 Marco Luciani
Marco Luciani Angela Gritti
Angela Gritti Vasco Meneghini
Vasco Meneghini
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Mol. Biosci. , 18 September 2020
Sec. Molecular Diagnostics and Therapeutics
Volume 7 - 2020 | https://doi.org/10.3389/fmolb.2020.00224
This article is part of the Research Topic Neurodegenerative Lysosomal Storage Diseases (LSDs): Between Biology and Innovative Therapeutic Approaches View all 8 articles
Lysosomal storage diseases (LSDs) are a group of rare genetic conditions. The absence or deficiency of lysosomal proteins leads to excessive storage of undigested materials and drives secondary pathological mechanisms including autophagy, calcium homeostasis, ER stress, and mitochondrial abnormalities. A large number of LSDs display mild to severe central nervous system (CNS) involvement. Animal disease models and post-mortem tissues partially recapitulate the disease or represent the final stage of CNS pathology, respectively. In the last decades, human models based on induced pluripotent stem cells (hiPSCs) have been extensively applied to investigate LSD pathology in several tissues and organs, including the CNS. Neural stem/progenitor cells (NSCs) derived from patient-specific hiPSCs (hiPS-NSCs) are a promising tool to define the effects of the pathological storage on neurodevelopment, survival and function of neurons and glial cells in neurodegenerative LSDs. Additionally, the development of novel 2D co-culture systems and 3D hiPSC-based models is fostering the investigation of neuron-glia functional and dysfunctional interactions, also contributing to define the role of neurodevelopment and neuroinflammation in the onset and progression of the disease, with important implications in terms of timing and efficacy of treatments. Here, we discuss the advantages and limits of the application of hiPS-NSC-based models in the study and treatment of CNS pathology in different LSDs. Additionally, we review the state-of-the-art and the prospective applications of NSC-based therapy, highlighting the potential exploitation of hiPS-NSCs for gene and cell therapy approaches in the treatment of neurodegenerative LSDs.
Lysosomal storage diseases (LSDs) are a group of inherited genetic disorders caused by the deficiency of lysosomal proteins that leads to the accumulation of undigested storage material. They are rare diseases if considered independently, with a prevalence ranging between 1:57,000 and 1:4,200,000 individuals. However, if taken as a group they are thought to affect up to 1:5,000 live births (Meikle et al., 1999; Poorthuis et al., 1999; Applegarth et al., 2000; Pinto et al., 2004; Poupetova et al., 2010; Al-Jasmi et al., 2013; Platt et al., 2018).
These disorders originate from defects in metabolic enzymes, hydrolases, channels or membrane proteins, cofactors, or components that traffic lysosomal proteins. The accumulation of storage material impairs lysosomal functions and affects several organelles and cellular activities (Ballabio and Gieselmann, 2009). Neurodegeneration is a prominent feature in the majority of LSDs (Wraith, 2002), suggesting an increased sensitivity of neural cells toward dysfunctional cellular clearance.
Enthusiasm has arisen for the possibility to generate neural stem/progenitor cells (NSCs) and differentiated progeny from human induced pluripotent stem cells (hiPSCs), a new source of stem cells obtained by reprogramming of somatic cells through the manipulation of a core set of transcription factors (Ma et al., 2013; Smith et al., 2016). The use of neural cells derived from patient-specific iPSCs to investigate the mechanisms of CNS pathology in these rare disorders has been extensively reviewed (Borger et al., 2017; De Filippis et al., 2017; Zunke and Mazzulli, 2019).
Here, we focus on the advantages and limitations in the use of hiPSC-derived NSCs (hiPS-NSCs) to develop novel therapeutics. In addition, we discuss the prospective application of novel hiPSC-based models to investigate the role of neurodevelopmental defects and to highlight the contribution of non-neural cells and neuroinflammation in the disease onset and progression of neurodegenerative LSDs.
Developmental studies in animal models and human embryonic stem cells (hESCs) have increased our knowledge of the key factors regulating the neuralization of pluripotent stem cells, the formation of neuroepithelial cells in neural tube-like structures, and the generation and regionalization of neural stem/progenitor cell populations. The combined inhibition of BMP signaling and Lefty/Activin/TGFβ pathways by co-treatment with Noggin and SB431542 during the first steps of neuroectodermal commitment has significantly increased the efficiency of neuralization from hiPSCs (Chambers et al., 2009). Dual-SMAD inhibition promotes the transient formation of early neuroepithelial cells (NEPs) that spontaneously convert into later-stage NEP populations growing in rosette-like structures (Elkabetz et al., 2008). NEPs can be expanded in presence of FGF2 and EGF for several passages, giving rise to an intermediate developmental stage between rosette-organized NEPs and radial glial cells displaying self-renewal, clonogenicity and responsiveness to instructive cues that promote the induction of distinct neuron subpopulations (Koch et al., 2009; Doerr et al., 2015). Despite displaying self-renewal and multipotency, single-seq RNA-seq analyses revealed the intrinsic heterogeneity hiPSC-derived NEP populations and their enrichment in neuronal and glial progenitors (Lam et al., 2019), with few cell clusters shared with somatic fetal brain-derived human NSCs (hfNSCs) (Lam et al., 2019). Subsequent amplification/selection/enrichment of hiPSC-derived NEPs are needed to obtain radial glial-like populations sharing transcriptional, phenotypic and functional identities with prospectively isolated (CD133+ CD34– CD45–) hfNSCs (Conti et al., 2005; Elkabetz et al., 2008; Meneghini et al., 2017; Rosati et al., 2018).
Both hiPS-derived NEPs and radial glial cells are generally defined as hiPS-NSCs being characterized by self-renewal, expression of NSC markers (e.g., PAX-6 and Nestin) and multipotency. These features make these cell populations suitable tools for disease modeling studies and the development of novel therapeutic approaches for several neurodegenerative disorders. Indeed, patient-derived hiPS-NSCs can be expanded at medium/large scale and differentiated in neurons, astrocytes and oligodendrocytes, thus recapitulating CNS-specific pathological mechanisms. Importantly, these human in vitro models harbor the natural disease-causing mutations in a genetic background which is permissive for the disorder. This feature allows to highlight the role of the genetic background in determining the disease progression and severity, which is particularly relevant in some LSDs (e.g., Metachromatic Leukodystrophy) characterized by high phenotypic variability in patient carrying similar or even identical mutations. Gene knock-out approaches in immortalized cell lines (e.g., SH-SY5Y cell line) cannot fully recapitulate the contribution of the genetic background, also considering that these cellular models are characterized by abnormal karyotypes and the expression of oncogenes that might hamper the identification of disease phenotypes. Additionally, knock-out strategies do not reproduce the many loss-of-function mutations that result in the generation of misfolded proteins potentially leading to LSD secondary pathological mechanisms, including stress pathways activated by the unfolded protein response (UPR) and impaired autophagy. Patient-derived skin fibroblasts harbor the disease-causing mutations and patient-specific genetic backgrounds, and they also display clinical hallmarks in some LSDs, like the accumulation of pathological macromolecules and/or dysfunction in the endosomal–autophagic–lysosomal trafficking (Platt et al., 2012). However, both fibroblasts and highly dividing immortalized cells have different baseline metabolic rates as compared to neural cells. Indeed, they are usually less vulnerable to the toxic effects of substrate accumulation, thus hampering the full investigation of CNS cell type-specific mechanisms.
Despite the recent progress in hiPS-NSC technologies allow the generation of engineered models for high-throughput drug screenings in neurological and psychiatric disease (Hunt et al., 2019), a major challenge in using hiPSC-based models is the potential intra- and inter-experimental variability between donors, clones derived from the same donor and different cell passages of the same clone. To reduce the clone-dependent variability, as well as to dissect the contribution of the genetic background in disease phenotypes, the experimental study might be carefully designed to include isogenic control lines and/or an appropriate number of lines derived from different donors, rather than multiple clones derived from a small number of donors (Germain and Testa, 2017). This is particularly relevant in the study of metabolic processes affected in LSDs (e.g., autophagic flux, lysosomal enzyme activity, and the turnover of macromolecules), which are usually characterized by high baseline variability already in the control cell lines. Considering that a metabolic reprogramming is occurring during neural commitment, in particular in the glycosphingolipid and sulfatide metabolisms (Frati et al., 2018; Russo et al., 2018), quality check-points during the differentiation protocol are critical to define the differentiation stages of patient-specific and control cells. Based on protocols developed in hESCs, the expression of several stage-specific markers can be analyzed to define the hiPSC-derived neural population, including SOX1, N-Cad, Forse-1 expressed in NEP cells, and RC2, SOX2, BLBP, GLAST, CD44 and CD133 expressed in more mature radial glial-like cells (reviewed in Conti and Cattaneo, 2010). The advantages and drawbacks of different 2D in vitro models for disease modeling studies and for the development of novel therapeutics are summarized in Figure 1.
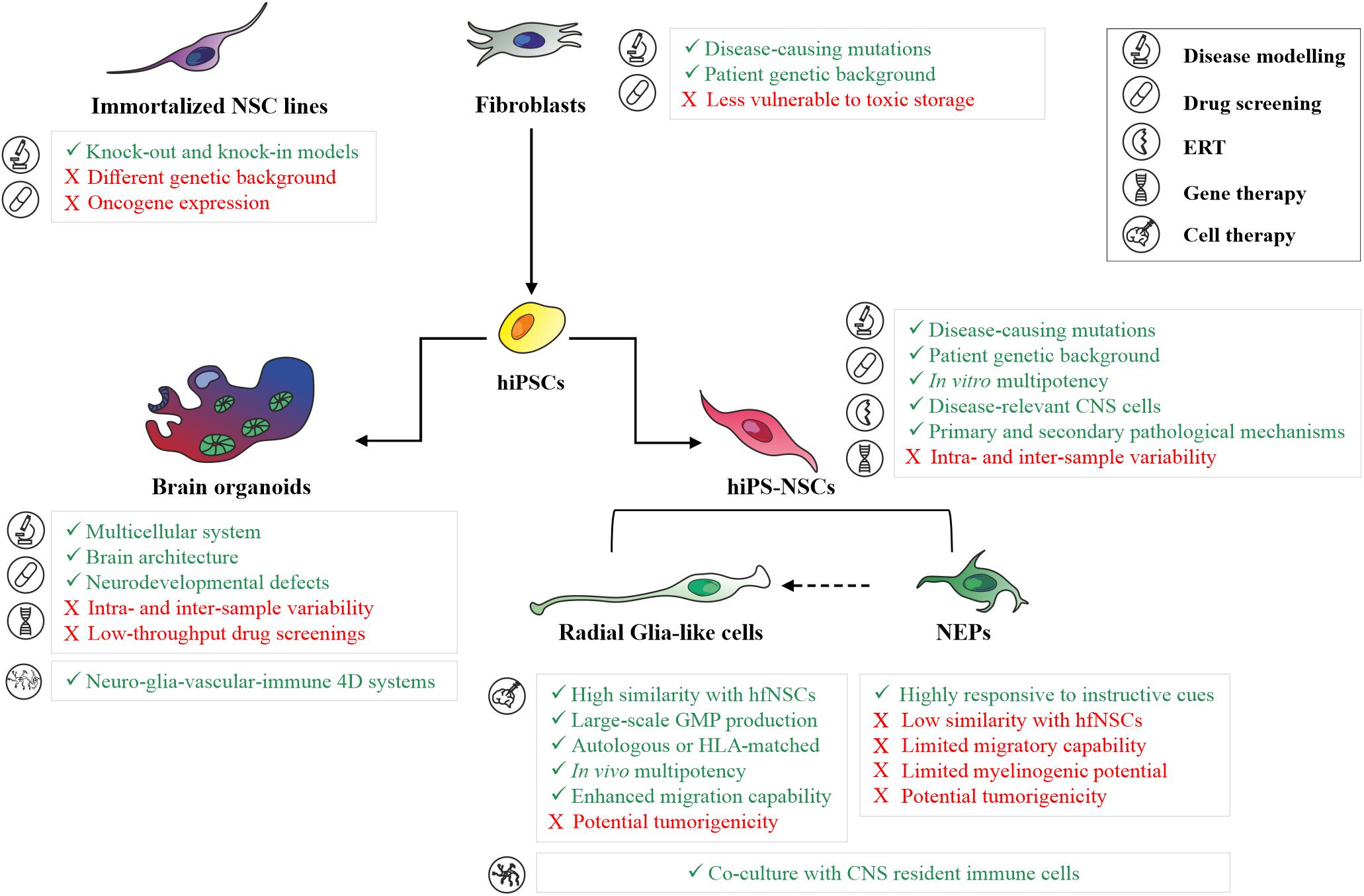
Figure 1. Human 2D and 3D in vitro models for basic and translation research in neurodegenerative LSDs. Summary of main advantages (green text) and limitations (red text) in the application of patient skin fibroblasts, immortalized neuronal cell lines, hiPS-NSC cell populations (neuroepithelial cells and radial glial-like cells), and brain organoids for disease modeling studies, development of novel therapeutics (drug screening, enzyme replacement therapy, gene therapy), and cell therapy in neurodegenerative LSDs. ERT, Enzyme Replacement Therapy; NEPs, Neuroepithelial cells.
Several studies highlighted the relevance of hiPS-NSCs and differentiated neuronal and glial progeny to test the efficacy and safety of therapeutic strategies targeting CNS pathology in LSDs (Table 1). The cell-specificity of LSD pathology has been confirmed in in vitro models of Niemann–Pick disease type C (NPC), where several compounds effective in restoring the cholesterol trafficking in patient NPC fibroblasts, including miglustat, suberoylanilide hydroxamic acid, curcumin, lovastatin, pravastatin, and rapamycin (Patterson et al., 2007; Madra and Sturley, 2010; Pipalia et al., 2011), did not display any therapeutic effects in hiPS-NSCs and differentiated progeny (Yu et al., 2014). Additionally, drugs that are effective in both patient’s fibroblasts and hiPSC-derived CNS cells could show a different dose-dependent safety profile. This is the case of the lipid chelators 2-hydroxypropyl-β-cyclodextrin (HPBCD) and 2-hydroxypropyl-γ-cyclodextrin (HPGCD), which favor the degradation/exocytosis of macromolecules through the generation of inclusion complexes with cholesterol, sphingomyelin, lipids and GM2 gangliosides in in vitro models of NPC (Soga et al., 2015), Niemann–Pick disease type A (NPA) (Long et al., 2016), Neuronal Ceroid Lipofucinosis (NCL) (Sima et al., 2018) and Tay-Sachs disease (TSD) (Vu et al., 2018). Studies on hiPSC-derived NPC neural cultures showed a dose-dependent neurotoxic effects of HPBCD (Long et al., 2016), which is currently used to treat NPC patients (Matsuo et al., 2013; Ory et al., 2017; Hastings et al., 2019; Ulloa et al., 2020), thus anticipating recent in vivo studies showing a region-specific alteration of the homeostasis of different lipid species in the brain of HPBCD-treated mice with potential detrimental effects that should to be carefully evaluated (Long et al., 2016; Glaser et al., 2020).
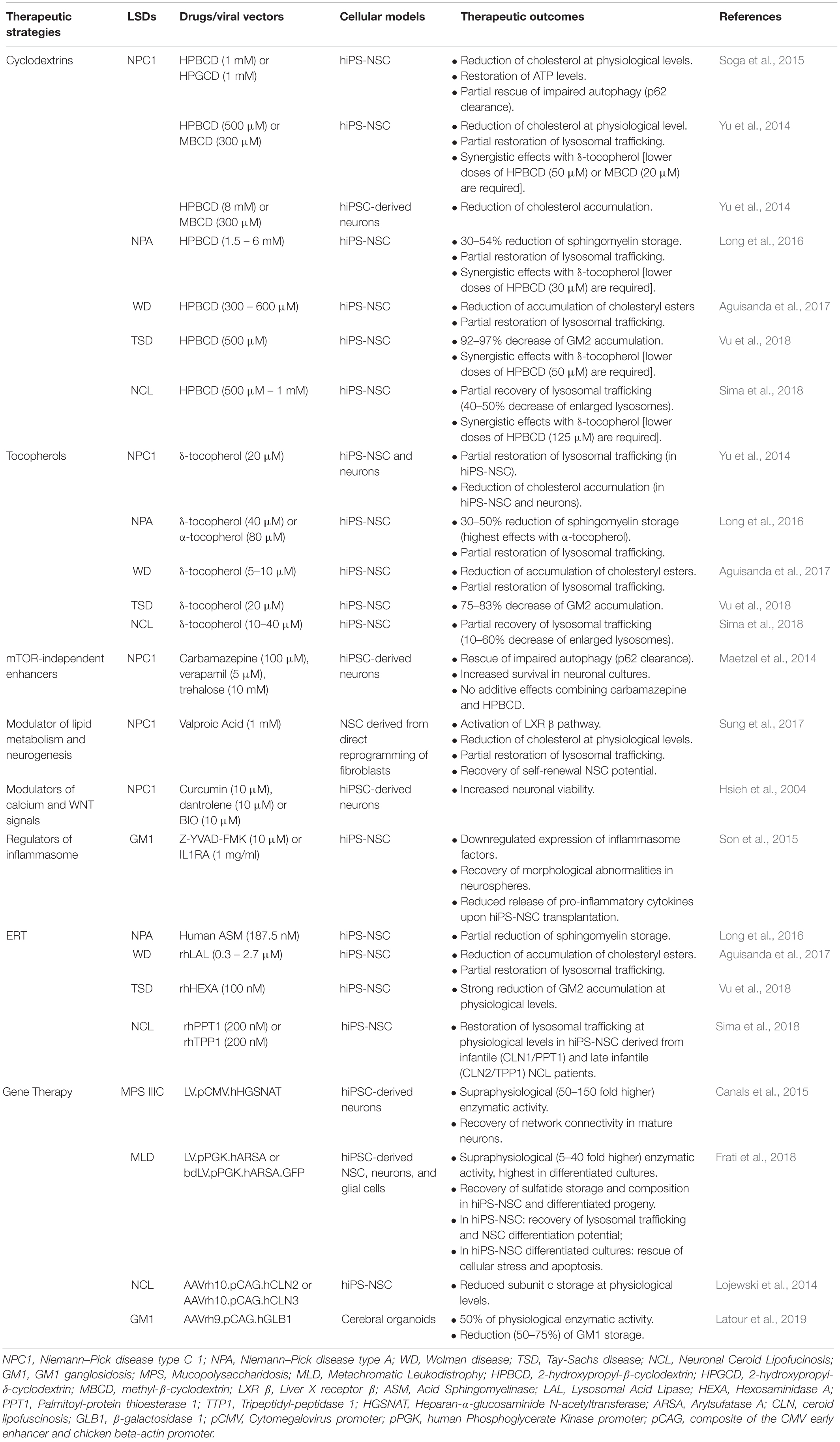
Table 1. List of therapeutic strategies applied in hiPSC-derived models to treat primary biochemical defects and secondary pathological dysfunctions in LSD.
Human iPS-NSCs can be exploited to define novel therapeutic strategies counteracting substrate accumulation in affected lysosomes. For example, studies on hiPS-NSCs proposed a complementary approach based on tocopherols to stimulate the exocytosis of macromolecules via the phagocytic, caveolae-, clathrin-, and cell adhesion molecule (CAM)-mediated pathways to restore the endosomal-lysosomal trafficking (Xu et al., 2012; Manthe et al., 2019). Both δ- and α-tocopherol significantly reduced the storage in NPC and NPA hiPSC-derived NPCs and neurons (Yu et al., 2014; Long et al., 2016), in agreement with previous observations showing that α-tocopherol-rich diet increases the survival of Purkinje neurons, reduces astrogliosis, and improves coordination and locomotor functions in NPC mouse models (Marin et al., 2014). Importantly, the use of hiPS-NSCs demonstrated that δ-tocopherol acted in synergy with low HPBCD doses, thus paving the way to future in vivo studies aimed at demonstrating that the co-administration of these drugs could be a safer and more effective therapeutic option for the treatment of several LSDs, including NPC, NPA, TSD and NCL (Yu et al., 2014; Long et al., 2016; Sima et al., 2018; Vu et al., 2018). These studies highlight that data collected in hiPSC-derived neural cells are not only accessory to in vivo studies in disease animal models, but could provide deeper insights on the metabolism, dose-response effects and toxicity of drugs designed for the treatment of LSDs.
The capability of hiPSC-derived neural cultures to recapitulate not only the primary biochemical defects but also secondary pathological mechanisms, stress the relevance of these cellular models in drug discovery processes aimed at identifying novel drugable pathways targeting common pathological mechanisms shared by several LSDs. Human iPSC-derived neurons from Gaucher disease (GD) patients show defects in the clearance of autophagosomes responsible of impaired calcium homeostasis and cytotoxicity (Schondorf et al., 2014; Awad et al., 2015). Similarly, the autophagic flux is impaired in NPC hiPS-NSCs and neurons (Trilck et al., 2013; Lee et al., 2014a; Maetzel et al., 2014; Soga et al., 2015; Trilck et al., 2017). Interestingly, mTOR-independent autophagy enhancers (Table 1) are cytoprotective (Maetzel et al., 2014). Also, recent studies suggest the contribution of Transcription Factor EB (TFEB) (Awad et al., 2015; Napolitano and Ballabio, 2016; Puertollano et al., 2018) and/or the VEGF/VEGFR2-mediated sphingosine kinase (SphK) activity (Lee et al., 2014a) to the impaired autophagy in hiPSC-derived neurons. In macrophages derived from peripheral monocytes of patients affected by type 1 Gaucher disease, inflammasome activation has been linked to impaired autophagy and increased IL-1β secretion (Aflaki et al., 2016). Similarly, accumulation of gangliosides, as well as other pathological substrates, leads to lysosomal disruption, inflammasome activation and altered autophagic flux responsible of reduced self-renewal potential and morphological abnormalities in hiPS-NSCs (Son et al., 2015). Upregulation of inflammatory caspases (e.g., CASP1) and IL-1β pathway are rescued by treatment with the CASP1 inhibitor Z-YVAD-FMK and the natural competitive IL1R antagonist Anakinra (Son et al., 2015) (Table 1). Despite the molecular mechanisms responsible of impaired autophagy in LSDs are still under investigation, autophagy is emerging as a relevant mechanisms involved in the clearance of pathological substrates, lysosomal trafficking and inflammation in different LSDs with important implications for the development of new therapeutic approaches (Lee et al., 2014a; Maetzel et al., 2014; Soga et al., 2015; Rabenstein et al., 2017; Seranova et al., 2017; Bayo-Puxan et al., 2018; Swaroop et al., 2018; Teinert et al., 2020).
Beside drug discovery, hiPS-NSCs have been exploited to investigate the efficacy and safety of therapies aimed at restoring the expression of the deficient lysosomal enzyme in disease-target cells (Table 1). The efficient uptake of recombinant human enzymes in enzyme replacement therapy (ERT) settings has been demonstrated in hiPSC-derived CNS cells in NPA, infantile and late infantile NCL, TSD and WD (Long et al., 2016; Aguisanda et al., 2017; Sima et al., 2018; Vu et al., 2018). Lentiviral vector (LV)-mediated overexpression of ARSA or heparan-α-glucosaminide N-acetyltransferase (HGSNAT) in MLD and MPSIIIC hiPS-NSCs, respectively, rescued both the primary storage and neurodevelopmental defects, without cytotoxic effects induced by transgene overexpression (Canals et al., 2015; Frati et al., 2018). These in vitro studies in relevant human models could provide deeper insights on the regulation of cross-correction mechanisms in neural cells, and on dose-dependent cytotoxicity of the delivered/over-expressed enzyme and genotoxicity of viral vectors in a human setting, which are relevant in perspective of the clinical translation of these approaches. In this view, they complement in vivo studies in small and large animal models, which are, however, indispensable to define the distribution of genetically modified cells and/or viral particles in GT approaches and the kinetics of enzyme distribution in both ERT and GT approaches (Lattanzi et al., 2010, 2014; Meneghini et al., 2016; Gigliobianco et al., 2019; Ohashi, 2019).
Patient-specific NSCs are relevant models to address the neurodevelopmental defects that could anticipate the disease onset and/or exacerbate disease progression in LSD patients (Figure 1). Progressive accumulation of undegraded substrates, alteration of endosomal-lysosomal and autophagic pathways, defective calcium homeostasis, and endoplasmic reticulum (ER)-mediated stress have been associated with impaired neural commitment, reduced self-renewal and differentiation potential, altered astroglial-neuronal interactions and synaptic transmission in hiPS-NSCs and differentiated progeny derived from LSD patients (Lojewski et al., 2014; Canals et al., 2015; Son et al., 2015; Bayo-Puxan et al., 2018; Frati et al., 2018; Kobolak et al., 2019; Matsushita et al., 2019). These neurodevelopmental defects are only partially recapitulated by LSD mouse models, due to the less vulnerability to substrate accumulations during fetal brain development and/or the presence of compensatory mechanisms decreasing the neurotoxic effects.
Studies on hiPS-NSCs suggested that treatments counteracting primary and secondary biochemical defects also exert independent pro-neurogenic effects (Table 1). Besides the effects of valproic acid (VPA) on Liver X receptor (LXR) β-dependent cholesterol metabolism (Sung et al., 2017) and on the folding and trafficking of mutated NPC1 protein (Subramanian et al., 2020), the pro-neurogenic effects of VPA (Hsieh et al., 2004; Yu et al., 2009; Vukicevic et al., 2015) are potentially contributing to rescue the defective self-renewal and neurogenic potential in NPC NSCs (Sung et al., 2017). Additionally, premature cell death in NPC neurons caused by impaired cholesterol homeostasis is rescued by restoring NSC biological processes regulated by calcium and WNT pathways through treatment with GSK3- or calcium-inhibitors (Hsieh et al., 2004; Efthymiou et al., 2015).
Most of these studies have been performed using 2D in vitro cultures, either enriched in a specific cell type or using mixed differentiated populations. While this is advantageous in defining cell-autonomous pathological mechanisms or neurodevelopmental defects affecting specific neuronal and/or glial subpopulations, this system does not faithfully mimic the high degree of morphological and functional complexity of the human brain. Recent advances in stem cell technologies may address and overcome these limitations through the generation of iPSC-based self-assembled 3D cultures (brain/cerebral organoids) composed of neural stem/progenitor cells, neurons and glial cells organized in tissue-like structures recapitulating relevant steps of the human fetal brain development, including the spatiotemporal dynamicity of neurogenesis, the formation of regional neural circuitries, and the integration of glial cells into neural networks (Di Lullo and Kriegstein, 2017; Quadrato and Arlotta, 2017). The first protocols developed to generate brain/cerebral organoids relied on the spontaneous capability of hiPSCs to differentiate in self-organized neuroepithelial structures giving rise to different brain regions (e.g., forebrain, midbrain and hindbrain, retina, choroid plexus) (Lancaster and Knoblich, 2014). The few studies reporting early neurodevelopmental defects in hiPSC-derived cerebral organoids of LSD patients are based on this “spontaneous” and “unguided” method. The use of this system in association to OMIC techniques (e.g., whole-transcriptome and metabolomics analyses) allowed the identification of early disease-specific signatures affecting CNS development, corticogenesis, synaptogenesis and neurotransmission in NCL-derived brain organoids, suggesting that disease-associated changes occur before disease onset (Gomez-Giro et al., 2019). 3D organoid models derived from Hexosaminidase B (HEXB) deficient hiPSCs or GLB1 knockout hiPSCs showed a progressive accumulation of GM2 and GM1 gangliosides during neurodevelopment (Allende et al., 2018; Latour et al., 2019). In particular, organoids derived from Sandhoff patients showed not only an impairment in pathways regulating CNS development and neuronal differentiation, but also increased cellular proliferation and organoid size (Allende et al., 2018). Interestingly, these human 3D brain models were successfully used to evaluate the efficacy of AAV-mediated GT (Table 1) (Allende et al., 2018; Latour et al., 2019), being not only a suitable model to investigate neurodevelopmental defects in a complex multicellular system mimicking the human brain architecture, but also a potential intermediate step between animal models and patients to evaluate vector tropism in a multicellular complex system mimicking human brain (Figure 1).
Despite the relevance of data collected in these studies, single-cell transcriptomic studies show that organoids generated using these protocols (from the same hiPSC lines or different donors) are characterized by high variability in cell and brain-region composition (Kanton et al., 2019). The recent development of “guided” methods based on small molecules and growth factors instructing the hiPSC differentiation processes allowed the generation cerebral organoids with reduced intra- and inter-sample variability (Qian et al., 2019; Velasco et al., 2019). These protocols are more suitable for disease modeling studies, in particular in the investigation of the dynamic metabolic processes affected in LSDs. Future studies combining the use of these advanced 3D models generated from engineered isogenic lines and single cell technologies could dissect the contribution of different cell subpopulations in neurodevelopmental defects and neurodegenerative mechanisms affecting LSD brains.
The majority of current 2D and 3D hiPSC-based models are designed to investigate pathological mechanisms in neurons. However, growing evidences show an important contribution of glial cells in LSD pathology. Indeed, the survival of astrocytes in 2D culture models of Hunter Syndrome is affected by the depletion of ER luminal Ca2+ storage and the increased cytoplasmic ion concentration induced by the accumulation of glycosaminoglycans (GAGs) during hiPS-NSC differentiation (Kobolak et al., 2019). Additionally, reduced protein kinase C-mediated phosphorylation of the intermediate filaments vimentin and GFAP leads to an astrogliosis-like phenotype in hiPSC-derived NPC astrocytes, suggesting that gliosis is a primary effect of the cholesterol accumulation within glial cells and not a secondary mechanism mediated by the neuronal loss in NPC (Peter et al., 2017). Finally, ARSA deficiency leads to impaired commitment/maturation of astrocytes and oligodendrocytes besides neurons in Metachromatic Leukodystrophy (MLD) (Frati et al., 2018). These data highlight the potential contribution of altered gliogenesis, astrogliosis and dysfunctional glia-neuronal interactions in LSD pathology which might precede or be concomitant to the appearance of pathological signs in the neuronal compartments. The development of protocols favoring the generation of astrocytes and mature myelinating oligodendrocytes in cerebral organoids (Madhavan et al., 2018; Marton et al., 2019) will improve the study of the neuron-glia interactions in neurodegenerative processes (Frati et al., 2018; Kobolak et al., 2019) and the definition of pathological mechanisms regulating oligodendrocyte degeneration in demyelinating LSDs (Frati et al., 2018).
Additionally, microglia and infiltrating macrophages are important players in the pathology and treatment of LSDs. Recent studies in GD, NPC1 and Sandhoff disease mice suggested that accumulation of macromolecules in the lysosomes of neurons results either directly or indirectly in the activation of neighboring microglia which generate a neuroinflammatory response contributing to the propagation of neuronal damage in affected brains (Cho et al., 2019). On the contrary, the role of infiltrating monocyte-derived macrophages is still controversial, with reports showing their contribution to the pathology in Sandhoff mice (Wu and Proia, 2004; Kyrkanides et al., 2008) and a recent study demonstrating the absence of infiltrating macrophages in the brain of GD, NPC1 and Sandhoff mice. In GT settings based on autologous transplantation of LV engineered hematopoietic stem cells, busulfan treatment induces myeloablation and the permeabilization of blood-brain barrier, thus favoring microglia reconstitution and infiltration of macrophages derived from myeloid cells over-expressing the lacking enzyme, which enable enzymatic cross-correction of deficient CNS cells (Biffi et al., 2013; Sessa et al., 2016). Therefore, human LSD models based on 3D human tri- and tetra-culture and brain-on-a-chip platforms integrating neuro-glia-vascular-immune system (Sharma et al., 2020) could be intriguing models to dissect the contribution of microglia and infiltrating macrophages in disease onset and progression, and optimize the molecular mechanisms regulating enzymatic cross-correction mediated by myeloid cells.
Since pioneer studies in rodent and human NSCs (Reynolds et al., 1992; Gritti et al., 1996; Vescovi et al., 1999), advances in methodology and protocols have allowed to efficiently isolate and expand somatic NSCs from fetal and adult tissues (Conti and Cattaneo, 2010) while preserving their self-renewal and multipotency capabilities. These cells represent an important source to test innovative cell-based approaches in pre-clinical studies and, ultimately, in patients.
Pre-clinical studies in animal models demonstrated efficient NSC engraftment, migration toward the damage areas and persistence in the perivascular “niches” upon intracerebral transplantation in neonatal and adult rodents (Martino and Pluchino, 2006; Bacigaluppi et al., 2009) and non-human primates (Pluchino et al., 2009; Kutikov et al., 2019). Increased evidences suggest that neurodegenerative disorders are associated with multicellular dysfunctions (Olivera-Bravo et al., 2016; Gleichman and Carmichael, 2020; Greenhalgh et al., 2020; Rodriguez-Gomez et al., 2020) that might be only partially rescued by transplantation of committed neuronal and glial progenitors or specific neuronal subpopulations. The “bystander effects” mediated by neurotrophic, neuroprotective, and immunomodulatory factors released by NSCs (Park et al., 2002; Lu et al., 2003; Pluchino et al., 2005; Richardson et al., 2005), and the NSC potentiality for cell replacement through the generation of both neuronal and glial progeny upon transplantation (Lee et al., 2014b) might potentially show improved clinical outcomes as compared to transplantation of committed progenitors or neurons. These features have been exploited for the treatment of acute and chronic neurodegenerative disorders, such as stroke (Bacigaluppi et al., 2016), Alzheimer disease (Ager et al., 2015), Parkinson disease (PD) (Lee et al., 2014b), Amyotrophic Lateral Sclerosis (ALS) (Xu et al., 2006; Park et al., 2009), and Multiple Sclerosis (MS) (Pluchino et al., 2009).
The development of consistent GMP-scale procedures for NSC isolation/enrichment (i.e., CD133 membrane-bound sorting) (Uchida et al., 2000), expansion and cryopreservation (Crook and Tomaskovic-Crook, 2017) paved the way to the clinical translation of NSC-based cell therapies for the treatment of neurodegenerative disorders. Phases I/II clinical trials based on the transplantation of human NSCs of fetal origin (human fetal NSCs, hfNSCs) have been authorized to treat ALS (NTC01640067; Mazzini et al., 2015), MS (NCT03282760, NCT03269071), PD (NCT03128450), Cerebral Palsy (NCT03005249), Ischemic Stroke (NCT03296618), Spinal cord injury (NCT02163876, NCT01725880, NCT01321333), infantile and late infantile Neuronal Ceroid Lipofuscinosis (Selden et al., 2013, NCT00337636, NCT01238315), and Pelizaeus–Merzbacher disease (NCT01005004, NCT01391637). These clinical trials documented the tolerability of the surgical procedures, the engraftment and persistence of transplanted NSCs and some evidence of stabilization of the clinical phenotypes (Selden et al., 2013; Mazzini et al., 2015; Tang et al., 2017). Despite these promising results, hfNSC-based cell therapy presents some drawbacks, including the requirement of a high number of donor cells that could be difficult to expand in large-scale GMP production, and immunosuppressive regiments to avoid the immune rejection of transplanted cells and graft-vs-host disease.
Some of these caveats might be overcome with the use of pluripotent-derived NSC. In fact, pluripotent stem cells could be: (i) isolated from human embryos (e.g., embryonic stem cells, ESC) or generated through highly efficient transgene-free reprogramming (e.g., hiPSC) from easily accessible cell sources (Haase et al., 2017); (ii) easily genetically modified through efficient and safe LV-mediated gene addition strategies (Doerr et al., 2015; Vanhee and Vandekerckhove, 2016; Meneghini et al., 2017) or site-specific genome-editing (Merkert et al., 2017); (iii) clonally expanded and differentiated for large-scale GMP production of transplantable cells (Zweigerdt et al., 2011) (Figure 1).
Several pluripotent stem cell-based clinical trials are ongoing or recruiting patients (Martin, 2017). ESCs-based therapies have been proposed for the treatment of PD (NCT03119636), ALS (NCT03482050) or Dry Age-related Macular Degeneration (NCT03046407, NCT03167203, NCT02590692). The prospective clinical application of iPSC-derived cells can overcome ethical and immunological issues related to the transplantation of ESC-derived cells. Retinal pigmented epithelium (RPE) cells derived from both autologous and human leukocyte antigen (HLA)-matched allogeneic hiPSCs are currently applied for the treatment of Macular Degeneration patients in Japan (Mandai et al., 2017) and in the United States. In 2018, a patient affected by PD has been transplanted with 2.4 million dopaminergic neuronal progenitors derived from allogenic hiPSC (Cyranoski, 2018) and a first clinical trial based on hiPS-NSCs started in 2019 (NCT03815071).
Prospective NSC-based cell therapy approaches for LSDs have to face an additional layer of complication, i.e., the coupling with GT strategies to provide the functional protein in the affected cells. In addition, pre-clinical studies and clinical observations highlight the need of supraphysiological expression of the deficient enzyme in transplanted cells to achieve therapeutic levels of functional enzyme in affected tissues, specifically in CNS (Gritti, 2011; Biffi, 2016; Penati et al., 2017) (Table 2). Human iPS-NSC-based gene and cell therapies could be exploited for the treatment of LSDs with prevalent CNS pathology [e.g., Batten disease (Jankowiak et al., 2015), and GM2 gangliosidoses (Lacorazza et al., 1996)] or in combined strategies with autologous transplantation of enzyme-overexpressing hematopoietic stem cells (HSC) to treat LSDs with central and peripheral neuropathies and affected visceral organs, i.e., GLD (Ricca et al., 2015).

Table 2. Summary of pre-clinical cell therapy approaches based on human and murine neural stem/progenitor cells for the treatment of neurodegenerative LSDs.
A proof-of-principle of the potential application of hiPS-NSCs in autologous ex vivo GT protocols, have been recently provided in mouse models of Sly disease and MLD. Bilateral striatal injections of hiPS-derived neural stem/progenitor cells overexpressing β-glucuronidase reduced neuroinflammation and microglia activation in the surrounding tissues, already at 4 weeks post-transplantation, suggesting that cross-correction mechanisms and bystander effects can cooperate in reducing neuroinflammation in LSD affected brains (Griffin et al., 2015). Intracerebral transplantation of LV-transduced MLD hiPS-NSC expressing supraphysiological ARSA levels into neonatal and adult immunodeficient MLD mice provided robust and long-lasting enzymatic supply in the whole CNS (Meneghini et al., 2017). Of note, reduction of the pathological sulfatide storage has been achieved also by transplanting hiPS-NSCs with a relatively low number of copies of integrated vector, due to their capabilities of repopulating the entire brain, in particular in neonatal settings, and of secreting therapeutically relevant levels of enzyme in the cerebrospinal fluid, thus inducing cross-correction in CNS regions far from the injection sites (Meneghini et al., 2017).
The stage of neural commitment of transplanted cells seems to be particularly relevant in cell therapy approaches because of its impact on their multipotent and migration capabilities. In the context of widespread demyelinating disorders, like MLD, transplantation of ARSA-overexpressing neuroepithelial (NEP) cells, with limited migration and myelinogenic potentials resulted in reduction of the sulfatide storage only in regions closed to the injection sites (Doerr et al., 2015). Conversely, hiPS-NSC sharing molecular, phenotypic and functional identity with human fetal NSCs showed an enhanced rostro-caudal distribution upon transplantation, in particular in the white matter areas, and the capability to differentiate in myelinating oligodendrocytes, thus potentially combining multiple therapeutic effects, i.e., increased production of ARSA from glial cells, widespread distribution of enzyme along CNS, and cell replacement (Meneghini et al., 2017).
Despite the justified enthusiasm, hiPS-NSC-based approaches have to face several challenges before reaching full clinical exploitation. The production of patient-specific hiPS-NSCs for personalized medicine is costly and time-consuming. The generation of HLA-typed hiPSC banks derived from healthy donors (50–150 HLA-typed donors could cover almost the 90% of the United Kingdom and Japan populations, Taylor et al., 2012) could overcome these limitations, favoring the broad application of hiPS-NSC-based cell therapy for the treatment of neurodegenerative disorders, including diseases characterized by a rapid progression, such as LSDs.
A major concern related to the application of hiPS-NSCs is their safety profile. Indeed, the potential spontaneous reactivation of master regulators of pluripotency in engrafted cells (Choi et al., 2014; Nori et al., 2015) or the transplantation of partially differentiated cells could give rise to the appearance of brain cancers or disrupt pre-existing neuronal circuits. In this perspective, the recent development of hiPSC lines with drug-inducible suicide genes (Caspase9-FKBPF36V fusion protein or herpes simplex virus-derived thymidine kinase) inserted immediately downstream to hiPSC-specific genes (e.g., NANOG) or constitutively expressed genes (e.g., ACTB) could provide methods to deplete teratoma-forming cells prior to transplantation or eliminate the entire populations of engrafted cells in presence of adverse events (Martin et al., 2020). To increase the quality of the transplanted hiPSC-based cell products, the validation of the transcriptional and epigenetic landscapes of hiPS-NSCs in comparison to clinically relevant human fetal NSCs could be important to ensure the silencing of genes and enhancers associated with pluripotency and tumorigenicity and to verify the maturation stages of transplanted cell populations. Additionally, single cell analyses of hiPS-NSC populations aimed to identify membrane-bound cell-specific markers could implement the prospective isolation of “pure” hiPS-NSCs or neuronal and glial progenitors with peculiar features (i.e., anti-inflammatory and neuroprotective effects in NSCs, and enhanced neurogenic or gliogenic potential in progenitors) that could be exploited to improve the recovery of disease-specific damages in neurodegenerative LSDs. The application of novel multi-omics technologies and editing platforms optimizing the production of hiPS-NSCs with a “bona fide neural transcriptional signature” coupled to in vitro and in vivo functional studies seems to be mandatory to make reliable the translation of hiPS-NSC transplantation in clinics.
Several neurodegenerative LSDs have been modeled by using hiPSC-derived NSCs and differentiated progeny, allowing the definition of cell type-specific pathological pathways downstream the primary biochemical defects (Borger et al., 2017; De Filippis et al., 2017; Zunke and Mazzulli, 2019). Early neurodevelopmental defects and impaired cellular crosstalk within the neuro-glia and neuro-immune systems seem to play a crucial role in the onset and development of CNS pathology in several LSDs. The collection of patient-specific hiPSCs in cell banks, the standardization of neural differentiation protocols, and the exploitation of novel 2D and 3D culture models will improve our basic knowledge on CNS pathology, favoring the identification of novel targets and the definition of the timing and efficacy of current and future therapeutic approaches.
The implementation and standardization of methodologies in basic research is parallel to the development of GMP-scale procedures to produce hiPS-NSCs with reliable NSC signatures and safety profile to be used in gene and cell therapy approaches. The possibility to produce limited numbers of iPSCs from HLA-matched donors or, ultimately, a genetically modified cell product derived from a single “universal donor” (Riolobos et al., 2013; Mattapally et al., 2018; Lanza et al., 2019) that could be easily expanded and cryopreserved with low manufacturing costs would represent a breakthrough in the field of cell therapy for neurodegenerative and demyelinating disease of different origin, including LSDs. However, crucial safety issues must be considered in the perspective of clinical development and application of these strategies in large cohorts of patients. Despite pre-clinical studies demonstrated the absence of engrafted cells expressing pluripotency markers or oncogenic events in mice analyzed 3- and 6-month after being transplanted with hiPS-NSCs (Doerr et al., 2015; Griffin et al., 2015; Meneghini et al., 2017), accurate long-term safety and efficacy assessments (i.e., toxicology, engraftment, long-term persistence) with GMP-scale cell products are required in small and large animals before moving to clinical development. Importantly, guidelines have to be discussed and defined in order to standardize protocols for hiPS-NSC production and transduction, and to define the efficient cell dosage and route of transplantation based on the target area and on the onset, progression and severity of the treated neurodegenerative LSD.
ML and VM wrote the manuscript. VM and AG revised the manuscript. VM conceived the review. All the authors contributed to the article and approved the submitted version.
This work was supported by grants from Fondazione Telethon (Tiget Core Grant 2016 2021 and Grant D2) and the Italian Ministry of Health (RF-2016-02362404) to AG., and from the European Union’s Horizon 2020 Research and Innovation Programme (Marie Skłodowska-Curie grant agreement no. 895111) to VM.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Aflaki, E., Moaven, N., Borger, D. K., Lopez, G., Westbroek, W., Chae, J. J., et al. (2016). Lysosomal storage and impaired autophagy lead to inflammasome activation in Gaucher macrophages. Aging Cell 15, 77–88. doi: 10.1111/acel.12409
Ager, R. R., Davis, J. L., Agazaryan, A., Benavente, F., Poon, W. W., LaFerla, F. M., et al. (2015). Human neural stem cells improve cognition and promote synaptic growth in two complementary transgenic models of Alzheimer’s disease and neuronal loss. Hippocampus 25, 813–826. doi: 10.1002/hipo.22405
Aguisanda, F., Yeh, C. D., Chen, C. Z., Li, R., Beers, J., Zou, J., et al. (2017). Neural stem cells for disease modeling of Wolman disease and evaluation of therapeutics. Orphanet. J. Rare Dis. 12:120. doi: 10.1186/s13023-017-0670-9
Ahmad, I., Hunter, R. E., Flax, J. D., Snyder, E. Y., and Erickson, R. P. (2007). Neural stem cell implantation extends life in Niemann-Pick C1 mice. J. Appl. Genet. 48, 269–272. doi: 10.1007/BF03195222
Al-Jasmi, F. A., Tawfig, N., Berniah, A., Ali, B. R., Taleb, M., Hertecant, J. L., et al. (2013). Prevalence and novel mutations of lysosomal storage disorders in united arab emirates : LSD in UAE. JIMD Rep. 10, 1–9. doi: 10.1007/8904_2012_182
Allende, M. L., Cook, E. K., Larman, B. C., Nugent, A., Brady, J. M., Golebiowski, D., et al. (2018). Cerebral organoids derived from Sandhoff disease-induced pluripotent stem cells exhibit impaired neurodifferentiation. J. Lipid Res. 59, 550–563. doi: 10.1194/jlr.M081323jlr.M081323
Applegarth, D. A., Toone, J. R., and Lowry, R. B. (2000). Incidence of inborn errors of metabolism in British Columbia, 1969-1996. Pediatrics 105:e10. doi: 10.1542/peds.105.1.e10
Awad, O., Sarkar, C., Panicker, L. M., Miller, D., Zeng, X., Sgambato, J. A., et al. (2015). Altered TFEB-mediated lysosomal biogenesis in Gaucher disease iPSC-derived neuronal cells. Hum. Mol. Genet. 24, 5775–5788. doi: 10.1093/hmg/ddv297ddv297
Bacigaluppi, M., Pluchino, S., Peruzzotti-Jametti, L., Kilic, E., Kilic, U., Salani, G., et al. (2009). Delayed post-ischaemic neuroprotection following systemic neural stem cell transplantation involves multiple mechanisms. Brain 132(Pt 8), 2239–2251. doi: 10.1093/brain/awp174
Bacigaluppi, M., Russo, G. L., Peruzzotti-Jametti, L., Rossi, S., Sandrone, S., Butti, E., et al. (2016). Neural stem cell transplantation induces stroke recovery by upregulating glutamate transporter GLT-1 in astrocytes. J. Neurosci. 36, 10529–10544. doi: 10.1523/jneurosci.1643-16.2016
Ballabio, A., and Gieselmann, V. (2009). Lysosomal disorders: from storage to cellular damage. Biochim. Biophys. Acta 1793, 684–696. doi: 10.1016/j.bbamcr.2008.12.001
Bayo-Puxan, N., Terrasso, A. P., Creyssels, S., Simao, D., Begon-Pescia, C., Lavigne, M., et al. (2018). Lysosomal and network alterations in human mucopolysaccharidosis type VII iPSC-derived neurons. Sci. Rep. 8:16644. doi: 10.1038/s41598-018-34523-310
Biffi, A. (2016). Gene therapy for lysosomal storage disorders: a good start. Hum. Mol. Genet. 25, R65–R75. doi: 10.1093/hmg/ddv457ddv457
Biffi, A., Montini, E., Lorioli, L., Cesani, M., Fumagalli, F., Plati, T., et al. (2013). Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 341:1233158. doi: 10.1126/science.1233158
Borger, D. K., McMahon, B., Roshan Lal, T., Serra-Vinardell, J., Aflaki, E., and Sidransky, E. (2017). Induced pluripotent stem cell models of lysosomal storage disorders. Dis. Model. Mech. 10, 691–704. doi: 10.1242/dmm.02900910/6/691
Buchet, D., Serguera, C., Zennou, V., Charneau, P., and Mallet, J. (2002). Long-term expression of β-glucuronidase by genetically modified human neural progenitor cells grafted into the mouse central nervous system. Mol. Cell Neurosci. 19, 389–401. doi: 10.1006/mcne.2001.1086
Canals, I., Soriano, J., Orlandi, J. G., Torrent, R., Richaud-Patin, Y., Jimenez-Delgado, S., et al. (2015). Activity and high-order effective connectivity alterations in sanfilippo c patient-specific neuronal networks. Stem Cell Rep. 5, 546–557. doi: 10.1016/j.stemcr.2015.08.016S2213-6711(15)00250-7
Chambers, S. M., Fasano, C. A., Papapetrou, E. P., Tomishima, M., Sadelain, M., and Studer, L. (2009). Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 27, 275–280. doi: 10.1038/nbt.1529
Cho, S. M., Vardi, A., Platt, N., and Futerman, A. H. (2019). Absence of infiltrating peripheral myeloid cells in the brains of mouse models of lysosomal storage disorders. J. Neurochem. 148, 625–638. doi: 10.1111/jnc.14483
Choi, H. W., Kim, J. S., Choi, S., Hong, Y. J., Kim, M. J., Seo, H. G., et al. (2014). Neural stem cells differentiated from iPS cells spontaneously regain pluripotency. Stem Cells 32, 2596–2604. doi: 10.1002/stem.1757
Clarke, D., Pearse, Y., Kan, S. H., Le, S. Q., Sanghez, V., Cooper, J. D., et al. (2018). Genetically corrected iPSC-derived neural stem cell grafts deliver enzyme replacement to affect CNS disease in sanfilippo B Mice. Mol. Ther. Methods Clin. Dev. 10, 113–127. doi: 10.1016/j.omtm.2018.06.005
Conti, L., and Cattaneo, E. (2010). Neural stem cell systems: physiological players or in vitro entities? Nat. Rev. Neurosci. 11, 176–187. doi: 10.1038/nrn2761
Conti, L., Pollard, S. M., Gorba, T., Reitano, E., Toselli, M., Biella, G., et al. (2005). Niche-independent symmetrical self-renewal of a mammalian tissue stem cell. PLoS Biol. 3:e283. doi: 10.1371/journal.pbio.0030283
Crook, J. M., and Tomaskovic-Crook, E. (2017). Culturing and cryobanking human neural stem cells. Methods Mol. Biol. 1590, 199–206. doi: 10.1007/978-1-4939-6921-0_15
Cyranoski, D. (2018). ‘Reprogrammed’ stem cells implanted into patient with Parkinson’s disease. Nature 563, 1–2. doi: 10.1038/d41586-018-07407-9
De Filippis, L., Zalfa, C., and Ferrari, D. (2017). Neural stem cells and human induced pluripotent stem cells to model rare CNS diseases. CNS Neurol. Disord. Drug Targets 16, 915–926.
Di Lullo, E., and Kriegstein, A. R. (2017). The use of brain organoids to investigate neural development and disease. Nat. Rev. Neurosci. 18, 573–584. doi: 10.1038/nrn.2017.107nrn.2017.107
Doerr, J., Bockenhoff, A., Ewald, B., Ladewig, J., Eckhardt, M., Gieselmann, V., et al. (2015). Arylsulfatase A overexpressing human iPSC-derived neural cells reduce CNS sulfatide storage in a mouse model of metachromatic leukodystrophy. Mol. Ther. 23, 1519–1531. doi: 10.1038/mt.2015.106
Efthymiou, A. G., Steiner, J., Pavan, W. J., Wincovitch, S., Larson, D. M., Porter, F. D., et al. (2015). Rescue of an in vitro neuron phenotype identified in Niemann-Pick disease, type C1 induced pluripotent stem cell-derived neurons by modulating the WNT pathway and calcium signaling. Stem Cells Transl. Med. 4, 230–238. doi: 10.5966/sctm.2014-0127sctm.2014-0127
Elkabetz, Y., Panagiotakos, G., Al Shamy, G., Socci, N. D., Tabar, V., and Studer, L. (2008). Human ES cell-derived neural rosettes reveal a functionally distinct early neural stem cell stage. Genes Dev. 22, 152–165. doi: 10.1101/gad.1616208
Frati, G., Luciani, M., Meneghini, V., De Cicco, S., Stahlman, M., Blomqvist, M., et al. (2018). Human iPSC-based models highlight defective glial and neuronal differentiation from neural progenitor cells in metachromatic leukodystrophy. Cell Death Dis. 9:698.
Germain, P. L., and Testa, G. (2017). Taming human genetic variability: transcriptomic meta-analysis guides the experimental design and interpretation of iPSC-based disease modeling. Stem Cell Rep. 8, 1784–1796. doi: 10.1016/j.stemcr.2017.05.012
Gigliobianco, M. R., Di Martino, P., Deng, S., Casadidio, C., and Censi, R. (2019). New advanced strategies for the treatment of lysosomal diseases affecting the central nervous system. Curr. Pharm. Des. 25, 1933–1950. doi: 10.2174/1381612825666190708213159
Givogri, M. I., Bottai, D., Zhu, H. L., Fasano, S., Lamorte, G., Brambilla, R., et al. (2008). Multipotential neural precursors transplanted into the metachromatic leukodystrophy brain fail to generate oligodendrocytes but contribute to limit brain dysfunction. Dev. Neurosci. 30, 340–357. doi: 10.1159/000150127
Glaser, A., Hammerl, F., Graler, M. H., Coldewey, S. M., Volkner, C., Frech, M. J., et al. (2020). Identification of brain-specific treatment effects in NPC1 disease by focusing on cellular and molecular changes of sphingosine-1-phosphate metabolism. Int. J. Mol. Sci. 21:502. doi: 10.3390/ijms21124502
Gleichman, A. J., and Carmichael, S. T. (2020). Glia in neurodegeneration: drivers of disease or along for the ride? Neurobiol. Dis. 142:104957. doi: 10.1016/j.nbd.2020.104957
Gomez-Giro, G., Arias-Fuenzalida, J., Jarazo, J., Zeuschner, D., Ali, M., Possemis, N., et al. (2019). Synapse alterations precede neuronal damage and storage pathology in a human cerebral organoid model of CLN3-juvenile neuronal ceroid lipofuscinosis. Acta Neuropathol. Commun. 7:222. doi: 10.1186/s40478-019-0871-710.1186/s40478-019-0871-7
Greenhalgh, A. D., David, S., and Bennett, F. C. (2020). Immune cell regulation of glia during CNS injury and disease. Nat. Rev. Neurosci. 21, 139–152. doi: 10.1038/s41583-020-0263-9
Griffin, T. A., Anderson, H. C., and Wolfe, J. H. (2015). Ex vivo gene therapy using patient iPSC-derived NSCs reverses pathology in the brain of a homologous mouse model. Stem Cell Rep. 4, 835–846. doi: 10.1016/j.stemcr.2015.02.022
Gritti, A. (2011). Gene therapy for lysosomal storage disorders. Expert. Opin. Biol. Ther. 11, 1153–1167. doi: 10.1517/14712598.2011.582036
Gritti, A., Parati, E. A., Cova, L., Frolichsthal, P., Galli, R., Wanke, E., et al. (1996). Multipotential stem cells from the adult mouse brain proliferate and self-renew in response to basic fibroblast growth factor. J. Neurosci. 16, 1091–1100. doi: 10.1523/jneurosci.16-03-01091.1996
Haase, A., Gohring, G., and Martin, U. (2017). Generation of non-transgenic iPS cells from human cord blood CD34(+) cells under animal component-free conditions. Stem Cell Res. 21, 71–73. doi: 10.1016/j.scr.2017.03.022
Hastings, C., Vieira, C., Liu, B., Bascon, C., Gao, C., Wang, R. Y., et al. (2019). Expanded access with intravenous hydroxypropyl-beta-cyclodextrin to treat children and young adults with Niemann-Pick disease type C1: a case report analysis. Orphanet. J. Rare Dis. 14:228. doi: 10.1186/s13023-019-1207-1
Hsieh, J., Nakashima, K., Kuwabara, T., Mejia, E., and Gage, F. H. (2004). Histone deacetylase inhibition-mediated neuronal differentiation of multipotent adult neural progenitor cells. Proc. Natl. Acad. Sci. U.S.A. 101, 16659–16664. doi: 10.1073/pnas.0407643101
Hunt, J., Li, M., Zhao, X., and Bhattacharyya, A. (2019). Using human neural progenitor cell models to conduct large-scale drug screens for neurological and Psychiatric diseases. Methods Mol. Biol. 1942, 79–88. doi: 10.1007/978-1-4939-9080-1_7
Jankowiak, W., Kruszewski, K., Flachsbarth, K., Skevas, C., Richard, G., Ruther, K., et al. (2015). Sustained neural stem cell-based intraocular delivery of CNTF attenuates photoreceptor loss in the nclf mouse model of neuronal ceroid lipofuscinosis. PLoS One 10:e0127204. doi: 10.1371/journal.pone.0127204
Jeyakumar, M., Lee, J. P., Sibson, N. R., Lowe, J. P., Stuckey, D. J., Tester, K., et al. (2009). Neural stem cell transplantation benefits a monogenic neurometabolic disorder during the symptomatic phase of disease. Stem Cells 27, 2362–2370. doi: 10.1002/stem.163
Kanton, S., Boyle, M. J., He, Z., Santel, M., Weigert, A., Sanchis-Calleja, F., et al. (2019). Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nature 574, 418–422. doi: 10.1038/s41586-019-1654-9
Kawabata, K., Migita, M., Mochizuki, H., Miyake, K., Igarashi, T., Fukunaga, Y., et al. (2006). Ex vivo cell-mediated gene therapy for metachromatic leukodystrophy using neurospheres. Brain Res. 1094, 13–23. doi: 10.1016/j.brainres.2006.03.116
Kobolak, J., Molnar, K., Varga, E., Bock, I., Jezso, B., Teglasi, A., et al. (2019). Modelling the neuropathology of lysosomal storage disorders through disease-specific human induced pluripotent stem cells. Exp. Cell Res. 380, 216–233. doi: 10.1016/j.yexcr.2019.04.021
Koch, P., Opitz, T., Steinbeck, J. A., Ladewig, J., and Brustle, O. (2009). A rosette-type, self-renewing human ES cell-derived neural stem cell with potential for in vitro instruction and synaptic integration. Proc. Natl. Acad. Sci. U.S.A. 106, 3225–3230. doi: 10.1073/pnas.0808387106
Kutikov, A. B., Moore, S. W., Layer, R. T., Podell, P. E., Sridhar, N., Santamaria, A. J., et al. (2019). Method and apparatus for the automated delivery of continuous neural stem cell trails into the spinal cord of small and large animals. Neurosurgery 85, 560–573. doi: 10.1093/neuros/nyy3795086664
Kyrkanides, S., Miller, A. W., Miller, J. N., Tallents, R. H., Brouxhon, S. M., Olschowka, M. E., et al. (2008). Peripheral blood mononuclear cell infiltration and neuroinflammation in the HexB-/- mouse model of neurodegeneration. J. Neuroimmunol. 203, 50–57. doi: 10.1016/j.jneuroim.2008.06.024
Lacorazza, H. D., Flax, J. D., Snyder, E. Y., and Jendoubi, M. (1996). Expression of human beta-hexosaminidase alpha-subunit gene (the gene defect of Tay-Sachs disease) in mouse brains upon engraftment of transduced progenitor cells. Nat. Med. 2, 424–429. doi: 10.1038/nm0496-424
Lam, M., Sanosaka, T., Lundin, A., Imaizumi, K., Etal, D., Karlsson, F. H., et al. (2019). Single-cell study of neural stem cells derived from human iPSCs reveals distinct progenitor populations with neurogenic and gliogenic potential. Genes Cells 24, 836–847. doi: 10.1111/gtc.12731
Lancaster, M. A., and Knoblich, J. A. (2014). Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 9, 2329–2340. doi: 10.1038/nprot.2014.158
Lanza, R., Russell, D. W., and Nagy, A. (2019). Engineering universal cells that evade immune detection. Nat. Rev. Immunol. 19, 723–733. doi: 10.1038/s41577-019-0200-1
Latour, Y. L., Yoon, R., Thomas, S. E., Grant, C., Li, C., Sena-Esteves, M., et al. (2019). Human GLB1 knockout cerebral organoids: a model system for testing AAV9-mediated GLB1 gene therapy for reducing GM1 ganglioside storage in GM1 gangliosidosis. Mol. Genet. Metab. Rep. 21:100513. doi: 10.1016/j.ymgmr.2019.100513
Lattanzi, A., Neri, M., Maderna, C., di Girolamo, I., Martino, S., Orlacchio, A., et al. (2010). Widespread enzymatic correction of CNS tissues by a single intracerebral injection of therapeutic lentiviral vector in leukodystrophy mouse models. Hum. Mol. Genet. 19, 2208–2227. doi: 10.1093/hmg/ddq099ddq099
Lattanzi, A., Salvagno, C., Maderna, C., Benedicenti, F., Morena, F., Kulik, W., et al. (2014). Therapeutic benefit of lentiviral-mediated neonatal intracerebral gene therapy in a mouse model of globoid cell leukodystrophy. Hum. Mol. Genet. 23, 3250–3268. doi: 10.1093/hmg/ddu034ddu034
Lee, H., Lee, J. K., Park, M. H., Hong, Y. R., Marti, H. H., Kim, H., et al. (2014a). Pathological roles of the VEGF/SphK pathway in Niemann-Pick type C neurons. Nat. Commun. 5:5514. doi: 10.1038/ncomms6514ncomms6514
Lee, H., Yun, S., Kim, I. S., Lee, I. S., Shin, J. E., Park, S. C., et al. (2014b). Human fetal brain-derived neural stem/progenitor cells grafted into the adult epileptic brain restrain seizures in rat models of temporal lobe epilepsy. PLoS One 9:e104092. doi: 10.1371/journal.pone.0104092
Lojewski, X., Staropoli, J. F., Biswas-Legrand, S., Simas, A. M., Haliw, L., Selig, M. K., et al. (2014). Human iPSC models of neuronal ceroid lipofuscinosis capture distinct effects of TPP1 and CLN3 mutations on the endocytic pathway. Hum. Mol. Genet. 23, 2005–2022. doi: 10.1093/hmg/ddt596ddt596
Long, Y., Xu, M., Li, R., Dai, S., Beers, J., Chen, G., et al. (2016). Induced pluripotent stem cells for disease modeling and evaluation of therapeutics for niemann-Pick disease Type A. Stem Cells Transl. Med. 5, 1644–1655. doi: 10.5966/sctm.2015-0373
Lu, P., Jones, L. L., Snyder, E. Y., and Tuszynski, M. H. (2003). Neural stem cells constitutively secrete neurotrophic factors and promote extensive host axonal growth after spinal cord injury. Exp. Neurol. 181, 115–129. doi: 10.1016/s0014-4886(03)00037-2
Ma, T., Xie, M., Laurent, T., and Ding, S. (2013). Progress in the reprogramming of somatic cells. Circ. Res. 112, 562–574. doi: 10.1161/CIRCRESAHA.111.249235
Madhavan, M., Nevin, Z. S., Shick, H. E., Garrison, E., Clarkson-Paredes, C., Karl, M., et al. (2018). Induction of myelinating oligodendrocytes in human cortical spheroids. Nat. Methods 15, 700–706. doi: 10.1038/s41592-018-0081-4
Madra, M., and Sturley, S. L. (2010). Niemann-Pick type C pathogenesis and treatment: from statins to sugars. Clin. Lipidol. 5, 387–395. doi: 10.2217/clp.10.19
Maetzel, D., Sarkar, S., Wang, H., Abi-Mosleh, L., Xu, P., Cheng, A. W., et al. (2014). Genetic and chemical correction of cholesterol accumulation and impaired autophagy in hepatic and neural cells derived from Niemann-Pick Type C patient-specific iPS cells. Stem Cell Rep. 2, 866–880. doi: 10.1016/j.stemcr.2014.03.014
Mandai, M., Kurimoto, Y., and Takahashi, M. (2017). Autologous induced stem-cell-derived retinal cells for macular degeneration. N. Engl. J. Med. 377, 792–793. doi: 10.1056/NEJMc1706274
Manthe, R. L., Rappaport, J. A., Long, Y., Solomon, M., Veluvolu, V., Hildreth, M., et al. (2019). Delta-tocopherol effect on endocytosis and its combination with enzyme replacement therapy for lysosomal disorders: a new type of drug interaction? J. Pharmacol. Exp. Ther. 370, 823–833. doi: 10.1124/jpet.119.257345
Marin, T., Contreras, P., Castro, J. F., Chamorro, D., Balboa, E., Bosch-Morato, M., et al. (2014). Vitamin E dietary supplementation improves neurological symptoms and decreases c-Abl/p73 activation in Niemann-Pick C mice. Nutrients 6, 3000–3017. doi: 10.3390/nu6083000
Martin, R. M., Fowler, J. L., Cromer, M. K., Lesch, B. J., Ponce, E., Uchida, N., et al. (2020). Improving the safety of human pluripotent stem cell therapies using genome-edited orthogonal safeguards. Nat. Commun. 11:2713. doi: 10.1038/s41467-020-16455-7
Martin, U. (2017). Therapeutic application of pluripotent stem cells: challenges and risks. Front. Med. 4:229. doi: 10.3389/fmed.2017.00229
Martino, G., and Pluchino, S. (2006). The therapeutic potential of neural stem cells. Nat. Rev. Neurosci. 7, 395–406. doi: 10.1038/nrn1908
Marton, R. M., Miura, Y., Sloan, S. A., Li, Q., Revah, O., Levy, R. J., et al. (2019). Differentiation and maturation of oligodendrocytes in human three-dimensional neural cultures. Nat. Neurosci. 22, 484–491. doi: 10.1038/s41593-018-0316-910.1038/s41593-018-0316-9
Matsuo, M., Togawa, M., Hirabaru, K., Mochinaga, S., Narita, A., Adachi, M., et al. (2013). Effects of cyclodextrin in two patients with Niemann-Pick Type C disease. Mol. Genet. Metab. 108, 76–81.
Matsushita, K., Numakawa, T., Odaka, H., Kajihara, R., Soga, M., Ozasa, S., et al. (2019). Presynaptic dysfunction in neurons derived from Tay-Sachs iPSCs. Neuroscience 414, 128–140. doi: 10.1016/j.neuroscience.2019.06.026
Mattapally, S., Pawlik, K. M., Fast, V. G., Zumaquero, E., Lund, F. E., Randall, T. D., et al. (2018). Human leukocyte antigen class I and II knockout human induced pluripotent stem cell-derived cells: universal donor for cell therapy. J. Am. Heart Assoc. 7:e010239. doi: 10.1161/JAHA.118.010239
Mazzini, L., Gelati, M., Profico, D. C., Sgaravizzi, G., Projetti Pensi, M., Muzi, G., et al. (2015). Human neural stem cell transplantation in ALS: initial results from a phase I trial. J. Transl. Med. 13:17. doi: 10.1186/s12967-014-0371-2
Meikle, P. J., Hopwood, J. J., Clague, A. E., and Carey, W. F. (1999). Prevalence of lysosomal storage disorders. JAMA 281, 249–254. doi: 10.1001/jama.281.3.249
Meneghini, V., Frati, G., Sala, D., De Cicco, S., Luciani, M., Cavazzin, C., et al. (2017). Generation of human induced pluripotent stem cell-derived bona fide neural stem cells for Ex vivo gene therapy of metachromatic leukodystrophy. Stem Cells Transl. Med. 6, 352–368. doi: 10.5966/sctm.2015-0414
Meneghini, V., Lattanzi, A., Tiradani, L., Bravo, G., Morena, F., Sanvito, F., et al. (2016). Pervasive supply of therapeutic lysosomal enzymes in the CNS of normal and Krabbe-affected non-human primates by intracerebral lentiviral gene therapy. EMBO Mol. Med. 8, 489–510. doi: 10.15252/emmm.201505850
Meng, X. L., Shen, J. S., Ohashi, T., Maeda, H., Kim, S. U., and Eto, Y. (2003). Brain transplantation of genetically engineered human neural stem cells globally corrects brain lesions in the mucopolysaccharidosis type VII mouse. J. Neurosci. Res. 74, 266–277. doi: 10.1002/jnr.10764
Merkert, S., Bednarski, C., Gohring, G., Cathomen, T., and Martin, U. (2017). Generation of a gene-corrected isogenic control iPSC line from cystic fibrosis patient-specific iPSCs homozygous for p.Phe508del mutation mediated by TALENs and ssODN. Stem Cell Res. 23, 95–97. doi: 10.1016/j.scr.2017.07.010
Napolitano, G., and Ballabio, A. (2016). TFEB at a glance. J. Cell Sci. 129, 2475–2481. doi: 10.1242/jcs.146365
Neri, M., Ricca, A., di Girolamo, I., Alcala’-Franco, B., Cavazzin, C., Orlacchio, A., et al. (2011). Neural stem cell gene therapy ameliorates pathology and function in a mouse model of globoid cell leukodystrophy. Stem Cells 29, 1559–1571. doi: 10.1002/stem.701
Nori, S., Okada, Y., Nishimura, S., Sasaki, T., Itakura, G., Kobayashi, Y., et al. (2015). Long-term safety issues of iPSC-based cell therapy in a spinal cord injury model: oncogenic transformation with epithelial-mesenchymal transition. Stem Cell Rep. 4, 360–373. doi: 10.1016/j.stemcr.2015.01.006
Ohashi, T. (2019). Gene therapy for lysosomal storage diseases and peroxisomal diseases. J. Hum. Genet. 64, 139–143. doi: 10.1038/s10038-018-0537-5
Olivera-Bravo, S., Isasi, E., Fernandez, A., Casanova, G., Rosillo, J. C., and Barbeito, L. (2016). Astrocyte dysfunction in developmental neurometabolic diseases. Adv. Exp. Med. Biol. 949, 227–243. doi: 10.1007/978-3-319-40764-7_11
Ory, D. S., Ottinger, E. A., Farhat, N. Y., King, K. A., Jiang, X., Weissfeld, L., et al. (2017). Intrathecal 2-hydroxypropyl-beta-cyclodextrin decreases neurological disease progression in Niemann-Pick disease, type C1: a non-randomised, open-label, phase 1-2 trial. Lancet 390, 1758–1768. doi: 10.1016/S0140-6736(17)31465-4
Park, K. I., Teng, Y. D., and Snyder, E. Y. (2002). The injured brain interacts reciprocally with neural stem cells supported by scaffolds to reconstitute lost tissue. Nat. Biotechnol. 20, 1111–1117. doi: 10.1038/nbt751
Park, S., Kim, H. T., Yun, S., Kim, I. S., Lee, J., Lee, I. S., et al. (2009). Growth factor-expressing human neural progenitor cell grafts protect motor neurons but do not ameliorate motor performance and survival in ALS mice. Exp. Mol. Med. 41, 487–500. doi: 10.3858/emm.2009.41.7.054
Patterson, M. C., Vecchio, D., Prady, H., Abel, L., and Wraith, J. E. (2007). Miglustat for treatment of Niemann-Pick C disease: a randomised controlled study. Lancet Neurol. 6, 765–772. doi: 10.1016/S1474-4422(07)70194-1
Penati, R., Fumagalli, F., Calbi, V., Bernardo, M. E., and Aiuti, A. (2017). Gene therapy for lysosomal storage disorders: recent advances for metachromatic leukodystrophy and mucopolysaccaridosis I. J. Inherit. Metab. Dis. 40, 543–554. doi: 10.1007/s10545-017-0052-410
Peter, F., Rost, S., Rolfs, A., and Frech, M. J. (2017). Activation of PKC triggers rescue of NPC1 patient specific iPSC derived glial cells from gliosis. Orphanet. J. Rare Dis. 12:145. doi: 10.1186/s13023-017-0697-y10
Pinto, R., Caseiro, C., Lemos, M., Lopes, L., Fontes, A., Ribeiro, H., et al. (2004). Prevalence of lysosomal storage diseases in Portugal. Eur. J. Hum. Genet. 12, 87–92. doi: 10.1038/sj.ejhg.52010445201044
Pipalia, N. H., Cosner, C. C., Huang, A., Chatterjee, A., Bourbon, P., Farley, N., et al. (2011). Histone deacetylase inhibitor treatment dramatically reduces cholesterol accumulation in Niemann-Pick type C1 mutant human fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 108, 5620–5625. doi: 10.1073/pnas.1014890108
Platt, F. M., Boland, B., and van der Spoel, A. C. (2012). The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J. Cell Biol. 199, 723–734. doi: 10.1083/jcb.201208152
Platt, F. M., d’Azzo, A., Davidson, B. L., Neufeld, E. F., and Tifft, C. J. (2018). Lysosomal storage diseases. Nat. Rev. Dis. Primers 4:27.
Pluchino, S., Gritti, A., Blezer, E., Amadio, S., Brambilla, E., Borsellino, G., et al. (2009). Human neural stem cells ameliorate autoimmune encephalomyelitis in non-human primates. Ann. Neurol. 66, 343–354. doi: 10.1002/ana.21745
Pluchino, S., Zanotti, L., Rossi, B., Brambilla, E., Ottoboni, L., Salani, G., et al. (2005). Neurosphere-derived multipotent precursors promote neuroprotection by an immunomodulatory mechanism. Nature 436, 266–271. doi: 10.1038/nature03889
Poorthuis, B. J., Wevers, R. A., Kleijer, W. J., Groener, J. E., de Jong, J. G., van Weely, S., et al. (1999). The frequency of lysosomal storage diseases in the Netherlands. Hum. Genet. 105, 151–156. doi: 10.1007/s004399900075
Poupetova, H., Ledvinova, J., Berna, L., Dvorakova, L., Kozich, V., and Elleder, M. (2010). The birth prevalence of lysosomal storage disorders in the Czech republic: comparison with data in different populations. J. Inherit. Metab. Dis. 33, 387–396. doi: 10.1007/s10545-010-9093-7
Puertollano, R., Ferguson, S. M., Brugarolas, J., and Ballabio, A. (2018). The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 37:8804. doi: 10.15252/embj.201798804
Qian, X., Song, H., and Ming, G. L. (2019). Brain organoids: advances, applications and challenges. Development 146:dev166074. doi: 10.1242/dev.166074
Quadrato, G., and Arlotta, P. (2017). Present and future of modeling human brain development in 3D organoids. Curr. Opin. Cell Biol. 49, 47–52. doi: 10.1016/j.ceb.2017.11.010
Rabenstein, M., Peter, F., Joost, S., Trilck, M., Rolfs, A., and Frech, M. J. (2017). Decreased calcium flux in Niemann-Pick type C1 patient-specific iPSC-derived neurons due to higher amount of calcium-impermeable AMPA receptors. Mol. Cell Neurosci. 83, 27–36. doi: 10.1016/j.mcn.2017.06.007
Reynolds, B. A., Tetzlaff, W., and Weiss, S. (1992). A multipotent EGF-responsive striatal embryonic progenitor cell produces neurons and astrocytes. J. Neurosci. 12, 4565–4574. doi: 10.1523/jneurosci.12-11-04565.1992
Ricca, A., Rufo, N., Ungari, S., Morena, F., Martino, S., Kulik, W., et al. (2015). Combined gene/cell therapies provide long-term and pervasive rescue of multiple pathological symptoms in a murine model of globoid cell leukodystrophy. Hum. Mol. Genet. 24, 3372–3389. doi: 10.1093/hmg/ddv086ddv086
Richardson, R. M., Broaddus, W. C., Holloway, K. L., and Fillmore, H. L. (2005). Grafts of adult subependymal zone neuronal progenitor cells rescue hemiparkinsonian behavioral decline. Brain Res. 1032, 11–22. doi: 10.1016/j.brainres.2004.10.043
Riolobos, L., Hirata, R. K., Turtle, C. J., Wang, P. R., Gornalusse, G. G., Zavajlevski, M., et al. (2013). HLA engineering of human pluripotent stem cells. Mol. Ther. 21, 1232–1241.
Rodriguez-Gomez, J. A., Kavanagh, E., Engskog-Vlachos, P., Engskog, M. K. R., Herrera, A. J., Espinosa-Oliva, A. M., et al. (2020). Microglia: agents of the CNS Pro-inflammatory response. Cells 9:1717. doi: 10.3390/cells9071717
Rosati, J., Ferrari, D., Altieri, F., Tardivo, S., Ricciolini, C., Fusilli, C., et al. (2018). Establishment of stable iPS-derived human neural stem cell lines suitable for cell therapies. Cell Death Dis. 9:937. doi: 10.1038/s41419-018-0990-2
Russo, D., Della Ragione, F., Rizzo, R., Sugiyama, E., Scalabri, F., Hori, K., et al. (2018). Glycosphingolipid metabolic reprogramming drives neural differentiation. EMBO J. 37:674. doi: 10.15252/embj.201797674
Schondorf, D. C., Aureli, M., McAllister, F. E., Hindley, C. J., Mayer, F., Schmid, B., et al. (2014). iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 5:4028. doi: 10.1038/ncomms5028ncomms5028
Selden, N. R., Al-Uzri, A., Huhn, S. L., Koch, T. K., Sikora, D. M., Nguyen-Driver, M. D., et al. (2013). Central nervous system stem cell transplantation for children with neuronal ceroid lipofuscinosis. J. Neurosurg. Pediatr. 11, 643–652. doi: 10.3171/2013.3.PEDS12397
Seranova, E., Connolly, K. J., Zatyka, M., Rosenstock, T. R., Barrett, T., Tuxworth, R. I., et al. (2017). Dysregulation of autophagy as a common mechanism in lysosomal storage diseases. Essays Biochem. 61, 733–749. doi: 10.1042/EBC20170055EBC20170055
Sessa, M., Lorioli, L., Fumagalli, F., Acquati, S., Redaelli, D., Baldoli, C., et al. (2016). Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: an ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet 388, 476–487. doi: 10.1016/S0140-6736(16)30374-9
Sharma, A., Sances, S., Workman, M. J., and Svendsen, C. N. (2020). Multi-lineage human iPSC-derived platforms for disease modeling and drug discovery. Cell Stem Cell 26, 309–329. doi: 10.1016/j.stem.2020.02.011
Shihabuddin, L. S., Numan, S., Huff, M. R., Dodge, J. C., Clarke, J., Macauley, S. L., et al. (2004). Intracerebral transplantation of adult mouse neural progenitor cells into the Niemann-Pick-A mouse leads to a marked decrease in lysosomal storage pathology. J. Neurosci. 24, 10642–10651. doi: 10.1523/JNEUROSCI.3584-04.2004
Sidman, R. L., Li, J., Stewart, G. R., Clarke, J., Yang, W., Snyder, E. Y., et al. (2007). Injection of mouse and human neural stem cells into neonatal Niemann-Pick A model mice. Brain Res. 1140, 195–204. doi: 10.1016/j.brainres.2007.01.011
Sima, N., Li, R., Huang, W., Xu, M., Beers, J., Zou, J., et al. (2018). Neural stem cells for disease modeling and evaluation of therapeutics for infantile (CLN1/PPT1) and late infantile (CLN2/TPP1) neuronal ceroid lipofuscinoses. Orphanet. J. Rare Dis. 13:54.
Smith, Z. D., Sindhu, C., and Meissner, A. (2016). Molecular features of cellular reprogramming and development. Nat. Rev. Mol. Cell Biol. 17, 139–154. doi: 10.1038/nrm.2016.6
Snyder, E. Y., Taylor, R. M., and Wolfe, J. H. (1995). Neural progenitor cell engraftment corrects lysosomal storage throughout the MPS VII mouse brain. Nature 374, 367–370. doi: 10.1038/374367a0
Soga, M., Ishitsuka, Y., Hamasaki, M., Yoneda, K., Furuya, H., Matsuo, M., et al. (2015). HPGCD outperforms HPBCD as a potential treatment for Niemann-Pick disease type C during disease modeling with iPS cells. Stem Cells 33, 1075–1088. doi: 10.1002/stem.1917
Son, M. Y., Kwak, J. E., Seol, B., Lee, D. Y., Jeon, H., and Cho, Y. S. (2015). A novel human model of the neurodegenerative disease GM1 gangliosidosis using induced pluripotent stem cells demonstrates inflammasome activation. J. Pathol. 237, 98–110. doi: 10.1002/path.4551
Strazza, M., Luddi, A., Carbone, M., Rafi, M. A., Costantino-Ceccarini, E., and Wenger, D. A. (2009). Significant correction of pathology in brains of twitcher mice following injection of genetically modified mouse neural progenitor cells. Mol. Genet. Metab 97, 27–34. doi: 10.1016/j.ymgme.2009.01.005
Subramanian, K., Hutt, D. M., Scott, S. M., Gupta, V., Mao, S., and Balch, W. E. (2020). Correction of Niemann-Pick type C1 trafficking and activity with the histone deacetylase inhibitor valproic acid. J. Biol. Chem. 295, 8017–8035. doi: 10.1074/jbc.RA119.010524
Sung, E. A., Yu, K. R., Shin, J. H., Seo, Y., Kim, H. S., Koog, M. G., et al. (2017). Generation of patient specific human neural stem cells from Niemann-Pick disease type C patient-derived fibroblasts. Oncotarget 8, 85428–85441. doi: 10.18632/oncotarget.1997619976
Swaroop, M., Brooks, M. J., Gieser, L., Swaroop, A., and Zheng, W. (2018). Patient iPSC-derived neural stem cells exhibit phenotypes in concordance with the clinical severity of mucopolysaccharidosis I. Hum. Mol. Genet. 27, 3612–3626. doi: 10.1093/hmg/ddy2595056151
Tang, Y., Yu, P., and Cheng, L. (2017). Current progress in the derivation and therapeutic application of neural stem cells. Cell Death Dis. 8:e3108. doi: 10.1038/cddis.2017.504cddis2017504
Taylor, C. J., Peacock, S., Chaudhry, A. N., Bradley, J. A., and Bolton, E. M. (2012). Generating an iPSC bank for HLA-matched tissue transplantation based on known donor and recipient HLA types. Cell Stem Cell 11, 147–152. doi: 10.1016/j.stem.2012.07.014
Teinert, J., Behne, R., Wimmer, M., and Ebrahimi-Fakhari, D. (2020). Novel insights into the clinical and molecular spectrum of congenital disorders of autophagy. J. Inherit. Metab. Dis. 43, 51–62. doi: 10.1002/jimd.12084
Trilck, M., Hubner, R., Seibler, P., Klein, C., Rolfs, A., and Frech, M. J. (2013). Niemann-Pick type C1 patient-specific induced pluripotent stem cells display disease specific hallmarks. Orphanet. J. Rare Dis. 8:144. doi: 10.1186/1750-1172-8-144
Trilck, M., Peter, F., Zheng, C., Frank, M., Dobrenis, K., Mascher, H., et al. (2017). Diversity of glycosphingolipid GM2 and cholesterol accumulation in NPC1 patient-specific iPSC-derived neurons. Brain Res. 1657, 52–61. doi: 10.1016/j.brainres.2016.11.031
Uchida, N., Buck, D. W., He, D., Reitsma, M. J., Masek, M., Phan, T. V., et al. (2000). Direct isolation of human central nervous system stem cells. Proc. Natl. Acad. Sci. U.S.A. 97, 14720–14725.
Ulloa, M. L., Froyshteter, A. B., Kret, L. N., Chang, D. P., Sarah, G. E., McCarthy, R. J., et al. (2020). Anesthetic management of pediatric patients with Niemann-Pick disease type C for intrathecal 2-hydroxypropyl-β-cyclodextrin injection. Paediatr. Anaesth. 30, 766–772. doi: 10.1111/pan.13902
Vanhee, S., and Vandekerckhove, B. (2016). Pluripotent stem cell based gene therapy for hematological diseases. Crit. Rev. Oncol. Hematol. 97, 238–246. doi: 10.1016/j.critrevonc.2015.08.022
Velasco, S., Kedaigle, A. J., Simmons, S. K., Nash, A., Rocha, M., Quadrato, G., et al. (2019). Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature 570, 523–527. doi: 10.1038/s41586-019-1289-x
Vescovi, A. L., Gritti, A., Galli, R., and Parati, E. A. (1999). Isolation and intracerebral grafting of nontransformed multipotential embryonic human CNS stem cells. J. Neurotraum. 16, 689–693. doi: 10.1089/neu.1999.16.689
Vu, M., Li, R., Baskfield, A., Lu, B., Farkhondeh, A., Gorshkov, K., et al. (2018). Neural stem cells for disease modeling and evaluation of therapeutics for Tay-Sachs disease. Orphanet. J. Rare Dis. 13:152.
Vukicevic, V., Qin, N., Balyura, M., Eisenhofer, G., Wong, M. L., Licinio, J., et al. (2015). Valproic acid enhances neuronal differentiation of sympathoadrenal progenitor cells. Mol. Psychiatry 20, 941–950. doi: 10.1038/mp.2015.3mp20153
Wu, Y. P., and Proia, R. L. (2004). Deletion of macrophage-inflammatory protein 1 alpha retards neurodegeneration in Sandhoff disease mice. Proc. Natl. Acad. Sci. U.S.A. 101, 8425–8430. doi: 10.1073/pnas.0400625101
Xu, L., Yan, J., Chen, D., Welsh, A. M., Hazel, T., Johe, K., et al. (2006). Human neural stem cell grafts ameliorate motor neuron disease in SOD-1 transgenic rats. Transplantation 82, 865–875. doi: 10.1097/01.tp.0000235532.00920.7a
Xu, M., Liu, K., Swaroop, M., Porter, F. D., Sidhu, R., Firnkes, S., et al. (2012). delta-Tocopherol reduces lipid accumulation in Niemann-Pick type C1 and Wolman cholesterol storage disorders. J. Biol. Chem. 287, 39349–39360. doi: 10.1074/jbc.m112.357707
Yu, D., Swaroop, M., Wang, M., Baxa, U., Yang, R., Yan, Y., et al. (2014). Niemann-pick disease Type C: induced pluripotent stem cell-derived neuronal cells for modeling neural disease and evaluating drug efficacy. J. Biomol. Screen. 19, 1164–1173. doi: 10.1177/1087057114537378
Yu, I. T., Park, J. Y., Kim, S. H., Lee, J. S., Kim, Y. S., and Son, H. (2009). Valproic acid promotes neuronal differentiation by induction of proneural factors in association with H4 acetylation. Neuropharmacology 56, 473–480. doi: 10.1016/j.neuropharm.2008.09.019
Zunke, F., and Mazzulli, J. R. (2019). Modeling neuronopathic storage diseases with patient-derived culture systems. Neurobiol. Dis. 127, 147–162. doi: 10.1016/j.nbd.2019.01.018
Keywords: lysosomal storage disorders, central nervous system, induced pluripotent stem cells, neural stem cells, organoids, drug discovery, gene therapy, cell therapy
Citation: Luciani M, Gritti A and Meneghini V (2020) Human iPSC-Based Models for the Development of Therapeutics Targeting Neurodegenerative Lysosomal Storage Diseases. Front. Mol. Biosci. 7:224. doi: 10.3389/fmolb.2020.00224
Received: 23 April 2020; Accepted: 10 August 2020;
Published: 18 September 2020.
Edited by:
Mario Hiroyuki Hirata, University of São Paulo, BrazilReviewed by:
Shikha Prasad, Feinberg School of Medicine, Northwestern University, United StatesCopyright © 2020 Luciani, Gritti and Meneghini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vasco Meneghini, bWVuZWdoaW5pLnZhc2NvQGhzci5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.