 Stephanie Nguyen
Stephanie Nguyen Jia Q. Truong
Jia Q. Truong John B. Bruning
John B. Bruning- 1Institute of Photonics and Advanced Sensing (IPAS), School of Biological Sciences, The University of Adelaide, Adelaide, SA, Australia
- 2School of Biological Sciences, The University of Adelaide, Adelaide, SA, Australia
The impact of invasive fungal infections on human health is a serious, but largely overlooked, public health issue. Commonly affecting the immunocompromised community, fungal infections are predominantly caused by species of Candida, Cryptococcus, and Aspergillus. Treatments are reliant on the aggressive use of pre-existing antifungal drug classes that target the fungal cell wall and membrane. Despite their frequent use, these drugs are subject to unfavorable drug-drug interactions, can cause undesirable side-effects and have compromised efficacy due to the emergence of antifungal resistance. Hence, there is a clear need to develop novel classes of antifungal drugs. A promising approach involves exploiting the metabolic needs of fungi by targeted interruption of essential metabolic pathways. This review highlights potential antifungal targets including enolase, a component of the enolase-plasminogen complex, and enzymes from the mannitol biosynthesis and purine nucleotide biosynthesis pathways. There has been increased interest in the enzymes that comprise these particular pathways and further investigation into their merits as antifungal targets and roles in fungal survival and virulence are warranted. Disruption of these vital processes by targeting unconventional pathways with small molecules or antibodies may serve as a promising approach to discovering novel classes of antifungals.
Introduction
Opportunistic fungal pathogens impose a significant societal and economic burden on the public health sector, particularly amongst immunocompromised patients. In the United States alone, it has been estimated that fungal infections incur hospitalization and outpatient costs that exceed $7.2 billion USD annually (Benedict et al., 2019). These costs are expected to rise in tandem with the expansion of the immunocompromised population and subsequent increase in the incidence of invasive mycoses. This predisposed population includes recipients of invasive surgery, chemotherapy, and immunosuppressive therapy, as well as sufferers of human immunodeficiency virus (HIV)/acquired immunodeficiency syndrome (AIDS) and cystic fibrosis (Nucci and Marr, 2005).
Global estimates suggest that over 150 million individuals are affected by serious fungal infections which culminate in over 1.6 million deaths per annum (Bongomin et al., 2017). Common fungal pathogens associated with these opportunistic infections include Aspergillus spp. (most commonly Aspergillus fumigatus), Candida albicans (and other non-albicans Candida species), Cryptococcus neoformans, Histoplasma capsulatum, Coccidioides imitis, and Pneumocystis jirovecii (Walsh and Dixon, 1996; Vandeputte et al., 2012). Amongst these fungal pathogens, species of Aspergillus, Candida, Pneumocystis, and Cryptococcus are responsible for over 90% of total global fungal mortalities and therefore, these fungal species pose the most prominent threat to human health (Brown et al., 2012). Aspergillosis, candidiasis, and cryptococcosis often manifest as deep tissue mycoses that occupy different niches within the mammalian host. A. fumigatus preferentially colonizes lung tissue, resulting in pulmonary aspergillosis whereas C. albicans primarily infects the blood, resulting in systemic candidiasis, and C. neoformans establishes an initial infection in the lungs, causing pulmonary cryptococcosis (Latgé, 1999; Wenzel and Gennings, 2005; Sabiiti and May, 2012). The severity of these diseases can escalate rapidly as the infection disseminates throughout the body and affects multiple organs. Infections caused by A. fumigatus and C. albicans can manifest as invasive aspergillosis and invasive candidiasis, respectively, both of which target the kidney and brain, whereas C. neoformans can cause cryptococcal meningitis which affects the central nervous system (Bicanic and Harrison, 2005; Schmiedel and Zimmerli, 2016; Shi and Mody, 2016).
In this advanced stage, treatment of invasive mycoses requires aggressive and expensive antifungal therapy. However, the success of treatment is often impeded by (i) fundamental issues in the diagnostic stage, (ii) unfavorable characteristics that are inherent to pre-existing antifungal drugs or the (iii) emergence of antifungal resistance, all of which ultimately lead to primary antifungal therapy failure (Nucci and Perfect, 2008). In patients suffering from invasive candidiasis or invasive aspergillosis, the rate of failure can be as high as 60 and 70%, respectively (Nucci and Perfect, 2008). To address these issues, there have been advancements in diagnostic techniques and several iterations of pre-existing drugs have been developed to improve their pharmacological properties (Houšt et al., 2020; Kidd et al., 2020). However, new classes of antifungal drugs that bypass existing resistance mechanisms by targeting alternate pathways are yet to be discovered.
In spite of the alarming rates of morbidity and mortality, the severity of invasive fungal infections remains underappreciated. Increases in disease incidence and prevalence of antifungal resistance highlights the need to identify novel targets and develop new classes of antifungals to manage mycoses amongst the immunocompromised population. There have been continual efforts to characterize enzymes involved in the biosynthesis of ergosterol or cell wall components, both of which are classic antifungal targets, to develop novel inhibitors (Urbina et al., 2000; Hata et al., 2010; Marshall et al., 2018). However, there has also been a notable shift in focus from these pathways exclusive to fungi to exploiting species-specific differences in shared pathways between fungi and humans (Rodriguez-Suarez et al., 2007; Marshall et al., 2017, 2019; Kummari et al., 2018). To effectively establish infection, the fungus must adapt to a different niche within the human host, combat, or circumvent the host immune response and obtain sufficient nutrients to reproduce and disseminate. Although these metabolic requirements may differ between fungal species, depending on their preferred infection site, disrupting shared metabolic pathways involved in these processes can impede fungal survival and pathogenesis. Targeting these pathways may present an elegant approach to develop novel classes of therapeutics with broad-spectrum activity.
In this article, we have focused primarily on emerging targets for the development of novel antifungal classes. We have identified key enzymes involved in several targetable metabolic pathways and consolidated extensive research to define their roles in fungal survival and virulence. Furthermore, we have discussed their merits as potential targets and provided practical discussions to drive future drug design efforts from bench to bedside.
Current Antifungal Classes and Treatments
Antifungal therapeutics are routinely administered to combat invasive mycoses. The four main classes of antifungals that currently exist function by targeting either the cell wall or cell membrane (Figure 1). They include azoles, allylamines, polyenes and echinocandins. The mechanism of action of each antifungal drug class and discussion of their biological targets have been extensively reviewed (Mazu et al., 2016). In this review, we have provided a brief overview of the currently available antifungals.
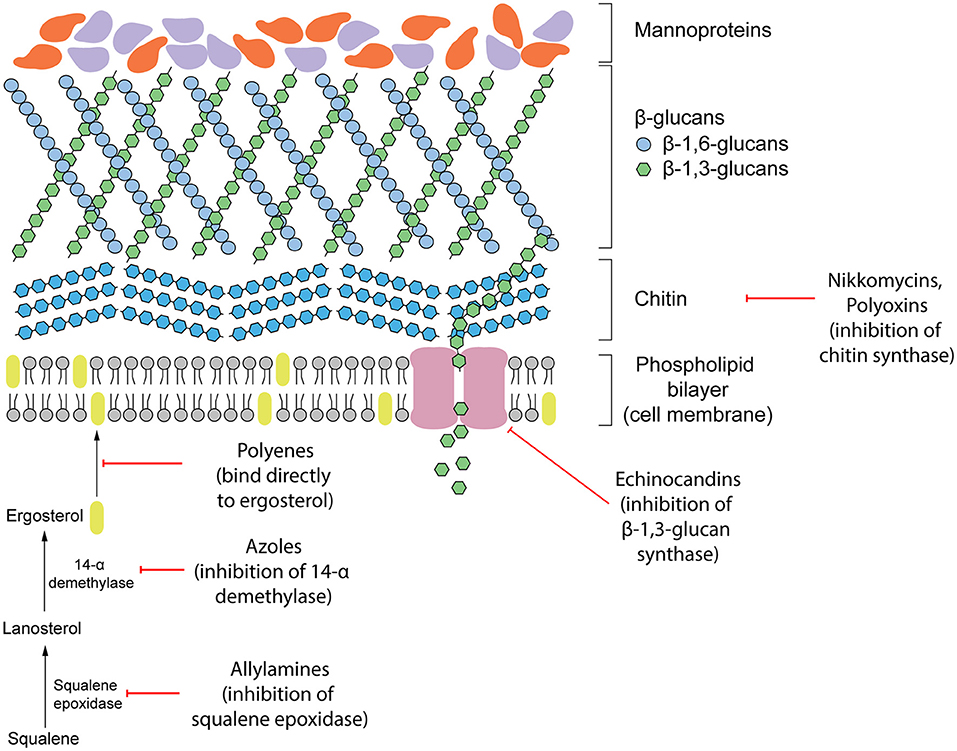
Figure 1. Existing antifungal drug classes that target the biosynthesis of the fungal cell wall components (nikkomycins, polyoxins, echinocandins), biosynthesis of the cell membrane (azoles, allylamines) or bind to ergosterol in the membrane directly (polyenes).
Targeting the Cell Membrane
Ergosterol is a steroid alcohol that forms a major component of the fungal cell membrane. It is responsible for the maintenance of membrane fluidity and structural integrity, as well as the regulation of membrane permeability (Gooday, 1995). Due to its implications in fungal survival, ergosterol biosynthesis enzymes have been the target of the allylamine and azole classes of antifungals, whereas the polyene class targets ergosterol function directly (Mazu et al., 2016). Allylamines target squalene epoxidase, an enzyme that catalyzes the conversion of squalene to lanosterol whereas azoles function as non-competitive inhibitors of lanosterol 14α-demethylase by complexing with the haem prosthetic group (Ryder, 1988; Allen et al., 2015; Mazu et al., 2016). Both of these enzymes are involved in the later stages of ergosterol biosynthesis. Disruption of this pathway leads to an accumulation of upstream sterol precursors and ultimately, the depletion of ergosterol. This deficiency results in impaired cell membrane function, growth inhibition, and cell death (Ghannoum and Rice, 1999; Allen et al., 2015). In comparison, polyenes induce fungal death by binding directly to ergosterol embedded in the cell membrane. Once incorporated into the membrane, this leads to the formation of pores which increase membrane permeability to water, ions and non-electrolytes (Ermishkin et al., 1976).
Targeting the Cell Wall
Due to the absence of a mammalian equivalent, several components of the fungal cell wall and the biosynthetic enzymes responsible for its construction have been the target for antifungal therapeutics. The cell wall, comprised of three main components including glucans, mannoproteins, and chitin, plays an important role in maintaining fungal survival by maintaining cell rigidity and resisting osmotic stress (Gow et al., 2017). Furthermore, there are several receptors displayed on the surface of the cell wall that can interact with the host during infection and contribute to fungal virulence. Echinocandins disrupt these fundamental functions of the cell wall by non-competitively inhibiting (1,3)-β-glucan synthase, the enzyme responsible for maintaining and synthesizing glucans. Insufficient biosynthesis of glucans leads to destabilization of the cell wall and a compromised ability to resist osmotic pressure that results in cell lysis (Wiederhold and Lewis, 2003).
Emergence of Antifungal Resistance and the Need for Novel Antifungal Classes
The extensive use of antifungals in both agricultural and clinical settings has led to the rise of widespread antifungal resistance with significant consequences to food security and human health. To control fungal phytopathogens in agricultural crops, a range of antifungal drugs specifically for agricultural use are used that target mitochondrial respiration processes, including succinate dehydrogenase and Qo inhibitors (Brauer et al., 2019). However, there is also significant overlap in the use of azoles to treat both human and plant fungal pathogens. In particular, they are frequently used in grain- and grass-growing environments, as well as in clinical settings, to control Candida spp., Cryptococcus spp. and Aspergillus spp. infections (Azevedo et al., 2015). As a result, bioactive azoles can persist in soil, water, and fresh produce (Hof, 2001). Ultimately, it has been suggested that these environmental factors contribute to the rise in azole resistance currently being observed in clinical settings.
The prominent use of azoles has resulted in a shift in the prevalence of infections caused by C. albicans toward less susceptible species of Candida, including C. glabrata, C. krusei, and C. guilliermondii (Hope et al., 2002; Pfaller et al., 2006; Perlin et al., 2015). Fungal strains can also acquire drug resistance during therapy which restricts the treatment options available to the patient and can ultimately cause antifungal therapy failure. Specifically, there are increasing concerns of echinocandin and azole resistance arising in species of Candida and A. fumigatus, respectively (Perlin, 2015). In the UK, there has been a consistent increase in the proportion of A. fumigatus isolates with resistance to azoles growing from 5% in 2004 to 14% in 2008 and 20% in 2009 (Bueid et al., 2010). Following a similar trend, the frequency of A. fumigatus clinical isolates with azole resistance in The Netherlands increased from 7.6% in 2013 to 14.7% in 2018 (Lestrade et al., 2020).
Several generations of antifungal drugs originating from these four classes have been developed to improve their pharmacokinetic profiles, increase potency, and combat resistance. However, treatment of invasive mycoses amongst immunocompromised patients remains challenging. Primary antifungal therapy failure is a prevalent issue, affecting 20–60% of invasive candidiasis patients, 40–60% of invasive aspergillosis patients and 30–100% of invasive fusariosis patients (Nucci and Perfect, 2008). Although the role of underlying host factors and immune status must be considered in antifungal therapy failure, several issues arise from inherent limitations in currently available drugs. Low bioavailability in target tissues, drug toxicity, unfavorable drug-drug interactions, and the emergence of drug resistance also contribute to the reduced efficacy of existing treatments (Nucci and Perfect, 2008).
As the number of antifungal classes available for clinical use is limited and the proportion of the population susceptible to invasive mycoses is expected to increase, there is a need to identify new targets for the design and development of novel classes of antifungals. Although cell membrane and cell wall biosynthesis enzymes have been traditional targets of antifungals, broadening our search to unconventional pathways can unlock an abundance of potential targets by exploiting the unique metabolic needs of fungi during pathogenesis. A summary of the proposed targets discussed in this review is outlined in Table 1.
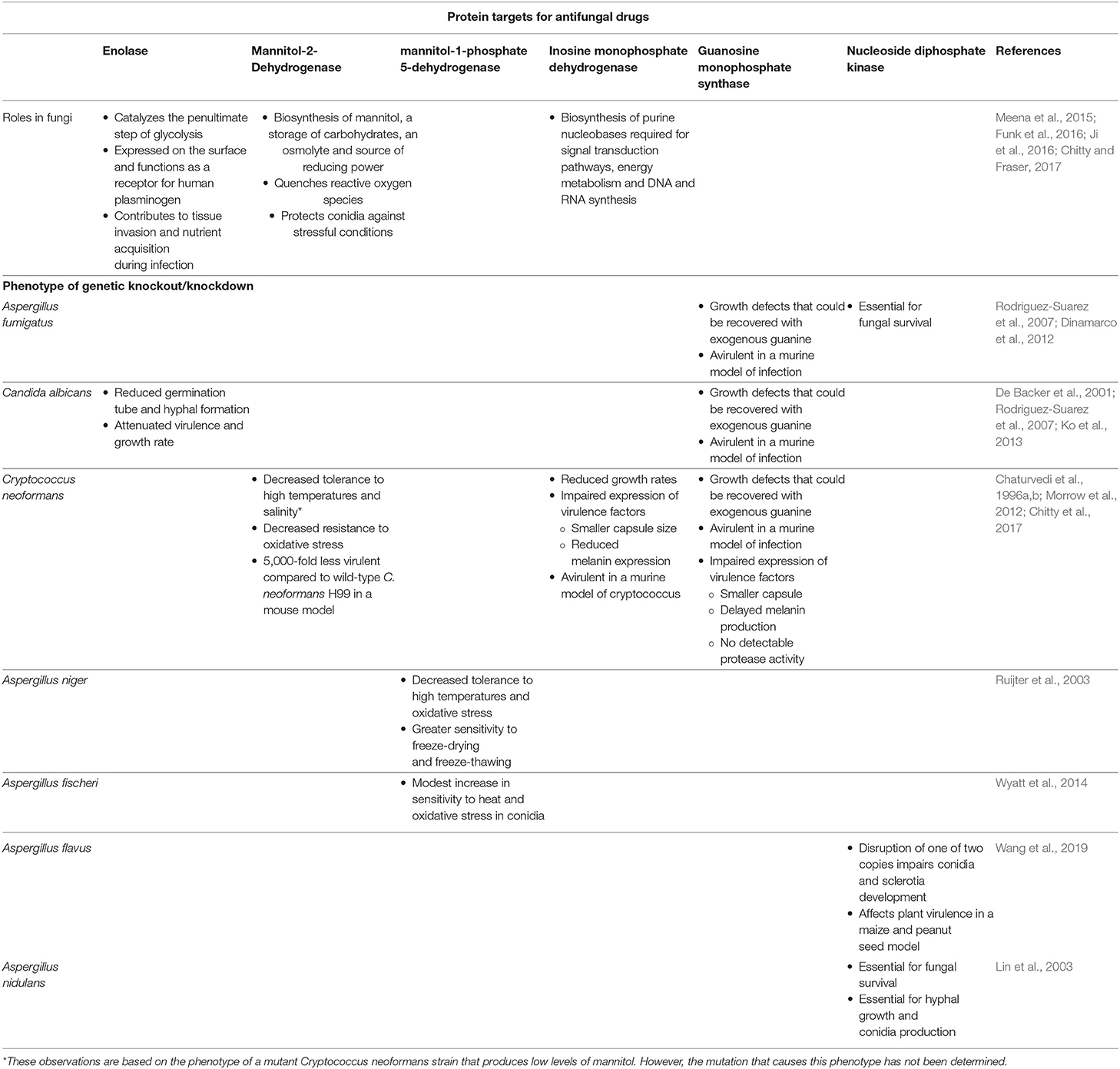
Table 1. Cellular function of the proposed antifungal protein targets and phenotypic characteristics of various fungi after genetic knockout/knockdown.
Emerging Targets for New Antifungal Development
Glycolysis Pathway
Glycolysis is a fundamental, multi-step metabolic process that occurs in most living organisms. The glycolysis pathway produces energy, in aerobic and anaerobic conditions, through the catabolism of sugars and provides useful intermediates for downstream biosynthetic pathways. The roles of these enzymes in glycolysis have been extensively studied but there has been growing interest in their additional “moonlighting” functions that deviate from their canonical function. These “moonlighting” functions may contribute to fungal survival and virulence mechanisms and have consequently been investigated as promising antifungal drug targets.
Enolase and Host Plasminogen
Enolase catalyzes the penultimate step of glycolysis, interconverting 2-phosphoglycerate (2-PG) and phosphoenolpyruvate (PEP) (Figure 2) (Ji et al., 2016). Consistent with its role in integral metabolic processes, a genetic knockout of enolase in C. albicans has shown to reduce germination tube and hyphal formation, resulting in attenuated virulence and growth rate (De Backer et al., 2001; Ko et al., 2013). Intriguingly, this enzyme fulfills its glycolytic roles in the cytoplasm but it is also expressed on the cell surface of many fungal and bacterial species. This has been observed in A. fumigatus and C. albicans, two of the most prominent etiological agents of invasive mycoses, as well as Aspergillus flavus, Aspergillus terrus, Aspergillus nidulans, Candida glabrata, Saccharomyces cerevisiae and bacterial Streptococci and Pneumococci species (Pancholi and Fischetti, 1998; Edwards et al., 1999; Bergmann et al., 2001; Funk et al., 2016). In S. cerevisiae, it has been suggested that enolase secretion is mediated by an N-terminal 28 amino acid translocation sequence and operates through a SNARE-dependent pathway (Miura et al., 2012). Alignment of homologous enolase protein sequences from S. cerevisiae with Aspergillus spp. and Candida spp. have identified a highly conserved motif in the N-terminus. This suggests that this SNARE-dependent mechanism of enolase secretion may be conserved amongst many fungal species (Funk et al., 2016).
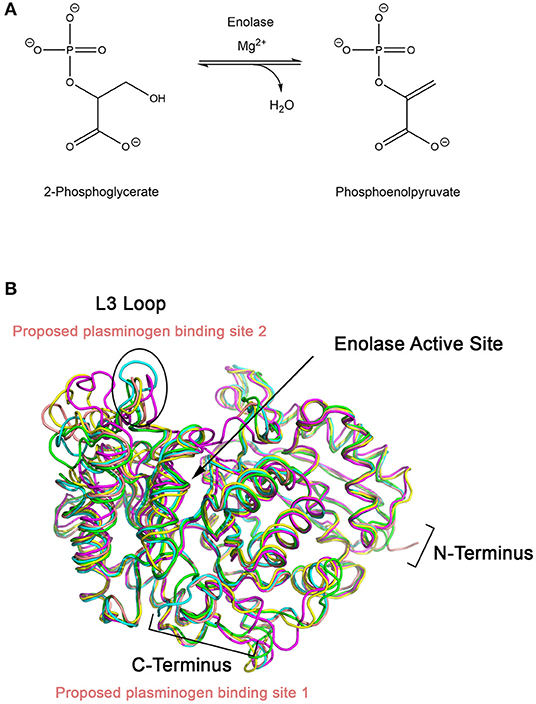
Figure 2. (A) Enolase catalyzes the penultimate step of glycolysis, interconverting 2-phosphoglycerate (2-PG) and phosphoenolpyruvate (PEP) using magnesium as a cofactor. (B) X-ray crystal structures of enolase from different species have similar global folds. Enolase structures from Escherichia coli (1E9I; green), Enterococcus hirae (1IYX; cyan), lobster (1PDZ; yellow), Streptococcus pneumoniae (1W6T; salmon), and Saccharomyces cerevisiae (1ONE; magenta) were superimposed using PyMOL (Stec and Lebioda, 1990; Duquerroy et al., 1995; Kuhnel and Luisi, 2001; Hosaka et al., 2003; Ehinger et al., 2004; Kang et al., 2008). The L3 loop, which shows the greatest structural deviation between the structures, is proposed to be the plasminogen binding loop for S. pneumoniae. Exposed lysine residues at the C-terminal end of enolase have also been proposed as a possible binding site.
The function of surface-expressed enolase deviates from its canonical role in the cytoplasm and instead contributes to tissue invasion and nutrient acquisition during pathogenesis. It functions as a receptor for plasminogen, a zymogen circulating in the host bloodstream. Plasminogen is cleaved by plasminogen activating proteins (including tissue-plasminogen activator and urokinase plasminogen activator) to form active plasmin which functions as a serine protease. The physiological function of the human plasminogen/plasmin system is to facilitate tissue remodeling, cell migration, hemostasis and wound healing, and induce inflammation and angiogenesis. It fulfills these roles by degrading proteins in the extracellular matrix, fibrin and fibrinogen in blood clots and targeting components of the complement system (Law et al., 2013). Inactive plasminogen is proposed to dock onto a lysine-rich motif located on the surface-expressed enolase of fungal pathogens. While bound, host plasminogen activators are capable of recognizing, binding, and cleaving plasminogen to produce the activated plasmin (Funk et al., 2016). The serine protease activity of plasmin becomes concentrated at the site of fungal infection and this can accelerate tissue invasion and disease progression. An in-depth study which focused on surface-expressed enolase from A. fumigatus confirmed its ability to interact with human immune regulators including factor H, factor-H-like protein 1 (FHL-1), C4b-binding protein (C4BP) and plasminogen (Dasari et al., 2019). Whilst bound to A. fumigatus enolase, factor H, FHL-1 and C4BP retained their normal cellular activity and plasminogen remained accessible to plasminogen activator proteins. When swollen A. fumigatus conidia coated with human plasminogen were exposed to human A549 epithelial cells or an epithelial monolayer, cellular metabolic activity was reduced by 41% and cell retraction was reduced (Dasari et al., 2019). These observations are consistent with the “moonlighting” role of surface-expressed enolase and their role in facilitating tissue invasion.
Immunogenic Activity of Enolase
The secretion and presentation of enolase on the cell surface is a common feature of many pathogenic fungal species (Edwards et al., 1999; Funk et al., 2016). As a surface-exposed receptor, it is capable of eliciting an immunogenic response that may be exploited for vaccine development. Preliminary studies have demonstrated the suitability of using enolase as an immunogenic agent to acquire modest protective effects against candidiasis in a murine model. When mice were challenged with C. albicans following subcutaneous vaccination with recombinant C. albicans enolase, an elevated antibody response was induced and fungal burden in major organs was reduced compared to non-immunized mice (Li et al., 2011). In an independent study, C. albicans enolase was expressed and presented on Lactobacillus casei cells, then orally administered to mice prior to challenging with a normally lethal dose of C. albicans. These immunized mice produced higher IgG antibody titers and had improved survival rates (Shibasaki et al., 2014).
Targeting Surface-Expressed Enolase
Designing a peptidomimetic or small molecule inhibitor that specifically targets the plasminogen docking site of enolase using a structure-guided approach is a possible avenue for new therapeutics. This approach requires intimate knowledge of the interactions between both proteins. Although high resolution crystal structures of the full-length human Type II plasminogen (4DUR) and fungal enolase from S. cerevisiae (3ENL) are already available, the interaction interface has not yet been defined (Stec and Lebioda, 1990; Law et al., 2012). There have been previous studies examining the molecular interaction between plasminogen and bacterial enolases. Studies conducted using enolase from S. pneumonia have suggested two possible binding sites—the L3 loop on the alpha-beta (αβ) barrel, a structurally flexible region, or the lysine-rich C-terminal tail of the protein (Pancholi and Fischetti, 1998; Bergmann et al., 2003; Ehinger et al., 2004). Superimposition of enolase from prokaryotic and eukaryotic sources including E. coli (1E9I), human (3B97), Enterococcus hirae (1IYX), lobster (1PDZ), Streptococcus pneumonia (1W6T), and S. cerevisiae (3ENL) reveal an overall similar fold and existence of a conserved, but structurally different, L3 loop (Figure 3) (Stec and Lebioda, 1990; Duquerroy et al., 1995; Kuhnel and Luisi, 2001; Hosaka et al., 2003; Ehinger et al., 2004; Kang et al., 2008). Therefore, it is likely that the plasminogen binding site is also conserved between homologous enolase enzymes but the mode of binding may differ between species. Building upon this work, efforts must be focused on elucidating the plasminogen binding interface of fungal enolase and structurally characterizing this site to guide rational inhibitor design.
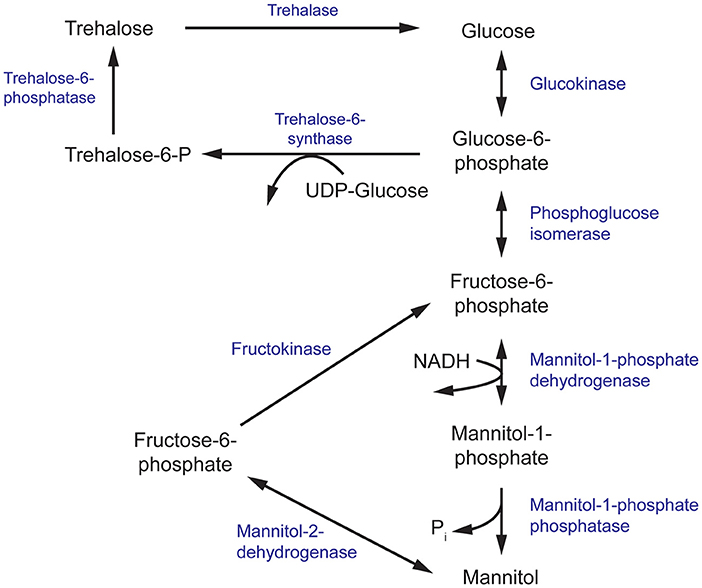
Figure 3. The mannitol and trehalose biosynthesis pathway in fungi. The primary biosynthesis enzymes of mannitol, and the focus of this review, are mannitol-1-phosphate dehydrogenase (M1P5DH) and mannitol-2-dehydrogenase (M2D).
Evidence of the immunogenicity of fungal enolase, particularly the C. albicans homolog, raises the possibility of using antibody therapy as an adjunct to antifungal therapy. Therapeutic antibodies specific for enolase could potentially reduce tissue invasion and fungal dissemination by interfering with interactions between fungal enolase and host plasminogen. Antibodies raised in mice against recombinant C. albicans or A. fumigatus enolase successfully recognized plasminogen-bound enolase from Candida spp. and Aspergillus spp., respectively. However, this antibody-enolase-plasminogen complex still allowed plasminogen activating proteins to access, cleave, and activate bound plasminogen (Funk et al., 2016). The viability of antibody therapy requires improved antibodies against enolase that recognize specific epitopes that interfere with this plasminogen activation process. Monoclonal antibodies that inhibit plasminogen activation have already been successfully raised against human α-enolase. During the process of fibrinolysis, plasminogen binds to surface-expressed α-enolase on leukocytic cells and this results in an accelerated rate of plasminogen activation. The plasminogen activation associated with binding to leukocytic cell lines was reduced by ~90% when monoclonal α-enolase antibodies (MAb 11G1) were introduced (López-Alemany et al., 2003). These experiments provide a proof-of-principle that disruption of an enolase-plasminogen interaction can affect plasmin activity and therefore, this approach may be feasible when applied to an antifungal therapeutic strategy.
Mannitol Biosynthesis Pathway
Mannitol biosynthesis is a crucial process that aids in fungal survivability and virulence. Mannitol-2-dehydrogenase (M2DH) and mannitol-1-phosphate 5-dehydrogenase (M1P5DH) are the primary biosynthesis enzymes that produce mannitol, an acyclic, six-carbon sterol alcohol (Figure 3). Present in all fungal structures, including mycelia, fruiting bodies and conidia, mannitol acts as a carbohydrate storage molecule, osmolyte, and source of reducing power (Meena et al., 2015). During pathogenesis, mannitol production is increased to exploit its ability to quench reactive oxygen species (ROS) in order to resist host defenses (Meena et al., 2015). The unique and diverse properties of mannitol, particularly in relation to human pathogenesis, have garnered considerable interest in the underlying mechanism of its biosynthesis and secretion during fungal infection.
Mannitol as a Biomarker of Fungal Infection
The efficacy of antifungal therapy is dependent on the early detection and accurate identification of the invading fungal species. Traditional methods include culturing from patient samples, direct microscopical examination of fungal samples and histopathology testing of infected tissue (Kozel and Wickes, 2014). Although culturing is considered as the gold standard of diagnosis, the utility of this technique can suffer from the slow growth rates of filamentous fungi, environmental contamination, and poor recovery of some fungal species from patient samples (Kozel and Wickes, 2014). Overall, these factors can delay adequate antifungal treatment which can compromise the efficacy of these drugs. Therefore, non-culture-based methods of diagnosis, such as screening patient samples for biomarkers of fungal infection, offer a faster and less invasive alternative.
Several fungal pathogens, including C. neoformans and Aspergillus spp., have been shown to produce high levels of mannitol in vitro and in animal disease models of infection, highlighting the suitability for its use as a biomarker. In liquid cultures of 12 human isolates of C. neoformans, extracellular mannitol levels increased ~30-fold over a 4 day period in which rapid mannitol biosynthesis and secretion occurred predominantly in the stationary growth phase (Wong et al., 1990). Similarly, several Aspergillus species have also demonstrated an ability to produce mannitol from glucose in vitro (Birkinshaw et al., 1931). Closely mirroring these studies, mannitol levels in the cerebrospinal fluid (CSF) of a cortisone-treated rabbit disease model of cryptococcal meningitis also increased over time in response to infection with C. neoformans strain H99. Furthermore, there was a positive correlation identified between mannitol CSF levels and CSF colony forming units as well as CSF cryptococcal antigen titers, both of which are markers of infection severity (Wong et al., 1990). In an experimental model of aspergillosis using rats infected with A. fumigatus, high levels of mannitol were detected in liver tissue 12 h post-infection and in serum 36 h post-infection (Wong et al., 1989). In addition, a singularly surviving rat 48 h post-infection also showed increased mannitol levels in lung, liver, and kidney tissue (Wong et al., 1989). From these animal models, the results indicate that infection of a human host with C. neoformans or A. fumigatus is also likely to induce upregulated mannitol production and secretion, localizing in the CSF or serum, respectively. Analysis of the CSF from patients suffering from AIDS and cryptococcal meningitis confirm that mannitol content increases to sufficiently high levels to be easily detected and quantified using gas-liquid chromatography-linked mass spectroscopy (Megson et al., 1996). As humans lack an equivalent mannitol biosynthesis pathway, the detected mannitol is likely of fungal origin. However, mannitol content in the CSF could not be significantly correlated to cryptococcal antigen titers in humans, contrary to observations made in the rabbit disease model. Taken together, these results provide valuable insights into the feasibility of using patient CSF and serum mannitol levels as a biomarker of fungal infection and presents possible methodologies that facilitate its use as a diagnostic tool.
Roles in Virulence and Protection Against Host Defenses
Fungal pathogens invading a human host experience multiple environmental stresses associated with occupying a new niche and chemical stresses invoked by the immune response. Sensing and adapting to abiotic stresses (temperature, pH), resisting phagocytosis from macrophages and neutralizing ROS produced by neutrophils is critical for survival (Cooney and Klein, 2008; Khanna et al., 2016). Mannitol has been connected to many of these host-pathogen interactions associated with fungal survival and virulence and this provides a basis to understand the function of its rapid biosynthesis and secretion during infection.
Phenotypic characterization of a C. neoformans mutant which produces reduced levels of mannitol (Cn MLP) exemplifies the multitude of roles adopted by mannitol. The Cn MLP mutant showed similar growth rates, morphology, and antifungal susceptibility as the wild-type strain, C. neoformans H99. However, Cn MLP had decreased tolerance to high temperatures and salinity (Chaturvedi et al., 1996a). These findings are consistent with the proposed role of mannitol as an osmolyte in fungi (Meena et al., 2015). Although the loss of thermotolerance in Cn MLP cannot be directly linked to mannitol function, resistance to thermal stress in the fungus Beauveria bassiana has been partially attributed to trehalose, a metabolite produced from mannitol (Figure 3) (Liu et al., 2008). The Cn MLP isolate also demonstrated decreased resistance to oxidative stress, a common killing mechanism of the human immune system (Chaturvedi et al., 1996b). Specifically, Cn MLP showed greater susceptibility to oxidative killing by polymorphonuclear neutrophils (PMNs) and ROS generated from an iron-hydrogen peroxide-iodide cytotoxic cell-free system, relative to the wild-type strain (Chaturvedi et al., 1996b). Protection against ROS improved in a dose-dependent manner when PMNs were incubated with mannitol. Similarly, fungicidal activity was completely abolished when mannitol was added simultaneously to the ROS cell free system (Chaturvedi et al., 1996b). In both scenarios, mannitol acted as an effective scavenger of ROS to confer protection against oxidative damage. In an experimental disease model, Cn MLP was 5,000-fold less virulent compared to C. neoformans H99 when intravenously injected into mice. Although both the mutant and wild-type strains proved to be lethal, 100% of mice succumbed to infection 51 days post-injection with C. neoformans H99 whereas 100% of mice injected with Cn MLP survived at the same time point (Chaturvedi et al., 1996a). Together, the osmotolerance and thermotolerance conferred by intracellular mannitol and the protection against ROS by extracellular mannitol may aid fungal survival and virulence by resisting environmental and immune-induced stresses in the human body.
Roles in Conidial Stress Response
Conidia are asexually-produced spores that are required for wide-spread fungal dispersal. Often dissipated through the air, conidia are inhaled into the respiratory system and at this developmental stage, fungal infection can be initiated (Dagenais and Keller, 2009). Conidia must be able to survive and adapt to this new environment to progress to subsequent germination stages and develop into a vegetative state. Mannitol has been implicated in the protection of conidia against these stress conditions in multiple species of fungi (Chaturvedi et al., 1996a; Ruijter et al., 2003; Wyatt et al., 2014).
High concentrations of intracellular mannitol have been detected in conidia from Aspergillus oryzae, Aspergillus clavatus and Aspergillus niger (Horikoshi et al., 1965; Corina and Munday, 1971; Witteveen and Visser, 1995). In order to assess mannitol function, a M1P5DH-null mutant of A. niger was generated. Disruption of M1P5DH resulted in a complete absence of mannitol in mycelia and a 30% reduction of intracellular mannitol content in conidia. Prolonged exposure to conditions that had no effect on the viability of wild-type conidia, such as high temperatures and oxidative stress induced by ROS, reduced the viability of M1P5DH-null conidia to 5 and 0.5%, respectively. To a lesser extent, these mutant conidia were more sensitive to freeze-drying and freeze-thawing processes. Supplementation of M1P5DH-null conidia with mannitol had a protective effect that reduced their sensitivity to heat and oxidative stress (Ruijter et al., 2003). These results indicate that mannitol is an important molecule that protects conidia against harsh environmental conditions.
In a separate study, the equivalent gene encoding M1P5DH in Aspergillus fischeri was deleted with unexpected effects. There was a severe reduction of mannitol in mycelia but an increase in mannitol and reduction of trehalose in its conidia (Wyatt et al., 2014). These conidia showed a modest increase in sensitivity to heat and oxidative stress, despite the higher mannitol content. Since trehalose and mannitol biosynthesis pathways are intrinsically linked and both sugars have been found in all stages of the fungal life cycle, it has been suggested that trehalose and mannitol may play overlapping roles in resistance to environmental stress (Figure 3) (Wyatt et al., 2014; Thammahong et al., 2017). There has also been growing interest in targeting components of the trehalose biosynthesis pathway for the development of novel antifungals (Thammahong et al., 2017). Thus, further investigation into the interplay between mannitol and trehalose in fungal survival is warranted to aid these efforts.
Targeting Mannitol Biosynthesis Enzymes
Several lines of evidence demonstrate the multifaceted roles of mannitol in the protection of fungi from stress conditions commonly experienced during infection of a host (Chaturvedi et al., 1996a; Ruijter et al., 2003; Wyatt et al., 2014). Accelerated mannitol production and secretion can confer thermotolerance and resistance to ROS generated from the human immune system needed to thrive in these harsh environments. Adding to the allure of targeting mannitol biosynthesis pathways in fungi is the fact that humans do not produce mannitol and therefore lack equivalent enzymes. This absence of a human homolog greatly simplifies the design and development of inhibitory compounds that specifically target these fungal enzymes. In turn, this will likely reduce the severity of off-target effects.
Efforts to target the mannitol biosynthesis enzyme should be focused on M1P5DH. In several species, knockout of the gene encoding M1P5DH (mpdA) and phenotypic characterization of these mutants have shown significant decreases in intracellular mannitol levels (Ruijter et al., 2003; Wyatt et al., 2014). Although mannitol can be synthesized in an alternate pathway using M2DH, it appears that M1P5DH features in the predominant pathway. Hence, inhibition of M1P5DH is likely to have a greater effect on conidia viability and can be useful to halt initial stages of infection and dissemination.
Targeting the mannitol biosynthesis pathway can also be beneficial to delay the progression of existing infections. As demonstrated in the animal models infected with Cn MLP, decreased mannitol levels were associated with reduced virulence and ultimately, greater survival. By inhibiting the activity of these biosynthesis enzymes, mannitol production is reduced and this may increase fungal sensitivity to oxidative killing by components of the immune system.
Purine Nucleotide Biosynthesis Pathway
Nucleobases, including purines and pyrimidines, are heterocyclic molecules that are found in all forms of life. They are obtained exogenously from the environment or can be synthesized endogenously. As they are involved in essential processes, including DNA and RNA synthesis, energy metabolism and signal transduction, the intracellular pool of nucleobases must be tightly maintained (Chitty and Fraser, 2017). Hence, disruption of the purine and pyrimidine biosynthesis pathway using small molecules has been explored as a potential approach to develop antimicrobials (Rodriguez-Suarez et al., 2007; Shu and Nair, 2008; Du Pré et al., 2018; Park et al., 2018; Trapero et al., 2018). De novo purine biosynthesis begins with phosphoribosylpyrophosphate (PRPP) which is ultimately converted to inosine monophosphate (IMP), a precursor to both adenine and guanine nucleobases, through a series of enzymatic reactions (Figure 4) (Chitty and Fraser, 2017). This section will focus on targeting enzymes responsible for the synthesis of guanine nucleobases.
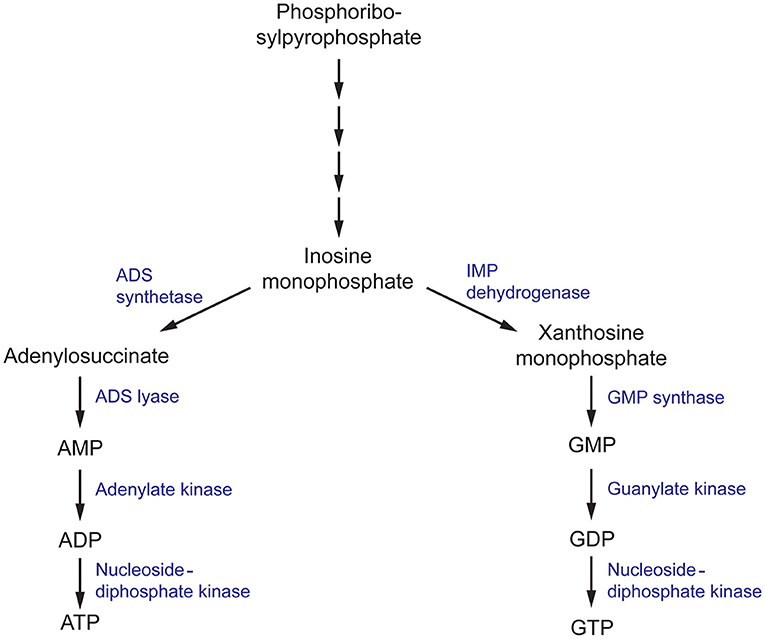
Figure 4. The purine metabolism pathway in fungi. ADS, (adenylosuccinate) synthetase; ADS, (adenylosuccinate) lyase; IMP (inosine monophosphate) dehydrogenase; GMP, (guanosine monophosphate) synthase.
Roles in Virulence
Inosine monophosphate dehydrogenase (IMPDH) is the first committed and rate-limiting step in the guanosine triphosphate (GTP) biosynthesis pathway. Using NAD+ as a cofactor, it catalyzes the conversion of IMP to xanthosine monophosphate (XMP) via a dehydrogenase and hydrolysis reaction (Hedstrom, 2009). IMPDH catalytic activity has been closely associated with microbial virulence, and has been the target of several drug discovery projects for antibiotics, antifungals, antivirals, and anticancer therapeutics with great success (Markland et al., 2000; Floryk and Thompson, 2010; Kummari et al., 2018; Trapero et al., 2018). In terms of antifungals, there has been substantial progress in elucidating the mechanisms in which IMPDH contributes to C. neoformans survival and virulence. To first validate the rationality of IMPDH as an antifungal target, an IMPDH-deficient mutant of C. neoformans was produced and phenotypically characterized. This mutant exhibited reduced growth rates and impaired expression of virulence factors, resulting in smaller capsule sizes and reduced melanin expression (Morrow et al., 2012). Importantly, these phenotypic defects could not be completely recovered with supplementation of exogenous guanine that may have been sourced from the human host during infection. Due to the direct involvement of IMPDH in virulence factor production, IMDPH-deficient C. neoformans was completely avirulent in a murine model of cryptoccoccus. As a result, fungal loads in the brain were cleared 3 days post-infection, total fungal clearance was achieved 2 weeks post-infection and ultimately, this resulted in a 100% survival rate 50 days post-infection (Morrow et al., 2012). These experiments confirm that IMPDH is involved in essential processes associated with C. neoformans virulence and this presents a promising avenue for the design of antifungal compounds.
In the following step of the purine biosynthesis pathway, XMP produced by IMPDH is converted to guanosine monophosphate (GMP) by guanosine monophosphate synthase (GMP synthase). In a similar manner to IMPDH, GMP synthase has been extensively studied in A. fumigatus, C. albicans, and C. neoformans and has been determined to be crucial in the survival and virulence of these fungal pathogens (Rodriguez-Suarez et al., 2007; Chitty et al., 2017). A conditional knock-out of the gene encoding GMP synthase (GUA1) in A. fumigatus and C. albicans, and deletion in C. neoformans, inhibited growth on minimal media but was recovered when supplemented with exogenous guanine. These growth defects are consistent with the essential role of purines as components of DNA and RNA. Similar to the in vivo experiments conducted with IMPDH-deficient C. neoformans, disruption of GUA1 in A. fumigatus, C. albicans, and C. neoformans also rendered these strains avirulent in a murine model of infection (Rodriguez-Suarez et al., 2007; Chitty et al., 2017). Phenotypic characterization of ΔGUA1 C. neoformans revealed significant differences in the production of key virulence factors that may account for the abolished virulence. Despite supplementation with exogenous guanine, ΔGUA1 C. neoformans mutants produced less capsule, showed delayed melanin production and lacked detectable protease activity relative to their wild-type counterparts. Collectively, these virulence factors function to aid survival by resisting host defenses and fungal dissemination. Specifically, the capsule protects against phagocytosis, melanin resists oxidative damage and secretion of proteases facilitates host tissue degradation for nutrient acquisition. Intriguingly, this effect was more pronounced at the optimum human body temperature of 37°C (Chitty et al., 2017). Since GUA1 is a determinant of virulence in several of these clinically-prominent fungal pathogens, inhibition of GMP synthase may be an ideal approach to develop antifungal drugs with broad-spectrum activity.
Roles in Survival
In the final steps of the purine biosynthesis pathway, GMP must be converted to an accessible form for its use in DNA and RNA synthesis and signal transduction. GMP is first converted to guanosine diphosphate and then nucleoside-diphosphate kinase (NDK) covalently attaches a phosphate moiety to form GTP (Chitty and Fraser, 2017). NDK is a promiscuous enzyme that can use all nucleoside diphosphates as substrates, with varying levels of selectivity, to generate their respective nucleoside triphosphates (Nguyen et al., in press). Hence it is also involved in the adenine nucleoside synthesis pathway to produce adenosine triphosphate (ATP) as well as the synthesis of the remaining building blocks of DNA and RNA. Consistent with these fundamental processes, in A. nidulans, NDK is involved in various aspects of the life cycle, including hyphal growth and conidia production which is necessary for its growth and dispersal (Lin et al., 2003). Similarly in A. flavus, disruption of one of the two copies of ndk impairs the development of conidia and sclerotia and affects plant virulence in a maize and peanut seed model (Wang et al., 2019). Identified as an essential gene for survival in A. fumigatus and A. nidulans, NDK has been identified as a promising target for antifungals against Aspergillus spp. (Lin et al., 2003; Dinamarco et al., 2012).
Targeting Purine Biosynthesis Pathways
Achieving species-selective targeting of de novo purine biosynthesis enzymes is an important consideration in the drug design process. This is especially important since bacteria, fungi and humans share commonality in many of these enzymes (Sperling, 1988; Kilstrup et al., 2005; Chitty and Fraser, 2017). Thus, intimate structural and kinetic knowledge of the fungal and human homologs of these enzymes will be invaluable in the design of inhibitors with high potency and selectivity. Currently, structures of human IMPDH, isoforms I (Risal et al. unpublished) and II (Colby et al., 1999), GMP synthase (Welin et al., 2013), and NDK (Chen et al., 2003) have been solved as well as structures of fungal IMPDH (C. neoformans) (Morrow et al., 2012) and NDK (A. fumigatus and A. flavus) (Wang and Wang, 2019; Nguyen et al., in press). Availability of these structures allows identification of key active site residues that differ between fungal and human enzymes and this may be integral in the design of highly selective inhibitors.
Knowledge of the enzyme structure and catalytic mechanism aids in identifying the most “druggable” binding pocket. In the case of IMPDH, there are three possible sites: (i) the natural substrate site, IMP, (ii) the co-factor site, NAD+, or (iii) an allosteric site (Shu and Nair, 2008). Ongoing drug discovery projects targeting IMPDH from Mycobacterium tuberculosis and Cryptosporidium parvum have shown success in focusing on the co-factor binding site due to its divergence from the mammalian equivalent (Umejiego et al., 2008; Trapero et al., 2018). In the case of C. parvum IMPDH, a high-throughput screening approach was employed to specifically target the NAD+ site. Ten compounds, from a pool of 134 preliminary hits, were determined to be reversible inhibitors that were 9- to 400-fold more selective for the parasitic IMPDH over the human IMPDH isoform II (Umejiego et al., 2008). These experiments provide a proof of concept that IMPDH can be effectively and selectively targeted by small molecule inhibitors. When considering IMPDH from C. neoformans, a similar screening approach targeting the co-factor binding site may also be feasible due to its structural and functional deviation from human IMPDH (Morrow et al., 2012). A structure of human IMPDH isoform II bound to selenazole-4-carboxyamide-adenine dinucleotide, an analog of NAD+, reveals differences in the residues that comprise the co-factor binding pocket between C. neoformans and human IMPDH. It can be inferred that the interaction between IMPDH and NAD+ is stabilized by a π-π stacking interaction between the adenine ring of NAD+ and R267 and Y296 of C. neoformans IMPDH which corresponds to R253 and Y282 in human isoform I or H253 and F282 in human isoform II, respectively (Figure 5A) (Morrow et al., 2012). Although many residues within the binding pocket are invariant between C. neoformans and human isoforms I and II, these differences are likely to be sufficient to achieve species-selective inhibition. A high-throughput screen against IMPDH from C. neoformans has identified promising candidates (Kummari et al., 2018). From a chemical library of 114,000 drug-like compounds, three 3-((5-substituted)-1,3,4-oxadiazol-2-yl)thio benzo[b]thiophene 1,1-dioxides were identified as having inhibitory activity against C. neoformans IMPDH. Through chemical modification, several analogs were then synthesized to assess the structure-activity relationships. Although many of these analogs inhibited C. neoformans IMPDH in vitro and had whole cell activity, they were also cytotoxic to human cell lines (Kummari et al., 2018). These results illustrate the complexities associated with targeting pathways shared between fungi and humans. However, there is also great opportunity to build upon this work by trialing modifications that improve selectivity that retain the antifungal properties of the compound whilst reducing cytotoxic effects.
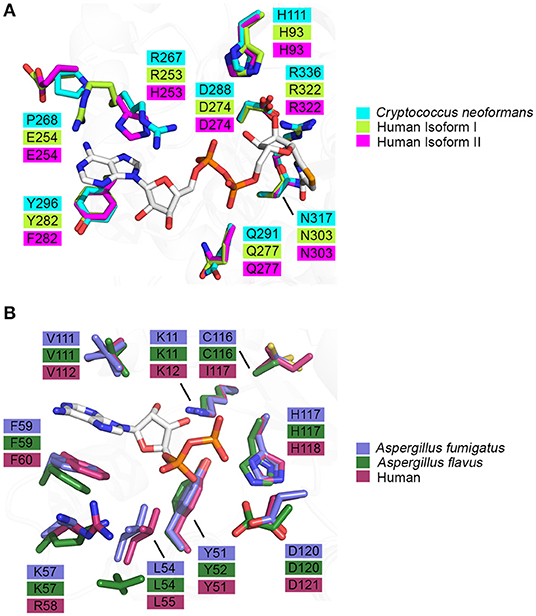
Figure 5. Binding pocket composition analysis reveals divergent residues between human and fungal homologs that may be exploited to achieve specificity. (A) Residues comprising the binding pocket of inosine monophosphate dehydrogenase from Cryptococcus neoformans (4AF0, cyan), human isoform I (1JCN, green), human isoform II (1B3O, magenta) and selenazole-4-carboxyamide-adenine dinucleotide, an NAD+ analog, are represented as sticks (Colby et al., 1999; Morrow et al., 2012) (Risal et al. unpublished). (B) Residues comprising the binding pocket of nucleoside di-phosphate kinase from Aspergillus fumigatus (6XP7, purple), Aspergillus flavus (6K3H, dark green) and human (2HVD, maroon) and adenosine diphosphate, a natural substrate, are represented as sticks (Giraud et al., 2006; Wang et al., 2019; Nguyen et al., in press).
Similarly to IMPDH, inhibition of fungal GMP synthase may be achieved effectively by high-throughput screening of chemical libraries or fragment screening. Since GMP synthase is a bifunctional protein with two catalytic domains working in tandem to fulfill its function, it is imperative to select the most feasible binding pocket to target. The glutamine amidotransferase (GAT) domain releases ammonia from glutamine which is shuttled to the ATP pyrophosphatase (ATPPase) domain where XMP is converted to GMP via an adenyl-XMP intermediate. There are pre-existing inhibitors that target the glutamine amidotransferase domain such as acivicin (ACI) and 6-diazo-5-oxo-L-norleucine (DON) that inadvertently inhibit GMP synthase activity (Ahluwalia et al., 1990; Chittur et al., 2001). However, targeting this catalytic module of GMP synthase would likely lead to non-specific and off-target effects due to the prevalence of GAT domains in other enzymes. Therefore, a more promising endeavor is to study the XMP- and ATP-binding pockets located within the ATPPase domain to achieve potent inhibition and high selectivity for the fungal enzyme over the mammalian equivalent. A novel compound, denoted ECC1385, identified from a synthetic compound library has been shown to inhibit C. albicans and C. neoformans GMP synthase activity in vitro and exhibits whole cell activity against a broad-spectrum of fungi and bacteria, including multiple species of Candida, A. fumigatus (MF5668), C. neoformans (MY2062), and Staphylococcus aureus (MB2865) (Rodriguez-Suarez et al., 2007). Mechanism-of-action studies of ECC1385 have revealed that inhibition of GMP synthase is achieved through a different mode of action from ACI and DON. These data suggest that small chemical compounds that target the activity of the ATPPase domain is possible and presents a promising avenue to pursue.
As NDK has maintained its essential role in NTP biosynthesis throughout all domains of life, there is high structural and functional conservation between different species. This conservation is reflected in the largely invariant composition of binding pocket residues between fungal and human homologs (Figure 5B). However, two key differences are revealed when the crystal structures of ADP-bound NDK from human (2HVD), A. fumigatus (6XP7), and A. flavus (6K3H) are superimposed (Figure 5B) (Giraud et al., 2006; Nguyen et al., in press). Specifically, in the nucleoside binding pocket, Lys57 and C116 in both fungal NDKs are replaced by Arg58 and I117 in the human structure, respectively. These discrepancies presents an opportunity to selectively target the catalytic site and this may be beneficial in the development of a pan inhibitor with broad-spectrum activity against multiple pathogenic fungi. Azidothymidine (AZT), also marketed as RETROVIR, is an antiretroviral medication currently used to treat HIV (Furman et al., 1986). It has been shown that AZT can inhibit NDK from A. flavus in vitro and exhibits antifungal activity in vivo (Wang et al., 2019). Docking of AZT to the crystal structure of A. flavus NDK predicts that it acts as a competitive inhibitor and interacts with residues R104, H117, and D120 in the catalytic site. Since AZT is an FDA-approved drug, there is potential to repurpose it as an antifungal drug or to use the AZT scaffold and improve upon its antimycotic properties. An alternate approach may be explored to inactivate fungal NDK preferentially over the human homolog using non-competitive inhibitors. Allosteric sites are likely to show more variation between species, compared to the catalytic site, and therefore, development of a non-competitive inhibitor may be a more effective approach. Thorough kinetic analysis has revealed that ebselen, a cysteine-modifying compound, acts as a non-competitive inhibitor of human NDK secreted by airway epithelial cells (Semianrio-Vidal et al., 2010). In this context, it is not used in an antifungal application. Instead, it is used to obtain stable and accurate measurements of nucleotides in airway surface liquid by disrupting nucleotide interconversion without the use of a nucleotide-derived inhibitor. Identifying allosteric sites in NDK from fungal pathogens, comparing them to the human homolog and exploiting their differences may be imperative to simultaneously achieve potent inhibition and high selectivity. Hence, it would be advantageous to explore non-competitive inhibitors of NDK that bind an allosteric site and influence catalytic activity distally.
Although not explicitly addressed in this review, there has been substantial progress made in targeting enzymes from the de novo pyrimidine biosynthesis pathway that is worth further discussion. Notably, a novel drug, known as F901318 or Olorofim), has shown antifungal activity against A. fumigatus and A. flavus by inhibiting the enzyme, dihydroorotate dehydrogenase (Du Pré et al., 2018). Furthermore, it has shown efficacy against A. fumigatus, both azole-sensitive and azole-resistant strains, in a murine model of pulmonary aspergillosis (Oliver et al., 2016). F901318 is currently being assessed for its use in treating invasive fungal infections caused by Lomentospora prolificans, Scedosporium spp., Aspergillus spp., and other resistant fungi. The development of F901318 and progression into phase 2 clinical trials highlights the untapped potential of targeting shared metabolic pathways between fungi and humans for the discovery of novel antifungal drug classes.
Conclusions
As the number of immunocompromised patients continues to grow, invasive fungal infections are becoming more pervasive. Due to inherent limitations in antifungal drug treatments and the emergence of resistance, the limited spectrum of currently available antifungals is a major barrier to effectively treat systemic fungal infections. Consequently, there is a need to identify and validate novel antifungal drug targets. Targeting eukaryotic proteins that are common to both fungi and humans is a difficult but necessary approach since the number of fungal enzymes that lack a human homolog and are essential for survival or virulence are scarce. Hence, there has been an increased interest in exploiting subtle structural and functional differences of metabolic enzymes shared by both fungi and humans. In the field, significant progress has been made in the characterization of enolase, an enzyme implicated in tissue invasion, as well as enzymes from the mannitol biosynthesis and de novo purine biosynthesis pathways in many pathogenic fungi. In addition to the pathways explored in this review, de novo pyrimidine biosynthesis enzymes, intracellular redox environment regulators and key phosphatases, kinases and transcription factors that form cellular signaling networks have been explored as potential antifungal drug targets (Oliver et al., 2016; Marshall et al., 2019; Jin et al., 2020). Altogether, these experiments lay the foundation for antifungal drug discovery projects that target unconventional, but promising, pathways.
Author Contributions
SN, JT, and JB: conceptualization, manuscript review, and editing. SN and JT: manuscript preparation. All authors contributed to the article and approved the submitted version.
Funding
SN and JT were recipients of an Australian Government Researching Training Program stipend scholarship.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to acknowledge the members of the Bruning Lab for their insightful discussions during the preparation of this manuscript.
References
Ahluwalia, G. S., Grem, J. L., Hao, Z., and Cooney, D. A. (1990). Metabolism and action of amino acid analog anti-cancer agents. Pharmacol. Therapeut. 46, 243–271. doi: 10.1016/0163-7258(90)90094-I
Allen, D., Wilson, D., Drew, R., and Perfect, J. R. (2015). Azole antifungals: 35 years of invasive fungal infection management. Expert Rev. Anti Infect. Ther. 13, 787–798. doi: 10.1586/14787210.2015.1032939
Azevedo, M.-M., Faria-Ramos, I., Cruz, L. C., Pina-Vaz, C., and Rodrigues, A. G. (2015). Genesis of azole antifungal resistance from agriculture to clinical settings. J. Agric. Food Chem. 63, 7463–7468. doi: 10.1021/acs.jafc.5b02728
Benedict, K., Jackson, B. R., Chiller, T., and Beer, K. D. (2019). Estimation of direct healthcare costs of fungal diseases in the United States. Clin. Infect. Dis. 68, 1791–1797. doi: 10.1093/cid/ciy776
Bergmann, S., Rohde, M., Chhatwal, G. S., and Hammerschmidt, S. (2001). α-Enolase of Streptococcus pneumoniae is a plasminogen-binding protein displayed on the bacterial cell surface. Mol. Microbiol. 40, 1273–1287. doi: 10.1046/j.1365-2958.2001.02448.x
Bergmann, S., Wild, D., Diekmann, O., Frank, R., Bracht, D., Chhatwal, G. S., et al. (2003). Identification of a novel plasmin(ogen)-binding motif in surface displayed α-enolase of Streptococcus pneumoniae. Mol. Microbiol. 49, 411–423. doi: 10.1046/j.1365-2958.2003.03557.x
Bicanic, T., and Harrison, T. S. (2005). Cryptococcal meningitis. Br. Med. Bull. 72, 99–118. doi: 10.1093/bmb/ldh043
Birkinshaw, J. H., Charles, J. H. V., Hetherington, A. C., and Raistrick, H. (1931). On the production of mannitol from glucose by species of Aspergillus. Philos. Trans. R. Soc. B 22, 153–171.
Bongomin, F., Gago, S., Oladele, R. O., and Denning, D. W. (2017). Global and multi-national prevalence of fungal disease - estimate precision. J. Fungi 3:57. doi: 10.3390/jof3040057
Brauer, V. S., Rezende, C. P., Pessoni, A. M., de Paula, R. G., Rangappa, K. S., Nayaka, S. C., et al. (2019). Antifungal agents in agriculture: friends and foes of public health. Biomolecules 9:521. doi: 10.3390/biom9100521
Brown, G. D., Denning, D. W., Gow, N. A. R., Levitz, S. M., Netea, M. G., White, T. C., et al. (2012). Hidden killers: human fungal infections. Med. Mycol. 4:165rv113. doi: 10.1126/scitranslmed.3004404
Bueid, A., Howard, S. J., Moore, C. B., Richardson, M. D., Harrison, E., Bowyer, P., et al. (2010). Azole antifungal resistance in Aspergillus fumigatus: 2008 and 2009. J. Antimicrob. Chemother. 65, 2116–2118. doi: 10.1093/jac/dkq279
Chaturvedi, V., Flynn, T., Neihaus, W. G., and Wong, B. (1996a). Stress tolerance and pathogenic potential of a mannitol mutant of Cryptococcus neoformans. Microbiology 142(Pt 4), 937–943. doi: 10.1099/00221287-142-4-937
Chaturvedi, V., Wong, B., and Newman, S. J. (1996b). Oxidative killing of Cryptococcus neoformans by human neutrophils. evidence that fungal mannitol protects by scavenging reactive oxygen intermediates. J. Immunol. 156, 3836–3840.
Chen, Y., Gallois-Montbrun, S., Schneider, B., Veron, M., Morera, S., Deville-Bonne, D., et al. (2003). Nucleotide binding to nucleoside diphosphate kinases: X-ray structure of human NDPK-A in complex with ADP and comparison to protein kinases. J. Mol. Biol. 332, 915–926. doi: 10.1016/j.jmb.2003.07.004
Chittur, S. V., Klem, T. J., Shafer, C. M., and Davisson, V. J. (2001). Mechanism for acivicin inactivation of triad glutamine amidotransferases. Biochemistry 40, 876–887. doi: 10.1021/bi0014047
Chitty, J. L., and Fraser, J. A. (2017). Purine acquisition and synthesis by human fungal pathogens. Microorganisms 5:33. doi: 10.3390/microorganisms5020033
Chitty, J. L., Tatzenko, T. L., Williams, S. J., Koh, Y. Q., Corfield, E. C., Butrler, M. S., et al. (2017). GMP synthase is required for virulence factor production and infection by Cryptococcus neoformans. J. Biol. Chem. 292, 3049–3059. doi: 10.1074/jbc.M116.767533
Colby, T. D., Vanderveen, K., Strickler, M. D., Markham, G. D., and Goldstein, B. M. (1999). Crystal structure of human type II inosine monophosphate dehydrogenase: implications for ligand binding and drug design. Proc. Natl. Acad. Sci. U.S.A. 96, 3531–3536. doi: 10.1073/pnas.96.7.3531
Cooney, N. M., and Klein, B. S. (2008). Fungal adaptation to the mammalian host: it's a new world, after all. Curr. Opin. Microbiol. 11, 511–516. doi: 10.1016/j.mib.2008.09.018
Corina, D. L., and Munday, K. A. (1971). Studies on polyol function in Aspergillus clavatus: a role for mannitol and ribitol. J. Gen. Microbiol. 69, 221–227. doi: 10.1099/00221287-69-2-221
Dagenais, T. R. T., and Keller, N. P. (2009). Pathogenesis of Aspergillus fumigatus in invasive aspergillosis. Clin. Microbiol. Rev. 22, 447–465. doi: 10.1128/CMR.00055-08
Dasari, P., Koleci, N., Shopova, I. A., Warternberg, D., Beyersdorf, N., Dietrich, S., et al. (2019). Enolase From Aspergillus fumigatus is a moonlighting protein that binds the human plasma complement proteins factor H, FHL-1, C4BP, and plasminogen. Front. Immunol. 10:2573. doi: 10.3389/fimmu.2019.02573
De Backer, M. D., Nelissen, B., Logghe, M., Viaene, J., Loonen, I., Vandoninck, S., et al. (2001). An antisense-based functional genomics approach for identification of genes critical for growth of Candida albicans. Nat. Biotechnol. 19, 235–241. doi: 10.1038/85677
Dinamarco, T. M., Brown, N. A., Couto De Almeida, R. S., Alves De Castro, P., Savoldi, M., De Souza Goldman, M. H., et al. (2012). Aspergillus fumigatus calcineurin interacts with a nucleoside diphosphate kinase. Microbes Infect. 14, 922–929. doi: 10.1016/j.micinf.2012.05.003
Du Pré, S., Beckmann, N., Almeida, M. C., Sibley, G. E. M., Law, D., Brand, A. C., et al. (2018). Effect of the novel antifungal drug F901318 (olorofim) on growth and viability of Aspergillus fumigatus. Antimicrob. Agents Chemother. 62:e00231–18. doi: 10.1128/AAC.00231-18
Duquerroy, S., Camus, C., and Janin, J. (1995). X-ray structure and catalytic mechanism of lobster enolase. Biochemistry 34, 12513–12523. doi: 10.1021/bi00039a005
Edwards, S. R., Braley, R., and Chaffin, W. L. (1999). Enolase is present in the cell wall of Saccharomyces cerevisiae. FEMS Microbiol. Lett. 177, 211–216. doi: 10.1111/j.1574-6968.1999.tb13734.x
Ehinger, S., Schubert, W.-D., Bergmann, S., Hammerschmidt, S., and Heinz, D. W. (2004). Plasmin(ogen)-binding alpha-enolase from Streptococcus pneumoniae: crystal structure and evaluation of plasming(ogen)-binding sites. J. Mol. Biol. 343, 997–1005. doi: 10.1016/j.jmb.2004.08.088
Ermishkin, L. N., Kasumov, K. M., and Potzeluyev, V. M. (1976). Single ionic channels induced in lipid bilayers by polyene antibiotics amphotericin B and nystatine. Nature 262, 698–699. doi: 10.1038/262698a0
Floryk, D., and Thompson, T. C. (2010). Antiproliferative effects of AVN944, a novel inosine 5-monophosphate dehydrogenase inhibitor, in prostate cancer cells. Int. J. Cancer 123, 2294–2302. doi: 10.1002/ijc.23788
Funk, J., Schaarschmidt, B., Slesiona, S., Hallström, T., Horn, U., and Brock, M. (2016). The glycolytic enzyme enolase represents a plasminogen-binding protein on the surface of a wide variety of medically important fungal species. Int. J. Med. Microbiol. 306, 59–68. doi: 10.1016/j.ijmm.2015.11.005
Furman, P. A., Fyfe, J. A., St Clair, M. H., Weinhold, K., Rideout, J. L., Greeman, G. A., et al. (1986). Phosphorylation of 3'-azido-3'-deoxythymidine and selective interaction of the 5'-triphosphate with human immunodeficiency virus reverse transcriptase. Proc. Natl. Acad. Sci. U.S.A. 83, 8333–8337. doi: 10.1073/pnas.83.21.8333
Ghannoum, M. A., and Rice, L. B. (1999). Antifungal agents: mode of action, mechanisms of resistance, and correlation of these mechanisms with bacterial resistance. Clin. Microbiol. Rev. 12, 510–517. doi: 10.1128/CMR.12.4.501
Giraud, M. F., Georgescauld, F., Lascu, I., and Dautant, A. (2006). Crystal structures of S120G mutant and wild type of human nucleoside diphosphate kinase A in complex with ADP. J. Bioenerg. Biomembr. 38, 261–264. doi: 10.1007/s10863-006-9043-0
Gooday, G. W. (1995). “Cell membrane,” in The Growing Fungus, eds N. A. R. Gow and G. M. Gadd (Dordrecht: Springer), 63–74.
Gow, N. A. R., Latgé, J. P., and Munro, C. A. (2017). The fungal cell wall: structure, biosynthesis, and function. Microbiol. Spectr. 5, 1–25. doi: 10.1128/9781555819583.ch12
Hata, M., Ishii, Y., Watanabe, E., Uoto, K., Kobayashi, S., Yoshida, K. I., et al. (2010). Inhibition of ergosterol synthesis by novel antifungal compounds targeting C-14 reductase. Med. Mycol. 48, 613–621. doi: 10.3109/13693780903390208
Hedstrom, L. (2009). IMP dehydrogenase: Structure, mechanism and inhibition. Chem. Rev. 109, 2903–2928. doi: 10.1021/cr900021w
Hof, H. (2001). Critical annotations to the use of azole antifungals for plant protection. Antimicrob. Agents Chemother. 45, 2987–2990. doi: 10.1128/AAC.45.11.2987-2990.2001
Hope, W., Morton, A., and Eisen, D. P. (2002). Increase in prevalence of nosocomial non-Candida albicans candidaemia and the association of Candida krusei with fluconazole use. J. Hosp. Infect. 50, 56–65. doi: 10.1053/jhin.2001.1131
Horikoshi, K., Iida, S., and Ikeda, Y. (1965). Mannitol and mannitol dehydrogenases in conidia of Aspergillus oryzae. J. Bacteriol. 89, 326–330. doi: 10.1128/JB.89.2.326-330.1965
Hosaka, T., Meguro, T., Yamato, I., and Shirakihara, Y. (2003). Crystal structure of Enterococcus hirae enolase at 2.8 Å resolution. J. Biochem. 133, 817–823. doi: 10.1093/jb/mvg104
Houšt, J., SpíŽek, J., and Havlíček, V. (2020). Antifungal drugs. Metabolites 10:106. doi: 10.3390/metabo10030106
Ji, H., Wang, J., Guo, J., Li, Y., Lian, S., Guo, W., et al. (2016). Progress in the biological function of alpha-enolase. Anim. Nutr. 2, 12–17. doi: 10.1016/j.aninu.2016.02.005
Jin, J.-H., Lee, K.-T., Hong, J., Lee, D., Jang, E.-H., Kim, J.-Y., et al. (2020). Genome-wide functional analysis of phosphatases in the pathogenic fungus Cryptococcus neoformans. Nat. Commun. 11:4212. doi: 10.1038/s41467-020-18028-0
Kang, H. J., Jung, S. K., Kim, S. J., and Chung, S. J. (2008). Structure of human alpha-enolase (hENO1), a multifunctional glycolytic enzyme. Acta Crystallogr. D Biol. Crystallogr. 64, 651–657. doi: 10.1107/S0907444908008561
Khanna, N., Stuehler, C., Lünemann, A., Wójtowicz, A., Bochud, P. Y., and Leibundgut-Landmann, S. (2016). Host response to fungal infections - how immunology and host genetics could help to identify and treat patients at risk. Swiss Med. Wkly. 146:w14350. doi: 10.4414/smw.2016.14350
Kidd, S. E., Chen, S. C.-A., Meyer, W., and Halliday, C. L. (2020). A new age in molecular diagnostics for invasive fungal disease: are we ready? Front. Microbiol. 10:2903. doi: 10.3389/fmicb.2019.02903
Kilstrup, M., Hammer, K., Jensen, P. R., and Martinussen, J. (2005). Nucleotide metabolism and its control in lactic acid bacteria. FEMS Microbiol. Rev. 29, 555–590. doi: 10.1016/j.fmrre.2005.04.006
Ko, H.-C., Hsiao, T.-Y., Chen, C.-T., and Yang, Y.-L. (2013). Candida albicans ENO1 null mutants exhibited altered drug susceptibility, hyphal formation, and virulence. J. Microbiol. 51, 345–351. doi: 10.1007/s12275-013-2577-z
Kozel, T. R., and Wickes, B. (2014). Fungal diagnostics. Cold Spring Harb. Perspect. Med. 4:a019299. doi: 10.1101/cshperspect.a019299
Kuhnel, K., and Luisi, B. (2001). Crystal structure of the Escherichia coli RNA degradosome component enolase. J. Mol. Biol. 313, 583–592. doi: 10.1006/jmbi.2001.5065
Kummari, L. K., Butler, M. S., Furlong, E., Blundell, R., Nouwens, A., Silva, A. B., et al. (2018). Antifungal benzo[b]thiophene 1,1-dioxide IMPDH inhibitors exhibit pan-assay interference (PAINS) profiles. Bioorganic Med. Chem. 26, 5408–5419. doi: 10.1016/j.bmc.2018.09.004
Latgé, J. P. (1999). Aspergillus fumigatus and Aspergillosis. Clin. Microbiol. Rev. 12, 310–350. doi: 10.1128/CMR.12.2.310
Law, R. H. P., Abu-Ssaydeh, D., and Whisstock, J. C. (2013). New insights into the structure and function of the plasminogen/plasmin system. Curr. Opin. Struct. Biol. 23, 836–841. doi: 10.1016/j.sbi.2013.10.006
Law, R. H. P., Caradoc-Davies, T., Cowieson, N. P., Horvath, A. J., Quek, A. J., Encanacao, J. A., et al. (2012). The X-ray crystal structure of full-length human plasminogen. Cell Rep. 1, 185–190. doi: 10.1016/j.celrep.2012.02.012
Lestrade, P., Buil, J. B., Van Der Beek, M. T., Kuijper, E. J., Van Dijk, K., Kampinga, G. A., et al. (2020). Paradoxal trends in azole-resistant Aspergillus fumigatus in a national multicenter surveillance program, the Netherlands, 2013–2018. Emerg. Infect. Dis. 26, 1447–1455. doi: 10.3201/eid2607.200088
Li, W. Q., Hu, X. C., Zhang, X., Ge, Y., Zhao, S., Hu, Y., et al. (2011). Immunisation with the glycolytic enzyme enolase confers effective protection against Candida albicans infection in mice. Vaccine 29, 5526–5533. doi: 10.1016/j.vaccine.2011.05.030
Lin, X., Momany, C., and Momany, M. (2003). SwoHp, a nucleoside diphosphate kinase, is essential in Aspergillus nidulans. Eukaryotic Cell 2, 1169–1177. doi: 10.1128/EC.2.6.1169-1177.2003
Liu, Q., Ying, S. H., Feng, M. G., and Jiang, X. H. (2008). Physiological implication of intracellular trehalose and mannitol changes in response of entomopathogenic fungus Beauveria bassiana to thermal stress. Antonie Van Leeuwenhoek 95, 65–75. doi: 10.1007/s10482-008-9288-1
López-Alemany, R., Longstaff, C., Hawley, S., Mirshahi, M., Fábregas, P., Jardí, M., et al. (2003). Inhibition of cell surface mediated plasminogen activation by a monoclonal antibody against alpha-Enolase. Am. J. Hematol. 72, 234–242. doi: 10.1002/ajh.10299
Markland, W., Mcquaid, T. J., Jain, J., and Kwong, A. D. (2000). Broad-spectrum antiviral activity of the IMP dehydrogenase inhibitor VX-497: a comparison with Ribavirin and demonstration of antiviral additivity with alpha interferon. Antimicrob. Agents Chemother. 44, 859–866. doi: 10.1128/AAC.44.4.859-866.2000
Marshall, A. C., Bond, C. S., and Bruning, J. B. (2018). Structure of Aspergillus fumigatus cytosolic thiolase: trapped tetrahedral reaction intermediates and activation by monovalent cations. ACS Catal. 8, 1973–1989. doi: 10.1021/acscatal.7b02873
Marshall, A. C., Kidd, S. E., Lamont-Friedrich, S. J., Arentz, G., Hoffman, P., Coad, B. R., et al. (2019). Structure, mechanism, and inhibition of Aspergillus fumigatus thioredoxin reductase. Antimicrob. Agents Chemother. 63:e02281–18. doi: 10.1128/AAC.02281-18
Marshall, A. C., Kroker, A. J., Murray, L. A. M., Gronthos, K., Rajapaksha, H., Wegener, K. L., et al. (2017). Structure of the sliding clamp from the fungal pathogen Aspergillus fumigatus (AfumPCNA) and interactions with Human p21. FEBS J. 284, 985–1002. doi: 10.1111/febs.14035
Mazu, T. K., Bricker, B. A., Flores-Rozas, H., and Ablordeppey, S. Y. (2016). The mechanistic targets of antifungal agents: an overview. Mini Rev. Med. Chem. 16, 555–578. doi: 10.2174/1389557516666160118112103
Meena, M., Prasad, V., Zehra, A., Gupta, V. K., and Upadhyay, R. S. (2015). Mannitol metabolism during pathogenic fungal-host interactions under stressed conditions. Front. Microbiol. 6:1019. doi: 10.3389/fmicb.2015.01019
Megson, G. M., Stevens, D. A., Hamilton, J. R., and Denning, D. W. (1996). D-mannitol in cerebrospinal fluid of patients with AIDS and cryptococcal meningitis. J. Clin. Microbiol. 34, 218–221. doi: 10.1128/JCM.34.1.218-221.1996
Miura, N., Kirino, A., Endo, S., Morisaka, H., Kuroda, K., Takagi, M., et al. (2012). Tracing putative trafficking of the glycolytic enzyme enolase via SNARE-driven unconventional secretion. Eukaryotic Cell 11, 1075–1082. doi: 10.1128/EC.00075-12
Morrow, C. A., Valkov, E., Stamp, A., Chow, E. W. L., Lee, I. R., Wronski, A., et al. (2012). De novo GTP biosynthesis is citical for virulence of the fungal pathogen Cryptococcus neoformans. PLoS Pathog. 8:e1002957. doi: 10.1371/journal.ppat.1002957
Nguyen, S., Jovcevski, B., Pukala, T. L., and Bruning, J. B. (in press). Nucleoside selectivity of Aspergillus fumigatus nucleoside-diphosphate kinase. FEBS J. doi: 10.1111/febs.15607. [Epub ahead of print].
Nucci, M., and Marr, K. A. (2005). Emerging fungal diseases. Clin. Infect. Dis. 41, 521–526. doi: 10.1086/432060
Nucci, M., and Perfect, J. R. (2008). When primary antifungal therapy fails. Clin. Infect. Dis. 46, 1426–1433. doi: 10.1086/587101
Oliver, J. D., Sibley, G. E. M., Beckmann, N., Dobb, K. S., Slater, M. J., Mcentee, L., et al. (2016). F901318 represents a novel class of antifungal drug that inhibits dihydroorotate dehydrogenase. Proc. Natl. Acad. Sci. U.S.A. 113, 12809–12814. doi: 10.1073/pnas.1608304113
Pancholi, V., and Fischetti, V. A. (1998). Alpha-enolase, a novel strong plasmin(ogen) binding protein on the surface of pathogenic Streptococci. J. Biol. Chem. 273, 14503–14515. doi: 10.1074/jbc.273.23.14503
Park, Y., Pacitto, A., Bayliss, T., Cleghorn, L. A., Wang, Z., Hartman, T., et al. (2018). Essential but not vulnerable: indazole sulfonamides targeting inosine monophosphate dehydrogenase as potential leads against Mycobacterium tuberculosis. ACS Infect Dis. 3, 18–33. doi: 10.1021/acsinfecdis.6b00103
Perlin, D. S. (2015). Echinocandin resistance in Candida. Clin. Infect. Dis. 61, S612–S617. doi: 10.1093/cid/civ791
Perlin, D. S., Shor, E., and Zhao, Y. (2015). Update on antifungal drug resistance. Curr. Clin. Microbiol. Rep. 2, 84–95. doi: 10.1007/s40588-015-0015-1
Pfaller, M. A., Diekema, D. J., Mendez, M., Kibbler, C., Erzsebet, P., Chang, S.-C., et al. (2006). Candida guilliermondii, an opportunist fungal pathogen with decrease susceptibility to fluconazole: geographic and temporal trends from the ARTEMIS DISK antifungal surveillance program. J. Clin. Microbiol. 44, 3551–3556. doi: 10.1128/JCM.00865-06
Rodriguez-Suarez, R., Xu, D., Veillette, K., Davison, J., Sillaots, S., Kauffman, S., et al. (2007). Mechanism-of-action determination of GMP synthase inhibitors and target validation in Candida albicans and Aspergillus fumigatus. Chem. Biol. 14, 1163–1175. doi: 10.1016/j.chembiol.2007.09.009
Ruijter, G. J. G., Bax, M., Patel, H., Flitter, S. J., Van De Vondervoort, P. J. I., De Vries, R. P., et al. (2003). Mannitol is required for stress tolerance in Aspergillus niger conidiospores. Eukaryotic Cell 2, 690–698. doi: 10.1128/EC.2.4.690-698.2003
Ryder, N. S. (1988). Mechanism of action and biochemical selectivity of allylamine antimycotic agents. Ann. N. Y. Acad. Sci. 544, 208–220. doi: 10.1111/j.1749-6632.1988.tb40405.x
Sabiiti, W., and May, R. C. (2012). Mechanisms of infection by the human fungal pathogen Cryptococcus neoformans. Future Microbiol. 7, 1297–1313. doi: 10.2217/fmb.12.102
Schmiedel, Y., and Zimmerli, S. (2016). Common invasive fungal diseases: an overview of invasive candidiasis, aspergillosis, cryptococcosis, and Pneumocystis pneumonia. Swiss Med. Wkly. 14:w14281. doi: 10.4414/smw.2016.14281
Semianrio-Vidal, L., Van Hesuden, C., Mugesh, G., and Lazarowski, E. R. (2010). Ebselen is a potent non-competitive inhibitor of extracellular nucleoside diphosphokinase. Purinerg. Signal 6, 383–391. doi: 10.1007/s11302-010-9203-x
Shi, M., and Mody, C. H. (2016). Fungal infection in the brain: what we learned from intravital imaging. Front. Immunol. 7:292. doi: 10.3389/fimmu.2016.00292
Shibasaki, S., Karasaki, M., Tafuku, S., Aoki, W., Sewaki, T., and Ueda, M. (2014). Oral immunization against candidiasis using Lactobacillus casei displaying enolase 1 from Candida albicans. Sci. Pharm. 82, 697–708. doi: 10.3797/scipharm.1404-07
Shu, Q., and Nair, V. (2008). Inosine monophosphate dehydrogenase (IMPDH) as a target in drug discovery. Med. Res. Rev. 28, 219–232. doi: 10.1002/med.20104
Sperling, O. (1988). “Human purine metabolism,” in Myocardial Energy Metabolism, ed J. W. De Jong (Dordrecht: Springer), 225–236.
Stec, B., and Lebioda, L. (1990). Refined structure of yeast apo-enolase at 2.25 Å resolution. J. Mol. Biol. 211, 235–248. doi: 10.1016/0022-2836(90)90023-F
Thammahong, A., Puttikamonkul, S., Perfect, J. R., Brennan, R. G., and Cramer, R. A. (2017). Central role of the trehalose biosynthesis pathway in the pathogenesis of human fungal infections: opportunities and challenges for therapeutic development. Microbiol. Mol. Biol. Rev. 81:e00053–16. doi: 10.1128/MMBR.00053-16
Trapero, A., Pacitto, A., Singh, V., Sabbah, M., Coyne, A. G., Mizrahi, V., et al. (2018). Fragment-based approach to targeting inosine-5'-monophosphate dehydrogenase (IMPDH) from Mycobacterium tuberculosis. J. Med. Chem. 61, 2806–2822. doi: 10.1021/acs.jmedchem.7b01622
Umejiego, N. N., Gollapalli, D., Sharling, L., Volftsun, A., Lu, J., Benjamin, N. N., et al. (2008). Targeting a prokaryotic protein in a eukaryotic pathogen: identification of lead compounds against Cryptosporidiosis. Chem. Biol. 15, 70–77. doi: 10.1016/j.chembiol.2007.12.010
Urbina, J. M., Cortés, J. C. G., Palma, A., López, S. N., Zacchino, S. A., Enriz, R. D., et al. (2000). Inhibitors of the fungal cell wall. Synthesis of 4-aryl-4-N-arylamine-1-butenes and related compounds with inhibitory activities on β(1–3) glucan and chitin synthases. Bioorganic Med. Chem. 8, 691–698. doi: 10.1016/S0968-0896(00)00003-1
Vandeputte, P., Ferrari, S., and Coste, A. T. (2012). Antifungal resistance and new strategies to control fungal infections. Int. J. Microbiol. 2012:713687. doi: 10.1155/2012/713687
Walsh, T. J., and Dixon, D. M. (1996). “sSpectrum of mycoses,” in Medical Microbiology, 4th Edn, eds S. Baron (Galveston, TX: University of Texas Medical Branch at Galveston).
Wang, Y., Wang, S., Nie, X., Yang, K., Xu, P., Wang, X., et al. (2019). Molecular and structural basis of nucleoside diphosphate kinase–mediated regulation of spore and sclerotia development in the fungus Aspergillus flavus. J. Biol. Chem. 294, 12415–12431. doi: 10.1074/jbc.RA119.007505
Wang, Y., Wang, S., Nie, X., Yang, K., Xu, P., Wang, X., et al. (2019). Molecular and structural basis of nucleoside diphosphate kinase-mediated regulation of spore and sclerotia development in the fungus Aspergillus flavus. J. Biol. Chem. 294, 12415–12431.
Welin, M., Lehtio, L., Johansson, A., Flodin, S., Nyman, T., Tresaugues, L., et al. (2013). Substrate specificty and oligomerization of human GMP synthetase. J. Mol. Biol. 425, 4323–4333. doi: 10.1016/j.jmb.2013.06.032
Wenzel, R. P., and Gennings, C. (2005). Bloodstream infections due to Candida species in the intensive care unit: identifying especially high-risk patients to determine prevention strategies. Clin. Infect. Dis. 41, S389–S393. doi: 10.1086/430923
Wiederhold, N. P., and Lewis, R. E. (2003). The echinocandin antifungals: an overview of the pharmacology, spectrum and clinical efficacy. Expert Opin. Investig. Drugs 12, 1313–1333. doi: 10.1517/13543784.12.8.1313
Witteveen, C. F. B., and Visser, J. (1995). Polyol pools in Aspergillus niger. FEMS Microbiol. Lett. 134, 57–62. doi: 10.1111/j.1574-6968.1995.tb07914.x
Wong, B., Brauer, K. L., Tsai, R. R., and Jayasimhulu, K. (1989). Increased amounts of the Aspergillus metabolite D-mannitol in serum of rats with experimental aspergillosis. J. Infect. Dis. 160, 95–103. doi: 10.1093/infdis/160.1.95
Wong, B., Perfect, J. R., Beggs, S., and Wright, K. A. (1990). Production of the hexitol D-mannitol by Cryptococcus neoformans in vitro and in rabbits with experimental meningitis. Infect. Immun. 58, 1664–1670. doi: 10.1128/IAI.58.6.1664-1670.1990
Keywords: Aspergillus fumigatus, Candida albicans, Cryptococcus neoformans, drug targets, drug discovery, antifungal
Citation: Nguyen S, Truong JQ and Bruning JB (2021) Targeting Unconventional Pathways in Pursuit of Novel Antifungals. Front. Mol. Biosci. 7:621366. doi: 10.3389/fmolb.2020.621366
Received: 26 October 2020; Accepted: 11 December 2020;
Published: 12 January 2021.
Edited by:
Amit Prasad, Indian Institute of Technology Mandi, IndiaReviewed by:
Praveen Rao Juvvadi, Duke University, United StatesVicente de Paulo Martins, University of Brasilia, Brazil
Copyright © 2021 Nguyen, Truong and Bruning. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John B. Bruning, am9obi5icnVuaW5nQGFkZWxhaWRlLmVkdS5hdQ==