 Bethany Carter1†
Bethany Carter1† Hannah S. Justin
Hannah S. Justin Danielle Gulick
Danielle Gulick Joshua J. Gamsby
Joshua J. Gamsby
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Biosci. , 26 March 2021
Sec. Molecular Diagnostics and Therapeutics
Volume 8 - 2021 | https://doi.org/10.3389/fmolb.2021.644747
This article is part of the Research Topic Rare Neurodegenerative Disorders: From Molecular Pathways to Therapeutic Strategies View all 6 articles
Circadian rhythm dysfunction occurs in both common and rare neurodegenerative diseases. This dysfunction manifests as sleep cycle mistiming, alterations in body temperature rhythms, and an increase in symptomatology during the early evening hours known as Sundown Syndrome. Disruption of circadian rhythm homeostasis has also been implicated in the etiology of neurodegenerative disease. Indeed, individuals exposed to a shifting schedule of sleep and activity, such as health care workers, are at a higher risk. Thus, a bidirectional relationship exists between the circadian system and neurodegeneration. At the heart of this crosstalk is the molecular circadian clock, which functions to regulate circadian rhythm homeostasis. Over the past decade, this connection has become a focal point of investigation as the molecular clock offers an attractive target to combat both neurodegenerative disease pathogenesis and circadian rhythm dysfunction, and a pivotal role for neuroinflammation and stress has been established. This review summarizes the contributions of molecular clock dysfunction to neurodegenerative disease etiology, as well as the mechanisms by which neurodegenerative diseases affect the molecular clock.
Circadian rhythms are daily cycles of biological processes in living organisms that are regulated endogenously and function to gate essential molecular, cellular, physiological, and behavioral activities to discrete times of the day. Circadian rhythms are defined by three properties: (1) They must have a period of approximately 24 h and continue to oscillate in the absence of external stimuli, (2) rhythmicity can be reset or “entrained” by external stimuli, and (3) they are temperature compensated or maintain periodicity regardless of temperature changes across the physiological permissible range (Dunlap et al., 2004). The central pacemaker in mammals, known as the suprachiasmatic nucleus (SCN), regulates the timing of circadian rhythms and resides in the anterior hypothalamus. The SCN controls output timing to peripheral oscillators, such as organ systems, to coordinate the daily cycling of numerous essential physiological processes. Examples of peripheral oscillators include the liver, heart and discrete regions of the brain, such as the hippocampus (Zhang and Sehgal, 2019). In healthy individuals this mechanism functions to regulate sleep–wake cycles, neuroendocrine activities, and memory (Moore, 2013; Sollars and Pickard, 2015).
As previously mentioned, an important feature of a circadian rhythm is that it can entrained by various external cues to allow an organism to adapt to changes in the environment. In humans, an example of this is light exposure when waking. Light, absorbed by intrinsically photosensitive retinal ganglion cells, signals through the retinohypothalamic tract to the SCN to reset the clock by adjusting the timing of the core molecular oscillator (reviewed below). The SCN then conveys this timing information to peripheral oscillators such as the heart, liver and lungs (Dibner et al., 2010; Richards and Gumz, 2012) via the immune, hormonal, and neurotransmitter signals to generate the proper timing of rhythmic physiological processes (Moore, 2013; Sollars and Pickard, 2015). When this system is perturbed, numerous adverse consequences can occur. For example, exposure to light at inappropriate times, such as prolonged light exposure to light-emitting electronic devices before bed, represses melatonin production and disrupts normal sleep patterns (Chang et al., 2015). Consistent perturbations of entrainment can lead to “circadian desynchrony,” a misalignment of the circadian clock with the external environment that is associated with an increased risk of numerous diseases and disorders such as cancer, diabetes, and Alzheimer’s disease (AD) (Sahar and Sassone-Corsi, 2009; Bass and Takahashi, 2010; Musiek and Holtzman, 2016; Logan and McClung, 2019). Additionally, extra-SCN clocks exist that can supersede or override the timing of the core clock (Anne-Marie Chang et al., 2014). For example, the hippocampus, which plays a central role in learning and memory function, has an entrainable circadian clock that can regulate the time-of-day changes in cognitive efficacy in the absence of descending signals from the SCN (Hasegawa et al., 2019).
Numerous excellent reviews have summarized our current understanding of the molecular circadian clock (Baker et al., 2012; Partch et al., 2014; Cohen and Golden, 2015; Takahashi, 2017; Patke et al., 2020). Therefore, this review will summarize the molecular components that are impacted by neurodegenerative disease. The molecular clock in humans is composed of a family of core clock genes that are involved in two interlocking feedback loops; a primary negative feedback loop, and a secondary stabilizing loop (Li et al., 2013; Patrick-Simon Welz, 2020). The primary is a transcription-translation feedback (TTFL) loop, which governs 24-h periodicity. Importantly, this aspect of the clock mechanism is conserved across almost all living organisms (Dunlap and Loros, 2017). The TTFL mechanism is initiated by the transcription factors Brain and Muscle Arnt-Like 1 (BMAL1) protein and Circadian Locomotor receptor Output Cycles Kaput (CLOCK), which heterodimerize then bind to enhancer box (e-box) consensus site sequences in the promoters of the Period (Per1-3) and Cryptochrome (Cry1-2) gene families. PER and CRY are then transcribed, translated, and ultimately shuttled back into the nucleus to bind to the heterodimeric BMAL1/CLOCK complex to repress their own transcription, as well as the transcription of any clock-controlled genes (CCGs) that also have e-box (or other clock-regulated) consensus sites. The secondary loop is centered on the production of BMAL1 and CLOCK. Here, rhythmic BMAL1 transcription is driven by the activation and repression of by the RAR-related orphan nuclear receptor (ROR) and Rev-Erb gene families, respectively, which competitively bind to retinoic acid response elements (RREs) within the BMAL1 promoter. Together, this system of feedback loops regulates periodicity, amplitude, and phase of the molecular clock (Figure 1) (Bae et al., 2001; Li et al., 2013). In addition to the roles of core clock genes, the molecular clock is regulated by rhythmic posttranslational modifications (PTMs). For example, isoforms of the casein kinase 1 (CK1) gene family phosphorylate PER in a time-of-day dependent manner to maintain proper clock timing (Etchegaray et al., 2009; Lee H. M. et al., 2011). An additional PTM that is characterized by the acetylation of CLOCK and BMAL1 mediates gene expression by interacting with histone acetyltransferases (HATs) (Masri and Sassone-Corsi, 2010; Zhang and Sehgal, 2019). All of these mechanisms function in concert to maintain the proper timing of the central and peripheral oscillators.
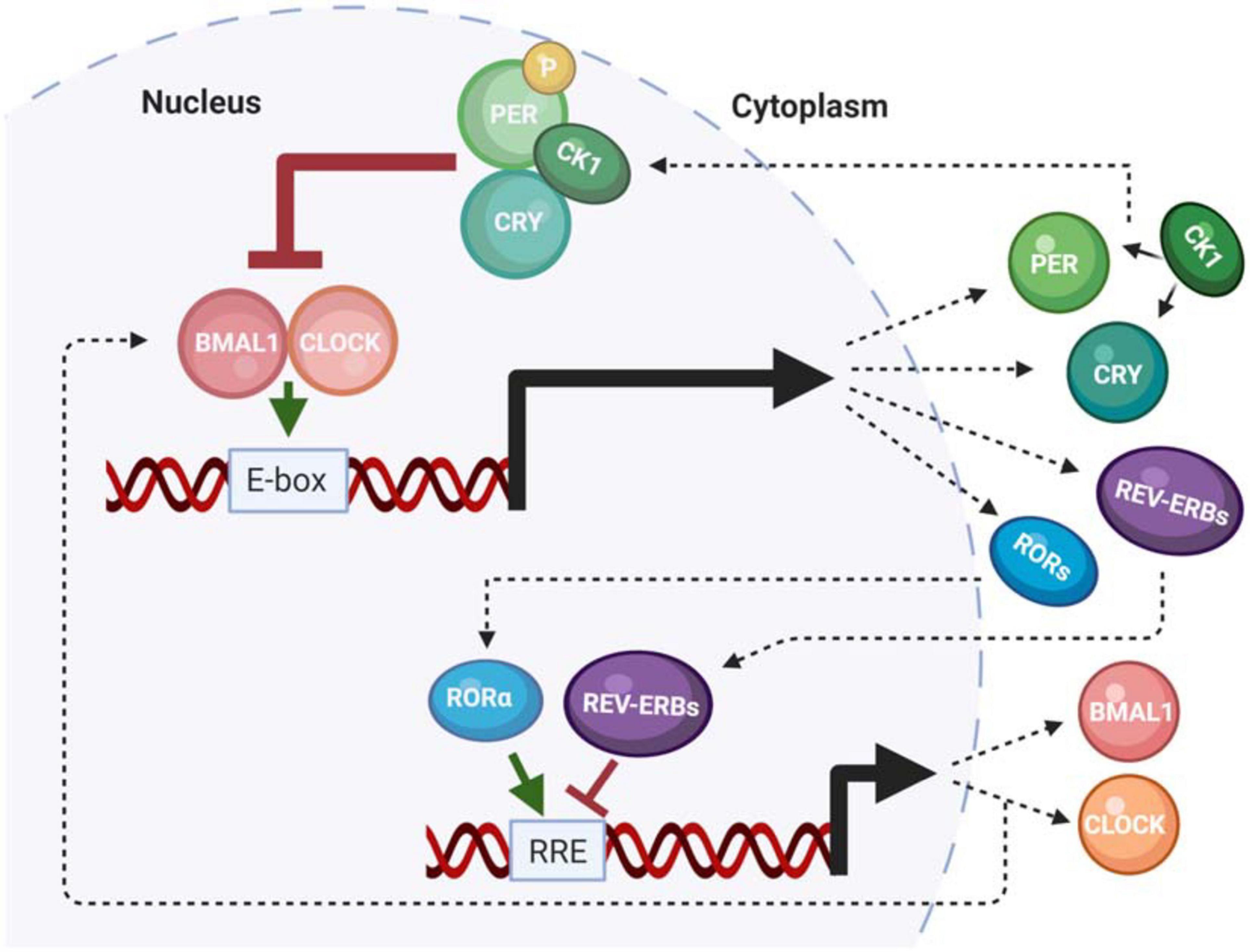
Figure 1. The mammalian molecular clock. A summary of the functionality of the mammalian core clock components that are impacted by neurodegenerative disease or play a role in pathogenesis.
Neurodegenerative diseases are characterized by the progressive loss of neuronal structure and function, usually through neuronal death, and are caused by both genetic and environmental factors. The symptoms of neurodegeneration can include loss of motor control, cognitive decline, and mood changes, depending on the affected area of the nervous system. One of the early indicators that the circadian system is impacted by neurodegenerative disease was the observation that some patients have aggravated symptomology during the early evening hours, which is known as Sundown Syndrome. Sundowning is characterized by greater cognitive deficits and psychotic breakdowns during the evening hours, disturbed sleep patterns, and difficulty thermoregulating (Bliwise et al., 1995; Canevelli et al., 2016). These functions are all controlled by the SCN, indicating a connection between the cognitive and effective symptoms of dementia and the molecular clock.
Recent evidence has established a clear relationship between neurodegenerative phenotypes and the molecular clock (Chang and Guarente, 2013; Musiek et al., 2013; Musiek and Holtzman, 2016; Singh et al., 2018; Lananna and Musiek, 2020). Additionally, chronic perturbations in normal circadian rhythmicity are also believed to exacerbate neurodegenerative disease progression (Musiek, 2015). Thus, a bidirectional relationship between the clock and neurodegeneration exists. An example of this is the relationship between the clock and the adrenal glucocorticoid (GC) stress responses. The SCN regulates the hypothalamus–pituitary–adrenal (HPA) axis, driving the rhythmic release of adrenocorticotropic hormone (ACTH). This rhythmicity of ACTH has downstream effects on ACTH and GC receptor sensitivity that disinhibit the HPA axis during the waking phase; this disinhibition drives time-of-day dependent sensitivity to stress exposure. In return, stress increases the activation of the HPA axis, and the subsequent production of cortisol. This increased activation of the HPA axis has downstream effects on the clock by way of increased distal corticosteroids disrupting sleep (Han et al., 2012). If continual sleep disruptions persist, rhythms in the sleep–wake cycle itself will be altered, causing irregularities in core clock timing or circadian desynchrony (CD). Thus, the clock functions to regulate the stress response, and chronic stress can also affect the timing of the clock, which could be impacted by neurodegenerative disease pathology.
One of the major output pathways of the molecular clock feeds into cellular metabolism, including processes such as redox homeostasis and neuroinflammation. Disruption of redox homeostasis drives oxidative stress (OS), which is a key nexus between neurodegeneration and the circadian clock (Lananna and Musiek, 2020). BMAL1 drives the transcription of redox genes NAD(P)H quinone dehydrogenase and aldehyde dehydrogenase 2 (QR2) in the brain (Musiek et al., 2013), which play vital roles in preventing OS accumulation and neuronal damage (Musiek et al., 2013; Musiek and Holtzman, 2016). TTFL factors also mediate antioxidant activity through their interactions with NAD-dependent deacetylase sirtuin 1 (SIRT1) (Asher et al., 2008) which is heavily involved in maintaining cellular redox homeostasis (Asher et al., 2008). SIRT1 activates the transcription of both BMAL1 and CLOCK in the SCN (Chang and Guarente, 2013). Its dysfunction is a hallmark of Parkinson’s disease (PD) (Singh et al., 2017), and emerging evidence also points to its role in AD pathogenesis. Together, these findings present a mechanism to explain the circadian impact on the onset and development of multiple neurodegenerative diseases and suggest a significant bidirectional relationship between neurodegeneration and circadian desynchrony that may provide a target to reduce the risk or severity of neurodegenerative diseases.
In this review, we will address the role of the circadian clock in the development and progression of both common and rare neurodegenerative diseases - specifically in AD, Lewy body dementia (LBD), PD, Huntington’s disease (HD), progressive supranuclear palsy (PSP), Pick’s disease (PiD), and frontotemporal lobar degeneration-tau (FTLD-tau). As the neuropathology of each disease covered is central to the circadian rhythm disorders observed in patients, we have chosen to group each neurodegenerative disease by their associated proteinopathies.
Lewy body dementia and PD are both characterized by neuronal α-synuclein inclusions known as Lewy bodies. The normal function of α-synuclein is not well-understood, but evidence suggests a role in synaptic vesicle mobility (Scott and Roy, 2012). Misfolded α-synuclein results in β-sheet aggregates, which are hypothesized to spread in a prion-like manner causing devastating neuronal damage (Ma et al., 2019). In prion diseases, misfolded proteins cause the misfolding of their normal protein counterpart (Walker et al., 2015).
Parkinson’s disease is a neurological motor disorder characterized by uncontrollable tremors and difficulty initiating voluntary movements. Presently, the precise etiology of PD is unclear, although genetics can play a central a role – specifically through the mutation of the Parkin gene (PRKN). During PD pathogenesis, significant cell death occurs among dopamine producing neurons of the substantia nigra, leading to this loss of motor function. Lewy bodies aggregate in the basal ganglia and neocortex (Walker et al., 2015). As it progresses, patients with PD develop cognitive decline, dementia, as well as mood and behavioral disorders. Additionally, circadian dysfunction also occurs in PD and is characterized by many sleep-related pathologies, such as excessive drowsiness, insomnia, night terrors, REM cycle disruptions, and sleep fragmentation (Petit et al., 2017; Trenkwalder et al., 2017). Furthermore, circadian dysfunction coupled with neuroinflammation is a risk factor for PD. Lauretti et al. (2017) subjected C57BL/6 mice to abnormal light-dark cycles (20:4 LD) prior to administering a PD-inducing neurotoxin. Circadian-disrupted mice had severe neuroinflammation and a greater degradation of motor skills, indicating that circadian dysfunction is a potential risk factor for PD. This effect was recapitulated in a rat model, where PD pathology was induced or exacerbated in one of three ways: by injection of lipopolysaccharide, injection of rotenone, or injection of both reagents (Li et al., 2019). All three groups exhibited similar loss of dopaminergic neurons and neuroinflammation. Of the nine core clock genes evaluated, six had decreased expression levels in all three groups. Circadian disturbances have also been shown in an α-synuclein overexpressing (ASO) mouse model of PD (Kudo et al., 2011). There were selective deficits in circadian activity such as lower waking activities and more fragmented sleep patterns. ASO mice had normal Per2 expression and lower daytime firing rates in the SCN neurons, which have also been observed in HD models (Smarr et al., 2019). Importantly, ASO mice showed no negative effects in their light adaptation responses, indicating that photic entrainment remained intact.
There is evidence that PD and the molecular clock are also connected at the genetic level (Gu et al., 2015). A screen of eight key core clock genes in sizable populations of PD patients and healthy controls revealed a significant association between PD risk and clock gene SNPs. Of all the screened genes, BMAL1 and Per1 had the greatest number of PD-associated SNPs. Different PD subtypes were also correlated with specific haplotypes in the identified BMAL1 SNPs. This indicates that SNPs in TTFL genes are risk factors for PD that could potentially serve as biomarkers to aid PD diagnosis and treatment. However, the core clock may be affected by the same factors that increase the risk of developing PD. For example, the PRKN mutation may influence crosstalk between the TTFL and mitochondrial bioenergetic pathways. Fibroblasts taken from two genetic PD patients exhibited dampened oscillations in bioenergetic activities, reduced rates of mitochondrial oxygen consumption, and dysregulated TTFL factor expression patterns - CLOCK, CRY1, and CRY2 were upregulated while PER2 was downregulated in these cells (Pacelli et al., 2019). Epigenetic modifications may also play a role in aberrant clock gene expressions. A screen of seven key TTFL genes in the leukocytes of PD patients showed that the Neuronal PAS Domain Protein 2 (NPAS2) promoter is hypomethylated compared to healthy controls. NPAS2 performs a similar function to CLOCK, as it can heterodimerize with BMAL1 to drive circadian rhythmicity. Cry1 conversely showed greater promoter methylation, while none of the remaining five genes showed any change in methylation (Lin et al., 2012).
In addition to increasing clinical evidence that human PD patients exhibit deregulation of BMAL1 (Breen et al., 2014; Gu et al., 2015), there is a decrease in expression of BMAL2 which is a paralog of BMAL1 (Ding et al., 2011). BMAL2 is not essential for normal clock functionality, but knockout of BMAL1 can be rescued by the constitutive expression of BMAL2 in mice, indicating paralog compensation. Additionally, Gu et al. (2015) speculate that lowered BMAL1 levels are a result of PD-induced dopamine deficiency. This postulation is supported by studies in a rat model of PD (Gu et al., 2015). Here, 6-hydroxydopamine (6-OHDA), a potent neurotoxin that destroys dopaminergic and noradrenergic neurons, induced a PD-like phenotype, which caused an increase in levels of acetylated SIRT1 and BMAL1, decreased Per and Cry expression, and an alteration of neuronal antioxidant activity (Wang et al., 2018). Furthermore, treatment with the antioxidant Resveratrol partially rescued this abnormal phenotype. Based on these results, it is possible that core clock abnormalities contribute to reduced antioxidant capacity in PD. To further outline the potential circadian component in PD, a study from Liu et al. (2020) shows inactivation of BMAL 1 in MPTP-treated mice results in significant reduction of dopaminergic neurons, and decreased levels of tyrosine hydroxylase, dopamine, and 3,4-dihydroxyhenylacetic acid content in experimental mice. Increased activation of the neuroinflammatory response was observed in vivo and in vitro, thereby suggesting BMAL1 may have a neuroprotective effect in the depletion of dopaminergic neurons and disrupted BMAL1 may accelerate PD pathology (Liu et al., 2020). “These studies taken as a whole suggest that selectively targeting TTFL factors with pharmaceuticals or other chronotherapies may succeed in alleviating symptoms in PD patients. However, inconsistencies concerning the impact of PD on the core clock components clouds our current understanding of the relationship between PD and the clock. Further investigation of this connection is needed before a therapeutic strategy can be developed.”
LBDs are the second most common type of degenerative dementia affecting the elderly (Petit et al., 2017). A pilot study investigating clock gene disturbances in dementia patients found that LBD patients had a higher frequency of aberrant methylation patterns of core clock genes compared to other dementia patients (Liu et al., 2008). Among the nine genes examined, Per1 and Cry1 were methylated in dementia patients but not in healthy controls. LBD patients had somewhat greater disturbances of locomotor circadian rhythmicity than AD patients (AD is discussed in further detail below); however, there was no association between behavioral disturbances and gene methylation in any of the dementia groups (Liu et al., 2008). Interestingly, the circadian sleep disturbances associated with LBD appear more severe than in AD, which was initially shown by Harper et al. (2004). This data was supported by more recent findings where a 70% increase in REM sleep behavior disorder (RBD)-like symptoms, including increased daytime drowsiness, was observed in LBD as compared to AD and controls (Cagnin et al., 2017). However, further research is necessary to understand the molecular mechanisms that underlie these behavioral observations. RBD has been proposed to be a precursor stage of synucleinopathies, some patients exhibiting signs of RBD full years prior to any neurodegenerative symptoms (Brown, 1994; Zhang et al., 2020). To evaluate the involvement of clock genes in RBD, Weissova et al. (2018) investigated 24-h melatonin blood profiles in RBD patients and healthy individuals. RBD patients did not have circadian rhythmicity for key clock genes Per2, BMAL1, and rev-Erbα, as compared to healthy individuals. Additionally, melatonin profiles of RBD patients were delayed by 2 h, and habitual sleep phases were delayed by 1 h. These results suggest that altered expression of Per2, BMAL1, and Rev-Erbα combined with delayed melatonin secretion could be responsible for RBD (Weissova et al., 2018). Interestingly, melatonin may also be involved in the progression of HD, discussed later in this review.
Tauopathies are a class of neurodegenerative diseases that arise when the protein tau, which functions to stabilize the microtubules of neurons, becomes misfolded and forms pathogenic intracellular aggregates that cause neurodegeneration (Irwin, 2016) and contribute to memory cognitive deficits (Lee V. M. et al., 2011). Genetic abnormalities such as missense mutations, SNPs, pathogenic haplotypes, and deregulated alternative splicing have all been implicated in tau pathogenesis (Caillet-Boudin et al., 2015; Irwin, 2016). These changes can induce structural instability, reduce tau’s microtubule binding capacity (Strang et al., 2019), increase intracellular concentrations of free-floating phosphorylated Tau (pTau), and contribute to the formation of neurofibrillary tangles (NFTs). Clinical evidence indicates that the SCN is also damaged by tau aggregation (Stopa et al., 1999) and circadian disturbances in the behavior of tauopathy patients suggest an imbalance in the TTFL (Anderson et al., 2009). In addition to tauopathy, a defining hallmark of some neurodegenerative diseases, like AD, is the presence of β-amyloid plaques, which are composed of toxic extracellular aggregates of amyloid beta (Aβ). The amyloid precursor protein (APP) is cleaved by both β-secretase (BACE1) and γ-secretase to generate Aβ and are associated with the secretion and aggregation of toxic Aβ (Luo et al., 2001; Steiner et al., 2018; Yuksel and Tacal, 2019). As amyloid plaques aggregate, they can also exacerbate tauopathy, driving further pathogenesis (Gan et al., 2019; Busche and Hyman, 2020).
Alzheimer’s disease is one of the most heavily researched neurodegenerative diseases due to its rapidly increasing prevalence, mostly because humans are living longer and AD is primarily a disease that affects those 65 years of age and older (Xia et al., 2018). In the United States alone, AD cases are projected to reach 14 million by 2050 (Hebert et al., 2013). Risk factors such as aging and OS are both tightly intertwined with the core clock. It is well known that aging leads to oxidative damage (Zhang et al., 2015; Gomes et al., 2017; Mecocci et al., 2018). In normal aging, several cellular processes are attenuated over time. This senescence is caused by a chronic state of low-level stress, induced by both genetic and environmental factors. However, AD patients generally have markedly higher OS levels, which are thought to contribute to more rapid degeneration (Chen and Zhong, 2014). In healthy individuals, TTFL factors mediate antioxidant processes in many organ systems, and the maintenance of redox homeostasis in the brain is essential in staving off neurodegeneration. It is unclear whether OS causally contributes to circadian dysfunction or vice versa. Most likely, feedback between the two pathologies contributes to both. Thus far, changes in the expression of PER and BMAL1 seem to have the strongest connections to aging and OS.
As previously mentioned, the TTFL is an evolutionarily conserved mechanism. Therefore, much insight into the relationship between the clock and neurodegenerative disease can be gained by evaluating this relationship in an organism with much simpler genetics and nervous system. For example, in the model organism Drosophila melanogaster there is only one Per gene, whereas in mammals there are three (Per1–3). This redundancy can lead to confounds such as the paralog compensation (Baggs et al., 2009). In Drosophila, Per regulates sleep patterns and working memory (Dissel et al., 2015). Furthermore, high expression of PER is associated with neuroprotective effects against oxidative damage (Krishnan et al., 2009). Ablation of Per accelerates neurodegeneration (Krishnan et al., 2009, 2012). When subjected to hyperoxia, Per knockout flies had greater oxidative damage, impaired antioxidant stress responses, neurodegeneration, and shortened lifespans. OS also induces aberrant sleep patterns such as those seen in aged AD patients. Over time, the sleep–wake cycle amplitudes decreased in OS-challenged flies, and the circadian rhythm loses its endogenous control, becoming temperature-dependent (Koh et al., 2006). Flies in a cooler environment also showed markedly slower neuronal degeneration during sleep cycles. This could be related to the lowering of core body temperature that occurs in healthy mammalian sleep cycles (Harding et al., 2019). Additionally, age-related losses of circadian rhythmicity in peripheral tissues have been recapitulated in other Drosophila studies (Chen et al., 2014; Long et al., 2014; Kuintzle et al., 2017). One such study by Buhl et al. (2019) demonstrates that the 0N4R isoform of Tau causes behavioral changes which match those of human AD patients. Additionally, a hyperactive circadian phenotype was observed, and then validated by whole-cell current clamp recordings of large lateral ventral neurons. These findings showed an especially depolarized membrane resting potential, and higher spontaneous firing rate among transgenic flies. Interestingly, input resistance, which is expected to cycle in wild type flies, maintained the same magnitude during the day and nighttime in the experimental group. These findings further outline the relationship between Tau isoforms and circadian disruption (Buhl et al., 2019).
Amyloid beta can also alter PER1 and PER2 expression in both the central oscillator (SCN) and peripheral cardiovascular tissues (Wang et al., 2016). Per is essential for maintaining healthy cardiac function and alterations in PER expression are strongly correlated with cardiac dysfunction (Young, 2006; Maemura et al., 2007). Similar patterns of PER expression may also exist in AD patients, as cardiovascular co-morbidities, such as atherosclerosis, are thought to be risk factors for AD because of the damage inflicted on the cerebrovascular system (Iadecola, 2003; Roher et al., 2011). Another potential player in these interactions is SIRT1 (Bellanti et al., 2017). A mouse model of AD that has both Aβ and tauopathy phenotypes (3xTG-AD) display altered hippocampal expression of SIRT1 after different patterns of light exposure (Bellanti et al., 2017) indicating circadian desynchrony influences OS pathways. SIRT1 even mediates transcriptional control of TTFL genes and this process is attenuated by aging (Chang and Guarente, 2013). These findings underscore the importance of OS pathways in circadian dysfunction (Song et al., 2015).
Evidence suggests that loss of rhythmicity contributes to Aβ production [by BACE1 (Ma et al., 2016)] and accumulation, and the reverse is also true - Aβ alters BMAL1 degradation (Song et al., 2015), deregulating the molecular clock. Exemplifying the bidirectional relationship between Aβ and BMAL, Kress et al. (2018), demonstrated that complete BMAL Knockout produces a rhythmic behavior in the deposition of AB plaque, and that local BMAL Knockout increases AB plaque burden. In addition, the circadian CK1 enzymes modulate Aβ activity by increasing the production of Aβ (Oumata et al., 2008; Ooms and Ju, 2016). BMAL1 levels are affected by Aβ as shown by Song et al. (2015) where in vitro Aβ induces degradation of BMAL1 and CBP via PTM SUMOylation. Consequently, PER2 expression was deregulated, corroborating the disrupted expression of BMAL1 and PER2 that was observed in vivo. Furthermore, knockdown of Sumo1 rescued the degradation of BMAL1. Given that Aβ over-accumulation increases ROS production (Smith et al., 2007) and that neuronal redox homeostasis is regulated by BMAL1/CLOCK (Musiek et al., 2013), the molecular dysfunction of the TTFL may contribute to Aβ-mediated neuronal cell death. On the other hand, circadian desynchrony may not be a direct product of pathogenic Aβ, but of some underlying mechanism that also increases Aβ production. Flies that overexpress the Drosophila homolog for APP in their central pacemaker neurons maintain robust circadian rhythms in spite of aging (Blake et al., 2015). However, overexpression of Drosophila β-secretase led to disturbed rest/activity rhythms, decreased PER expression and dampened oscillations of PER in the central pacemaker neurons. These effects were most prominent in aging flies, suggesting that β-secretase mediated circadian dysfunction is exacerbated in an age-dependent manner.
BMAL1 is neuroprotective against aging-induced neuronal decline, and yet BMAL1 is itself affected by aging and oxidative damage. BMAL1 knockout mice display an early aging phenotype characterized by sarcopenia, decreased hair regrowth, and increased ROS levels in the heart, kidneys, and spleen (Kondratova et al., 2010). BMAL1 deficiency also impairs PER and CRY expression, habituation, neuronal ROS homeostasis, and causes hyperactivity (Kondratova et al., 2010). Additionally, BMAL1 knockout mice also exhibit severe reactive astrogliosis in cortical and hippocampal tissue, while single knockouts of clock genes Per1, Per2, CLOCK, or Npas2 produced no astrogliosis (Musiek et al., 2013; Musiek and Holtzman, 2016). Astrogliosis is an increase in the number of astrocytes, which are important cellular mediators of the neuroimmune response. Both global and neuronal deletion of BMAL1 produced astrogliosis, indicating a role for neuronal BMAL1 in this effect on astrocytes. Interestingly, SCN astrocytes have a significant impact upon neuronal activity during the circadian rest phase by inhibiting neuronal activity (Brancaccio et al., 2017). SCN astrocyte rhythmicity is also suspected to determine period length of locomotor activity. In an experiment conducted by Tso et al. (2017), it was found that knocking out astrocyte-specific BMAL1 lengthened the period of wheel running activity. Furthermore, combined knockout of CLOCK and Npas2 produced similar astrocyte activation to the BMAL1 KO mice, suggesting that dysfunction in the positive arm of the TTFL plays a vital role in the development of this age-dependent neuropathology. An aged hamster model also demonstrated attenuated BMAL1 expression across many regions of the brain, particularly in extra-SCN tissues (Duncan et al., 2013). Researchers postulated that this subsequent weakening of clock gene expression in regions of the brain might contribute to age-related cognitive deficits. Taken together, these findings suggest that circadian dysfunction has a clear impact on memory deficits and that natural aging processes are associated with decreased BMAL1 expression. In addition to BMAL1’s role in astrogliosis, a recent report has revealed further evidence for the relationship between the clock and astrocyte function. Lananna et al. (2020) show that the clock regulates the production of Chi3l1, which encodes for YKL-40, a glycoprotein biomarker for neuroinflammation in CSF that is increased in AD patients. The authors observed that Chi3l1 is not only regulated by the core clock, but that deletion of Chi3l1/YKL-40 reduced amyloid plaque formation and Aβ phagocytosis (Lananna et al., 2020). This exciting result suggests that deregulation of a CCG such as Chi2l1 might lead to AD pathogenesis.
In addition to role of core clock genes involved in transcriptional activation and transcriptional repression, core clock genes involved in post translational modifications are also impacted by AD. CK1δ may contribute to the aggregation of hyperphosphorylated tau inclusions (Li et al., 2004) and associates with NFTs in several neurodegenerative diseases, such as AD (Ghoshal et al., 1999; Schwab et al., 2000) PD, PSP, and PiD (Schwab et al., 2000). Among the dementia patients studied by Schwab et al. (2000), CK1δ did not associate with tau-negative pathogenic inclusions (e.g., Lewy bodies, Marinesco bodies) of non-tauopathy neurodegenerative diseases, whereas it had variable immunostaining intensity amongst dystrophic neurites, NFTs, and neuropil threads in AD brains (Schwab et al., 2000). Thus, it is possible that CK1δ accumulation could serve as a clinical biomarker for tau-specific neurodegenerative lesions. CK1δ isozymes may also directly influence protein turnover in the autophagic and phagocytotic processes that are essential in maintaining neuronal health. The dysfunction of these processes is widely implicated in neurodegeneration (Menzies et al., 2015, 2017; Martini-Stoica et al., 2016; Mecocci et al., 2018). Future research is needed to clarify the mechanisms of CK1δ-related accumulation in pathogenic tau plaques, although some work suggests a possible interaction with transactive response DNA-binding protein (TDP-43), another hallmark of neurodegeneration. CK1δ phosphorylates TDP-43 and promote its mislocalization (Nonaka et al., 2014; Gu et al., 2020), aggregating with TDP-43 and tau in granulovacuolar degeneration bodies (GVBs) of the cytoplasm, to promote tau hyperphosphorylation (Ghoshal et al., 1999; Yamazaki et al., 2011). Of note, CK1δ also contributes to neurodegenerative diseases through amyloidosis. We have previously studied this role in APP-PS1 mice, which manifest an abnormal sleep–wake rhythm and impaired cognition for tasks that rely on the prefrontal cortex and hippocampus (Sundaram et al., 2019). Treatment of this model with a selective CK1 δ/ε inhibitor resulted in a dose-dependent reduction of Aβ burden in the hippocampus and prefrontal cortex compared to vehicle-treated littermates. Additionally, the CK1 inhibitor reduced Aβ plaque size in the prefrontal cortex and hippocampus, increased period length and rescued some cognitive function. Since CK1 indirectly controls Aβ formation by phosphorylating APP and its subsequent proteases β and γ secretase (Chen et al., 2017), targeting its activity with pharmaceuticals may be a promising avenue of treatment for tauopathies that co-occur with amyloidosis. The Drosophila ortholog of CK1δ, Doubletime (Dblt), can shorten period length when mutated. Findings from Drosophila suggest a further interaction between CK1δ and tau. Means et al. (2015) knocked down Spaghetti, an HSP-90 co-chaperone that upregulates Dblt, and observed both defects in dClk expression, and upregulation of the initiator caspase Dronc that cleaves tau and accelerates neurodegeneration.
Less studied, but essential to this discussion is the influence of aberrant epigenetic modifications on TTFL factors. For example, oscillatory DNA methylation contributes to the regulation of BMAL1 (Cronin et al., 2017). Researchers increased methylation in NIH3T3 fibroblasts using S-adenosyl-methionine, a methyl donor. This led to decreased amplitude, longer periods, and phase delays in circadian rhythmicity, while treatment with a DNA methyltransferase inhibitor produced the exact opposite effects (Cronin et al., 2017). A subsequent postmortem analysis of midfrontal cortical tissue from deceased AD patients revealed abnormal oscillatory methylation of the BMAL1 promoter compared to healthy controls (Cronin et al., 2017).
Frontotemporal lobar degeneration-tau is a less common dementia that is nonetheless characterized by hyperphosphorylated tau deposits, often secondary to tau mutations (Bodea et al., 2016). This is subcategory of frontotemporal dementia (FTD), which is a broad term to designate neurodegeneration that produces pathology in discrete regions of the brain as well as distinct behavioral, emotional, language, and motor impairments (Bang et al., 2015). Although FTDs encompass a wide range of neurodegenerative diseases, there is clear clinical evidence of associated circadian dysfunction across this spectrum. FTD patients have comparable circadian symptoms to AD patients, such as increased difficulty falling and staying asleep, persistent daytime fatigue (Sani et al., 2019), and declines in sleep continuity (Kundermann et al., 2011). Additionally, our own research demonstrates impaired circadian functionality in a transgenic tau mouse model of FTLD-tau, which expresses a human mutant tau allele and is known as the Tg4510 line (Stevanovic et al., 2017). Tg4510 mice display a long free-running period in activity patterns as well as alterations in the circadian timing of BMAL1 expression in the hippocampus, and PER2 expression in the hippocampus and hypothalamus (Stevanovic et al., 2017). These defects may be due to the presence of elevated pTau in the SCN, which was also noted in this study. Furthermore, recent evidence demonstrates impaired cytoplasmic homeostasis, caused by tauopathy (i.e., elevated pTau), might impede PER trafficking into the nucleus of neurons of the SCN, ultimately leading to circadian rhythm dysfunction (Beesley et al., 2017). Clinical evidence also indicates that the SCN is also damaged by tau aggregation (Stopa et al., 1999) and circadian disturbances in the behavior of tauopathy patients suggest an imbalance in the TTFL (Anderson et al., 2009). Additionally, human tissue studies have proposed a link between dipeptide repeat (DPR) protein inclusions and sleep–wake disturbances in FTLD patients, as well as severe sleep disturbances and possible phase delay indicated by sleep diary data in human FTD cohorts (Anderson et al., 2009; Dedeene et al., 2019). However, a clear understanding as to how the clock is impacted by FTD is obscured by the paucity of information on this topic.
Similar to other tauopathies, CK1δ was found in NFTs in PSP and associated with Pick bodies in PiD (Schwab et al., 2000). PSP patients share similar clinical phenotypes with PD patients, including poor sleep quality, obstructive sleep apnea, restless leg syndrome and difficulty falling asleep and staying asleep (Gama et al., 2010; Walsh et al., 2017). These conditions may exacerbate underlying circadian dysfunction, as restless leg syndrome was associated with sleep deficits in PSP. The circadian activity rhythms in PSP patients also have decreased amplitude and inter-daily stability compared to healthy controls (Walsh et al., 2016). Additionally, there is a positive correlation between PSP severity and lower relative and absolute amplitudes of circadian activity rhythms. However, there was no association between intra-daily rhythm variability or inter-daily rhythm stability and disease severity (Walsh et al., 2016). Therefore, it is unclear whether or not circadian dysfunction influences PSP severity.
Huntington’s disease is a rare genetic neurodegenerative disease in which the Huntingtin (HTT) has an abnormal expansion of 40 or more CAG trinucleotide repeats (Orr and Zoghbi, 2007). HD causes severe cognitive decline into dementia, locomotor impairments, circadian dysfunction, and psychotic episodes (Morton, 2013). Onset occurs during middle age and is generally fatal within 10 years of diagnosis. Like other neurodegenerative diseases, HD has no cure and shares similar sleep and circadian rhythm disturbances, which associate with greater cognitive impairment and depression (Aziz et al., 2010).
In Drosophila, TTFL proteins can affect the aggregation of mutant Huntingtin (mHTT) protein. A partial knockdown of Drosophila CLOCK (dClk), an ortholog of the mammalian Clock gene, is suppressed by mHTT aggregation. However, this neuroprotective effect was lost in the absence of Per, which was drastically reduced during its normal peak hours (Xu et al., 2019). Additionally, preclinical HD models show improvements with chronotherapy. For example, light therapy improves some forms of circadian desynchrony in several different HD mouse models (Wood et al., 2013; Ouk et al., 2017; Wang et al., 2017). After receiving 6 h of daily blue light exposure, two mouse models of HD, Q175 and BACHD, had improved locomotor activity, although sleep patterns were unaffected (Wang et al., 2017). The R6/2 mouse also has a positive response to light therapy. HD-induced circadian dysfunction was considerably delayed phase shifts (Wood et al., 2013). In one case, a prolonged photoperiod (16:8 LD) extended lifespan and improved nocturnal behavior rhythms while the opposite pattern (8:16 LD) decreased survival (Ouk et al., 2017). Interestingly, the mice were still able to adapt to 4-h phase advances and delays (Wood et al., 2013), indicating increased reliance on external cues during entrainment. However, a Drosophila model of HD demonstrated conflicting evidence. Mutant flies had abnormally prolonged expression of PER and TIMELESS (another component of the Drosophila negative arm) and hallmark circadian activity disturbances (Farago et al., 2019) indicating a disrupted TTFL In mammals, PER1 and PER2 are also important players in photic entrainment., and both are deregulated in symptomatic HD mice with retinal degeneration (Morton et al., 2005). As a result, mice have reduced light sensitivity. Furthermore, benzodiazepine treatment rescues abnormal PER2 expression in R2/6 mice, thus improving circadian function (Pallier et al., 2007).
The rhythmicity of peripheral oscillators is also affected by HD. Q175 mice have blunted cardiac and body temperature rhythms, impaired autonomic nervous system function, and a completely ablated sympathetic nervous system (Smarr et al., 2019). A newly emerging chronotherapy is time restricted feeding (TRF), in which eating is restricted to a 6- to 8-h time window (Manoogian and Panda, 2017). This method has seen some success in Q175 mice in the form of improved locomotor rhythms and restored autonomic nervous system functionality (Wang et al., 2018). TRF also restores peripheral clock gene expression and ameliorates metabolic dysfunction in the livers of R2/6 mice (Maywood et al., 2010). This HD model is characterized by irregular Cry and Per rhythms and the preliminary success of TRF indicates that these genes are positively affected by treatment.
Recently, melatonin has been found to be produced in neuronal mitochondria (Suofu et al., 2017). Melatonin is well known to be circadian regulated, and signals darkness to the SCN. In a study conducted by Jauhari et al. (2020), it is demonstrated that HD (AANAT knockout) mice exhibit increased levels of mtDNA release, cGAS activation, and inflammation, all of which can be modulated by exogenous melatonin. Melatonin deficient mice exhibited accelerated aging and neurodegenerative pathology. This mtDNA activation of neuronal proinflammatory response provides potential insight to the circadian involvement in HD (Suofu et al., 2017; Jauhari et al., 2020). Although the precise molecular relationship between CD and HD remains unclear, the association between the onset of HD and dysfunction of the molecular clock is not. CD likely contributes to the pathology of HD by way of chronic inflammation, mitochondrial dysfunction, and DNA damage. This suggests a codependent relationship between HD and CD, where HD onset may present with symptoms of CD, and CD can accelerate the development of HD (Kuljis et al., 2012).
As previously described, one important function of the clock is to mediate the timing of specific activities of the immune system. These essential activities include leukocyte recruitment, cytokine production, and cell proliferation (Scheiermann et al., 2013). The reverse is also true. In an excellent review, Segal et al. (2018) demonstrate that immune mediators can also alter clock gene expression. Several excellent sources have outlined the circadian influence over multiple cytokines, chemokines, and hormones linked to the pro-inflammatory response. CLOCK and CRY both activate protein complexes essential to the production of cytokines and chemokines, and BMAL1 can both inhibit CLOCK’s activation of these complexes and bind to e-boxes in promoter regions of immune mediator genes to produce rhythmic levels of cytokines and chemokines (Segal et al., 2018). These changes produce higher levels of cytokines and chemokines important to inflammatory responses during the active phase (Segal et al., 2018).
Neuroinflammation is integral to the onset and progression of several neurodegenerative diseases. In the healthy brain, neuroinflammation is kept in check by anti-inflammatory signals. This process is essential because there is little neurogenesis to replace damaged neurons in the adult brain. Thus, aging and disease-related processes that shift the balance toward pro-inflammatory signaling will increase neuron loss. A strong association between the neuroinflammatory response and the core clock has been established through Rev-Erbα. Recent studies have shown that Rev-Erbα is essential to microglial activation and neuroinflammation and is directly mediated by BMAL1. Microglia are the specialized immune cells of the central nervous system; in a healthy brain, they are primarily anti-inflammatory. Furthermore, microglia have a functional molecular clock and express PER1, PER2, Rev-Erbα, and BMAL1 in a rhythmic manner that is disrupted by aging (Fonken et al., 2016). In a study conducted by Griffin et al. (2019), Rev-Erbα had a strong connection to neuroinflammation by a series of in vivo and in vitro experiments. Rev-Erbα deletion induced spontaneous hippocampal microgliosis (microglia activation). Rev-Erbα deletion also exacerbated LPS-induced neuroinflammation. However, when injected twice daily with the pharmacological Rev-Erbα activator SR9009, wild type (WT) mice showed reduced levels of pro-inflammatory transcripts. Additionally, in a glial cell model containing microglia and astrocytes from postnatal WT mice, Rev-Erbα knockdown increased neuronal death when OS was induced with hydrogen peroxide (Griffin et al., 2019). However, results in the 5xFAD mouse model of β-amyloidosis show a conflicting role for REV-ERBs in microglial activation. Here, a dramatic decrease in Aβ plaque burden is shown in REV-ERBα deficient mice, with no impact on APP expression when compared to WT. Additionally, inhibition of REV-ERBα in vitro not only showed an overall increase in Aβ uptake, but that uptake is increased in a CT-dependent manner which correlates with upregulated BMAL1 expression (Lee et al., 2020). Together, these findings suggest that Rev-Erbα plays a pivotal neuroprotective role against OS and an excessive neuroinflammatory response, although this relationship is confounded by conflicting results concerning REV-ERB’s regulation of microglial activation.
There is a toxic cycle of protein accumulation and pro-inflammatory signaling that is exacerbated by clock dysfunction in neurodegenerative diseases. For example, tau NFTs are one source of the persistent pro-inflammatory response in neurodegeneration, as tau misfolding produces axon degeneration and eventual neuron death. It is postulated that protein unfolding events in several neurodegenerative disorders can also be triggered by microglial activation (Guzman-Martinez et al., 2019; Maccioni et al., 2020). This pro-inflammatory response serves as positive feedback for overstimulation of glial cells, which are thought to be involved in the excessive phosphorylation of tau (Guzman-Martinez et al., 2019). This increased neuroinflammatory stress accelerates tau pathology, and also gives rise to an alternative AD-specific conformation of astrocytes (Habib et al., 2020). Typically, astrocytes offer a neuroprotective function against excessive inflammation. However, altered functionality may have an effect on the function of the astrocytes, including toxic gains of function, or loss of function in neuronal support and Aβ plaque uptake and clearance (Garwood et al., 2017; Gonzalez-Reyes et al., 2017). Similar cycles have been demonstrated in all other proteinopathies discussed – toxic a-synuclein, TDP-43, and huntingtin proteins all drive neuroinflammation and are, in turn, exacerbated by the inflammatory response (Figure 2) (Fellner et al., 2011; Shi et al., 2010; Correia et al., 2015; Crotti and Glass, 2015; Lopes da Fonseca et al., 2015; Pearce et al., 2015; Jung and Chung, 2018; Spiller et al., 2018; Tremblay et al., 2019). Independent of which proteinopathy is being examined, the protein accumulation-driven neuroinflammatory response stimulates microglia to release cytokines that further drive a pro-inflammatory response, resulting in chronic inflammation and eventual neuronal death (Guzman-Martinez et al., 2019). It is also important to note the proposed circadian rhythmicity observed in glymphatic influx and clearance in mice, where drainage and influx of CSF from the cisterna magna appear to be time-of-day dependent (Hablitz et al., 2020). As previously demonstrated, the molecular clock exerts tight control over immune activity (Segal et al., 2018). The activation and inhibition mechanisms described by Segal et al. (2018), combined with the interactions of neuroinflammation, toxic protein aggregation and neuronal damage, suggest an overarching relationship between proteinopathy, neuroinflammatory stress, and the molecular clock. It is also important to remember that the clock is impacted by stress, as discussed earlier in this review. In a self-perpetuating cascade, circadian dysfunction both accelerates tau pathogenesis and is negatively impacted by tau toxicity and the chronic stress response. This feedback could explain the steep acceleration of cognitive decline often seen in laboratory models of proteinopathy-associated dementias (Baron et al., 2014).
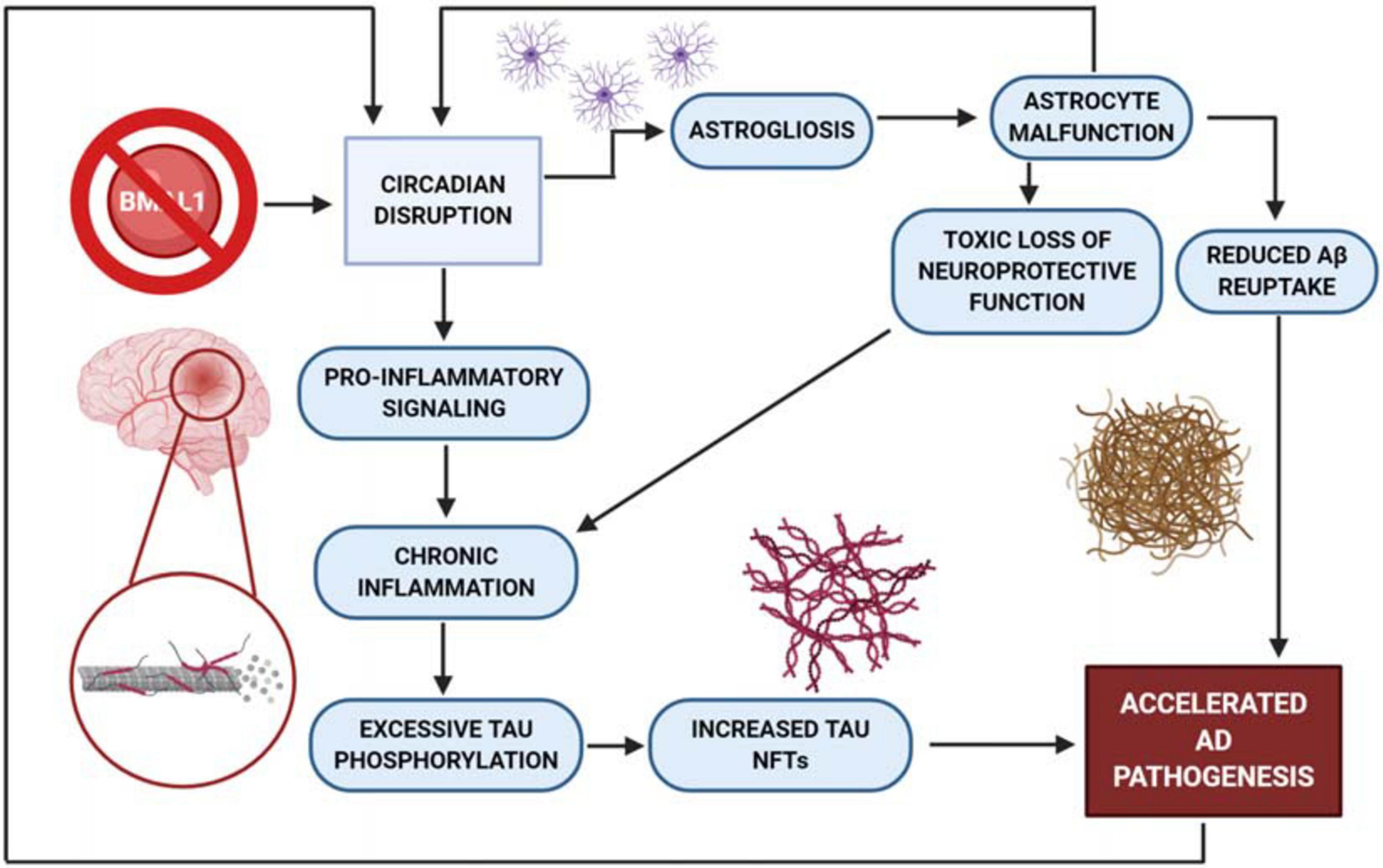
Figure 2. BMAL1 knockout mouse model produces increased astrogliosis. Knock out of BMAL1 drives excess astrogliosis, contributing to the acceleration of AD pathogenesis by way of astrocyte malfunction. This malfunction produces a loss of neuroprotection and reduces the astrocyte’s ability to transport Aβ out of the cell. Without protection from astrocytes, neuroinflammation then drives excessive pro-inflammatory signaling and tau phosphorylation, while producing an increased quantity of NFT’s. AD pathogenesis has also been shown to increase circadian dysfunction which further perpetuates neuronal damage and death.
This review summarizes findings from studies examining the effects of neurodegeneration and its associated pathology on the core clock (Table 1). Numerous findings indicate stress and neuroinflammation as a central process that links both circadian dysfunction and neurodegenerative disease (Figure 3). Perhaps the most compelling findings are the association of CK1δ with pathogenic tau deposits and the roles of Rev-Erbα in the neuroinflammatory response. Rev-Erbα presents itself as an interesting pharmacological treatment target to rescue neuronal damage, which would be an incredible step toward slowing neurodegenerative disease pathogenesis. The Rev-Erbα activator SR90009 has exhibited promising results in decreasing Aβ levels in the brain, improving synaptic health and axonal structure, as well as reversing cognitive defects in an AD mouse model (Roby et al., 2019). Additionally, our work demonstrating rescue of pathology, memory deficits, and circadian dysfunction in a mouse model of beta-amyloidosis by inhibiting CK1 activity suggests that targeting post-translational mechanisms is also a viable strategy – especially since kinases, such as CK1, that work on both pathogenic and clock mechanisms act as a nexus between the two processes, which potentially addresses the neurodegenerative disease pathogenesis and circadian dysfunction simultaneously.
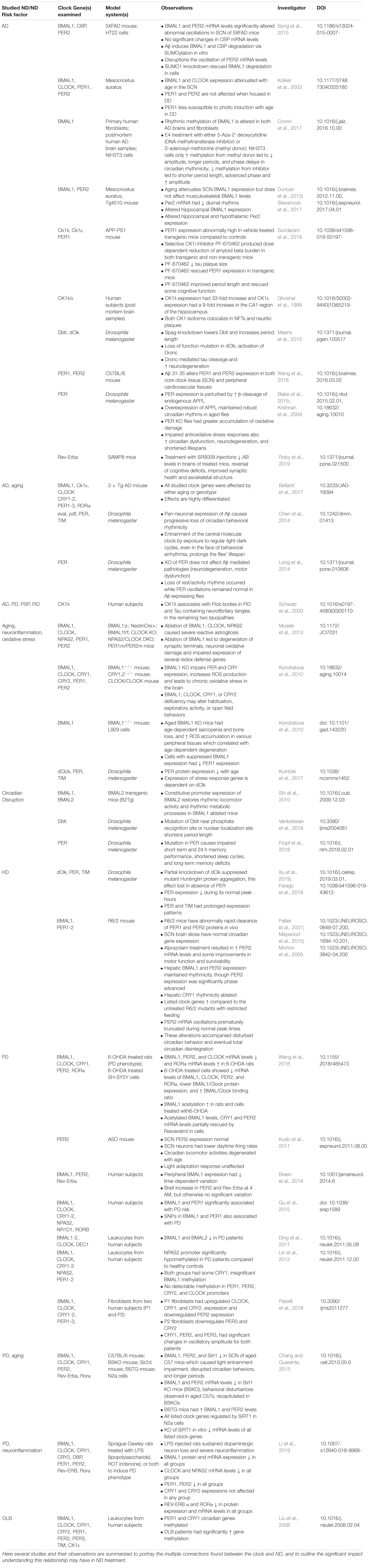
Table 1. ND’s and ND Risk factors have been associated with the molecular clock in numerous studies.
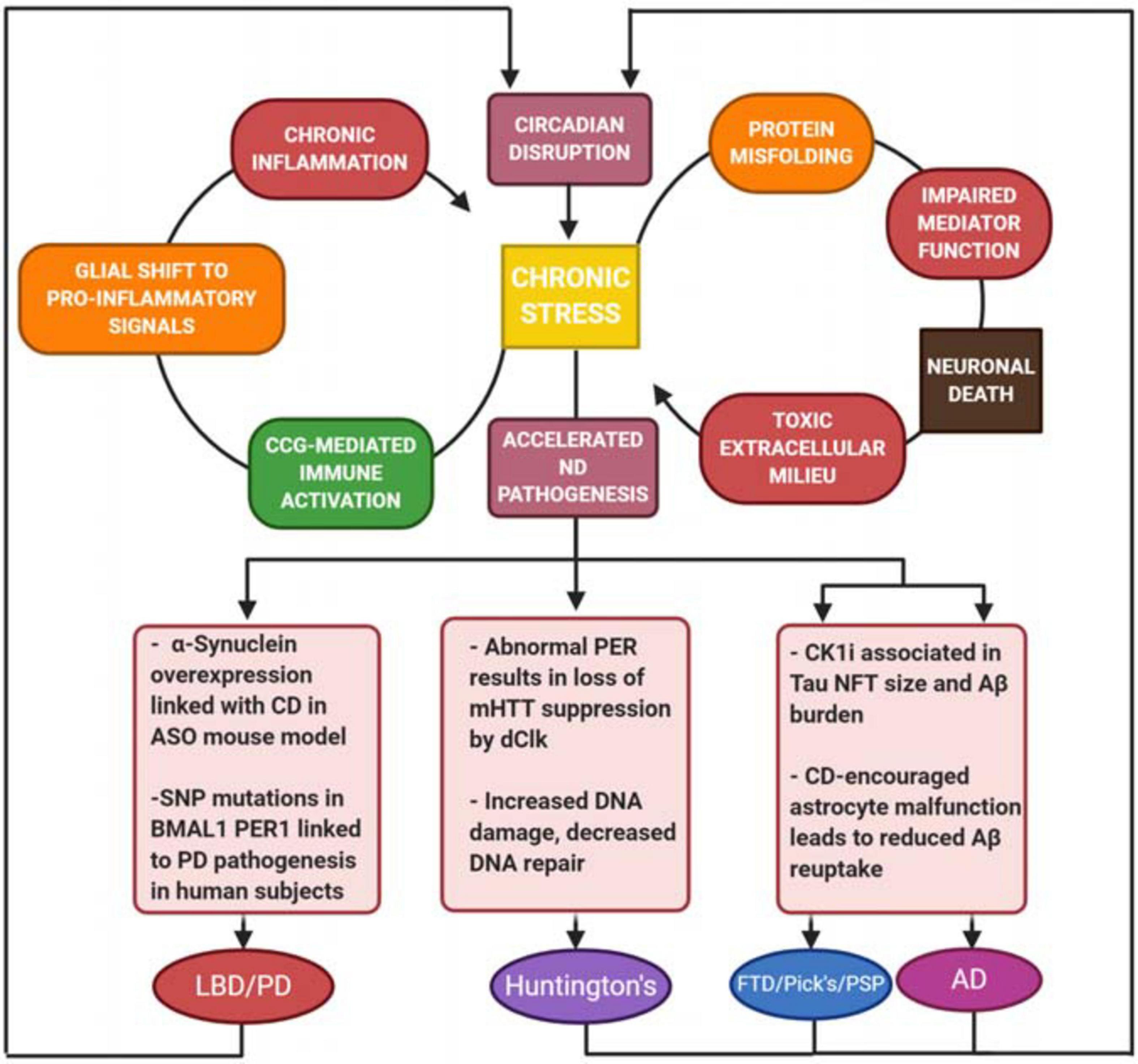
Figure 3. Harmful positive feedback as a driver for neurodegeneration. Stress and toxic extracellular milieu produced by neuronal death drives a chronic inflammatory response, as well as further breakdown of circadian function in cells. There is a common link between neurodegenerative disease (ND) and overactivity of the immune system, which plays a key role in each of the ND’s described. These conditions result in not only increased cell death, but deregulation of protein folding, increased DNA damage, astrocyte malfunction, and increased Aβ burden. There is a common link between ND and overactivity of the immune system, which plays a key role in each of the ND’s described. Additionally, symptoms of circadian dysfunction are often reported in ND patients, suggesting that the pathology of the disease also drives further CD, creating a devastating circular relationship between CD and neuronal damage.
Although significant progress has been made over the past decade, there are many lingering questions about the complex relationship between the molecular clock and the pathogenic mechanisms of neurodegeneration. Answers to these questions may lead to improved diagnosis, treatment, and management of these debilitating diseases.
HJ and BC wrote the early drafts and designed the figures. DG and JG were responsible for writing and editing the text and figures. All authors contributed to the article and approved the submitted version.
This work was supported by the Florida Department of Health Ed and Ethel Moore Alzheimer’s Disease Program (JG: 9AZ31; DG: 7AZ13).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank David Gonzalez for his critical review of our manuscript. All figures were generated using Biorender.com.
Anderson, K. N., Hatfield, C., Kipps, C., Hastings, M., and Hodges, J. R. (2009). Disrupted sleep and circadian patterns in frontotemporal dementia. Eur. J. Neurol. 16, 317–323. doi: 10.1111/j.1468-1331.2008.02414.x
Anne-Marie Chang, D. A., Duffy, J. F., and Czeisler, C. A. (2014). Evening use of light-emitting eReaders negatively affects sleep, circadian timing, and next-morning alertness. Proc. Natl. Acad. Sci. U S A. 112, 1232–1237. doi: 10.1073/pnas.1418490112
Asher, G., Gatfield, D., Stratmann, M., Reinke, H., Dibner, C., Kreppel, F., et al. (2008). SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell 134:4.
Aziz, N. A., Anguelova, G. V., Marinus, J., Lammers, G. J., and Roos, R. A. (2010). Sleep and circadian rhythm alterations correlate with depression and cognitive impairment in Huntington’s disease. Parkinsonism Relat. Disord. 16, 345–350. doi: 10.1016/j.parkreldis.2010.02.009
Bae, K., Jin, X., Maywood, E. S., Hastings, M. H., Reppert, S. M., and Weaver, D. R. (2001). Differential functions of mPer1, mPer2, and mPer3 in the SCN circadian clock. Neuron 30, 525–536. doi: 10.1016/s0896-6273(01)00302-6
Baggs, J. E., Price, T. S., DiTacchio, L., Panda, S., Fitzgerald, G. A., and Hogenesch, J. B. (2009). Network features of the mammalian circadian clock. PLoS Biol. 7:e52. doi: 10.1371/journal.pbio.1000052
Baker, C. L., Loros, J. J., and Dunlap, J. C. (2012). The circadian clock of Neurospora crassa. FEMS Microbiol. Rev. 36, 95–110.
Baron, R., Babcock, A. A., Nemirovsky, A., Finsen, B., and Monsonego, A. (2014). Accelerated microglial pathology is associated with Abeta plaques in mouse models of Alzheimer’s disease. Aging Cell 13, 584–595. doi: 10.1111/acel.12210
Bass, J., and Takahashi, J. S. (2010). Circadian integration of metabolism and energetics. Science 330, 1349–1354. doi: 10.1126/science.1195027
Beesley, S., Kim, D. W., D’Alessandro, M., Jin, Y., Lee, K., Joo, H., et al. (2017). Wake-sleep cycles are severely disrupted by diseases affecting cytoplasmic homeostasis. Proc. Natl. Acad. Sci. U S A. 117, 28402–28411. doi: 10.1073/pnas.2003524117
Bellanti, F., Iannelli, G., Blonda, M., Tamborra, R., Villani, R., Romano, A., et al. (2017). Alterations of clock gene RNA expression in brain regions of a triple transgenic model of Alzheimer’s Disease. J. Alzheimers. Dis. 59, 615–631. doi: 10.3233/jad-160942
Blake, M. R., Holbrook, S. D., Kotwica-Rolinska, J., Chow, E. S., Kretzschmar, D., and Giebultowicz, J. M. (2015). Manipulations of amyloid precursor protein cleavage disrupt the circadian clock in aging Drosophila. Neurobiol. Dis. 77, 117–126. doi: 10.1016/j.nbd.2015.02.012
Bliwise, D. L., Watts, R. L., Watts, N., Rye, D. B., Irbe, D., and Hughes, M. (1995). Disruptive nocturnal behavior in Parkinson’s disease and Alzheimer’s disease. J. Geriatr. Psychiatry Neurol. 8, 107–110.
Bodea, L. G., Eckert, A., Ittner, L. M., Piguet, O., and Gotz, J. (2016). Tau physiology and pathomechanisms in frontotemporal lobar degeneration. J. Neurochem. 138(Suppl. 1), 71–94. doi: 10.1111/jnc.13600
Brancaccio, M., Patton, A. P., Chesham, J. E., Maywood, E. S., and Hastings, M. H. (2017). Astrocytes control circadian timekeeping in the suprachiasmatic nucleus via glutamatergic signaling. Neuron 93, 1420–1435.e5.
Breen, D. P., Vuono, R., Nawarathna, U., Fisher, K., Shneerson, J. M., Reddy, A. B., et al. (2014). Sleep and circadian rhythm regulation in early Parkinson disease. JAMA Neurol. 71, 589–595. doi: 10.1001/jamaneurol.2014.65
Brown, G. M. (1994). Light, melatonin and the sleep-wake cycle. J. Psychiatry Neurosci. 19, 345–353.
Buhl, E., Higham, J. P., and Hodge, J. J. L. (2019). Alzheimer’s disease-associated tau alters Drosophila circadian activity, sleep and clock neuron electrophysiology. Neurobiol. Dis. 130:104507. doi: 10.1016/j.nbd.2019.104507
Busche, M. A., and Hyman, B. T. (2020). Synergy between amyloid-β and tau in Alzheimer’s disease. Nat. Neurosci. 23, 1183–1193. doi: 10.1038/s41593-020-0687-6
Cagnin, A., Fragiacomo, F., Camporese, G., Turco, M., Busse, C., Ermani, M., et al. (2017). Sleep-Wake profile in dementia with lewy bodies, Alzheimer’s Disease, and normal aging. J. Alzheimers. Dis. 55, 1529–1536. doi: 10.3233/jad-160385
Caillet-Boudin, M. L., Buee, L., Sergeant, N., and Lefebvre, B. (2015). Regulation of human MAPT gene expression. Mol. Neurodegener. 10:28.
Canevelli, M., Valletta, M., Trebbastoni, A., Sarli, G., D’Antonio, F., Tariciotti, L., et al. (2016). Sundowning in dementia: clinical relevance, pathophysiological determinants, and therapeutic approaches. Front. Med. (Lausanne) 3:73. doi: 10.3389/fmed.2016.00073
Chang, H. C., and Guarente, L. (2013). SIRT1 mediates central circadian control in the SCN by a mechanism that decays with aging. Cell 153:1448. doi: 10.1016/j.cell.2013.05.027
Chang, Y. G., Cohen, S. E., Phong, C., Myers, W. K., Kim, Y. I., Tseng, R., et al. (2015). Circadian rhythms. a protein fold switch joins the circadian oscillator to clock output in cyanobacteria. Science 349, 324–328. doi: 10.1126/science.1260031
Chen, C., Gu, J., Basurto-Islas, G., Jin, N., Wu, F., Gong, C. X., et al. (2017). Up-regulation of casein kinase 1epsilon is involved in tau pathogenesis in Alzheimer’s disease. Sci. Rep. 7:13478.
Chen, K. F., Possidente, B., Lomas, D. A., and Crowther, D. C. (2014). The central molecular clock is robust in the face of behavioural arrhythmia in a Drosophila model of Alzheimer’s disease. Dis. Model. Mech. 7, 445–458. doi: 10.1242/dmm.014134
Chen, Z., and Zhong, C. (2014). Oxidative stress in Alzheimer’s disease. Neurosci. Bull. 30, 271–281.
Cohen, S. E., and Golden, S. S. (2015). Circadian rhythms in cyanobacteria. Microbiol. Mol. Biol. Rev. 79, 373–385.
Correia, A. S., Patel, P., Dutta, K., and Julien, J.-P. (2015). Inflammation induces TDP-43 mislocalization and aggregation. PLoS One 10:e0140248. doi: 10.1371/journal.pone.0140248
Cronin, P., McCarthy, M. J., Lim, A. S. P., Salmon, D. P., Galasko, D., Masliah, E., et al. (2017). Circadian alterations during early stages of Alzheimer’s disease are associated with aberrant cycles of DNA methylation in BMAL1. Alzheimers Dement 13, 689–700. doi: 10.1016/j.jalz.2016.10.003
Crotti, A., and Glass, C. K. (2015). The choreography of neuroinflammation in Huntington’s disease. Trends Immunol. 36, 364–373. doi: 10.1016/j.it.2015.04.007
Dedeene, L., Van Schoor, E., Vandenberghe, R., Van Damme, P., Poesen, K., and Thal, D. R. (2019). Circadian sleep/wake-associated cells show dipeptide repeat protein aggregates in C9orf72-related ALS and FTLD cases. Acta Neuropathol. Commun. 7:189.
Dibner, C., Schibler, U., and Albrecht, U. (2010). The mammalian circadian timing system: organization and coordination of central and peripheral clocks. Annu. Rev. Physiol. 72, 517–549. doi: 10.1146/annurev-physiol-021909-135821
Ding, H., Liu, S., Yuan, Y., Lin, Q., Chan, P., and Cai, Y. (2011). Decreased expression of Bmal2 in patients with Parkinson’s disease. Neurosci. Lett. 499, 186–188. doi: 10.1016/j.neulet.2011.05.058
Dissel, S., Melnattur, K., and Shaw, P. J. (2015). Sleep, performance, and memory in flies. Curr. Sleep Med. Rep. 1, 47–54. doi: 10.1007/s40675-014-0006-4
Duncan, M. J., Prochot, J. R., Cook, D. H., Tyler Smith, J., and Franklin, K. M. (2013). Influence of aging on Bmal1 and Per2 expression in extra-SCN oscillators in hamster brain. Brain Res. 1491, 44–53. doi: 10.1016/j.brainres.2012.11.008
Dunlap, J. C., and Loros, J. J. (2017). Making time: conservation of biological clocks from fungi to animals. Microbiol. Spectr. 5:1128/microbiolspec.FUNK-0039-2016.
Dunlap, J. C., Loros, J. J., and DeCoursey, P. J. (2004). Chronobiology : Biological Timekeeping Sinauer Associates. Sunderland: University of Sunderland.
Etchegaray, J. P., Machida, K. K., Noton, E., Constance, C. M., Dallmann, R., Di Napoli, M. N., et al. (2009). Casein kinase 1 delta regulates the pace of the mammalian circadian clock. Mol. Cell. Biol. 29, 3853–3866. doi: 10.1128/mcb.00338-09
Farago, A., Zsindely, N., and Bodai, L. (2019). Mutant huntingtin disturbs circadian clock gene expression and sleep patterns in Drosophila. Sci. Rep. 9:7174.
Fellner, L., Jellinger, K. A., Wenning, G. K., and Stefanova, N. (2011). Glial dysfunction in the pathogenesis of α-synucleinopathies: emerging concepts. Acta Neuropathol. 121:675. doi: 10.1007/s00401-011-0833-z
Fonken, L. K., Kitt, M. M., Gaudet, A. D., Barrientos, R. M., Watkins, L. R., and Maier, S. F. (2016). Diminished circadian rhythms in hippocampal microglia may contribute to age-related neuroinflammatory sensitization. Neurobiol. Aging 47, 102–112. doi: 10.1016/j.neurobiolaging.2016.07.019
Fropf, R., Zhou, H., and Yin, J. C. P. (2018). The clock gene period differentially regulates sleep and memory in Drosophila. Neurobiol. Learn. Mem. 153, 2–12. doi: 10.1016/j.nlm.2018.02.016
Gama, R. L., Tavora, D. G., Bomfim, R. C., Silva, C. E., de Bruin, V. M., and de Bruin, P. F. (2010). Sleep disturbances and brain MRI morphometry in Parkinson’s disease, multiple system atrophy and progressive supranuclear palsy - a comparative study. Parkinsonism Relat. Disord. 16, 275–279. doi: 10.1016/j.parkreldis.2010.01.002
Gan, Q., Yao, H., Na, H., Ballance, H., Tao, Q., Leung, L., et al. (2019). Effects of amylin against amyloid-β-induced tauopathy and synapse loss in primary neurons. J. Alzheimer’s Dis. 70, 1025–1040. doi: 10.3233/jad-190161
Garwood, C. J., Ratcliffe, L. E., Simpson, J. E., Heath, P. R., Ince, P. G., and Wharton, S. B. (2017). Review: astrocytes in Alzheimer’s disease and other age-associated dementias: a supporting player with a central role. Neuropathol. Appl. Neurobiol. 43, 281–298. doi: 10.1111/nan.12338
Ghoshal, N., Smiley, J. F., DeMaggio, A. J., Hoekstra, M. F., Cochran, E. J., Binder, L. I., et al. (1999). A new molecular link between the fibrillar and granulovacuolar lesions of Alzheimer’s disease. Am. J. Pathol. 155, 1163–1172. doi: 10.1016/s0002-9440(10)65219-4
Gomes, M. J., Martinez, P. F., Pagan, L. U., Damatto, R. L., Cezar, M. D. M., Lima, A. R. R., et al. (2017). Skeletal muscle aging: influence of oxidative stress and physical exercise. Oncotarget 8, 20428–20440.
Gonzalez-Reyes, R. E., Nava-Mesa, M. O., Vargas-Sanchez, K., Ariza-Salamanca, D., and Mora-Munoz, L. (2017). Involvement of astrocytes in Alzheimer’s Disease from a neuroinflammatory and oxidative stress perspective. Front. Mol. Neurosci. 10:427. doi: 10.3389/fnmol.2017.00427
Griffin, P., Dimitry, J. M., Sheehan, P. W., Lananna, B. V., Guo, C., Robinette, M. L., et al. (2019). Circadian clock protein Rev-erbalpha regulates neuroinflammation. Proc. Natl. Acad. Sci. U S A. 116, 5102–5107.
Gu, J., Hu, W., Tan, X., Qu, S., Chu, D., Gong, C. X., et al. (2020). Elevation of casein kinase 1ε associated with TDP-43 and tau pathologies in Alzheimer’s disease. Brain Pathol. (Zurich, Switzerland) 30, 283–297. doi: 10.1111/bpa.12775
Gu, Z., Wang, B., Zhang, Y. B., Ding, H., Zhang, Y., Yu, J., et al. (2015). Association of ARNTL and PER1 genes with Parkinson’s disease: a case-control study of Han Chinese. Sci. Rep. 5:15891.
Guzman-Martinez, L., Maccioni, R. B., Andrade, V., Navarrete, L. P., Pastor, M. G., and Ramos-Escobar, N. (2019). Neuroinflammation as a common feature of neurodegenerative disorders. Front. Pharmacol. 10:1008. doi: 10.3389/fphar.2019.01008
Habib, N., McCabe, C., Medina, S., Varshavsky, M., Kitsberg, D., Dvir-Szternfeld, R., et al. (2020). Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci. 23, 701–706.
Hablitz, L. M., Pla, V., Giannetto, M., Vinitsky, H. S., Staeger, F. F., Metcalfe, T., et al. (2020). Circadian control of brain glymphatic and lymphatic fluid flow. Nat. Commun. 11:4411.
Harding, E. C., Franks, N. P., and Wisden, W. (2019). The temperature dependence of sleep. Front. Neurosci. 13:336. doi: 10.3389/fnins.2019.00336
Harper, D. G., Stopa, E. G., McKee, A. C., Satlin, A., Fish, D., and Volicer, L. (2004). Dementia severity and Lewy bodies affect circadian rhythms in Alzheimer disease. Neurobiol. Aging 25, 771–781. doi: 10.1016/j.neurobiolaging.2003.04.009
Hasegawa, S., Fukushima, H., Hosoda, H., Serita, T., Ishikawa, R., Rokukawa, T., et al. (2019). Hippocampal clock regulates memory retrieval via dopamine and PKA-induced GluA1 phosphorylation. Nat. Commun. 10:5766.
Hebert, L. E., Weuve, J., Scherr, P. A., and Evans, D. A. (2013). Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology 80, 1778–1783. doi: 10.1212/wnl.0b013e31828726f5
Iadecola, C. (2003). Atherosclerosis and neurodegeneration: unexpected conspirators in Alzheimer’s dementia. Arterioscler. Thromb. Vasc. Biol. 23, 1951–1953. doi: 10.1161/01.atv.0000102660.99744.85
Irwin, D. J. (2016). Tauopathies as clinicopathological entities. Parkinsonism Relat. Disord. 22(Suppl. 1), S29–S33.
Jauhari, A., Baranov, S. V., Suofu, Y., Kim, J., Singh, T., Yablonska, S., et al. (2020). Melatonin inhibits cytosolic mitochondrial DNA-induced neuroinflammatory signaling in accelerated aging and neurodegeneration. J. Clin. Invest. 130, 3124–3136. doi: 10.1172/jci135026
Jung, Y.-J., and Chung, W.-S. (2018). Phagocytic roles of glial cells in healthy and diseased brains. Biomol. Therapeut. 26:350. doi: 10.4062/biomolther.2017.133
Koh, K., Evans, J. M., Hendricks, J. C., and Sehgal, A. (2006). A Drosophila model for age-associated changes in sleep:wake cycles. Proc. Natl. Acad. Sci. U S A. 103, 13843–13847.
Kolker, D. E., Fukuyama, H., Huang, D. S., Takahashi, J. S., Horton, T. H., and Turek, F. W. (2003). Aging alters circadian and light-induced expression of clock genes in golden hamsters. J. Biol. Rhythms 18, 159–169. doi: 10.1177/0748730403251802
Kondratova, A. A., Dubrovsky, Y. V., Antoch, M. P., and Kondratov, R. V. (2010). Circadian clock proteins control adaptation to novel environment and memory formation. Aging (Albany NY) 2, 285–297. doi: 10.18632/aging.100142
Kress, G. J., Liao, F., Dimitry, J., Cedeno, M. R., FitzGerald, G. A., Holtzman, D. M., et al. (2018). Regulation of amyloid-beta dynamics and pathology by the circadian clock. J. Exp. Med. 215, 1059–1068. doi: 10.1084/jem.20172347
Krishnan, N., Kretzschmar, D., Rakshit, K., Chow, E., and Giebultowicz, J. M. (2009). The circadian clock gene period extends healthspan in aging Drosophila melanogaster. Aging (Albany NY) 1, 937–948. doi: 10.18632/aging.100103
Krishnan, N., Rakshit, K., Chow, E. S., Wentzell, J. S., Kretzschmar, D., and Giebultowicz, J. M. (2012). Loss of circadian clock accelerates aging in neurodegeneration-prone mutants. Neurobiol. Dis. 45, 1129–1135. doi: 10.1016/j.nbd.2011.12.034
Kudo, T., Loh, D. H., Truong, D., Wu, Y., and Colwell, C. S. (2011). Circadian dysfunction in a mouse model of Parkinson’s disease. Exp. Neurol. 232, 66–75. doi: 10.1016/j.expneurol.2011.08.003
Kuintzle, R. C., Chow, E. S., Westby, T. N., Gvakharia, B. O., Giebultowicz, J. M., and Hendrix, D. A. (2017). Circadian deep sequencing reveals stress-response genes that adopt robust rhythmic expression during aging. Nat. Commun. 8:14529.
Kuljis, D., Schroeder, A. M., Kudo, T., Loh, D. H., Willison, D. L., and Colwell, C. S. (2012). Sleep and circadian dysfunction in neurodegenerative disorders: insights from a mouse model of Huntington’s disease. Minerva Pneumol. 51, 93–106.
Kundermann, B., Thum, A., Rocamora, R., Haag, A., Krieg, J. C., and Hemmeter, U. (2011). Comparison of polysomnographic variables and their relationship to cognitive impairment in patients with Alzheimer’s disease and frontotemporal dementia. J. Psychiatr. Res. 45, 1585–1592. doi: 10.1016/j.jpsychires.2011.07.008
Lananna, B. V., McKee, C. A., King, M. W., Del-Aguila, J. L., Dimitry, J. M., Farias, F. H. G., et al. (2020). Chi3l1/YKL-40 is controlled by the astrocyte circadian clock and regulates neuroinflammation and Alzheimer’s disease pathogenesis. Sci. Transl. Med. 12:eaax3519. doi: 10.1126/scitranslmed.aax3519
Lananna, B. V., and Musiek, E. S. (2020). The wrinkling of time: aging, inflammation, oxidative stress, and the circadian clock in neurodegeneration. Neurobiol. Dis. 139:104832. doi: 10.1016/j.nbd.2020.104832
Lauretti, E., Di Meco, A., Merali, S., and Pratico, D. (2017). Circadian rhythm dysfunction: a novel environmental risk factor for Parkinson’s disease. Mol. Psychiatry 22, 280–286. doi: 10.1038/mp.2016.47
Lee, H. M., Chen, R., Kim, H., Etchegaray, J. P., Weaver, D. R., and Lee, C. (2011). The period of the circadian oscillator is primarily determined by the balance between casein kinase 1 and protein phosphatase 1. Proc. Natl. Acad. Sci. U S A. 108, 16451–16456. doi: 10.1073/pnas.1107178108
Lee, J., Kim, D. E., Griffin, P., Sheehan, P. W., Kim, D. H., Musiek, E. S., et al. (2020). Inhibition of REV-ERBs stimulates microglial amyloid-beta clearance and reduces amyloid plaque deposition in the 5XFAD mouse model of Alzheimer’s disease. Aging Cell 19, e13078.
Lee, V. M., Brunden, K. R., Hutton, M., and Trojanowski, J. Q. (2011). Developing therapeutic approaches to tau, selected kinases, and related neuronal protein targets. Cold Spring Harb. Perspect. Med. 1:a006437. doi: 10.1101/cshperspect.a006437
Li, G., Yin, H., and Kuret, J. (2004). Casein kinase 1 delta phosphorylates tau and disrupts its binding to microtubules. J. Biol. Chem. 279, 15938–15945. doi: 10.1074/jbc.m314116200
Li, H., Song, S., Wang, Y., Huang, C., Zhang, F., Liu, J., et al. (2019). Low-Grade inflammation aggravates rotenone neurotoxicity and disrupts circadian clock gene expression in rats. Neurotox. Res. 35, 421–431. doi: 10.1007/s12640-018-9968-1
Li, J. Z., Bunney, B. G., Meng, F., Hagenauer, M. H., Walsh, D. M., and Vawter, M. P. (2013). Circadian patterns of gene expression in the human brain and disruption in major depressive disorder. PNAS 110, 9950–9955.
Lin, Q., Ding, H., Zheng, Z., Gu, Z., Ma, J., Chen, L., et al. (2012). Promoter methylation analysis of seven clock genes in Parkinson’s disease. Neurosci. Lett. 507, 147–150. doi: 10.1016/j.neulet.2011.12.007
Liu, H. C., Hu, C. J., Tang, Y. C., and Chang, J. G. (2008). A pilot study for circadian gene disturbance in dementia patients. Neurosci. Lett. 435, 229–233. doi: 10.1016/j.neulet.2008.02.041
Liu, W. W., Wei, S. Z., Huang, G. D., Liu, L. B., Gu, C., Shen, Y., et al. (2020). BMAL1 regulation of microglia-mediated neuroinflammation in MPTP-induced Parkinson’s disease mouse model. FASEB J. 34, 6570–6581. doi: 10.1096/fj.201901565rr
Logan, R. W., and McClung, C. A. (2019). Rhythms of life: circadian disruption and brain disorders across the lifespan. Nat. Rev. Neurosci. 20, 49–65. doi: 10.1038/s41583-018-0088-y
Long, D. M., Blake, M. R., Dutta, S., Holbrook, S. D., Kotwica-Rolinska, J., Kretzschmar, D., et al. (2014). Relationships between the circadian system and Alzheimer’s disease-like symptoms in Drosophila. PLoS One 9:e106068. doi: 10.1371/journal.pone.0106068
Lopes da Fonseca, T., Villar-Piqué, A., and Outeiro, T. F. (2015). The interplay between alpha-synuclein clearance and spreading. Biomolecules 5, 435–471. doi: 10.3390/biom5020435
Luo, Y., Bolon, B., Kahn, S., Bennett, B. D., Babu-Khan, S., Denis, P., et al. (2001). Mice deficient in BACE1, the Alzheimer’s β-secretase, have normal phenotype and abolished β-amyloid generation. Nat. Neurosci. 4, 231–232. doi: 10.1038/85059
Ma, J., Gao, J., Wang, J., and Xie, A. (2019). Prion-Like mechanisms in Parkinson’s Disease. Front. Neurosci. 13:552. doi: 10.3389/fnins.2019.00552
Ma, Z., Jiang, W., and Zhang, E. E. (2016). Orexin signaling regulates both the hippocampal clock and the circadian oscillation of Alzheimer’s disease-risk genes. Sci. Rep. 6:36035.
Maccioni, R. B., Navarrete, L. P., Gonzalez, A., Gonzalez-Canacer, A., Guzman-Martinez, L., and Cortes, N. (2020). Inflammation: a major target for compounds to control Alzheimer’s Disease. J. Alzheimers. Dis. 76, 1199–1213.
Maemura, K., Takeda, N., and Nagai, R. (2007). Circadian rhythms in the CNS and peripheral clock disorders: role of the biological clock in cardiovascular diseases. J. Pharmacol. Sci. 103, 134–138. doi: 10.1254/jphs.fmj06003x2
Manoogian, E. N. C., and Panda, S. (2017). Circadian rhythms, time-restricted feeding, and healthy aging. Ageing Res. Rev. 39, 59–67. doi: 10.1016/j.arr.2016.12.006
Martini-Stoica, H., Xu, Y., Ballabio, A., and Zheng, H. (2016). The autophagy-lysosomal pathway in neurodegeneration: a TFEB perspective. Trends Neurosci. 39, 221–234. doi: 10.1016/j.tins.2016.02.002
Masri, S., and Sassone-Corsi, P. (2010). Plasticity and specificity of the circadian epigenome. Nat. Neurosci. 13, 1324–1329. doi: 10.1038/nn.2668
Maywood, E. S., Fraenkel, E., McAllister, C. J., Wood, N., Reddy, A. B., Hastings, M. H., et al. (2010). Disruption of peripheral circadian timekeeping in a mouse model of Huntington’s disease and its restoration by temporally scheduled feeding. J. Neurosci. 30, 10199–10204. doi: 10.1523/jneurosci.1694-10.2010
Means, J. C., Venkatesan, A., Gerdes, B., Fan, J. Y., Bjes, E. S., and Price, J. L. (2015). Drosophila spaghetti and doubletime link the circadian clock and light to caspases, apoptosis and tauopathy. PLoS Genet. 11:e1005171. doi: 10.1371/journal.pgen.1005171
Mecocci, P., Boccardi, V., Cecchetti, R., Bastiani, P., Scamosci, M., Ruggiero, C., et al. (2018). A long journey into aging, brain aging, and Alzheimer’s Disease following the oxidative stress tracks. J. Alzheimers. Dis. 62, 1319–1335. doi: 10.3233/jad-170732
Menzies, F. M., Fleming, A., Caricasole, A., Bento, C. F., Andrews, S. P., Ashkenazi, A., et al. (2017). Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron 93, 1015–1034. doi: 10.1016/j.neuron.2017.01.022
Menzies, F. M., Fleming, A., and Rubinsztein, D. C. (2015). Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 16, 345–357. doi: 10.1038/nrn3961
Moore, R. Y. (2013). The suprachiasmatic nucleus and the circadian timing system. Prog. Mol. Biol. Transl. Sci. 119, 1–28. doi: 10.1016/b978-0-12-396971-2.00001-4
Morton, A. J. (2013). Circadian and sleep disorder in Huntington’s disease. Exp. Neurol. 243, 34–44.
Morton, A. J., Wood, N. I., Hastings, M. H., Hurelbrink, C., Barker, R. A., and Maywood, E. S. (2005). Disintegration of the sleep-wake cycle and circadian timing in Huntington’s disease. J. Neurosci. 25, 157–163. doi: 10.1523/jneurosci.3842-04.2005
Musiek, E. S. (2015). Circadian clock disruption in neurodegenerative diseases: cause and effect? Front. Pharmacol. 6:29. doi: 10.3389/fphar.2015.00029
Musiek, E. S., and Holtzman, D. M. (2016). Mechanisms linking circadian clocks, sleep, and neurodegeneration. Science 354, 1004–1008. doi: 10.1126/science.aah4968
Musiek, E. S., Lim, M. M., Yang, G., Bauer, A. Q., Qi, L., Lee, Y., et al. (2013). Circadian clock proteins regulate neuronal redox homeostasis and neurodegeneration. J. Clin. Invest. 123, 5389–5400. doi: 10.1172/jci70317
Nonaka, T., Masai, H., and Hasegawa, M. (2014). Phosphorylation of tdp-43 by casein kinase 1 delta facilitates mislocalization and intracellular aggregate formation of TDP-43. Alzheimer’s Dement. 10:790.
Ooms, S., and Ju, Y.-E. (2016). Treatment of sleep disorders in dementia. Curr. Treatment Options Neurol. 18:40.
Orr, H. T., and Zoghbi, H. Y. (2007). Trinucleotide repeat disorders. Annu. Rev. Neurosci. 30, 575–621.
Ouk, K., Aungier, J., and Morton, A. J. (2017). Prolonged day length exposure improves circadian deficits and survival in a transgenic mouse model of Huntington’s disease. Neurobiol. Sleep Circadian Rhythms 2, 27–38. doi: 10.1016/j.nbscr.2016.11.004
Oumata, N., Bettayeb, K., Ferandin, Y., Demange, L., Lopez-Giral, A., Goddard, M. L., et al. (2008). Roscovitine-derived, dual-specificity inhibitors of cyclin-dependent kinases and casein kinases 1. J. Med. Chem. 51, 5229–5242. doi: 10.1021/jm800109e
Pacelli, C., Rotundo, G., Lecce, L., Menga, M., Bidollari, E., Scrima, R., et al. (2019). Parkin mutation affects clock gene-dependent energy metabolism. Int. J. Mol. Sci. 20:2772. doi: 10.3390/ijms20112772
Pallier, P. N., Maywood, E. S., Zheng, Z., Chesham, J. E., Inyushkin, A. N., Dyball, R., et al. (2007). Pharmacological imposition of sleep slows cognitive decline and reverses dysregulation of circadian gene expression in a transgenic mouse model of Huntington’s disease. J. Neurosci. 27, 7869–7878. doi: 10.1523/jneurosci.0649-07.2007
Partch, C. L., Green, C. B., and Takahashi, J. S. (2014). Molecular architecture of the mammalian circadian clock. Trends Cell Biol. 24, 90–99. doi: 10.1016/j.tcb.2013.07.002
Patke, A., Young, M. W., and Axelrod, S. (2020). Molecular mechanisms and physiological importance of circadian rhythms. Nat. Rev. Mol. Cell Biol. 21, 67–84. doi: 10.1038/s41580-019-0179-2
Patrick-Simon Welz, S. A. B. (2020). Molecular connections between circadian clocks and aging. J. Mol. Biol. 432, 3661–3679. doi: 10.1016/j.jmb.2019.12.036
Pearce, M. M., Spartz, E. J., Hong, W., Luo, L., and Kopito, R. R. (2015). Prion-like transmission of neuronal huntingtin aggregates to phagocytic glia in the Drosophila brain. Nat. Commun. 6:6768.
Petit, D., Montplaisir, J., Louis, E. K. S., and Boeve, B. F. (2017). Principles and Practice of Sleep Medicine. Amsterdam: Elsevier Inc.,.
Richards, J., and Gumz, M. L. (2012). Advances in understanding the peripheral circadian clocks. FASEB J. 26, 3602–3613. doi: 10.1096/fj.12-203554
Roby, D. A., Ruiz, F., Kermath, B. A., Voorhees, J. R., Niehoff, M., Zhang, J., et al. (2019). Pharmacological activation of the nuclear receptor REV-ERB reverses cognitive deficits and reduces amyloid-beta burden in a mouse model of Alzheimer’s disease. PLoS One 14:e0215004. doi: 10.1371/journal.pone.0215004
Roher, A. E., Garami, Z., Tyas, S. L., Maarouf, C. L., Kokjohn, T. A., Belohlavek, M., et al. (2011). Transcranial doppler ultrasound blood flow velocity and pulsatility index as systemic indicators for Alzheimer’s disease. Alzheimers Dement 7, 445–455. doi: 10.1016/j.jalz.2010.09.002
Sahar, S., and Sassone-Corsi, P. (2009). Metabolism and cancer: the circadian clock connection. Nat. Rev. Cancer 9, 886–896. doi: 10.1038/nrc2747
Sani, T. P., Bond, R. L., Marshall, C. R., Hardy, C. J. D., Russell, L. L., Moore, K. M., et al. (2019). Sleep symptoms in syndromes of frontotemporal dementia and Alzheimer’s disease: a proof-of-principle behavioural study. eNeurologicalSci 17:100212. doi: 10.1016/j.ensci.2019.100212
Scheiermann, C., Kunisaki, Y., and Frenette, P. S. (2013). Circadian control of the immune system. Nat. Rev. Immunol. 13, 190–198. doi: 10.1038/nri3386
Schwab, C., DeMaggio, A. J., Ghoshal, N., Binder, L. I., Kuret, J., and McGeer, P. L. (2000). Casein kinase 1 delta is associated with pathological accumulation of tau in several neurodegenerative diseases. Neurobiol. Aging 21, 503–510. doi: 10.1016/s0197-4580(00)00110-x
Scott, D., and Roy, S. (2012). alpha-Synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. J. Neurosci. 32, 10129–10135. doi: 10.1523/jneurosci.0535-12.2012
Segal, J. P., Tresidder, K. A., Bhatt, C., Gilron, I., and Ghasemlou, N. (2018). Circadian control of pain and neuroinflammation. J. Neurosci. Res. 96, 1002–1020. doi: 10.1002/jnr.24150
Serio, A., Bilican, B., Barmada, S. J., Ando, D. M., Zhao, C., Siller, R., et al. (2013). Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of TDP-43 proteinopathy. Proc. Natl. Acad. Sci. U S A. 110, 4697–4702. doi: 10.1073/pnas.1300398110
Shi, S., Hida, A., McGuinness, O. P., Wasserman, D. H., Yamazaki, S., and Johnson, C. H. (2010). Circadian clock gene Bmal1 is not essential; functional replacement with its paralog, Bmal2. Curr. Biol. 20, 316–321. doi: 10.1016/j.cub.2009.12.034
Singh, C. K., Chhabra, G., Ndiaye, M. A., Garcia-Peterson, L. M., Mack, N. J., and Ahmad, N. (2018). The role of sirtuins in antioxidant and redox signaling. Antioxid. Redox. Signal. 28, 643–661. doi: 10.1089/ars.2017.7290
Singh, P., Hanson, P. S., and Morris, C. M. (2017). SIRT1 ameliorates oxidative stress induced neural cell death and is down-regulated in Parkinson’s disease. BMC Neurosci. 18:46. doi: 10.1186/s12868-017-0364-1
Smarr, B., Cutler, T., Loh, D. H., Kudo, T., Kuljis, D., Kriegsfeld, L., et al. (2019). Circadian dysfunction in the Q175 model of Huntington’s disease: network analysis. J. Neurosci. Res. 97, 1606–1623.
Smith, D. G., Cappai, R., and Barnham, K. J. (2007). The redox chemistry of the Alzheimer’s disease amyloid beta peptide. Biochim. Biophys. Acta 1768, 1976–1990.
Sollars, P. J., and Pickard, G. E. (2015). The neurobiology of circadian rhythms. Psychiatr. Clin. North Am. 38, 645–665.
Song, H., Moon, M., Choe, H. K., Han, D. H., Jang, C., Kim, A., et al. (2015). Abeta-induced degradation of BMAL1 and CBP leads to circadian rhythm disruption in Alzheimer’s disease. Mol. Neurodegener. 10:13.
Spiller, K. J., Restrepo, C. R., Khan, T., Dominique, M. A., Fang, T. C., Canter, R. G., et al. (2018). Microglia-mediated recovery from ALS-relevant motor neuron degeneration in a mouse model of TDP-43 proteinopathy. Nat. Neurosci. 21, 329–340. doi: 10.1038/s41593-018-0083-7
Steiner, H., Fukumori, A., Tagami, S., and Okochi, M. (2018). Making the final cut: pathogenic amyloid-β peptide generation by γ-secretase. Cell Stress 2:292. doi: 10.15698/cst2018.11.162
Stevanovic, K., Yunus, A., Joly-Amado, A., Gordon, M., Morgan, D., Gulick, D., et al. (2017). Disruption of normal circadian clock function in a mouse model of tauopathy. Exp. Neurol. 294, 58–67. doi: 10.1016/j.expneurol.2017.04.015
Stopa, E. G., Volicer, L., Kuo-Leblanc, V., Harper, D., Lathi, D., Tate, B., et al. (1999). Pathologic evaluation of the human suprachiasmatic nucleus in severe dementia. J. Neuropathol. Exp. Neurol. 58, 29–39. doi: 10.1097/00005072-199901000-00004
Strang, K. H., Golde, T. E., and Giasson, B. I. (2019). MAPT mutations, tauopathy, and mechanisms of neurodegeneration. Lab. Invest. 99, 912–928. doi: 10.1038/s41374-019-0197-x
Sundaram, S., Nagaraj, S., Mahoney, H., Portugues, A., Li, W., Millsaps, K., et al. (2019). Inhibition of casein kinase 1delta/epsilonimproves cognitive-affective behavior and reduces amyloid load in the APP-PS1 mouse model of Alzheimer’s disease. Sci. Rep. 9:13743.
Suofu, Y., Li, W., Jean-Alphonse, F. G., Jia, J., Khattar, N. K., Li, J., et al. (2017). Dual role of mitochondria in producing melatonin and driving GPCR signaling to block cytochrome c release. Proc. Natl. Acad. Sci. U S A. 114, E7997–E8006.
Takahashi, J. S. (2017). Transcriptional architecture of the mammalian circadian clock. Nat. Rev. Genet. 18, 164–179. doi: 10.1038/nrg.2016.150
Tremblay, M.-E., Cookson, M. R., and Civiero, L. (2019). Glial phagocytic clearance in Parkinson’s disease. Mol. Neurodegenerat. 14:16.
Trenkwalder, C., Arnulf, I., and Postuma, R. (2017). Principles and Practice of Sleep Medicine. Amsterdam: Elsevier Inc.,.
Tso, C. F., Simon, T., Greenlaw, A. C., Puri, T., Mieda, M., and Herzog, E. D. (2017). Astrocytes regulate daily rhythms in the suprachiasmatic nucleus and behavior. Curr. Biol. 27, 1055–1061. doi: 10.1016/j.cub.2017.02.037
Venkatesan, A., Fan, J.-Y., Bouyain, S., and Price, J. L. (2019). The circadian tau mutation in casein kinase 1 is part of a larger domain that can be mutated to shorten circadian period. Int. J. Mol. Sci. 20:813. doi: 10.3390/ijms20040813
Walker, Z., Possin, K. L., Boeve, B. F., and Aarsland, D. (2015). Lewy body dementias. Lancet 386, 1683–1697.
Walsh, C. M., Ruoff, L., Varbel, J., Walker, K., Grinberg, L. T., Boxer, A. L., et al. (2016). Rest-activity rhythm disruption in progressive supranuclear palsy. Sleep Med. 22, 50–56. doi: 10.1016/j.sleep.2016.05.002
Walsh, C. M., Ruoff, L., Walker, K., Emery, A., Varbel, J., Karageorgiou, E., et al. (2017). Sleepless night and day, the plight of progressive supranuclear palsy. Sleep 40:zsx154.
Wang, H. B., Loh, D. H., Whittaker, D. S., Cutler, T., Howland, D., and Colwell, C. S. (2018). Time-Restricted feeding improves circadian dysfunction as well as motor symptoms in the Q175 mouse model of Huntington’s Disease. eNeuro 5:ENEURO.0431-17.2017.
Wang, H. B., Whittaker, D. S., Truong, D., Mulji, A. K., Ghiani, C. A., Loh, D. H., et al. (2017). Blue light therapy improves circadian dysfunction as well as motor symptoms in two mouse models of Huntington’s disease. Neurobiol. Sleep Circadian Rhythms 2, 39–52. doi: 10.1016/j.nbscr.2016.12.002
Wang, X., Wang, L., Yu, Q., Xu, Y., Zhang, L., Zhao, X., et al. (2016). Alterations in the expression of Per1 and Per2 induced by Abeta31-35 in the suprachiasmatic nucleus, hippocampus, and heart of C57BL/6 mouse. Brain Res. 1642, 51–58. doi: 10.1016/j.brainres.2016.03.026
Weissova, K., Skrabalova, J., Skalova, K., Cervena, K., Bendova, Z., Miletinova, E., et al. (2018). Circadian rhythms of melatonin and peripheral clock gene expression in idiopathic REM sleep behavior disorder. Sleep Med. 52, 1–6. doi: 10.1016/j.sleep.2018.07.019
Wood, N. I., McAllister, C. J., Cuesta, M., Aungier, J., Fraenkel, E., and Morton, A. J. (2013). Adaptation to experimental jet-lag in R6/2 mice despite circadian dysrhythmia. PLoS One 8:e55036. doi: 10.1371/journal.pone.0055036
Xia, X., Jiang, Q., McDermott, J., and Han, J. J. (2018). Aging and Alzheimer’s disease: comparison and associations from molecular to system level. Aging Cell 17:e12802. doi: 10.1111/acel.12802
Xu, F., Kula-Eversole, E., Iwanaszko, M., Hutchison, A. L., Dinner, A., and Allada, R. (2019). Circadian clocks function in concert with heat shock organizing protein to modulate mutant huntingtin aggregation and toxicity. Cell Rep. 27, 59–70.e4.
Yamazaki, Y., Matsubara, T., Takahashi, T., Kurashige, T., Dohi, E., Hiji, M., et al. (2011). Granulovacuolar degenerations appear in relation to hippocampal phosphorylated tau accumulation in various neurodegenerative disorders. PLoS One 6:e26996. doi: 10.1371/journal.pone.0026996
Young, M. E. (2006). The circadian clock within the heart: potential influence on myocardial gene expression, metabolism, and function. Am. J. Physiol. Heart Circ. Physiol. 290, H1–H16.
Yuksel, M., and Tacal, O. (2019). Trafficking and proteolytic processing of amyloid precursor protein and secretases in Alzheimer’s disease development: an up-to-date review. Eur. J. Pharmacol. 856:172415. doi: 10.1016/j.ejphar.2019.172415
Zhang, F., Niu, L., Liu, X., Liu, Y., Li, S., Yu, H., et al. (2020). Rapid eye movement sleep behavior disorder and neurodegenerative diseases: an update. Aging Dis. 11, 315–326. doi: 10.14336/ad.2019.0324
Zhang, H., Davies, K. J. A., and Forman, H. J. (2015). Oxidative stress response and Nrf2 signaling in aging. Free Radic. Biol. Med. 88, 314–336. doi: 10.1016/j.freeradbiomed.2015.05.036
Keywords: circadian, neurodegeneration, dementia, proteinopathy, stress
Citation: Carter B, Justin HS, Gulick D and Gamsby JJ (2021) The Molecular Clock and Neurodegenerative Disease: A Stressful Time. Front. Mol. Biosci. 8:644747. doi: 10.3389/fmolb.2021.644747
Received: 21 December 2020; Accepted: 01 March 2021;
Published: 26 March 2021.
Edited by:
Maj-Linda B. Selenica, University of Kentucky, United StatesReviewed by:
Erik Steven Musiek, Saint Louis University, United StatesCopyright © 2021 Carter, Justin, Gulick and Gamsby. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joshua J. Gamsby, Z2Ftc2J5QHVzZi5lZHU=
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.