 Gabriela Canalli Kretzschmar1†
Gabriela Canalli Kretzschmar1† Valéria Bumiller-Bini1†
Valéria Bumiller-Bini1† Miguel Angelo Gasparetto Filho1
Miguel Angelo Gasparetto Filho1 Yohan Ricci Zonta2
Yohan Ricci Zonta2 Kaio Shu Tsyr Yu2
Kaio Shu Tsyr Yu2 Ricardo Lehtonen R. de Souza3
Ricardo Lehtonen R. de Souza3 Luciane Alarcão Dias-Melicio2,4
Luciane Alarcão Dias-Melicio2,4 Angelica Beate Winter Boldt1*
Angelica Beate Winter Boldt1*- 1Laboratory of Human Molecular Genetics, Postgraduate Program in Genetics, Department of Genetics, Federal University of Paraná (UFPR), Belém, Brazil
- 2Medical School of Botucatu, Laboratory of Immunopathology and Infectious Agents–LIAI, UNIPEX–Experimental Research Unity, Sector 5, São Paulo State University (UNESP), Botucatu, Brazil
- 3Laboratory of Polymorphism and Linkage, Department of Genetics, Federal University of Paraná, Curitiba, Brazil
- 4Medical School of Botucatu, Department of Pathology, São Paulo State University (UNESP), Botucatu, Brazil
Complement system (CS) components are associated with Alzheimer’s disease (AD), the commonest cause of dementia in the world. Neutrophils can be attracted to amyloid-β plaques by several pro-inflammatory factors, including the complement anaphylatoxin C5a. They may release neutrophil extracellular traps (NETs), which are chromatin nets associated with myeloperoxidase, elastase, and other enzymes. Some CS molecules, such as C5a, C1q, and CR1, are associated with increased neutrophil recruitment and NETs release. However, the relationship between CS molecules and NETs in AD is poorly understood. In this work, we detected higher NET concentrations in plasma and serum of Brazilian AD patients, than in elderly controls (medians = 2.78 [2.07–6.19] vs. 2.23 [0.33–4.14] ng/mL, p = 0.0005). We discussed these results within the context of our former findings on complement and AD and the context of the literature on complement and NET release, suggesting both as possible therapeutic targets to prevent the progress of the disease.
Introduction
Neuroinflammation is a well-established phenomenon in AD (Czirr and Wyss-Coray, 2012; Wyss-Coray and Rogers, 2012; Dansokho and Heneka, 2018; Kloske and Wilcock, 2020) whose mechanisms are still poorly understood. They are related to the accumulation of amyloid-β (Aβ) plaques and neurofibrillary tangles (NFTs), characteristic AD biomarkers. The first present damage-associated molecular patterns (DAMPs) (Wyss-Coray and Rogers, 2012; Heppner et al., 2015) which are recognized by the complement system (CS) (McGeer et al., 1989; Veerhuis et al., 2011; Tenner et al., 2018). They also induce the expression of endothelial adhesion molecules and the release of pro-inflammatory cytokines by stimulated glial cells (reviewed in Pietronigro et al., 2017). Indeed, the CS appears to play a relevant role in AD, as judged by the strong association of complement genetic polymorphisms with this disease (Morgan, 2018; Krance et al., 2019; Kretzschmar et al., 2020; Tenner, 2020). Besides that, the CS has already been correlated with the formation of neutrophil extracellular traps (NETs) (Palmer et al., 2016; de Bont et al., 2019). NETs are composed of chromatin fibers, citrullinated histones, and cytoplasmic enzymes as myeloperoxidase (MPO) and neutrophil elastase (NE), which altogether operate as an extracellular platform for trapping and killing bacteria (Brinkmann et al., 2004; Yousefi et al., 2009). They have been observed in AD patients and an AD animal model (Zenaro et al., 2015; Dong et al., 2018). However, the possible role of the CS in NET release within AD has never been discussed before. In this study, we focused on reported interactions of the CS with Aβ plaques and their possible role in neutrophil recruitment and NETs release in AD. We also confirmed the presence of higher NET levels in an AD Brazilian cohort.
Alzheimer’s Disease and the Complement System
AD is a neurodegenerative disease responsible for the largest number of dementia cases globally (Alzheimer’s Association, 2020). Several authors sought to understand the disease’s etiology by analyzing different pathways and metabolic processes, most of them causing or being influenced by neuroinflammation (Zlokovic, 2011; Heppner et al., 2015; Du et al., 2018). In AD, the production of Aβ plaques and NFTs are exacerbated. The accumulation of Aβ plaques in the extracellular environment causes the loss of interneural communication (synapses) and activates the local and systemic immunological responses since Aβ plaques can be recognized as DAMPs by phagocytic cells (Czirr and Wyss-Coray, 2012; Wyss-Coray and Rogers, 2012; Heppner et al., 2015) and activate the CS (McGeer et al., 2016).
The CS is considered a key element in innate immunity, playing an essential role in regulating and protecting the central nervous system. The CS consists of an enzymatic cascade with the participation of more than 50 circulating proteins, as well as soluble or membrane receptors and regulators (Lee et al., 2019). It can be activated by three different pathways: classical (CP), lectin (LP), and alternative (AP). For more information on activation and participating CS elements, consult the review of Ricklin et al., 2016. Although the complement pathways are activated in different ways, they all enhance phagocytic activity and may progress to the formation of membrane attack complexes (MAC), leading to cell lysis. In addition, the activation of the complement cascade also results in anaphylatoxin production and recruitment of inflammatory cells. The CS must be tightly regulated. If out of control, the cascade may become offensive, permanently injuring surrounding tissues (Ricklin et al., 2016).
It is not a novelty that CS components and genetic polymorphisms are associated with AD (McGeer et al., 1989; Webster et al., 1997; Loeffler et al., 2008), as complement component 3 (Stoltzner et al., 2000; reviewed by Wyss-Coray and Rogers, 2012; Goetzl et al., 2018), complement component 4 (reviewed by Wyss-Coray and Rogers, 2012; Goetzl et al., 2018), complement membrane complex C5b-C9 (Goetzl et al., 2018), complement component 3a (C3a) and its receptor C3aR (Lian et al., 2015; Litvinchuk et al., 2018), complement component 5a (C5a) and its receptor C5aR1 (An et al., 2018), complement receptor 1 (CR1) (Lambert et al., 2009; Kretzschmar et al., 2020), clusterin (CLU) (Lambert et al., 2009) and complement component 1q (C1q) (Stoltzner et al., 2000; reviewed by; Wyss-Coray and Rogers, 2012; Lian et al., 2016; McGeer et al., 2016; Dejanovic et al., 2018; Goetzl et al., 2018), complement component 9 (reviewed by Wyss-Coray and Rogers, 2012), factor B and factor D (Goetzl et al., 2018). In this work, we will focus only on the CS components associated with AD, which may be related to the recruitment of neutrophils and the formation of NETs (Figure 1).
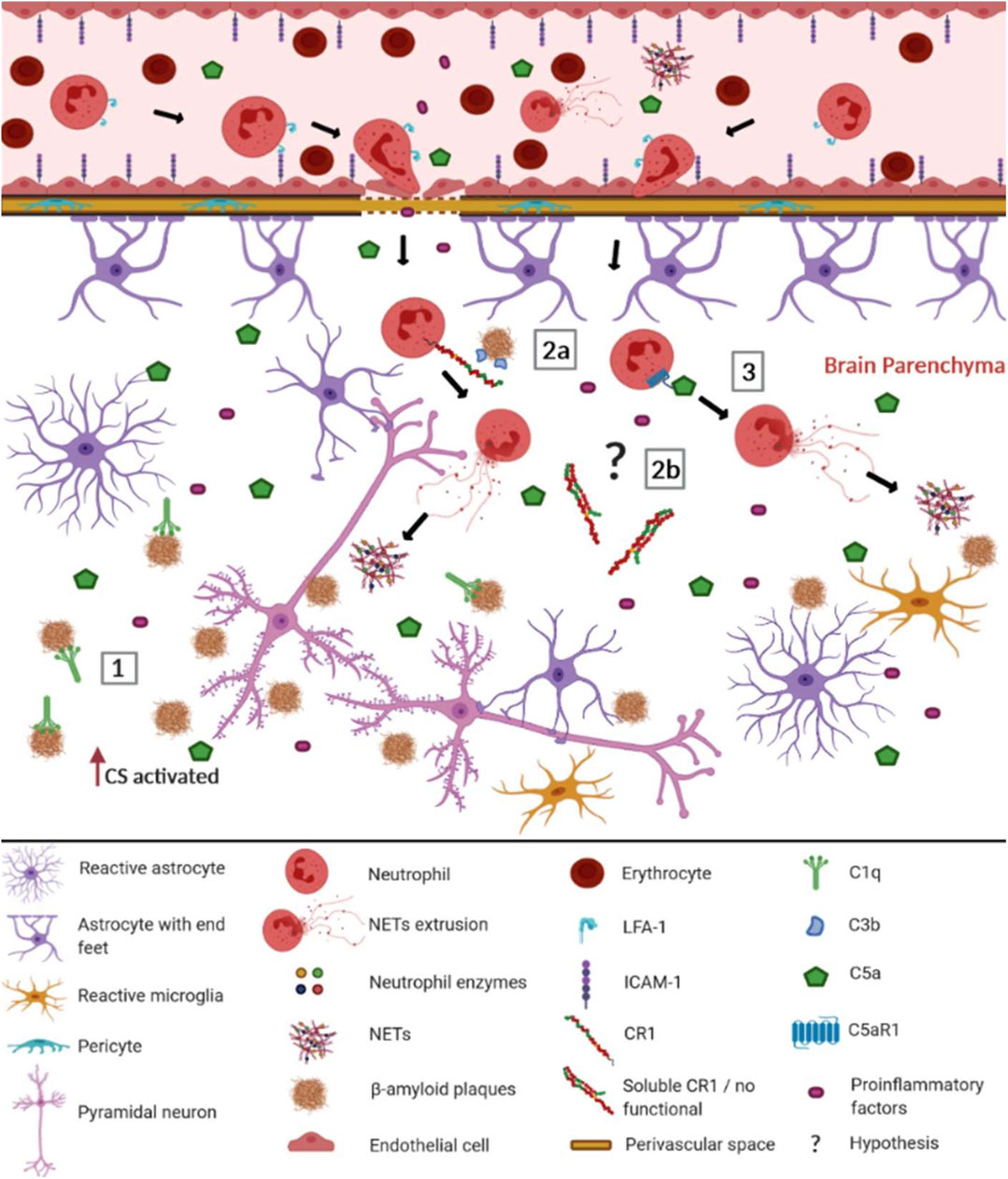
FIGURE 1. Possible involvement of the complement system (CS) in the recruitment of neutrophils and the formation of NETs in Alzheimer's disease. When the blood-brain barrier (BBB) is intact, most complement molecules do not pass through it but can be produced constitutively by brain cells. In AD, the BBB is compromised, facilitating the passage of complement elements, among other pro-inflammatory elements (Alexander, 2018). Aβ plaques are recognized as DAMPs by CS components and other innate immunity receptors, leading to the activation of BBB endothelial cells. They up-regulate ICAM-1 adhesion molecules, allowing neutrophils to adhere and interact with LFA-1, invading the cerebral parenchyma (Pietronigro et al., 2017). There the neutrophils worsen neuroinflammation, performing NET extrusion. In this context, the CS may be involved trough: (1) C1q binding to the Aβ plates, activating the classical complement pathway; (2a) CR1 (CR1*A) molecules present in the neutrophil membrane, that recognize the Aβ plates opsonized with C3b fragments, preferentially leading to NET extrusion; (2b) soluble CR1 formed by non-functional isoforms (CR1*B) that do not inhibit the CS (3) Potent anaphylatoxins such as C5a, which recruit neutrophils from the periphery to the Aβ plaque, being recognized by C5aR1 neutrophil receptors, promoting the release of NETs. Although other elements of the inflammatory reaction do occur, in this figure, we focus on elements of the CS discussed throughout this study. The complement cascades’ reactions were not represented due to space and number of elements participating in the pathways. The role of cytokines and endothelial adhesion molecules in neutrophil recruitment for Aβ plaques was extensively reviewed by Pietronigro et al. (2017) ICAM-1- Intercellular adhesion molecule 1; LFA-1- Lymphocyte function-associated antigen 1; NET- extracellular neutrophil traps; CS- complement system; C1q- complement component 1q; CR1- complement receptor 1; C3b- complement component 3b; C5a- complement component 5a; C5aR1- complement component 5a receptor. This figure was created with BioRender.com.
Alzheimer’s Disease and Neutrophil Traps
Neutrophils have an essential role in inflammation, acting through many mechanisms: phagocytosis, degranulation, and extrusion of NETs (reviewed by Shen et al., 2001; Rossi et al., 2020). NETs are involved in host tissue injury and inflammation associated with autoimmune diseases, acute injuries, atherosclerosis, vasculitis, and cancer (Jorch and Kubes, 2017). In 2015, Zenaro and colleagues observed NETs adjacent to Aβ plaque deposits in the cerebral vascular and intraparenchymal region of an AD animal model and European AD patients. They suggested that Aβ plaques may play an essential role in the recruitment and movement of neutrophils, which Baik et al. (2014) also observed in an AD animal model. Furthermore, NET extrusion was also detected in high concentrations in European AD patients’ serum (Dong et al., 2018).
C5a-C5aR1 Axis and the Relation With Neutrophil Traps and Alzheimer’s Disease
After complement activation (McGeer et al., 1989; Veerhuis et al., 2011), C3 and C5 are cleaved, generating the anaphylatoxins C3a and C5a (reviewed by de Bont et al., 2019). C3a lacks chemotactic activity (Ehrengruber et al., 1994), but C5a generates a potent chemotactic response and induces neutrophil migration (Ehrengruber et al., 1994), as well as the release of NETs (reviewed by de Bont et al., 2019). Therapeutic inhibitors blocking C5a and/ or its receptor C5aR1 have been proposed to treat AD (Fonseca et al., 2013; Landlinger et al., 2015; An et al., 2018). Although the functional connection between high NET levels and C5a has been well described (Yousefi et al., 2009; Huang et al., 2015; Yuen et al., 2016; Fattahi et al., 2018; de Bont et al., 2019), it has never been explored in AD.
The CS seems to be, at least through C5a, attracting neutrophils to the brain and inducing NETs extrusion. Neutrophil activation by C5a extrudes the mitochondrial DNA (Yousefi et al., 2009), which suggests that in AD, after NETs extrusion, the neutrophils do not die, at least not through C5a stimulation. NETs deposition triggers CS activation via the alternative pathway and properdin binding (Yuen et al., 2016). The neutrophils probably recognize the Aβ plaques and trap them, resulting in increased inflammation and tissue injury. After triggering the neutrophils to the tissue, the CS may not degrade the NETs, resulting in tissue NET accumulation and CS over-activation (de Bont et al., 2019). Thus, the use of C5a inhibitors may decrease NETs activation in AD.
CR1 and Its Relationship With Neutrophil Traps and Alzheimer’s Disease
Even before NET’s discovery, several authors sought to understand the mechanisms of interaction between the complement system and neutrophils. Cytokines as tumor necrosis factor-alpha (TNFα), granulocyte-monocyte and granulocyte colony stimulating factors (GM-CSF, G-CSF), interleukin 1(IL-1), platelet activating factor (PAF), and lymphotoxin-beta (LTB) up-regulate phagocytic complement 1 receptor (CR1, also known as CD35) in neutrophils, increasing its association with C3b-opsonized microspheres. However, only TNFα, G-CSF, and PAF increased their phagocytic uptake (Ogle et al., 1990). Besides that, CR1 recognizing C3b-IgG complexes on the neutrophil membrane led to changes in signal transduction events associated with Fc receptors, resulting in myeloperoxidase's release and generation of hypochlorous acid (Sambandam and Chatham, 1998). CR1 blockage is followed by a decrease in NET concentration, revealing an essential role of this molecule in the extrusion process (Palmer et al., 2016). Thus, recognition of C3b-opsonized particles by CR1 on neutrophil membranes may preferentially lead to the release of NETs instead of phagocytosis, although more studies are needed to corroborate this hypothesis.
Interestingly, increased AD susceptibility has been repeatedly associated with CR1 polymorphisms (Lambert et al., 2009). Nowadays, the hypothesis proposed for this association is that some polymorphisms (such as rs6656401*A) facilitate non-homologous recombination resulting in the preferential expression of dysfunctional CR1*B isoform (Brouwers et al., 2012; Mahmoudi et al., 2015). Although CR1*B has an additional C3b/C4b binding site, its neuronal expression occurs in vesicular form (Hazrati et al., 2012). It is also expressed in lower amounts in erythrocytes than the CR1*A functional protein (Mahmoudi et al., 2018). Thus, CR1*B probably impairs the process of removing Aβ plates and regulating the CS (Mahmoudi et al., 2018). Heterozygote CR1*A/CR1*B individuals express both isoforms. In this case, neutrophil recognition of C3b-opsonized Aβ plates may occur by CR1*A, with consequent release of NETs. CR1 can also be found in a soluble form (sCR1). AD patients have higher levels of sCR1 in serum (Mahmoudi et al., 2018), plasma (Kretzschmar et al., 2020), and cerebrospinal fluid (CFS) (Daborg et al., 2012). sCR1 is a potent local inhibitor of the complement system and is formed through vesiculation or proteolysis of the membrane-bound CR1 (Pascual et al., 1993; Danielsson et al., 1994; Dervillez et al., 1997; Hamer et al., 1998), inhibiting the CS by dissociating C3 convertases, and targeting C3b and C4b for degradation (Zhu et al., 2015). It is possible that large sCR1 quantities would inhibit complement’s beneficial role of removing Aβ plaques, contributing to its accumulation. However, no studies to date demonstrated whether individuals who have only the CR1*B isoform present functional sCR1. If sCR1 is generated from CR1*B, CS inhibition probably will not occur properly, recruiting more neutrophils to the affected region, with higher extrusion of NETs, ultimately contributing to chronic neuroinflammation. Although CR1’s participation in the increase of NETs release seems to be evident, it is not yet clear how this may be related to the different isoforms of the molecule and its association with AD. Still, it raises an exciting possibility of a new role for CR1 in the disease, which needs to be investigated in further functional studies.
C1q and the Relation With Neutrophil Traps and Alzheimer’s Disease
The C1q molecule of the CP participates within an essential process in brain homeostasis. In periods when synapse pruning happens, C1q tags inappropriate connections between neurons for removal by the microglia (Presumey et al., 2017). In neurodegenerative diseases, C1q may lead to aberrant synapse loss (Dejanovic et al., 2018). Curiously, Aβ binds and activates C1q in the absence of immunoglobulins (Rogers et al., 1992), starting the CP and probably promoting synapse loss. A study with a mouse model of AD lacking C1q demonstrated a significant reduction in inflammation and neuropathological features (Presumey et al., 2017).
Some researchers already analyzed the relationship between C1q and NETs. Increased C1q deposition inhibits DNase activity, resulting in NET accumulation (Leffler et al., 2012). When C1q is inhibited, the complement cascade does not progress, and NETs do not appear (Hair et al., 2018). NETs are mainly degraded by endonuclease DNase1 (Hakkim et al., 2010) and then cleared by macrophages (Farrera and Fadeel, 2013). DNases have already been used to successfully treat AD in a case report (Tetz and Tetz, 2016). Genetic DNASE1 variants have been investigated in systemic lupus erythematosus (Pruchniak et al., 2019), however, its role in AD has never been investigated. DNase has been used as an efficient drug to degrade NET structure in breast cancer, lung injury, and lupus mouse models (reviewed in Jorch and Kubes, 2017).
High Neutrophil Traps Levels in a Brazilian Cohort
Recently, we confirmed the genetic association of complement receptor 1 (CR1) polymorphisms in an AD Brazilian cohort (Kretzschmar et al., 2020). Based on the association between molecules of the complement system and NETs, we aimed to investigate if NET levels are also increased within the same Brazilian cohort. We quantified NETs in plasma of 22 AD patients and 20 elderly controls (EC), and serum of another 11 EC (considering that NET levels are similar in serum and plasma of the same individual (Abrams et al., 2019)). The study was approved by the local ethics committee (CAAE 55965316.1.0000.0102). All participating individuals were older than 65 years (AD median = 82.5 [70–88] years old; EC median = 76 [69–99] years old). AD were recruited from the Clinical Hospital of the Federal University of Paraná. AD and EC were diagnosed or confirmed to be neurologically normal based on clinical history and cognitive tests (Frota et al., 2011). Diabetes and systemic arterial hypertension (SAH) are common pathologies in the elderly that may cause NET release (Soongsathitanon et al., 2019; Parackova et al., 2020). Both diseases were not associated with NETs in our study and the association of Alzheimer's with NETs was also independent of HAS (OR = [95%CI = 1.75–6.91], p = 0.003) (data available in Supplementary Table S1). The NE-DNA concentrations in serum and plasma samples were quantified using an adapted ELISA test with immunofluorescence (Czaikoski et al., 2016; Colón et al., 2019) (Supplementary Figure S1). Anti-elastase antibodies were used for capturing these NE-DNA complexes, and dsDNA fluorescent reagent was used for detection and quantification. The data was tested for normality (D’Agostino and Pearson test). We compared patients and controls in two ways: 1) using the absolute values (ng/mL), and 2) establishing the 3rd quartile in controls as a threshold for defining high and low NET levels (2.548 ng/mL). We also used this threshold for defining the theoretical median in the Wilcoxon test. The groups were compared using unpaired T-test and two-way ANOVA. All the analyses were done using GraphPad Prism v.6 software. The p-values were corrected for multiple testing using the false discovery rate (FDR) method (Benjamini and Hochberg, 1995), performed in R language 3.6.1, through the Stats package (R Development Core Team, 2011). Corrected p-values lower than 0.05 were considered significant. The data used is available in the Supplementary Table S1.
Our study brings, for the first time, higher circulating NET levels in AD Brazilian patients. In this work, we detected higher NET concentrations in plasma and serum of Brazilian AD patients, than in elderly controls (medians = 2.78 [2.07–6.19] vs. 2.23 [0.33–4.14] ng/mL, p = 0.0005) (Figure 2A). The difference remained after dichotomizing AD and EC into high- and low-NET producers. The median NET concentration of high-NET producers was 3.95 (3.09–6.19 ng/mL) in AD, compared to 3.0 (2.586–4.14 ng/mL) in EC (p = 0.012). For low-NET producers, the median level of AD was 2.28 (2.07–2.46 ng/mL), compared to 2.0 in EC (0.33–2.46 ng/mL) (p = 0.042) (Figure 2B). Although NETs extrusion can lead to cell death by NETosis, we did not perform the assays to evaluate it. Despite the small number of samples used here, few studies investigating NETs in AD patients were published. All of them confirm the increased NETs in AD (Zenaro et al., 2015; Dong et al., 2018).
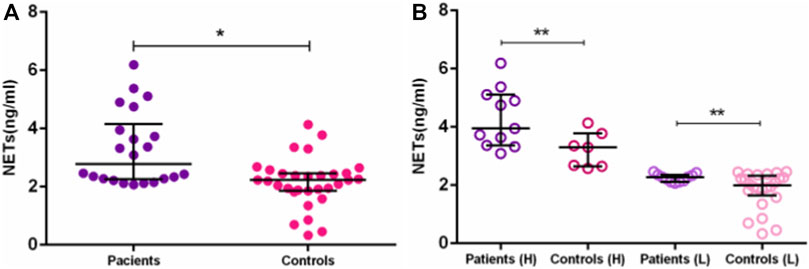
FIGURE 2. NETs concentration between AD patients and controls. Differences in NETs concentration between: (A)—AD patients and elderly controls. (B)—AD patients and elderly controls in High (H) and Low (L) groups. *p ≤ 0.01; **p ≤ 0.05.
Concluding Remarks
NETs seem to be promising as new therapeutic targets for AD treatment. We propose more investigations into the connection between C5a, C1q, and CR1 with NETs in AD, as well as genetic associations studies to investigate variants in DNase genes (DNASE1, DNASE2, and DNASE1L3) that can result in a down-regulation of DNase expression in AD.
Data Availability Statement
The original data presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Research Ethics Committee of the Health Sciences Sector (Federal University of Paraná) (CAAE:55965316.1.0000.0102), according to Resolution 466/2012 of the National Health Council and and the 1964 Helsinki declaration and its later amendments or comparable ethical standards. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
AB, LD-M, and GK contributed to the conception of the work. AB, MG, VB-B, and GK designed the study. RS provided the samples. LD-M made available the infrastructure and material for the ELISA assay. MG, YZ, and KY performed ELISA assay. AB, VB-B, and GK did the statistical analysis. GK and MG made the illustrations. AB, MG, VB-B, and GK drafted the manuscript. All authors revised the work critically for intellectual content and approved the final version of the work.
Funding
This work was supported by grants of Coordenação de Aperfeiçoamento de Pessoal de Nível Superior–Brasil (CAPES-40001016006P1 GCK and VBB scholarships, and CAPES/PROAP—Finance Code 001); Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP–2018/09706-7). AB is a recipient of CNPq fellowship (CNPq-314288/2018-0). The funding agencies had no role in study design, in the collection, analysis, and interpretation of data, in the writing of the letter, and in the decision to submit it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank all study participants, especially the patients and the staff of the Laboratory of Polymorphism and Linkage for sample collection.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2021.630869/full#supplementary-material.
References
Abrams, S. T., Morton, B., Alhamdi, Y., Alsabani, M., Lane, S., Welters, I. D., et al. (2019). A novel assay for neutrophil extracellular trap formation independently predicts disseminated intravascular coagulation and mortality in critically ill patients. Am. J. Respir. Crit. Care Med. 200, 869–880. doi:10.1164/rccm.201811-2111OC
Alexander, J. J. (2018). Blood-brain barrier (BBB) and the complement landscape. Mol. Immunol. 102, 26–31. doi:10.1016/j.molimm.2018.06.267
Alzheimer’s-Association(2020). Alzheimer’s disease facts and figures. Alzheimer’s Dement. 16, 391–460. doi:10.1002/alz.12068
An, X.-Q., Xi, W., Gu, C.-Y., and Huang, X. (2018). Complement protein C5a enhances the β-amyloid-induced neuro-inflammatory response in microglia in Alzheimer's disease. Med. Sci. (Paris) 34, 116–120. doi:10.1051/medsci/201834f120
Baik, S. H., Cha, M.-Y., Hyun, Y.-M., Cho, H., Hamza, B., Kim, D. K., et al. (2014). Migration of neutrophils targeting amyloid plaques in Alzheimer’s disease mouse model. Neurobiol. Aging 35, 1286–1292. doi:10.1016/j.neurobiolaging.2014.01.003
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. J.R. Stat. Soc. B. 57, 289–300. doi:10.1111/j.2517-6161.1995.tb02031.x
Brinkmann, V., Reichard, U., Goosmann, C., Fauler, B., Uhlemann, Y., Weiss, D. S., et al. (2004). Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535. doi:10.1126/science.1092385
Brouwers, N., Van Cauwenberghe, C., Engelborghs, S., Lambert, J.-C., Bettens, K., Le Bastard, N., et al. (2012). Alzheimer risk associated with a copy number variation in the complement receptor 1 increasing C3b/C4b binding sites. Mol. Psychiatry 17, 223–233. doi:10.1038/mp.2011.24
Colón, D. F., Wanderley, C. W., Franchin, M., Silva, C. M., Hiroki, C. H., Castanheira, F. V. S., et al. (2019). Neutrophil extracellular traps (NETs) exacerbate severity of infant sepsis. Crit. Care 23, 1–13. doi:10.1186/s13054-019-2407-8
Czaikoski, P. G., Mota, J. M. S. C., Nascimento, D. C., Sônego, F., Castanheira, F. V. E. S., Melo, P. H., et al. (2016). Neutrophil extracellular traps induce organ damage during experimental and clinical sepsis. PLoS One 11, e0148142. doi:10.1371/journal.pone.0148142
Czirr, E., and Wyss-Coray, T. (2012). The immunology of neurodegeneration. J. Clin. Invest. 122, 1156–1163. doi:10.1172/JCI58656
Daborg, J., Andreasson, U., Pekna, M., Lautner, R., Hanse, E., Minthon, L., et al. (2012). Cerebrospinal fluid levels of complement proteins C3, C4 and CR1 in Alzheimer’s disease. J. Neural Transm. 119, 789–797. doi:10.1007/s00702-012-0797-8
Danielsson, C., Pascual, M., French, L., Steiger, G., and Schifferli, J.-A. (1994). Soluble complement receptor type 1 (CD35) is released from leukocytes by surface cleavage. Eur. J. Immunol. 24, 2725–2731. doi:10.1002/eji.1830241123
Dansokho, C., and Heneka, M. T. (2018). Neuroinflammatory responses in Alzheimer's disease. J. Neural Transm. 125, 771–779. doi:10.1007/s00702-017-1831-7
de Bont, C. M., Boelens, W. C., and Pruijn, G. J. M. (2019). NETosis, complement, and coagulation: a triangular relationship. Cell. Mol. Immunol. 16, 19–27. doi:10.1038/s41423-018-0024-0
Dejanovic, B., Huntley, M. A., De Mazière, A., Meilandt, W. J., Wu, T., Srinivasan, K., et al. (2018). Changes in the synaptic proteome in tauopathy and rescue of tau-induced synapse loss by C1q antibodies. Neuron 100, 1322–1336. doi:10.1016/j.neuron.2018.10.014
Dervillez, X., Oudin, S., Libyh, M. T., Tabary, T., Reveil, B., Philbert, F., et al. (1997). Catabolism of the human erythrocyte C3b/C4b receptor (CR1, CD35) : vesiculation and/or proteolysis?. Immunopharmacol. 38, 129–140. doi:10.1016/S0162-3109(97)00066-0
Dong, Y., Lagarde, J., Xicota, L., Corne, H., Chantran, Y., Chaigneau, T., et al. (2018). Neutrophil hyperactivation correlates with Alzheimer’s disease progression. Ann. Neurol. 83, 387–405. doi:10.1002/ana.25159
Du, X., Wang, X., and Geng, M. (2018). Alzheimer’s disease hypothesis and related therapies. Transl. Neurodegener. 7, 2. doi:10.1186/s40035-018-0107-y
Ehrengruber, M. U., Geiser, T., and Deranleau, D. A. (1994). Activation of human neutrophils by C3a and C5A: Comparison of the effects on shape changes, chemotaxis, secretion, respiratory burst. FEBS Lett. 346, 181–184. doi:10.1016/0014-5793(94)00463-3
Farrera, C., and Fadeel, B. (2013). Macrophage clearance of neutrophil extracellular traps is a silent process. J. Immunol. 191, 2647–2656. doi:10.4049/jimmunol.1300436
Fattahi, F., Frydrych, L. M., Bian, G., Kalbitz, M., Herron, T. J., Malan, E. A., et al. (2018). Role of complement C5a and histones in septic cardiomyopathy. Mol. Immunol. 102, 32–41. doi:10.1016/j.molimm.2018.06.006
Fonseca, M. I., McGuire, S. O., Counts, S. E., and Tenner, A. J. (2013). Complement activation fragment C5a receptors, CD88 and C5L2, are associated with neurofibrillary pathology. J. Neuroinflammation 10, 1–12. doi:10.1186/1742-2094-10-25
Frota, N. A. F., Nitrini, R., Damasceno, B. P., Forlenza, O., Dias-Tosta, E., Silva, A. B. d., et al. (2011). Critérios para diagnóstico de doença de Alzheimer. Dement. Neuropsychol. 5, 5–10. doi:10.1590/S1980-57642011DN05030002
Goetzl, E. J., Schwartz, J. B., Abner, E. L., Jicha, G. A., and Kapogiannis, D. (2018). High complement levels in astrocyte-derived exosomes of Alzheimer disease. Ann. Neurol. 83, 544–552. doi:10.1002/ana.25172
Hair, P. S., Enos, A. I., Krishna, N. K., and Cunnion, K. M. (2018). Inhibition of immune complex complement activation and neutrophil extracellular trap formation by peptide inhibitor of complement C1. Front. Immunol. 9, 1–12. doi:10.3389/fimmu.2018.00558
Hakkim, A., Furnrohr, B. G., Amann, K., Laube, B., Abed, U. A., Brinkmann, V., et al. (2010). Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. U.S.A. 107, 9813–9818. doi:10.1073/pnas.0909927107
Hamer, I., Paccaud, J.-P., Belin, D., Maeder, C., and Carpentier, J.-L. (1998). Soluble form of complement C3b/C4b receptor (CR1) results from a proteolytic cleavage in the C-terminal region of CR1 transmembrane domain. Biochem. J. 329, 183–190. doi:10.1042/bj3290183
Hazrati, L.-N., Van Cauwenberghe, C., Brooks, P. L., Brouwers, N., Ghani, M., Sato, C., et al. (2012). Genetic association of CR1 with Alzheimer’s disease: A tentative disease mechanism. Neurobiol. Aging 33, 2949.e5–2949.e12. doi:10.1016/j.neurobiolaging.2012.07.001
Heppner, F. L., Ransohoff, R. M., and Becher, B. (2015). Immune attack: the role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 16, 358–372. doi:10.1038/nrn3880
Huang, Y.-M., Wang, H., Wang, C., Chen, M., and Zhao, M.-H. (2015). Promotion of hypercoagulability in antineutrophil cytoplasmic antibody-associated vasculitis by C5a-induced tissue factor-expressing microparticles and neutrophil extracellular traps. Arthritis Rheumatol. 67, 2780–2790. doi:10.1002/art.39239
Jorch, S. K., and Kubes, P. (2017). An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 23, 279–287. doi:10.1038/nm.4294
Kloske, C. M., and Wilcock, D. M. (2020). The important interface between apolipoprotein E and neuroinflammation in Alzheimer’s disease. Front. Immunol. 11, 1–12. doi:10.3389/fimmu.2020.00754
Krance, S. H., Wu, C.-Y., Zou, Y., Mao, H., Toufighi, S., He, X., et al. (2019). The complement cascade in Alzheimer’s disease: A systematic review and meta-analysis. Mol. Psychiatry [Epub ahead of print]. doi:10.1038/s41380-019-0536-8
Kretzschmar, G. C., Antoniazzi, A. A. H., Oliveira, L. C., Nisihara, R. M., Petzl-Erler, M. L., de Souza, R. L. R., et al. (2020). First report of CR1 polymorphisms and soluble CR1 levels associated with late onset Alzheimer’s disease (LOAD) in Latin America. J. Mol. Neurosci. 70, 1338–1344. doi:10.1007/s12031-020-01547-2
Lambert, J.-C., Heath, S., Even, G., Campion, D., Sleegers, K., Hiltunen, M., et al. (2009). Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 41, 1094–1099. doi:10.1038/ng.439
Landlinger, C., Oberleitner, L., Gruber, P., Noiges, B., Yatsyk, K., Santic, R., et al. (2015). Active immunization against complement factor C5a: a new therapeutic approach for Alzheimer’s disease. J. Neuroinflammation 12, 1–13. doi:10.1186/s12974-015-0369-6
Lee, J. D., Coulthard, L. G., and Woodruff, T. M. (2019). Complement dysregulation in the central nervous system during development and disease. Semin. Immunol. 45, 101340. doi:10.1016/j.smim.2019.101340
Leffler, J., Martin, M., Gullstrand, B., Tydén, H., Lood, C., Truedsson, L., et al. (2012). Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J. Immunol. 188, 3522–3531. doi:10.4049/jimmunol.1102404
Lian, H., Litvinchuk, A., Chiang, A. C.-A., Aithmitti, N., Jankowsky, J. L., and Zheng, H. (2016). Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of Alzheimer’s disease. J. Neurosci. 36, 577–589. doi:10.1523/JNEUROSCI.2117-15.2016
Lian, H., Yang, L., Cole, A., Sun, L., Chiang, A. C.-A., Fowler, S. W., et al. (2015). NFκB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron 85, 101–115. doi:10.1016/j.neuron.2014.11.018
Litvinchuk, A., Wan, Y.-W., Swartzlander, D. B., Chen, F., Cole, A., Propson, N. E., et al. (2018). Complement C3aR inactivation attenuates tau pathology and reverses an immune network deregulated in tauopathy models and Alzheimer’s disease. Neuron 100, 1337–1353. doi:10.1016/j.neuron.2018.10.031
Loeffler, D. A., Camp, D. M., and Bennett, D. A. (2008). Plaque complement activation and cognitive loss in Alzheimer’s disease. J. Neuroinflammation 5, 1–8. doi:10.1186/1742-2094-5-9
Mahmoudi, R., Feldman, S., Kisserli, A., Duret, V., Tabary, T., Bertholon, L.-A., et al. (2018). Inherited and acquired decrease in complement receptor 1 (CR1) density on red blood cells associated with high levels of soluble CR1 in Alzheimer’s disease. Int. J. Mol. Sci. 19, 2175. doi:10.3390/ijms19082175
Mahmoudi, R., Kisserli, A., Novella, J.-L., Donvito, B., Dramé, M., Réveil, B., et al. (2015). Alzheimer’s disease is associated with low density of the long CR1 isoform. Neurobiol. Aging 36, 1766.e5–1766.e12. doi:10.1016/j.neurobiolaging.2015.01.006
McGeer, P. L., Akiyama, H., Itagaki, S., and McGeer, E. G. (1989). Activation of the classical complement pathway in brain tissue of Alzheimer patients. Neurosci. Lett. 107, 341–346. doi:10.1016/0304-3940(89)90843-4
McGeer, P. L., Rogers, J., and McGeer, E. G. (2016). Inflammation, antiinflammatory agents, and Alzheimer’s disease: The last 22 years. J. Alzheimer’s Dis. 54, 853–857. doi:10.3233/JAD-160488
Morgan, B. P. (2018). Complement in the pathogenesis of Alzheimer’s disease. Semin. Immunopathol. 40, 113–124. doi:10.1007/s00281-017-0662-9
Ogle, J. D., Noel, J. G., Sramkoski, R. M., Ogle, C. K., and Alexander, J. W. (1990). The effects of cytokines, platelet activating factor, and arachidonate metabolites on C3B receptor (CR1, CD35) expression and phagocytosis by neutrophils. Cytokine 2, 447–455. doi:10.1016/1043-4666(90)90054-W
Palmer, L. J., Damgaard, C., Holmstrup, P., and Nielsen, C. H. (2016). Influence of complement on neutrophil extracellular trap release induced by bacteria. J. Periodontal Res. 51, 70–76. doi:10.1111/jre.12284
Parackova, Z., Zentsova, I., Vrabcova, P., Klocperk, A., Sumnik, Z., Pruhova, S., et al. (2020). Neutrophil extracellular trap induced dendritic cell activation leads to Th1 polarization in type 1 diabetes. Front. Immunol. 11, 1–9. doi:10.3389/fimmu.2020.00661
Pascual, M., Lutz, H. U., Steiger, G., Stamer, P., and Schifferli, J. A. (1993). Release of vesicles enriched in complement receptor 1 from human erythrocytes. J. Immunol. 151, 397–404.
Pietronigro, E. C., Della Bianca, V., Zenaro, E., and Constantin, G. (2017). NETosis in Alzheimer’s disease. Front. Immunol. 8, 211. doi:10.3389/fimmu.2017.00211
Presumey, J., Bialas, A. R., and Carroll, M. C. (2017). Complement system in neural synapse elimination in development and disease. Adv Immunol. 135, 53–79. doi:10.1016/bs.ai.2017.06.004
Pruchniak, M. P., Ostafin, M., Wachowska, M., Jakubaszek, M., Kwiatkowska, B., Olesinska, M., et al. (2019). Neutrophil extracellular traps generation and degradation in patients with granulomatosis with polyangiitis and systemic lupus erythematosus. Autoimmunity 52, 126–135. doi:10.1080/08916934.2019.1631812
R Development Core Team(2011). R: A language and environment for statistical computing. R. Found. Stat. Comput. 1, 409. doi:10.1007/978-3-540-74686-7
Ricklin, D., Reis, E. S., and Lambris, J. D. (2016). Complement in disease: a defence system turning offensive. Nat. Rev. Nephrol. 12, 383–401. doi:10.1038/nrneph.2016.70
Rogers, J., Cooper, N. R., Webster, S., Schultz, J., McGeer, P. L., Styren, S. D., et al. (1992). Complement activation by β-amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 89, 10016–10020. doi:10.1073/pnas.89.21.10016
Rossi, B., Constantin, G., and Zenaro, E. (2020). The emerging role of neutrophils in neurodegeneration. Immunobiology 225, 151865. doi:10.1016/j.imbio.2019.10.014
Sambandam, T., and Chatham, W. W. (1998). Ligation of CR1 attenuates Fc receptor-mediated myeloperoxidase release and HOCl production by neutrophils. J. Leukoc. Biol. 63, 477–485. doi:10.1002/jlb.63.4.477
Shen, Y., Lue, L. F., Yang, L. B., Roher, A., Kuo, Y. M., Strohmeyer, R., et al. (2001). Complement activation by neurofibrillary tangles in Alzheimer’s disease. Neurosci. Lett. 305, 165–168. doi:10.1016/S0304-3940(01)01842-0
Soongsathitanon, J., Umsa-Ard, W., and Thongboonkerd, V. (2019). Proteomic analysis of peripheral blood polymorphonuclear cells (PBMCs) reveals alteration of neutrophil extracellular trap (NET) components in uncontrolled diabetes. Mol. Cel. Biochem. 461, 1–14. doi:10.1007/s11010-019-03583-y
Stoltzner, S. E., Grenfell, T. J., Mori, C., Wisniewski, K. E., Wisniewski, T. M., Selkoe, D. J., et al. (2000). Temporal accrual of complement proteins in amyloid plaques in down’s syndrome with Alzheimer’s disease. Am. J. Pathol. 156, 489–499. doi:10.1016/S0002-9440(10)64753-0
Tenner, A. J. (2020). Complement-Mediated events in Alzheimer’s disease: mechanisms and potential therapeutic targets. J. Immunol. 204, 306–315. doi:10.4049/jimmunol.1901068
Tenner, A. J., Stevens, B., and Woodruff, T. M. (2018). New tricks for an ancient system: Physiological and pathological roles of complement in the CNS. Mol. Immunol. 102, 3–13. doi:10.1016/j.molimm.2018.06.264
Tetz, V., and Tetz, G. (2016). Effect of deoxyribonuclease I treatment for dementia in end-stage Alzheimer’s disease: a case report. J. Med. Case Rep. 10, 131. doi:10.1186/s13256-016-0931-6
Veerhuis, R., Nielsen, H. M., and Tenner, A. J. (2011). Complement in the brain. Mol. Immunol. 48, 1592–1603. doi:10.1016/j.molimm.2011.04.003
Webster, S., Bradt, B., Rogers, J., and Cooper, N. (1997). Aggregation state-dependent activation of the classical complement pathway by the amyloid β peptide. J. Neurochem. 69, 388–398. doi:10.1046/j.1471-4159.1997.69010388.x
Wyss-Coray, T., and Rogers, J. (2012). Inflammation in alzheimer disease--A brief review of the basic science and clinical literature. Cold Spring Harb. Perspect. Med. 2, a006346. doi:10.1101/cshperspect.a006346
Yousefi, S., Mihalache, C., Kozlowski, E., Schmid, I., and Simon, H. U. (2009). Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 16, 1438–1444. doi:10.1038/cdd.2009.96
Yuen, J., Pluthero, F. G., Douda, D. N., Riedl, M., Cherry, A., Ulanova, M., et al. (2016). NETosing neutrophils activate complement both on their own NETs and bacteria via alternative and non-alternative pathways. Front. Immunol. 7, 1–14. doi:10.3389/fimmu.2016.00137
Zenaro, E., Pietronigro, E., Bianca, V. Della., Piacentino, G., Marongiu, L., Budui, S., et al. (2015). Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 21, 880–886. doi:10.1038/nm.3913
Zhu, X. C., Yu, J. T., Jiang, T., Wang, P., Cao, L., and Tan, L. (2015). CR1 in Alzheimer’s disease. Mol. Neurobiol. 51, 753–765. doi:10.1007/s12035-014-8723-8
Keywords: Alzheimer's Disease, neutrophil extracellular traps, inflammation, complement system, CR1, C5a, C1q
Citation: Kretzschmar GC, Bumiller-Bini V, Gasparetto Filho MA, Zonta YR, Yu KST, de Souza RLR, Dias-Melicio LA and Boldt ABW (2021) Neutrophil Extracellular Traps: A Perspective of Neuroinflammation and Complement Activation in Alzheimer’s Disease. Front. Mol. Biosci. 8:630869. doi: 10.3389/fmolb.2021.630869
Received: 18 November 2020; Accepted: 18 February 2021;
Published: 08 April 2021.
Edited by:
Grazia Daniela Femminella, University of Naples Federico II, ItalyReviewed by:
Carlos Romá-Mateo, University of Valencia, SpainTheocharis Konstantinidis, Democritus University of Thrace, Greece
Copyright © 2021 Kretzschmar, Bumiller-Bini, Gasparetto Filho, Zonta, Yu, de Souza, Dias-Melicio and Boldt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angelica Beate Winter Boldt, YW5nZWxpY2Fib2xkdEBnbWFpbC5jb20=
†These authors have contributed equally to this work