liss, B., and Roeper, J. (2001). Molecular Physiology of Neuronal K-ATP Channels (Review). Mol. Membr. Biol. 18 (2), 117–127. doi:10.1080/09687680110047373
Liss, B., Haeckel, O., Wildmann, J., Miki, T., Seino, S., and Roeper, J. (2005). K-ATP Channels Promote the Differential Degeneration of Dopaminergic Midbrain Neurons. Nat. Neurosci. 8 (12), 1742–1751. doi:10.1038/nn1570
Liu, J. C., Liu, J., Holmström, K. M., Menazza, S., Parks, R. J., Fergusson, M. M., et al. (2016a). MICU1 Serves as a Molecular Gatekeeper to Prevent In Vivo Mitochondrial Calcium Overload. Cel Rep. 16 (6), 1561–1573. doi:10.1016/j.celrep.2016.07.011
Liu, J., Xin, L., Benson, V. L., Allen, D. G., and Ju, Y. K. (2015). Store-operated Calcium Entry and the Localization of STIM1 and Orai1 Proteins in Isolated Mouse Sinoatrial Node Cells. Front. Physiol. 6, 69. doi:10.3389/fphys.2015.00069
Liu, W. J., Ye, L., Huang, W. F., Guo, L. J., Xu, Z. G., Wu, H. L., et al. (2016b). p62 Links the Autophagy Pathway and the Ubiqutin-Proteasome System upon Ubiquitinated Protein Degradation. Cell Mol Biol Lett 21, 29. doi:10.1186/s11658-016-0031-z
Liu, W., Yamashita, T., Tian, F., Morimoto, N., Ikeda, Y., Deguchi, K., et al. (2013). Mitochondrial Fusion and Fission Proteins Expression Dynamically Change in a Murine Model of Amyotrophic Lateral Sclerosis. Cnr 10 (3), 222–230. doi:10.2174/15672026113109990060
Lonnqvist, T., Paetau, A., Valanne, L., and Pihko, H. (2009). Recessive Twinkle Mutations Cause Severe Epileptic Encephalopathy. Brain 132 (Pt 6), 1553–1562. doi:10.1093/brain/awp045
Luoma, P., Melberg, A., Rinne, J. O., Kaukonen, J. A., Nupponen, N. N., Chalmers, R. M., et al. (2004). Parkinsonism, Premature Menopause, and Mitochondrial DNA Polymerase γ Mutations: Clinical and Molecular Genetic Study. The Lancet 364 (9437), 875–882. doi:10.1016/s0140-6736(04)16983-3
MacDonald, M. E., Ambrose, C. A., Duyao, M. P., Myers, R. H., Lin, C., Srinidhi, L., et al. (1993). A Novel Gene Containing a Trinucleotide Repeat that Is Expanded and Unstable on Huntington's Disease Chromosomes. The Huntington's Disease Collaborative Research Group. Cell 72 (6), 971–983. doi:10.1016/0092-8674(93)90585-e
Madruga, E., Maestro, I., and Martinez, A. (2021). Mitophagy Modulation, a New Player in the Race against ALS. Int. J. Mol. Sci. 22 (2). doi:10.3390/ijms22020740
Magrané, J., Cortez, C., Gan, W.-B., and Manfredi, G. (2014). Abnormal Mitochondrial Transport and Morphology Are Common Pathological Denominators in SOD1 and TDP43 ALS Mouse Models. Hum. Mol. Genet. 23 (6), 1413–1424. doi:10.1093/hmg/ddt528
Maltecca, F., Baseggio, E., Consolato, F., Mazza, D., Podini, P., Young, S. M., et al. (2015). Purkinje Neuron Ca2+ Influx Reduction Rescues Ataxia in SCA28 Model. J. Clin. Invest. 125 (1), 263–274. doi:10.1172/jci74770
Manczak, M., Calkins, M. J., and Reddy, P. H. (2011). Impaired Mitochondrial Dynamics and Abnormal Interaction of Amyloid Beta with Mitochondrial Protein Drp1 in Neurons from Patients with Alzheimer's Disease: Implications for Neuronal Damage. Hum. Mol. Genet. 20 (13), 2495–2509. doi:10.1093/hmg/ddr139
Mao, P., Manczak, M., Calkins, M. J., Truong, Q., Reddy, T. P., Reddy, A. P., et al. (2012). Mitochondria-targeted Catalase Reduces Abnormal APP Processing, Amyloid Production and BACE1 in a Mouse Model of Alzheimer's Disease: Implications for Neuroprotection and Lifespan Extension. Hum. Mol. Genet. 21 (13), 2973–2990. doi:10.1093/hmg/dds128
Marchi, S., Patergnani, S., Missiroli, S., Morciano, G., Rimessi, A., Wieckowski, M. R., et al. (2018). Mitochondrial and Endoplasmic Reticulum Calcium Homeostasis and Cell Death. Cell Calcium 69, 62–72. doi:10.1016/j.ceca.2017.05.003
Marland, J. R. K., Hasel, P., Bonnycastle, K., and Cousin, M. A. (2016). Mitochondrial Calcium Uptake Modulates Synaptic Vesicle Endocytosis in Central Nerve Terminals. J. Biol. Chem. 291 (5), 2080–2086. doi:10.1074/jbc.m115.686956
Matsuda, N., Sato, S., Shiba, K., Okatsu, K., Saisho, K., Gautier, C. A., et al. (2010). PINK1 Stabilized by Mitochondrial Depolarization Recruits Parkin to Damaged Mitochondria and Activates Latent Parkin for Mitophagy. J. Cel Biol 189 (2), 211–221. doi:10.1083/jcb.200910140
Matsuda, W., Furuta, T., Nakamura, K. C., Hioki, H., Fujiyama, F., Arai, R., et al. (2009). Single Nigrostriatal Dopaminergic Neurons Form Widely Spread and Highly Dense Axonal Arborizations in the Neostriatum. J. Neurosci. 29 (2), 444–453. doi:10.1523/jneurosci.4029-08.2009
McCormack, J. G., and Denton, R. M. (1979). The Effects of Calcium Ions and Adenine Nucleotides on the Activity of Pig Heart 2-oxoglutarate Dehydrogenase Complex. Biochem. J. 180 (3), 533–544. doi:10.1042/bj1800533
McGregor, M. M., and Nelson, A. B. (2019). Circuit Mechanisms of Parkinson's Disease. Neuron 101 (6), 1042–1056. doi:10.1016/j.neuron.2019.03.004
McLelland, G.-L., and Fon, E. A. (2018). MFN2 Retrotranslocation Boosts Mitophagy by Uncoupling Mitochondria from the ER. Autophagy 14 (9), 1658–1660. doi:10.1080/15548627.2018.1505154
McLelland, G. L., Soubannier, V., Chen, C. X., McBride, H. M., and Fon, E. A. (2014). Parkin and PINK1 Function in a Vesicular Trafficking Pathway Regulating Mitochondrial Quality Control. EMBO J. 33 (4), 282–295. doi:10.1002/embj.201385902
McLelland, G. L., Goiran, T., Yi, W., Dorval, G., Chen, C. X., Lauinger, N. D., et al. (2018). Mfn2 Ubiquitination by PINK1/parkin gates the P97-dependent Release of ER from Mitochondria to Drive Mitophagy. Elife 7. doi:10.7554/elife.32866
McManus, M. J., Murphy, M. P., and Franklin, J. L. (2011). The Mitochondria-Targeted Antioxidant MitoQ Prevents Loss of Spatial Memory Retention and Early Neuropathology in a Transgenic Mouse Model of Alzheimer's Disease. J. Neurosci. 31 (44), 15703–15715. doi:10.1523/jneurosci.0552-11.2011
Melkov, A., and Abdu, U. (2018). Regulation of Long-Distance Transport of Mitochondria along Microtubules. Cell. Mol. Life Sci. 75 (2), 163–176. doi:10.1007/s00018-017-2590-1
Melov, S., Adlard, P. A., Morten, K., Johnson, F., Golden, T. R., Hinerfeld, D., et al. (2007). Mitochondrial Oxidative Stress Causes Hyperphosphorylation of Tau. PLoS One 2 (6), e536. doi:10.1371/journal.pone.0000536
Michel, P. P., Hirsch, E. C., and Hunot, S. (2016). Understanding Dopaminergic Cell Death Pathways in Parkinson Disease. Neuron 90 (4), 675–691. doi:10.1016/j.neuron.2016.03.038
Misgeld, T., and Schwarz, T. L. (2017). Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron 96 (3), 651–666. doi:10.1016/j.neuron.2017.09.055
Misko, A., Jiang, S., Wegorzewska, I., Milbrandt, J., and Baloh, R. H. (2010). Mitofusin 2 Is Necessary for Transport of Axonal Mitochondria and Interacts with the Miro/Milton Complex. J. Neurosci. 30 (12), 4232–4240. doi:10.1523/jneurosci.6248-09.2010
Mizuno, Y., Amari, M., Takatama, M., Aizawa, H., Mihara, B., and Okamoto, K. (2006). Immunoreactivities of P62, an Ubiqutin-Binding Protein, in the Spinal Anterior Horn Cells of Patients with Amyotrophic Lateral Sclerosis. J. Neurol. Sci. 249 (1), 13–18. doi:10.1016/j.jns.2006.05.060
Molla, B., Munoz-Lasso, D. C., Riveiro, F., Bolinches-Amoros, A., Pallardo, F. V., Fernandez-Vilata, A., et al. (2017). Reversible Axonal Dystrophy by Calcium Modulation in Frataxin-Deficient Sensory Neurons of YG8R Mice. Front. Mol. Neurosci. 10, 264. doi:10.3389/fnmol.2017.00264
Monaco, A., and Fraldi, A. (2020). Protein Aggregation and Dysfunction of Autophagy-Lysosomal Pathway: A Vicious Cycle in Lysosomal Storage Diseases. Front. Mol. Neurosci. 13, 37. doi:10.3389/fnmol.2020.00037
Montagne, A., Nation, D. A., Pa, J., Sweeney, M. D., Toga, A. W., and Zlokovic, B. V. (2016). Brain Imaging of Neurovascular Dysfunction in Alzheimer's Disease. Acta Neuropathol. 131 (5), 687–707. doi:10.1007/s00401-016-1570-0
Moore, A. S., and Holzbaur, E. L. F. (2016). Dynamic Recruitment and Activation of ALS-Associated TBK1 with its Target Optineurin Are Required for Efficient Mitophagy. Proc. Natl. Acad. Sci. USA 113 (24), E3349–E3358. doi:10.1073/pnas.1523810113
Morotz, G. M., De Vos, K. J., Vagnoni, A., Ackerley, S., Shaw, C. E., and Miller, C. C. J. (2012). Amyotrophic Lateral Sclerosis-Associated Mutant VAPBP56S Perturbs Calcium Homeostasis to Disrupt Axonal Transport of Mitochondria. Hum. Mol. Genet. 21 (9), 1979–1988. doi:10.1093/hmg/dds011
Mortiboys, H., Johansen, K. K., Aasly, J. O., and Bandmann, O. (2010). Mitochondrial Impairment in Patients with Parkinson Disease with the G2019S Mutation in LRRK2. Neurology 75 (22), 2017–2020. doi:10.1212/wnl.0b013e3181ff9685
Mossmann, D., Vögtle, F.-N., Taskin, A. A., Teixeira, P. F., Ring, J., Burkhart, J. M., et al. (2014). Amyloid-β Peptide Induces Mitochondrial Dysfunction by Inhibition of Preprotein Maturation. Cel Metab. 20 (4), 662–669. doi:10.1016/j.cmet.2014.07.024
Muto, V., Flex, E., Kupchinsky, Z., Primiano, G., Galehdari, H., Dehghani, M., et al. (2018). Biallelic SQSTM1 Mutations in Early-Onset, Variably Progressive Neurodegeneration. Neurology 91 (4), e319–e330. doi:10.1212/wnl.0000000000005869
Nam, T., Han, J. H., Devkota, S., and Lee, H. W. (2017). Emerging Paradigm of Crosstalk between Autophagy and the Ubiquitin-Proteasome System. Mol. Cell 40 (12), 897–905. doi:10.14348/molcells.2017.0226
Napoli, E., Wong, S., Hung, C., Ross-Inta, C., Bomdica, P., and Giulivi, C. (2013). Defective Mitochondrial Disulfide Relay System, Altered Mitochondrial Morphology and Function in Huntington's Disease. Hum. Mol. Genet. 22 (5), 989–1004. doi:10.1093/hmg/dds503
Neuhaus, J. F., Baris, O. R., Hess, S., Moser, N., Schroder, H., Chinta, S. J., et al. (2014). Catecholamine Metabolism Drives Generation of Mitochondrial DNA Deletions in Dopaminergic Neurons. Brain 137 (Pt 2), 354–365. doi:10.1093/brain/awt291
Neuhaus, J. F. G., Baris, O. R., Kittelmann, A., Becker, K., Rothschild, M. A., and Wiesner, R. J. (2017). Catecholamine Metabolism Induces Mitochondrial DNA Deletions and Leads to Severe Adrenal Degeneration during Aging. Neuroendocrinology 104 (1), 72–84. doi:10.1159/000444680
Ngo, H. B., Kaiser, J. T., and Chan, D. C. (2011). The Mitochondrial Transcription and Packaging Factor Tfam Imposes a U-Turn on Mitochondrial DNA. Nat. Struct. Mol. Biol. 18 (11), 1290–1296. doi:10.1038/nsmb.2159
Nijssen, J., Comley, L. H., and Hedlund, E. (2017). Motor Neuron Vulnerability and Resistance in Amyotrophic Lateral Sclerosis. Acta Neuropathol. 133 (6), 863–885. doi:10.1007/s00401-017-1708-8
Nishimura, A. L., Mitne-Neto, M., Silva, H. C. A., Richieri-Costa, A., Middleton, S., Cascio, D., et al. (2004). A Mutation in the Vesicle-Trafficking Protein VAPB Causes Late-Onset Spinal Muscular Atrophy and Amyotrophic Lateral Sclerosis. Am. J. Hum. Genet. 75 (5), 822–831. doi:10.1086/425287
Nixon, R. A. (2013). The Role of Autophagy in Neurodegenerative Disease. Nat. Med. 19 (8), 983–997. doi:10.1038/nm.3232
Novak, M. J. U., Seunarine, K. K., Gibbard, C. R., McColgan, P., Draganski, B., Friston, K., et al. (2015). Basal Ganglia-Cortical Structural Connectivity in Huntington's Disease. Hum. Brain Mapp. 36 (5), 1728–1740. doi:10.1002/hbm.22733
Nozaki, M., Otomo, A., Mitsui, S., Ono, S., Shirakawa, R., Chen, Y., et al. (2021). SQSTM1L341V Variant that Is Linked to Sporadic ALS Exhibits Impaired Association with MAP1LC3 in Cultured Cells. eNeurologicalSci 22, 100301. doi:10.1016/j.ensci.2020.100301
Obeso, J. A., Stamelou, M., Goetz, C. G., Poewe, W., Lang, A. E., Weintraub, D., et al. (2017). Past, Present, and Future of Parkinson's Disease: A Special Essay on the 200th Anniversary of the Shaking Palsy. Mov Disord. 32 (9), 1264–1310. doi:10.1002/mds.27115
Ochaba, J., Lukacsovich, T., Csikos, G., Zheng, S., Margulis, J., Salazar, L., et al. (2014). Potential Function for the Huntingtin Protein as a Scaffold for Selective Autophagy. Proc. Natl. Acad. Sci. USA 111 (47), 16889–16894. doi:10.1073/pnas.1420103111
Oliveira, J. M. A., Jekabsons, M. B., Chen, S., Lin, A., Rego, A. C., Gonçalves, J., et al. (2007). Mitochondrial Dysfunction in Huntington's Disease: the Bioenergetics of Isolated and In Situ Mitochondria from Transgenic Mice. J. Neurochem. 101 (1), 241–249. doi:10.1111/j.1471-4159.2006.04361.x
Onesto, E., Colombrita, C., Gumina, V., Borghi, M. O., Dusi, S., Doretti, A., et al. (2016). Gene-specific Mitochondria Dysfunctions in Human TARDBP and C9ORF72 Fibroblasts. Acta Neuropathol. Commun. 4 (1), 47. doi:10.1186/s40478-016-0316-5
Ordureau, A., Sarraf, S. A., Duda, D. M., Heo, J.-M., Jedrychowski, M. P., Sviderskiy, V. O., et al. (2014). Quantitative Proteomics Reveal a Feedforward Mechanism for Mitochondrial PARKIN Translocation and Ubiquitin Chain Synthesis. Mol. Cel 56 (3), 360–375. doi:10.1016/j.molcel.2014.09.007
Orr, A. L., Li, S., Wang, C.-E., Li, H., Wang, J., Rong, J., et al. (2008). N-terminal Mutant Huntingtin Associates with Mitochondria and Impairs Mitochondrial Trafficking. J. Neurosci. 28 (11), 2783–2792. doi:10.1523/jneurosci.0106-08.2008
Ortner, N. J., Bock, G., Dougalis, A., Kharitonova, M., Duda, J., Hess, S., et al. (2017). Lower Affinity of Isradipine for L-type Ca2+ Channels during Substantia Nigra Dopamine Neuron-like Activity: Implications for Neuroprotection in Parkinson's Disease. J. Neurosci. 37 (28), 6761–6777. doi:10.1523/jneurosci.2946-16.2017
Pacelli, C., Giguère, N., Bourque, M.-J., Lévesque, M., Slack, R. S., and Trudeau, L.-É. (2015). Elevated Mitochondrial Bioenergetics and Axonal Arborization Size Are Key Contributors to the Vulnerability of Dopamine Neurons. Curr. Biol. 25 (18), 2349–2360. doi:10.1016/j.cub.2015.07.050
Pack-Chung, E., Meyers, M. B., Pettingell, W. P., Moir, R. D., Brownawell, A. M., Cheng, I., et al. (2000). Presenilin 2 Interacts with Sorcin, a Modulator of the Ryanodine Receptor. J. Biol. Chem. 275 (19), 14440–14445. doi:10.1074/jbc.m909882199
Padman, B. S., Nguyen, T. N., Uoselis, L., Skulsuppaisarn, M., Nguyen, L. K., and Lazarou, M. (2019). LC3/GABARAPs Drive Ubiquitin-independent Recruitment of Optineurin and NDP52 to Amplify Mitophagy. Nat. Commun. 10 (1), 408. doi:10.1038/s41467-019-08335-6
Paillusson, S., Gomez-Suaga, P., Stoica, R., Little, D., Gissen, P., Devine, M. J., et al. (2017). α-Synuclein Binds to the ER-Mitochondria Tethering Protein VAPB to Disrupt Ca2+ Homeostasis and Mitochondrial ATP Production. Acta Neuropathol. 134 (1), 129–149. doi:10.1007/s00401-017-1704-z
Palin, E. J., Paetau, A., and Suomalainen, A. (2013). Mesencephalic Complex I Deficiency Does Not Correlate with Parkinsonism in Mitochondrial DNA Maintenance Disorders. Brain 136 (Pt 8), 2379–2392. doi:10.1093/brain/awt160
Panov, A. V., Gutekunst, C.-A., Leavitt, B. R., Hayden, M. R., Burke, J. R., Strittmatter, W. J., et al. (2002). Early Mitochondrial Calcium Defects in Huntington's Disease Are a Direct Effect of Polyglutamines. Nat. Neurosci. 5 (8), 731–736. doi:10.1038/nn884
Parkinson Study Group, S.-P. D. I. I. I. I. (2020). Isradipine versus Placebo in Early Parkinson Disease: A Randomized Trial. Ann. Intern. Med. 172 (9), 591–598. doi:10.7326/M19-2534
Pass, T., Assfalg, M., Tolve, M., Blaess, S., Rothermel, M., Wiesner, R. J., et al. (2020). The Impact of Mitochondrial Dysfunction on Dopaminergic Neurons in the Olfactory Bulb and Odor Detection. Mol. Neurobiol. 57 (9), 3646–3657. doi:10.1007/s12035-020-01947-w
Patron, M., Sprenger, H.-G., and Langer, T. (2018). m-AAA Proteases, Mitochondrial Calcium Homeostasis and Neurodegeneration. Cell Res 28 (3), 296–306. doi:10.1038/cr.2018.17
Paulson, H. L., Shakkottai, V. G., Clark, H. B., and Orr, H. T. (2017). Polyglutamine Spinocerebellar Ataxias - from Genes to Potential Treatments. Nat. Rev. Neurosci. 18 (10), 613–626. doi:10.1038/nrn.2017.92
Pellman, J. J., Hamilton, J., Brustovetsky, T., and Brustovetsky, N. (2015). Ca2+ Handling in Isolated Brain Mitochondria and Cultured Neurons Derived from the YAC128 Mouse Model of Huntington's Disease. J. Neurochem. 134 (4), 652–667. doi:10.1111/jnc.13165
Perez Carrion, M., Pischedda, F., Biosa, A., Russo, I., Straniero, L., Civiero, L., et al. (2018). The LRRK2 Variant E193K Prevents Mitochondrial Fission upon MPP+ Treatment by Altering LRRK2 Binding to DRP1. Front. Mol. Neurosci. 11, 64. doi:10.3389/fnmol.2018.00064
Perez, F. A., and Palmiter, R. D. (2005). Parkin-deficient Mice Are Not a Robust Model of Parkinsonism. Proc. Natl. Acad. Sci. 102 (6), 2174–2179. doi:10.1073/pnas.0409598102
Perreault, S., Bousquet, O., Lauzon, M., Paiement, J., and Leclerc, N. (2009). Increased Association between Rough Endoplasmic Reticulum Membranes and Mitochondria in Transgenic Mice that Express P301L Tau. J. Neuropathol. Exp. Neurol. 68 (5), 503–514. doi:10.1097/nen.0b013e3181a1fc49
Pham, A. H., Meng, S., Chu, Q. N., and Chan, D. C. (2012). Loss of Mfn2 Results in Progressive, Retrograde Degeneration of Dopaminergic Neurons in the Nigrostriatal Circuit. Hum. Mol. Genet. 21 (22), 4817–4826. doi:10.1093/hmg/dds311
Philippart, F., Destreel, G., Merino-Sepulveda, P., Henny, P., Engel, D., and Seutin, V. (2016). Differential Somatic Ca2+ Channel Profile in Midbrain Dopaminergic Neurons. J. Neurosci. 36 (27), 7234–7245. doi:10.1523/jneurosci.0459-16.2016
Picard, M., Vincent, A. E., and Turnbull, D. M. (2016). Expanding Our Understanding of mtDNA Deletions. Cel Metab. 24 (1), 3–4. doi:10.1016/j.cmet.2016.06.024
Pickford, F., Masliah, E., Britschgi, M., Lucin, K., Narasimhan, R., Jaeger, P. A., et al. (2008). The Autophagy-Related Protein Beclin 1 Shows Reduced Expression in Early Alzheimer Disease and Regulates Amyloid Beta Accumulation in Mice. J. Clin. Invest. 118 (6), 2190–2199. doi:10.1172/JCI33585
Pickrell, A. M., Huang, C.-H., Kennedy, S. R., Ordureau, A., Sideris, D. P., Hoekstra, J. G., et al. (2015). Endogenous Parkin Preserves Dopaminergic Substantia Nigral Neurons Following Mitochondrial DNA Mutagenic Stress. Neuron 87 (2), 371–381. doi:10.1016/j.neuron.2015.06.034
Pla-Martín, D., Calpena, E., Lupo, V., Márquez, C., Rivas, E., Sivera, R., et al. (2015). Junctophilin-1 Is a Modifier Gene of GDAP1-Related Charcot-Marie-Tooth Disease. Hum. Mol. Genet. 24 (1), 213–229. doi:10.1093/hmg/ddu440
Pla-Martín, D., Rueda, C. B., Estela, A., Sánchez-Piris, M., González-Sánchez, P., Traba, J., et al. (2013). Silencing of the Charcot-Marie-Tooth Disease-Associated Gene GDAP1 Induces Abnormal Mitochondrial Distribution and Affects Ca2+ Homeostasis by Reducing Store-Operated Ca2+ Entry. Neurobiol. Dis. 55, 140–151. doi:10.1016/j.nbd.2013.03.010
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the -Synuclein Gene Identified in Families with Parkinson's Disease. Science 276 (5321), 2045–2047. doi:10.1126/science.276.5321.2045
Poston, C. N., Krishnan, S. C., and Bazemore-Walker, C. R. (2013). In-depth Proteomic Analysis of Mammalian Mitochondria-Associated Membranes (MAM). J. Proteomics 79, 219–230. doi:10.1016/j.jprot.2012.12.018
Postuma, R. B., Aarsland, D., Barone, P., Burn, D. J., Hawkes, C. H., Oertel, W., et al. (2012). Identifying Prodromal Parkinson's Disease: Pre-motor Disorders in Parkinson's Disease. Mov. Disord. 27 (5), 617–626. doi:10.1002/mds.24996
Quintanilla, R. A., Jin, Y. N., von Bernhardi, R., and Johnson, G. V. (2013). Mitochondrial Permeability Transition Pore Induces Mitochondria Injury in Huntington Disease. Mol. Neurodegeneration 8, 45. doi:10.1186/1750-1326-8-45
Quintanilla, R. A., Matthews-Roberson, T. A., Dolan, P. J., and Johnson, G. V. W. (2009). Caspase-cleaved Tau Expression Induces Mitochondrial Dysfunction in Immortalized Cortical Neurons. J. Biol. Chem. 284 (28), 18754–18766. doi:10.1074/jbc.m808908200
Ramachandran, P. S., Boudreau, R. L., Schaefer, K. A., La Spada, A. R., and Davidson, B. L. (2014). Nonallele Specific Silencing of Ataxin-7 Improves Disease Phenotypes in a Mouse Model of SCA7. Mol. Ther. 22 (9), 1635–1642. doi:10.1038/mt.2014.108
Rangaraju, V., Calloway, N., and Ryan, T. A. (2014). Activity-driven Local ATP Synthesis Is Required for Synaptic Function. Cell 156 (4), 825–835. doi:10.1016/j.cell.2013.12.042
Raymond, L. A., André, V. M., Cepeda, C., Gladding, C. M., Milnerwood, A. J., and Levine, M. S. (2011). Pathophysiology of Huntington's Disease: Time-dependent Alterations in Synaptic and Receptor Function. Neuroscience 198, 252–273. doi:10.1016/j.neuroscience.2011.08.052
Rcom-H'cheo-Gauthier, A., Goodwin, J., and Pountney, D. (2014). Interactions between Calcium and Alpha-Synuclein in Neurodegeneration. Biomolecules 4 (3), 795–811. doi:10.3390/biom4030795
Reddy, P. H., Manczak, M., and Kandimalla, R. (2017). Mitochondria-targeted Small Molecule SS31: a Potential Candidate for the Treatment of Alzheimer's Disease. Hum. Mol. Genet. 26 (8), 1597. doi:10.1093/hmg/ddx129
Reeve, A. K., Krishnan, K. J., Elson, J. L., Morris, C. M., Bender, A., Lightowlers, R. N., et al. (2008). Nature of Mitochondrial DNA Deletions in Substantia Nigra Neurons. Am. J. Hum. Genet. 82 (1), 228–235. doi:10.1016/j.ajhg.2007.09.018
Reilly, M. M., Shy, M. E., Muntoni, F., and Pareyson, D. (2010). 168th ENMC International Workshop: Outcome Measures and Clinical Trials in Charcot-Marie-Tooth Disease (CMT). Neuromuscul. Disord. 20 (12), 839–846. doi:10.1016/j.nmd.2010.08.001
Rice, A. C., Keeney, P. M., Algarzae, N. K., Ladd, A. C., Thomas, R. R., and Bennett Jr., J. P. (2014). Mitochondrial DNA Copy Numbers in Pyramidal Neurons Are Decreased and Mitochondrial Biogenesis Transcriptome Signaling Is Disrupted in Alzheimer's Disease Hippocampi. Jad 40 (2), 319–330. doi:10.3233/jad-131715
Richter, B., Sliter, D. A., Herhaus, L., Stolz, A., Wang, C., Beli, P., et al. (2016). Phosphorylation of OPTN by TBK1 Enhances its Binding to Ub Chains and Promotes Selective Autophagy of Damaged Mitochondria. Proc. Natl. Acad. Sci. USA 113 (15), 4039–4044. doi:10.1073/pnas.1523926113
Ricke, K. M., Pass, T., Kimoloi, S., Fährmann, K., Jüngst, C., Schauss, A., et al. (2020). Mitochondrial Dysfunction Combined with High Calcium Load Leads to Impaired Antioxidant Defense Underlying the Selective Loss of Nigral Dopaminergic Neurons. J. Neurosci. 40 (9), 1975–1986. doi:10.1523/jneurosci.1345-19.2019
Rizzo, F., Ronchi, D., Salani, S., Nizzardo, M., Fortunato, F., Bordoni, A., et al. (2016). Selective Mitochondrial Depletion, Apoptosis Resistance, and Increased Mitophagy in Human Charcot-Marie-Tooth 2A Motor Neurons. Hum. Mol. Genet. 25 (19), 4266–4281. doi:10.1093/hmg/ddw258
Rizzuto, R., De Stefani, D., Raffaello, A., and Mammucari, C. (2012). Mitochondria as Sensors and Regulators of Calcium Signalling. Nat. Rev. Mol. Cel Biol 13 (9), 566–578. doi:10.1038/nrm3412
Rizzuto, R., Pinton, P., Carrington, W., Fay, F. S., Fogarty, K. E., Lifshitz, L. M., et al. (1998). Close Contacts with the Endoplasmic Reticulum as Determinants of Mitochondrial Ca2+ Responses. Science 280 (5370), 1763–1766. doi:10.1126/science.280.5370.1763
Rocha, A. G., Franco, A., Krezel, A. M., Rumsey, J. M., Alberti, J. M., Knight, W. C., et al. (2018). MFN2 Agonists Reverse Mitochondrial Defects in Preclinical Models of Charcot-Marie-Tooth Disease Type 2A. Science 360 (6386), 336–341. doi:10.1126/science.aao1785
Rodríguez, L. R., Calap-Quintana, P., Lapeña-Luzón, T., Pallardó, F. V., Schneuwly, S., Navarro, J. A., et al. (2020). Oxidative Stress Modulates Rearrangement of Endoplasmic Reticulum-Mitochondria Contacts and Calcium Dysregulation in a Friedreich's Ataxia Model. Redox Biol. 37, 101762. doi:10.1016/j.redox.2020.101762
Rosencrans, W. M., Aguilella, V. M., Rostovtseva, T. K., and Bezrukov, S. M. (2021). α-Synuclein Emerges as a Potent Regulator of VDAC-Facilitated Calcium Transport. Cell Calcium 95, 102355. doi:10.1016/j.ceca.2021.102355
Roussarie, J.-P., Yao, V., Rodriguez-Rodriguez, P., Oughtred, R., Rust, J., Plautz, Z., et al. (2020). Selective Neuronal Vulnerability in Alzheimer's Disease: A Network-Based Analysis. Neuron 107 (5), 821–835. doi:10.1016/j.neuron.2020.06.010
Rubino, E., Rainero, I., Chio, A., Rogaeva, E., Galimberti, D., Fenoglio, P., et al. (2012). SQSTM1 Mutations in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Neurology 79 (15), 1556–1562. doi:10.1212/wnl.0b013e31826e25df
Rubinsztein, D. C., Mariño, G., and Kroemer, G. (2011). Autophagy and Aging. Cell 146 (5), 682–695. doi:10.1016/j.cell.2011.07.030
Rugarli, E. I., and Langer, T. (2012). Mitochondrial Quality Control: a Matter of Life and Death for Neurons. EMBO J. 31 (6), 1336–1349. doi:10.1038/emboj.2012.38
Rui, Y.-N., Xu, Z., Patel, B., Chen, Z., Chen, D., Tito, A., et al. (2015). Huntingtin Functions as a Scaffold for Selective Macroautophagy. Nat. Cel Biol 17 (3), 262–275. doi:10.1038/ncb3101
Sahu, G., Wazen, R.-M., Colarusso, P., Chen, S. R. W., Zamponi, G. W., and Turner, R. W. (2019). Junctophilin Proteins Tether a Cav1-RyR2-KCa3.1 Tripartite Complex to Regulate Neuronal Excitability. Cel Rep. 28 (9), 2427–2442. doi:10.1016/j.celrep.2019.07.075
Sanz-Blasco, S., Valero, R. A., Rodriguez-Crespo, I., Villalobos, C., and Nunez, L. (2008). Mitochondrial Ca2+ Overload Underlies Abeta Oligomers Neurotoxicity Providing an Unexpected Mechanism of Neuroprotection by NSAIDs. PLoS One 3 (7), e2718. doi:10.1371/journal.pone.0002718
Saporta, A. S. D., Sottile, S. L., Miller, L. J., Feely, S. M. E., Siskind, C. E., and Shy, M. E. (2011). Charcot-Marie-Tooth Disease Subtypes and Genetic Testing Strategies. Ann. Neurol. 69 (1), 22–33. doi:10.1002/ana.22166
Sarraf, S. A., Raman, M., Guarani-Pereira, V., Sowa, M. E., Huttlin, E. L., Gygi, S. P., et al. (2013). Landscape of the PARKIN-dependent Ubiquitylome in Response to Mitochondrial Depolarization. Nature 496 (7445), 372–376. doi:10.1038/nature12043
Sasaki, S. (2011). Autophagy in Spinal Cord Motor Neurons in Sporadic Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol. 70 (5), 349–359. doi:10.1097/nen.0b013e3182160690
Sasaki, S., and Iwata, M. (2007). Mitochondrial Alterations in the Spinal Cord of Patients with Sporadic Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol. 66 (1), 10–16. doi:10.1097/nen.0b013e31802c396b
Sattler, R., and Tymianski, M. (2000). Molecular Mechanisms of Calcium-dependent Excitotoxicity. J. Mol. Med. 78 (1), 3–13. doi:10.1007/s001090000077
Schaefer, A. M., Sanes, J. R., and Lichtman, J. W. (2005). A Compensatory Subpopulation of Motor Neurons in a Mouse Model of Amyotrophic Lateral Sclerosis. J. Comp. Neurol. 490 (3), 209–219. doi:10.1002/cne.20620
Schapira, A. H. (2012). Mitochondrial Diseases. The Lancet 379 (9828), 1825–1834. doi:10.1016/s0140-6736(11)61305-6
Scorziello, A., Savoia, C., Sisalli, M. J., Adornetto, A., Secondo, A., Boscia, F., et al. (2013). NCX3 Regulates Mitochondrial Ca(2+) Handling through the AKAP121-Anchored Signaling Complex and Prevents Hypoxia-Induced Neuronal Death. J. Cel Sci 126 (Pt 24), 5566–5577. doi:10.1242/jcs.129668
Seaman, M. N. (2012). The Retromer Complex - Endosomal Protein Recycling and beyond. J. Cel Sci 125 (Pt 20), 4693–4702. doi:10.1242/jcs.103440
Sekine, S., and Youle, R. J. (2018). PINK1 Import Regulation; a fine System to Convey Mitochondrial Stress to the Cytosol. BMC Biol. 16 (1), 2. doi:10.1186/s12915-017-0470-7
Shaltouki, A., Hsieh, C.-H., Kim, M. J., and Wang, X. (2018). Alpha-synuclein Delays Mitophagy and Targeting Miro Rescues Neuron Loss in Parkinson's Models. Acta Neuropathol. 136 (4), 607–620. doi:10.1007/s00401-018-1873-4
Sharma, M., and Subramaniam, S. (2019). Rhes Travels from Cell to Cell and Transports Huntington Disease Protein via TNT-like Protrusion. J. Cel Biol 218 (6), 1972–1993. doi:10.1083/jcb.201807068
Shi, Y., Dierckx, A., Wanrooij, P. H., Wanrooij, S., Larsson, N.-G., Wilhelmsson, L. M., et al. (2012). Mammalian Transcription Factor A Is a Core Component of the Mitochondrial Transcription Machinery. Proc. Natl. Acad. Sci. 109 (41), 16510–16515. doi:10.1073/pnas.1119738109
Shirendeb, U., Reddy, A. P., Manczak, M., Calkins, M. J., Mao, P., Tagle, D. A., et al. (2011). Abnormal Mitochondrial Dynamics, Mitochondrial Loss and Mutant Huntingtin Oligomers in Huntington's Disease: Implications for Selective Neuronal Damage. Hum. Mol. Genet. 20 (7), 1438–1455. doi:10.1093/hmg/ddr024
Sickmann, A., Reinders, J., Wagner, Y., Joppich, C., Zahedi, R., Meyer, H. E., et al. (2003). The Proteome of Saccharomyces cerevisiae Mitochondria. Proc. Natl. Acad. Sci. 100 (23), 13207–13212. doi:10.1073/pnas.2135385100
Siklós, L., Engelhardt, J., Harati, Y., Smith, R. G., Joó, F., and Appel, S. H. (1996). Ultrastructural Evidence for Altered Calcium in Motor Nerve Terminals in Amyotrophc Lateral Sclerosis. Ann. Neurol. 39 (2), 203–216. doi:10.1002/ana.410390210
Siklós, L., Engelhardt, J. I., Alexianu, M. E., Gurney, M. E., Siddique, T., and Appel, S. H. (1998). Intracellular Calcium Parallels Motoneuron Degeneration in SOD-1 Mutant Mice. J. Neuropathol. Exp. Neurol. 57 (6), 571–587. doi:10.1097/00005072-199806000-00005
Smith, G. A., Jansson, J., Rocha, E. M., Osborn, T., Hallett, P. J., and Isacson, O. (2016). Fibroblast Biomarkers of Sporadic Parkinson's Disease and LRRK2 Kinase Inhibition. Mol. Neurobiol. 53 (8), 5161–5177. doi:10.1007/s12035-015-9435-4
Snow, B. J., Rolfe, F. L., Lockhart, M. M., Frampton, C. M., O'Sullivan, J. D., Fung, V., et al. (2010). A Double-Blind, Placebo-Controlled Study to Assess the Mitochondria-Targeted Antioxidant MitoQ as a Disease-Modifying Therapy in Parkinson's Disease. Mov. Disord. 25 (11), 1670–1674. doi:10.1002/mds.23148
Song, W., Chen, J., Petrilli, A., Liot, G., Klinglmayr, E., Zhou, Y., et al. (2011). Mutant Huntingtin Binds the Mitochondrial Fission GTPase Dynamin-Related Protein-1 and Increases its Enzymatic Activity. Nat. Med. 17 (3), 377–382. doi:10.1038/nm.2313
Song, W., Song, Y., Kincaid, B., Bossy, B., and Bossy-Wetzel, E. (2013). Mutant SOD1G93A Triggers Mitochondrial Fragmentation in Spinal Cord Motor Neurons: Neuroprotection by SIRT3 and PGC-1α. Neurobiol. Dis. 51, 72–81. doi:10.1016/j.nbd.2012.07.004
Spillantini, M. G., Schmidt, M. L., Lee, V. M.-Y., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). α-Synuclein in Lewy Bodies. Nature 388 (6645), 839–840. doi:10.1038/42166
Sprenger, H.-G., MacVicar, T., Bahat, A., Fiedler, K. U., Hermans, S., Ehrentraut, D., et al. (2021). Cellular Pyrimidine Imbalance Triggers Mitochondrial DNA-dependent Innate Immunity. Nat. Metab. 3 (5), 636–650. doi:10.1038/s42255-021-00385-9
Stafa, K., Tsika, E., Moser, R., Musso, A., Glauser, L., Jones, A., et al. (2014). Functional Interaction of Parkinson's Disease-Associated LRRK2 with Members of the Dynamin GTPase Superfamily. Hum. Mol. Genet. 23 (8), 2055–2077. doi:10.1093/hmg/ddt600
Stefanatos, R., and Sanz, A. (2018). The Role of Mitochondrial ROS in the Aging Brain. FEBS Lett. 592 (5), 743–758. doi:10.1002/1873-3468.12902
Sterky, F. H., Lee, S., Wibom, R., Olson, L., and Larsson, N.-G. (2011). Impaired Mitochondrial Transport and Parkin-independent Degeneration of Respiratory Chain-Deficient Dopamine Neurons In Vivo. Proc. Natl. Acad. Sci. 108 (31), 12937–12942. doi:10.1073/pnas.1103295108
Stifani, N. (2014). Motor Neurons and the Generation of Spinal Motor Neuron Diversity. Front. Cel Neurosci 8, 293. doi:10.3389/fncel.2014.00293
Stiles, A. R., Simon, M. T., Stover, A., Eftekharian, S., Khanlou, N., Wang, H. L., et al. (2016). Mutations in TFAM, Encoding Mitochondrial Transcription Factor A, Cause Neonatal Liver Failure Associated with mtDNA Depletion. Mol. Genet. Metab. 119 (1-2), 91–99. doi:10.1016/j.ymgme.2016.07.001
Stoica, R., De Vos, K. J., Paillusson, S., Mueller, S., Sancho, R. M., Lau, K. F., et al. (2014). ER-mitochondria Associations Are Regulated by the VAPB-PTPIP51 Interaction and Are Disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 5, 3996. doi:10.1038/ncomms4996
Stoica, R., Paillusson, S., Gomez‐Suaga, P., Mitchell, J. C., Lau, D. H., Gray, E. H., et al. (2016). ALS/FTD ‐associated FUS Activates GSK ‐3β to Disrupt the VAPB - PTPIP 51 Interaction and ER -mitochondria Associations. EMBO Rep. 17 (9), 1326–1342. doi:10.15252/embr.201541726
Su, Y.-C., and Qi, X. (2013). Inhibition of Excessive Mitochondrial Fission Reduced Aberrant Autophagy and Neuronal Damage Caused by LRRK2 G2019S Mutation. Hum. Mol. Genet. 22 (22), 4545–4561. doi:10.1093/hmg/ddt301
Subramaniam, S., Sixt, K. M., Barrow, R., and Snyder, S. H. (2009). Rhes, a Striatal Specific Protein, Mediates Mutant-Huntingtin Cytotoxicity. Science 324 (5932), 1327–1330. doi:10.1126/science.1172871
Sugiura, A., McLelland, G. L., Fon, E. A., and McBride, H. M. (2014). A New Pathway for Mitochondrial Quality Control: Mitochondrial‐derived Vesicles. EMBO J. 33 (19), 2142–2156. doi:10.15252/embj.201488104
Suomalainen, A., Majander, A., Wallin, M., Setala, K., Kontula, K., Leinonen, H., et al. (1997). Autosomal Dominant Progressive External Ophthalmoplegia with Multiple Deletions of mtDNA: Clinical, Biochemical, and Molecular Genetic Features of the 1Oq-Linked Disease. Neurology 48 (5), 1244–1253. doi:10.1212/wnl.48.5.1244
Surmeier, D. J., Guzman, J. N., Sanchez, J., and Schumacker, P. T. (2012). Physiological Phenotype and Vulnerability in Parkinson's Disease. Cold Spring Harbor Perspect. Med. 2 (7), a009290. doi:10.1101/cshperspect.a009290
Surmeier, D. J., Guzman, J. N., and Sanchez-Padilla, J. (2010). Calcium, Cellular Aging, and Selective Neuronal Vulnerability in Parkinson's Disease. Cell Calcium 47 (2), 175–182. doi:10.1016/j.ceca.2009.12.003
Suzuki, M., Nagai, Y., Wada, K., and Koike, T. (2012). Calcium Leak through Ryanodine Receptor Is Involved in Neuronal Death Induced by Mutant Huntingtin. Biochem. Biophys. Res. Commun. 429 (1-2), 18–23. doi:10.1016/j.bbrc.2012.10.107
Swarnkar, S., Chen, Y., Pryor, W. M., Shahani, N., Page, D. T., and Subramaniam, S. (2015). Ectopic Expression of the Striatal-Enriched GTPase Rhes Elicits Cerebellar Degeneration and an Ataxia Phenotype in Huntington's Disease. Neurobiol. Dis. 82, 66–77. doi:10.1016/j.nbd.2015.05.011
Swerdlow, R. H., Burns, J. M., and Khan, S. M. (2014). The Alzheimer's Disease Mitochondrial cascade Hypothesis: Progress and Perspectives. Biochim. Biophys. Acta (Bba) - Mol. Basis Dis. 1842 (8), 1219–1231. doi:10.1016/j.bbadis.2013.09.010
Synofzik, M. (2019). Parkinsonism in Neurodegenerative Diseases Predominantly Presenting with Ataxia. Int. Rev. Neurobiol. 149, 277–298. doi:10.1016/bs.irn.2019.10.019
Synofzik, M., Srulijes, K., Godau, J., Berg, D., and Schöls, L. (2012). Characterizing POLG Ataxia: Clinics, Electrophysiology and Imaging. Cerebellum 11 (4), 1002–1011. doi:10.1007/s12311-012-0378-2
Tabata, Y., Imaizumi, Y., Sugawara, M., Andoh-Noda, T., Banno, S., Chai, M., et al. (2018). T-Type Calcium Channels Determine the Vulnerability of Dopaminergic Neurons to Mitochondrial Stress in Familial Parkinson Disease. Stem Cel Rep. 11 (5), 1171–1184. doi:10.1016/j.stemcr.2018.09.006
Tanaka, A., Cleland, M. M., Xu, S., Narendra, D. P., Suen, D.-F., Karbowski, M., et al. (2010). Proteasome and P97 Mediate Mitophagy and Degradation of Mitofusins Induced by Parkin. J. Cel Biol 191 (7), 1367–1380. doi:10.1083/jcb.201007013
Tang, F.-L., Liu, W., Hu, J.-X., Erion, J. R., Ye, J., Mei, L., et al. (2015). VPS35 Deficiency or Mutation Causes Dopaminergic Neuronal Loss by Impairing Mitochondrial Fusion and Function. Cel Rep. 12 (10), 1631–1643. doi:10.1016/j.celrep.2015.08.001
Tang, T.-S., Slow, E., Lupu, V., Stavrovskaya, I. G., Sugimori, M., Llinas, R., et al. (2005). Disturbed Ca2+ Signaling and Apoptosis of Medium Spiny Neurons in Huntington’s Disease. Proc. Natl. Acad. Sci. 102 (7), 2602–2607. doi:10.1073/pnas.0409402102
Tang, T.-S., Tu, H., Chan, E. Y. W., Maximov, A., Wang, Z., Wellington, C. L., et al. (2003). Huntingtin and Huntingtin-Associated Protein 1 Influence Neuronal Calcium Signaling Mediated by Inositol-(1,4,5) Triphosphate Receptor Type 1. Neuron 39 (2), 227–239. doi:10.1016/s0896-6273(03)00366-0
Thal, D. R., Rüb, U., Orantes, M., and Braak, H. (2002). Phases of Aβ-Deposition in the Human Brain and its Relevance for the Development of AD. Neurology 58 (12), 1791–1800. doi:10.1212/wnl.58.12.1791
Thau, N., Knippenberg, S., Körner, S., Rath, K. J., Dengler, R., and Petri, S. (2012). Decreased mRNA Expression of PGC-1α and PGC-1α-Regulated Factors in the SOD1G93AALS Mouse Model and in Human Sporadic ALS. J. Neuropathol. Exp. Neurol. 71 (12), 1064–1074. doi:10.1097/nen.0b013e318275df4b
Tian, J., Yan, Y.-P., Zhou, R., Lou, H.-F., Rong, Y., and Zhang, B.-R. (2014). Soluble N-Terminal Fragment of Mutant Huntingtin Protein Impairs Mitochondrial Axonal Transport in Cultured Hippocampal Neurons. Neurosci. Bull. 30 (1), 74–80. doi:10.1007/s12264-013-1393-0
Toonen, L. J., Schmidt, I., Luijsterburg, M. S., van Attikum, H., and van Roon-Mom, W. M. (2016). Antisense Oligonucleotide-Mediated Exon Skipping as a Strategy to Reduce Proteolytic Cleavage of Ataxin-3. Sci. Rep. 6, 35200. doi:10.1038/srep35200
Trottier, Y., Devys, D., Imbert, G., Saudou, F., An, I., Lutz, Y., et al. (1995). Cellular Localization of the Huntington's Disease Protein and Discrimination of the normal and Mutated Form. Nat. Genet. 10 (1), 104–110. doi:10.1038/ng0595-104
Truban, D., Hou, X., Caulfield, T. R., Fiesel, F. C., and Springer, W. (2017). PINK1, Parkin, and Mitochondrial Quality Control: What Can We Learn about Parkinson's Disease Pathobiology? Jpd 7 (1), 13–29. doi:10.3233/jpd-160989
Tsai, M. F., Phillips, C. B., Ranaghan, M., Tsai, C. W., Wu, Y., Willliams, C., et al. (2016). Dual Functions of a Small Regulatory Subunit in the Mitochondrial Calcium Uniporter Complex. Elife 5. doi:10.7554/elife.15545
Tyynismaa, H., Mjosund, K. P., Wanrooij, S., Lappalainen, I., Ylikallio, E., Jalanko, A., et al. (2005). Mutant Mitochondrial Helicase Twinkle Causes Multiple mtDNA Deletions and a Late-Onset Mitochondrial Disease in Mice. Proc. Natl. Acad. Sci. 102 (49), 17687–17692. doi:10.1073/pnas.0505551102
Tzoulis, C., Tran, G. T., Schwarzlmuller, T., Specht, K., Haugarvoll, K., Balafkan, N., et al. (2013). Severe Nigrostriatal Degeneration without Clinical Parkinsonism in Patients with Polymerase Gamma Mutations. Brain 136 (Pt 8), 2393–2404. doi:10.1093/brain/awt103
Ulfhake, B., and Cullheim, S. (1981). A Quantitative Light Microscopic Study of the Dendrites of Cat Spinal γ-motoneurons after Intracellular Staining with Horseradish Peroxidase. J. Comp. Neurol. 202 (4), 585–596. doi:10.1002/cne.902020410
Vaccaro, V., Devine, M. J., Higgs, N. F., and Kittler, J. T. (2017). Miro1‐dependent Mitochondrial Positioning Drives the Rescaling of Presynaptic Ca 2+ Signals during Homeostatic Plasticity. EMBO Rep. 18 (2), 231–240. doi:10.15252/embr.201642710
Van Den Bosch, L., Vandenberghe, W., Klaassen, H., Van Houtte, E., and Robberecht, W. (2000). Ca(2+)-permeable AMPA Receptors and Selective Vulnerability of Motor Neurons. J. Neurol. Sci. 180 (1-2), 29–34. doi:10.1016/s0022-510x(00)00414-7
Van Goethem, G., Luoma, P., Rantamaki, M., Al Memar, A., Kaakkola, S., Hackman, P., et al. (2004). POLG Mutations in Neurodegenerative Disorders with Ataxia but No Muscle Involvement. Neurology 63 (7), 1251–1257. doi:10.1212/01.wnl.0000140494.58732.83
Vande Velde, C., McDonald, K. K., Boukhedimi, Y., McAlonis-Downes, M., Lobsiger, C. S., Bel Hadj, S., et al. (2011). Misfolded SOD1 Associated with Motor Neuron Mitochondria Alters Mitochondrial Shape and Distribution Prior to Clinical Onset. PLoS One 6 (7), e22031. doi:10.1371/journal.pone.0022031
Vanstone, J. R., Smith, A. M., Smith, A. M., McBride, S., Naas, T., Holcik, M., et al. (2016). DNM1L-related Mitochondrial Fission Defect Presenting as Refractory Epilepsy. Eur. J. Hum. Genet. 24 (7), 1084–1088. doi:10.1038/ejhg.2015.243
Vedartham, V., Sundaram, S., Nair, S. S., Ganapathy, A., Mannan, A., and Menon, R. (2019). Homozygous Sequestosome 1 (SQSTM1) Mutation: a Rare Cause for Childhood-Onset Progressive Cerebellar Ataxia with Vertical Gaze Palsy. Ophthalmic Genet. 40 (4), 376–379. doi:10.1080/13816810.2019.1666414
Verhoeven, K., Claeys, K. G., Zuchner, S., Schroder, J. M., Weis, J., Ceuterick, C., et al. (2006). MFN2 Mutation Distribution and Genotype/phenotype Correlation in Charcot-Marie-Tooth Type 2. Brain 129 (Pt 8), 2093–2102. doi:10.1093/brain/awl126
Vilariño-Güell, C., Wider, C., Ross, O. A., Dachsel, J. C., Kachergus, J. M., Lincoln, S. J., et al. (2011). VPS35 Mutations in Parkinson Disease. Am. J. Hum. Genet. 89 (1), 162–167. doi:10.1016/j.ajhg.2011.06.001
Villa, E., Marchetti, S., and Ricci, J.-E. (2018). No Parkin Zone: Mitophagy without Parkin. Trends Cel Biol. 28 (11), 882–895. doi:10.1016/j.tcb.2018.07.004
Vonsattel, J. P. (2008). Huntington Disease Models and Human Neuropathology: Similarities and Differences. Acta Neuropathol. 115 (1), 55–69. doi:10.1007/s00401-007-0306-6
Wang, R., and Reddy, P. H. (2017). Role of Glutamate and NMDA Receptors in Alzheimer's Disease. Jad 57 (4), 1041–1048. doi:10.3233/jad-160763
Wang, W., Li, L., Lin, W.-L., Dickson, D. W., Petrucelli, L., Zhang, T., et al. (2013). The ALS Disease-Associated Mutant TDP-43 Impairs Mitochondrial Dynamics and Function in Motor Neurons. Hum. Mol. Genet. 22 (23), 4706–4719. doi:10.1093/hmg/ddt319
Wang, W., Wang, X., Fujioka, H., Hoppel, C., Whone, A. L., Caldwell, M. A., et al. (2016). Parkinson's Disease-Associated Mutant VPS35 Causes Mitochondrial Dysfunction by Recycling DLP1 Complexes. Nat. Med. 22 (1), 54–63. doi:10.1038/nm.3983
Wang, W., Yin, J., Ma, X., Zhao, F., Siedlak, S. L., Wang, Z., et al. (2017). Inhibition of Mitochondrial Fragmentation Protects against Alzheimer's Disease in Rodent Model. Hum. Mol. Genet. 26 (21), 4118–4131. doi:10.1093/hmg/ddx299
Wang, X., Becker, K., Levine, N., Zhang, M., Lieberman, A. P., Moore, D. J., et al. (2019). Pathogenic Alpha-Synuclein Aggregates Preferentially Bind to Mitochondria and Affect Cellular Respiration. Acta Neuropathol. Commun. 7 (1), 41. doi:10.1186/s40478-019-0696-4
Wang, X., Su, B., Fujioka, H., and Zhu, X. (2008). Dynamin-like Protein 1 Reduction Underlies Mitochondrial Morphology and Distribution Abnormalities in Fibroblasts from Sporadic Alzheimer's Disease Patients. Am. J. Pathol. 173 (2), 470–482. doi:10.2353/ajpath.2008.071208
Wang, X., Su, B., Lee, H.-g., Li, X., Perry, G., Smith, M. A., et al. (2009). Impaired Balance of Mitochondrial Fission and Fusion in Alzheimer's Disease. J. Neurosci. 29 (28), 9090–9103. doi:10.1523/jneurosci.1357-09.2009
Wang, X., Yan, M. H., Fujioka, H., Liu, J., Wilson-Delfosse, A., Chen, S. G., et al. (2012). LRRK2 Regulates Mitochondrial Dynamics and Function through Direct Interaction with DLP1. Hum. Mol. Genet. 21 (9), 1931–1944. doi:10.1093/hmg/dds003
Wang, Y.-C., Lee, C.-M., Lee, L.-C., Tung, L.-C., Hsieh-Li, H.-M., Lee-Chen, G.-J., et al. (2011). Mitochondrial Dysfunction and Oxidative Stress Contribute to the Pathogenesis of Spinocerebellar Ataxia Type 12 (SCA12). J. Biol. Chem. 286 (24), 21742–21754. doi:10.1074/jbc.m110.160697
Wang, J.-Q., Chen, Q., Wang, X., Wang, Q.-C., Wang, Y., Cheng, H.-P., et al. (2013). Dysregulation of Mitochondrial Calcium Signaling and Superoxide Flashes Cause Mitochondrial Genomic DNA Damage in Huntington Disease. J. Biol. Chem. 288 (5), 3070–3084. doi:10.1074/jbc.m112.407726
Ward, J. M., Stoyas, C. A., Switonski, P. M., Ichou, F., Fan, W., Collins, B., et al. (2019). Metabolic and Organelle Morphology Defects in Mice and Human Patients Define Spinocerebellar Ataxia Type 7 as a Mitochondrial Disease. Cel Rep. 26 (5), 1189–1202. doi:10.1016/j.celrep.2019.01.028
Weber, B., and Barros, L. F. (2015). The Astrocyte: Powerhouse and Recycling Center. Cold Spring Harb Perspect. Biol. 7 (12). doi:10.1101/cshperspect.a020396
Wei, W., Keogh, M. J., Wilson, I., Coxhead, J., Ryan, S., Rollinson, S., et al. (2017). Mitochondrial DNA point Mutations and Relative Copy Number in 1363 Disease and Control Human Brains. Acta Neuropathol. Commun. 5 (1), 13. doi:10.1186/s40478-017-0419-7
Wong, Y. C., and Holzbaur, E. L. F. (2014). Optineurin Is an Autophagy Receptor for Damaged Mitochondria in Parkin-Mediated Mitophagy that Is Disrupted by an ALS-Linked Mutation. Proc. Natl. Acad. Sci. USA 111 (42), E4439–E4448. doi:10.1073/pnas.1405752111
Xie, W., and Chung, K. K. K. (2012). Alpha-synuclein Impairs normal Dynamics of Mitochondria in Cell and Animal Models of Parkinson's Disease. J. Neurochem. 122 (2), 404–414. doi:10.1111/j.1471-4159.2012.07769.x
Xu, Y.-F., Gendron, T. F., Zhang, Y.-J., Lin, W.-L., D'Alton, S., Sheng, H., et al. (2010). Wild-type Human TDP-43 Expression Causes TDP-43 Phosphorylation, Mitochondrial Aggregation, Motor Deficits, and Early Mortality in Transgenic Mice. J. Neurosci. 30 (32), 10851–10859. doi:10.1523/jneurosci.1630-10.2010
Yakubovskaya, E., Guja, K. E., Eng, E. T., Choi, W. S., Mejia, E., Beglov, D., et al. (2014). Organization of the Human Mitochondrial Transcription Initiation Complex. Nucleic Acids Res. 42 (6), 4100–4112. doi:10.1093/nar/gkt1360
Yang, Y., Ouyang, Y., Yang, L., Beal, M. F., McQuibban, A., Vogel, H., et al. (2008). Pink1 Regulates Mitochondrial Dynamics through Interaction with the Fission/fusion Machinery. Proc. Natl. Acad. Sci. 105 (19), 7070–7075. doi:10.1073/pnas.0711845105
Yao, J., Irwin, R. W., Zhao, L., Nilsen, J., Hamilton, R. T., and Brinton, R. D. (2009). Mitochondrial Bioenergetic Deficit Precedes Alzheimer's Pathology in Female Mouse Model of Alzheimer's Disease. Proc. Natl. Acad. Sci. 106 (34), 14670–14675. doi:10.1073/pnas.0903563106
Ye, X., Sun, X., Starovoytov, V., and Cai, Q. (2015). Parkin-mediated Mitophagy in Mutant hAPP Neurons and Alzheimer's Disease Patient Brains. Hum. Mol. Genet. 24 (10), 2938–2951. doi:10.1093/hmg/ddv056
Yilmaz, R., Muller, K., Brenner, D., Volk, A. E., Borck, G., Hermann, A., et al. (2020). SQSTM1/p62 Variants in 486 Patients with Familial ALS from Germany and Sweden. Neurobiol. Aging 87, 139–139 e115. doi:10.1016/j.neurobiolaging.2019.10.018
Youle, R. J., and van der Bliek, A. M. (2012). Mitochondrial Fission, Fusion, and Stress. Science 337 (6098), 1062–1065. doi:10.1126/science.1219855
Yue, M., Hinkle, K. M., Davies, P., Trushina, E., Fiesel, F. C., Christenson, T. A., et al. (2015). Progressive Dopaminergic Alterations and Mitochondrial Abnormalities in LRRK2 G2019S Knock-In Mice. Neurobiol. Dis. 78, 172–195. doi:10.1016/j.nbd.2015.02.031
Zanna, C., Ghelli, A., Porcelli, A. M., Karbowski, M., Youle, R. J., Schimpf, S., et al. (2008). OPA1 Mutations Associated with Dominant Optic Atrophy Impair Oxidative Phosphorylation and Mitochondrial Fusion. Brain 131 (Pt 2), 352–367. doi:10.1093/brain/awm335
Zhang, Y., Chen, K., Sloan, S. A., Bennett, M. L., Scholze, A. R., O'Keeffe, S., et al. (2014). An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J. Neurosci. 34 (36), 11929–11947. doi:10.1523/jneurosci.1860-14.2014
Zhao, C., Takita, J., Tanaka, Y., Setou, M., Nakagawa, T., Takeda, S., et al. (2001). Charcot-Marie-Tooth Disease Type 2A Caused by Mutation in a Microtubule Motor KIF1Bβ. Cell 105 (5), 587–597. doi:10.1016/s0092-8674(01)00363-4
Zhou, C., Huang, Y., Shao, Y., May, J., Prou, D., Perier, C., et al. (2008). The Kinase Domain of Mitochondrial PINK1 Faces the Cytoplasm. Proc. Natl. Acad. Sci. 105 (33), 12022–12027. doi:10.1073/pnas.0802814105
Zhou, Y., Carmona, S., Muhammad, A. K. M. G., Bell, S., Landeros, J., Vazquez, M., et al. (2019). Restoring Mitofusin Balance Prevents Axonal Degeneration in a Charcot-Marie-Tooth Type 2A Model. J. Clin. Invest. 129 (4), 1756–1771. doi:10.1172/jci124194
Zilocchi, M., Finzi, G., Lualdi, M., Sessa, F., Fasano, M., and Alberio, T. (2018). Mitochondrial Alterations in Parkinson's Disease Human Samples and Cellular Models. Neurochem. Int. 118, 61–72. doi:10.1016/j.neuint.2018.04.013
Zimmermann, L., Morado-Diaz, C. J., Davis-Lopez de Carrizosa, M. A., de la Cruz, R. R., May, P. J., Streicher, J., et al. (2013). Axons Giving Rise to the Palisade Endings of Feline Extraocular Muscles Display Motor Features. J. Neurosci. 33 (7), 2784–2793. doi:10.1523/jneurosci.4116-12.2013
Zimon, M., Baets, J., Fabrizi, G. M., Jaakkola, E., Kabzinska, D., Pilch, J., et al. (2011). Dominant GDAP1 Mutations Cause Predominantly Mild CMT Phenotypes. Neurology 77 (6), 540–548. doi:10.1212/wnl.0b013e318228fc70
Zimprich, A., Benet-Pagès, A., Struhal, W., Graf, E., Eck, S. H., Offman, M. N., et al. (2011). A Mutation in VPS35, Encoding a Subunit of the Retromer Complex, Causes Late-Onset Parkinson Disease. Am. J. Hum. Genet. 89 (1), 168–175. doi:10.1016/j.ajhg.2011.06.008
Zimprich, A., Biskup, S., Leitner, P., Lichtner, P., Farrer, M., Lincoln, S., et al. (2004). Mutations in LRRK2 Cause Autosomal-Dominant Parkinsonism with Pleomorphic Pathology. Neuron 44 (4), 601–607. doi:10.1016/j.neuron.2004.11.005
Zou, Z.-Y., Zhou, Z.-R., Che, C.-H., Liu, C.-Y., He, R.-L., and Huang, H.-P. (2017). Genetic Epidemiology of Amyotrophic Lateral Sclerosis: a Systematic Review and Meta-Analysis. J. Neurol. Neurosurg. Psychiatry 88 (7), 540–549. doi:10.1136/jnnp-2016-315018
Zuccato, C., Ciammola, A., Rigamonti, D., Leavitt, B. R., Goffredo, D., Conti, L., et al. (2001). Loss of Huntingtin-Mediated BDNF Gene Transcription in Huntington's Disease. Science 293 (5529), 493–498. doi:10.1126/science.1059581
Keywords: mitochondria, neurodegeneration, selective vulnerability, mitochondrial DNA maintenance, mitochondrial dynamics, quality control, Ca2+ homeostasis
Citation: Paß T, Wiesner RJ and Pla-Martín D (2021) Selective Neuron Vulnerability in Common and Rare Diseases—Mitochondria in the Focus. Front. Mol. Biosci. 8:676187. doi: 10.3389/fmolb.2021.676187
Received: 04 March 2021; Accepted: 08 June 2021;
Published: 30 June 2021.
Edited by:
Mahendra Pratap Kashyap, University of Alabama at Birmingham, United StatesReviewed by:
Soumyabrata Munshi, Tulane University, United StatesAbhishek Jauhari, University of Pittsburgh, United States
Copyright © 2021 Paß, Wiesner and Pla-Martín. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas Paß, dGhvbWFzLnBhc3NAdWsta29lbG4uZGU=