 Ziqi Chen
Ziqi Chen Martin Renqiang Min
Martin Renqiang Min Xia Ning
Xia Ning- 1Computer Science and Engineering Department, The Ohio State University, Columbus, OH, United States
- 2Machine Learning Department, NEC Labs America, Princeton, NJ, United States
- 3Biomedical Informatics Department, The Ohio State University, Columbus, OH, United States
- 4Translational Data Analytics Institute, The Ohio State University, Columbus, OH, United States
T-cell receptors can recognize foreign peptides bound to major histocompatibility complex (MHC) class-I proteins, and thus trigger the adaptive immune response. Therefore, identifying peptides that can bind to MHC class-I molecules plays a vital role in the design of peptide vaccines. Many computational methods, for example, the state-of-the-art allele-specific method
1 Introduction
Immunotherapy, an important treatment of cancers, treats the disease by boosting patients’ immune systems to kill cancer cells (Mellman et al., 2011; Couzin-Frankel, 2013; Esfahani et al., 2020; Waldman et al., 2020). To trigger patients’ adaptive immune responses, Cytotoxic T cells, also known as CD8+ T-cells, have to recognize peptides presented on the cancer cell surface (Valitutti et al., 1995; Blum et al., 2013). These peptides are fragments derived from self-proteins or pathogens by proteasomal proteolysis within the cell. To have the peptides presented on the cell surface to be recognized by CD8 receptors, they need to be brought from inside the cells to the cell surface, typically through binding with and transported by major histocompatibility complex (MHC) class-I molecules. To mimic natural occurring proteins from pathogens, synthetic peptide vaccines are developed for therapeutic purposes (Purcell et al., 2007). Therefore, to design successful peptide vaccines, it is critical to identify and study peptides that can bind with MHC class-I molecules.
Many computational methods have been developed to predict the binding affinities between peptides and MHC class-I molecules (Han and Kim, 2017; O’Donnell et al., 2018). These existing computational methods can be categorized into two types: allele-specific methods and pan methods. Allele-specific methods train one model for one allele such that the model can capture binding patterns specific to the allele, and thus it is better customized to that allele (Lundegaard et al., 2008; O’Donnell et al., 2018). Pan methods train one model for all the alleles at the same time, and thus the information across different alleles can be shared and integrated into a general model (Jurtz et al., 2017; Hu et al., 2018). These existing methods can achieve significant performance on the prediction of binding affinities. However, most existing methods formulate the prediction problem as to predict the exact binding affinity values (e.g., IC50 values) via regression. Such formulations may suffer from two potential issues. First of all, they tend to be sensitive to the measurement errors when the measured IC50 values are not accurate. In addition, many of these methods use ranking-based measurement such as Kendall’s Tau correlations to measure the performance of regression-based methods (Bhattacharya et al., 2017; O’Donnell et al., 2020). This could lead to sub-optimal solution as small regression errors do not necessarily correlate to large Kendall’s Tau. Therefore, these methods are limited in their capability of prioritizing the most possible peptide-MHC pairs of high binding affinities.
In this study, we formulate the problem as to prioritize the most possible peptide-MHC binding pairs via ranking based learning. We propose three ranking-based learning objectives such that through optimizing these objectives, we impose peptide-MHC pairs of high binding affinities ranked higher than those of low binding affinities. Coupled with these objectives, we develop two allele-specific Convolutional Neural Network (CNN)-based methods with attention mechanism, denoted as
We summarize our contributions below:
• We formulate the problem as to optimize the rankings of peptide-MHC pairs instead of predicting the exact binding affinity values. Our experimental results demonstrate that our ranking-based learning is able to significantly improve the performance of identifying the most possible peptide-MHC binding pairs.
• We develop two allele-specific methods
• We incorporate both global and local features in
• Our methods outperform the state-of-the-art baseline
2 Literature Review
The existing computational methods for peptide-MHC binding prediction can be generally classified into two categories: linear regression-based methods and deep learning (DL)-based methods. Below, we present a literature review for each of the categories, including the key ideas and the representative work.
2.1 Peptide Binding Prediction Via Linear Regression
Many early developed methods on peptide-MHC binding prediction are based on linear regression. For example, Peters and Sette (2005) proposed a method named Stabilized Matrix Method (
2.2 Peptide Binding Prediction Via Deep Learning
The DL-based models can be categorized into allele-specific methods and pan methods. Allele-specific methods train a model for each allele and learn the binding patterns of each allele separately. Instead, pan methods train a model for all alleles to learn all the binding patterns together within one model. Both the methods use similar encoding methods such
2.2.1 Allele-specific Deep Learning Methods
Among these allele-specific methods, Lundegaard et al. (2008) proposed
2.2.2 Pan Deep Learning Methods
Nielsen and Andreatta (2016) developed a DL-based pan method named
3 Materials
3.1 Peptide-MHC Binding Data
The dataset is collected from the Immune Epitope Database (IEDB) (Vita et al., 2018). Each peptide-MHC entry m in the dataset measures the binding affinity between a peptide and an allele. These binding affinity entries could be of either quantitative values (e.g., IC50) or qualitative levels indicating levels of binding strength. The mapping between quantitative values and qualitative levels is shown in Table 1. Note that higher IC50 values indicate lower binding affinities.
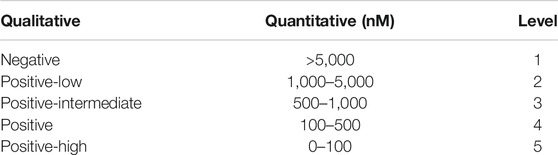
TABLE 1. Binding affinity measurement mapping.
We combined the widely used IEDB benchmark dataset curated by Kim et al. (2014) and the latest data added to IEDB (downloaded from the IEDB website on Jun. 24, 2019). The benchmark dataset contains two datasets BD2009 and BD2013 compiled in 2009 and 2013, respectively. BD2009 consists of 137,654 entries, and BD2013 consists of 179,692 entries. The latest dataset consists of 189,063 peptide-MHC entries. Specifically, we excluded those entries with non-specific, mutant or unparseable allele names such as HLA-A2. We then combined the datasets by processing the duplicated entries and entries with conflicting affinities as follows. We first mapped the quantitative values of all these duplicated or conflicting entries into qualitative levels based on Table 1, and used majority voting to identify the major binding level of the peptide-MHC pairs. If such binding levels cannot be identified, we simply removed all the conflicting entires; otherwise, we assigned the average quantitative values in the identified major binding level to the peptide-MHC pairs. The combined dataset consists of 202,510 entries across 128 alleles and 53,253 peptides as in Table 2. We further normalized the binding affinity values ranging from 0 to 107 to [0, 1] via formula
where x is the measured binding affinity value, and

TABLE 2. Data statistics.
4 Definitions and Notations
All the key definitions and notations are listed in Table 3.

TABLE 3. Notations.
5 Methods
We developed two new models:
5.1 Convolutional Neural Networks with Attention Layers (
In this section, we introduce our new model
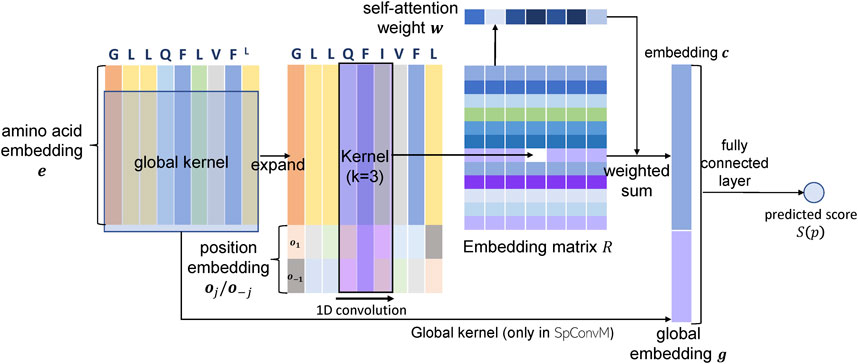
FIGURE 1. Architectures of
5.1.1 Peptide Representation in
In
5.1.2 Model Architecture of
The
where
The embedding vector
5.2 Convolutional Neural Networks with Global Kernels and Attention Layers (
We further develop
Given the input FG, the convolution using dg global kernels will generate a vector
5.3 Loss Functions
We propose three pair-wise hinge loss functions, denoted as Hv, Hl and Hi, respectively. We will compare these loss functions with the widely used mean-square loss function (O’Donnell et al., 2018), denoted as MS, in learning peptide bindings.
5.3.1 Hinge Loss Functions for Peptide Binding Ranking
We first evaluate the hinge loss as the loss function in conjunction with various model architectures. The use of hinge loss is inspired by the following observation. We noticed that in literature, peptide-MHC binding prediction is often formulated into either a regression problem, in which the binding affinities between peptides and alleles are predicted, or a classification problem, in which whether the peptide will bind to the allele is the target to predict. However, in practice, it is also important to first prioritize the most promising peptides with acceptable binding affinities for further assessment, whereas regression and classification are not optimal for prioritization. Besides, recent work has already employed several evaluation metrics on top ranked peptides, for example, (Zeng and Gifford, 2019) evaluated the performance through the true positive rate at 5% false positive rate, which suggests the importance of top-ranked peptides in addition to accurate affinity prediction. All of these inspire us to consider ranking based formulation for peptide prioritization.
Given two normalized binding affinity values bi and bj of any two peptides pi and pj with respect to an allele, the allele-specific pair-wise ranking problem can be considered as to learn a scoring function S(·), such that
Please note that S(pi) is a score for peptide pi, which is not necessarily close to the binding affinity bi, as long as it reconstructs the ranking structures among all peptides. This allows the ranking based formulation more flexibility to identify the most promising peptides without accurately estimating their binding affinities. To learn such scoring functions, hinge loss is widely used, and thus we develop three hinge loss functions to emphasize different aspects during peptide ranking.
5.3.1.1 Value-Based Hinge Loss Function
The first hinge loss function, denoted as Hv, aims to well rank peptides with significantly different binding affinities. Given two peptides pi and pj, this hinge loss function is defined as follows:
where li denotes the binding level of peptide pi according to the Table 1; li > lj denotes that the binding level of peptide pi is higher than the peptide pj; bi and bj are the ground-truth normalized binding affinities of pi and pj, respectively; c > 0 is a pre-specified constant to increase the difference between two predicted scores. Hv learns from two peptides of different binding levels and defines a margin value between two peptides as the difference of their ground-truth binding affinities bi − bj plus a constant c. If two peptides pi and pj are on different binding levels li > lj, and the difference of their predicted scores is smaller than the margin c + (bi − bj), this pair of peptides will contribute to the overall loss; otherwise, the loss of this pair will be 0. Note that Hv is only defined on peptides of different binding levels. For the peptides with the same or similar binding affinities, Hv allows incorrect ranking among them.
5.3.1.2 Level-Based Hinge Loss Function
Instead of ranking with respect to the margin as in Hv, we relax the ranking criterion and use a margin according to the difference of binding levels (Table 1). Thus, the second hinge loss, denoted as Hl, is defined as follows:
where r > 0 is a constant. Given a pair of peptides in two different binding levels, similar to Hv, Hl requires that if the difference of their predicted scores is smaller than a margin, this pair of peptides will contribute to the overall loss; otherwise, the loss of these two peptides will be 0. However, unlike Hv, the margin defined in Hl depends on the difference of binding levels between two peptides (i.e.,
5.3.1.3 Constrained Level-Based Hinge Loss Function
The third hinge loss function Hi extends Hl by adding a constraint that two peptides of a same binding level can have similar predicted scores. This hinge loss is defined as follows:
Given a pair of peptides on a same binding level, the added constraint (the case if li = lj) requires that if the absolute difference
5.3.2 Mean-Squares Loss
We also compare a mean-squares loss function, denoted as MS, proposed in (O’Donnell et al., 2018; Paul et al., 2019), to fit the entries without exact binding affinity values as below:
where “
Note that in MS, the predicted score S(p) needs to be normalized into range [0,1]. This is because b is in range [0,1] (Eq. 1) so that S(p) needs to be in the same range and thus neither S(p) nor b will dominate the squared errors due to substantially large or small values. However, in the three hinge loss functions (Eqs. 5–7), the potential different range between S(p) and b or l could be accommodated by the constant c (Eq. 5) or r (Eqs. 6, 7), respectively. In MS, we use sigmoid function to normalize S(p).
6 Experimental Settings
6.1 Baseline Methods
6.1.1 Encoding Methods
Encoding methods represent each amino acid with a vector. Popular encoding methods used by the previous works include
6.1.2 Baseline Method: Local Connected Neural Networks
Note that we did not compare with other methods including
6.2 Batch Generation
For models with MS as the loss function, we randomly sample a batch of peptides as the training batch. For models with the proposed pair-wise hinge loss functions (Hv, Hl, Hi), to reduce computational costs, we construct pairs of peptides for each training batch from a sampled batch of peptides. Specifically, for Hv and Hl, each pair consists of two peptides from different binding levels; and for Hi, the constructed pairs can consist of two peptides from the same or different binding levels.
6.3 Model Training
We use 5-fold cross validation (Bishop, 2006) to tune the hyper-parameters of all methods through a grid search approach. We use 10% of the training data as a validation set and explicitly ensure that the training set, validation set and testing set do not overlap. This validation set is applied to adjust the learning rate dynamically and determine the early stopping of training process. If the loss on the validation set does not decrease in 5 epochs, we will decrease the learning rate by 10%. The learning rate is initialized as 0.05. If the loss does not decrease on the validation set for continuous 20 epochs, we stop the training process. For each allele, we run the grid search algorithm to find the optimal hyper-parameters for the allele-specific model through the above cross validation process. We apply stochastic gradient descent (SGD) to optimize the loss functions. We set the dimension of
6.4 Evaluation Metrics
We use 4 types of evaluation metrics, including average rank (
where
The hit rate HRh (e.g., HR500) is defined as follows,
where
We use h = 500 nM as the threshold to distinguish positive peptides and negative peptides, and apply two metrics for classification to evaluate the model performance. The first classification metric
where
Larger values of
In order to compare the models with respect to one single metric in a holistic way, we define a hybrid metric
where
7 Experimental Results
We present the experimental results in this section. All the parameters used in the experiments are reported in the Appendix.
7.1 Model Architecture Comparison
We evaluate all the 12 possible combinations of the 3 model architectures (
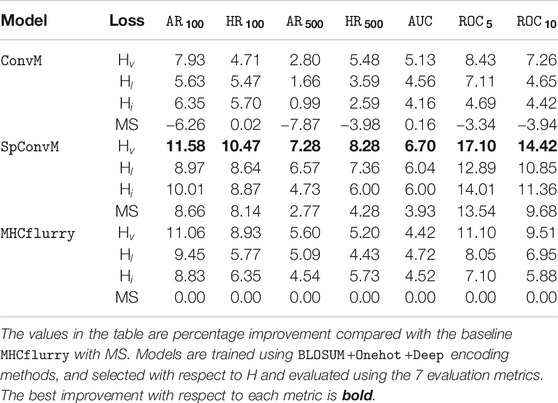
TABLE 4. Overall performance comparison (H;
Table 4 shows that as for the model architectures, on average,
We also report the results of our methods on 34 HLA-A molecules and 35 HLA-B molecules with the optimal hyperparameters determined by hybrid metric H separately in Table 5. HLA-A and HLA-B are two groups of the human leukocyte antigen (HLA) complex that are important to the immune system. The results of HLA-A and HLA-B molecules show the same trend as that in Table 4, that is, for both HLA-A and HLA-B groups, on average,
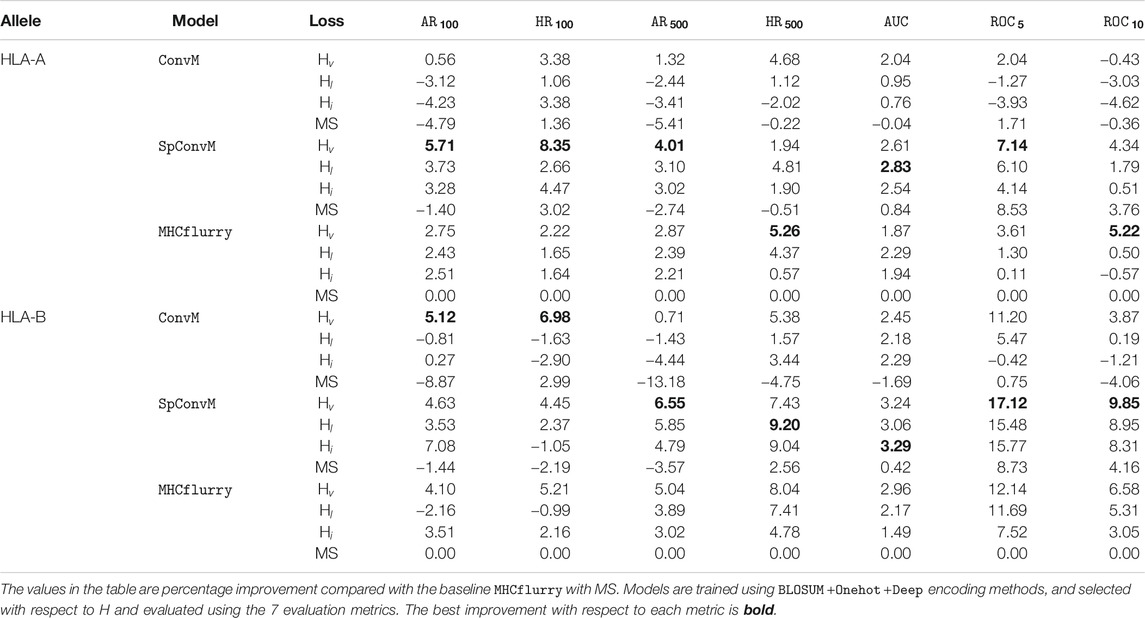
TABLE 5. Overall performance comparison across HLA-A and HLA-B alleles (
In addition to using the hybrid metric
Figure 2 show the distributions of performance improvement among all the alleles from
FIGURE 2. Performance improvement compared with
7.2 Loss Function Comparison
Table 4 also demonstrates that Hv is the most effective loss function in combination with each of the learning architectures, and all three hinge loss functions Hv, Hl and Hi can outperform the MS loss function. For example, for
All the three hinge loss functions Hv, Hl and Hi outperform MS across all the model architectures. This might be due to two reasons. First, the pairwise hinge loss functions are less sensitive to the imbalance of different amounts of peptides, either strongly binding or weakly/non-binding, by sampling and constructing pairs from respective peptides. Thus, the learning is not biased by one type of peptides, and the models can better learn the difference among different types of peptides, and accordingly produce better ranking orders of peptides. Second, the pairwise hinge loss functions can tolerate insignificant measurement errors to some extent. All the three hinge loss functions do not consider pairs of peptides with similar binding affinities. This enables our models to be more robust and tolerant to noisy data due to the measurement inaccuracy of binding affinities.
7.3 Encoding Method Comparison
We evaluate three encoding methods (
TABLE 6. Encoding performance comparison on
We also select the optimal set of hyperparameters with respect to
7.4 Attention Weights
Figures 3A,B, 4A,B present the attention weights over peptides of allele HLA-A*02:01, HLA-A*24:02, HLA-B*27:05 and HLA-B*58:01, respectively, learned by the attention layer of
FIGURE 3. Attention weights and motifs for HLA-A*02:01 and HLA-A*24:02.

FIGURE 4. Attention weights and motifs for HLA-B*27:05 and HLA-B*58:01.
Figures 3C,D, 4C,D present the allele-specific binding motifs of the peptides with high affinity for allele HLA-A*02:01, HLA-A*24:02, HLA-B*27:05 and HLA-B*58:01, respectively. Comparing with Figures 3A,B, 4A,B respectively, we noticed that for sequence positions that have higher weights, there are a few preferred amino acids; for positions with lower weights, the amino acids can be diverse. We further check whether the learned attention weights correlate with amino acid conservation. We first calculate a matrix, denoted as
where
Figure 5 presents the attention weights learned from

FIGURE 5. Attention weights of three peptides for HLA-A*02:01 learned from
We do not present the attention weights learned by
7.5 Position Embeddings
We conduct an ablation study to verify the effect of position embedding in the
FIGURE 6. Performance comparison on
Literature Chorowski et al. (2015) shows that a main limitation of weighted-sum operation in attention layer is that the same k-mers with position-unaware embeddings will be associated with the exactly same attention weights regardless of their position in the peptide sequence. Such position-unaware weights are against our knowledge, that is, amino acids in specific positions have been known to be important for binding events O’Donnell et al. (2018); O’Donnell et al. (2020). Hence, encoding position information into the embeddings of k-mers is necessary for the self-attention layer to learn meaningful position-specific binding patterns.
7.6 Training and Inference Time
We implemented the models using Python-3.6.9, Pytorch-1.3.1 and Numpy-1.18.1. We trained the models on machines with Intel Xeon E5-2680 v4 CPUs and NVIDIA Tesla P100 (Pascal) GPU 16 GB memory with Red Hat Enterprise 7.7. With different hyperparameters, the average training time for each allele with 5-fold cross validation (i.e., 5 models for each allele) is 3.81 min [(3.41, 4.28), standard deviation (±0.34)] with a single CPU core and a single GPU. We also calculated the average inference time for three alleles HLA-A-2402, HLA-A-0201, HLA-A-0301. On average, the inference with our
8 Discussions
8.1 Experiments on Mass Spectrometry Benchmark Dataset
We evaluated the performance of our methods with the Mass Spectrometry benchmark dataset curated by O’Donnell et al. (2018). This MS benchmark dataset contains 23,653 sequences of MHC-displayed ligands eluted from B cell lines expressing 15 MHC class I alleles. For each eluted ligand, 100 decoys will be sampled from the protein-coding transcripts that contained this eluted ligand. Specifically, they sampled an equal number of decoys of each length 8–15. After removing all the entries present in the IEDB dataset, the yielded Mass Spectrometry benchmark dataset contains 23,653 positive peptides and 2,377,037 randomly sampled negative peptides.
To compare with other methods on the Mass Spectrometry benchmark dataset, we follow the idea of model ensemble that
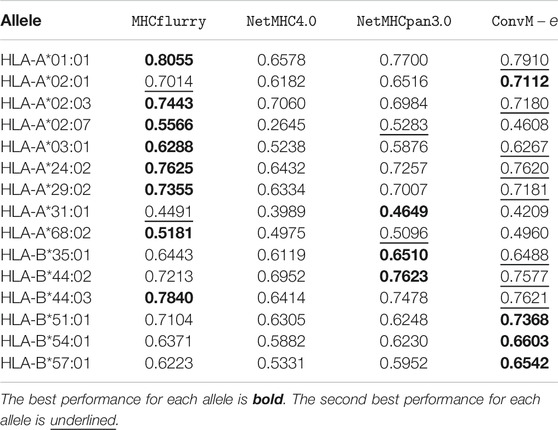
TABLE 7. Performance comparison over mass spectrometry dataset in
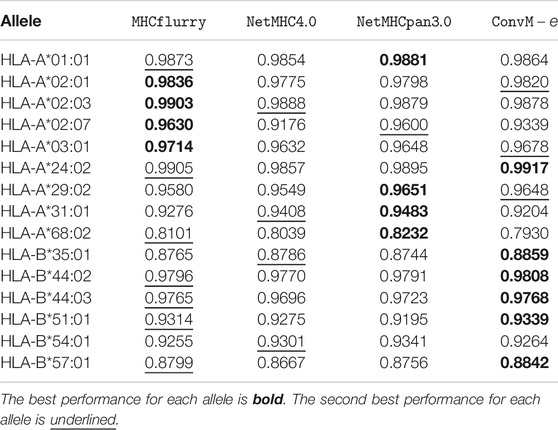
TABLE 8. Performance comparison over mass spectrometry dataset in
Table 7 shows that in terms of PPV (positive predictive value, a popular metric using on the Mass Spectrometry dataset), our ensemble methods achieve either the best or the second best performance on 12 out of 15 alleles among all the methods. When our ensemble achieves the second best performance on an allele, it is very comparable to the best performance—on average, the difference is 0.0112. For HLA-B alleles, our ensemble methods are also the best or the second best. Table 8 shows a similar trend in terms of AUC, that is, our ensemble methods achieve either the best or the second best performance among out 9 of 15 alleles among all the methods; when it is the second best method, its performance is very comparable to the best method. In particular, for HLA-B alleles, our ensemble methods achieve the best performance on 5 out of 6 alleles. The results in the above two tables demonstrate that our methods either outperform the other methods, or are very comparable to the other methods.
8.1.1 Discussion on Using an Independent Test Set
One concern with this benchmark dataset is that the random negative sampling method creates a data distribution that is different from that of real data. Figures 7A,B present the distributions of binding ligands and the randomly sampled negative samples for two different alleles in the Mass Spectrometry benchmark dataset, respectively; Figures 7C,D present the corresponding distributions in the IEDB dataset. Both the benchmark dataset and the IEDB dataset have similar distributions on the binding peptides, that is, most of the binding peptides have sequence length 9 to 11. In the IEDB dataset, the negative peptides have a similar distribution over sequence lengths as the positive peptides, and the dataset is balanced in terms of positive and negative sample size. However, in the benchmark dataset, the negative samples are uniformly distributed over sequence lengths and the distribution is significantly different from that of the positive peptides. In addition, the dataset is highly unbalanced with significantly more negative samples. Given the different distributions between the IEDB dataset and the Mass Spectrometry benchmark dataset, models trained using IEDB data without altering its negative sample distribution will not work well on the Mass Spectrometry benchmark dataset.
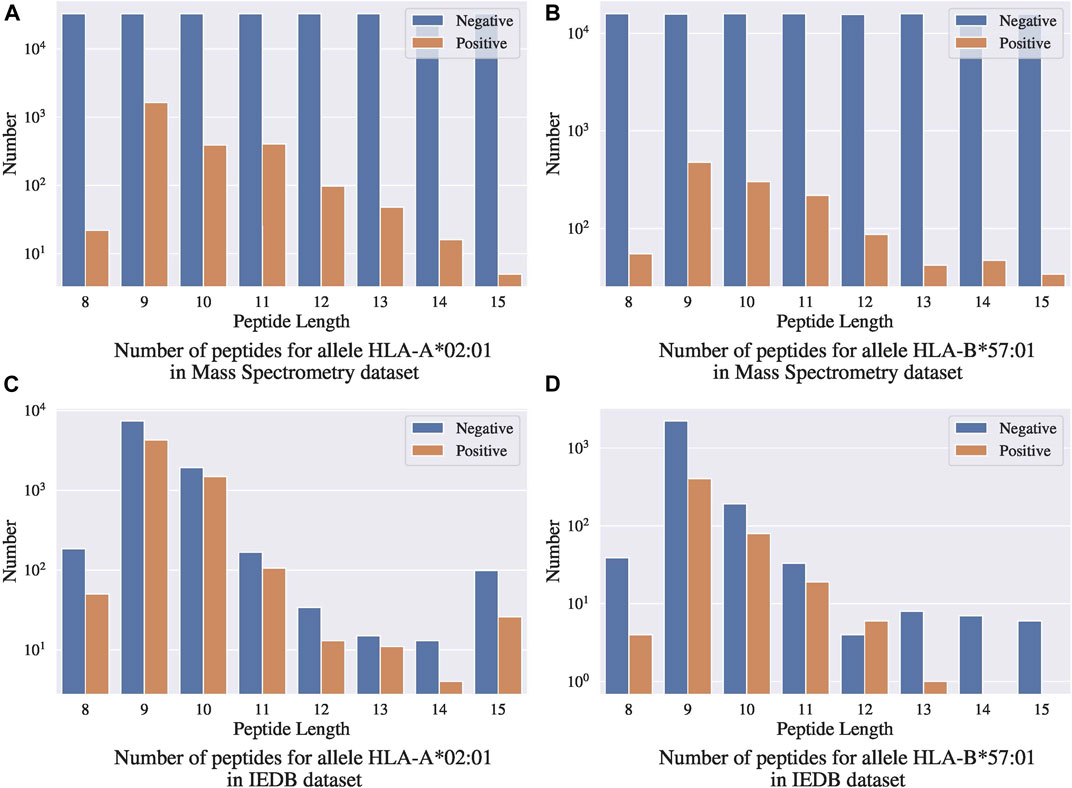
FIGURE 7. Comparison between mass spectrometry dataset and IEDB dataset.
Even though the Mass Spectrometry benchmark dataset has a data distribution that is different from IEDB, it is still valid to be used as an independent test set. However, to ensure it is truly “independent”, its label information including the distributions of positive and negative samples must not be available or used during the model training process. If a method does the same negative sampling and constructs a similar training data distribution as in the benchmark dataset during its training process, implicitly it uses the label information from test set. Thus, such training process violates the “independence” of the test set and the model performance could be over-estimated. We noticed that
8.2 Reproducibility
We published our data and code at https://github.com/ziqi92/peptide-binding-prediction.
9 Conclusion
Our methods contribute to the study of peptide-MHC binding prediction problem in two ways. First, instead of predicting the exact binding affinities values as in the existing methods, we formulate the problem as to prioritize most possible peptide-MHC binding pairs via a ranking-based learning. We developed three pairwise ranking-based learning objectives for such prioritization, and the corresponding learning methods that impose the peptide-MHC pairs of higher binding affinities ranked above those with lower binding affinities with a certain margin. Our experimental results in comparison with the state-of-the-art regression based methods demonstrate the superior prediction performance of our methods in prioritizing and identifying the most likely binding peptides. In addition to the learning objectives, we also developed two convolutional neural network-based model architectures
Data Availability Statement
Publicly available datasets were analyzed in this study. This data can be found here: The Immune Epitope Database (IEDB) https://www.iedb.org/home_v3.php.
Author Contributions
ZC: Data curation, Formal analysis, Methodology, Visualization, Writing—original draft, review, editing. RM: Conceptualization, Investigation, Validation, Writing—review, editing. XN: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Supervision, Project administration, Writing—original draft, review, editing.
Funding
This project was made possible, in part, by support from the National Science Foundation under Grant Number IIS-1855501 and IIS-1827472, the National Institute of General Medical Sciences under Grant Number 2R01GM118470–05, the National Library of Medicine under Grant Numbers 1R01LM012605-01A1 and 1R21LM013678-01, and AWS Machine Learning Research Award. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the funding agencies.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2021.634836/full#supplementary-material
References
Andreatta, M., and Nielsen, M. (2015). Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics 32, 4511–4517. doi:10.1093/bioinformatics/btv639
Bhattacharya, R., Sivakumar, A., Tokheim, C., Guthrie, V. B., Anagnostou, V., Velculescu, V. E., et al. (2017). Evaluation of machine learning methods to predict peptide binding to MHC Class I proteins. Berlin, Heidelberg: Springer-Verlag. doi:10.1101/154757
Bishop, C. M. (2006). Pattern recognition and machine learning (information science and statistics). Berlin, Heidelberg: Springer-Verlag.
Blum, J. S., Wearsch, P. A., and Cresswell, P. (2013). Pathways of antigen processing. Annu. Rev. Immunol. 31, 1443–1473. doi:10.1146/annurev-immunol-032712-095910
Bonsack, M., Hoppe, S., Winter, J., Tichy, D., Zeller, C., Küpper, M. D., et al. (2019). Performance Evaluation of MHC Class-I Binding Prediction tools Based on an Experimentally validated MHC–Peptide Binding data set. Cancer Immunol. Res. 7, 5719–5736. doi:10.1158/2326-6066.cir-18-0584
Chorowski, J., Bahdanau, D., Serdyuk, D., Cho, K., and Bengio, Y. (2015). Attention-based models for speech recognition. arXiv:cs.CL/ http://arxiv.org/abs/1506.07503v1
Couzin-Frankel, J. (2013). Cancer immunotherapy. Science 342, 61651432–6161433. doi:10.1126/science.342.6165.1432arXiv
Esfahani, K., Roudaia, L., Buhlaiga, N., Del Rincon, S. V., Papneja, N., and Miller, W. H. (2020). A review of cancer immunotherapy: from the past, to the present, to the future. Curr. Oncol. 27 (Suppl. 2), S87–S97. doi:10.3747/co.27.5223
Goldberg, Y., and Levy, O. (2014). word2vec Explained: deriving Mikolov et al.’s negative-sampling word-embedding method. arXiv:cs.CL/ http://arxiv.org/abs/1402.3722v1
Han, Y., and Kim, D. (2017). Deep convolutional neural networks for pan-specific peptide-MHC class I binding prediction. BMC Bioinform. 18, 1. doi:10.1186/s12859-017-1997-x
Henikoff, S., and Henikoff, J. G. (1992). Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. 89, 2210915–2210919. doi:10.1073/pnas.89.22.10915
Hu, Y., Wang, Z., Hu, H., Wan, F., Chen, L., Xiong, Y., et al. (2018). ACME: Pan-specific peptide-MHC class I binding prediction through attention-based deep neural networks. Berlin: Springer. doi:10.1101/468363
Jurtz, V., Paul, S., Andreatta, M., Marcatili, P., Peters, B., and Nielsen, M. (2017). NetMHCpan-4.0: improved Peptide–MHC Class I interaction Predictions integrating Eluted ligand and Peptide Binding Affinity data. J. Immunol. 199, 93360–3368. doi:10.4049/jimmunol.1700893
Kim, Y., Sidney, J., Buus, S., Alessandro, S., Nielsen, M., and Peters, B. (2014). Dataset size and composition impact the reliability of performance benchmarks for peptide-MHC binding predictions. BMC Bioinform. 15, 1. doi:10.1186/1471-2105-15-241
Kim, Y., Sidney, J., Pinilla, C., Alessandro, S., and Peters, B. (2009). Derivation of an amino acid similarity matrix for peptide:MHC binding and its application as a Bayesian prior. BMC Bioinform. 10, 1. doi:10.1186/1471-2105-10-394
Kuksa, P. P., Martin, R., Dugar, R., and Gerstein, M. (2015). High-order neural networks and kernel methods for peptide-MHC binding prediction. Bioinformatics 12, btv371. doi:10.1093/bioinformatics/btv371
Lundegaard, C., Lamberth, K., Harndahl, M., Buus, S., Lund, O., and Nielsen, M. (2008). NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res. 36, W509–W512. doi:10.1093/nar/gkn202
Mellman, I., George, C., and Glenn, D. (2011). Cancer immunotherapy comes of age. Nature 480, 7378480–7378489. doi:10.1038/nature10673
Michael Boehm, K., Bhinder, B., Joseph Raja, V., Dephoure, N., and Elemento, O. (2019). Predicting peptide presentation by major histocompatibility complex class I: an improved machine learning approach to the immunopeptidome. BMC Bioinform. 20, 1. doi:10.1186/s12859-018-2561-z
Nielsen, M., and Andreatta, M. (2016). NetMHCpan-3.0: improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med. 8, 1. doi:10.1186/s13073-016-0288-x
O’Donnell, T. J., Rubinsteyn, A., Bonsack, M., Riemer, A. B., Laserson, U., and Hammerbacher, J. (2018). MHCflurry: open-source Class I MHC binding affinity prediction. Cell Syst. 7, 1129–132. doi:10.1016/j.cels.2018.05.014
O’Donnell, T. J., Rubinsteyn, A., and Laserson, U. (2020). MHCflurry 2.0: improved Pan-Allele prediction of MHC Class I-presented peptides by incorporating antigen processing. Cell Syst. 11, 1–48. doi:10.1016/j.cels.2020.06.010
Paul, S., Croft, N. P., Purcell, A. W., and Tscharke, D. C. (2019). Benchmarking predictions of MHC class I restricted T cell epitopes. Berlin: Springer. doi:10.1101/694539
Peters, B., and Sette, A. (2005). Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinform. 6, 1. doi:10.1186/1471-2105-6-132
Phloyphisut, P., Pornputtapong, N., Sriswasdi, S., and Chuangsuwanich, E. (2019). MHCSeqNet: a deep neural network model for universal MHC binding prediction. BMC Bioinform 20, 1. doi:10.1186/s12859-019-2892-4
Purcell, A. W., McCluskey, J., and Rossjohn, J. (2007). More than one reason to rethink the use of peptides in vaccine design. Nat. Rev. Drug Discov. 6, 5404–5414. doi:10.1038/nrd2224
Valitutti, S., Müller, S., Cella, M., Padovan, E., and Lanzavecchia, A. (1995). Serial triggering of many T-cell receptors by a few peptide–MHC complexes. Nature 375, 6527148–6527151.
Vang, Y. S., and Xie, X. (2017). HLA class I binding prediction via convolutional neural networks. Bioinformatics 33, 172658–172665. doi:10.1093/bioinformatics/btx264
Venkatesh, G., Grover, A., Srinivasaraghavan, G., and Rao, S. (2020). MHCAttnNet: predicting MHC-peptide bindings for MHC alleles classes I and II using an attention-based deep neural model. Bioinformatics 36, i399–i406. doi:10.1093/bioinformatics/btaa479
Vita, R., Mahajan, S., James, A. O. K., Martini, S., Cantrell, J. R., Wheeler, D. K., et al. (2018). The immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 47, D1D339–DID343. doi:10.1093/nar/gky1006
Waldman, A. D., Jill, M., and Lenardo, M. J. (2020). A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat. Rev. Immunol. 20, 11651–11668. doi:10.1038/s41577-020-0306-5
Zeng, H., and Gifford, D. K. (2019). DeepLigand: accurate prediction of MHC class I ligands using peptide embedding. Bioinformatics 35, 14i278–i283. doi:10.1093/bioinformatics/btz330
Keywords: deep learning, prioritization, peptide vaccine design, convolutional neural networks, attention
Citation: Chen Z, Min MR and Ning X (2021) Ranking-Based Convolutional Neural Network Models for Peptide-MHC Class I Binding Prediction. Front. Mol. Biosci. 8:634836. doi: 10.3389/fmolb.2021.634836
Received: 29 November 2020; Accepted: 16 February 2021;
Published: 17 May 2021.
Edited by:
Shuai Cheng Li, City University of Hong Kong, Hong KongReviewed by:
Haicang Zhang, Columbia University, United StatesEsam Abualrous, Freie Universität Berlin, Germany
Copyright © 2021 Chen, Min and Ning. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xia Ning, bmluZy4xMDRAb3N1LmVkdQ==