 Qingxin Li
Qingxin Li- Guangdong Provincial Engineering Laboratory of Biomass High Value Utilization, Guangdong Provincial Bioengineering Institute, Guangzhou Sugarcane Industry Research Institute, Guangdong Academy of Sciences, Guangzhou, China
Fragment-based drug discovery (FBDD) is a powerful method to develop potent small-molecule compounds starting from fragments binding weakly to targets. As FBDD exhibits several advantages over high-throughput screening campaigns, it becomes an attractive strategy in target-based drug discovery. Many potent compounds/inhibitors of diverse targets have been developed using this approach. Methods used in fragment screening and understanding fragment-binding modes are critical in FBDD. This review elucidates fragment libraries, methods utilized in fragment identification/confirmation, strategies applied in growing the identified fragments into drug-like lead compounds, and applications of FBDD to different targets. As FBDD can be readily carried out through different biophysical and computer-based methods, it will play more important roles in drug discovery.
Introduction
Fragment-based drug design (FBDD) is an approach to develop potent compounds from fragments. FBDD usually generates a compound starting from a chemical fragment with a low binding affinity to the target, low complexity in chemical structures and low molecular weight (less than 300 Da) (Murray and Rees, 2009; Doak et al., 2016). These starting hits are usually identified from a compound library using sensitive biophysical methods. The identified hit is then grown into drug-like molecules through different strategies. Although FBDD cannot replace high-throughput screening (HTS) campaigns in drug discovery, it has some attractive advantages such as saving experimental cost, offering diverse hits, and exhibiting multiple ways to develop novel compounds (Erlanson et al., 2016). These advantages have encouraged researchers to adopt this method to develop inhibitors of different types of targets. With the development of new approaches in screening and progress made in structural biology, FBDD has been readily carried on and playing important roles in target-based drug discovery (Bollag et al., 2010, 2012; Harner et al., 2013). Several drugs such as vemurafenib-an inhibitor of oncogenic B-RAF kinase activity derived from fragment-based approach have been approved by FDA (Erlanson, 2012). With more and more compounds derived from FBDD entering different stages of clinical studies, this method has been highly recognized in drug discovery. To carry out a fragment screening experiment, following procedures are usually required, namely selecting a compound library, setting up a method for hits identification, determining structures of fragment-target complexes, developing an assay for analyzing structure-activity relationship (SAR) and designing a strategy to grow the fragment into a potent inhibitor (Figure 1). In this review, fragment library, methods utilized in fragment screening, strategies applied in fragment optimization and targets that have been studied using FBDD are summarized. With more and more compounds developed through this method, FBDD will be playing essential roles in target-based drug discovery (Whittaker et al., 2010; Jacquemard and Kellenberger, 2019).
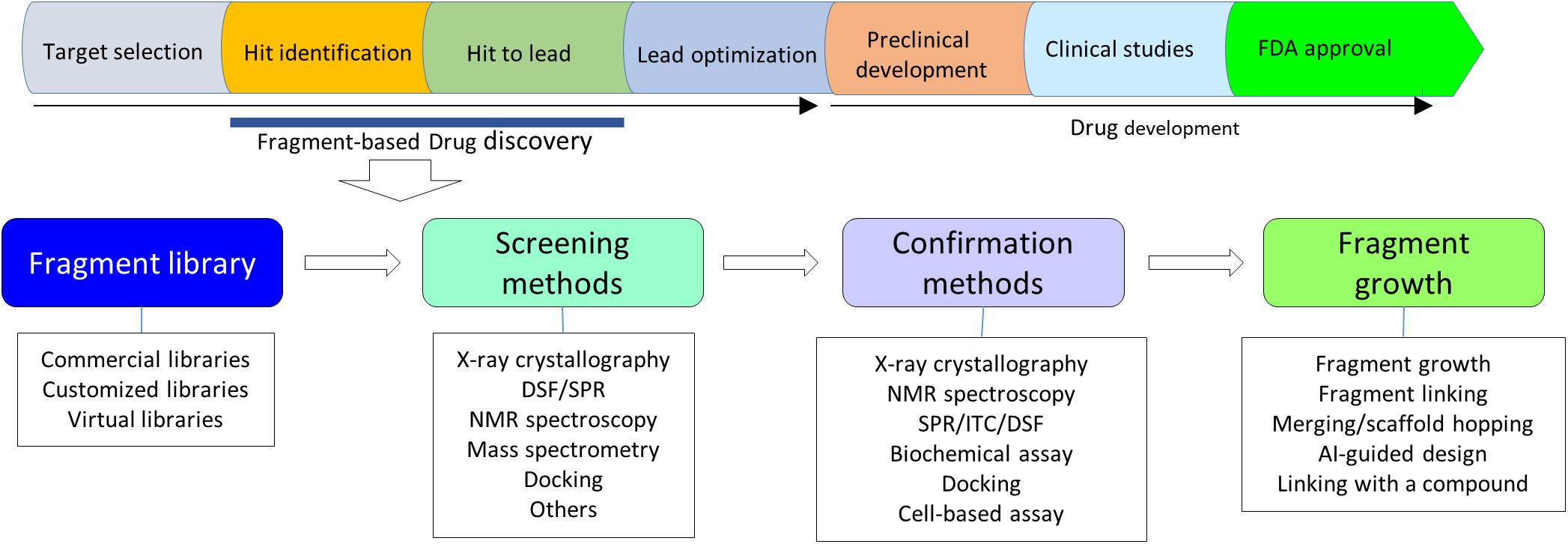
Figure 1. A flowchart of FBDD. Upper panel shows steps in a target-based drug discovery. Lower panel shows a flowchart in FBDD. X-ray crystallography should be always considered in FBDD. NMR plays critical roles in fragment screening.
Fragment Library
There are no strict rules for the size and the number of compounds in a library. The term of fragment indicates that the molecular weight of compounds is relatively small, which gives rise to high ligand efficiency and provides more opportunities for growing the hits. It is suggested that fragments should follow the rules-of-three in which compounds have a molecular weight less than 300 Da, ClogP value less than three, and less than three hydrogen donors and acceptors (Congreve et al., 2003). Recent studies indicated that a fragment does not have to follow the rule-of-three as the fragment in screening utilizes simple organic compounds that can be modified efficiently (Jhoti et al., 2013). Researchers usually have their own customized libraries in FBDD and molecular weight of a fragment can be above 300 Da. In a virtual screening, the fragment library can be expanded with increasing diversity as the screening can be accomplished in a short time. As fragments can provide diverse compounds for optimization, the number of compounds in the fragment library is not a limitation factor. A potent compound was able to be developed through FBDD in which a library of about 800 fragments was utilized (Sabbah et al., 2020). Quite a few fragment libraries are commercially available (Singh et al., 2018). Many researchers have built up their own fragment libraries based on their respective experience (Garner et al., 2019; Heidrich et al., 2019). The customized library usually does not contain molecules that are reactive to targets, bind to proteins un-specifically, form aggregate or form covalent bonds with proteins. One of the fragment libraries in Fesik group consists of approximately 14,000 compounds with a molecular weight of 100–250 Da. Some compounds are following the rule-of-three while some compounds have four hydrogen donors and ClogP value up to 3.5 (Harner et al., 2013). A different library with scaffold-like compounds was utilized for screening kinase inhibitors. This library contains approximately 20,000 compounds with selected chemical properties and molecular weight of 150–350 Da (Bollag et al., 2012). The availability of diverse compound libraries makes FBDD possible to be applied to various targets (Kidd et al., 2018).
Fragment Screening Methods
Binding affinities between fragments and their targets are normally very low in micromolar to millimolar range. Probing fragment and target interactions through biochemical methods which are based on spectrophotometric and fluorescence assays is very challenging. Therefore, other sensitive approaches that are able to determine low binding affinities are very useful in hit identification. These approaches such as differential scanning fluorimetry (DSF), isothermal titration calorimetry (ITC), nuclear magnetic resonance (NMR), surface plasmon resonance (SPR), and X-ray crystallography have been widely used in FBDD.
Differential Scanning Fluorimetry (DSF)
Differential scanning fluorimetry is to measure thermally induced protein denature in the presence of a fluorescence dye such as Synpro Orange that binds to hydrophobic regions of a protein. The method is based on a phenomenon that stability of most proteins decreases when the environmental temperature (Tm) is increased. The Tm at which the amounts of folded and unfolded proteins are equal is termed as melting Tm (Niesen et al., 2007). The compound binding to a protein enhances the Tm of a protein and such a compound is then considered as a positive hit. DSF is a sensitive method and also utilized to understand the effect of point mutations on protein stability (Gayen et al., 2011). DSF experiments can be performed at a medium or high throughput level, making this method more attractive in fragment screening. In the assay mixture, protein concentration is very low and only a small amount of sample is required, which is especially useful for some proteins with low yields or unstable at high concentrations (Niesen et al., 2007). The protein concentration is normally in μM range and the compound concentration is in mM range. Such a high ligand-to-protein ratio will give rise to significant shifts in Tm values. It has been noted that the shift of Tm is proportional to the concentration or affinity of fragments in most cases, but it is not straightforward to correlate the shifts in Tms of compounds with their binding affinities. It is always a good strategy to confirm the identified hits through other biophysical methods (Cramer et al., 2017; Hassaan et al., 2020; Figure 2). It has been noted that other factors such as protein dynamics might influence Tm changes induced by ligand binding. The ligand binding to a protein might not always result in a positive shift. Both positive and negative shifts in Tm values could be observed in a screening. Although DSF is a very powerful tool in FBDD, this approach also has some limitations such as low hit rate due to exhibiting no shift or negative shift of Tm for the ligand binding. Therefore, other methods to confirm the identified hits are required.
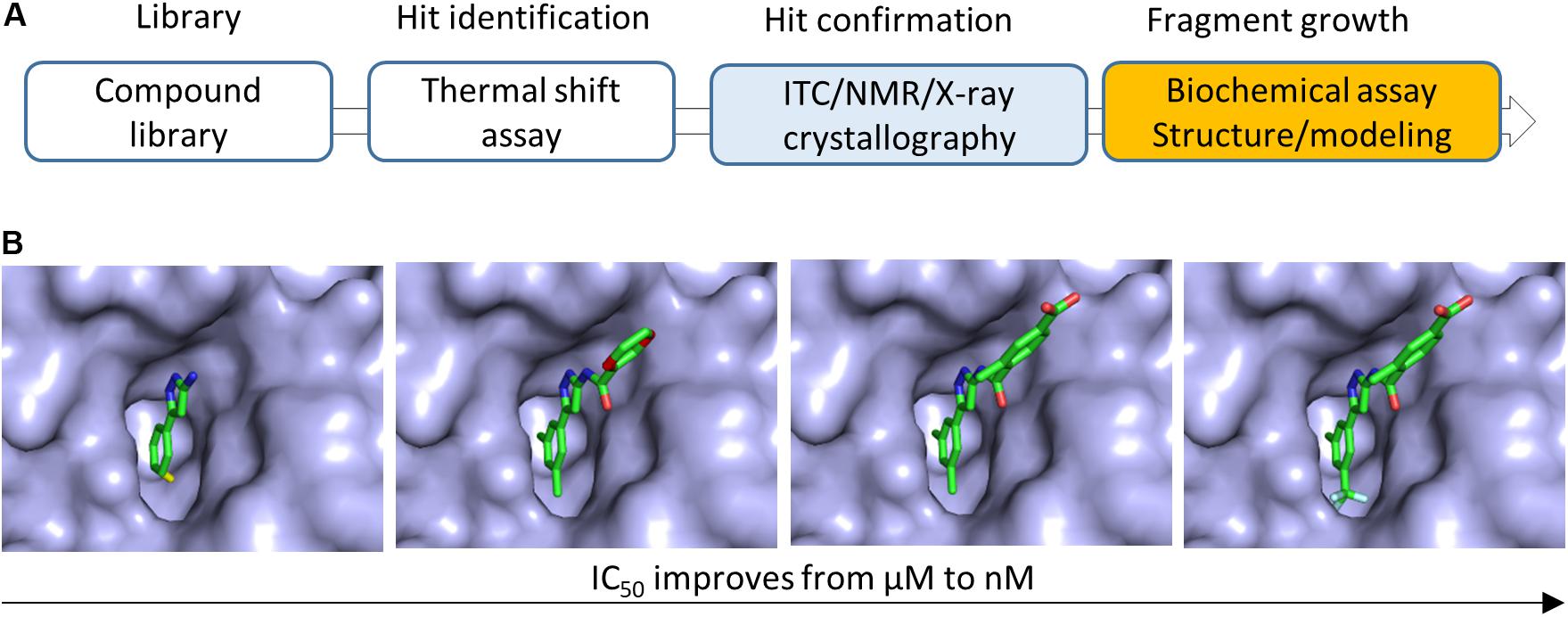
Figure 2. Fragment growing in FBDD. (A) Flowchart of FBDD in developing inhibitors of bacterial Gyrase B. Methods utilized for growing fragments are indicated. (B) Fragment growing strategy was applied to grow a fragment to a potent compound. The IC50 of the compound was improved from 628 μM to 160 nM. This figure illustrated the modification of the compound (Chen et al., 2015). Details should be referred to the original publication (Chen et al., 2015).
Isothermal Titration Calorimetry (ITC)
Isothermal titration calorimetry is a powerful technique to measure binding affinity, binding stoichiometry and enthalpy changes of molecular interactions between a protein and a protein/ligand in solution. ITC experiment is usually carried out in following steps (Du et al., 2016). The first step is to titrate one molecule into a solution containing another molecule (protein). The second step is to monitor heat changes in the mixture. The final step is data analysis to obtain the required parameters. ITC has been routinely applied to determine binding affinities. Therefore, it is very useful to rank binding capabilities of molecules against a protein, making it very attractive in selecting hits for further development. ITC can also be applied in hit-to-lead and lead optimization steps in which SAR can be interpreted. Although ITC is very useful to characterize protein and ligand interactions, there are some limitations which hinder its application in some drug discovery projects. It is not an efficient tool in fragment screening due to the following drawbacks. Firstly, fragments with low binding affinities might not give measurable results easily, making this method unable to identify weak binders. Secondly, it is a time-consuming technique and a large amount of protein sample is required in comparison with other techniques such as DSF. Lastly, not every protein is suitable for ITC studies as protein aggregation and dynamics might affect the results. Nonetheless, ITC is still a powerful tool in drug discovery furnishing useful information for protein–ligand interactions while experimental conditions need to be optimized.
Surface Plasmon Resonance (SPR)
Surface plasmon resonance has been widely applied in probing protein–protein, protein–ligand, protein–DNA/RNA, and DNA–DNA interactions in real time (Bakhtiar, 2013). In addition to measuring binding specificity, binding affinity and thermodynamic parameters, SPR is able to determine dissociation and association rate constants, which provides additional information to understand molecular interactions. This information is particularly useful in lead-optimization step as it offers information to understand the relationship between binding affinity and activity. The dissociation and association rates will guide medicinal chemists to better understand SAR. Immobilizing samples on a biosensor chip is a critical step in the measurement. There are various chips available for immobilizing protein samples. For example, samples can be attached on the surface of chips through covalent bond formation and non-covalent reaction via a high affinity molecule (Arslan Yildiz et al., 2013; Bakhtiar, 2013). Another advantage of SPR is that the amount of sample required for immobilization is very low and the sample immobilized on the chips can be reutilized. Therefore, proteins with low yields are applicable to this method. SPR is able to measure the off rates of ligands, which becomes prevalent in characterizing and ranking the identified hits in drug discovery (Murray et al., 2014).
Surface plasmon resonance has been shown to play important roles in screening and guiding cell-based assays. In the development of regulatory phosphatase PPP1R15B, SPR was utilized to rank small molecules binding to R15A and R15B as other available assays did not have sufficient sensitivity (Krzyzosiak et al., 2018). Based on the measured steady-state affinity of inhibitors for R15A-PP1, the authors were able to define a concentration for inhibitors used in cell-based assays (Krzyzosiak et al., 2018). This study provides an evidence that SPR can be performed in complicated systems in which multiple proteins are present. SPR has advantages in fragment screening over other biophysical methods as this approach consumes very little amount of protein samples and furnishes kinetics and thermodynamics for molecular interactions (Navratilova and Hopkins, 2010). SPR-based fragment screening has been successfully applied to different targets such as carbonic anhydrase II (Navratilova and Hopkins, 2010), thrombin, carbonic anhydrase, glutathione-S-transferase (Hämäläinen et al., 2008).
NMR Spectroscopy
Nuclear magnetic resonance spectroscopy is a powerful tool in drug discovery especially in FBDD. This technique is sensitive enough to identify fragments with different binding affinities (from nanomolar to millimolar). Compared with other methods, NMR screening gives rise to less false positive hits and a mixture of fragments can be screened. A number of NMR experiments have been utilized in FBDD by identifying various hits binding to a specific site on targets (Gossert and Jahnke, 2016; Norton et al., 2016; Li and Kang, 2017; Sugiki et al., 2018; Kang, 2019a, b). As shown in Table 1, several methods can be applied in fragment screening. All these experiments can be summarized as two methodologies. One is to monitor signal changes from fragments (ligands) and the other is to monitor signal changes from targets (proteins). Monitoring signals from ligands in the absence and presence of the target protein is an economic strategy in screening (Mayer and Meyer, 1999). In this method, the amount of the target protein required for screening is less than that utilized in protein-observed NMR studies. Saturation transfer difference spectroscopy (STD) (Mayer and Meyer, 1999; Munawar et al., 2018) and Water-LOGSY (Dalvit et al., 2001) are frequently applied in hit identification. As signal changes from compounds can be monitored through these two methods, there is no limitation for the size of the target protein. Although compound mixtures can be utilized in screening, the number of compounds is limited due to signal overlap. These methods can be also applied to rank binding affinities of the screened hits and determine which groups of the hits are critical for binding (Aretz and Rademacher, 2019).
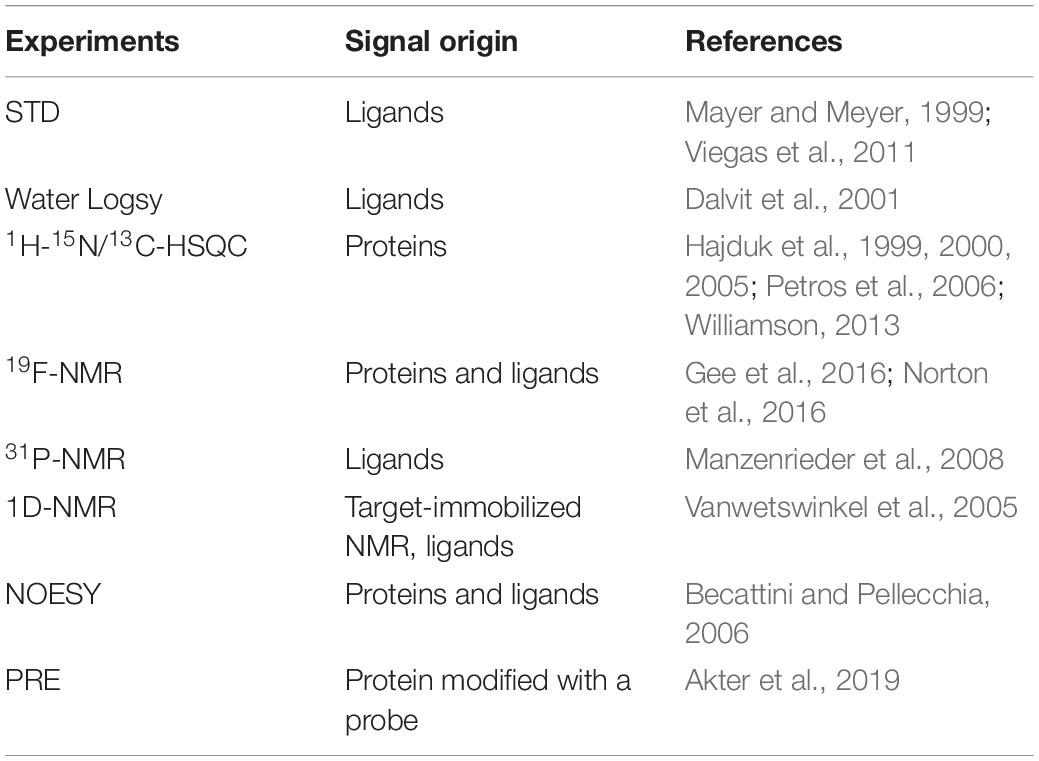
Table 1. Some NMR methods frequently used in screening.
19F-NMR is an efficient approach when it is applied in fragment screening (Norton et al., 2016). Fluorine atom is not present in biological molecules while it is commonly used in drug discovery as it can improve the property of compounds. Therefore, 19F-NMR has no background for biological samples, giving rise to clear signals. Like proton atoms, 19F nucleus has 100% natural abundance, making it measured easily in NMR experiments. The high signal sensitivity (83% of protons) makes it attractive in drug discovery as samples with low concentrations can be measured. The wide dispersion of 19F signals make it possible to use fragment mixtures in screening, which saves the data acquisition time. The availability of 19F-labeled compound libraries makes 19F-NMR more powerful in FBDD (Kang, 2019b; Lingel et al., 2020). 19F-NMR is most attractive in fragment screening for the reason that a mixture of compounds can be screened and the correct hit can be readily picked out, making it become a high-throughput method.
Another frequently used method in probing protein–ligand interactions is 1H-15N-HSQC (hetero-nuclear single quantum coherence spectroscopy) experiment in which chemical shifts of amino acids of a protein are compared in the absence and presence of a ligand. This method is able to confirm molecular interactions between a target and a ligand, determine the binding affinity and map the ligand binding site. It has been noted that this method has been widely utilized to confirm interactions of ligands with different binding affinities to a target. Based on docking software such as HADDOCK (High Ambiguity Driven biomolecular DOCKing), the protein–ligand complex can be obtained according to the chemical shift perturbation induced by ligand binding (Proudfoot et al., 2017). This approach is particularly useful for a target which is difficult to be crystallized (Li et al., 2018a). Unlike proton or 19F-NMR experiments, 1H-15N-HSQC experiment requires the target protein to be isotopically labeled. Since SAR by NMR was proposed in 1996 (Shuker et al., 1996), this method has been widely applied in FBDD. The cost in protein production can be reduced by using sensitive probes, low-volume samples, more sensitive or faster data acquisition strategies, application of compound mixtures in screening and recycle of the protein sample (Hajduk et al., 1997).
X-Ray Crystallography
X-ray crystallography is a powerful tool to obtain structures of proteins and complexes at high resolutions. It plays essential roles in structure-based drug discovery (Hartshorn et al., 2005; Thomas et al., 2019). Many potent inhibitors were developed based on the structural information obtained through X-ray crystallography (Salah et al., 2011). There is no doubt that co-crystal structures offer direct and clear information to understand SAR and mechanism of action of the developed compounds (Carvalho et al., 2010). X-ray structures furnish structural information to understand binding modes of various inhibitors that bind to the active site of a target, inhibit the target through allosteric mechanisms and form covalent bonds with the target (Li et al., 2017a, b, 2018b; Anantharajan et al., 2019; Zhong et al., 2019). X-ray crystallography is a robust method that can be applied in fragment hit identification and confirmation (Schiebel et al., 2016a; Glockner et al., 2020). The bottleneck in X-ray structural studies is to obtain the crystals of targets and complexes (Carpenter et al., 2008). X-ray crystallography plays important roles in FBDD as fragments can be soaked into crystals to obtain their binding modes at a high resolution (Böttcher et al., 2011). Combination of X-ray structures with other biophysical methods is commonly used in drug discovery (Wyss et al., 2012; Munzker et al., 2020). It has been noted that not all targets can be crystallized for X-ray studies. Sometimes, the initial fragments soaked into crystals of the target might not generate high-resolution structures. Under such conditions, other biophysical methods have to be applied to guide fragment growth (Erlanson et al., 2019).
Computational-Based Methods
Virtual screening has been applied in fragment screening (Erlanson et al., 2004a; Behnen et al., 2012; Abdulmalek et al., 2020; De Souza Neto et al., 2020), furnishing diverse chemical structures as the number of libraries in a virtual screening is not a limitation factor (Hoffer et al., 2013). This strategy usually includes structure determination of the target, virtual library preparation, docking, and hit confirmation through docking and MD simulation (Bian and Xie, 2018; De Souza Neto et al., 2020). A library with a large number of fragments can be screened, which offers a high hit rate (Zoete et al., 2009). Artificial intelligence (AI) will furnish a rational design in the hit-to-lead step. An example has been cited in the development of inhibitors of the main protease of SARS-CoV-2 (Choudhury, 2020).
Other Methods
While aforementioned methods are useful in FBDD, other methods that are able to probe protein–ligand interactions have been utilized in FBDD. An anchoring approach was applied to develop protease inhibitors (Hassaan et al., 2020; Konstantinidou et al., 2020). Capillary electrophoresis was successfully applied to identify fragment hits binding to heat shock protein 90 ATPase (Austin et al., 2012). Weak affinity chromatography was developed as a tool to screen hits in FBDD (Duong-Thi et al., 2011). Fragment-based screening was also carried out using cell-based assays (Szõllõsi et al., 2015; Schulze et al., 2018). A study showed that fragments reacting with cysteine residues were able to identify proteins that formed interactions with these compounds (Backus et al., 2016). Mass spectrometry is particular useful for identifying fragments that form covalent interactions with targets (Pedro and Quinn, 2016). In practice, any methods that can probe protein–ligand interactions can be utilized in fragment screening while experimental cost and time have to be considered. Whenever possible, application of X-ray crystallography in FBDD should be considered, which will offer a clear guidance for fragment growth (Schiebel et al., 2016b). In a FBDD, the following strategy can be considered (Figure 1). Bioinformatics analysis of a target should be carried on and its ligand binding site could be analyzed upon an available structure/model. Crystallization trials will be first applied to the target, if the fragment could be soaked into the crystals (Schiebel et al., 2016a, b). Virtual screening, DSA and NMR will be considered in fragment identification when a protein structure is available and the target protein can be purified easily (Figure 1).
Compound Optimization
As fragments usually bind weakly to targets and exhibit no potent inhibitory effect on the activity of the targets, further chemical modification of the hits is required in hit-to-lead step. In this procedure, hits will be developed into leads which bind to the target with higher affinities and exhibit potent activities against the target (Erlanson, 2006; Erlanson et al., 2016; Lamoree and Hubbard, 2017). Three strategies namely fragment growing, fragment hopping, and fragment linking are frequently utilized.
Growing of Fragment Hits
Fragment growing is the most commonly used strategy to grow fragments into compounds with higher molecular weights and higher potencies. Various chemical groups can be added to the building block (hit) to improve its potency (Mondal et al., 2016; Lamoree and Hubbard, 2017). Co-crystal structures of hits with the target are very important for fragment growing (Tao et al., 2015). It has been noted that a fragment can be grown into a potent compound even without structural information (Erlanson et al., 2019). Availability of biochemical or biophysical assays to understand SAR is critical in hit-to-lead step. This strategy has been proven to be successful in numerous targets such as developing bacterial Gyrase B inhibitors (Figure 2; Chen et al., 2015). It has been noted that the four approved drugs were developed by this strategy (Figure 3; Tsai et al., 2008; Souers et al., 2013; Zhang et al., 2013; Murray C.W. et al., 2019). The details of these drugs and other compounds in clinical studies are listed in the Practical Fragments blog website1.
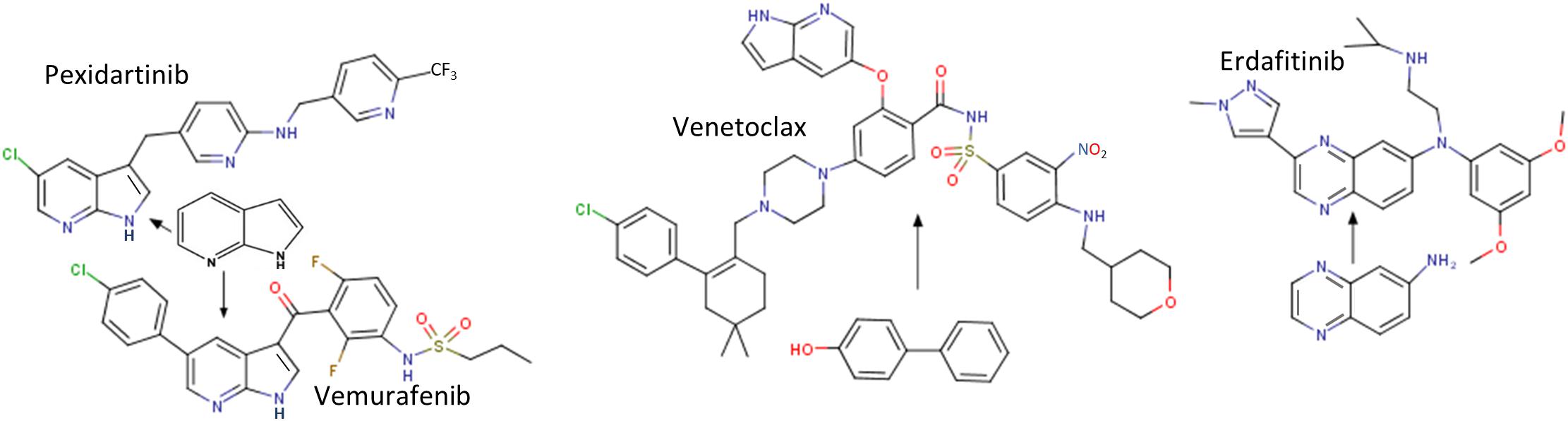
Figure 3. Structures of four approved drugs derived from FBDD. PLX3397 (Pexidartinib) was started from 7-azaindole (Zhang et al., 2013). Vemurafenib was built from the same fragment (Tsai et al., 2008). Structures of Venetoclax (an inhibitor of Bcl2) (Souers et al., 2013) and Erdafitinib (Murray C.W. et al., 2019) are shown. More information about FBDD can be obtained from the blog website http://practicalfragments.blogspot.com/.
Merging/Scaffold Hopping of Fragment Hits
Fragment merging or scaffold hopping is another strategy to grow fragments into potent compounds (Miyake et al., 2019). This strategy is based on condition that the identified fragments have an overlapped binding site. Potent compounds can be developed by combining/merging chemical features of two or more fragments (Temple et al., 2019). To carry out fragment merging of the identified hits, binding modes of fragments should be determined through X-ray crystallography, NMR spectroscopy, or docking methods. This strategy is very useful for replacing non-drug like core structures in the hits with suitable scaffolds to generate more drug-like molecules (Harner et al., 2013). In addition, it offers a chance to generate more patentable compounds (Lamberth, 2018). Unlike the fragment growth strategy, fragment merging requires structural information to understand the binding mode or certain type of experiments such as STD–NMR to determine which part of the ligand is critical for binding. This strategy has been successfully applied in several targets (Fradera et al., 2019; Li et al., 2019; Zhang et al., 2020) such as the development of inhibitors of cytochrome P450 of Mycobacterium tuberculosis by which fragment merging of the hits resulted in a compound with 15–60 fold improvement in binding affinity comparing to its origins (Hudson et al., 2012).
Fragment Linking
Fragment linking is considered as the most powerful way to develop potent inhibitors from fragments. A lead compound can be developed by linking two or more fragments together (Mondal et al., 2016). This is an attractive strategy as binding affinities can be improved dramatically (Mondal et al., 2016). Based on the calculated binding free energy, linking two fragments with binding affinities in mM range will result in a compound with an affinity in nM range (Ichihara et al., 2011; Harner et al., 2013). The challenge in this strategy is to identify fragments that are in close proximity and the introduced linker has no negative effect on the activity of the fragment. The target usually should have a relatively large binding pocket to enable identification of hits binding to different regions in the pocket. Extensive structural information is helpful to understand such molecular interactions. Ideally, this strategy can be carried on when the ligand binding pocket contains two sites with different binding affinities to the fragment (Harner et al., 2013). The first binding site favors identifying fragments with higher binding affinities. To identify fragments binding to the second binding site, the first binding site should be blocked using identified compounds. This can be achieved by growing a fragment into a more potent fragment or inducing a Cys residue at the first site to form a covalent bond with the fragment (Erlanson et al., 2004b). Then the target with the first site occupied can be utilized for screening another type of fragments (Erlanson et al., 2004b). Fragments binding to different sites can also be selectively screened by designing a spy molecule which is fully characterized through different methods (Skora and Jahnke, 2017). Screening can be carried out to monitor release of the spy molecule, which is able to identify the required fragments. 19F-based NMR is very useful for achieving this goal (Li and Kang, 2017; Kang, 2019b). 1H-15N-HSQC based screening plays important roles in screening fragments binding to different sites. This strategy has been successfully applied in the development of compounds binding to replication protein A (RPA70) (Frank et al., 2013). In the study, the N-terminal region of RPA70 composed of approximately 110 amino acids was labeled and utilized for fragment screening (Frank et al., 2013). To analyze the identified hits, the potential ligand binding sites were analyzed carefully. Two residues S55 and T60 from two sites in close proximity were selected to identify the required fragments. Fragments affecting chemical shift of S55, T60 or both were classified (Figure 4). By linking the identified fragments, a potent inhibitor was developed.
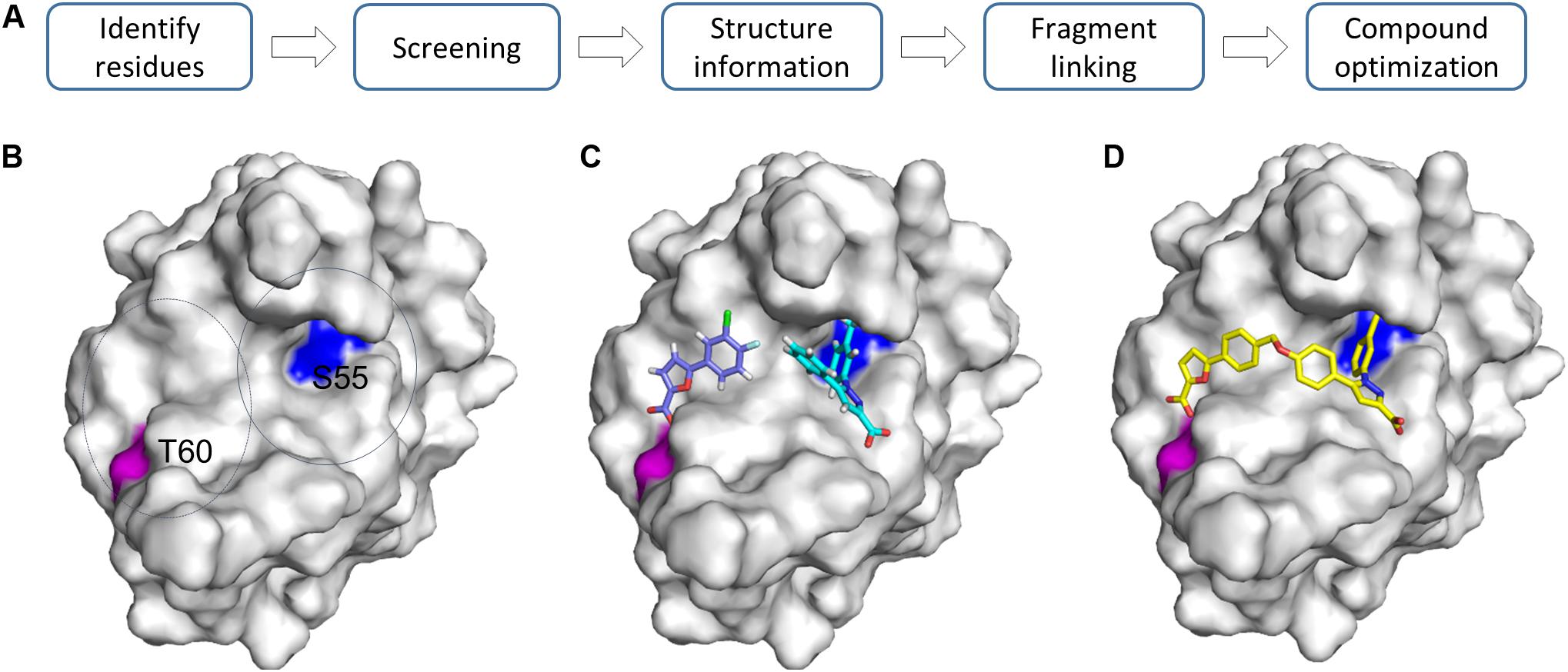
Figure 4. Growing fragments through fragment-linking. (A) Strategy for FBDD. (B) Two representative residues are identified in the same pocket. (C) Two fragments were identified and selected for linking experiments. (D) A linked compound was developed. The structures (PDB id 4LUZ and 4LUV) of protein–ligand complexes are shown. The protein is shown as a surface and compounds are shown as sticks. For more information, please refer to the original publication (Frank et al., 2013).
Growing fragment hits into potent leads can be performed in different strategies, which depends on the hit rate, available structural information, availability of assays for SAR, and assays used for screening (Sommer et al., 2019). Identified fragments can also be linked with hits from HTS, which is seen in a recent study and provides an efficient way to develop potent inhibitors of undruggable targets (Hillig et al., 2019). With the development of docking methods, these computation-based strategies will play a role in guiding fragment growth (Korczynska et al., 2016).
Targets for FBDD
Fragment-based drug discovery is mainly applicable to target-based drug discovery. Druggability of a target is always analyzed in target-based drug discovery projects and is utilized to predict possibility of developing drugs by identifying a pocket favoring binding to small-molecule compounds (Aretz et al., 2016; Gee et al., 2016). In most cases, druggable targets are of great interests as the probability to develop small molecule drugs is very high (Dang et al., 2017). FBDD has been applied to quite a few druggable and undruggable targets (Stamford and Strickland, 2013; Kessler et al., 2019). Some examples are listed in Table 2.
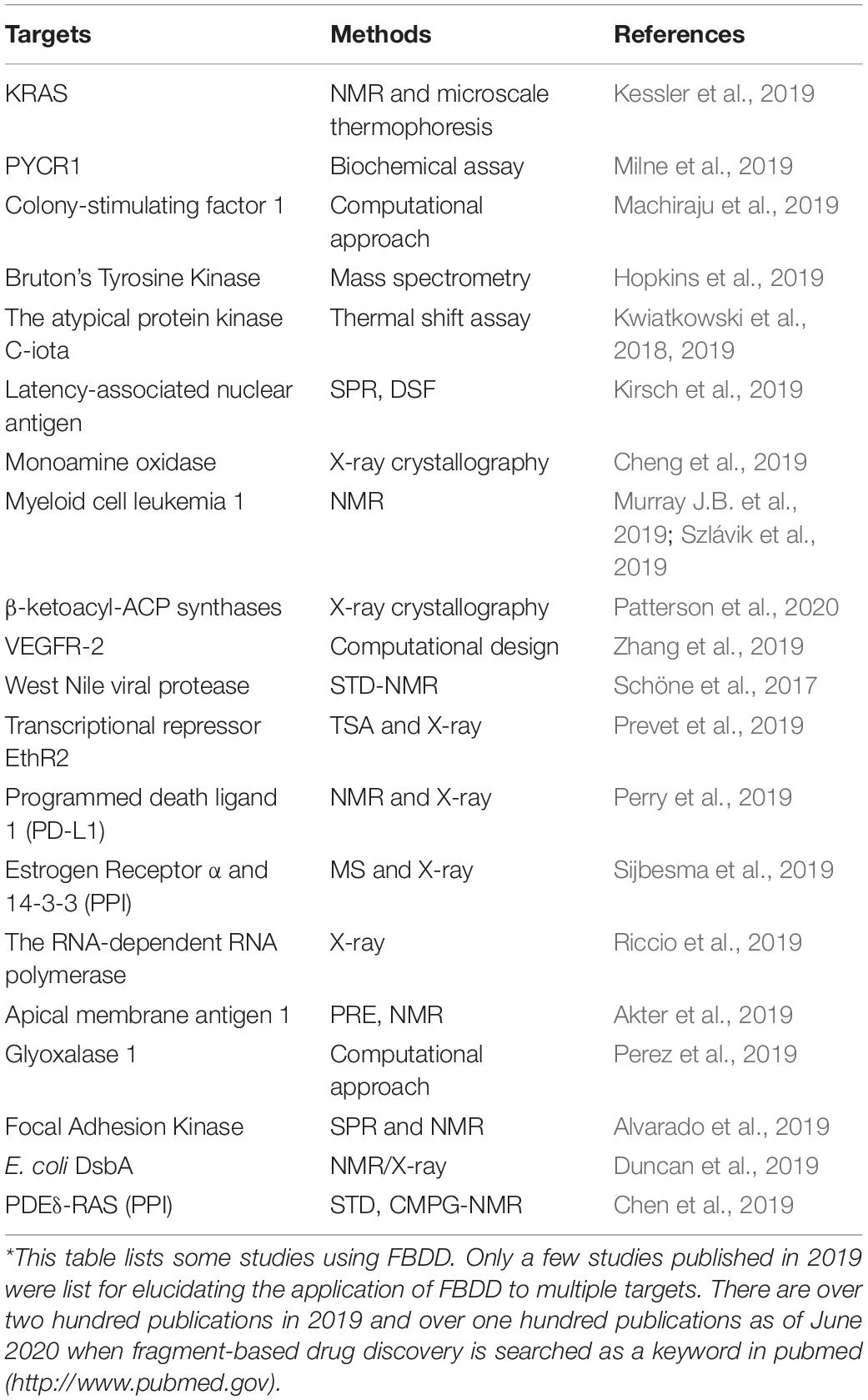
Table 2. List of some targets with inhibitors designed using FBDD*.
Targets With a Well-Defined Pocket
Druggability is utilized to evaluate whether a small-molecule drug can be developed to affect the biological function of a protein (Owens, 2007). Several methods are applied to determine the druggability of targets (Cheng et al., 2007), which is important in drug discovery. A druggable target usually contains a hydrophobic pocket favoring its binding to hydrophobic compounds. However, undruggable targets do not have a pocket or the pocket is highly hydrophilic and shallow. This type of targets includes unstructured proteins that play important roles in disease regulation. A high hit rate can be obtained when FBDD is applied to these druggable targets, furnishing more candidates for further development. For example, several research groups have carried out FBDD against bacterial Gyrase B, and they have developed compounds with different scaffolds (Basarab et al., 2013; Chen et al., 2015).
Targets With a Shallow Pocket
An undruggable target refers to those proteins with shallow pockets un-favoring small molecule interactions (Dang et al., 2017). Many undruggable targets are important for cancer development. These targets include protein–protein interactions (PPIs), transcription factors, phosphatases, Ras proteins, and others (Arkin et al., 2014; Dang et al., 2017). These undruggable targets are usually not considered in small-molecule drug discovery due to challenges in hit identification and lead optimization. Accumulated studies have shown that it is still feasible to develop small molecules binding to these targets (Macalino et al., 2018; Das et al., 2019). One strategy is to develop allosteric inhibitors which induce a new binding site to affect function of the target (Anantharajan et al., 2019). FBDD is successful in the development of KRAS inhibitors. KRAS is a validated target due to its association with cancer initiation and progression (Kano et al., 2019). It was considered as an undruggable target due to lacking of a pocket that is suitable for small molecule binding (Welsch et al., 2017). Potent KRAS inhibitors have been developed using FBDD, proving that this method is very powerful in drug discovery.
Fragment-based drug discovery has been applied for developing compounds targeting PPIs (Si et al., 2019; Figure 5). Several strategies such as hot spot and allosteric site identification can be pursued for this type of targets (Turnbull et al., 2014). It is known that many PPIs are important in drug discovery while the development of small-molecule inhibitors disrupting such targets is challenging (Laraia et al., 2015; Goncearenco et al., 2017). Although HTS campaign is a strategy to identify hits, FBDD is playing important roles in developing various types of compounds disrupting or initiating PPIs as assays for hit screening can be set up easily (Patrone et al., 2013; Turnbull et al., 2014). Compounds affecting PPIs will be achieved through the following mechanisms. Firstly, compounds bind to one protein to generate a protein–ligand complex that does not favor the interaction with another protein. Secondly, compounds are able to stabilize PPIs to affect certain signaling pathways (Andrei et al., 2017). Disulfide screening paradigm was utilized to screen fragments that affect the interaction between 14-3-3σ and a peptide derived from Estrogen Receptor α, offering a strategy to develop PPI stabilizers (Sijbesma et al., 2019). Lastly, compound binding to proteins induces PPIs in which the two proteins are not physically interacting under physiological conditions. A proteolysis targeting chimera (PROTAC) is a multi-functional compound that can link a target of interest with an E3 ligase resulting in protein degradation (Lu et al., 2015; Gadd et al., 2017; Pettersson and Crews, 2019).
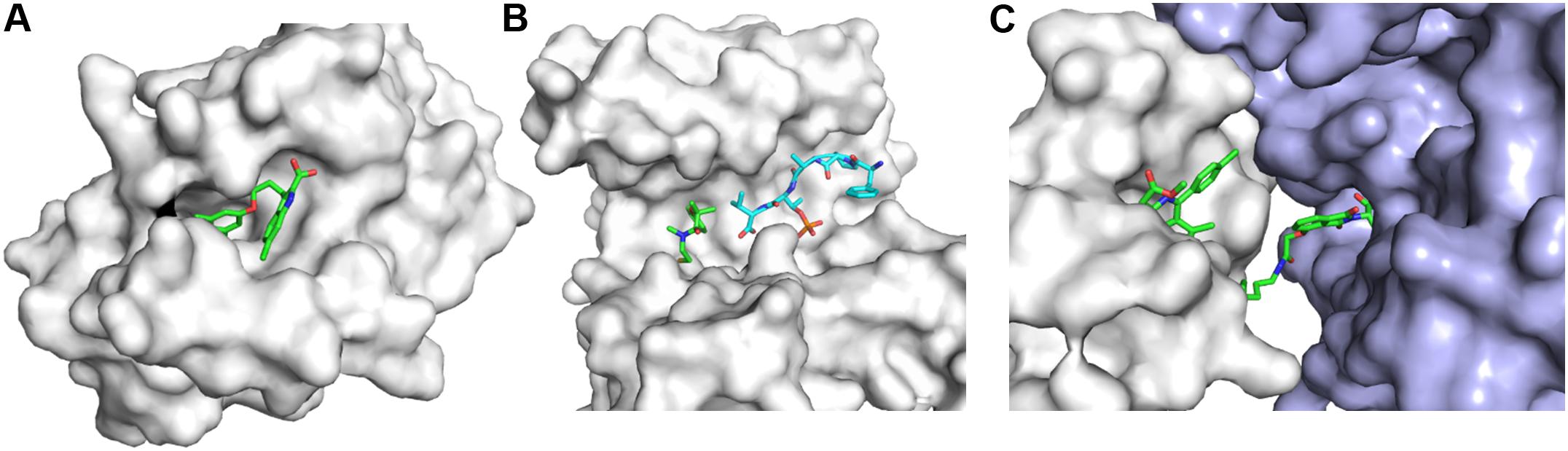
Figure 5. Three types of compounds targeting PPIs. (A) An inhibitor binding to PPI interface to disrupt PPIs. The structure of the complex (PDB id 4HW2) is shown. The development of myeloid cell leukemia inhibitors was described in the reference (Friberg et al., 2013). (B) A PPI stabilizer. The structure (PDB id 6HHP) is shown. A fragment forms a disulfide bond with the target and a fragment from another protein is shown in cyan (Sijbesma et al., 2019). (C) Ligand-induced PPIs. The structure (PDB id 6BN7) is shown. BRD4 and CRBN are shown in white and light blue, respectively. Proteins in this figure are shown in surface representation and compounds are shown as sticks.
Perspectives
Fragment-based drug discovery has been applied to various targets and plays important roles in target-based drug discovery. This method is also very important in chemical biology by developing high-quality chemical probes for diverse targets (Scott et al., 2012). FBDD can be pursued by considering the following steps (Figure 6). Firstly, when a target is defined, bioinformatics will be applied to understand the structure which can be obtained from X-ray crystallography or other methods such as homology modeling. Secondly, the target protein will be overexpressed. If the isotopically labeled protein can be easily purified and the purified protein exhibited dispersed cross-peaks in 1H-15N-HSQC spectrum, this protein-based NMR can be considered in screening. Otherwise, DSF, or 19F-NMR will be utilized in fragment screening. X-ray crystallography will be the first method in screening if the target can be crystallized easily. Virtual screening can be always carried out when a structure of the target is available. Thirdly, a suitable library will be selected from many sources, which is not a limitation factor. Fourthly, hit confirmation will be performed through structural, biophysical and biochemical methods. Lastly, fragment growth can be utilized via suitable strategies. Medicinal chemists will play key roles in this step.

Figure 6. A flowchart of FBDD. The steps required in FBDD are listed. Structural information of the target with a fragment is always helpful for fragment growth. X-ray, DSF, and NMR are commonly used methods in fragment screening.
Conclusion
Fragment-based drug discovery should be applied in drug discovery projects. FBDD is applicable to diverse targets and the hit rate of fragment screening can be also utilized to assess druggability of a target, which can further guide HTS activities to offer an evidence to make Go or No-Go decision for a project. In addition, FBDD is very useful for developing potent binders of a protein which does not have enzymatic activity.
Author Contributions
QL conceived the study and wrote, reviewed, and edited the manuscript.
Funding
This research was supported by funds from the “Hundred-Talent Program” (Grant Numbers: 2020GDASYL-20200102010 and 2020GDASYL-20200102009), Guangdong Academy of Sciences, China.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
QL appreciates the support from Guangdong Bioengineering Institute (Guangzhou Sugarcane Industry Research Institute), Guangdong Academy of Sciences, Guangdong, China.
Footnotes
References
Abdulmalek, A. B., Sufyan, M. A., Abdulrahman, K. A., and Mohammed, A. K. (2020). Fragment-based discovery of potential anticancer lead: computational and in vitro studies. Curr. Comput. Aid. Drug Design 16, 1–9. doi: 10.2174/1573409916666200620195025
Akter, M., Drinkwater, N., Devine, S. M., Drew, S. C., Krishnarjuna, B., Debono, C. O., et al. (2019). Identification of the binding site of apical membrane antigen 1 (AMA1) inhibitors using a paramagnetic probe. ChemMedChem 14, 603–612. doi: 10.1002/cmdc.201800802
Alvarado, C., Stahl, E., Koessel, K., Rivera, A., Cherry, B. R., Pulavarti, S. V. S. R. K., et al. (2019). Development of a fragment-based screening assay for the focal adhesion targeting domain using SPR and NMR. Molecules 24, 3352. doi: 10.3390/molecules24183352
Anantharajan, J., Zhou, H., Zhang, L., Hotz, T., Vincent, M. Y., Blevins, M. A., et al. (2019). Structural and Functional analyses of an allosteric EYA2 phosphatase inhibitor that has on-target effects in human lung cancer cells. Mol. Cancer Therapeut. 18, 1484–1496. doi: 10.1158/1535-7163.mct-18-1239
Andrei, S. A., Sijbesma, E., Hann, M., Davis, J., O’Mahony, G., Perry, M. W. D., et al. (2017). Stabilization of protein-protein interactions in drug discovery. Expert Opin. Drug Discovery 12, 925–940. doi: 10.1080/17460441.2017.1346608
Aretz, J., Kondoh, Y., Honda, K., Anumala, U. R., Nazaré, M., Watanabe, N., et al. (2016). Chemical fragment arrays for rapid druggability assessment. Chem. Commun. 52, 9067–9070. doi: 10.1039/C5CC10457B
Aretz, J., and Rademacher, C. (2019). Ranking Hits From saturation transfer difference nuclear magnetic resonance–based fragment screening. Front. Chem. 7:215. doi: 10.3389/fchem.2019.00215
Arkin, M. R., Tang, Y., and Wells, J. A. (2014). Small-molecule inhibitors of protein-protein interactions: progressing toward the reality. Chem. Biol. 21, 1102–1114. doi: 10.1016/j.chembiol.2014.09.001
Arslan Yildiz, A., Kang, C., and Sinner, E. K. (2013). Biomimetic membrane platform containing hERG potassium channel and its application to drug screening. Analyst 138, 2007–2012. doi: 10.1039/c3an36159d
Austin, C., Pettit, S. N., Magnolo, S. K., Sanvoisin, J., Chen, W., Wood, S. P., et al. (2012). Fragment Screening Using capillary electrophoresis (CEfrag) for hit identification of heat shock protein 90 ATPase inhibitors. J. Biomol. Screening 17, 868–876. doi: 10.1177/1087057112445785
Backus, K. M., Correia, B. E., Lum, K. M., Forli, S., Horning, B. D., González-Páez, G. E., et al. (2016). Proteome-wide covalent ligand discovery in native biological systems. Nature 534, 570–574. doi: 10.1038/nature18002
Bakhtiar, R. (2013). Surface plasmon resonance spectroscopy: a versatile technique in a biochemist’s toolbox. J. Chem. Educ. 90, 203–209. doi: 10.1021/ed200549g
Basarab, G. S., Manchester, J. I., Bist, S., Boriack-Sjodin, P. A., Dangel, B., Illingworth, R., et al. (2013). Fragment-to-hit-to-lead discovery of a novel pyridylurea scaffold of ATP competitive dual targeting type II topoisomerase inhibiting antibacterial agents. J. Med. Chem. 56, 8712–8735. doi: 10.1021/jm401208b
Becattini, B., and Pellecchia, M. (2006). SAR by ILOEs: an NMR-based approach to reverse chemical genetics. Chemistry 12, 2658–2662. doi: 10.1002/chem.200500636
Behnen, J., Köster, H., Neudert, G., Craan, T., Heine, A., and Klebe, G. (2012). Experimental and computational active site mapping as a starting point to fragment-based lead discovery. ChemMedChem 7, 248–261. doi: 10.1002/cmdc.201100490
Bian, Y., and Xie, X.-Q. S. (2018). Computational fragment-based drug design: current trends, strategies, and applications. AAPS J. 20:59. doi: 10.1208/s12248-018-0216-7
Bollag, G., Hirth, P., Tsai, J., Zhang, J., Ibrahim, P. N., Cho, H., et al. (2010). Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 467:596. doi: 10.1038/nature09454
Bollag, G., Tsai, J., Zhang, J., Zhang, C., Ibrahim, P., Nolop, K., et al. (2012). Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discovery 11:873. doi: 10.1038/nrd3847
Böttcher, J., Jestel, A., Kiefersauer, R., Krapp, S., Nagel, S., Steinbacher, S., et al. (2011). “Chapter three–key factors for successful generation of protein–fragment structures: requirement on protein, crystals, and technology,” in Methods in Enzymology, ed. L. C. Kuo (Cambridge, MA: Academic Press), 61–89. doi: 10.1016/b978-0-12-381274-2.00003-0
Carpenter, E. P., Beis, K., Cameron, A. D., and Iwata, S. (2008). Overcoming the challenges of membrane protein crystallography. Curr. Opin. Struct. Biol. 18, 581–586. doi: 10.1016/j.sbi.2008.07.001
Carvalho, A. L., Trincão, J., and Romão, M. J. (2010). “X-ray crystallography in drug discovery,” in Ligand-Macromolecular Interactions in Drug Discovery: Methods and Protocols, ed. A. C. A. Roque (Totowa, NJ: Humana Press), 31–56.
Chen, D., Chen, Y., Lian, F., Chen, L., Li, Y., Cao, D., et al. (2019). Fragment-based drug discovery of triazole inhibitors to block PDEδ-RAS protein-protein interaction. Eur. J. Med. Chem. 163, 597–609. doi: 10.1016/j.ejmech.2018.12.018
Chen, G.-Y., Ng, F. M., Tan, Y. W., Poulsen, A., Seetoh, W., Lin, G., et al. (2015). Application of fragment-based drug discovery against DNA Gyrase B. ChemPlusChem 80, 1250–1254. doi: 10.1002/cplu.201500197
Cheng, A. C., Coleman, R. G., Smyth, K. T., Cao, Q., Soulard, P., Caffrey, D. R., et al. (2007). Structure-based maximal affinity model predicts small-molecule druggability. Nat. Biotechnol. 25, 71–75. doi: 10.1038/nbt1273
Cheng, K., Li, S., Lv, X., Tian, Y., Kong, H., Huang, X., et al. (2019). Design, synthesis and biological evaluation of novel human monoamine oxidase B inhibitors based on a fragment in an X-ray crystal structure. Bioorgan. Med. Chem. Lett. 29, 1012–1018. doi: 10.1016/j.bmcl.2019.02.008
Choudhury, C. (2020). Fragment tailoring strategy to design novel chemical entities as potential binders of novel corona virus main protease. J. Biomol. Struct. Dynam. 20, 1–14. doi: 10.1080/07391102.2020.1771424
Congreve, M., Carr, R., Murray, C., and Jhoti, H. (2003). A ‘Rule of Three’ for fragment-based lead discovery? Drug Discovery Today 8, 876–877. doi: 10.1016/S1359-6446(03)02831-9
Cramer, J., Schiebel, J., Wulsdorf, T., Grohe, K., Najbauer, E. E., Ehrmann, F. R., et al. (2017). A false-positive screening hit in fragment-based lead discovery: watch out for the red herring. Angew. Chem. Int. Ed. 56, 1908–1913. doi: 10.1002/anie.201609824
Dalvit, C., Fogliatto, G., Stewart, A., Veronesi, M., and Stockman, B. (2001). WaterLOGSY as a method for primary NMR screening: practical aspects and range of applicability. J. Biomol. NMR 21, 349–359.
Dang, C. V., Reddy, E. P., Shokat, K. M., and Soucek, L. (2017). Drugging the ‘undruggable’ cancer targets. Nat. Rev. Cancer 17, 502–508. doi: 10.1038/nrc.2017.36
Das, S. K., Sarkar, D., Cavenee, W. K., Emdad, L., and Fisher, P. B. (2019). Rethinking glioblastoma therapy: MDA-9/syntenin targeted small molecule. ACS Chem. Neurosc. 10, 1121–1123. doi: 10.1021/acschemneuro.9b00016
De Souza Neto, L. R., Moreira-Filho, J. T., Neves, B. J., Maidana, R. L. B. R., Guimarães, A. C. R., Furnham, N., et al. (2020). In silico strategies to support fragment-to-lead optimization in drug discovery. Front. Chem. 8:93. doi: 10.3389/fchem.2020.00093
Doak, B. C., Norton, R. S., and Scanlon, M. J. (2016). The ways and means of fragment-based drug design. Pharmacol. Therapeut. 167, 28–37. doi: 10.1016/j.pharmthera.2016.07.003
Du, X., Li, Y., Xia, Y.-L., Ai, S.-M., Liang, J., Sang, P., et al. (2016). Insights into protein–ligand interactions: mechanisms, models, and methods. Int. J. Mol. Sci. 17:144. doi: 10.3390/ijms17020144
Duncan, L. F., Wang, G., Ilyichova, O. V., Scanlon, M. J., Heras, B., and Abbott, B. M. (2019). The fragment-based development of a benzofuran hit as a new class of Escherichia coli DsbA inhibitors. Molecules 24:3756. doi: 10.3390/molecules24203756
Duong-Thi, M.-D., Meiby, E., Bergström, M., Fex, T., Isaksson, R., and Ohlson, S. (2011). Weak affinity chromatography as a new approach for fragment screening in drug discovery. Anal. Biochem. 414, 138–146. doi: 10.1016/j.ab.2011.02.022
Erlanson, D. A. (2006). Fragment-based lead discovery: a chemical update. Curr. Opin. Biotechnol. 17, 643–652. doi: 10.1016/j.copbio.2006.10.007
Erlanson, D. A. (2012). “Introduction to fragment-based drug discovery,” in Fragment-Based Drug Discovery and X-Ray Crystallography, eds T. G. Davies and M. Hyvönen (Berlin: Springer), 1–32. doi: 10.1007/128_2011_180
Erlanson, D. A., Davis, B. J., and Jahnke, W. (2019). Fragment-based drug discovery: advancing fragments in the absence of crystal structures. Cell Chem. Biol. 26, 9–15. doi: 10.1016/j.chembiol.2018.10.001
Erlanson, D. A., Fesik, S. W., Hubbard, R. E., Jahnke, W., and Jhoti, H. (2016). Twenty years on: the impact of fragments on drug discovery. Nat. Rev. Drug Discov. 15, 605–619. doi: 10.1038/nrd.2016.109
Erlanson, D. A., McDowell, R. S., and O’Brien, T. (2004a). Fragment-based drug discovery. J. Med. Chem. 47, 3463–3482. doi: 10.1021/jm040031v
Erlanson, D. A., Wells, J. A., and Braisted, A. C. (2004b). Tethering: fragment-based drug discovery. Ann. Rev. Biophys. Biomol. Struct. 33, 199–223. doi: 10.1146/annurev.biophys.33.110502.140409
Fradera, X., Methot, J. L., Achab, A., Christopher, M., Altman, M. D., Zhou, H., et al. (2019). Design of selective PI3Kδ inhibitors using an iterative scaffold-hopping workflow. Bioorgan. Med. Chem. Lett. 29, 2575–2580. doi: 10.1016/j.bmcl.2019.08.004
Frank, A. O., Feldkamp, M. D., Kennedy, J. P., Waterson, A. G., Pelz, N. F., Patrone, J. D., et al. (2013). Discovery of a potent inhibitor of replication protein a protein–protein interactions using a fragment-linking approach. J. Med. Chem. 56, 9242–9250. doi: 10.1021/jm401333u
Friberg, A., Vigil, D., Zhao, B., Daniels, R. N., Burke, J. P., Garcia-Barrantes, P. M., et al. (2013). Discovery of Potent myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods and structure-based design. J. Med. Chem. 56, 15–30. doi: 10.1021/jm301448p
Gadd, M. S., Testa, A., Lucas, X., Chan, K.-H., Chen, W., Lamont, D. J., et al. (2017). Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 13:514. doi: 10.1038/nchembio.2329
Garner, P., Cox, P. B., Rathnayake, U., Holloran, N., and Erdman, P. (2019). Design and synthesis of pyrrolidine-based fragments that sample three-dimensional molecular space. ACS Med. Chem. Lett. 10, 811–815. doi: 10.1021/acsmedchemlett.9b00070
Gayen, S., Li, Q., Chen, A. S., Nguyen, T. H., Huang, Q., Hill, J., et al. (2011). An NMR study of the N-terminal domain of wild-type hERG and a T65P trafficking deficient hERG mutant. Proteins 79, 2557–2565. doi: 10.1002/prot.23089
Gee, C. T., Arntson, K. E., Urick, A. K., Mishra, N. K., Hawk, L. M. L., Wisniewski, A. J., et al. (2016). Protein-observed 19F-NMR for fragment screening, affinity quantification and druggability assessment. Nat. Protoc. 11:1414. doi: 10.1038/nprot.2016.079
Glockner, S., Heine, A., and Klebe, G. (2020). A Proof-of-concept fragment screening of a hit-validated 96-compounds library against human carbonic anhydrase II. Biomolecules 10:518. doi: 10.3390/biom10040518
Goncearenco, A., Li, M., Simonetti, F. L., Shoemaker, B. A., and Panchenko, A. R. (2017). “Exploring protein-protein interactions as drug targets for anti-cancer therapy with in silico workflows,” in Proteomics for Drug Discovery: Methods and Protocols, eds I. M. Lazar, M. Kontoyianni, and A. C. Lazar (New York, NY: Springer), 221–236. doi: 10.1007/978-1-4939-7201-2_15
Gossert, A. D., and Jahnke, W. (2016). NMR in drug discovery: a practical guide to identification and validation of ligands interacting with biological macromolecules. Prog. Nuclear Magnet. Resona. Spectros. 97, 82–125. doi: 10.1016/j.pnmrs.2016.09.001
Hajduk, P. J., Augeri, D. J., Mack, J., Mendoza, R., Yang, J., Betz, S. F., et al. (2000). NMR-based screening of proteins containing 13C-labeled Methyl Groups. J. Am. Chem. Soc. 122, 7898–7904. doi: 10.1021/ja000350l
Hajduk, P. J., Huth, J. R., and Fesik, S. W. (2005). Druggability indices for protein targets derived from NMR-based screening data. J. Med. Chem. 48, 2518–2525. doi: 10.1021/jm049131r
Hajduk, P. J., Meadows, R. P., and Fesik, S. W. (1997). Discovering high-affinity ligands for proteins. Science 278, 497–499. doi: 10.1126/science.278.5337.497
Hajduk, P. J., Meadows, R. P., and Fesik, S. W. (1999). NMR-based screening in drug discovery. Q. Rev. Biophys. 32, 211–240.
Hämäläinen, M. D., Zhukov, A., Ivarsson, M., Fex, T., Gottfries, J., Karlsson, R., et al. (2008). Label-free primary screening and affinity ranking of fragment libraries using parallel analysis of protein panels. J. Biomol. Screen. 13, 202–209. doi: 10.1177/1087057108314651
Harner, M. J., Frank, A. O., and Fesik, S. W. (2013). Fragment-based drug discovery using NMR spectroscopy. J. Biomol. NMR 56, 65–75. doi: 10.1007/s10858-013-9740-z
Hartshorn, M. J., Murray, C. W., Cleasby, A., Frederickson, M., Tickle, I. J., and Jhoti, H. (2005). Fragment-based lead discovery using X-ray crystallography. J. Med. Chem. 48, 403–413. doi: 10.1021/jm0495778
Hassaan, E., Hohn, C., Ehrmann, F. R., Goetzke, F. W., Movsisyan, L., Hüfner-Wulsdorf, T., et al. (2020). Fragment screening hit draws attention to a novel transient pocket adjacent to the recognition site of the tRNA-modifying enzyme TGT. J. Med. Chem. 13, 6802–6820. doi: 10.1021/acs.jmedchem.0c00115
Heidrich, J., Sperl, L. E., and Boeckler, F. M. (2019). Embracing the diversity of halogen bonding motifs in fragment-based drug discovery—construction of a diversity-optimized halogen-enriched fragment library. Front. Chem. 7:9. doi: 10.3389/fchem.2019.00009
Hillig, R. C., Sautier, B., Schroeder, J., Moosmayer, D., Hilpmann, A., Stegmann, C. M., et al. (2019). Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS–SOS1 interaction. Proc. Natl. Acad. Sci. U.S.A. 116, 2551–2560. doi: 10.1073/pnas.1812963116
Hoffer, L., Renaud, J.-P., and Horvath, D. (2013). In Silico fragment-based drug discovery: setup and validation of a fragment-to-lead computational protocol using S4MPLE. J. Chem. Inform. Model. 53, 836–851. doi: 10.1021/ci4000163
Hopkins, B. T., Bame, E., Bell, N., Bohnert, T., Bowden-Verhoek, J. K., Bui, M., et al. (2019). Optimization of novel reversible Bruton’s tyrosine kinase inhibitors identified using tethering-fragment-based screens. Bioorg. Med. Chem. 27, 2905–2913. doi: 10.1016/j.bmc.2019.05.021
Hudson, S. A., McLean, K. J., Surade, S., Yang, Y.-Q., Leys, D., Ciulli, A., et al. (2012). Application of fragment screening and merging to the discovery of inhibitors of the mycobacterium tuberculosis cytochrome P450 CYP121. Angew. Chem. Int. Ed. 51, 9311–9316. doi: 10.1002/anie.201202544
Ichihara, O., Barker, J., Law, R. J., and Whittaker, M. (2011). Compound design by fragment-linking. Mol. Inform. 30, 298–306. doi: 10.1002/minf.201000174
Jacquemard, C., and Kellenberger, E. (2019). A bright future for fragment-based drug discovery: what does it hold? Exp. Opin. Drug Discov. 14, 413–416. doi: 10.1080/17460441.2019.1583643
Jhoti, H., Williams, G., Rees, D. C., and Murray, C. W. (2013). The ‘rule of three’ for fragment-based drug discovery: where are we now? Nat. Rev. Drug Discov. 12, 644–644. doi: 10.1038/nrd3926-c1
Kang, C. B. (2019a). Applications of in-cell NMR in structural biology and drug discovery. Int. J. Mol. Sci. 20:139. doi: 10.3390/ijms20010139
Kang, C. B. (2019b). 19F-NMR in target-based drug discovery. Curr. Med. Chem. 26, 4964–4983. doi: 10.2174/0929867326666190610160534
Kano, Y., Gebregiworgis, T., Marshall, C. B., Radulovich, N., Poon, B. P. K., St-Germain, J., et al. (2019). Tyrosyl phosphorylation of KRAS stalls GTPase cycle via alteration of switch I and II conformation. Nat. Commun. 10:224. doi: 10.1038/s41467-018-08115-8
Kessler, D., Gmachl, M., Mantoulidis, A., Martin, L. J., Zoephel, A., Mayer, M., et al. (2019). Drugging an undruggable pocket on KRAS. Proc. Natl. Acad. Sci. U.S.A. 116, 15823–15829. doi: 10.1073/pnas.1904529116
Kidd, S. L., Osberger, T. J., Mateu, N., Sore, H. F., and Spring, D. R. (2018). Recent applications of diversity-oriented synthesis toward novel, 3-dimensional fragment collections. Front. Chem. 6:460. doi: 10.3389/fchem.2018.00460
Kirsch, P., Jakob, V., Oberhausen, K., Stein, S. C., Cucarro, I., Schulz, T. F., et al. (2019). Fragment-based discovery of a qualified hit targeting the latency-associated nuclear antigen of the oncogenic kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8. J. Med. Chem. 62, 3924–3939. doi: 10.1021/acs.jmedchem.8b01827
Konstantinidou, M., Magari, F., Sutanto, F., Haupenthal, J., Jumde, V. R., Ünver, M. Y., et al. (2020). Rapid discovery of aspartyl protease inhibitors using an anchoring approach. ChemMedChem 15, 680–684. doi: 10.1002/cmdc.202000024
Korczynska, M., Le, D. D., Younger, N., Gregori-Puigjané, E., Tumber, A., Krojer, T., et al. (2016). Docking and linking of fragments to discover jumonji histone demethylase inhibitors. J. Med. Chem. 59, 1580–1598. doi: 10.1021/acs.jmedchem.5b01527
Krzyzosiak, A., Sigurdardottir, A., Luh, L., Carrara, M., Das, I., Schneider, K., et al. (2018). Target-based discovery of an inhibitor of the regulatory phosphatase PPP1R15B. Cell 174, 1216–1228.e1219. doi: 10.1016/j.cell.2018.06.030
Kwiatkowski, J., Baburajendran, N., Poulsen, A., Liu, B., Tee, D. H. Y., Wong, Y. X., et al. (2019). Fragment-based discovery of a small-molecule protein kinase c-iota inhibitor binding post-kinase domain residues. ACS Med. Chem. Lett. 10, 318–323. doi: 10.1021/acsmedchemlett.8b00546
Kwiatkowski, J., Liu, B., Tee, D. H. Y., Chen, G., Ahmad, N. H. B., Wong, Y. X., et al. (2018). Fragment-based drug discovery of potent protein kinase C iota inhibitors. J. Med. Chem. 61, 4386–4396. doi: 10.1021/acs.jmedchem.8b00060
Lamberth, C. (2018). Agrochemical lead optimization by scaffold hopping. Pest Manag. Sci. 74, 282–292. doi: 10.1002/ps.4755
Lamoree, B., and Hubbard, R. E. (2017). Current perspectives in fragment-based lead discovery (FBLD). Essays Biochem. 61, 453–464. doi: 10.1042/ebc20170028
Laraia, L., McKenzie, G., Spring, D. R., Venkitaraman, A. R., and Huggins, D. J. (2015). Overcoming chemical, biological, and computational challenges in the development of inhibitors targeting protein-protein interactions. Chem. Biol. 22, 689–703. doi: 10.1016/j.chembiol.2015.04.019
Li, Q., Meng, L., Zhou, S., Deng, X., Wang, N., Ji, Y., et al. (2019). Rapid generation of novel benzoic acid–based xanthine derivatives as highly potent, selective and long acting DPP-4 inhibitors: scaffold-hopping and prodrug study. Eur. J. Med. Chem. 180, 509–523. doi: 10.1016/j.ejmech.2019.07.045
Li, Y., and Kang, C. (2017). Solution NMR spectroscopy in target-based drug discovery. Molecules 22:1399. doi: 10.3390/molecules22091399
Li, Y., Liu, S., Ng, E. Y., Li, R., Poulsen, A., Hill, J., et al. (2018a). Structural and ligand-binding analysis of the YAP-binding domain of transcription factor TEAD4. Biochem. J. 475, 2043–2055. doi: 10.1042/BCJ20180225
Li, Y., Zhang, Z., Phoo, W. W., Loh, Y. R., Li, R., Yang, H. Y., et al. (2018b). Structural insights into the inhibition of zika virus NS2B-NS3 protease by a small-molecule inhibitor. Structure 26, 555–564.e553. doi: 10.1016/j.str.2018.02.005
Li, Y., Phoo, W. W., Loh, Y. R., Zhang, Z., Ng, E. Y., Wang, W., et al. (2017a). Structural characterization of the linked NS2B-NS3 protease of Zika virus. FEBS Lett. 591, 2338–2347. doi: 10.1002/1873-3468.12741
Li, Y., Zhang, Z., Phoo, W. W., Loh, Y. R., Wang, W., Liu, S., et al. (2017b). Structural dynamics of zika virus NS2B-NS3 protease binding to dipeptide inhibitors. Structure 25, 1242–1250.e1243. doi: 10.1016/j.str.2017.06.006
Lingel, A., Vulpetti, A., Reinsperger, T., Proudfoot, A., Denay, R., Frommlet, A., et al. (2020). Comprehensive and high-throughput exploration of chemical space using broadband 19F NMR-based screening. Angew. Chem. Int. Ed. doi: 10.1002/anie.202002463
Lu, J., Qian, Y., Altieri, M., Dong, H., Wang, J., Raina, K., et al. (2015). Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Biol. 22, 755–763. doi: 10.1016/j.chembiol.2015.05.009
Macalino, S. J. Y., Basith, S., Clavio, N. A. B., Chang, H., Kang, S., and Choi, S. (2018). Evolution of in silico strategies for protein-protein interaction drug discovery. Molecules 23:963. doi: 10.3390/molecules23081963
Machiraju, P. K., Yedla, P., Gubbala, S. P., Bohari, T., Abdul, J. K. V., Xu, S., et al. (2019). Identification, synthesis and evaluation of CSF1R inhibitors using fragment based drug design. Comput. Biol. Chem. 80, 374–383. doi: 10.1016/j.compbiolchem.2019.04.015
Manzenrieder, F., Frank, A. O., and Kessler, H. (2008). Phosphorus NMR spectroscopy as a versatile tool for compound library screening. Angew. Chem. Int. Ed. 47, 2608–2611. doi: 10.1002/anie.200705256
Mayer, M., and Meyer, B. (1999). Characterization of ligand binding by saturation transfer difference NMR spectroscopy. Angew. Chem. Int. Ed. 38, 1784–1788. doi: 10.1002/(SICI)1521-3773(19990614)38:12<1784::AID-ANIE1784>3.0.CO;2-Q
Milne, K., Sun, J., Zaal, E. A., Mowat, J., Celie, P. H. N., Fish, A., et al. (2019). A fragment-like approach to PYCR1 inhibition. Bioorgan. Med. Chem. Lett. 29, 2626–2631. doi: 10.1016/j.bmcl.2019.07.047
Miyake, Y., Itoh, Y., Hatanaka, A., Suzuma, Y., Suzuki, M., Kodama, H., et al. (2019). Identification of novel lysine demethylase 5-selective inhibitors by inhibitor-based fragment merging strategy. Bioorg. Med. Chem. 27, 1119–1129. doi: 10.1016/j.bmc.2019.02.006
Mondal, M., Radeva, N., Fanlo-Virgós, H., Otto, S., Klebe, G., and Hirsch, A. K. H. (2016). Fragment Linking and optimization of inhibitors of the aspartic protease endothiapepsin: fragment-based drug design facilitated by dynamic combinatorial chemistry. Angew. Chem. Int. Ed. 55, 9422–9426. doi: 10.1002/anie.201603074
Munawar, A., Beelen, S., Munawar, A., Lescrinier, E., and Strelkov, S. V. (2018). Discovery of novel druggable sites on zika virus NS3 helicase using x-ray crystallography-based fragment screening. Int. J. Mol. Sci. 19:3664. doi: 10.3390/ijms19113664
Munzker, L., Petrick, J., Schleberger, C., Clavel, D., Cornaciu, I., Wilcken, R., et al. (2020). Fragment-based discovery of non-bisphosphonate binders of Trypanosoma brucei farnesyl pyrophosphate synthase. Chembiochem doi: 10.1002/cbic.202000246
Murray, C. W., Newell, D. R., and Angibaud, P. (2019). A successful collaboration between academia, biotech and pharma led to discovery of erdafitinib, a selective FGFR inhibitor recently approved by the FDA. MedChemComm 10, 1509–1511. doi: 10.1039/C9MD90044F
Murray, J. B., Davidson, J., Chen, I., Davis, B., Dokurno, P., Graham, C. J., et al. (2019). Establishing drug discovery and identification of hit series for the anti-apoptotic proteins, Bcl-2 and Mcl-1. ACS Omega 4, 8892–8906. doi: 10.1021/acsomega.9b00611
Murray, C. W., and Rees, D. C. (2009). The rise of fragment-based drug discovery. Nat. Chem. 1, 187–192. doi: 10.1038/nchem.217
Murray, J. B., Roughley, S. D., Matassova, N., and Brough, P. A. (2014). Off-rate screening (ORS) by surface plasmon resonance. an efficient method to kinetically sample hit to lead chemical space from unpurified reaction products. J. Med. Chem. 57, 2845–2850. doi: 10.1021/jm401848a
Navratilova, I., and Hopkins, A. L. (2010). Fragment screening by surface plasmon resonance. ACS Med. Chem. Lett. 1, 44–48. doi: 10.1021/ml900002k
Niesen, F. H., Berglund, H., and Vedadi, M. (2007). The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2:2212. doi: 10.1038/nprot.2007.321
Norton, R. S, Leung, E. W, Chandrashekaran, I. R, and MacRaild, C. A (2016). Applications of 19F-NMR in fragment-based drug discovery. Molecules 21:860. doi: 10.3390/molecules21070860
Patrone, J. D., Kennedy, J. P., Frank, A. O., Feldkamp, M. D., Vangamudi, B., Pelz, N. F., et al. (2013). Discovery of protein–protein interaction inhibitors of replication protein a. ACS Med. Chem. Lett. 4, 601–605. doi: 10.1021/ml400032y
Patterson, E. I, Nanson, J. D., Abendroth, J., Bryan, C., Sankaran, B., Myler, P. J., et al. (2020). Structural characterization of β-ketoacyl ACP synthase I bound to platencin and fragment screening molecules at two substrate binding sites. Proteins 88:47. doi: 10.1002/prot.25765
Pedro, L., and Quinn, R. J. (2016). Native mass spectrometry in fragment-based drug discovery. Molecules 21:984. doi: 10.3390/molecules21080984
Perez, C., Barkley-Levenson, A. M., Dick, B. L., Glatt, P. F., Martinez, Y., Siegel, D., et al. (2019). Metal-Binding pharmacophore library yields the discovery of a glyoxalase 1 inhibitor. J. Med. Chem. 62, 1609–1625. doi: 10.1021/acs.jmedchem.8b01868
Perry, E., Mills, J. J., Zhao, B., Wang, F., Sun, Q., Christov, P. P., et al. (2019). Fragment-based screening of programmed death ligand 1 (PD-L1). Bioorg. Med. Chem. Lett. 29, 786–790. doi: 10.1016/j.bmcl.2019.01.028
Petros, A. M., Dinges, J., Augeri, D. J., Baumeister, S. A., Betebenner, D. A., Bures, M. G., et al. (2006). Discovery of a potent inhibitor of the antiapoptotic protein Bcl-xL from NMR and parallel synthesis. J. Med. Chem. 49, 656–663. doi: 10.1021/jm0507532
Pettersson, M., and Crews, C. M. (2019). PROteolysis TArgeting Chimeras (PROTACs) — Past, present and future. Drug Discov. Today 31, 15–27. doi: 10.1016/j.ddtec.2019.01.002
Prevet, H., Moune, M., Tanina, A., Kemmer, C., Herledan, A., Frita, R., et al. (2019). A fragment-based approach towards the discovery of N-substituted tropinones as inhibitors of Mycobacterium tuberculosis transcriptional regulator EthR2. Eur. J. Med. Chem. 167, 426–438. doi: 10.1016/j.ejmech.2019.02.023
Proudfoot, A., Bussiere, D. E., and Lingel, A. (2017). High-confidence protein–ligand complex modeling by nmr-guided docking enables early hit optimization. J. Am. Chem. Soc. 139, 17824–17833. doi: 10.1021/jacs.7b07171
Riccio, F., Talapatra, S. K., Oxenford, S., Angell, R., Mazzon, M., and Kozielski, F. (2019). Development and validation of RdRp Screen, a crystallization screen for viral RNA-dependent RNA polymerases. Biol. Open 8:bio037663. doi: 10.1242/bio.037663
Sabbah, M., Mendes, V., Vistal, R. G., Dias, D. M. G., Záhorszká, M., Mikušová, K., et al. (2020). Fragment-based design of mycobacterium tuberculosis InhA inhibitors. J. Med. Chem. 63, 4749–4761. doi: 10.1021/acs.jmedchem.0c00007
Salah, E., Ugochukwu, E., Barr, A. J., von Delft, F., Knapp, S., and Elkins, J. M. (2011). Crystal Structures of ABL-related gene (ABL2) in complex with imatinib, tozasertib (VX-680), and a type i inhibitor of the triazole carbothioamide class. J. Med. Chem. 54, 2359–2367. doi: 10.1021/jm101506n
Schiebel, J., Krimmer, Stefan, G., Röwer, K., Knörlein, A., Wang, X., et al. (2016a). High-throughput crystallography: reliable and efficient identification of fragment hits. Structure 24, 1398–1409. doi: 10.1016/j.str.2016.06.010
Schiebel, J., Radeva, N., Krimmer, S. G., Wang, X., Stieler, M., Ehrmann, F. R., et al. (2016b). Six biophysical screening methods miss a large proportion of crystallographically discovered fragment hits: a case study. ACS Chem. Biol. 11, 1693–1701. doi: 10.1021/acschembio.5b01034
Schöne, T., Grimm, L. L., Sakai, N., Zhang, L., Hilgenfeld, R., and Peters, T. (2017). STD-NMR experiments identify a structural motif with novel second-site activity against West Nile virus NS2B-NS3 protease. Antiv. Res. 146, 174–183. doi: 10.1016/j.antiviral.2017.09.008
Schulze, J., Baukmann, H., Wawrzinek, R., Fuchsberger, F. F., Specker, E., Aretz, J., et al. (2018). CellFy: a cell-based fragment screen against C-Type lectins. ACS Chem. Biol. 13, 3229–3235. doi: 10.1021/acschembio.8b00875
Scott, D. E., Coyne, A. G., Hudson, S. A., and Abell, C. (2012). Fragment-based approaches in drug discovery and chemical biology. Biochemistry 51, 4990–5003. doi: 10.1021/bi3005126
Shuker, S. B., Hajduk, P. J., Meadows, R. P., and Fesik, S. W. (1996). Discovering high-affinity ligands for proteins: SAR by NMR. Science 274, 1531–1534. doi: 10.1126/science.274.5292.1531
Si, Y., Xu, D., Bum-Erdene, K., Ghozayel, M. K., Yang, B., Clemons, P. A., et al. (2019). Chemical space overlap with critical protein–protein interface residues in commercial and specialized small-molecule libraries. ChemMedChem 14, 119–131. doi: 10.1002/cmdc.201800537
Sijbesma, E., Hallenbeck, K. K., Leysen, S., de Vink, P. J., Skóra, L., Jahnke, W., et al. (2019). Site-directed fragment-based screening for the discovery of protein–protein interaction stabilizers. J. Am. Chem. Soc. 141, 3524–3531. doi: 10.1021/jacs.8b11658
Singh, M., Tam, B., and Akabayov, B. (2018). NMR-fragment based virtual screening: a brief overview. Molecules 23:233. doi: 10.3390/molecules23020233
Skora, L., and Jahnke, W. (2017). 19F-NMR-based dual-site reporter assay for the discovery and distinction of catalytic and allosteric kinase inhibitors. ACS Med. Chem. Lett. 8, 632–635. doi: 10.1021/acsmedchemlett.7b00084
Sommer, K., Flachsenberg, F., and Rarey, M. (2019). NAOMInext–synthetically feasible fragment growing in a structure-based design context. Eur. J. Med. Chem. 163, 747–762. doi: 10.1016/j.ejmech.2018.11.075
Souers, A. J., Leverson, J. D., Boghaert, E. R., Ackler, S. L., Catron, N. D., Chen, J., et al. (2013). ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 19, 202–208. doi: 10.1038/nm.3048
Stamford, A., and Strickland, C. (2013). Inhibitors of BACE for treating Alzheimer’s disease: a fragment-based drug discovery story. Curr. Opin. Chem. Biol. 17, 320–328. doi: 10.1016/j.cbpa.2013.04.016
Sugiki, T., Furuita, K., Fujiwara, T., and Kojima, C. (2018). Current NMR techniques for structure-based drug discovery. Molecules 23:148. doi: 10.3390/molecules23010148
Szlávik, Z., Ondi, L., Csékei, M., Paczal, A., Szabó, Z. B., Radics, G., et al. (2019). Structure-guided discovery of a selective Mcl-1 inhibitor with cellular activity. J. Med. Chem. 62, 6913–6924. doi: 10.1021/acs.jmedchem.9b00134
Szõllõsi, E., Bobok, A., Kiss, L., Vass, M., Kurkó, D., Kolok, S., et al. (2015). Cell-based and virtual fragment screening for adrenergic α2C receptor agonists. Bioorg. Med. Chem. 23, 3991–3999. doi: 10.1016/j.bmc.2015.01.013
Tao, W., Mian-Bin, W., Zheng-Jie, C., Hua, C., Jian-Ping, L., and Li-Rong, Y. (2015). Fragment-based drug discovery and molecular docking in drug design. Curr. Pharmaceut. Biotechnol. 16, 11–25. doi: 10.2174/1389201015666141122204532
Temple, K. J., Engers, J. L., Long, M. F., Gregro, A. R., Watson, K. J., Chang, S., et al. (2019). Discovery of a novel 3,4-dimethylcinnoline carboxamide M4 positive allosteric modulator (PAM) chemotype via scaffold hopping. Bioorg. Med. Chem. Lett. 29:126678. doi: 10.1016/j.bmcl.2019.126678
Thomas, S. E., Collins, P., James, R. H., Mendes, V., Charoensutthivarakul, S., Radoux, C., et al. (2019). Structure-guided fragment-based drug discovery at the synchrotron: screening binding sites and correlations with hotspot mapping. Philos. Transact. R. Soc. A 377:20180422. doi: 10.1098/rsta.2018.0422
Tsai, J., Lee, J. T., Wang, W., Zhang, J., Cho, H., Mamo, S., et al. (2008). Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. U.S.A. 105, 3041–3046. doi: 10.1073/pnas.0711741105
Turnbull, A. P, Boyd, S. M, and Walse, B. (2014). Fragment-based drug discovery and protein-protein interactions. Res. Rep. Biochem. 4, 13–26.
Vanwetswinkel, S., Heetebrij, R. J., van Duynhoven, J., Hollander, J. G., Filippov, D. V., Hajduk, P. J., et al. (2005). TINS, target immobilized NMR screening: an efficient and sensitive method for ligand discovery. Chem. Biol. 12, 207–216. doi: 10.1016/j.chembiol.2004.12.004
Viegas, A., Manso, J., Nobrega, F. L., and Cabrita, E. J. (2011). Saturation-transfer difference (STD) NMR: a simple and fast method for ligand screening and characterization of protein binding. J. Chem. Educat. 88, 990–994. doi: 10.1021/ed101169t
Welsch, M. E., Kaplan, A., Chambers, J. M., Stokes, M. E., Bos, P. H., Zask, A., et al. (2017). Multivalent small-molecule pan-RAS inhibitors. Cell 168, 878–889.e829. doi: 10.1016/j.cell.2017.02.006
Whittaker, M., Law, R. J., Ichihara, O., Hesterkamp, T., and Hallett, D. (2010). Fragments: past, present and future. Drug Discov. Today 7, e163–e171. doi: 10.1016/j.ddtec.2010.11.007
Williamson, M. P. (2013). Using chemical shift perturbation to characterise ligand binding. Prog. Nuclear Magnet. Res. Spectr. 73, 1–16. doi: 10.1016/j.pnmrs.2013.02.001
Wyss, D. F., Wang, Y.-S., Eaton, H. L., Strickland, C., Voigt, J. H., Zhu, Z., et al. (2012). “Combining NMR and X-ray crystallography in fragment-based drug discovery: discovery of highly potent and selective BACE-1 inhibitors,” in Fragment-Based Drug Discovery and X-Ray Crystallography, eds T. G. Davies and M. Hyvönen (Berlin: Springer), 83–114. doi: 10.1007/128_2011_183
Zhang, C., Ibrahim, P. N., Zhang, J., Burton, E. A., Habets, G., Zhang, Y., et al. (2013). Design and pharmacology of a highly specific dual FMS and KIT kinase inhibitor. Proc. Natl. Acad. Sci. U.S.A. 110, 5689–5694. doi: 10.1073/pnas.1219457110
Zhang, P., Jia, L., Tian, Y., Xi, L., Duan, R., Chen, X., et al. (2020). Discovery of potential Toxoplasma gondii CDPK1 inhibitors with new scaffolds based on the combination of QSAR and scaffold-hopping method with in-vitro validation. Chem. Biol. Drug Design 95, 476–484. doi: 10.1111/cbdd.13603
Zhang, Y., Zhang, M., Wang, Y., Fan, Y., Chen, X., Yang, Y., et al. (2019). Protein–ligand interaction-guided discovery of novel VEGFR-2 inhibitors. J. Biomol. Struct. Dynam. 19, 1–16. doi: 10.1080/07391102.2019.1635915
Zhong, W., Koay, A., Ngo, A., Li, Y., Nah, Q., Wong, Y. H., et al. (2019). Targeting the bacterial epitranscriptome for antibiotic development: discovery of novel tRNA-(N(1)G37) methyltransferase (TrmD) inhibitors. ACS Infect Dis. 5, 326–335. doi: 10.1021/acsinfecdis.8b00275
Keywords: fragment-based drug discovery, screening, drug discovery, structural biology, lead design
Citation: Li Q (2020) Application of Fragment-Based Drug Discovery to Versatile Targets. Front. Mol. Biosci. 7:180. doi: 10.3389/fmolb.2020.00180
Received: 16 April 2020; Accepted: 10 July 2020;
Published: 05 August 2020.
Edited by:
Rajarshi Chakrabarti, Indian Institute of Technology Bombay, IndiaReviewed by:
Ruchi Anand, Indian Institute of Technology Bombay, IndiaManfred S. Weiss, Helmholtz-Gemeinschaft Deutscher Forschungszentren (HZ), Germany
Copyright © 2020 Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qingxin Li, cWluZ3hpbl9saUBvdXRsb29rLmNvbQ==