 Anne E. J. Hillen
Anne E. J. Hillen Vivi M. Heine
Vivi M. Heine- 1Pediatric Neurology, Emma Children’s Hospital, Amsterdam UMC, Amsterdam Neuroscience, Vrije Universiteit Amsterdam, Amsterdam, Netherlands
- 2Child and Youth Psychiatry, Emma Children’s Hospital, Amsterdam UMC, Amsterdam Neuroscience, Vrije Universiteit Amsterdam, Amsterdam, Netherlands
- 3Department of Complex Trait Genetics, Center for Neurogenomics and Cognitive Research, Amsterdam Neuroscience, Vrije Universiteit Amsterdam, Amsterdam, Netherlands
Glutamate homeostasis is an important determinant of health of the central nervous system (CNS). Mitochondria play crucial roles in glutamate metabolism, especially in processes with a high energy demand such as action potential generation. Mitochondrial glutamate carriers (GCs) and aspartate-GCs (AGCs) regulate the transport of glutamate from the cytoplasm across the mitochondrial membrane, which is needed to control energy demand, lipid metabolism, and metabolic activity including oxidative phosphorylation and glycolysis. Dysfunction in these carriers are associated with seizures, spasticity, and/or myelin deficits, all of which are associated with inherited metabolic disorders. Since solute carrier functioning and associated processes are cell type- and context-specific, selective vulnerability to glutamate excitotoxicity and mitochondrial dysfunctioning is expected. Understanding this could offer important insights into the pathomechanisms of associated disorders. This perspective aims to explore the link between functions of both AGCs and GCs and their role in metabolic disorders, with a focus on a subclass of lysosomal storage disorders called leukodystrophies (LDs).
A Role for Mitochondrial Glutamate Carriers in Inherited Metabolic Disorders
Mitochondrial dysfunction can lead to affected myelin metabolism (Carvalho, 2013), glutamatergic neurotransmission deficits (Vos et al., 2010), and excitotoxicity (Schinder et al., 1996; Wang and Thayer, 1996), and is associated with inherited metabolic disorders (Saffari et al., 2017). Mitochondria dynamically regulate energy homeostasis by controlling mitochondrial numbers (Ploumi et al., 2017) and by continuously relocating to subcellular locations with high energy demands (Alshaabi et al., 2019). They rely on active glutamate metabolism in order to partake in multiple metabolic pathways (Mahmoud et al., 2019). Glutamate is the most abundant amino acid and neurotransmitter in the central nervous system (CNS), and levels are meticulously regulated (Mahmoud et al., 2019). Excessive concentrations can cause excitotoxicity, leading to calcium influx, mitochondrial dysfunction, and subsequent cell death (Kritis et al., 2015). Glutamate homeostasis is regulated by cytoplasmic as well as mitochondrial carriers (Fiermonte et al., 2002; Berkich et al., 2007; Amoedo et al., 2016; Goubert et al., 2017; Mahmoud et al., 2019). Here, we will discuss the glutamate carriers (GCs) and aspartate-GCs (AGCs), which transport glutamate over the mitochondrial membrane, in the context of leukodystrophies (LDs), a subclass of lysosomal storage disorders. Considering the central role of astrocytes in glutamate metabolism, we will highlight mitochondrial defects in the astrocytopathies Alexander disease (AxD) and Vanishing White Matter (VWM) disease. These illustrate the close link between white matter pathology and astrocytic dysfunction, and will be used to look into how mitochondrial glutamate exchange might be coupled to cell type-specific pathology.
Mitochondrial Aspartate-Glutamate Carriers
Aspartate-GC 1 [AGC1, SLC25A12, Aralar(1)] and 2 (AGC2, SLC25A13, Citrin) transport glutamate plus a proton into the mitochondrion in exchange for aspartate, and in this way support Malate-Aspartate Shuttle (MAS) activity. As cytosolic NADH cannot pass the inner mitochondrial membrane, NAD+ is transported instead together with malate, which is formed from aspartate that is transported by AGCs. Once inside the mitochondrial matrix, the NAD+ is reduced and the resulting NADH donates electrons to the mitochondrial electron transport chain, supporting ATP production (Satrustegui et al., 2007). Mitochondrial NADH levels are also increased due to additional NAD+ reduction upon conversion of malate to oxaloacetate, while the reverse takes place in the cytosol. Such AGC-mediated regulation of NADH/NAD+ ratios aids glycolysis, which requires cytosolic NAD+ molecules (Amoedo et al., 2016; Alkan et al., 2018). By transporting mitochondrial NH3-derived aspartate into the cytoplasm (Meijer et al., 1972), AGCs facilitate the urea cycle and associated NADH levels in the liver. In accordance, an increased cytosolic NADH/NAD+ ratio caused by AGC2 dysfunction disrupts urea cycle metabolism (Moriyama et al., 2006).
In neurons, aspartate that is transported into the cytoplasm by AGCs is converted into N-acetylaspartate (NAA) and shuttled to oligodendrocytes (Dahlin et al., 2015). In oligodendrocytes, NAA is metabolized into acetate, which in turn serves as a substrate for acetyl-CoA, forming fatty acids and ultimately myelin lipids (Dahlin et al., 2015). As AGCs play essential roles in regulating glutamate and aspartate homeostasis in mitochondrial and cytoplasmic compartments, they are involved in mitochondrial oxidative phosphorylation, lipid metabolism, the urea cycle, and glycolysis in the cytoplasm (Lasorsa et al., 2003; Amoedo et al., 2016).
In the CNS, AGC1 is the most highly expressed isoform (Iijima et al., 2001). AGC1 expression is high in neurons compared to oligodendrocytes (Profilo et al., 2017) and astrocytes (Ramos et al., 2003; McKenna et al., 2006; Berkich et al., 2007; Juaristi et al., 2019). While still debated, some report astrocytic AGC1 expression that increased with maturity (Li et al., 2012) and could support MAS function (Hertz, 2011). AGC1 is activated by an external regulatory binding site for calcium (Contreras et al., 2007; Satrustegui et al., 2007), stimulating the MAS and subsequent ATP production (Lasorsa et al., 2003; Gellerich et al., 2012). AGC2 is predominantly expressed in the liver, kidneys, and heart (Begum et al., 2002), and its neuronal expression is much more spatially restricted than AGC1s (Contreras et al., 2010; Profilo et al., 2017). Tissues expressing more AGC2 than AGC1 generally show high expression of genes involved in the urea cycle (Begum et al., 2002). The kinetics and calcium sensitivity of the two carriers also differ (Palmieri et al., 2001; Contreras et al., 2007). AGC2 dysfunction by mutations in the SLC25A13 gene leads to citrullinemia type II (Kobayashi et al., 1999; Saheki et al., 2002). This disease primarily shows dysfunction in the liver where no AGC1 is present, suggesting some compensatory function between the two isoforms occurs when co-expressed. However, the role of AGC2 in the CNS has not been studied extensively and its exact function there remains elusive.
AGC1 deficiency is associated with epilepsy (Falk et al., 2014), hypomyelination (Wibom et al., 2009), and reduced NAA (Jalil et al., 2005; Falk et al., 2014; Profilo et al., 2017), which is an important substrate for myelin lipids. Furthermore, oligodendrocyte precursor cell (OPC)-specific AGC1 deficiency resulted in inhibited proliferation and increased maturation, suggesting an interactive pathway between AGC1 and oligodendrocytes in addition to the provision of NAA (Petralla et al., 2019).
Mitochondrial Glutamate Carriers
Glutamate carrier 1 (GC1; SLC25A22) and 2 (GC2; SLC25A18) also transport glutamate over the mitochondrial inner membrane. The glutamate is again co-transported with a proton, allowing glutamate dehydrogenase inside the mitochondrial matrix to generate NH3 for ureogenesis (Meijer et al., 1972), and α-ketoglutarate to be used in the tricarboxylic acid (TCA) cycle (Hudson and Daniel, 1993; Li et al., 2017) or for gluconeogenesis (Stumvoll et al., 1997). Calcium signaling induces a preferential transamination of glutamate with oxaloacetate to form aspartate and subsequent aspartate efflux by AGCs, curtailing the input of AGCs to the TCA cycle (see Satrustegui et al., 2007) as compared to that of GCs. Different GC kinetics result in faster glutamate transport by GC1 than by GC2, suggesting a division of workload into basal and on-demand glutamate metabolism (Fiermonte et al., 2002). GC1 and GC2 are present in oligodendrocytes (Sun et al., 2015) and in a subset of neurons (Scifo et al., 2013; Llavero Hurtado et al., 2017). Studies also suggest a relatively high expression of GC1 and GC2 in astrocytes, as GC1 levels are higher in cortical astrocytes than in whole cortex-samples (Berkich et al., 2007) and GC2 is enriched in astrocyte endfeet containing an abundance of mitochondria (Mathiisen et al., 2010; Boulay et al., 2017). Others showed that GC2 expression is higher in the cortex than in the cerebellum and increases with age in both regions (Orre et al., 2014; Boulay et al., 2017; Boisvert et al., 2018). These data indicate GC expression levels differs based on neural cell type, brain region, and age, likely resulting in functional heterogeneity.
Glutamate carrier dysfunction is directly associated with epilepsy (Molinari et al., 2009; Poduri et al., 2013), among other neural defects. GC1 is an interaction partner of Battenin, encoded by neuronal ceroid lipofuscinosis-3 (CLN3; Scifo et al., 2013), which has been linked to intracellular trafficking and autophagy (Behrends et al., 2010). CLN3 mutations lead to neuronal ceroid lipofuscinosis (NCL; Luiro et al., 2006), a family of lysosomal storage diseases collectively referred to as Batten disease. Neural pathology presents with lipopigment deposits, and with mitochondrial dysfunction in interneurons in the cortex, thalamus, and hippocampus, and in cerebellar Purkinje cells (see Nelvagal et al., 2019). Primary NCL cortical microglia and astrocyte cultures show impaired glutamate clearing and desynchronized calcium waves (Parviainen et al., 2017). Experimental inactivation of GC1 in astrocytes resulted in decreased NAD+ and ATP levels, as well as an intracellular accumulation of glutamate that the authors proposed (Goubert et al., 2017) may lead to the altered neuronal synchronicity and epilepsy seen in GC1-deficient patients (Molinari et al., 2009). GC2 expression appears linked to inflammation levels, as GC2 levels significantly decreased upon spinal cord injury (Anderson et al., 2016) whereas increased GC2 expression was shown after inflammatory compound treatment of macrophage cultures (Hans et al., 2019). Because glial scar formation protects against inflammation (Sofroniew, 2015), GC2 could be involved in modulating inflammatory conditions. It is clear that GCs are crucial for glutamate metabolism and energy supply to the cell. Links to autophagy and inflammation also suggest a potential role for GC1 in white matter health and merit further studies in the context of clinical presentations.
Mitochondrial Dysfunctioning and (A)GC Expression in White Matter Disorders
(A)GC deficiency is associated with hypomyelination (Wibom et al., 2009), epilepsy (Falk et al., 2014), spasticity (Molinari et al., 2005), and disruption of metabolic pathways, including oxidative phosphorylation, the urea cycle, and the MAS. These functions are regulated by different mechanisms, depending on cell type, brain region, and cellular state. First, neurons and astrocytes share similar machinery for glutamate metabolism but utilize it differently under basal conditions (Frigerio et al., 2008; Llorente-Folch et al., 2016). Second, glutamate levels in white matter areas are about half of that in gray matter areas (Hassel et al., 2003), caused by more effective glutamate-to-glutamine metabolism in white compared to gray matter astrocytes (Lundgaard et al., 2014). Third, cellular conditions such as stress, low energy supply, and altered calcium levels can affect intra- and extracellular glutamate levels. Thus, the relationship between mitochondrial metabolism and glutamate differs across cell types, is highly interactive, and supports various brain functions. It is therefore expected that certain neural structures and conditions show increased vulnerability to (A)GC dysfunction (Figure 1). We explore a role for (A)GCs in mitochondrial dysfunction in two LDs that present with symptoms similar to (A)GC deficiency. Before we discuss how cell types affected in these LDs might be vulnerable due to downstream effects of dysregulated (A)GC functioning, a short introduction into the mitochondrial symptoms of the two astrocytopathic LDs is given.
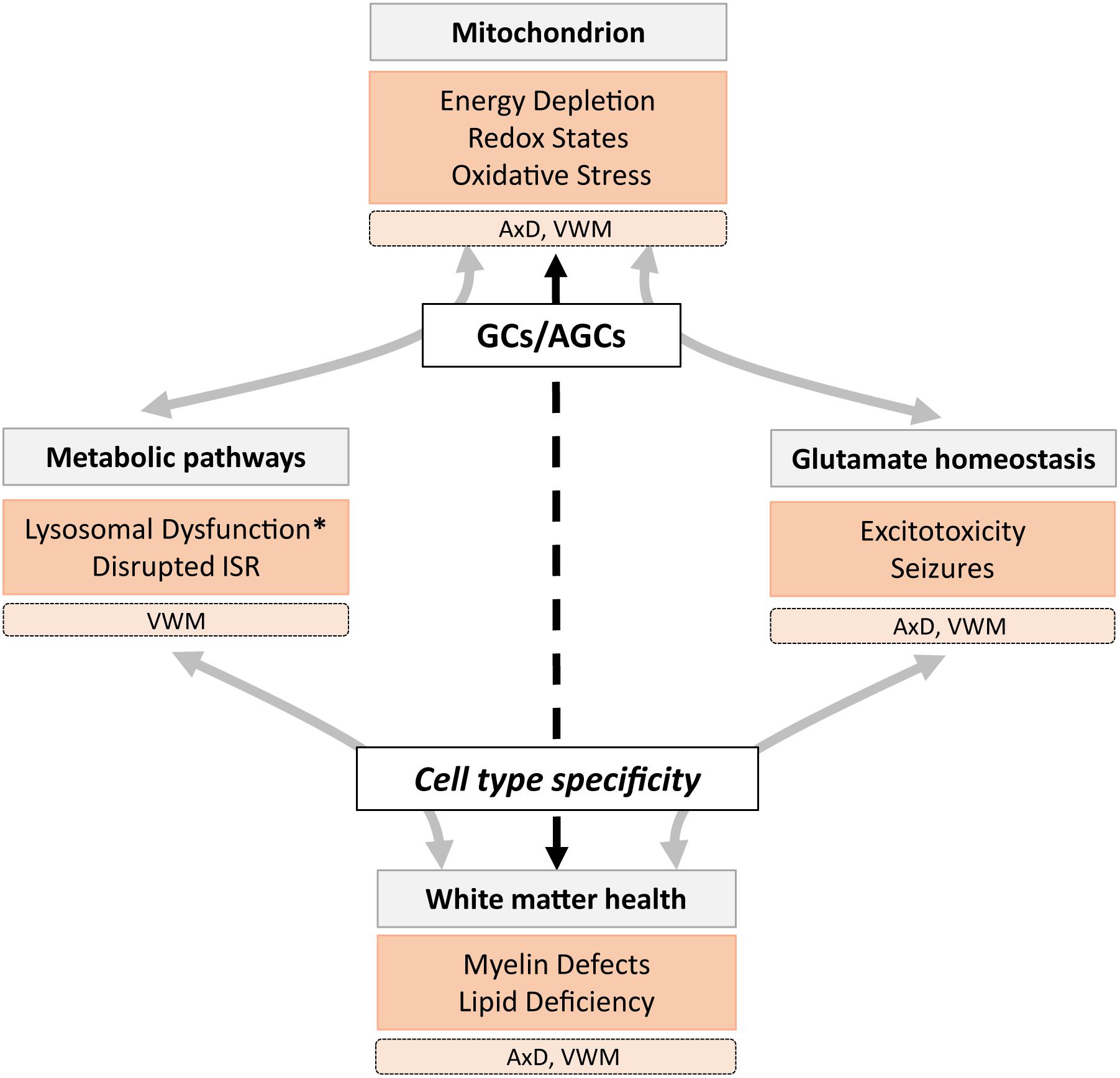
Figure 1. A schematic overview of the role of mitochondrial glutamate carriers GCs and AGCs in metabolic and glutamate homeostasis mechanisms involved in healthy (gray) and pathological (orange) states associated with lysosomal storage diseases (LSDs), including the leukodystrophies AxD and VWM. We hypothesize that (A)GC (dys)function has cell- and region-specific effects (dashed arrow) that affect white matter health directly and indirectly. Pathological states marked with an asterisk are not observed in AxD and VWM, but are present in many LSDs.
Mitochondrial Dysfunctioning in Alexander Disease
Alexander disease is caused by mutations in the GFAP gene, which encodes for intermediate filaments in astrocytes. The AxD brain shows ultrastructurally abnormal mitochondria closely located to Rosenthal fibers, a hallmark of AxD pathology (Herndon et al., 1970; Caceres-Marzal et al., 2006), and shows an oxidative stress response (Castellani et al., 1998; Wang et al., 2011) in astrocytes of the white matter (Hagemann et al., 2006). Perturbed mitochondrial transfer was confirmed in models of AxD (Gao et al., 2019), and GFAP mutations were directly linked to reduced glial glutamate buffering (Wang et al., 2011). Interestingly, AxD patient iPSC-derived astrocytes showed downregulation of both GCs and both AGCs (Table 1), as well as less glycolysis, high oxidative respiration rates, lowered calcium release, and decreased extracellular ATP (Jones et al., 2018). Others showed that GFAP mutations are associated with decreased GLT-1 expression in astrocytes, concurrent with a decreased ability to rescue neurons from glutamate-induced excitotoxicity (Tian et al., 2010). These studies support the notion of secretion-related pathology of AxD astrocytes and involvement of affected glutamate homeostasis by mitochondrial dysfunctioning.

Table 1. Overview of findings of altered (A)GC expression in leukodystrophies.
Mitochondrial Dysfunctioning in Vanishing White Matter Disease
Vanishing White Matter (VWM) disease is caused by mutations in the genes encoding for eukaryotic translation initiation factor EIF2B subunits. Astrocytes are primary affected by VWM mutations in both mice and patient tissues (Dooves et al., 2016). A hallmark of its white matter pathology is foamy oligodendrocytes with membranous vacuoles composed of mitochondrial membranes and myelin lamellae. They contain mitochondrial numbers of up to five times the amount in healthy appearing oligodendrocytes (Wong et al., 2000). Various models of VWM implicate mitochondrial and glutamate dysfunction. Raini et al. (2017) report that primary astrocytes obtained from VWM mice showed decreased oxidative phosphorylation, which was partially compensated by increased mitochondrial numbers and glycolysis. Primary VWM mouse embryonic fibroblasts (MEF) showed GC1 protein upregulation as well as a trend toward decreased AGC1 levels (Raini et al., 2017; Table 1). In severely affected VWM mice, a significant decrease in Gc1 and Agc1 transcripts was observed in the cerebellum (Wong et al., 2019; Table 1). Also, using VWM patient iPSCs, various differentially expressed mitochondrion-related genes genes were found in white matter astrocytes (Leferink et al., 2019). Reduced Sigma-1 receptor expression in VWM mouse astrocytes furthermore points to disrupted mitochondria-ER communication in VWM (Atzmon et al., 2018). Taken together, the mitochondrial dysfunction in VWM pathomechanisms suggests a role for (A)GCs.
Mitochondrial Glutamate Carrier Involvement in White Matter Defects
Deficient Mitochondrial Calcium Buffering Leads to Glutamate Excitotoxicity
Upon increased cytosolic calcium, mitochondrial carriers are activated to adjust glutamate supply to the mitochondria and to regulate ATP production via oxidative phosphorylation (Gellerich et al., 2012). High calcium levels within mitochondria lead to depolarization of the mitochondrial membrane (Duchen, 2000), increased production of radical oxygen species, and cell death (Orrenius et al., 2003). Because glutamate buffering in the brain is most pronounced in astrocytes (Danbolt et al., 2016), perturbed astrocytic glutamate uptake due to mitochondrial dysfunction contributes to a great degree to excitotoxic states and overall brain dysfunctioning. While the exact mechanism is unknown, GC1 dysfunction in astrocytes resulted in decreased ATP levels and accumulation of intracellular glutamate (Goubert et al., 2017), which can cause reversed glutamate transport into the extracellular space (Longuemare et al., 1999) and promote excitotoxicity. In neurons, it has been shown that glutamate-induced calcium buffering in mitochondria modulates NMDA glutamate receptor activity (Kannurpatti et al., 2000) and prevents excitotoxicity (Wang and Thayer, 1996). In astrocytic processes, mitochondria, AGC1, and GC1, are co-compartmentalized with cytosolic GLT-1 glutamate receptors (Genda et al., 2011; Ugbode et al., 2014), suggesting a coupling of energy demand of cytoplasmic glutamate transport and energy supply by mitochondrial GCs. Indeed, AxD presents with downregulated GLT-1 levels (Tian et al., 2010), alongside diminished calcium wave propagation and ATP export (Jones et al., 2018) and increased sensitivity to excitotoxicity and seizure development (Hagemann et al., 2012). The lineage of oligodendrocytic cells shows high vulnerability to glutamatergic excitotoxicity (Domercq et al., 2011), which could contribute to white matter damage in metabolic and lysosomal storage disorders. Glutamate excitotoxicity during white matter dysfunction is mainly studied in context of cytosolic GCs. However, considering the central role of (A)GCs in regulating calcium homeostasis upon glutamate level changes, mitochondrial carrier (dys)function should receive more attention.
MAS Dysfunction Affects Oligodendrocyte Maturation and Myelin Production
Myelination requires mitochondrial production of acetyl-CoA, metabolized from neuronal aspartate (Dahlin et al., 2015), which is supported by oligodendrocyte lineage cell activity that increases mitochondrial ATP production and mitochondrial transcript levels (Silva et al., 2009; Schoenfeld et al., 2010). In line with this, AGC1 deficiency leads to decreased OPC proliferation (Petralla et al., 2019) and also to myelin deficits considering its function in the MAS (Wibom et al., 2009). The OPC maturation defects in VWM and AxD (Li et al., 2018; Leferink et al., 2019) could be associated to dysfunction of mitochondria, and specifically of AGC1 (Petralla et al., 2019). The MAS function of AGCs furthermore regulates the balance of glycolysis in the cytosol and oxidative phosphorylation in the mitochondrion (Lasorsa et al., 2003; Kasai et al., 2019). AGC1 expression is coupled to increased glycolysis (Lasorsa et al., 2003), increased glutamate oxidation (Herbst and Holloway, 2016), and is activated by calcium (Menga et al., 2015). These qualities of AGC1 suggest a link between neural and mitochondrial activity on the one hand, and oligodendrocyte differentiation and myelin metabolism on the other. Interestingly, both AxD and VWM astrocytes showed dysregulated oxidative phosphorylation, increased glycolysis and altered AGC1 expression (Raini et al., 2017; Jones et al., 2018), suggesting that these white matter disorders could share a common affected pathway. However, other neural subtypes could be affected as well, as a perturbed MAS is further determined by the different metabolic needs across cell types. Taken together, disrupted MAS activity likely plays a role in pathomechanisms of white matter deficiencies.
Signaling Pathways Interact With Mitochondrial Functioning in a Cell Type-Dependent Manner
Of additional interest are metabolic pathways that interact with mitochondrial metabolism and that are regulated in a cell type-dependent manner. While the mitochondrial genome consists of only 37 genes, its transcription is modulated by many nuclear factors (Nunnari and Suomalainen, 2012). Conversely, mitochondrial dysfunction can regulate nuclear transcription (Suomalainen and Battersby, 2018), depending on cell type-specific differences (Bolea et al., 2019). Several mitochondrial genes contain cAMP response element (CRE) sequences. The associated transcription factor CRE-binding protein (CREB) regulates differential functional pathways in astrocytes and neurons, with more pronounced effects on mitochondrial metabolism in astrocytes (Pardo et al., 2017). The AGC1 promotor regions contain a CRE site, which in neurons leads to increased AGC1 expression after binding of CREB upon its calcium-induced phosphorylation (Menga et al., 2015). CREB-dependent transcription additionally upregulates Sigma-1 receptor expression, and leads to lowered astrocytic excitability by decreasing ATP-dependent subcellular calcium waves (Eraso-Pichot et al., 2017). ATF4 is another CREB protein. It is part of the Integrated Stress Response (ISR) and is upregulated following mitochondrial disease or inhibition (Silva et al., 2009). An ATF4–CRE interaction is interesting with respect to VWM, considering the constitutive ISR activation in this disease (Abbink et al., 2019; Wong et al., 2019). In addition, Sigma-1 receptor expression is decreased in VWM (Atzmon et al., 2018). Interestingly, basal activation of the ISR in two forebrain astrocyte populations and in Bergmann glia of the cerebellum was significantly higher compared to other cell types (Wong et al., 2019), pointing toward an astrocyte-specific vulnerability to ISR disruption. Differential effects across cell types are further complicated by the interaction of ISR activation and mitochondrial functioning through Transcription factor EB (TFEB) and Transcription Factor A, Mitochondrial (TFAM). TFEB regulates autophagy and lysosomal biogenesis (Settembre et al., 2011) and is activated in response to ER stress and starvation (Martina et al., 2016; Yoneshima et al., 2016). Tfam knockout upregulated Tfeb and lowered NAD+ levels (Baixauli et al., 2015). It caused apoptosis in neurons (Beckervordersandforth et al., 2017) but not in astrocytes, although it did abolish their neuroprotective qualities (Fiebig et al., 2019). These data illustrate the complexity and importance of taking cell type-specific pathway regulation into account when investigating functional effects of (A)GCs.
Conclusion
A prominent role for mitochondrial dysfunction and in particular mitochondrial glutamate pathways has been explored in inherited metabolic disorders characterized by white matter abnormalities. The role of (A)GCs in MAS function, (myelin) metabolism, and glutamate homeostasis, paired with the importance of astrocytes in glutamatergic homeostasis, align well with the central role of glial cells in many metabolic diseases. A more detailed interrogation of GC and AGC (in)activation, in various cell types within a single model, would be of interest in order to elucidate underlying disease mechanisms in white matter disorders.
Author Contributions
AH and VH wrote the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abbink, T. E. M., Wisse, L. E., Jaku, E., Thiecke, M. J., Voltolini-Gonzalez, D., Fritsen, H., et al. (2019). Vanishing white matter: deregulated integrated stress response as therapy target. Ann. Clin. Transl. Neurol. 6, 1407–1422. doi: 10.1002/acn3.50826
Alkan, H. F., Walter, K. E., Luengo, A., Madreiter-Sokolowski, C. T., Stryeck, S., Lau, A. N., et al. (2018). Cytosolic aspartate availability determines cell survival when glutamine is limiting. Cell Metab. 28, 706.e6–720.e6. doi: 10.1016/j.cmet.2018.07.021
Alshaabi, H., Heininger, M., and Cunniff, B. (2019). Dynamic regulation of subcellular mitochondrial position for localized metabolite levels. J. Biochem. 167, 109–117. doi: 10.1093/jb/mvz058
Amoedo, N. D., Punzi, G., Obre, E., Lacombe, D., De Grassi, A., Pierri, C. L., et al. (2016). AGC1/2, the mitochondrial aspartate-glutamate carriers. Biochim. Biophys. Acta 1863, 2394–2412. doi: 10.1016/j.bbamcr.2016.04.011
Anderson, M. A., Burda, J. E., Ren, Y., Ao, Y., O’Shea, T. M., Kawaguchi, R., et al. (2016). Astrocyte scar formation aids central nervous system axon regeneration. Nature 532, 195–200. doi: 10.1038/nature17623
Atzmon, A., Herrero, M., Sharet-Eshed, R., Gilad, Y., Senderowitz, H., and Elroy-Stein, O. (2018). Drug screening identifies sigma-1-receptor as a target for the therapy of VWM leukodystrophy. Front. Mol. Neurosci. 11:336. doi: 10.3389/fnmol.2018.00336
Baixauli, F., Acin-Perez, R., Villarroya-Beltri, C., Mazzeo, C., Nunez-Andrade, N., Gabande-Rodriguez, E., et al. (2015). Mitochondrial respiration controls lysosomal function during inflammatory T cell responses. Cell. Metab. 22, 485–498. doi: 10.1016/j.cmet.2015.07.020
Beckervordersandforth, R., Ebert, B., Schaffner, I., Moss, J., Fiebig, C., Shin, J., et al. (2017). Role of mitochondrial metabolism in the control of early lineage progression and aging phenotypes in adult hippocampal neurogenesis. Neuron 93:1518. doi: 10.1016/j.neuron.2017.03.008
Begum, L., Jalil, M. A., Kobayashi, K., Iijima, M., Li, M. X., Yasuda, T., et al. (2002). Expression of three mitochondrial solute carriers, citrin, aralar1 and ornithine transporter, in relation to urea cycle in mice. Biochim. Biophys. Acta 1574, 283–292. doi: 10.1016/s0167-4781(01)00376-1
Behrends, C., Sowa, M. E., Gygi, S. P., and Harper, J. W. (2010). Network organization of the human autophagy system. Nature 466, 68–76. doi: 10.1038/nature09204
Berkich, D. A., Ola, M. S., Cole, J., Sweatt, A. J., Hutson, S. M., and LaNoue, K. F. (2007). Mitochondrial transport proteins of the brain. J. Neurosci. Res. 85, 3367–3377. doi: 10.1002/jnr.21500
Boisvert, M. M., Erikson, G. A., Shokhirev, M. N., and Allen, N. J. (2018). The aging astrocyte transcriptome from multiple regions of the mouse brain. Cell. Rep. 22, 269–285. doi: 10.1016/j.celrep.2017.12.039
Bolea, I., Gella, A., Sanz, E., Prada-Dacasa, P., Menardy, F., Bard, A. M., et al. (2019). Defined neuronal populations drive fatal phenotype in a mouse model of Leigh syndrome. eLife 8:e47163. doi: 10.7554/eLife.47163
Boulay, A. C., Saubamea, B., Adam, N., Chasseigneaux, S., Mazare, N., Gilbert, A., et al. (2017). Translation in astrocyte distal processes sets molecular heterogeneity at the gliovascular interface. Cell Discov. 3:17005. doi: 10.1038/celldisc.2017.5
Caceres-Marzal, C., Vaquerizo, J., Galan, E., and Fernandez, S. (2006). Early mitochondrial dysfunction in an infant with Alexander disease. Pediatr. Neurol. 35, 293–296. doi: 10.1016/j.pediatrneurol.2006.03.010
Carvalho, K. S. (2013). Mitochondrial dysfunction in demyelinating diseases. Semin. Pediatr. Neurol. 20, 194–201. doi: 10.1016/j.spen.2013.09.001
Castellani, R. J., Perry, G., Harris, P. L., Cohen, M. L., Sayre, L. M., Salomon, R. G., et al. (1998). Advanced lipid peroxidation end-products in Alexander’s disease. Brain Res. 787, 15–18. doi: 10.1016/s0006-8993(97)01224-9
Choquet, K., Forget, D., Meloche, E., Dicaire, M. J., Bernard, G., Vanderver, A., et al. (2019). Leukodystrophy-associated POLR3A mutations down-regulate the RNA polymerase III transcript and important regulatory RNA BC200. J. Biol. Chem. 294, 7445–7459. doi: 10.1074/jbc.RA118.006271
Contreras, L., Gomez-Puertas, P., Iijima, M., Kobayashi, K., Saheki, T., and Satrustegui, J. (2007). Ca2+ Activation kinetics of the two aspartate-glutamate mitochondrial carriers, aralar and citrin: role in the heart malate-aspartate NADH shuttle. J. Biol. Chem. 282, 7098–7106. doi: 10.1074/jbc.M610491200
Contreras, L., Urbieta, A., Kobayashi, K., Saheki, T., and Satrustegui, J. (2010). Low levels of citrin (SLC25A13) expression in adult mouse brain restricted to neuronal clusters. J. Neurosci. Res. 88, 1009–1016. doi: 10.1002/jnr.22283
Dahlin, M., Martin, D. A., Hedlund, Z., Jonsson, M., von Dobeln, U., and Wedell, A. (2015). The ketogenic diet compensates for AGC1 deficiency and improves myelination. Epilepsia 56, e176–e181. doi: 10.1111/epi.13193
Danbolt, N. C., Furness, D. N., and Zhou, Y. (2016). Neuronal vs glial glutamate uptake: resolving the conundrum. Neurochem. Int. 98, 29–45. doi: 10.1016/j.neuint.2016.05.009
Domercq, M., Alberdi, E., Sanchez-Gomez, M. V., Ariz, U., Perez-Samartin, A., and Matute, C. (2011). Dual-specific phosphatase-6 (Dusp6) and ERK mediate AMPA receptor-induced oligodendrocyte death. J. Biol. Chem. 286, 11825–11836. doi: 10.1074/jbc.M110.153049
Dooves, S., Bugiani, M., Postma, N. L., Polder, E., Land, N., Horan, S. T., et al. (2016). Astrocytes are central in the pathomechanisms of vanishing white matter. J. Clin. Invest. 126, 1512–1524. doi: 10.1172/JCI83908
Duchen, M. R. (2000). Mitochondria and calcium: from cell signalling to cell death. J. Physiol. 529(Pt 1), 57–68. doi: 10.1111/j.1469-7793.2000.00057.x
Elitt, M. S., Shick, H. E., Madhavan, M., Allan, K. C., Clayton, B. L. L., Weng, C., et al. (2018). Chemical screening identifies enhancers of mutant oligodendrocyte survival and unmasks a distinct pathological phase in pelizaeus-merzbacher disease. Stem Cell Reports, 11, 711–726. doi: 10.1016/j.stemcr.2018.07.015
Eraso-Pichot, A., Larramona-Arcas, R., Vicario-Orri, E., Villalonga, R., Pardo, L., Galea, E., et al. (2017). CREB decreases astrocytic excitability by modifying subcellular calcium fluxes via the sigma-1 receptor. Cell Mol. Life. Sci. 74, 937–950. doi: 10.1007/s00018-016-2397-5
Falk, M. J., Li, D., Gai, X., McCormick, E., Place, E., Lasorsa, F. M., et al. (2014). AGC1 deficiency causes infantile epilepsy, abnormal myelination, and reduced N-acetylaspartate. JIMD Rep. 14, 77–85. doi: 10.1007/8904_2013_287
Fiebig, C., Keiner, S., Ebert, B., Schaffner, I., Jagasia, R., Lie, D. C., et al. (2019). Mitochondrial dysfunction in astrocytes impairs the generation of reactive astrocytes and enhances neuronal cell death in the cortex upon photothrombotic lesion. Front. Mol. Neurosci. 12:40. doi: 10.3389/fnmol.2019.00040
Fiermonte, G., Palmieri, L., Todisco, S., Agrimi, G., Palmieri, F., and Walker, J. E. (2002). Identification of the mitochondrial glutamate transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution of two human isoforms. J. Biol. Chem. 277, 19289–19294. doi: 10.1074/jbc.M201572200
Frigerio, F., Casimir, M., Carobbio, S., and Maechler, P. (2008). Tissue specificity of mitochondrial glutamate pathways and the control of metabolic homeostasis. Biochim. Biophys. Acta 1777, 965–972. doi: 10.1016/j.bbabio.2008.04.031
Gao, L., Zhang, Z., Lu, J., and Pei, G. (2019). Mitochondria are dynamically transferring between human neural cells and alexander disease-associated GFAP mutations impair the astrocytic transfer. Front. Cell. Neurosci. 13:316. doi: 10.3389/fncel.2019.00316
Gellerich, F. N., Gizatullina, Z., Trumbekaite, S., Korzeniewski, B., Gaynutdinov, T., Seppet, E., et al. (2012). Cytosolic Ca2+ regulates the energization of isolated brain mitochondria by formation of pyruvate through the malate-aspartate shuttle. Biochem. J. 443, 747–755. doi: 10.1042/BJ20110765
Genda, E. N., Jackson, J. G., Sheldon, A. L., Locke, S. F., Greco, T. M., O’Donnell, J. C., et al. (2011). Co-compartmentalization of the astroglial glutamate transporter, GLT-1, with glycolytic enzymes and mitochondria. J. Neurosci. 31, 18275–18288. doi: 10.1523/JNEUROSCI.3305-11.2011
Goubert, E., Mircheva, Y., Lasorsa, F. M., Melon, C., Profilo, E., Sutera, J., et al. (2017). Inhibition of the mitochondrial glutamate carrier SLC25A22 in astrocytes leads to intracellular glutamate accumulation. Front. Cell Neurosci. 11:149. doi: 10.3389/fncel.2017.00149
Hagemann, T. L., Connor, J. X., and Messing, A. (2006). Alexander disease-associated glial fibrillary acidic protein mutations in mice induce Rosenthal fiber formation and a white matter stress response. J. Neurosci. 26, 11162–11173. doi: 10.1523/JNEUROSCI.3260-06.2006
Hagemann, T. L., Jobe, E. M., and Messing, A. (2012). Genetic ablation of Nrf2/antioxidant response pathway in Alexander disease mice reduces hippocampal gliosis but does not impact survival. PLoS One 7:e37304. doi: 10.1371/journal.pone.0037304
Hans, C. P., Sharma, N., Sen, S., Zeng, S., Dev, R., Jiang, Y., et al. (2019). Transcriptomics analysis reveals new insights into the roles of notch1 signaling on macrophage polarization. Sci. Rep. 9:7999. doi: 10.1038/s41598-019-44266-4
Hassel, B., Boldingh, K. A., Narvesen, C., Iversen, E. G., and Skrede, K. K. (2003). Glutamate transport, glutamine synthetase and phosphate-activated glutaminase in rat CNS white matter. A quantitative study. J. Neurochem. 87, 230–237. doi: 10.1046/j.1471-4159.2003.01984.x
Herbst, E. A., and Holloway, G. P. (2016). Exercise increases mitochondrial glutamate oxidation in the mouse cerebral cortex. Appl. Physiol. Nutr. Metab. 41, 799–801. doi: 10.1139/apnm-2016-0033
Herndon, R. M., Rubinstein, L. J., Freeman, J. M., and Mathieson, G. (1970). Light and electron microscopic observations on Rosenthal fibers in Alexander’s disease and in multiple sclerosis. J. Neuropathol. Exp. Neurol. 29, 524–551. doi: 10.1097/00005072-197010000-00002
Hertz, L. (2011). Brain glutamine synthesis requires neuronal aspartate: a commentary. J. Cereb. Blood Flow Metab. 31, 384–387. doi: 10.1038/jcbfm.2010.199
Hudson, R. C., and Daniel, R. M. (1993). L-glutamate dehydrogenases: distribution, properties and mechanism. Comp. Biochem. Physiol. B 106, 767–792. doi: 10.1016/0305-0491(93)90031-y
Iijima, M., Jalil, A., Begum, L., Yasuda, T., Yamaguchi, N., Xian Li, M., et al. (2001). Pathogenesis of adult-onset type II citrullinemia caused by deficiency of citrin, a mitochondrial solute carrier protein: tissue and subcellular localization of citrin. Adv. Enzyme Regul. 41, 325–342. doi: 10.1016/s0065-2571(00)00022-4
Jalil, M. A., Begum, L., Contreras, L., Pardo, B., Iijima, M., Li, M. X., et al. (2005). Reduced N-acetylaspartate levels in mice lacking aralar, a brain- and muscle-type mitochondrial aspartate-glutamate carrier. J. Biol. Chem. 280, 31333–31339. doi: 10.1074/jbc.M505286200
Jones, J. R., Kong, L., Hanna, M. G. T., Hoffman, B., Krencik, R., Bradley, R., et al. (2018). Mutations in GFAP disrupt the distribution and function of organelles in human astrocytes. Cell Rep. 25, 947.e4–958.e4. doi: 10.1016/j.celrep.2018.09.083
Juaristi, I., Contreras, L., Gonzalez-Sanchez, P., Perez-Liebana, I., Gonzalez-Moreno, L., Pardo, B., et al. (2019). The response to stimulation in neurons and astrocytes. Neurochem. Res. 44, 2385–2391. doi: 10.1007/s11064-019-02803-7
Kannurpatti, S. S., Joshi, P. G., and Joshi, N. B. (2000). Calcium sequestering ability of mitochondria modulates influx of calcium through glutamate receptor channel. Neurochem. Res. 25, 1527–1536. doi: 10.1023/a:1026602100160
Kasai, S., Yamazaki, H., Tanji, K., Engler, M. J., Matsumiya, T., and Itoh, K. (2019). Role of the ISR-ATF4 pathway and its cross talk with Nrf2 in mitochondrial quality control. J. Clin. Biochem. Nutr. 64, 1–12. doi: 10.3164/jcbn.18-37
Kobayashi, K., Sinasac, D. S., Iijima, M., Boright, A. P., Begum, L., Lee, J. R., et al. (1999). The gene mutated in adult-onset type II citrullinaemia encodes a putative mitochondrial carrier protein. Nat. Genet. 22, 159–163. doi: 10.1038/9667
Kritis, A. A., Stamoula, E. G., Paniskaki, K. A., and Vavilis, T. D. (2015). Researching glutamate - induced cytotoxicity in different cell lines: a comparative/collective analysis/study. Front. Cell Neurosci. 9:91. doi: 10.3389/fncel.2015.00091
Lasorsa, F. M., Pinton, P., Palmieri, L., Fiermonte, G., Rizzuto, R., and Palmieri, F. (2003). Recombinant expression of the Ca(2+)-sensitive aspartate/glutamate carrier increases mitochondrial ATP production in agonist-stimulated Chinese hamster ovary cells. J. Biol. Chem. 278, 38686–38692. doi: 10.1074/jbc.M304988200
Leferink, P. S., Dooves, S., Hillen, A. E. J., Watanabe, K., Jacobs, G., Gasparotto, L., et al. (2019). Astrocyte subtype vulnerability in stem cell models of vanishing white matter. Ann. Neurol. 86, 780–792. doi: 10.1002/ana.25585
Li, B., Hertz, L., and Peng, L. (2012). Aralar mRNA and protein levels in neurons and astrocytes freshly isolated from young and adult mouse brain and in maturing cultured astrocytes. Neurochem. Int. 61, 1325–1332. doi: 10.1016/j.neuint.2012.09.009
Li, L., Tian, E., Chen, X., Chao, J., Klein, J., Qu, Q., et al. (2018). GFAP mutations in astrocytes impair oligodendrocyte progenitor proliferation and myelination in an hiPSC model of alexander disease. Cell Stem Cell 23, 239.e6–251.e6. doi: 10.1016/j.stem.2018.07.009
Li, X., Chung, A. C. K., Li, S., Wu, L., Xu, J., Yu, J., et al. (2017). LC-MS-based metabolomics revealed SLC25A22 as an essential regulator of aspartate-derived amino acids and polyamines in KRAS-mutant colorectal cancer. Oncotarget 8, 101333–101344. doi: 10.18632/oncotarget.21093
Llavero Hurtado, M., Fuller, H. R., Wong, A. M. S., Eaton, S. L., Gillingwater, T. H., Pennetta, G., et al. (2017). Proteomic mapping of differentially vulnerable pre-synaptic populations identifies regulators of neuronal stability in vivo. Sci. Rep. 7:12412. doi: 10.1038/s41598-017-12603-0
Llorente-Folch, I., Rueda, C. B., Perez-Liebana, I., Satrustegui, J., and Pardo, B. (2016). L-Lactate-mediated neuroprotection against glutamate-induced excitotoxicity requires ARALAR/AGC1. J. Neurosci. 36, 4443–4456. doi: 10.1523/JNEUROSCI.3691-15.2016
Longuemare, M. C., Rose, C. R., Farrell, K., Ransom, B. R., Waxman, S. G., and Swanson, R. A. (1999). K(+)-induced reversal of astrocyte glutamate uptake is limited by compensatory changes in intracellular Na+. Neuroscience 93, 285–292. doi: 10.1016/s0306-4522(99)00152-9
Luiro, K., Kopra, O., Blom, T., Gentile, M., Mitchison, H. M., Hovatta, I., et al. (2006). Batten disease (JNCL) is linked to disturbances in mitochondrial, cytoskeletal, and synaptic compartments. J. Neurosci. Res. 84, 1124–1138. doi: 10.1002/jnr.21015
Lundgaard, I., Osorio, M. J., Kress, B. T., Sanggaard, S., and Nedergaard, M. (2014). White matter astrocytes in health and disease. Neuroscience 276, 161–173. doi: 10.1016/j.neuroscience.2013.10.050
Mahmoud, S., Gharagozloo, M., Simard, C., and Gris, D. (2019). Astrocytes maintain glutamate homeostasis in the CNS by controlling the balance between glutamate uptake and release. Cells 8:184. doi: 10.3390/cells8020184
Martina, J. A., Diab, H. I., Brady, O. A., and Puertollano, R. (2016). TFEB and TFE3 are novel components of the integrated stress response. EMBO J. 35, 479–495. doi: 10.15252/embj.201593428
Mathiisen, T. M., Lehre, K. P., Danbolt, N. C., and Ottersen, O. P. (2010). The perivascular astroglial sheath provides a complete covering of the brain microvessels: an electron microscopic 3D reconstruction. Glia 58, 1094–1103. doi: 10.1002/glia.20990
McKenna, M. C., Waagepetersen, H. S., Schousboe, A., and Sonnewald, U. (2006). Neuronal and astrocytic shuttle mechanisms for cytosolic-mitochondrial transfer of reducing equivalents: current evidence and pharmacological tools. Biochem. Pharmacol. 71, 399–407. doi: 10.1016/j.bcp.2005.10.011
Meijer, A. J., Brouwer, A., Reijngoud, D. J., Hoek, J. B., and Tager, J. M. (1972). Transport of glutamate in rat-liver mitochondria. Biochim. Biophys. Acta 283, 421–429. doi: 10.1016/0005-2728(72)90259-9
Menga, A., Iacobazzi, V., Infantino, V., Avantaggiati, M. L., and Palmieri, F. (2015). The mitochondrial aspartate/glutamate carrier isoform 1 gene expression is regulated by CREB in neuronal cells. Int. J. Biochem. Cell Biol. 60, 157–166. doi: 10.1016/j.biocel.2015.01.004
Molinari, F., Raas-Rothschild, A., Rio, M., Fiermonte, G., Encha-Razavi, F., Palmieri, L., et al. (2005). Impaired mitochondrial glutamate transport in autosomal recessive neonatal myoclonic epilepsy. Am. J. Hum. Genet. 76, 334–339. doi: 10.1086/427564
Molinari, F., Kaminska, A., Fiermonte, G., Boddaert, N., Raas-Rothschild, A., Plouin, P., et al. (2009). Mutations in the mitochondrial glutamate carrier SLC25A22 in neonatal epileptic encephalopathy with suppression bursts. Clin. Genet. 76, 188–194. doi: 10.1111/j.1399-0004.2009.01236.x
Moriyama, M., Li, M. X., Kobayashi, K., Sinasac, D. S., Kannan, Y., Iijima, M., et al. (2006). Pyruvate ameliorates the defect in ureogenesis from ammonia in citrin-deficient mice. J. Hepatol. 44, 930–938. doi: 10.1016/j.jhep.2005.09.018
Nelvagal, H. R., Lange, J., Takahashi, K., Tarczyluk-Wells, M. A., and Cooper, J. D. (2019). Pathomechanisms in the neuronal ceroid lipofuscinoses. Biochim. Biophys. Acta Mol. Basis Dis. 2019:165570. doi: 10.1016/j.bbadis.2019.165570
Nunnari, J., and Suomalainen, A. (2012). Mitochondria: in sickness and in health. Cell 148, 1145–1159. doi: 10.1016/j.cell.2012.02.035
Orre, M., Kamphuis, W., Osborn, L. M., Melief, J., Kooijman, L., Huitinga, I., et al. (2014). Acute isolation and transcriptome characterization of cortical astrocytes and microglia from young and aged mice. Neurobiol. Aging 35, 1–14. doi: 10.1016/j.neurobiolaging.2013.07.008
Orrenius, S., Zhivotovsky, B., and Nicotera, P. (2003). Regulation of cell death: the calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 4, 552–565. doi: 10.1038/nrm1150
Palmieri, L., Pardo, B., Lasorsa, F. M., del Arco, A., Kobayashi, K., Iijima, M., et al. (2001). Citrin and aralar1 are Ca(2+)-stimulated aspartate/glutamate transporters in mitochondria. EMBO J. 20, 5060–5069. doi: 10.1093/emboj/20.18.5060
Pardo, L., Valor, L. M., Eraso-Pichot, A., Barco, A., Golbano, A., Hardingham, G. E., et al. (2017). CREB regulates distinct adaptive transcriptional programs in astrocytes and neurons. Sci. Rep. 7:6390. doi: 10.1038/s41598-017-06231-x
Parviainen, L., Dihanich, S., Anderson, G. W., Wong, A. M., Brooks, H. R., Abeti, R., et al. (2017). Glial cells are functionally impaired in juvenile neuronal ceroid lipofuscinosis and detrimental to neurons. Acta Neuropathol. Commun. 5:74. doi: 10.1186/s40478-017-0476-y
Petralla, S., Pena-Altamira, L. E., Poeta, E., Massenzio, F., Virgili, M., Barile, S. N., et al. (2019). Deficiency of mitochondrial aspartate-glutamate carrier 1 leads to oligodendrocyte precursor cell proliferation defects both in vitro and in vivo. Int. J. Mol. Sci. 20:4486. doi: 10.3390/ijms20184486
Ploumi, C., Daskalaki, I., and Tavernarakis, N. (2017). Mitochondrial biogenesis and clearance: a balancing act. FEBS J. 284, 183–195. doi: 10.1111/febs.13820
Poduri, A., Heinzen, E. L., Chitsazzadeh, V., Lasorsa, F. M., Elhosary, P. C., LaCoursiere, C. M., et al. (2013). SLC25A22 is a novel gene for migrating partial seizures in infancy. Ann. Neurol. 74, 873–882. doi: 10.1002/ana.23998
Profilo, E., Pena-Altamira, L. E., Corricelli, M., Castegna, A., Danese, A., Agrimi, G., et al. (2017). Down-regulation of the mitochondrial aspartate-glutamate carrier isoform 1 AGC1 inhibits proliferation and N-acetylaspartate synthesis in Neuro2A cells. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 1422–1435. doi: 10.1016/j.bbadis.2017.02.022
Raini, G., Sharet, R., Herrero, M., Atzmon, A., Shenoy, A., Geiger, T., et al. (2017). Mutant eIF2B leads to impaired mitochondrial oxidative phosphorylation in vanishing white matter disease. J. Neurochem. 141, 694–707. doi: 10.1111/jnc.14024
Ramos, M., del Arco, A., Pardo, B., Martinez-Serrano, A., Martinez-Morales, J. R., Kobayashi, K., et al. (2003). Developmental changes in the Ca2+-regulated mitochondrial aspartate-glutamate carrier aralar1 in brain and prominent expression in the spinal cord. Brain Res. Dev. Brain Res. 143, 33–46. doi: 10.1016/s0165-3806(03)00097-x
Saffari, A., Kolker, S., Hoffmann, G. F., and Ebrahimi-Fakhari, D. (2017). Linking mitochondrial dysfunction to neurodegeneration in lysosomal storage diseases. J. Inherit. Metab. Dis. 40, 631–640. doi: 10.1007/s10545-017-0048-0
Saheki, T., Kobayashi, K., Iijima, M., Nishi, I., Yasuda, T., Yamaguchi, N., et al. (2002). Pathogenesis and pathophysiology of citrin (a mitochondrial aspartate glutamate carrier) deficiency. Metab. Brain Dis. 17, 335–346. doi: 10.1023/a:1021961919148
Satrustegui, J., Pardo, B., and Del Arco, A. (2007). Mitochondrial transporters as novel targets for intracellular calcium signaling. Physiol. Rev. 87, 29–67. doi: 10.1152/physrev.00005.2006
Schinder, A. F., Olson, E. C., Spitzer, N. C., and Montal, M. (1996). Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J. Neurosci. 16, 6125–6133. doi: 10.1523/jneurosci.16-19-06125.1996
Schoenfeld, R., Wong, A., Silva, J., Li, M., Itoh, A., Horiuchi, M., et al. (2010). Oligodendroglial differentiation induces mitochondrial genes and inhibition of mitochondrial function represses oligodendroglial differentiation. Mitochondrion 10, 143–150. doi: 10.1016/j.mito.2009.12.141
Scifo, E., Szwajda, A., Debski, J., Uusi-Rauva, K., Kesti, T., Dadlez, M., et al. (2013). Drafting the CLN3 protein interactome in SH-SY5Y human neuroblastoma cells: a label-free quantitative proteomics approach. J. Proteome Res. 12, 2101–2115. doi: 10.1021/pr301125k
Settembre, C., Di Malta, C., Polito, V. A., Garcia Arencibia, M., Vetrini, F., Erdin, S., et al. (2011). TFEB links autophagy to lysosomal biogenesis. Science 332, 1429–1433. doi: 10.1126/science.1204592
Silva, J. M., Wong, A., Carelli, V., and Cortopassi, G. A. (2009). Inhibition of mitochondrial function induces an integrated stress response in oligodendroglia. Neurobiol. Dis. 34, 357–365. doi: 10.1016/j.nbd.2009.02.005
Sofroniew, M. V. (2015). Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci. 16, 249–263. doi: 10.1038/nrn3898
Stumvoll, M., Meyer, C., Mitrakou, A., Nadkarni, V., and Gerich, J. E. (1997). Renal glucose production and utilization: new aspects in humans. Diabetologia 40, 749–757. doi: 10.1007/s001250050745
Sun, S., Sun, Y., Ling, S. C., Ferraiuolo, L., McAlonis-Downes, M., Zou, Y., et al. (2015). Translational profiling identifies a cascade of damage initiated in motor neurons and spreading to glia in mutant SOD1-mediated ALS. Proc. Natl. Acad. Sci. U.S.A. 112, E6993–E7002. doi: 10.1073/pnas.1520639112
Suomalainen, A., and Battersby, B. J. (2018). Mitochondrial diseases: the contribution of organelle stress responses to pathology. Nat. Rev. Mol. Cell Biol. 19, 77–92. doi: 10.1038/nrm.2017.66
Tian, R., Wu, X., Hagemann, T. L., Sosunov, A. A., Messing, A., McKhann, G. M., et al. (2010). Alexander disease mutant glial fibrillary acidic protein compromises glutamate transport in astrocytes. J. Neuropathol. Exp. Neurol. 69, 335–345. doi: 10.1097/NEN.0b013e3181d3cb52
Ugbode, C. I., Hirst, W. D., and Rattray, M. (2014). Neuronal influences are necessary to produce mitochondrial co-localization with glutamate transporters in astrocytes. J. Neurochem. 130, 668–677. doi: 10.1111/jnc.12759
Vos, M., Lauwers, E., and Verstreken, P. (2010). Synaptic mitochondria in synaptic transmission and organization of vesicle pools in health and disease. Front. Synaptic Neurosci. 2:139. doi: 10.3389/fnsyn.2010.00139
Wang, G. J., and Thayer, S. A. (1996). Sequestration of glutamate-induced Ca2+ loads by mitochondria in cultured rat hippocampal neurons. J. Neurophysiol. 76, 1611–1621. doi: 10.1152/jn.1996.76.3.1611
Wang, L., Colodner, K. J., and Feany, M. B. (2011). Protein misfolding and oxidative stress promote glial-mediated neurodegeneration in an Alexander disease model. J. Neurosci. 31, 2868–2877. doi: 10.1523/JNEUROSCI.3410-10.2011
Wibom, R., Lasorsa, F. M., Tohonen, V., Barbaro, M., Sterky, F. H., Kucinski, T., et al. (2009). AGC1 deficiency associated with global cerebral hypomyelination. N. Engl. J. Med. 361, 489–495. doi: 10.1056/NEJMoa0900591
Wong, K., Armstrong, R. C., Gyure, K. A., Morrison, A. L., Rodriguez, D., Matalon, R., et al. (2000). Foamy cells with oligodendroglial phenotype in childhood ataxia with diffuse central nervous system hypomyelination syndrome. Acta Neuropathol. 100, 635–646. doi: 10.1007/s004010000234
Wong, Y. L., LeBon, L., Basso, A. M., Kohlhaas, K. L., Nikkel, A. L., Robb, H. M., et al. (2019). eIF2B activator prevents neurological defects caused by a chronic integrated stress response. eLife 8:e42940. doi: 10.7554/eLife.42940
Keywords: mitochondria, glutamate carriers, aspartate-glutamate carriers, white matter, leukodystrophy, excitotoxicity, cell type specificity, cell metabolism
Citation: Hillen AEJ and Heine VM (2020) Glutamate Carrier Involvement in Mitochondrial Dysfunctioning in the Brain White Matter. Front. Mol. Biosci. 7:151. doi: 10.3389/fmolb.2020.00151
Received: 14 April 2020; Accepted: 18 June 2020;
Published: 21 July 2020.
Edited by:
Caroline Sevin, Bicêtre Hospital, FranceReviewed by:
Uma Gaur, University of Macau, ChinaSergio Akira Uyemura, University of São Paulo, Brazil
Copyright © 2020 Hillen and Heine. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vivi M. Heine, dm0uaGVpbmVAYW1zdGVyZGFtdW1jLm5s