 Yekatarina Gadasheva
Yekatarina Gadasheva Claudia Grossmann
Claudia Grossmann
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Biosci. , 28 May 2021
Sec. Cellular Biochemistry
Volume 8 - 2021 | https://doi.org/10.3389/fmolb.2021.667990
This article is part of the Research Topic Dysregulated Protein Homeostasis in the Aging Organism View all 6 articles
During aging, the cardiovascular system is especially prone to a decline in function and to life-expectancy limiting diseases. Cardiovascular aging is associated with increased arterial stiffness and vasoconstriction as well as left ventricular hypertrophy and reduced diastolic function. Pathological changes include endothelial dysfunction, atherosclerosis, fibrosis, hypertrophy, inflammation, and changes in micromilieu with increased production of reactive oxygen and nitrogen species. The renin-angiotensin-aldosterone-system is an important mediator of electrolyte and blood pressure homeostasis and a key contributor to pathological remodeling processes of the cardiovascular system. Its effects are partially conveyed by the mineralocorticoid receptor (MR), a ligand-dependent transcription factor, whose activity increases during aging and cardiovascular diseases without correlating changes of its ligand aldosterone. There is growing evidence that the MR can be enzymatically and non-enzymatically modified and that these modifications contribute to ligand-independent modulation of MR activity. Modifications reported so far include phosphorylation, acetylation, ubiquitination, sumoylation and changes induced by nitrosative and oxidative stress. This review focuses on the different posttranslational modifications of the MR, their impact on MR function and degradation and the possible implications for cardiovascular aging and diseases.
The MR is a ligand-dependent transcription factor with aldosterone as an endogenous ligand in humans. Cloning of the MR occurred relatively late compared to the other steroid receptors (Arriza et al., 1987; Morrison et al., 1990). The human MR gene comprises ten exons, of which eight are translated (Zennaro et al., 1995). Exon 1a and 1b act as functional promoters, and alternative transcription of the two 5′-UTRs generates the isoforms hMRa and hMRb, which are co-expressed in MR target tissues (Zennaro et al., 1997). The translational start site of the MR is located within exon 2 so that both isoforms lead to the same 107 kDa hMR protein. Exon 2 gives rise to the N-terminal domain (NTD), which is the longest and most variable domain compared to other steroid hormone receptors. Exon 3 and 4 each encode one of the two zinc fingers of the DNA-binding domain (DBD) while the last five exons encode the ligand-binding domain of the MR (LBD).
The N-terminal A/B domain is highly evolutionarily conserved but possesses less than 15% homology with the other steroid receptors. It contains an autonomous activation functional domain (AF-1) consisting of two parts, AF-1a and AF-1b, separated by an inhibitory domain (ID) (Figure 1). AF-1 is constitutively active but suppressed by the C-terminal E/F domain in the absence of ligand. Furthermore, the NTD is responsible for modulating selectivity of the MR for ligand and cofactor interactions (Jausonsloffreda et al., 1994). It also contains a serine/threonine-rich nuclear localization signal NLS0 involved in ligand-independent nuclear localization of MR (Walther et al., 2005). Adjacent to the A/B domain is the C domain containing the DBD. The DBD is highly conserved among steroid receptors, contains two zinc fingers, and binds to GRE hormone response elements of the DNA. The N-terminal zinc finger is responsible for the protein-DNA interaction and contains a P box that leads to interaction with a half-sites of a GRE by intercalating into the major groove. The second zinc finger contains a D box and is responsible for weak MR dimerization (Sartorato et al., 2004). A bipartite basic motif-containing nuclear localization site 1 (NLS1) is localized at the C terminus of the DBD and stretches into the hinge region. The short hinge region connects the DBD and the LBD and contains multiple prolines, which lead to a twist that allows the receptor to contact the transcriptional machinery (Tsai and O'Malley, 1994). The LBD of the MR consists of twelve α-helices and one beta-sheet formation in three antiparallel layers (Hellal-Levy et al., 2000; Farman and Rafestin-Oblin, 2001). In its unliganded state, the MR is bound to chaperone proteins, including heat shock proteins and immunophilins. Besides a ligand-binding pocket, the LBD also possesses several chaperone interaction points, a nuclear localization signal, an area involved in heterodimerization with the GR, and the transcriptional activation functional domain 2 (AF-2) (Savory et al., 2001; Bain et al., 2007). Binding of aldosterone causes conformational changes, which lead to activation of AF-2 and released suppression of the AF-1 function in the NTD (Hong et al., 1997; Fuse et al., 2000). Although AF-1 and AF-2 can act independently, both are required for full ligand-dependent MR transactivation. Additionally, cofactor binding sites are exposed, leading to the recruitment of coactivators, for example members of the steroid receptor coactivator family or GRIP1 (Bain et al., 2007; Hong et al., 1997; Tsai and O'Malley, 1994). The conformational change also was reported to unmask the nuclear localization signal 2 (NLS2) (Sanchez et al., 1985; Walther et al., 2005; Galigniana et al., 2010).
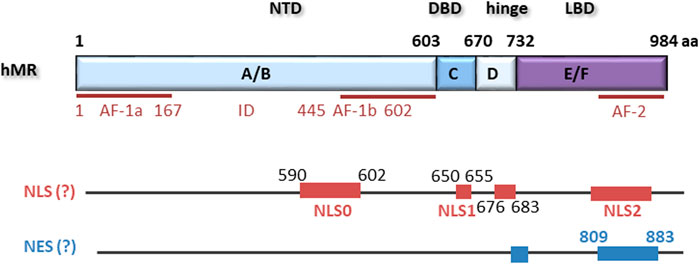
FIGURE 1. hMR comprises 984 amino acids with an N-terminal domain (NTD), a DNA-binding domain (DBD), a hinge region and a ligand binding domain (LBD). Located within the NTD is an activation function 1 (AF-1) that is divided by an inhibitory domain (ID) into two parts, AF-1a and AF-1b. AF-2 is located in the LBD. Several nuclear localization signals (NLS) and putative nuclear export signals (NES) have been described.
The MR and its closest relative the glucocorticoid receptor (GR) evolved from a common corticoid receptor. Evolutionwise, a separation into MR and GR was first found in cartilaginous fish. However, these fish do not possess distinct ligands for the two receptors, so that 11-desoxycorticosterone, glucocorticoids but also progesterone act as natural ligands for both receptors (Sturm et al., 2005; Baker and Katsu, 2017). In lungfish, aldosterone occurs for the first time, and in terrestrial vertebrates, aldosterone typically is the specific ligand for the MR (Joss et al., 1994; Rossier et al., 2015). In humans, the enzyme 11β hydroxysteroid dehydrogenase 2 (11β -HSD2) is located near MR and inactivates glucocorticoids to prevent them from accessing the MR. In tissues with low 11β-HSD2 activity, glucocorticoids bind to MR with comparable affinity to aldosterone.
In the absence of ligand, the MR is primarily located in the cytosol (Nishi et al., 2004; Piwien Pilipuk et al., 2007), or it is equally distributed between cytosol and nucleus depending on cell type and cellular context (Fejes-Toth et al., 1998; Nishi et al., 2001; Tanaka et al., 2005; Walther et al., 2005). In cells with low HSP90 content like cardiomyocytes, it has been demonstrated to be constitutively in the nucleus (Hernandez-Diaz et al., 2010). Nevertheless, MR without ligand is transcriptionally inert. In the cytosol, the MR is associated with chaperones including HSP90, HSP70, and p23 and various immunophilins like FKBP51 (Sanchez et al., 1985; Galigniana et al., 2010) that prevent degradation and ensures a conformation that allows binding of ligand (Caamano et al., 1993; Binart et al., 1995). Upon binding of ligand, a change in associated proteins occurs, and the MR rapidly translocates into the nucleus, where it becomes transcriptionally active (Piwien Pilipuk et al., 2007; Grossmann et al., 2012). Nuclear import of MR is controlled through three nuclear localization signals (Walther et al., 2005). Nuclear localization of naive MR is mediated primarily through NLS0 in the NTD. Specific amino acid substitutions that mimicked phosphorylation selectively enhanced or repressed NLS0 activity suggesting potential regulation sites (Walther et al., 2005). Nuclear transfer through NLS2 within the LBD is dependent on steroid agonists. The third NLS (NLS1) acts together with NLS0 and NLS2 to stimulate nuclear uptake of the agonist-treated receptor, but also directs the complete nuclear localization of MR in response to treatment with steroid antagonist. Tanaka et al. demonstrate that intact NLS1 is necessary for the trafficking of MR together with importin alpha to the nucleus in COS1 cells (Tanaka et al., 2005). However, inhibition of HSP90 strongly delayed nuclear translocation without preventing complete nuclear accumulation of MR. Blocking of NLS1 also did not fully inhibit MR nuclear translocation. Therefore, HSP90- and NLS1-dependent and independent nuclear translocation mechanisms seem to exist (Walther et al., 2005; Piwien Pilipuk et al., 2007; Galigniana et al., 2010). Previously, it was hypothesized that binding of ligand causes dissociation of MR from HSP90 and thereby unmasking of NLS with associated nuclear translocation (Picard and Yamamoto, 1987; Walther et al., 2005). This finding was challenged by experiments showing that mutants without NLS but associated with NLS-containing-HSP90 also are transported into the nucleus (Kang et al., 1994). Overall, the conformational change after ligand binding seems to initiate a shift in the composition of attached chaperone molecules, allowing FKBP52 to replace FKBP51. FKBP52 then links the MR complex to dynein/dynactin motor proteins which propagate nuclear translocation (Banerjee et al., 2008; Galigniana et al., 2010). Simultaneously, several investigators found that HSP90 is not exclusively located in the cytoplasm but also in the nucleus (Galigniana et al., 2010; Hernandez-Diaz et al., 2010; Grossmann et al., 2012) and that HSP90 inhibitors can affect nuclear translocation and transcriptional activity. Additionally, HSP90 enhances DNA binding of MR without being bound to DNA itself, probably by stabilization of receptor structure (Bain et al., 2007). No transactivation of GR or MR occurs without ligand or after incubation with an HSP90 inhibitor, but the exact function of HSP90 is still under debate (Nemoto et al., 1993; Grossmann et al., 2012).
In the nucleus, the MR binds to hormone response elements on the DNA as a homodimer (Tsai et al., 1988; Wrange et al., 1989; Drouin et al., 1992) but may also be able to form heterodimers with the GR, although the relevance of these still is not clear (Liu et al., 1995; Savory et al., 2001; Nishi et al., 2004). Homodimerization of MR and other steroid receptors seems to occur as soon as MR is released from HSP90 and therefore at least in vitro independently of DNA or ligand (Savory et al., 2001; Grossmann et al., 2012). There are several indications that dimerization occurs in the nucleus before DNA binding. Binding of steroid receptors to their hormone response elements involves rapid cycling with the rate of exchange influencing the transcriptional activity. HSP90 has been shown to modulate receptor cycling at DNA and possibly also receptor degradation (Kang et al., 1999; Freeman and Yamamoto, 2002; Stavreva et al., 2004). However, only homodimers formed in the nucleus seem to be able to regulate gene expression.
Nuclear export, on the other hand, has not been well characterized for steroid receptors in general. For the androgen receptor, an untypical, not leucine-rich nuclear export signal was found and characterized in the LBD, which functioned independently of CRM1/exportin. Corresponding structural elements were also found in the MR, but the sequence homology is only moderate (Saporita et al., 2003). For the GR, also a CRM1-independent nuclear export that takes several hours after steroid withdrawal was described. It relies on 15 amino acids of the DBD between the two zinc fingers (Madan and DeFranco, 1993; Black et al., 2001; Walther et al., 2003). However, for the MR no export was detected, and it has been suggested that it is an atypical nuclear hormone receptor that moves unidirectionally from the cytoplasm to the nucleus (Walther et al., 2005). Of all the steroid receptors, MR signaling is the least well explored, and it is not clear how MR inactivation and degradation occurs.
The MR is part of the renin-angiotensin-aldosterone-system (RAAS), and angiotensin II stimulates aldosterone secretion. Classical epithelial MR effects occur in kidney, colon and sweat glands and include sodium and water reabsorption as well as proton and potassium secretion. MR effects in the kidney regulate overall volume and electrolyte homeostasis and thereby control systemic blood pressure. The MR is also expressed in various non-epithelial tissues. For example, in the vascular wall, the MR was detected in vascular smooth muscle cells (VSMCs), in endothelial cells but also fibroblasts and immune cells.
MR antagonists have proven beneficial for different cardiovascular diseases going clearly beyond a lowering of blood pressure and optimization of water-electrolyte-homeostasis. Several large clinical trials indicate that MR antagonists effectively treat heart failure with reduced ejection fraction, a typical cardiovascular disease of the elderly (Zannad et al., 2010; Pitt, 2012; Pitt et al., 2014). Overall, a reduction in mortality and morbidity was registered. Additional clinical benefits of MR antagonists were reported in patients with left ventricular hypertrophy and hypertension (Pitt et al., 2003) and in patients with atrial fibrillation (Dabrowski et al., 2010; Fudim et al., 2018). For patients with heart failure with preserved ejection fraction, the picture is less clear with no significant reduction in the primary composite outcome of death due to cardiovascular causes or hospitalization (Pitt et al., 2014). However, when looking at subgroups, some benefits of MR antagonists were found depending on the geographical origin of the patients, sex and age (Pitt et al., 2014; Pfeffer et al., 2015; Merrill et al., 2019). Recently published results show that new MR antagonists reduce the risk of chronic kidney disease progression and cardiovascular events in patients with chronic kidney disease and type 2 diabetes regardless of pre-existing cardiovascular problems (Bakris et al., 2020; Filippatos et al., 2021). Therefore, results from clinical trials suggest that an overactive RAAS is accountable for some of the cardiovascular changes occurring in cardiovascular diseases and in an attenuated form also during healthy aging.
During aging, plasma renin activity and aldosterone levels decrease, so it is not clear at first glance why RAAS should contribute to cardiovascular changes during aging (Weidmann et al., 1975; Noth et al., 1977; Tzunoda et al., 1986; Bauer, 1993). However, 11β-HSD2 activity required for preventing glucocorticoids from occupying the MR also declines with age, so that binding of cortisol to MR increases (Henschkowski et al., 2008; Campino et al., 2013). Under normal circumstances in non-epithelial tissues with low 11β-HSD2 expression, this does not necessarily activate the MR. This may change in the presence of a permissive micro-milieu found in older patients where oxidative stress and pro-inflammation as well as hypoxia may be more prevalent. Consequently, posttranslational modification of MR or associated factors may occur and affect MR activity. Nanba et al. report a decrease in physiological aldosterone secretion but an increase in autonomous aldosterone production in aged individuals, which might also explain how the RAAS contributes to cardiovascular diseases during aging (Nagarajan et al., 2017). Furthermore, the expression of MR in the vascular wall is increased during aging (Krug et al., 2010; DuPont et al., 2016). MR activation was shown to enhance angiotensin II receptor 1 and angiotensin converting enzyme expression, thus leading to a vicious cycle (Keidar et al., 2005; Pitt, 2012). Typically, the effects of the MR in the cardiovascular tissue are very versatile and context-dependent, indicating additional levels of regulation beyond straightforward gene expression.
For instance, in vessels and in vivo, MR activation can lead to both vasodilation or vasoconstriction depending on the experimental setup (Schmidt et al., 2003; Uhrenholt et al., 2003; Nagata et al., 2006; Schmidt et al., 2006; Skott et al., 2006). This suggests that MR effects may be regulated cell-type and context-dependent by external micro-milieu factors. In pathological experimental animal models, MR leads to a series of pathophysiological alterations in the cardiovascular system that mimic aging-dependent changes like an increase in blood pressure, vasoconstriction, oxidative stress or inflammation. Furthermore, an inverse correlation between white blood cell telomere length, a biomarker for oxidative stress and inflammation, and aldosterone levels has been found in normotensive men (Benetos et al., 2005). In patients with cardiovascular disease, the telomere length was reduced, suggesting aldosterone promotes cardiovascular diseases.
To investigate the underlying molecular mechanisms, the MR was studied further in isolated vessels, VSMCs and endothelial cells. In rat aorta and in isolated rat VSMCs, MR expression increased with aging and enhanced the expression of pro-inflammatory genes like ICAM, TGFβ and pro-collagen1 via epidermal growth factor receptor (EGFR) signaling (Krug et al., 2010). MR expression was also increased with aging in mesenteric resistance arteries of mice (DuPont et al., 2016). A VSMC specific MR-KO mouse model demonstrated that VSMC-MR leads to increased vascular remodeling after an injury as well as to increased myogenic tone, vasoconstriction and oxidative stress and thereby may promote the rise in blood pressure with aging (McCurley et al., 2012; DuPont et al., 2016). Additionally, the aging-associated increase in vascular fibrosis and stiffness was prevented by the VSMC-specific MR-KO model (Kim et al., 2018).
Healthy endothelial cells play an important anti-inflammatory and antithrombotic role in the vasculature and convey vasorelaxation. Under physiological conditions, the role of the MR seems to be vasoprotective, but it is not as well characterized as in models with cardiovascular risk factors (Nietlispach et al., 2007; Hwang et al., 2016). In these pathophysiological models, MR aggravated cardiovascular diseases and MR antagonists were beneficial in helping to restore normal endothelial and vascular function (Farquharson Colin et al., 2000; Garg et al., 2015; Chen et al., 2016). Again, this suggests, external factors can regulate MR function to confer the switch from physiological to pathophysiological functions. Pathological MR effects include initiation of endothelial dysfunction, inflammation and oxidative stress by activating NADPH oxidase, inhibiting NO activity and reducing the expression of protective glucose-6-phosphate dehydrogenase (Hashikabe et al., 2006; Nagata et al., 2006; Leopold et al., 2007; Iwashima et al., 2008; Fels et al., 2010; Maron et al., 2012). Additionally, increased uncoupling of NO synthase has been described, again leading to an increase in ROS (Taylor et al., 2003). Besides enhancing reactive oxygen species (ROS) formation, MR also augmented the expression of cell adhesion molecules like ICAM and VCAM that support inflammation (Caprio et al., 2008; Deuchar et al., 2011). In an atherosclerosis model, endothelial MR was involved in plaque inflammation, leukocyte-endothelial interaction and vascular inflammation, and a specific endothelial cell MR-KO could prevent these changes (Moss et al., 2019). Furthermore, the expression of pro-inflammatory cytokines was enhanced by MR activation (Jia et al., 2016). On the one hand, in models of vascular injury, MR was reported to be pro-thrombotic while, on the other hand, overexpression of MR in endothelial cells increased protein C expression and thereby attenuated thrombin generation and vascular thrombosis (Bodary et al., 2006; Gromotowicz et al., 2011; Lagrange et al., 2014). Aldosterone also induced changes in the expression of epithelial sodium channels, which led to enhanced sodium transport and changes in mechanical cell properties, which contribute to vascular stiffness and fibrosis (Kusche-Vihrog et al., 2008; Drueppel et al., 2013; DuPont et al., 2014; Jia et al., 2016). Substantial sex-dependent differences were reported (Moss et al., 2019). Taken together, MR has diverse effects in the vasculature, which seem to be highly dependent on cellular context and micro-milieu factors, cell-type, sex and experimental setup. Because detrimental effects of the MR are especially pronounced in aged or pre-injured tissues or in the presence of risk factors, we hypothesize that changes in micro-milieu during aging or diseases may influence MR activity by affecting its signaling through induction of posttranslational modifications.
Posttranslational modification of steroid receptors allows fine-tuning of receptors stability, ligand-binding, transformation, co-regulator binding, DNA-binding and transactivation (Figure 2). Early studies have shown that the glucocorticoid and estrogen receptor exist as phosphoproteins (Migliaccio et al., 1982; Sanchez et al., 1985; Bodwell et al., 1991). For MR first evidence comes from MR overexpressed in Sf9 cells, where Western blots of precipitated hMR possessed three major protein bands. Pulse-labeling with 32Pi indicated that the recombinant hMR is highly phosphorylated (Alnemri et al., 1991). Galigniana confirmed that native rat kidney MR is a phosphoprotein by showing that incubation with active phosphatase diminished aldosterone binding but accelerated the receptor transformation to a DNA binding form. Endogenous phosphatase could be inhibited by okadaic acid and tautomycin, indicating that it is a serine/threonine phosphatase belonging to the PP1/PP2A subgroup (Galigniana, 1998; Piwien-Pilipuk and Galigniana, 1998). A compact disk (CD)-based microfluidic method for selective detection of phosphopeptides by mass spectrometry identified two phosphorylation sites, Thr 735 and Ser 737, in the ligand-binding domain of the human mineralocorticoid receptor (Hirschberg et al., 2004). Next, in MR-overexpressing COS-7 cells, phospho-peptides were purified and analyzed by LC-MS/MS. This approach revealed sixteen phosphorylation sites of the MR under basal conditions, fourteen of which were located in the NTD, one in the hinge region and one in the LBD (Shibata et al., 2013). Predicted kinases for these sites are proline-directed kinases, glycogen synthase kinase-2, casein kinase II (CK2) and calmodulin kinase II (Shibata et al., 2013).
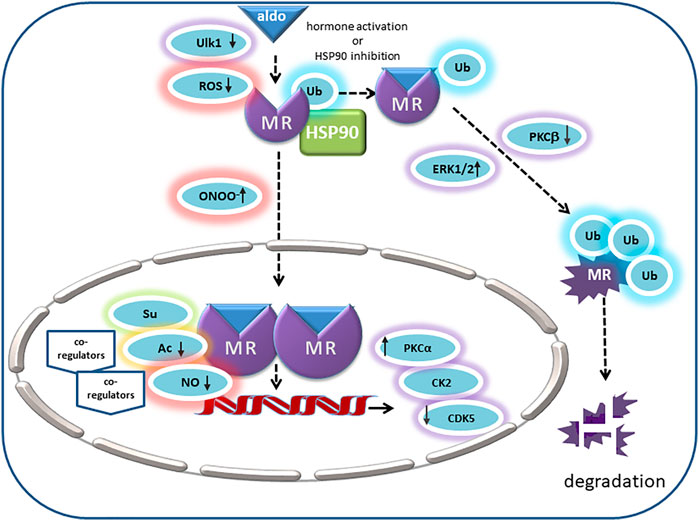
FIGURE 2. In its unliganded state the MR is monoubiquitinated and located in the cytosol with chaperone molecules like HSP90. Its endogenous ligand aldosterone binds to MR in the cytosol and thereby triggers nuclear translocation. Ligand binding is inhibited by MR oxidation. In intercalated cells, autophagy activating kinase 1 (Ulk1) phosphorylates MR S843 in the LBD, mitigates its affinity for agonists and reducing its transcriptional activity. Peroxynitrite (ONOO-) can cause ligand-independent nuclear translocation and activation of MR. Inhibition of HSP90 or activation by aldosterone causes polyubiquitination of MR and degradation. ERK1/2 and PKC also modulate MR polyubiquitination and degradation. Positive regulators of MR transactivation activity are PKCa and CK2. In the prescence of cytokines, CK2 inhibits GRE signaling but promotes NFKB signaling and transcription of pro-inflammatory cytokines. CDK5 inhibits MR transactivation in neuronal cells. Acetylation (Ac) and SUMOylation (Su) lead to modulation of aldosterone-activated MR transactivation by direct and indirect effects. NO attenuates binding of MR to DNA, consequently reducing the MR’s transcriptional activity (↑ = stimulatory effect; ↓ = inhibitory effect).
Of these phosphorylation sites, S843 in the LBD of the MR has been studied in most detail (Shibata et al., 2013). Phosphorylation of MR-S843 resulted in MR inactivation and reduced its ligand binding affinity. It occurred only in intercalated cells of the distal nephron of the kidney and controlled the response to volume depletion and hyperkalemia. In volume depletion, angiotensin II and WNK4 signaling decreased MR-S843 phosphorylation levels, resulting in an increase in apical proton pumps and Cl−/HCO3− exchangers, which prevented a lumen-negative potential from forming. Consequently, adjacent principal cells promoted an increase in sodium and water reabsorption but no K+ secretion. Hyperkalemia, on the other hand, increased phosphorylation of MR-S843 and thereby inactivated aldosterone effects in intercalated cells so that a lumen-negative potential with resulting K+ secretion occurred in the renal collecting duct. This mechanism serves as a switch allowing aldosterone to exert distinct effects in different physiological contexts (Shibata et al., 2013). With a high-throughput screening assay and confirmed by in vitro and in vivo analyses, ULK1 was identified as the principal kinase responsible for the phosphorylation of MR-S843. It was shown to be highly abundant in the intercalated cells of the collecting duct and could be inhibited by angiotensin II via mTOR signaling (Shibata et al., 2018). The group of Alvarez de la Rosa investigated this phosphorylation site with the help of phospho-deficient and -mimetic mutants of mouse MR-839 corresponding to human MR-S843. A phospho-mimetic mutant of hMR-S843 lowered its affinity for agonists, slowed down its ligand-induced translocation and abrogated its transcriptional activity. Co-factor recruitment was impaired as shown for SRC-1 by proximity ligation assay. MR stability and dimerization were not affected (Jimenez-Canino et al., 2016a). A phospho-mimetic MR mutant displayed a dominant-negative effect when forming heterodimers with wild type MR, indicating that phosphorylation of one MR per homodimer is sufficient to impair transcriptional activity so that the overall effect is amplified.
As another functionally relevant kinase, ERK1/2 and its effect on MR signaling were studied in renal epithelial cells. Aldosterone led to a rapid phosphorylation of MR that was detected by a phosphatase-sensitive shift in gel band size and that could be abrogated by an inhibitor of ERK1/2 phosphorylation. There are six conserved predicted ERK1/2 phosphorylation sites in the NTD of the MR and only mutation of all six diminished the shift in band size and abolished the signal detected with an anti-phosphoserine antibody. Functionally, MR phosphorylation led to the disruption of the interaction between MR and tumor susceptibility gene 101 (Tsg101) found under basal conditions. Subsequently, removal of a monoubiquitin, followed by polyubiquitination and degradation of the MR took place (Faresse et al., 2012). Since aldosterone via MR can non-genomically induce ERK1/2 phosphorylation, this may be a negative feedback system involved in limiting for example aldosterone-induced sodium reabsorption (Grossmann et al., 2004; Grossmann et al., 2005; Faresse et al., 2012).
Additionally, there are some indications that different PKC isoforms can affect MR signaling through direct phosphorylation of the receptor. Aldosterone triggered both an early nongenomic and a late genomic increase in sodium transport in rat cortical collecting duct cells dependent on PKCα. The early aldosterone-induced increase in short-circuit current involved rapid phosphorylation of MR on serine and threonine residues, which could be abolished by a PKCα inhibitor. In contrast, the late aldosterone-induced increase in ion transport and gene expression required protein synthesis but could also be inhibited by blocking the PKCα pathway so that a crosstalk between rapid and prolonged MR effects was suggested (Le Moellic et al., 2004). In contrast, stimulation of PKCβ signaling, for example, by high glucose concentrations, led to a reduction in MR ubiquitination and degradation, which caused elevated MR levels and transcriptional activity (Hayashi et al., 2017). For PKCδ, angiotensin II led to MR-PKCδ complex formation in association with increased serine phosphorylation of the MR-NTD in vascular cells. AngII also activated MR transcriptional activity, target gene expression, and SMC proliferation in a PKCδ-dependent manner that may involve phosphorylation of MR-NTD (Lu et al., 2019). Although MR phosphorylation by PKC isoforms has been demonstrated by several groups, it is not always clear if the effects described evolve from a direct MR phosphorylation or indirectly through phosphorylation of co-regulators or a combination of both.
Another potential kinase for MR phosphorylation is the ubiquitously expressed serine/threonine kinase CK2. Inhibition of CK2 reduced the shift in gel band size of MR caused by aldosterone and inhibited MR transcriptional activity under control conditions (Ruhs et al., 2017). Peptide microarrays and site-directed mutagenesis experiments identified the highly conserved S459 as a functionally relevant CK2 phosphorylation site for MR signaling (Ruhs et al., 2017). Consequently, CK2 seems to act as a positive modulator of MR transcriptional activity by phosphorylating the MR. Further investigations unveiled that CK2 facilitates the MR-DNA interaction with subsequent rapid MR degradation. In the presence of pro-inflammatory cytokines, MR and CK2 expression were enhanced, and MR activation by aldosterone augmented CK2-dependent NF-κB signaling and enhanced the expression of pro-inflammatory genes. This may be a mechanism to explain MR over-activation in a pathological micro-milieu.
In neuronal cells, MR has an impact on neuronal viability, synaptic plasticity and emotional changes. There, CDK5 interacted with the LBD of the MR and aldosterone-dependently phosphorylated the MR-NTD residues S128, S250, and Th159, identified by mass spectrometry analysis and mutation experiments. Phosphorylation of MR reduced its transcriptional activity in an MMTV-reporter assay but did not affect nuclear accumulation. On the other hand, aldosterone and CDK5 increased the expression of brain-derived neurotrophic factor in rat cortical neuronal cells (Kino et al., 2010). This suggests that CDK5 through phosphorylation mediates the interaction of the MR with co-regulators and thereby modulates its transcriptional activity without affecting hormone binding or receptor translocation, similar as has been demonstrated for the GR (Kino et al., 2007).
Other kinases for which a regulatory role on MR signaling through phosphorylation has been postulated include PKA and G protein-coupled receptor kinases (GRK). Activation of cAMP-PKA signaling stimulated GRE-containing promoters in a ligand-independent manner and also synergistically after aldosterone activation. Protein kinase-inhibiting peptide (PKI) prevented both cAMP and aldosterone induction, which indicates that a functional cAMP pathway is required for MR transactivation activity. Binding of the MR to a GRE-containing oligonucleotide in a gel shift assay was enhanced by PKA and required the NTD of the MR. However, purified MR NTD protein was not directly phosphorylated by PKA in vitro so that PKA seems to act indirectly, probably by relieving the effect of an MR repressor (Massaad et al., 1999). Conversely, MR-S601, predicted to be a phosphorylation site for PKA or for casein kinase I, has been identified as a phosphorylation site by mass spectrometry under basal conditions by another group (Shibata et al., 2013). It is located within the NLS0 of the NTD and consequently affects nuclear translocation. Phospho-mimetic mutants of MR-S601 restricted the MR to the cytosol while phospho-deficient mutants shifted the MR to the nucleus (Walther et al., 2005). In vivo implications of these findings have not been studied so far. Last, GRK2 and -5 were also shown to phosphorylate aldosterone receptors. While GRK5 was reported to phosphorylate and thereby inhibit cardiac MR, GRK2 phosphorylated and desensitized the aldosterone membrane receptor GPER. In H9C2 cardiomyocytes and adult rat ventricular myocytes, β2-adrenergic receptor activation led to phosphorylation of MR by GRK5, which inhibited the transcriptional MR activity. GRK5 was necessary for the cardioprotective effects of MR antagonists against aldosterone-induced apoptosis and oxidative stress. Conversely, GRK2 seemed to oppose the beneficial effects of aldosterone mediated by GPER signaling (Maning et al., 2020). However, the role of GPER for overall pathophysiological aldosterone effects is still under debate. Supplementary Table S1 summarizes phosphorylation sites, kinases and effects.
Acetylation is a general epigenetic modification responsible for controlling the activity of various cytosolic and nuclear proteins involved in gene expression like histones, nuclear receptors and chaperones (Kovacs et al., 2005; Murphy et al., 2005; Minucci and Pelicci, 2006). It is a reversible modification depending on the balance between lysine acetyltransferases (KAT) and lysine deacetylases (KDAC) also known as histone acetyltransferases and histone deacetylases (HDAC). Aberrant acetylation of lysine residues has previously been linked to diseases, and inhibitors of HDAC exhibit antifibrotic, anti-inflammatory, anti-hypertrophic, and anti-hypertensive effects (Minucci and Pelicci, 2006; Cardinale et al., 2010; Iyer et al., 2010; Schneider et al., 2013; Seo et al., 2015). Acetylation of the N-terminus of histones is essential to expose promoter DNA to transcription factors and RNA polymerase II for initiating transcription. Acetylation of nuclear receptors controls a wide variety of cellular functions, including nuclear receptor activity, DNA binding affinity, ligand sensitivity, receptor stability, and subcellular distribution (Wang et al., 2011). GR, progesterone receptor, androgen receptor and MR share a KXKK acetylation motif in the hinge region within NLS1 (Faus and Haendler, 2006), suggesting a common regulatory mechanism. GR deacetylation in this region facilitates NF-κB suppression (Ito et al., 2006) and regulates the expression of genes involved in generating circadian rhythm (Nader et al., 2009). For the MR, acetylation at K677 in the NLS1 of the hinge region has been demonstrated. Mutated MR without the lysine residues in the hinge region showed less acetylation and more transcriptional activity (Lee et al., 2013). Overexpression of CBP and p300 in the presence of aldosterone led to increased acetylation of MR in the nucleus, suggesting they are the responsible lysine acetyltransferases (Pascual-Le Tallec and Lombes, 2005; Seo et al., 2015). Although aldosterone treatment enhanced the nuclear availability of the MR, it did not affect the subcellular localization of p300 or CBP (Seo et al., 2015). Increased acetylation led to decreased MR transcriptional activity with reduced expression of MR target genes and attenuated MR and Pol II recruitment to specific hormone response elements. No effect on MR expression or translocation was found (Lee et al., 2013; Kang et al., 2015; Lee et al., 2015; Seo et al., 2015; Seok et al., 2016). Besides having lysine acetyltransferase activity, CBP and p300 can also function as transcriptional adaptors and thereby MR co-activators to enhance MR activity (Fuse et al., 2000; Peter et al., 2017). Therefore, CBP and p300 context-dependently seem to act as co-regulators of the MR. Only inhibition of HDACI but not of HDACII activity increased MR acetylation and attenuated MR target gene expression (Lee et al., 2015). Of the lysine deacetylases in group I, HDAC3 was shown to interact with MR and to reduce its transcriptional activity. Accordingly, HDAC3 knockdown increased MR acetylation and decreased MR target gene expression by inhibiting the MR interaction with hormone-responsive-elements and RNA polymerase (Lee et al., 2013). Furthermore, HDAC4 was identified as a necessary scaffolding protein between MR and HDAC3. Non-genomic effect of MR via PKA and PP1/2 induced nuclear translocation of HDAC4 to facilitate the interaction between MR and HDAC3. Lower levels of HDAC4 expression led to a decreased interaction between MR and HDAC3, promoting increased MR acetylation and decreased transcriptional activity (Lee et al., 2015).
Accordingly, the HDACI inhibitor valproic acid (VPA) increased MR acetylation and decreased MR transcriptional activity in reporter gene experiments. This corresponded to reduced MR target gene expression as well as reduced pol II recruitment to the respective promoters. Pathophysiological relevance of MR acetylation was also demonstrated with HDAC inhibitors. In mCCD cells, aldosterone augmented ENaC-induced Na+ absorption and increased SGK1 activity and abundance. HDAC inhibition by trichostatin A could inhibit aldosterone/MR dependent regulation of SGK1/ENAC signaling leading to sodium reabsorption. Insulin-dependent activation of SGK/ENAC signaling was not modified. Consequently, the SGK1-ENAC pathway was not affected by pan HDAC inhibition, but the control of MR over this pathway was perturbed, which again supports that MR acetylation inhibits MR activity (Mansley et al., 2019). Furthermore, HDAC inhibition attenuated pathophysiological MR effects like hypertension, hypertrophy, inflammation and fibrosis in vivo in animal models of spontaneous hypertension and hyperaldosteronism (Cardinale et al., 2010; Iyer et al., 2010; Lee et al., 2013; Kang et al., 2015; Seok et al., 2016). Although treatment with VPA not only enhanced MR acetylation but also increased histone-3-acetylation (H3Ac) and trimethylation (H3K4me3) in the promoter regions of MR target genes, expression of MR target genes was decreased, and the development of hypertension in deoxycorticosterone acetate-induced hypertensive rats and spontaneously hypertensive rats (SHR) was prevented (Seok et al., 2016). These results suggest that HDAC inhibition attenuates the development of hypertension in SHR through acetylation of MR and independent of histone effects. Treatment with VPA also histologically attenuated cardiac hypertrophy and fibrosis through acetylation of MR in spontaneously hypertensive rats (Kang et al., 2015). An indirect effect of acetylation on MR signaling was also reported for its chaperone HSP90. Increasing acetylation of residue K295 of HSP90 by inhibiting HDAC6 weakened HSP90′s interaction with the MR and supported MR nuclear shuttling. However, HSP90-K295 acetylation did not alter MR expression or transactivation activity (Jimenez-Canino et al., 2016b).
Ubiquitin is covalently conjugated to lysine residues of substrate proteins through an ATP-dependent reaction that involves successive action of ubiquitin-activating enzyme E1, ubiquitin-conjugating enzyme E2, and ubiquitin ligases E3. Either monoubiquitination with a single ubiquitin molecule or polyubiquitination with ubiquitin chains is possible. Ubiquitin itself can be ubiquitinated further or ubiquitin-like molecules like SUMO or NEDD8 can modify it. Additional posttranslational modification through acetylation or phosphorylation is possible, and each modification may lead to an altered signaling outcome. For example, covalent attachment of ubiquitin polypeptide often promotes protein turnover by targeting the protein to the proteasome for degradation. The ubiquitin-proteasome pathway regulates the turnover of many nuclear hormone receptors and decreases their ligand-mediated transcriptional activity (Wallace and Cildlowski, 2001; Yokota et al., 2004). For the GR, proteasome activity is required for rapid GR exchange at promoters (Stavreva et al., 2004). Treatment with proteasome inhibitor enhanced GR activity and increased in vivo DNA occupancy time, indicating that ubiquitination is involved in regulating GR stability (Deroo et al., 2002; Stavreva et al., 2004). Furthermore, ubiquitination of GR at Lys419 was shown to stimulate GR nuclear export and subsequent degradation (Kino et al., 2004; Wallace et al., 2010). Yokota et al. first showed that treatment with aldosterone leads to reduced MR expression and targeting of the MR to the proteasome (Yokota et al., 2004). Accordingly, proteasomal inhibition prevented ligand-dependent degradation of MR and thereby enhanced its transcriptional activity. However, mutation of the lysines of two PEST motifs predicted as candidate ubiquitination sites, K715 and K367, failed to prevent degradation and therefore other residues or indirect effects seem to be responsible (Yokota et al., 2004). Later it was demonstrated that under basal conditions, MR is monoubiquitinated, and this state is stabilized by Tsg101 (Burgdorf et al., 2004; Ismaili et al., 2005; La Rosa et al., 2011). In the cytosol, polyubiquitination of unstimulated MR is prevented by its interaction with HSP90. Inhibition of HSP90 promoted polyubiquitination through interaction with the ubiquitin-protein ligase CHIP, which initiated proteasomal degradation of unliganded MR and reduced MR protein levels as well as aldosterone-dependent transcription and sodium transport (Faresse et al., 2010). In renal M1 cells, USP2-45, a ubiquitin-specific protease, was responsible for removal of monoubiquitin. USP2-45 down-regulation or USP-2 deficiency led to increased MR protein expression in vitro and in vivo. In vivo this increase in MR protein did not lead to physiological disturbances (Faresse et al., 2013). Aldosterone also led to disruption of MR/Tsg100 association and removal of monoubiquitin followed by polyubiquitination and MR degradation. Nevertheless, polyubiquitination and proteasome activity seemed essential for nuclear mobility and MR transcriptional activity (Tirard et al., 2007). The exact target lysines and ligases are not fully clear. Phosphorylation of MR via ERK1/2, which can be induced by non-genomic MR effects, also promoted monoubiquitin removal and MR degradation (Yokota et al., 2004; Tirard et al., 2007; Faresse et al., 2012). Conversely, aldosterone-mediated MR degradation could be prevented by ERK1/2 inhibition or mutation of target serines (Faresse et al., 2012), indicating a possible negative feedback loop. On the other hand, in vitro and in vivo experiments showed that ERK activation in the kidney through EGFR activation counteracted MR ubiquitination and increased MR protein expression and transcriptional activity. EGFR inhibition lowered systolic blood pressure and MR activity in DOCA/salt-treated mice (Mitsuishi et al., 2018). Another cytoplasmic binding partner of MR that promotes receptor accumulation and aldosterone-induced sodium uptake in renal cells due to less proteasomal degradation of MR is protein phosphatase 1 alpha (PP1α). To achieve this, PP1α dephosphorylates and thereby inhibits the ubiquitin ligase Mdm2 and thereby prevents its interaction with MR (Nagarajan et al., 2017). As mentioned previously, activation of PKCβ by high glucose concentrations also increased MR expression and transcriptional activity by increasing phosphorylation and decreasing ubiquitination of MR. Accordingly, in kidneys of db/db mice, a type 2 diabetes mouse model with PKCβ activation, levels of MR and Sgk-1 were elevated and could be reversed by PKC inhibition. PKC inhibition also lowered systolic blood pressure (Hayashi et al., 2017).
Sumoylation involves covalent attachment of small proteins called small ubiquitin modifiers (SUMO), which possess four isoforms SUMO1-4 in humans. SUMOs are preferentially attached to lysine in the consensus sequence ΨKD/E and share a similar structure with ubiquitin. As with ubiquitination, attachment requires three enzymes E1-E3. The MR contains five consensus SUMO-1 binding sites, four in the AF1 domain of the NTD and one in the LBD. PIAS1 and some of its family members can function as E3 ligases and are able to interact with the MR. They can directly sumoylate the MR as demonstrated by multiple shifted bands in Western blot analyses, which are missing after mutating respective amino acids. Overexpression of PIAS-1, which led to increased MR sumoylation, had no effect on MR expression or subcellular localization but reduced its transcriptional activity upon ligand binding. The inhibition was equally strong after deleting the first 453 amino acids of the NTD but was not present for GR and dexamethasone (Pascual-Le Tallec et al., 2003). Mutation of all four NTD-SUMO-1 attachment sites abolished MR sumoylation and enhanced transcriptional activity of aldosterone-stimulated MR at compound GRE elements while nuclear mobility was decreased. However, transcriptional activity at MMTV promoter elements was unchanged after aldosterone treatment (Tirard et al., 2007). Because SUMO-1 overexpression also led to inhibition of the activity of sumoylation-deficient MR at a compound GRE element, this suggests that additional MR cofactors also get sumoylated and modulate transcriptional MR activity (Pascual-Le Tallec et al., 2003; Tirard et al., 2007). The NTD of the MR not only interacted with PIAS but also with SUMO-1 and Ubc9, a SUMO-E2 conjugating enzyme (Pascual-Le Tallec et al., 2003; Tirard et al., 2007). Overexpression of Ubc9 increased MR transactivation at MRE, ENaC, or MMTV promoter elements in a hormone-sensitive manner, and knockdown reduced expression of MR target genes. However, Ubc9 showed similar effects when applying a sumoylation-inactive Ubc9 mutant or an MR with four mutated sumoylation motifs, indicating that Ubc9 acts as a transcriptional co-activator beyond its SUMO E2-conjugating function. Accordingly, MR, Ubc9, and the steroid receptor co-activator SRC-1 formed a complex and were recruited in an aldosterone dependent manner to endogenous ENaC gene promoters, where they synergistically enhanced MR transactivation activity (Yokota et al., 2007). Another factor interacting with Ubc9, SUMO1 and MR is FAF1, an MR co-repressor, which contains two SUMO interacting motifs with which it binds to sumoylated MR and thereby represses its aldosterone-activated transactivation. Silencing FAF1 expression augmented MR target gene expression. Mechanistically, FAF1-mediated MR transrepression involved inhibition of MR N/C terminal interactions and promotion of MR polyubiquitination and degradation (Wang et al., 2019). Additionally, an indirect effect on MR signaling was demonstrated by sumoylation of 11β-HSD2 at lysine 266. Despite having no effect on protein stability or subcellular localization and only a slight effect on enzyme kinetics, sumoylation decrease translocation of MR upon cortisol treatment without affecting transcriptional activity (Jimenez-Canino et al., 2017). Overall, sumoylation of MR and of interaction partners seems to be important for regulating the interaction of MR with co-regulators again in a context- and promoter-dependent manner. For the GR it is also known that it contains two sumoylation sites in the NTD and one in the LBD. Sumoylation in the NTD leads to a similar context-dependent inhibition of receptor transcriptional activity with no clear effect at MMTV promoters. The third sumoylation site is involved in stimulating GR activity under conditions of cellular stress. Furthermore, FKBP51 was sumoylated under these conditions and supported GR-FKBP51 complex formation and interaction with HSP90, which promoted cytosolic, inactive GR. A similar interaction seems likely for the MR. Other steroid receptors show a similar sumoylation of the NTD, which modulates their respective transcriptional activity (Poukka et al., 2000). The data suggest that the overall transcriptional activity of MR can be modulated by direct sumoylation as well as the sumoylation of MR-interacting proteins (Tirard et al., 2007).
During aging, oxidized proteins accumulate in tissues because of increased ROS generation and a reduced antioxidative capacity (Stadtman, 2006). Overall, the expression of the MR in target tissues is low, its HSP90 heterocomplex easily dissociates, and it is highly susceptible to proteolysis and oxidation (Galigniana and Piwien-Pilipuk, 1999a). Oxidation of MR has been shown to prevent aldosterone and DNA binding of the receptor and to attenuate biological MR effects, all of which can be reversed or attenuated by reducing agents (Galigniana, 1996; Piwien-Pilipuk and Galigniana, 2000; Piwien-Pilipuk et al., 2002; Ruhs et al., 2012). Reduced aldosterone- and DNA-binding of MR caused by H2O2 was enhanced by iron and inhibited by sulfhydryl reducing reagents, suggesting that disulfide bonds of cysteines are responsible (Galigniana, 1996). Treatments with metals in vitro and in vivo in adrenalectomized rats inhibited both steroid-binding capacity and aldosterone-dependent sodium-retaining properties (Galigniana and Piwien-Pilipuk, 1999a). As a mechanism leading to loss of steroid binding capacity and MR function, receptor oxidation at essential cysteines was identified because a decreased number of reactive thiols was found on immunopurified receptors after in vivo glutathione depletion (Piwien-Pilipuk and Galigniana, 2000). Furthermore, treatment of cytosolic MR of Sf9 cells with a cysteine modifying agent inhibited the binding of hMR to [3H]aldosterone. Agonist-bound MR was resistant to cysteine modifying agents while free hMR and antagonist-bound hMR was sensitive. Therefore, sulfhydryl groups are involved in binding of ligands to the MR, and their accessibility varies depending on the ligand-status of the MR and the receptor conformation (Souque et al., 1996). Another mechanism leading to reduced ligand-binding and MR function in a glutathione depletion model was enhanced MR and EF-2 carbonylation with inhibited protein translation and MR effects (Piwien-Pilipuk and Galigniana, 2000; Piwien-Pilipuk et al., 2002). Association with chaperone complex was not affected by MR oxidation (Piwien-Pilipuk et al., 2002). Overall, depletion of glutathione in kidney cells was shown to mimic the cumulative effect of aging (Piwien-Pilipuk et al., 2002).
Not only oxidative stress but also nitrosative stress occurs during aging and affects MR signaling. For the GR it has been shown that NO can react with SH groups to form S-nitrosothiols, which inhibit binding of GR to ligand without affecting MR-HSP90 interaction (Galigniana et al., 1999b). For the MR, NO led to a reduction of MR and GR transactivation activity upon corticosteroid treatment (Ruhs et al., 2012). In contrast, peroxynitrite administration induced ligand-independent MR transactivation, whereas genomic GR activity remained unchanged. This effect was mediated by ligand-independent nuclear translocation of peroxynitrite-treated MR (Ruhs et al., 2012). On the other hand, aldosterone via the MR can also activate NADPH oxidases in vascular cells and thereby increase ROS levels, leading to posttranslational modification of other proteins (Hashikabe et al., 2006). For instance, posttranslational modification of Cys-122 of the guanylyl cyclase can inhibit GC activity by modulating NO sensing. A C122A-GC-mutant exposed to aldosterone or hydrogen peroxide did not show increased cGMP levels like WT (Maron et al., 2009). In hPAEC, endothelin-1 increased aldosterone levels via upregulation of aldosterone synthase. Aldosterone then increases ROS production, which oxidatively modified cysteinyl thiols in the endothelial NO synthase-activating region of the endothelin-B-receptor, thereby reducing endothelin-1-mediated endothelial NO synthase activity. An MR antagonist restored Endothelin-B-receptor-mediated NO production. In vivo in two rat pulmonary arterial hypertension models, MR antagonists prevented or reversed pulmonary vascular remodeling. There elevated endothelin-1 levels were associated with elevated aldosterone levels in plasma and decreased lung NO metabolites (Maron et al., 2012).
Besides exerting classical epithelial effects on volume-and electrolyte homeostasis, the MR has a large impact on cardiovascular pathophysiology with cell-type-specific and context-dependent functions. While ensuring homeostasis in young and healthy tissues, the MR tends to aggravate pathological changes in aged or pre-injured tissues exposed to cardiovascular risk factors. It is unclear what causes the switch from physiological to pathological functions, but one possibility is posttranslational modification induced by a pathological micro-milieu. Signaling of the MR resembles that of its steroid receptor relatives and involves cytosolic association to chaperones, binding of ligand, nuclear translocation and function as a transcription factor. For the MR, many variants are known and have contributed to the understanding of MR structure and function. Loss of function mutations, which in the extreme cause pseudohypoaldosteronism type 1, and gain-of function-mutations, which give rise to inherited forms of hypertension, usually lead to structural alterations of the MR that affect its fundamental functions like binding of ligand or DNA (Zennaro and Fernandes-Rosa, 2017). Over 380 rare coding variants have been described in the major MR transcript in the Exome Aggregation Consortium (ExAC) (Lek et al., 2016). These data indicate that MR is intolerant for missense and nonsense mutations, suggesting that rare variants and common single nucleotide polymorphisms may have functional consequences in the general population and modulate disease susceptibility. Much evidence indicates that MR can be posttranslationally modified, affecting MR signaling and function. The possible impact of MR variants for its posttranslational modifications has not been assessed. Modifications described include enzymatic and non-enzymatic modifications, including phosphorylation (p), acetylation (ac), sumoylation (su), ubiquitination (ub) and oxidation (Figure 3). A direct modification of the MR was proposed for each one, although the exact molecular mechanisms, modifying enzymes and how PTMs are regulated during physiological and pathological situations is not clear. Especially, the in vivo relevance of posttranslational modifications for MR function needs further exploration for understanding the mechanisms of cardiovascular diseases and healthy aging.
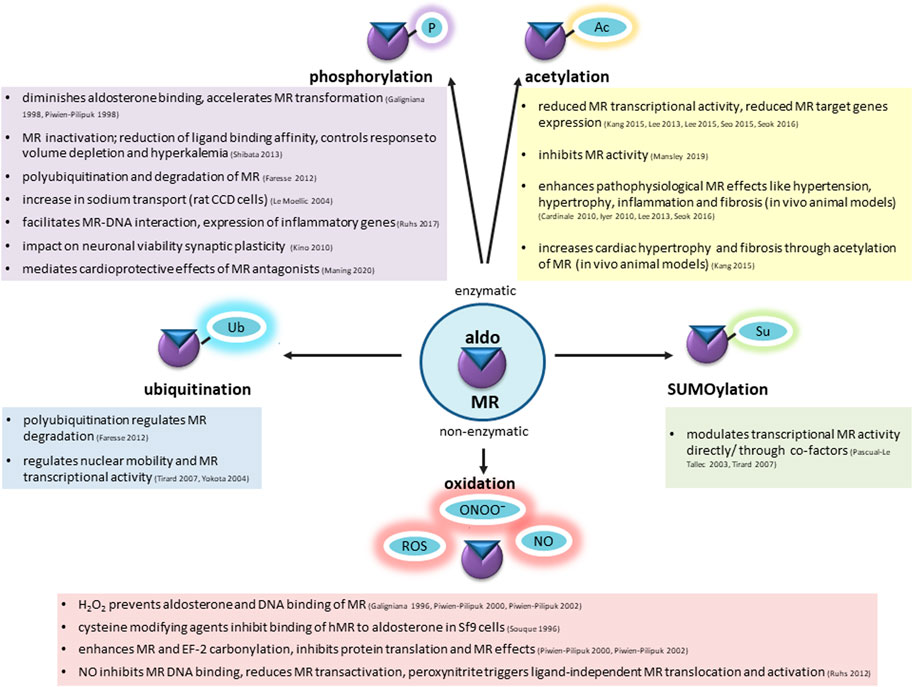
FIGURE 3. Mineralocorticoid receptor undergoes enzymatic and non-enzymatic posttranslational modifications, affecting the basal condition of the receptor, ligand binding, shuttling to the nucleus, DNA interaction and transactivation activity. Among enzymatic modifications phosphorylation (P), acetylation (Ac), ubiquitination (Ub), SUMOylation (Su) has been well-described. Increased ROS and RNS production during aging causes non-enzymatic alterations. Possible consequences are summarized in this figure.
CG wrote the manuscript. YG and AN contributed to reviewing and editing of the manuscript. YG designed the figures.
The work was supported by the Deutsche Forschungsgemeinschaft (DFG): grant GR 3415/1-5 and RTG 2155 ProMoAge.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2021.667990/full#supplementary-material
Alnemri, E. S., Maksymowych, A. B., Robertson, N. M., and Litwack, G. (1991). Overexpression and Characterization of the Human Mineralocorticoid Receptor. J. Biol. Chem. 266, 18072–18081. doi:10.1016/s0021-9258(18)55238-8
Arriza, J., Weinberger, C., Cerelli, G., Glaser, T., Handelin, B., Housman, D., et al. (1987). Cloning of Human Mineralocorticoid Receptor Complementary DNA: Structural and Functional Kinship with the Glucocorticoid Receptor. Science 237, 268–275. doi:10.1126/science.3037703
Bain, D. L., Heneghan, A. F., Connaghan-Jones, K. D., and Miura, M. T. (2007). Nuclear Receptor Structure: Implications for Function. Annu. Rev. Physiol. 69, 201–220. doi:10.1146/annurev.physiol.69.031905.160308
Baker, M. E., and Katsu, Y. (2017). 30 YEARS of the MINERALOCORTICOID RECEPTOR: Evolution of the Mineralocorticoid Receptor: Sequence, Structure and Function. J. Endocrinol. 234, T1–T16. doi:10.1530/joe-16-0661
Bakris, G. L., Agarwal, R., Anker, S. D., Pitt, B., Ruilope, L. M., Rossing, P., et al. (2020). Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N. Engl. J. Med. 383, 2219–2229. doi:10.1056/nejmoa2025845
Banerjee, A., Periyasamy, S., Wolf, I. M., Hinds, T. D., Yong, W., Shou, W., et al. (2008). Control of Glucocorticoid and Progesterone Receptor Subcellular Localization by the Ligand-Binding Domain Is Mediated by Distinct Interactions with Tetratricopeptide Repeat Proteins†. Biochemistry 47, 10471–10480. doi:10.1021/bi8011862
Bauer, J. H. (1993). Age-Related Changes in the Renin-Aldosterone System. Drugs & Aging 3, 238–245. doi:10.2165/00002512-199303030-00005
Benetos, A., Gardner, J. P., Kimura, M., Labat, C., Nzietchueng, R., Dousset, B., et al. (2005). Aldosterone and Telomere Length in White Blood Cells. Journals Gerontol. Ser. A: Biol. Sci. Med. Sci. 60, 1593–1596. doi:10.1093/gerona/60.12.1593
Binart, N., Lombès, M., and Baulieu, E. E. (1995). Distinct Functions of the 90 kDa Heat-Shock Protein (Hsp90) in Oestrogen and Mineralocorticosteroid Receptor Activity: Effects of Hsp90 Deletion Mutants. Biochem. J. 311, 797–804. doi:10.1042/bj3110797
Black, B. E., Holaska, J. M., Rastinejad, F., and Paschal, B. M. (2001). DNA Binding Domains in Diverse Nuclear Receptors Function as Nuclear export Signals. Curr. Biol. 11, 1749–1758. doi:10.1016/s0960-9822(01)00537-1
Bodary, P. F., Sambaziotis, C., Wickenheiser, K. J., Rajagopalan, S., Pitt, B., and Eitzman, D. T. (2006). Aldosterone Promotes Thrombosis Formation after Arterial Injury in Mice. Atvb 26, 233. doi:10.1161/01.atv.0000195782.07637.44
Bodwell, J. E., Ortí, E., Coull, J. M., Pappin, D. J., Smith, L. I., and Swift, F. (1991). Identification of Phosphorylated Sites in the Mouse Glucocorticoid Receptor. J. Biol. Chem. 266, 7549–7555. doi:10.1016/s0021-9258(20)89482-4
Burgdorf, S., Leister, P., and Scheidtmann, K. H. (2004). TSG101 Interacts with Apoptosis-Antagonizing Transcription Factor and Enhances Androgen Receptor-Mediated Transcription by Promoting its Monoubiquitination. J. Biol. Chem. 279, 17524–17534. doi:10.1074/jbc.m313703200
Caamano, C. A., Morano, M. I., Patel, P. D., Watson, S. J., and Akil, H. (1993). A Bacterially Expressed Mineralocorticoid Receptor Is Associated In Vitro with the 90-kilodalton Heat Shock Protein and Shows Typical Hormone- and DNA-Binding Characteristics. Biochemistry 32, 8589–8595. doi:10.1021/bi00084a028
Campino, C., Martinez-Aguayo, A., Baudrand, R., Carvajal, C. A., Aglony, M., Garcia, H., et al. (2013). Age-Related Changes in 11 -Hydroxysteroid Dehydrogenase Type 2 Activity in Normotensive Subjects. Am. J. Hypertens. 26, 481–487. doi:10.1093/ajh/hps080
Caprio, M., Newfell, B. G., la Sala, A., Baur, W., Fabbri, A., Rosano, G., et al. (2008). Functional Mineralocorticoid Receptors in Human Vascular Endothelial Cells Regulate Intercellular Adhesion Molecule-1 Expression and Promote Leukocyte Adhesion. Circ. Res. 102, 1359–1367. doi:10.1161/circresaha.108.174235
Cardinale, J. P., Sriramula, S., Pariaut, R., Guggilam, A., Mariappan, N., Elks, C. M., et al. (2010). HDAC Inhibition Attenuates Inflammatory, Hypertrophic, and Hypertensive Responses in Spontaneously Hypertensive Rats. Hypertension 56, 437–444. doi:10.1161/hypertensionaha.110.154567
Chen, L., Ding, M.-L., Wu, F., He, W., Li, J., Zhang, X.-Y., et al. (2016). Impaired Endothelial Repair Capacity of Early Endothelial Progenitor Cells in Hypertensive Patients with Primary Hyperaldosteronemia. Hypertension 67, 430–439. doi:10.1161/hypertensionaha.115.06597
Dabrowski, R., Borowiec, A., Smolis-Bak, E., Kowalik, I., Sosnowski, C., Kraska, A., et al. (2010). Effect of Combined Spironolactone-β-Blocker ± Enalapril Treatment on Occurrence of Symptomatic Atrial Fibrillation Episodes in Patients with a History of Paroxysmal Atrial Fibrillation (SPIR-AF Study). Am. J. Cardiol. 106, 1609–1614. doi:10.1016/j.amjcard.2010.07.037
Deroo, B. J., Rentsch, C., Sampath, S., Young, J., DeFranco, D. B., and Archer, T. K. (2002). Proteasomal Inhibition Enhances Glucocorticoid Receptor Transactivation and Alters its Subnuclear Trafficking. Mcb 22, 4113–4123. doi:10.1128/mcb.22.12.4113-4123.2002
Deuchar, G. A., McLean, D., Hadoke, P. W. F., Brownstein, D. G., Webb, D. J., Mullins, J. J., et al. (2011). 11β-Hydroxysteroid Dehydrogenase Type 2 Deficiency Accelerates Atherogenesis and Causes Proinflammatory Changes in the Endothelium in Apoe−/− Mice. Endocrinology 152, 236–246. doi:10.1210/en.2010-0925
Drouin, J., Sun, Y. L., Tremblay, S., Lavender, P., Schmidt, T. J., de Léan, A., et al. (1992). Homodimer Formation Is Rate-Limiting for High Affinity DNA Binding by Glucocorticoid Receptor. Mol. Endocrinol. 6, 1299–1309. doi:10.1210/mend.6.8.1406707
Drüppel, V., Kusche-Vihrog, K., Grossmann, C., Gekle, M., Kasprzak, B., Brand, E., et al. (2013). Long-term Application of the Aldosterone Antagonist Spironolactone Prevents Stiff Endothelial Cell Syndrome. FASEB J. 27, 3652–3659. doi:10.1096/fj.13-228312
DuPont, J. J., Hill, M. A., Bender, S. B., Jaisser, F., and Jaffe, I. Z. (2014). Aldosterone and Vascular Mineralocorticoid Receptors. Hypertension 63, 632–637. doi:10.1161/hypertensionaha.113.01273
DuPont, J. J., McCurley, A., Davel, A. P., McCarthy, J., Bender, S. B., Hong, K., et al. (2016). Vascular Mineralocorticoid Receptor Regulates microRNA-155 to Promote Vasoconstriction and Rising Blood Pressure with Aging. JCI Insight 1, e88942. doi:10.1172/jci.insight.88942
Faresse, N., Ruffieux-Daidie, D., Salamin, M., Gomez-Sanchez, C. E., and Staub, O. (2010). Mineralocorticoid Receptor Degradation Is Promoted by Hsp90 Inhibition and the Ubiquitin-Protein Ligase CHIP. Am. J. Physiology-Renal Physiol. 299, F1462–F1472. doi:10.1152/ajprenal.00285.2010
Faresse, N., Vitagliano, J. J., and Staub, O. (2012). Differential Ubiquitylation of the Mineralocorticoid Receptor Is Regulated by Phosphorylation. FASEB j. 26, 4373–4382. doi:10.1096/fj.12-209924
Faresse, N., Debonneville, A., and Staub, O. (2013). USP2-45 Represses Aldosterone Mediated Responses by Decreasing Mineralocorticoid Receptor Availability. Cell Physiol. Biochem. 31, 462–472. doi:10.1159/000343382
Farman, N., and Rafestin-Oblin, M.-E. (2001). Multiple Aspects of Mineralocorticoid Selectivity. Am. J. Physiology-Renal Physiol. 280, F181–F192. doi:10.1152/ajprenal.2001.280.2.f181
Farquharson, C. A. J., and Struthers, A. D. (2000). Spironolactone Increases Nitric Oxide Bioactivity, Improves Endothelial Vasodilator Dysfunction, and Suppresses Vascular Angiotensin I/Angiotensin II Conversion in Patients with Chronic Heart Failure. Circulation 101, 594–597. doi:10.1161/01.cir.101.6.594
Faus, H., and Haendler, B. (2006). Post-translational Modifications of Steroid Receptors. Biomed. Pharmacother. 60, 520–528. doi:10.1016/j.biopha.2006.07.082
Fejes-Toth, G., Pearce, D., and Naray-Fejes-Toth, A. (1998). Subcellular Localization of Mineralocorticoid Receptors in Living Cells: Effects of Receptor Agonists and Antagonists. Proc. Natl. Acad. Sci. 95, 2973–2978. doi:10.1073/pnas.95.6.2973
Fels, J., Callies, C., Kusche-Vihrog, K., and Oberleithner, H. (2010). Nitric Oxide Release Follows Endothelial Nanomechanics and Not Vice Versa. Pflugers Arch. - Eur. J. Physiol. 460, 915–923. doi:10.1007/s00424-010-0871-8
Filippatos, G., Anker, S. D., Agarwal, R., Pitt, B. G., Ruilope, L. M., Rossing, P., et al. (2021). Finerenone and Cardiovascular Outcomes in Patients With Chronic Kidney Disease and Type 2 Diabetes. Circulation 540–552. doi:10.1161/CIRCULATIONAHA.120.051898
Freeman, B. C., and Yamamoto, K. R. (2002). Disassembly of Transcriptional Regulatory Complexes by Molecular Chaperones. Science 296, 2232–2235. doi:10.1126/science.1073051
Fudim, M., Liu, P. R., Shrader, P., Blanco, R. G., Allen, L. A., Fonarow, G. C., et al. (2018). Mineralocorticoid Receptor Antagonism in Patients with Atrial Fibrillation: Findings from the ORBIT-AF (Outcomes Registry for Better Informed Treatment of Atrial Fibrillation) Registry. J. Am. Heart Assoc. 7, e007987. doi:10.1161/jaha.117.007987
Fuse, H., Kitagawa, H., and Kato, S. (2000). Characterization of Transactivational Property and Coactivator Mediation of Rat Mineralocorticoid Receptor Activation Function-1 (AF-1). Mol. Endocrinol. 14, 889–899. doi:10.1210/mend.14.6.0467
Galigniana, M. D., and Piwien-Pilipuk, G. (1999a). Comparative Inhibition by Hard and Soft Metal Ions of Steroid-Binding Capacity of Renal Mineralocorticoid Receptor Cross-Linked to the 90-kDa Heat-Shock Protein Heterocomplex. Biochem. J. 341 (Pt 3), 585–592. doi:10.1042/0264-6021:3410585
Galigniana, M. D., Piwien-Pilipuk, G., and Assreuy, J. (1999b). Inhibition of Glucocorticoid Receptor Binding by Nitric Oxide. Mol. Pharmacol. 55, 317–323. doi:10.1124/mol.55.2.317
Galigniana, M. D., Erlejman, A. G., Monte, M., Gomez-Sanchez, C., and Piwien-Pilipuk, G. (2010). The Hsp90-FKBP52 Complex Links the Mineralocorticoid Receptor to Motor Proteins and Persists Bound to the Receptor in Early Nuclear Events. Mcb 30, 1285–1298. doi:10.1128/mcb.01190-09
Galigniana, M. D. (1996). Stability Study on Renal Type I Mineralocorticoid Receptor. Life Sci. 59, 511–521. doi:10.1016/0024-3205(96)00331-1
Galigniana, M. D. (1998). Native Rat Kidney Mineralocorticoid Receptor Is a Phosphoprotein Whose Transformation to a DNA-Binding Form Is Induced by Phosphatases. Biochem. J. 333 (Pt 3), 555–563. doi:10.1042/bj3330555
Garg, R., Rao, A. D., Baimas-George, M., Hurwitz, S., Foster, C., Shah, R. V., et al. (2015). Mineralocorticoid Receptor Blockade Improves Coronary Microvascular Function in Individuals with Type 2 Diabetes. Diabetes 64, 236–242. doi:10.2337/db14-0670
Gromotowicz, A., Szemraj, J., Stankiewicz, A., Zakrzeska, A., Mantur, M., Jaroszewicz, E., et al. (2011). Study of the Mechanisms of Aldosterone Prothrombotic Effect in Rats. J. Renin Angiotensin Aldosterone Syst. 12, 430–439. doi:10.1177/1470320310397405
Grossmann, C., Freudinger, R., Mildenberger, S., Krug, A. W., and Gekle, M. (2004). Evidence for Epidermal Growth Factor Receptor as Negative-Feedback Control in Aldosterone-Induced Na+ Reabsorption. Am. J. Physiology-Renal Physiol. 286, F1226–F1231. doi:10.1152/ajprenal.00378.2003
Grossmann, C., Benesic, A., Krug, A. W., Freudinger, R., Mildenberger, S., Gassner, B., et al. (2005). Human Mineralocorticoid Receptor Expression Renders Cells Responsive for Nongenotropic Aldosterone Actions. Mol. Endocrinol. 19, 1697–1710. doi:10.1210/me.2004-0469
Grossmann, C., Ruhs, S., Langenbruch, L., Mildenberger, S., Strätz, N., Schumann, K., et al. (2012). Nuclear Shuttling Precedes Dimerization in Mineralocorticoid Receptor Signaling. Chem. Biol. 19, 742–751. doi:10.1016/j.chembiol.2012.04.014
Hashikabe, Y., Suzuki, K., Jojima, T., Uchida, K., and Hattori, Y. (2006). Aldosterone Impairs Vascular Endothelial Cell Function. J. Cardiovasc. Pharmacol. 47, 609–613. doi:10.1097/01.fjc.0000211738.63207.c3
Hayashi, T., Shibata, H., Kurihara, I., Yokota, K., Mitsuishi, Y., Ohashi, K., et al. (2017). High Glucose Stimulates Mineralocorticoid Receptor Transcriptional Activity through the Protein Kinase C β Signaling. Int. Heart J. 58, 794–802. doi:10.1536/ihj.16-649
Hellal-Levy, C., Fagart, J., Souque, A., Wurtz, J.-M., Moras, D., and Rafestin-Oblin, M.-E. (2000). Crucial Role of the H11-H12 Loop in Stabilizing the Active Conformation of the Human Mineralocorticoid Receptor. Mol. Endocrinol. 14, 1210–1221. doi:10.1210/mend.14.8.0502
Henschkowski, J., Stuck, A. E., Frey, B. M., Gillmann, G., Dick, B., Frey, F. J., et al. (2008). Age-dependent Decrease in 11 -Hydroxysteroid Dehydrogenase Type 2 (11 -HSD2) Activity in Hypertensive Patients. Am. J. Hypertens. 21, 644–649. doi:10.1038/ajh.2008.152
Hernández-Díaz, I., Giraldez, T., Arnau, M. R., Smits, V. A., Jaisser, F., Farman, N., et al. (2010). The Mineralocorticoid Receptor Is a Constitutive Nuclear Factor in Cardiomyocytes Due to Hyperactive Nuclear Localization Signals. Endocrinology 151, 3888–3899. doi:10.1210/en.2010-0099
Hirschberg, D., Jägerbrink, T., Samskog, J., Gustafsson, M., Ståhlberg, M., Alvelius, G., et al. (2004). Detection of Phosphorylated Peptides in Proteomic Analyses Using Microfluidic Compact Disk Technology. Anal. Chem. 76, 5864–5871. doi:10.1021/ac040044g
Hong, H., Kohli, K., Garabedian, M. J., and Stallcup, M. R. (1997). GRIP1, a Transcriptional Coactivator for the AF-2 Transactivation Domain of Steroid, Thyroid, Retinoid, and Vitamin D Receptors. Mol. Cel. Biol. 17, 2735–2744. doi:10.1128/mcb.17.5.2735
Hwang, M.-H., Yoo, J.-K., Luttrell, M., Kim, H.-K., Meade, T. H., English, M., et al. (2016). Acute Effect of Mineralocorticoid Receptor Antagonism on Vascular Function in Healthy Older Adults. Exp. Gerontol. 73, 86–94. doi:10.1016/j.exger.2015.11.017
Ismaili, N., Blind, R., and Garabedian, M. J. (2005). Stabilization of the Unliganded Glucocorticoid Receptor by TSG101. J. Biol. Chem. 280, 11120–11126. doi:10.1074/jbc.m500059200
Ito, K., Yamamura, S., Essilfie-Quaye, S., Cosio, B., Ito, M., Barnes, P. J., et al. (2006). Histone Deacetylase 2-mediated Deacetylation of the Glucocorticoid Receptor Enables NF-Κb Suppression. J. Exp. Med. 203, 7–13. doi:10.1084/jem.20050466
Iwashima, F., Yoshimoto, T., Minami, I., Sakurada, M., Hirono, Y., and Hirata, Y. (2008). Aldosterone Induces Superoxide Generation via Rac1 Activation in Endothelial Cells. Endocrinology 149, 1009–1014. doi:10.1210/en.2007-0864
Iyer, A., Fenning, A., Lim, J., Le, G. T., Reid, R. C., Halili, M. A., et al. (2010). Antifibrotic Activity of an Inhibitor of Histone Deacetylases in DOCA-Salt Hypertensive Rats. Br. J. Pharmacol. 159, 1408–1417. doi:10.1111/j.1476-5381.2010.00637.x
Jausonsloffreda, N., Chabret, C., and Pons, M. (1994). Role of the A/B Region of the Human Mineralocorticoid Receptor in Aldosterone Response Selectivity. Biochem. Biophysical Res. Commun. 205, 1610–1616. doi:10.1006/bbrc.1994.2851
Jia, G., Habibi, J., Aroor, A. R., Martinez-Lemus, L. A., Demarco, V. G., Ramirez-Perez, F. I., et al. (2016). Endothelial Mineralocorticoid Receptor Mediates Diet-Induced Aortic Stiffness in Females. Circ. Res. 118, 935–943. doi:10.1161/circresaha.115.308269
Jiménez-Canino, R., Fernandes, M. X., and de la Rosa, D. A. (2016a). Phosphorylation of Mineralocorticoid Receptor Ligand Binding Domain Impairs Receptor Activation and Has a Dominant Negative Effect over Non-phosphorylated Receptors. J. Biol. Chem. 291, 19068–19078. doi:10.1074/jbc.m116.718395
Jiménez-Canino, R., Lorenzo-Díaz, F., Jaisser, F., Farman, N., Giraldez, T., and Alvarez de la Rosa, D. (2016b). Histone Deacetylase 6-Controlled Hsp90 Acetylation Significantly Alters Mineralocorticoid Receptor Subcellular Dynamics but Not its Transcriptional Activity. Endocrinology 157, 2515–2532. doi:10.1210/en.2015-2055
Jiménez-Canino, R., Lorenzo-Díaz, F., Odermatt, A., Bailey, M. A., Livingstone, D. E. W., Jaisser, F., et al. (2017). 11β-HSD2 SUMOylation Modulates Cortisol-Induced Mineralocorticoid Receptor Nuclear Translocation Independently of Effects on Transactivation. Endocrinology 158, 4047–4063. doi:10.1210/en.2017-00440
Joss, J. M. P., Arnold-Reed, D. E., and Balment, R. J. (1994). The Steroidogenic Response to Angiotensin II in the Australian Lungfish, Neoceratodus forsteri. J. Comp. Physiol. B 164, 378–382. doi:10.1007/bf00302553
Kang, K. I., Devin, J., Cadepond, F., Jibard, N., Guiochon-Mantel, A., Baulieu, E. E., et al. (1994). In Vivo functional Protein-Protein Interaction: Nuclear Targeted Hsp90 Shifts Cytoplasmic Steroid Receptor Mutants into the Nucleus. Proc. Natl. Acad. Sci. 91, 340–344. doi:10.1073/pnas.91.1.340
Kang, K. I., Meng, X., Devin-Leclerc, J., Bouhouche, I., Chadli, A., Cadepond, F., et al. (1999). The Molecular Chaperone Hsp90 Can Negatively Regulate the Activity of a Glucocorticosteroid-dependent Promoter. Proc. Natl. Acad. Sci. 96, 1439–1444. doi:10.1073/pnas.96.4.1439
Kang, S.-H., Seok, Y. M., Song, M.-j., Lee, H.-A., Kurz, T., and Kim, I. (2015). Histone Deacetylase Inhibition Attenuates Cardiac Hypertrophy and Fibrosis through Acetylation of Mineralocorticoid Receptor in Spontaneously Hypertensive Rats. Mol. Pharmacol. 87, 782–791. doi:10.1124/mol.114.096974
Keidar, S., Gamliel-Lazarovich, A., Kaplan, M., Pavlotzky, E., Hamoud, S., Hayek, T., et al. (2005). Mineralocorticoid Receptor Blocker Increases Angiotensin-Converting Enzyme 2 Activity in Congestive Heart Failure Patients. Circ. Res. 97, 946–953. doi:10.1161/01.res.0000187500.24964.7a
Kim, S. K., McCurley, A. T., DuPont, J. J., Aronovitz, M., Moss, M. E., Stillman, I. E., et al. (2018). Smooth Muscle Cell-Mineralocorticoid Receptor as a Mediator of Cardiovascular Stiffness with Aging. Hypertension 71, 609–621. doi:10.1161/hypertensionaha.117.10437
Kino, T., Liou, S.-H., Charmandari, E., and Chrousos, G. P. (2004). Glucocorticoid Receptor Mutants Demonstrate Increased Motility inside the Nucleus of Living Cells: Time of Fluorescence Recovery after Photobleaching (FRAP) Is an Integrated Measure of Receptor Function. Mol. Med. 10, 80–88. doi:10.2119/2005-00026.kino
Kino, T., Ichijo, T., Amin, N. D., Kesavapany, S., Wang, Y., Kim, N., et al. (2007). Cyclin-Dependent Kinase 5 Differentially Regulates the Transcriptional Activity of the Glucocorticoid Receptor through Phosphorylation: Clinical Implications for the Nervous System Response to Glucocorticoids and Stress. Mol. Endocrinol. 21, 1552–1568. doi:10.1210/me.2006-0345
Kino, T., Jaffe, H., Amin, N. D., Chakrabarti, M., Zheng, Y.-L., Chrousos, G. P., et al. (2010). Cyclin-dependent Kinase 5 Modulates the Transcriptional Activity of the Mineralocorticoid Receptor and Regulates Expression of Brain-Derived Neurotrophic Factor. Mol. Endocrinol. 24, 941–952. doi:10.1210/me.2009-0395
Kovacs, J. J., Murphy, P. J. M., Gaillard, S., Zhao, X., Wu, J.-T., Nicchitta, C. V., et al. (2005). HDAC6 Regulates Hsp90 Acetylation and Chaperone-Dependent Activation of Glucocorticoid Receptor. Mol. Cel 18, 601–607. doi:10.1016/j.molcel.2005.04.021
Krug, A. W., Allenhofer, L., Monticone, R., Spinetti, G., Gekle, M., Wang, M., et al. (2010). Elevated Mineralocorticoid Receptor Activity in Aged Rat Vascular Smooth Muscle Cells Promotes a Proinflammatory Phenotype via Extracellular Signal-Regulated Kinase 1/2 Mitogen-Activated Protein Kinase and Epidermal Growth Factor Receptor-dependent Pathways. Hypertension 55, 1476–1483. doi:10.1161/hypertensionaha.109.148783
Kusche-Vihrog, K., Sobczak, K., Bangel, N., Wilhelmi, M., Nechyporuk-Zloy, V., Schwab, A., et al. (2008). Aldosterone and Amiloride Alter ENaC Abundance in Vascular Endothelium. Pflugers Arch. - Eur. J. Physiol. 455, 849–857. doi:10.1007/s00424-007-0341-0
La Rosa, P., Pesiri, V., Marino, M., and Acconcia, F. (2011). 17β-Estradiol-induced Cell Proliferation Requires Estrogen Receptor (ER) α Monoubiquitination. Cell Signal. 23, 1128–1135. doi:10.1016/j.cellsig.2011.02.006
Lagrange, J., Li, Z., Fassot, C., Bourhim, M., Louis, H., Cat, A. N. D., et al. (2014). Endothelial Mineralocorticoid Receptor Activation Enhances Endothelial Protein C Receptor and Decreases Vascular Thrombosis in Mice. FASEB j. 28, 2062–2072. doi:10.1096/fj.13-238188
Le Moëllic, C., Ouvrard-Pascaud, A., Capurro, C., Cluzeaud, F., Fay, M., Jaisser, F., et al. (2004). Early Nongenomic Events in Aldosterone Action in Renal Collecting Duct Cells: PKCalpha Activation, Mineralocorticoid Receptor Phosphorylation, and Cross-Talk with the Genomic Response. J. Am. Soc. Nephrol. 15, 1145–1160.
Lee, H.-A., Lee, D.-Y., Cho, H.-M., Kim, S.-Y., Iwasaki, Y., and Kim, I. K. (2013). Histone Deacetylase Inhibition Attenuates Transcriptional Activity of Mineralocorticoid Receptor through its Acetylation and Prevents Development of Hypertension. Circ. Res. 112, 1004–1012. doi:10.1161/circresaha.113.301071
Lee, H. A., Song, M. J., Seok, Y. M., Kang, S. H., Kim, S. Y., and Kim, I. (2015). Histone Deacetylase 3 and 4 Complex Stimulates the Transcriptional Activity of the Mineralocorticoid Receptor. PLOS ONE 10, e0136801. doi:10.1371/journal.pone.0136801
Lek, M., Karczewski, K. J., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., et al. (2016). Analysis of Protein-Coding Genetic Variation in 60,706 Humans. Nature 536, 285–291. doi:10.1038/nature19057
Leopold, J. A., Dam, A., Maron, B. A., Scribner, A. W., Liao, R., Handy, D. E., et al. (2007). Aldosterone Impairs Vascular Reactivity by Decreasing Glucose-6-Phosphate Dehydrogenase Activity. Nat. Med. 13, 189–197. doi:10.1038/nm1545
Liu, W., Wang, J., Sauter, N. K., and Pearce, D. (1995). Steroid Receptor Heterodimerization Demonstrated In Vitro and In Vivo. Proc. Natl. Acad. Sci. 92, 12480–12484. doi:10.1073/pnas.92.26.12480
Lu, Q., Davel, A. P., McGraw, A. P., Rao, S. P., Newfell, B. G., and Jaffe, I. Z. (2019). PKCdelta Mediates Mineralocorticoid Receptor Activation by Angiotensin II to Modulate Smooth Muscle Cell Function. Endocrinology 160, 2101–2114. doi:10.1210/en.201
Madan, A. P., and DeFranco, D. B. (1993). Bidirectional Transport of Glucocorticoid Receptors across the Nuclear Envelope. Proc. Natl. Acad. Sci. 90, 3588–3592. doi:10.1073/pnas.90.8.3588
Maning, J., McCrink, K. A., Pollard, C. M., Desimine, V. L., Ghandour, J., Perez, A., et al. (2020). Antagonistic Roles of GRK2 and GRK5 in Cardiac Aldosterone Signaling Reveal GRK5-Mediated Cardioprotection via Mineralocorticoid Receptor Inhibition. Ijms 21, 2868. doi:10.3390/ijms21082868
Mansley, M. K., Roe, A. J., Francis, S. L., Gill, J. H., Bailey, M. A., and Wilson, S. M. (2019). TrichostatinAblocks Aldosterone‐induced Na+transport and Control of Serum‐ and Glucocorticoid‐inducible Kinase 1 in Cortical Collecting Duct Cells. Br. J. Pharmacol. 176, 4708–4719. doi:10.1111/bph.14837
Maron, B. A., Zhang, Y.-Y., Handy, D. E., Beuve, A., Tang, S.-S., Loscalzo, J., et al. (2009). Aldosterone Increases Oxidant Stress to Impair Guanylyl Cyclase Activity by Cysteinyl Thiol Oxidation in Vascular Smooth Muscle Cells. J. Biol. Chem. 284, 7665–7672. doi:10.1074/jbc.m809460200
Maron, B. A., Zhang, Y.-Y., White, K., Chan, S. Y., Handy, D. E., Mahoney, C. E., et al. (2012). Aldosterone Inactivates the Endothelin-B Receptor via a Cysteinyl Thiol Redox Switch to Decrease Pulmonary Endothelial Nitric Oxide Levels and Modulate Pulmonary Arterial Hypertension. Circulation 126, 963–974. doi:10.1161/circulationaha.112.094722
Massaad, C., Houard, N., Lombès, M., and Barouki, R. (1999). Modulation of Human Mineralocorticoid Receptor Function by Protein Kinase A. Mol. Endocrinol. 13, 57–65. doi:10.1210/mend.13.1.0226
McCurley, A., Pires, P. W., Bender, S. B., Aronovitz, M., Zhao, M. J., Metzger, D., et al. (2012). Direct Regulation of Blood Pressure by Smooth Muscle Cell Mineralocorticoid Receptors. Nat. Med. 18, 1429–1433. doi:10.1038/nm.2891
Merrill, M., Sweitzer, N. K., Lindenfeld, J., and Kao, D. P. (2019). Sex Differences in Outcomes and Responses to Spironolactone in Heart Failure with Preserved Ejection Fraction. JACC: Heart Fail. 7, 228–238. doi:10.1016/j.jchf.2019.01.003
Migliaccio, A., Lastoria, S., Moncharmont, B., Rotondi, A., and Auricchio, F. (1982). Phosphorylation of Calf Uterus 17β-Estradiol Receptor by Endogenous Ca2+-Stimulated Kinase Activating the Hormone Binding of the Receptor. Biochem. Biophysical Res. Commun. 109, 1002–1010. doi:10.1016/0006-291x(82)92039-3
Minucci, S., and Pelicci, P. G. (2006). Histone Deacetylase Inhibitors and the Promise of Epigenetic (And More) Treatments for Cancer. Nat. Rev. Cancer 6, 38–51. doi:10.1038/nrc1779
Mitsuishi, Y., Shibata, H., Kurihara, I., Kobayashi, S., Yokota, K., Murai-Takeda, A., et al. (2018). Epidermal Growth Factor Receptor/extracellular Signal-Regulated Kinase Pathway Enhances Mineralocorticoid Receptor Transcriptional Activity through Protein Stabilization. Mol. Cell Endocrinol. 473, 89–99. doi:10.1016/j.mce.2018.01.007
Morrison, N., Harrap, S. B., Arriza, J. L., Boyd, E., and Connor, J. M. (1990). Regional Chromosomal Assignment of the Human Mineralocorticoid Receptor Gene to 4q31.1. Hum. Genet. 85, 130–132. doi:10.1007/bf00276340
Moss, M. E., Lu, Q., Iyer, S. L., Engelbertsen, D., Marzolla, V., Caprio, M., et al. (2019). Endothelial Mineralocorticoid Receptors Contribute to Vascular Inflammation in Atherosclerosis in a Sex-Specific Manner. Atvb 39, 1588–1601. doi:10.1161/atvbaha.119.312954
Murphy, P. J. M., Morishima, Y., Kovacs, J. J., Yao, T.-P., and Pratt, W. B. (2005). Regulation of the Dynamics of Hsp90 Action on the Glucocorticoid Receptor by Acetylation/Deacetylation of the Chaperone. J. Biol. Chem. 280, 33792–33799. doi:10.1074/jbc.m506997200
Nader, N., Chrousos, G. P., and Kino, T. (2009). Circadian Rhythm Transcription Factor CLOCK Regulates the Transcriptional Activity of the Glucocorticoid Receptor by Acetylating its Hinge Region Lysine Cluster: Potential Physiological Implications. FASEB j. 23, 1572–1583. doi:10.1096/fj.08-117697
Nagarajan, S., Vohra, T., Loffing, J., and Faresse, N. (2017). Protein Phosphatase 1α Enhances Renal Aldosterone Signaling via Mineralocorticoid Receptor Stabilization. Mol. Cell Endocrinol. 450, 74–82. doi:10.1016/j.mce.2017.04.020
Nagata, D., Takahashi, M., Sawai, K., Tagami, T., Usui, T., Shimatsu, A., et al. (2006). Molecular Mechanism of the Inhibitory Effect of Aldosterone on Endothelial NO Synthase Activity. Hypertension 48, 165–171. doi:10.1161/01.hyp.0000226054.53527.bb
Nemoto, T., Ohara-Nemoto, Y., Sato, N., and Ota, M. (1993). Dual Roles of 90-kDa Heat Shock Protein in the Function of the Mineralocorticoid Receptor. J. Biochem. 113, 769–775.
Nietlispach, F., Julius, B., Schindler, R., Bernheim, A., Binkert, C., Kiowski, W., et al. (2007). Influence of Acute and Chronic Mineralocorticoid Excess on Endothelial Function in Healthy Men. Hypertension 50, 82–88. doi:10.1161/hypertensionaha.107.088955
Nishi, M., Ogawa, H., Ito, T., Matsuda, K.-I., and Kawata, M. (2001). Dynamic Changes in Subcellular Localization of Mineralocorticoid Receptor in Living Cells: In Comparison with Glucocorticoid Receptor Using Dual-Color Labeling with Green Fluorescent Protein Spectral Variants. Mol. Endocrinol. 15, 1077–1092. doi:10.1210/mend.15.7.0659
Nishi, M., Tanaka, M., Matsuda, K. i., Sunaguchi, M., and Kawata, M. (2004). Visualization of Glucocorticoid Receptor and Mineralocorticoid Receptor Interactions in Living Cells with GFP-Based Fluorescence Resonance Energy Transfer. J. Neurosci. 24, 4918–4927. doi:10.1523/jneurosci.5495-03.2004
Noth, R. H., Lassman, M. N., Tan, S. Y., Fernandez-Cruz, A., and Mulrow, P. J. (1977). Age and the Renin-Aldosterone System. Arch. Intern. Med. 137, 1414–1417. doi:10.1001/archinte.137.10.1414
Pascual-Le Tallec, L., and Lombès, M. (2005). The Mineralocorticoid Receptor: A Journey Exploring its Diversity and Specificity of Action. Mol. Endocrinol. 19, 2211–2221. doi:10.1210/me.2005-0089
Pascual-Le Tallec, L., Kirsh, O., Lecomte, M. C., Viengchareun, M.-C., Zennaro, M. C., Dejean, M.-C., et al. (2003). Protein Inhibitor of Activated Signal Transducer and Activator of Transcription 1 Interacts with the N-Terminal Domain of Mineralocorticoid Receptor and Represses its Transcriptional Activity: Implication of Small Ubiquitin-Related Modifier 1 Modification. Mol. Endocrinol. 17, 2529–2542. doi:10.1210/me.2003-0299
Peter, J. F., Jun, Y., and Morag, J. Y. (2017). 30 YEARS of the MINERALOCORTICOID RECEPTOR: Coregulators as Mediators of Mineralocorticoid Receptor Signalling Diversity. J. Endocrinol. 234, T23–T34. doi:10.1530/JOE-17-0060
Pfeffer, M. A., Claggett, B., Assmann, S. F., Boineau, R., Anand, I. S., Clausell, N., et al. (2015). Regional Variation in Patients and Outcomes in the Treatment of Preserved Cardiac Function Heart Failure with an Aldosterone Antagonist (TOPCAT) Trial. Circulation 131, 34–42. doi:10.1161/circulationaha.114.013255
Picard, D., and Yamamoto, K. R. (1987). Two Signals Mediate Hormone-dependent Nuclear Localization of the Glucocorticoid Receptor. EMBO J. 6, 3333–3340. doi:10.1002/j.1460-2075.1987.tb02654.x
Pitt, B., Reichek, N., Willenbrock, R., Zannad, F., Phillips, R. A., Roniker, B., et al. (2003). Effects of Eplerenone, Enalapril, and Eplerenone/Enalapril in Patients with Essential Hypertension and Left Ventricular Hypertrophy. Circulation 108, 1831–1838. doi:10.1161/01.cir.0000091405.00772.6e
Pitt, B., Pfeffer, M. A., Assmann, S. F., Boineau, R., Anand, I. S., Claggett, B., et al. (2014). Spironolactone for Heart Failure with Preserved Ejection Fraction. N. Engl. J. Med. 370, 1383–1392. doi:10.1056/nejmoa1313731
Pitt, B. (2012). The Role of Mineralocorticoid Receptor Antagonists (MRAs) in Very Old Patients with Heart Failure. Heart Fail. Rev. 17, 573–579. doi:10.1007/s10741-011-9286-7
Piwien Pilipuk, G., Vinson, G. P., Gomez Sanchez, C., and Galigniana, M. D. (2007). Evidence for NL1-independent Nuclear Translocation of the Mineralocorticoid Receptor†. Biochemistry 46, 1389–1397. doi:10.1021/bi0621819
Piwien-Pilipuk, G., and Galigniana, M. D. (1998). Tautomycin Inhibits Phosphatase-dependent Transformation of the Rat Kidney Mineralocorticoid Receptor. Mol. Cell Endocrinol. 144, 119–130. doi:10.1016/s0303-7207(98)00142-7
Piwien-Pilipuk, G., and Galigniana, M. D. (2000). Oxidative Stress Induced by L-buthionine-(S,R)-sulfoximine, a Selective Inhibitor of Glutathione Metabolism, Abrogates Mouse Kidney Mineralocorticoid Receptor Function. Biochim. Biophys. Acta (Bba) - Mol. Cel Res. 1495, 263–280. doi:10.1016/s0167-4889(99)00166-4
Piwien-Pilipuk, G., Ayala, A., Machado, A., and Galigniana, M. D. (2002). Impairment of Mineralocorticoid Receptor (MR)-dependent Biological Response by Oxidative Stress and Aging. J. Biol. Chem. 277, 11896–11903. doi:10.1074/jbc.m109530200
Poukka, H., Karvonen, U., Janne, O. A., and Palvimo, J. J. (2000). Covalent Modification of the Androgen Receptor by Small Ubiquitin-like Modifier 1 (SUMO-1). Proc. Natl. Acad. Sci. 97, 14145–14150. doi:10.1073/pnas.97.26.14145
Rossier, B. C., Baker, M. E., and Studer, R. A. (2015). Epithelial Sodium Transport and its Control by Aldosterone: The Story of Our Internal Environment Revisited. Physiol. Rev. 95, 297–340. doi:10.1152/physrev.00011.2014
Ruhs, S., Strätz, N., Schlör, K., Meinel, S., Mildenberger, S., Rabe, S., et al. (2012). Modulation of Transcriptional Mineralocorticoid Receptor Activity by Nitrosative Stress. Free Radic. Biol. Med. 53, 1088–1100. doi:10.1016/j.freeradbiomed.2012.06.028
Ruhs, S., Straetz, N., Quarch, K., Masch, A., Schutkowski, M., Gekle, M., et al. (2017). Modulation of Transcriptional Mineralocorticoid Receptor Activity by Casein Kinase 2. Sci. Rep. 7, 15340. doi:10.1038/s41598-017-15418-1
Sanchez, E. R., Toft, D. O., Schlesinger, M. J., and Pratt, W. B. (1985). Evidence that the 90-kDa Phosphoprotein Associated with the Untransformed L-Cell Glucocorticoid Receptor Is a Murine Heat Shock Protein. J. Biol. Chem. 260, 12398–12401. doi:10.1016/s0021-9258(17)38886-5
Saporita, A. J., Zhang, Q., Navai, N., Dincer, Z., Hahn, J., Cai, X., et al. (2003). Identification and Characterization of a Ligand-Regulated Nuclear Export Signal in Androgen Receptor. J. Biol. Chem. 278, 41998–42005. doi:10.1074/jbc.m302460200
Sartorato, P., Cluzeaud, F., Fagart, J., Viengchareun, S., Lombes, M., and Zennaro, M.-C. (2004). New Naturally Occurring Missense Mutations of the Human Mineralocorticoid Receptor Disclose Important Residues Involved in Dynamic Interactions with Deoxyribonucleic Acid, Intracellular Trafficking, and Ligand Binding. Mol. Endocrinol. 18, 2151–2165. doi:10.1210/me.2003-0408
Savory, J. G. A., Prefontaine, G. G., Lamprecht, C., Liao, M., Walther, R. F., Lefebvre, Y. A., et al. (2001). Glucocorticoid Receptor Homodimers and Glucocorticoid-Mineralocorticoid Receptor Heterodimers Form in the Cytoplasm through Alternative Dimerization Interfaces. Mol. Cel. Biol. 21, 781–793. doi:10.1128/mcb.21.3.781-793.2001
Schmidt, B. M. W., Oehmer, S., Delles, C., Bratke, R., Schneider, M. P., Klingbeil, A., et al. (2003). Rapid Nongenomic Effects of Aldosterone on Human Forearm Vasculature. Hypertension 42, 156–160. doi:10.1161/01.hyp.0000083298.23119.16
Schmidt, B. M. W., Sammer, U., Fleischmann, I., Schlaich, M., Delles, C., and Schmieder, R. E. (2006). Rapid Nongenomic Effects of Aldosterone on the Renal Vasculature in Humans. Hypertension 47, 650–655. doi:10.1161/01.hyp.0000205224.58715.cc
Schneider, A., Chatterjee, S., Bousiges, O., Selvi, B. R., Swaminathan, A., Cassel, R., et al. (2013). Acetyltransferases (HATs) as Targets for Neurological Therapeutics. Neurotherapeutics 10, 568–588. doi:10.1007/s13311-013-0204-7
Seo, M., Song, M., Seok, Y. M., Kang, S. H., Lee, H. A., Sohn, U. D., et al. (2015). Lysine Acetyltransferases Cyclic Adenosine Monophosphate Response Element-Binding Binding Protein and Acetyltransferase P300 Attenuate Transcriptional Activity of the Mineralocorticoid Receptor through its Acetylation. Clin. Exp. Pharmacol. Physiol. 42, 559–566. doi:10.1111/1440-1681.12377
Seok, Y. M., Lee, H. A., Park, K. M., Hwangbo, M.-H., and Kim, I. K. (2016). Lysine Deacetylase Inhibition Attenuates Hypertension and Is Accompanied by Acetylation of Mineralocorticoid Receptor Instead of Histone Acetylation in Spontaneously Hypertensive Rats. Naunyn-schmiedeberg's Arch. Pharmacol. 389, 799–808. doi:10.1007/s00210-016-1246-2
Shibata, S., Rinehart, J., Zhang, J., Moeckel, G., Castañeda-Bueno, M., Stiegler, A. L., et al. (2013). Mineralocorticoid Receptor Phosphorylation Regulates Ligand Binding and Renal Response to Volume Depletion and Hyperkalemia. Cel Metab. 18, 660–671. doi:10.1016/j.cmet.2013.10.005
Shibata, S., Ishizawa, K., Wang, Q., Xu, N., Fujita, T., Uchida, S., et al. (2018). ULK1 Phosphorylates and Regulates Mineralocorticoid Receptor. Cel Rep. 24, 569–576. doi:10.1016/j.celrep.2018.06.072
Skøtt, O., Uhrenholt, T. R., Schjerning, J., Hansen, P. B., Rasmussen, L. E., and Jensen, B. L. (2006). Rapid Actions of Aldosterone in Vascular Health and Disease-Ffriend or Foe? Pharmacol. Ther. 111, 495–507. doi:10.1016/j.pharmthera.2005.10.010
Souque, A., Fagart, J., Couette, B., and Rafestin-Oblin, M.-E. (1996). Sulfhydryl Groups Are Involved in the Binding of Agonists and Antagonists to the Human Mineralocorticoid Receptor. J. Steroid Biochem. Mol. Biol. 57, 315–321. doi:10.1016/0960-0760(95)00278-2
Stadtman, E. R. (2006). Protein Oxidation and Aging. Free Radic. Res. 40, 1250–1258. doi:10.1080/10715760600918142
Stavreva, D. A., Muller, W. G., Hager, G. L., Smith, C. L., and McNally, J. G. (2004). Rapid Glucocorticoid Receptor Exchange at a Promoter Is Coupled to Transcription and Regulated by Chaperones and Proteasomes. Mcb 24, 2682–2697. doi:10.1128/mcb.24.7.2682-2697.2004
Sturm, A., Bury, N., Dengreville, L., Fagart, J., Flouriot, G., Rafestin-Oblin, M. E., et al. (2005). 11-Deoxycorticosterone Is a Potent Agonist of the Rainbow Trout (Oncorhynchus mykiss) Mineralocorticoid Receptor. Endocrinology 146, 47–55. doi:10.1210/en.2004-0128
Tanaka, M., Nishi, M., Morimoto, M., Sugimoto, T., and Kawata, M. (2005). Imaging Analysis of Mineralocorticoid Receptor and Importins in Single Living Cells by Using GFP Color Variants. Cell Tissue Res 320, 447–453. doi:10.1007/s00441-004-0984-5
Taylor, T. A., Pollock, J. S., and Pollock, D. M. (2003). Down-regulation of Soluble Guanylyl Cyclase in the Inner Medulla of DOCA-Salt Hypertensive Rats. Vasc. Pharmacol. 40, 155–160. doi:10.1016/s1537-1891(03)00048-x
Tirard, M., Almeida, O. F. X., Hutzler, P., Melchior, F., and Michaelidis, T. M. (2007). Sumoylation and Proteasomal Activity Determine the Transactivation Properties of the Mineralocorticoid Receptor. Mol. Cell Endocrinol. 268, 20–29. doi:10.1016/j.mce.2007.01.010
Tsai, M., and O'Malley, B. W. (1994). Molecular Mechanisms of Action of Steroid/thyroid Receptor Superfamily Members. Annu. Rev. Biochem. 63, 451–486. doi:10.1146/annurev.bi.63.070194.002315
Tsai, S. Y., Carlstedt-Duke, J., Weigel, N. L., Dahlman, K., Gustafsson, J.-Å., Tsai, M.-J., et al. (1988). Molecular Interactions of Steroid Hormone Receptor with its Enhancer Element: Evidence for Receptor Dimer Formation. Cell 55, 361–369. doi:10.1016/0092-8674(88)90059-1
Tzunoda, K., Abe, K., Goto, T., Yasujima, M., Sato, M., Omata, K., et al. (1986). Effect of Age on the Renin-Angiotensin-Aldosterone System in Normal Subjects: Simultaneous Measurement of Active and Inactive Renin, Renin Substrate, and Aldosterone in Plasma. J. Clin. Endocrinol. Metab. 62, 384–389. doi:10.1210/jcem-62-2-384
Uhrenholt, T. R., Schjerning, J., Hansen, P. B., Nørregaard, R., Jensen, B. L., Sorensen, G. L., et al. (2003). Rapid Inhibition of Vasoconstriction in Renal Afferent Arterioles by Aldosterone. Circ. Res. 93, 1258–1266. doi:10.1161/01.res.0000106135.02935.e1
Wallace, A. D., and Cidlowski, J. A. (2001). Proteasome-mediated Glucocorticoid Receptor Degradation Restricts Transcriptional Signaling by Glucocorticoids. J. Biol. Chem. 276, 42714–42721. doi:10.1074/jbc.m106033200
Wallace, A. D., Cao, Y., Chandramouleeswaran, S., and Cidlowski, J. A. (2010). Lysine 419 Targets Human Glucocorticoid Receptor for Proteasomal Degradation. Steroids 75, 1016–1023. doi:10.1016/j.steroids.2010.06.015
Walther, R. F., Lamprecht, C., Ridsdale, A., Groulx, I., Lee, S., Lefebvre, Y. A., et al. (2003). Nuclear Export of the Glucocorticoid Receptor Is Accelerated by Cell Fusion-dependent Release of Calreticulin. J. Biol. Chem. 278, 37858–37864. doi:10.1074/jbc.m306356200
Walther, R. F., Atlas, E., Carrigan, A., Rouleau, Y., Edgecombe, A., Visentin, L., et al. (2005). A Serine/Threonine-Rich Motif Is One of Three Nuclear Localization Signals that Determine Unidirectional Transport of the Mineralocorticoid Receptor to the Nucleus. J. Biol. Chem. 280, 17549–17561. doi:10.1074/jbc.m501548200
Wang, C., Tian, L., Popov, V. M., and Pestell, R. G. (2011). Acetylation and Nuclear Receptor Action. J. Steroid Biochem. Mol. Biol. 123, 91–100. doi:10.1016/j.jsbmb.2010.12.003
Wang, C.-H., Hung, P.-W., Chiang, C.-W., Lombès, M., Chen, C.-H., Lee, K.-H., et al. (2019). Identification of Two Independent SUMO-Interacting Motifs in Fas-Associated Factor 1 (FAF1): Implications for Mineralocorticoid Receptor (MR)-mediated Transcriptional Regulation. Biochim. Biophys. Acta (Bba) - Mol. Cel Res. 1866, 1282–1297. doi:10.1016/j.bbamcr.2019.03.014
Weidmann, P., De Myttenaere-Bursztein, S., Maxwell, M. H., and de Lima, J. (1975). Effect of Aging on Plasma Renin and Aldosterone in normal Man. Kidney Int. 8, 325–333. doi:10.1038/ki.1975.120
Wrange, O., Eriksson, P., and Perlmann, T. (1989). The Purified Activated Glucocorticoid Receptor Is a Homodimer. J. Biol. Chem. 264, 5253–5259. doi:10.1016/s0021-9258(18)83726-7
Yokota, K., Shibata, H., Kobayashi, S., Suda, N., Murai, A., Kurihara, I., et al. (2004). Proteasome‐Mediated Mineralocorticoid Receptor Degradation Attenuates Transcriptional Response to Aldosterone. Endocr. Res. 30, 611–616. doi:10.1081/erc-200043783
Yokota, K., Shibata, H., Kurihara, I., Kobayashi, S., Suda, N., Murai-Takeda, A., et al. (2007). Coactivation of the N-Terminal Transactivation of Mineralocorticoid Receptor by Ubc9. J. Biol. Chem. 282, 1998–2010. doi:10.1074/jbc.m607741200
Zannad, F., McMurray, J. J., Krum, H., van Veldhuisen, D. J., Swedberg, K., Shi, H., et al. (2011). Eplerenone in Patients with Systolic Heart Failure and Mild Symptoms. N. Engl. J. Med. 364, 11–21. doi:10.1056/NEJMoa1009492
Zennaro, M.-C., and Fernandes-Rosa, F. (2017). 30 YEARS of the MINERALOCORTICOID RECEPTOR: Mineralocorticoid Receptor Mutations. J. Endocrinol. 234, T93–T106. doi:10.1530/joe-17-0089
Zennaro, M.-C., Keightley, M.-C., Kotelevtsev, Y., Conway, G. S., Soubrier, F., and Fuller, P. J. (1995). Human Mineralocorticoid Receptor Genomic Structure and Identification of Expressed Isoforms. J. Biol. Chem. 270, 21016–21020. doi:10.1074/jbc.270.36.21016
Keywords: mineralocorticoid receptor, cardiovascular aging, posttranslational modification, phosphorylation, acetylation, ubiquitination, sumoylation, oxidation
Citation: Gadasheva Y, Nolze A and Grossmann C (2021) Posttranslational Modifications of the Mineralocorticoid Receptor and Cardiovascular Aging. Front. Mol. Biosci. 8:667990. doi: 10.3389/fmolb.2021.667990
Received: 15 February 2021; Accepted: 07 May 2021;
Published: 28 May 2021.
Edited by:
Thorsten Pfirrmann, Health and Medical University Potsdam, GermanyReviewed by:
Ritu Chakravarti, University of Toledo, United StatesCopyright © 2021 Gadasheva, Nolze and Grossmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claudia Grossmann, Y2xhdWRpYS5ncm9zc21hbm5AbWVkaXppbi51bmktaGFsbGUuZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.