 María Moreno-del Alamo1
María Moreno-del Alamo1 Rubén Torres1
Rubén Torres1 Candela Manfredi1†
Candela Manfredi1† José A. Ruiz-Masó2
José A. Ruiz-Masó2 Gloria del Solar2
Gloria del Solar2 Juan Carlos Alonso1*
Juan Carlos Alonso1*- 1Department of Microbial Biotechnology, Centro Nacional de Biotecnología, CNB-CSIC, Madrid, Spain
- 2Centro de Investigaciones Biológicas Margarita Salas, CIB-CSIC, Madrid, Spain
Bacillus subtilis PcrA abrogates replication-transcription conflicts in vivo and disrupts RecA nucleoprotein filaments in vitro. Inactivation of pcrA is lethal. We show that PcrA depletion lethality is suppressed by recJ (involved in end resection), recA (the recombinase), or mfd (transcription-coupled repair) inactivation, but not by inactivating end resection (addAB or recQ), positive and negative RecA modulators (rarA or recX and recU), or genes involved in the reactivation of a stalled RNA polymerase (recD2, helD, hepA, and ywqA). We also report that B. subtilis mutations previously designated as recL16 actually map to the recO locus, and confirm that PcrA depletion lethality is suppressed by recO inactivation. The pcrA gene is epistatic to recA or mfd, but it is not epistatic to addAB, recJ, recQ, recO16, rarA, recX, recU, recD2, helD, hepA, or ywqA in response to DNA damage. PcrA depletion led to the accumulation of unsegregated chromosomes, and this defect is increased by recQ, rarA, or recU inactivation. We propose that PcrA, which is crucial to maintain cell viability, is involved in different DNA transactions.
Introduction
Homologous recombination is the major pathway to circumvent a replicative stress, a replication fork collapse and for the elimination of DNA double-strand breaks (DSBs) induced by endogenous or exogenous stress. Super-family 1 (SF1) DNA helicases, which are conserved motor proteins that couple nucleoside triphosphate hydrolysis to the unwinding of duplex DNA, play crucial roles in repair-by-recombination and in coping with replication-transcription conflicts (RTCs) (Wu and Hickson, 2006; Singleton et al., 2007). The prototype of bacterial SF1 helicases that translocate with 3′ → 5′ direction is UvrD (Singleton et al., 2007; Dillingham, 2011). This enzyme shares a significant degree of structural similarity with Rep, which is restricted to the γ-Proteobacteria Class, PcrA and yeast Srs2 DNA helicases (Wu and Hickson, 2006; Marini and Krejci, 2010). Rep and PcrA play essential roles in the replication of extrachromosomal elements, whereas UvrD and PcrA participate in the resolution of RTCs by poorly understood mechanisms (Boubakri et al., 2010; Bruning et al., 2014; Epshtein et al., 2014; Merrikh et al., 2015). In vitro studies reveal that PcrA and UvrD interact with the RNA polymerase (RNAP), Rep interacts with the replicative DNA helicase (DnaB in Proteobacteria) and Srs2 physically interacts with Rad51 and with the PCNA sliding clamp (ortholog of bacterial DnaN) among other proteins (Antony et al., 2009; Guy et al., 2009; Kaniecki et al., 2017; Sanders et al., 2017). Absence of Escherichia coli Rep and UvrD renders cells inviable when grown in rich medium (Taucher-Scholtz et al., 1983), but lack of Bacillus subtilis PcrA renders cells inviable even when grown in minimal medium (Petit et al., 1998; Merrikh et al., 2015). To gain insight into the crucial steps carried on by PcrA, a comparative analysis with UvrD and Rep was undertaken. From the comparative analysis of E. coli (best-characterized representative of the Proteobacteria Phylum) and B. subtilis (best-characterized from the Firmicutes Phylum), which are evolutionarily separated by more than 2,000 million years, a genetic divergence larger than that between human and paramecium, we expect to understand the role of the SF1 UvrD-like DNA helicases.
When the DNA of a single inert mature haploid non-replicating chromosome of a B. subtilis spore is damaged, the RTCs of the single spore genome are compounded during the rapid outgrowth. In the absence of end resection (AddAB and/or RecJ-RecQ [RecS]) cells remain recombination proficient and apparently are as capable of repairing damaged template bases as the wild type (wt) control (Vlasic et al., 2014). These spores, which lack an intact homologous template, require RecA mediators (RecO, RecR), RecA itself, and positive (RecF) and negative (RecX, RecU) RecA modulators, with PcrA facilitating DNA replication through transcription units (Vlasic et al., 2014; Merrikh et al., 2015; Raguse et al., 2017). It is likely that PcrA is implicated in the processing of damaged replication forks and/or recombination intermediates formed at damaged forks and in circumventing DNA lesions in the absence of an intact homologous template, without generating a fork breakage that should be lethal for the revival of a haploid spore (Raguse et al., 2017). Then, a nucleoprotein RecA filament aids to overcome RTCs via different DNA damage tolerance pathways and to reactivate replication by recruiting the damage checkpoint DisA and pre-primosome DnaD proteins (Million-Weaver et al., 2015; Torres et al., 2019). This is consistent with the observation that the lethality of B. subtilis ΔpcrA cells is suppressed by recF17, recL16, ΔrecO, or ΔrecR mutations (Petit and Ehrlich, 2002).
In E. coli cells, the synthetic lethality of ΔuvrD Δrep is partially suppressed by recJ, recQ, recO, recR, or recF inactivation in minimal medium, but it is only marginally suppressed by recA inactivation (Lestini and Michel, 2008; Guy et al., 2009). Inactivation of recJ or recQ provides very limited suppression of Δrep ΔuvrD rich medium lethality, but recA inactivation does not (Petit and Ehrlich, 2002; Veaute et al., 2005; Lestini and Michel, 2008; Guy et al., 2009). in vivo assays reveal that PcrA expression in E. coli can substitute several functions of UvrD, but antagonizes the function of Rep, providing a heterologous dominant negative phenotype (Petit et al., 1998). These data suggest that in the absence of UvrD and Rep, toxic RecA nucleoprotein filaments and/or RecA-mediated recombination intermediates or DNA structures can accumulate. This hypothesis is supported by the following observations: (i) in budding yeast, suppressors of Srs2 mutations map in the Rad51 gene (Aboussekhra et al., 1992); and (ii) RecA- or Rad51-mediated DNA strand exchange is actively prevented by the PcrA, UvrD, or Srs2 DNA helicase in vitro, considered as a paradigmatic anti-recombinase activity (Krejci et al., 2003; Veaute et al., 2003, 2005; Anand et al., 2007; Park et al., 2010; Fagerburg et al., 2012; Petrova et al., 2015; Kaniecki et al., 2017).
Other studies have demonstrated that the rich-medium synthetic lethality of ΔuvrD Δrep cells is caused primarily by RTCs, with partial reduction of transcription or translation rates across heavily transcribed genes in the opposite orientation relative to the replication forks, to compensate the impact of transcription on DNA replication (Guy et al., 2009; Baharoglu et al., 2010; Kamarthapu et al., 2016; Myka et al., 2017). Indeed, rich-medium synthetic lethality of ΔuvrD Δrep cells is fully suppressed by reducing transcription, as mutations in different RNAP subunits (rpo* [rpoB and rpoC point mutants]), or reducing translation elongation, such as by mutations in a tRNA gene (AspRS, aspT), in an aminoacyl tRNA synthetase, in a translation factor needed for efficient formation of proline-proline bonds (EF-P), and spoT1 mutation [encoding a (p)ppGpp pyrophosphorylase-defective SpoT] (Guy et al., 2009; Baharoglu et al., 2010; Kamarthapu et al., 2016; Myka et al., 2017). Transcription and translation are coupled with the leading ribosome pushing RNAP forward; however, when these two processes become uncoupled, RNAP rpo* mutants are prone to pausing with (p)ppGpp promoting UvrD-mediated RNAP backtracking (Kamarthapu et al., 2016; Myka et al., 2017). These observations altogether open the question about the primary cause of PcrA lethality. (Unless stated otherwise, indicated genes and products are of B. subtilis origin. The nomenclature used to denote the origin of proteins from other bacteria is based on the bacterial genus and species abbreviation [e.g., E. coli UvrD is referred to as UvrDEco]).
The physiological causes of PcrA/UvrD-RepEco lethality is/are poorly understood. To understand the role of PcrA in rich medium exponentially growing B. subtilis cells, we have studied the genetic linkage of PcrA depletion (Merrikh et al., 2015) with mutations in genes acting at the presynaptic (ΔrecJ, ΔrecQ, ΔaddAB, recL16, ΔrarA, ΔrecX, ΔrecU) and synaptic (ΔrecA) stages, as well as in genes that contribute to bypass RTCs or facilitate RNAP backtracking or removal (ΔhelD, ΔrecD2 [absent in E. coli], ΔhepA [also termed yqhHor rapA], ΔywqA or Δmfd). We show that PcrA depletion reduced cell viability by >4,000-fold, and survival in the presence of limiting H2O2 or methyl methanesulfonate (MMS) concentrations, suggesting that PcrA is involved in repair-by-recombination. The recL16 mutations were mapped in the recO gene and the mutation was termed recO16. The PcrA depletion lethality is suppressed by recO16, recJ, recA, or mfd inactivation, but not by recQ, rarA, recX, recU, addAB, helD, hepA, or ywqA inactivation. The pcrA gene is not epistatic to recO16, recJ, rarA, recQ, recX, recU, recD2, addAB, helD, hepA, or ywqA in response to MMS-induced DNA damage, but it is epistatic to recA or mfd. Absence of PcrA promotes a net accumulation of unsegregated nucleoids, and this defect is increased in the absence of RarA, RecQ, and RecU. We conclude that PcrA contributes to untangle branched intermediates and works at the interface of DNA replication, transcription, recombination and segregation.
Materials and Methods
Bacterial Strains and Plasmids
All B. subtilis strains used derived from BG214, and are listed in Table 1. The gene to be characterized was deleted by gene replacement with the six-cat-six (SCS) cassette flanked by appropriate homologous regions up- and downstream. The SCS cassette, composed of two directly oriented β-recombinase cognate sites (six sites) and the cat gene, confers chloramphenicol resistance (CmR) (Rojo and Alonso, 1995). Natural competent cells were transformed with the SCS cassette flanked by homologous regions to the gene to be deleted with selection for CmR. Integration of the SCS cassette, through double crossover recombination, replaced the gene under characterization. The β recombinase promoter, which maps within the six site, can read the downstream gene to overcome any potential polar effect (Rojo et al., 1994). A plasmid-borne β-recombinase gene was moved into the background, and followed by β site-specific recombinase-mediated excision between the two directly oriented six sites, leading to the deletion of the cat gene and one six site (Rojo et al., 1994). The final outcome of this strategy is that the gene to be characterized is replaced by a single six site (Sanchez et al., 2005). Accuracy of deletions was confirmed by complementation with a plasmid-borne gene (Sanchez et al., 2005). The IPTG-inducible sspB cassette encoding the SspB adaptor protein (a gift from Houra and Christopher Merrikh, Vanderbilt University, USA) was ectopically integrated into the amy locus by natural transformation (Alonso et al., 1988; Torres et al., 2017). The null hepA or ywqA mutations (a gift from Marie-Agnès Petit, Université Paris-Saclay, France) were moved into the BG214 background (Table 1) by SPP1-mediated transduction (Valero-Rello et al., 2017).
Table 1. Bacillus subtilis strains.
The ssrA degradation tag fused to the 3′-end of the pcrA gene (pcrA-ssrA) replaces the pcrA gene (Merrikh et al., 2015; Torres et al., 2017). The pcrA-ssrA gene was moved by SPP1-mediated transduction into recO16 sspB, ΔrecJ sspB, ΔrecD2 sspB, ΔrecQ sspB, ΔrecX sspB, ΔrecU sspB, ΔrarA sspB, ΔaddAB sspB, ΔhelD sspB, ΔhepA sspB, ΔywqA sspB, or Δmfd sspB strain as well as in the otherwise wt background (rec+ sspB strain). The ΔrecA mutation was mobilized by SPP1-mediated transduction into the pcrA-ssrA sspB (pcrAT) context.
To segregate the recO mutations and to reconstruct a new BG107 strain (BG107-1), limiting concentrations of chromosomal DNA of the original BG107 (recL16) strain and 0.1 μg/ml of pHP14 DNA were added to competent BG214 cells, with selection for the plasmid-borne CmR marker. From those transformants it was expected that by co-transformation of any unlinked markers (congression) 0.1 to 1% of the CmR cells should receive the recL16 MMS sensitive (MMSS) phenotype (Alonso et al., 1988). Selection of MMSS was performed by streaking colonies on agar plate containing or lacking MMS (Alonso et al., 1988). Five selected MMSS clones were sequenced by high-throughput sequence analyzer (Illumina) technology using standard sequencing libraries and filtered sequence data (Beijing Genomics Institute [BGI]), of ~1 gigabases per sample, followed by whole-genome comparison with the nucleotide sequencing of the MMSR parental BG214 strain.
The K37A, T65I, or Q254A pcrA variants were generated by means of mutation site directed mutagenesis (QuickChange Kit, Stratagene) using the pQE-1-borne wt pcrA plasmid as a template. Unexpectedly, all the Q254A mutants analyzed also contained the unselected E224V mutation. K37A, T65I, or Q254A-E224V pcrA genes were amplified by PCR and were cloned into XmaI-BamHI-cleaved pHP14. The resultant recombinant plasmids were used to transform competent B. subtilis BG214 cells.
Survival Assays and Colony Size
Plating exponentially growing pcrA-ssrA sspB (pcrAT) cells in rich medium onto agar plates containing isopropyl-β-D thiogalactopyranoside (IPTG) induced SspB expression from a regulated promoter, which then bound the SsrA peptide tag and rapidly delivered the tagged PcrA-SsrA protein to the B. subtilis ClpXP protease for degradation (PcrA degron [pcrAT] strain) (Keiler et al., 1996; Griffith and Grossman, 2008; Merrikh et al., 2015). PcrA degron cultures were grown to OD560 = 0.4. The cultures were divided and aliquots plated in LB agar plates alone or with 500 μM IPTG (Calbiochem). The percentage of colony forming units (CFUs) in LB agar plates containing IPTG was measured. The mean and SEM were calculated using Prism 6 software (GraphPad), and a Student's t-test, with P < 0.01, was performed to denote the threshold of significance.
Cell sensitivity to chronic MMS (Sigma Aldrich) or H2O2 (Sigma Aldrich) exposure was determined by growing cultures to OD560 = 0.4 and plating appropriate dilutions on rich LB agar plates containing IPTG (500 μM) and MMS (1.3 mM) or H2O2 (0.2 mM) as described (Sánchez et al., 2007). Cells grew in rich LB medium with a doubling time of 28–35 min. Plates were incubated overnight (16–18 h, 37°C) and the number of CFUs determined. Experiments were conducted independently at least four times. Fractional survival data are shown as mean ± SEM. Statistical analysis was performed with a two-tailed Student's t-test. For experiments involving more than two groups, one-way analysis of variance (ANOVA) was performed. For all tests, a P < 0.1 was considered significant (Supplementary Material). All statistical analyses were performed using Prism 6 software.
B. subtilis cells form round smooth colonies that raised above the agar. Colony size on Petri dishes was calculated via the diameter of a hypothetical circular colony. After overnight incubation, Petri dishes pictures were acquired and analyzed with the aid of a BioRad ChemiDocTM imaging system equipped with the QuantityOne software (BioRad). The relative mean colony diameter of ~50 isolated blind scoring colonies from pcrAT vs. pcrAT cells bearing a second mutation were measured from the pictures using ImageJ software (NIH). Upon colony magnification, the relative mean colony size was calculated using the formula for the area of a circle. Average and standard deviation were calculated using Prism 6 software. The colony area was compared by analysis of variance or Student's t-test, with P < 0.01 as the threshold of significance.
Nucleotide Sequence Analysis
The samples of genomic DNA from B. subtilis wt (BG214, Reference strain) and the Test recL16 (BG107) strains were analyzed using the first step of comparative genome sequencing, a service provided by NimbleGen Systems, Inc. as described earlier (Albert et al., 2005). Briefly, the genome of B. subtilis wt was tiled on custom-designed “mapping” microarrays with 29 base oligonucleotide pieces (probes) and a 7 or 8 base spacing from the start of one probe to the start of the next. The genomic DNA from the Reference BG214 strain was labeled with Cy5 and of the Test BG107 strain with Cy3. The ratios of hybridization intensities (reference/test) were calculated and plotted against genome position. If the test sample contains no mutation, the reference/test ratio equals 1. This analysis gives a high-resolution map of possible mutation sites, in which each mutation is localized in a window of 29 bases (the length of the reporting probe). A custom algorithm, based on hybridization intensity ratios significantly above background that have good agreement between results from corresponding probes from both strands, allows the identification of likely sites of mutation. The data were analyzed graphically using the SignalMap software provided by NimbleGen. The regions around the probes with a reference/test ratio significantly >1 were tested for mutations by direct DNA sequencing. Sequencing was performed with Big Dye (from Operon Technologies Inc.) using the protocols of the University of Wisconsin Biotechnology Center, after PCR amplification of the target genomic DNA sequence.
The samples of genomic DNA from B. subtilis wt (BG214, Reference strain) and five MMSS clones from the newly constructed. Test strain were re-sequenced by high-throughput sequence analyzer (Illumina) technology using standard sequencing libraries and filtered sequence data, a service provided by BGI, of ~1 gigabase per sample. The sequencing data were used to conduct paired-end nucleotide sequencing with the rec+ BG214 reference strain and the five MMSS clones from the newly constructed BG107-1 sample as described (Quail et al., 2008).
Fluorescence Microscopy and Data Analysis
For chromosome segregation analyses (Figure 2), cells were fixed and stained as described (Carrasco et al., 2004). To obtain exponentially growing cells, overnight cultures were inoculated in LB rich medium. The recO16, ΔrecA, ΔrarA, ΔrecJ, ΔrecQ, ΔrecX, or ΔrecU mutants in the pcrA-ssrA sspB (pcrAT) context (Table 1) were grown unperturbed in LB medium to OD560 = 0.2 (37°C). IPTG (500 μM) was added to half of the culture, and both cultures were incubated (60 min, 37°C). Then, cells were collected, subjected to fixation with 2% formaldehyde, and finally stained with 4′,6′-diamino-2-phenylindole (DAPI) (1 μg/ml). Samples were visualized and photographed by fluorescence microscopy with a Hamamatsu 3CCD Digital Camera C7780 coupled to a BX61 Olympus fluorescence microscope, equipped with an 100x immersion oil lens and a DAPI filter (U-MNU2).
The ImageJ software (NIH) was used to merge the phase contrast and DAPI-fluorescence images, which allowed us to distinguish the septum, and thus determine the filamentation event and was also used to determine the cell length. Blind scoring was performed on captured images as described (Carrasco et al., 2004).
Results and Discussion
Experimental Rationale
The phenotypes associated with the absence of PcrA are exceedingly complex and reflect an involvement in several aspects of DNA metabolism, including DNA replication, transcription, RTCs, repair-by-recombination and chromosomal segregation. The PcrA depletion lethality was attributed to the accumulation of toxic RecA-mediated intermediates or SOS toxicity. In fact, inactivation of positive RecA mediators (recO, recR), modulator (recF) (Table S1) or an unknown function (recL) suppresses the ΔpcrA lethality of cells grown in minimal medium (Petit and Ehrlich, 2002). In addition, a mutation in recO, recR, recF, or recL reduces and delays the SOS induction (Gassel and Alonso, 1989). However, the lethality in the ΔpcrA recA1 (a leaky recA mutation) challenged such hypothesis (Petit and Ehrlich, 2002). This observation was revisited and extended using rec-deficient strains (Table 1).
PcrA-like enzymes contribute to release stalled RTCs with the subsequent recruitment of repair factors or to modulate the re-initiation of DNA replication and transcription (Guy et al., 2009; Boubakri et al., 2010; Merrikh et al., 2015). Several B. subtilis translocases, namely the SF1 DNA helicases (as PcrA, HelD, RecD2) and the SF2 enzymes (Mfd, HepA [also termed YqhH] and YwqA) have been shown to interact with the RNAP (Selby and Sancar, 1993; Muzzin et al., 1998; Sukhodolets et al., 2001; Shaw et al., 2008; Yawn et al., 2009; Wiedermannova et al., 2014; Sanders et al., 2017; Le et al., 2018). The poorly characterized HepA and YwqA enzymes, which belong to the Swi2/Snf2 family of translocases, share ~30% identity to HepAEco. These translocases actively process a RNAP backwards as in the case of a RNAP stalled elongation complex (backtracking) or remove RNAP from the DNA template to resolve RTCs (Table S2) (Muzzin et al., 1998; Sukhodolets et al., 2001; Shaw et al., 2008; Yawn et al., 2009; Wiedermannova et al., 2014; Sanders et al., 2017; Le et al., 2018). The contribution of these functions to the inviability of PcrA depletion will be tested.
Finally, the UvrDEco or PcrA helicase/translocase plays a crucial role in nucleotide excision repair (Mendonca et al., 1993; Petit and Ehrlich, 2002; Epshtein, 2015), but only the former contributes to DNA mismatch repair (Lenhart et al., 2016). In this report, we show that PcrA also contributes to the repair of non-bulky lesions of oxidative nature generated upon exposure to MMS or H2O2 (Figure 1). Upon PcrA depletion, cells were exposed to limiting MMS or H2O2 concentrations, whose lesions are specifically removed by base excision repair (Sedgwick, 2004; Fu et al., 2012). If damaged template bases escape specialized repair, because they are in ssDNA regions, the offending lesion halts DNA polymerase (DNAP) or RNAP progression, and the lesion-containing gap is circumvented via damage avoidance pathways (template switching, fork reversal, translesion synthesis). When the damage is bypassed and the lesion is present in duplex DNA, it is removed by specialized pathways (Sedgwick, 2004; Fu et al., 2012).
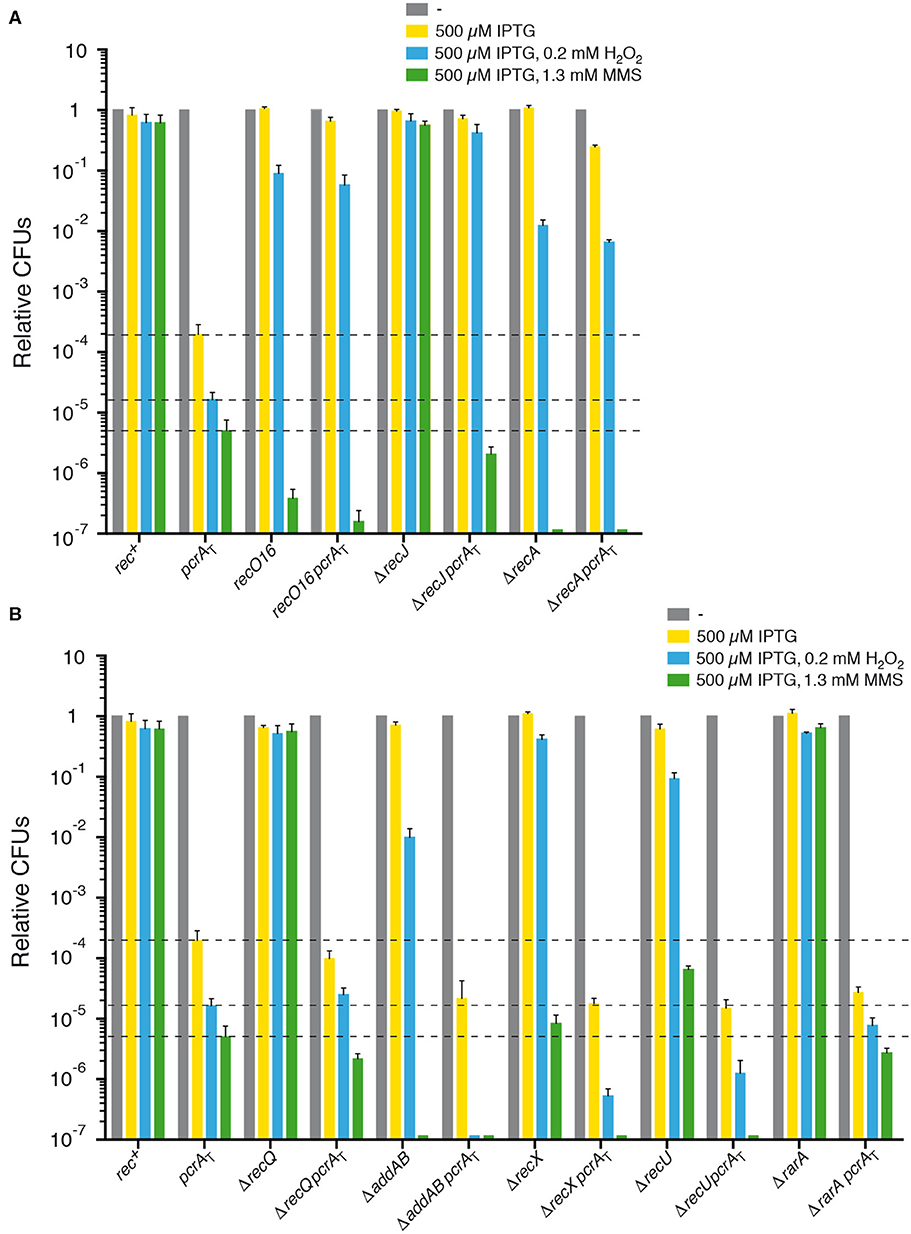
Figure 1. PcrA lethality is suppressed by recJ, recO, or recA inactivation, but not by addAB, recQ, rarA, recX, or recU inactivation. Log phase cultures of wt, single and double mutant strains were diluted and plated on LB agar containing 500 μM IPTG (yellow bars) or lacking it (gray bars). Lethality assays showing cell viability upon PcrA depletion in recJ, recO, or recA cells (A) and in addAB, recQ, rarA, recX, or recU cells (B). Log phase cultures of indicated strains were diluted and plated on LB agar containing IPTG and 0.2 mM H2O2 (blue bars) or IPTG and 1.3 mM MMS (green bars). Experiments were performed at least four times. The dotted lines mark the survival rate upon PcrA depletion. Data are shown as mean fractional survival ± SEM.
PcrA ATP Binding Mutants Render Cells Unviable
PcrA3Sau (T61I), defective in ATPase and helicase activities, and the PcrASau (PcrAH−) mutant variant (K33A Q250A), which lacks ATPase and helicase activities, can displace RecAEco from ssDNA, and inhibit RecAEco-mediated DNA strand exchange in vitro (Anand et al., 2007), suggesting that ATP binding and hydrolysis and the DNA helicase activity of PcrA are dispensable for its anti-recombinase activity. To assess whether the ATPase and/or translocase activity of B. subtilis PcrA are essential for cell proliferation, plasmid-borne pcrA genes with T65I, K37A, or Q254A mutations, equivalent to S. aureus mutations, were constructed and used to replace the wt pcrA gene. The single pcrA Q254A mutant gene also contained the unselected E224V mutation. The crystal structure of PcrABst DNA helicase with ADP suggests that residue Q254 is in hydrogen bonding distance to the γ-phosphate group of ATP, the E224 and a water molecule at the nucleotide binding pocket (Subramanya et al., 1996; Dillingham et al., 1999). The Q254 residue directly contacts the γ-phosphate of ATP, and the E224 forms hydrogen bonds with two water molecules that are hydrogen bonded to γ-phosphate oxygen atoms (Dillingham et al., 1999). Residue E224 is in an ideal position for activating the nucleophilic water molecule during hydrolysis. Mutations in residue Q254 alter the coupling between ATPase and helicase activities, and a transient interaction of Q254 with the γ-phosphate of ATP modifies the protein DNA binding site. Same mutations in residue Q254 are toxic for E. coli cells (Dillingham et al., 1999), and the untargeted E224V mutation might overcome such toxicity. The plasmid-borne pcrA Q254A (E224V) gene was also tested.
Monomeric plasmid DNA is inactive for transformation, but if it shares homology with recipient is activated upon interaction with the homologous region in the chromosome via RecA-mediated gene conversion, and the information present in the plasmid is transferred to the host chromosome [see Canosi et al. (1981)]. Monomeric DNA of plasmid-borne pcrA T65I (pCB1225), pcrA K37A (pCB1133), or pcrA Q254A (E224V) (pCB1119) was used to transform B. subtilis BG214 cells with selection for the plasmid marker (Table 1). Transformants carrying the established low copy unstable pCB1225, pCB1133, or pCB1119 plasmids were grown in the absence of selective pressure, and the plasmid-less segregants were subjected to nucleotide sequence analysis. We confirmed that in all transformants that lost pCB1225, the pcrA T65I gene had replaced the chromosomal wt pcrA gene by gene conversion (Petit et al., 1998). In only ~20% of the transformants that lost pCB1119, pcrA Q254A (E224V) mutant gene had replaced the wt gene, but all sequenced pcrA Q254A clones also contained the unselected E224V mutation. However, in all transformants that loss pCB1133, nucleotide sequence analyses revealed the presence of the wt gene, thus we have failed in the attempt to observe the replacement of the wt gene by the pcrA K37A mutant gene, suggesting that the K37A mutation, which impairs ATP binding, renders cells non-viable. This failure, however, could also be attributed to different reasons, for example that the mutation indirectly impacts in the expression of the downstream essential ligA gene (http://www.subtiwiki.uni-goettingen.de). Since the PcrAH−Sau mutant variant can promote disassembly of heterologous RecAEco from ssDNA (Anand et al., 2007), but PcrAH− fails to overcome RTCs, and depletion of wt PcrA in a background expressing PcrAH− renders cell inviable (Merrikh et al., 2015), we have dropped the plasmid segregation approach and moved our analysis to the condition in which PcrA is selectively depleted by the pcrA-ssrA sspB degron (pcrAT) strain (Merrikh et al., 2015) to answer these puzzling observations.
PcrA Depletion Inviability Requires RecJ and RecO, but Not RecQ, RarA, RecU, RecX, or AddAB
First, to confirm the reduction of cell survival following PcrA depletion, the pcrA-ssrA sspB degron (pcrAT) strain (Table 1) was exponentially grown to an OD560 = 0.4 (at 37° C) in LB medium and then, appropriate dilutions were plated on LB agar plates lacking or containing 500 μM IPTG (Materials and methods). In the absence of IPTG, the viability of the pcrA-ssrA sspB (pcrAT) degron strain was slightly compromised (1.3-fold) (Figure 1A, gray bar [- IPTG condition]) under the experimental conditions used (see below), perhaps due to noise introduced by sspB gene expression (see below). The plating efficiency of the pcrAT strain was reduced ~5,000-fold upon plating onto LB agar plates containing IPTG when compared to the pcrA+ control (rec+) (Figure 1A, yellow bar [+ IPTG condition]). This is consistent with the earlier observation that pcrAT cell viability was reduced by >1,000-fold when plated onto 100 μM IPTG-containing plates (Merrikh et al., 2015). We have observed a linear decrease in the number of viable cells with increasing IPTG concentration, but it saturates above 500 μM IPTG (data not shown). Upon IPTG addition the expression of the SspB adaptor increases, and SspB interacts with the SsrA moiety of the PcrA-SsrA protein to deliver it to the ClpXP protease for PcrA degradation [see Keiler et al. (1996), Griffith and Grossman (2008)]. It is worth mentioning that no fitness cost to B. subtilis cells was observed at IPTG concentrations as high as 5 mM IPTG. After 15 min of IPTG addition to the pcrAT degron strain, 60 to 90% of PcrA is degraded (Merrikh et al., 2015).
To gain insight into PcrA contribution to repair-by-recombination, the pcrAT degron strain was exposed to limiting H2O2 (0.2 mM) or MMS (1.3 mM) concentrations. The survival of pcrA-ssrA sspB cells in the presence of 500 μM IPTG and 0.2 mM H2O2 (blue bar [+ IPTG and H2O2]) or 1.3 mM MMS (green bar [+ IPTG and MMS]) was significantly decreased (by ~12- and ~40-fold, respectively) when compared to the absence of H2O2 or MMS (Figures 1A,B), suggesting that depletion of PcrA renders cells sensitive to both DNA damaging agents.
To elucidate whether PcrA prevents unscheduled RecA loading or dismantles its cognate recombinase assembled at or behind a stalled fork and which function(s) may counteract the PcrA antirecombinase activity, null mutants in presynaptic functions, namely end resection (ΔaddAB, ΔrecJ, ΔrecQ), RecA mediation (recO16), and positive (ΔrarA) or negative (ΔrecX, ΔrecU) modulators (Table S1), were assessed in the pcrA-ssrA sspB (pcrAT) context (Table 1) (Sanchez et al., 2006; Cárdenas et al., 2012; Romero et al., 2020).
B. subtilis ΔpcrA lethality is suppressed in the recL16 background (Petit and Ehrlich, 2002). Before testing the causes of the suppression of B. subtilis ΔpcrA lethality in the recL16 context, we must understand the function(s) impaired in the recL16 strain. As described in Supplementary Material, Annex 1, the B. subtilis mutations previously designated as recL16 actually map to the recO locus (C. M, Marielle C. Gruenig, Michael M. Cox and J.C.A., to be published elsewhere). To simplify the analysis, the MMSS phenotype was transferred by gene congression to competent BG214 cells. Five of the resulting MMSS clones were selected for whole genome sequencing along with the isogenic BG214 rec+ isogenic strain (Supplementary Material, Annex 1). One of the MMSS clones showed a TGA Opal stop triplet at codon 37 of recO and was designated recO16 (BG107-1 strain) and selected for further analysis (Table 1).
In the presence of IPTG, the survival of the recO16, ΔaddAB, ΔrecX, or ΔrecU strain decreased ~12-, ~65-, ~3-, and ~7-fold upon addition of 0.2 mM H2O2 (blue bar [+ IPTG and H2O2]), and ~2.5 × 106-, ~6 × 106-, ~1 × 105,- and ~1 × 104-fold upon addition of 1.3 mM MMS (green bar [+ IPTG and MMS]), respectively (Figure 1). Cell survival when comparing the single ΔrecJ, ΔrecQ, and ΔrarA mutant strains and the rec+ strain was not statistically significant decreased under the H2O2 and MMS concentrations used in this assay (Figure 1), although cell survival was significantly decreased in the ΔrecJ and ΔrecQ background at higher H2O2 and MMS concentrations, and in the ΔrarA context at higher H2O2 concentrations (Sanchez et al., 2006; Romero et al., 2019a). No significant differences were observed when the survival of rec+ and rec–deficient strains in the absence of IPTG was compared to the presence of IPTG.
Next, the contribution of the absence of the AddAB helicase-nucleases complex (counterpart of RecBCDEco), the RecJ 5′ → 3′ ssDNA exonuclease, or the RecQ DNA helicase (Table S1) in the pcrAT context was analyzed. In E. coli cells, the RecBCD complex during the end resection process loads RecA onto naked ssDNA, to generate a 3'-overhang coated by RecA (Kowalczykowski, 2015). If AddAB works in a similar fashion it is expected that PcrA depletion inviability requires AddAB. The presence of IPTG in rich medium agar plates did not significantly affected cell viability of the ΔrecJ pcrAT (Figure 1A), and thus the lethality upon PcrA depletion was suppressed by inactivating recJ, but the colony area was ~9-fold smaller than in the absence of IPTG. In contrast, PcrA depletion was not suppressed by addAB or ΔrecQ inactivation (Figure 1B). It is likely that the inviability upon PcrA depletion required the RecJ, but not the AddAB or RecQ functions. Furthermore, the viability of ΔaddAB pcrAT cells was significantly decreased (~10-fold), but not that of ΔrecQ pcrAT cells (~3-fold) when compared with the pcrAT degron strain (Figure 1B).
Exponentially growing ΔrecJ pcrAT, ΔaddAB pcrAT, or ΔrecQ pcrAT cells were then plated on LB agar plates containing IPTG and H2O2 or MMS. The ΔrecJ mutation significantly increased the survival of pcrAT cells on plates containing IPTG and H2O2 (P < 0.001). Unexpectedly, cell survival decreased by ~3-fold on plates containing IPTG and MMS when compared with pcrAT cells (Figure 1A), suggesting that addition of IPTG and MMS renders ΔrecJ pcrAT cells extremely sensitive (P < 0.001) when compared to the only IPTG condition (Figure 1A, yellow vs. green bar). Addition of IPTG and H2O2 or MMS to the ΔrecQ pcrAT strain did not significantly affect cell survival, when compared with pcrAT cells (Figure 1B). Cell survival in the ΔaddAB pcrAT strain was significantly decreased (by ~150- and ~50-fold) on plates containing IPTG and H2O2 or MMS, respectively, when compared with pcrAT cells (Figure 1B). It is likely that pcrA is not epistatic to recJ or addAB in response to MMS-induced DNA damage, and that PcrA is important possibly for backup pathways for single-strand gap and DSB repair.
Here, we have observed that there are different host requirements for the suppression of lethality between E. coli and B. subtilis cells impaired in end resection: first, the minimal medium synthetic lethality of E. coli Δrep ΔuvrD cells is suppressed by recJ or recQ inactivation (Lestini and Michel, 2008), although lack of RecQ or RecJ provides very limited suppression of Δrep ΔuvrD rich medium lethality (Guy et al., 2009). On the other hand, in B. subtilis, the lethality upon PcrA depletion is suppressed by recJ inactivation of cells grown in rich medium (Figure 1A) and it will be of significant interest to understand the molecular basis of the small colony size of ΔrecJ pcrAT upon PcrA depletion. Second, the lethality upon PcrA depletion is not suppressed by recQ inactivation, and survival of the ΔrecQ pcrAT cells was not significantly affected and reduced in the presence of H2O2- and MMS-induced DNA damage, respectively (Figure 1B). Unlike E. coli, two RecQ-like enzymes (RecQ and RecS) are present in B. subtilis (Fernández et al., 1998). RecQ, which is 591 amino acid long, shares ~43% identity with RecS (496 amino acid in length) if the first 346 residues containing the DExH helicase domains are used for the alignment; in short, RecS lacks the zinc-finger, the winged-helix, and the RNaseD C-terminal domains (Fernández et al., 1998; Bernstein et al., 2003). We cannot rule out that the partial genetic redundancy, exerted by RecS and RecQ, might mask the phenotype. However, since the viability of the ΔrecJ pcrAT or ΔrecQ pcrAT strain in the presence of IPTG and MMS was similar (Figures 1A,B), we have to assume that PcrA depletion and ΔrecJ or ΔrecQ inactivation might have different host requirements.
Next, the contribution of the recO16 mutation in the survival of the recO16 pcrAT strain was evaluated. The presence of IPTG did not significantly affect cell viability of the recO16 pcrAT strain grown in rich medium (Figure 1A). This is in good agreement with the observation that pcrA inactivation inviability requires RecO when grown in synthetic minimal medium (Petit and Ehrlich, 2002). To test whether PcrA works in a similar or different pathway than RecO, exponentially growing recO16 pcrAT cells were plated on LB agar plates containing 500 μM IPTG and H2O2 or MMS. The survival of the recO16 pcrAT strain significantly increased (>3,500-fold), when compared to the pcrAT strain upon plating in IPTG and H2O2 containing plates. Unexpectedly, the survival of the recO16 pcrAT strain was significantly decreased (>30-fold) when compared to the pcrAT strain on plates containing IPTG and MMS (Figure 1A). Addition of IPTG and MMS rendered recO16 pcrAT cells extremely sensitive (P < 0.001) when compared to just the addition of IPTG alone (Figure 1A, yellow vs. green bar). RecO has two activities: to load RecA onto SsbA-coated ssDNA in concert with RecR and to mediate DNA strand annealing independently of RecR (Kidane et al., 2004; Manfredi et al., 2008, 2010; Lenhart et al., 2014). Since ΔpcrA inviability requires RecO and RecR (Petit and Ehrlich, 2002), we assumed that the inactivation of recO compromises RecA loading at or behind a stalled fork, and thereby avoids unscheduled or unwanted RecA-mediated recombination during DNA replication in the context of PcrA depletion.
The contribution of mutants in the modulation of RecA filament growth in the inviability of PcrA depleted cells was also assessed. RarA has at least two activities: to control the loading of pre-primosomal proteins at a stalled fork and to positively modulate RecA filament growth (Carrasco et al., 2018; Romero et al., 2019a, 2020). PcrA depletion lethality was not suppressed by rarA inactivation (Figure 1B). After IPTG addition, the viability of PcrA depleted cells was significantly decreased (by ~7-fold) in the ΔrarA pcrAT context when compared with pcrAT cells (Figure 1B).
The absence of RarA renders cells sensitive to H2O2, but not to MMS (Romero et al., 2019a). Cell survival in the ΔrarA pcrAT background was not significantly reduced on plates containing IPTG and H2O2 or MMS, when compared to the pcrAT strain (Figure 1B). However, this observation is not consistent with the proposal that in E. coli, UvrD prevents RecA binding to ssDNA, possibly by counteracting RarA (Lestini and Michel, 2007).
RecX is a negative modulator of RecA filament growth (Cárdenas et al., 2012; Le et al., 2017), whereas RecU has two activities: to negatively modulate RecA filament growth and to cleave Holliday junctions (HJs) at a cognate site in concert with the RuvAB branch migration translocase (Ayora et al., 2004; Carrasco et al., 2005; Cárdenas et al., 2012; Cañas et al., 2014; Serrano et al., 2018). The absence of RecX or RecU did not restore viability of PcrA depleted cells grown in rich medium agar plates containing IPTG. The viability of PcrA depleted cells was significantly reduced (by ~12- and ~13-fold) in the ΔrecX pcrAT or ΔrecU pcrAT context, respectively, when compared to the pcrAT strain (Figure 1B). The presence of IPTG and H2O2 or MMS strongly reduced cell survival in the ΔrecX pcrAT (~30- and ~40-fold) and ΔrecU pcrAT (~13- and ~40-fold) backgrounds, respectively, when compared to the pcrAT strain (Figure 1B), suggesting that the pcrA gene is likely not epistatic to recX or recU in response to non-bulky DNA lesions of oxidative nature. In E. coli, ΔuvrD ΔruvC (counterpart of B. subtilis RecU) cells are inviable (Magner et al., 2007).
PcrA Depletion Inviability Requires RecA
Rich medium synthetic lethality of E. coli ΔuvrD Δrep cells is not suppressed by recA inactivation (Veaute et al., 2005; Guy et al., 2009). Similarly, B. subtilis pcrA lethality is not suppressed when the leaky recA1 (formerly termed recE1) mutation is moved into the background (Petit and Ehrlich, 2002), but the RecA1 activities present in the background may mask the outcome (e.g., competent recA1 cells are marginally affected, whereas null recA cells are blocked in chromosomal transformation) (Alonso et al., 1988). To address whether recA inactivation suppresses cell inviability upon PcrA depletion, a recA null mutant allele (ΔrecA) was moved, via SPP1-mediated generalized transduction, onto the pcrAT strain (see Table 1). Absence of RecA reduced survival of the parental strain by ~80-fold in response to H2O2, and strongly reduced cell survival (~6 ×106-fold) in response to MMS, when compared to the wt strain (Figure 1A).
The lethality of PcrA depleted cells was significantly suppressed (P < 0.001) by recA inactivation upon IPTG addition to rich LB agar plates (Figure 1A), but the colony area was ~17-fold smaller than in the absence of IPTG. Addition of IPTG and H2O2 or MMS did not significantly reduce cell survival in the ΔrecA pcrAT background when compared to the ΔrecA strain (Figure 1A, blue vs. green bar). To re-evaluate the results in the ΔrecA pcrAT background, the ΔrecA pcrAT strain was reconstructed, and similar results were observed. The apparent contradiction between PcrA depleted cells in the ΔrecA context (Figure 1A) with the ΔpcrA recA1 condition (Petit and Ehrlich, 2002), can be attributed either to the leaky recA1 mutation or to background differences, since there are extrachromosomal elements (as the conjugative element ICEBs1 and the prophage SPβ) in the ΔpcrA recA1 context (Petit and Ehrlich, 2002) that are absent in the ΔrecA pcrAT background (see Table 1).
It is likely, therefore, that: (i) inviability upon PcrA depletion could not be associated with the inability to promote auto-proteolysis of the transcriptional repressor LexA, because inactivation of recA prevents SOS induction, but suppresses pcrAT lethality in the presence of IPTG (Figure 1A); and (ii) PcrA depletion inviability requires RecA, and pcrA is epistatic to recA in response to non-bulky DNA lesions of oxidative nature.
PcrA Depletion Leads to Unsegregated Chromosomes
As previously proposed for eukaryotic Srs2 (Marini and Krejci, 2010), the role of PcrA might be to prevent the accumulation of crossovers (CO) by promoting synthesis-dependent strand annealing (SDSA), leading to the exclusive accumulation of non-crossover (NCO) products. In circular chromosomes, the outcome of CO and NCO will be a dimeric or two monomeric chromosomes, respectively. Dimeric chromosomes are deleterious and need to be processed before cell division. To test whether PcrA prevents CO accumulation, the chromosome segregation of PcrA depleted cells was studied.
Branched DNA structures can be processed by different pathways. First, the RecU HJ resolvase, in concert with the RuvAB translocase, cleaves the outside or the inside strands of a HJ, followed by religation to produce CO and NCO products, respectively (Carrasco et al., 2004; Cañas et al., 2014). Second, a HJ can be dissolved by the RecQ helicase in concert with a Type I DNA topoisomerase to produce NCO products (Kowalczykowski, 2015). Finally, PcrA may dismantle RecA nucleoprotein filaments from branched structures and may promote SDSA by unwinding the elongated invading strand, a step that is followed by annealing to the ssDNA of the other end of the break, an event associated with NCO products (Marini and Krejci, 2010).
To evaluate whether PcrA depletion provokes a chromosomal segregation defect, the nucleoid (supercoiled and compacted chromosome bound by proteins) of the pcrAT, recO16 pcrAT, ΔrecJ pcrAT, ΔrecQ pcrAT, ΔrarA pcrAT, ΔrecX pcrAT, ΔrecU pcrAT, or ΔrecA pcrAT cells was DAPI-stained and analyzed by fluorescence microscopy. As controls we have used the ΔrecU and ΔrecA strains. In the absence of any external DNA damage and at mid-exponential phase, ΔrecU or ΔrecA mutations reduce the number of CFUs by ~5- and ~10-fold (Figure 1), and ~30 and ~40% of cells are filamented, respectively (Figure 2) (Carrasco et al., 2004), suggesting that in the absence of RecU or RecA, a cell subpopulation undergoes a death-by-recombination phenotype.
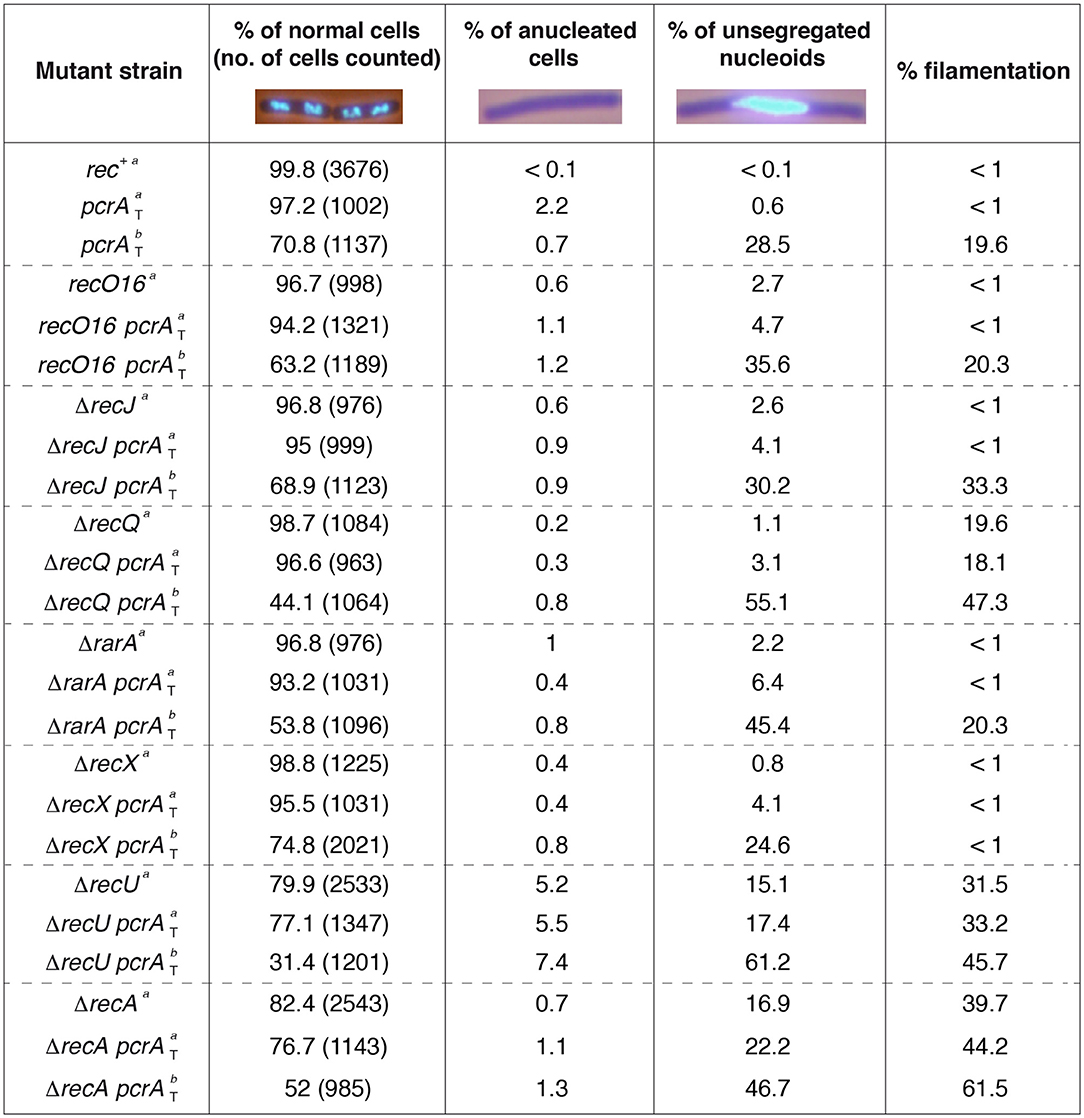
Figure 2. Chromosome segregation in the absence of presynaptic or synaptic functions. Cells were grown in LB medium to OD560 = 0.2; after 60 min, cells were harvested, prepared for DAPI DNA-fluorescence microscopy, and the percentage of anucleate and unsegregated nucleoids determined (condition a). In parallel, at OD560 = 0.2, IPTG (500 μM) was added and after 60 min, cells were harvested, DAPI-stained, and the percentage of anucleate and unsegregated nucleoids determined (condition b). Representative fluorescent images of two dividing DAPI-treated cells (DNA stain, light blue) are shown. The pictures are taken at the same amplification, two none separated cells (four nucleoids) are presented under normal conditions. The mean of at least three independent experiments is shown.
Previously it has been shown that after 15 min (37°C) of 100 μM IPTG addition to B. subtilis cells 60 to 90% of PcrA is degraded (Merrikh et al., 2015). In this study, cells were grown in rich medium under unperturbed conditions until they reached OD560 = 0.2 (37°C). IPTG (500 μM) was added to half of the culture, and both cultures were incubated (60 min, 37°C) before harvesting, fixing and staining the cells with DAPI. During vegetative growth, net accumulation of anucleated cells, unsegregated chromosomes and filamented cells was rare in rec+ cells in the absence (Figure 2) or presence of IPTG (data not shown). In this scenario, cells displayed an average length of 4–6 μm, and exhibited a bimodal distribution of nucleoid positioning with ~35% of total cells having two nucleoids, and ~65% of total cells containing only one nucleoid with about twice the fluorescence signal as judged by eye (Carrasco et al., 2004). This suggests that the former class were replicated cells with segregated chromosomes and the latter were replicated cells with yet-unsegregated chromosomes.
In the absence of IPTG, ~97% of pcrA-ssrA cells appeared normal compared to the rec+ control (~100%) (Figure 2). Absence of DAPI staining (anucleated cells) significantly increased (by ~20-fold) and the fraction of cells with aberrant chromosomes by ~6-fold when compared to the rec+ control, whereas upon IPTG addition absence of DAPI staining was significantly decreased (Figure 2), suggesting that the pcrA-ssrA fusion or noise from sspB gene expression affects chromosomal segregation of unperturbed exponentially growing pcrAT cells. In the presence of IPTG, PcrA dropped, the proportion of cells with an incompletely separated nucleoid or aberrant chromosomes increased by ~50-fold when compared to the condition without IPTG, and the average cell length was >8 μm in ~20% of total pcrAT cells (Figure 2). Since those elongated cells contained a single nucleoid it was assumed that they were filamented cells. It is likely that, upon PcrA depletion, NCO shifted toward CO products. In other words, PcrA might suppress COs or might directly contribute to the formation of NCO products, as was shown for its yeast homolog Srs2 (Marini and Krejci, 2010).
In the absence of IPTG, the proportion of anucleated cells and cells with aberrant chromosomes was marginally affected in the recO16 pcrAT, ΔrecJ pcrAT, and ΔrecA pcrAT strains when compared to the single mutant strain, but in the ΔrecA or ΔrecA pcrAT condition 40–45% of total cells were filamented (Figure 2). In the presence of IPTG, the absence of DAPI staining and the proportion of unsegregated nucleoids were not significantly affected (<2-fold) when compared to the parental control (rec+ pcrAT) strain (Figure 2). Likewise, in the recO16 pcrAT, ΔrecJ pcrAT, and ΔrecA pcrAT conditions a significant proportion of cells formed filaments, with <2-fold increase in the proportion of filamented cells when compared to the most affected parental strain (Figure 2). It is likely that the PcrA pro-SDSA function requires RecJ, RecO, or RecA.
The remaining strains were classified into three different classes. First, for RarA and RecQ, which have two activities each (see above): inactivation of recQ or rarA revealed a marginal chromosome segregation defect, but ~20% of total ΔrecQ cells formed filaments (Figure 2). In the presence of IPTG, the proportion of unsegregated nucleoids significantly increased in ΔrecQ pcrAT or ΔrarA pcrAT when compared to the pcrAT control, but absence of DAPI staining was not significantly affected (Figure 2). Upon PcrA depletion the proportion of filamentous cells was similar in pcrAT and ΔrarA pcrAT cells, but significantly increased in the ΔrecQ pcrAT context. Second, RecX negatively modulates RecA filament growth (Cárdenas et al., 2012; Le et al., 2017). In the presence of IPTG, the proportion of unsegregated nucleoids in ΔrecX pcrAT was similar to the pcrAT control, but counteracted the formation of filamented cells (Figure 2). Third, for RecU, which has two activities (see above): in the absence of RecU, ~5% of total cells were anucleated as previously described (Carrasco et al., 2004), suggesting that cell division occurred in regions that had not received a nucleoid, and unsegregated nucleoids accounted up to ~15% of total cells. In the presence of IPTG, most of ΔrecU pcrAT cells (~60%) had unsegregated nucleoids, and ~45% of cells were present as “filaments” (Figure 2), suggesting that PcrA may promote SDSA prior to the formation of a double-HJ that can be resolved to NCO and CO by the RecU HJ resolvase in concert with the RuvAB branch migration translocase (Ayora et al., 2004; Cañas et al., 2014).
Altogether, the data presented in Figures 1, 2 revealed certain paradoxes. First, the inviability of PcrA depletion requires RecJ, RecO, and RecA, but under these conditions a chromosomal segregation defect was observed, suggesting that PcrA processes branched DNA structures formed at replication or at replication-transcription stalled forks, but with the help of accessory proteins (e.g., RecJ, RecO) a formed RecA nucleoprotein filament may be dismantled by PcrA. Alternatively, PcrA removes proteins bound to stalled forks to indirectly allow the formation of branched structures. Second, PcrA depletion halts cell proliferation, initiates accumulation of unprocessed branched intermediates, and additively reduces repair-by-recombination in the ΔrecQ or ΔrarA context. Finally, PcrA depletion exacerbates the segregation defect of ΔrecU cells, with only ~30% having normal chromosomal segregation, and in the absence of both negative RecA modulators there is a synergistic repair-by-recombination defect.
PcrA Inviability Requires Mfd, but Not RecD2, HelD, HepA, or YwqA
Enzymes of the UvrD family of translocases provide different solutions to cope with a replicative stress and/or RTCs. UvrDEco and PcrA can interact with and backtrack RNAP in vivo and in vitro, that is a crucial step for minimizing RTCs and for the repair of lesions occluded by a stalled RNAP, which become a major obstacle to DNA replication (Epshtein et al., 2014). Other DNA helicases/translocases of SF1, namely HelD and RecD2, and of SF2, such as Mfd, HepA (YqhH), and YwqA, also interact with RNAP via a conserved domain (Table S2) (Muzzin et al., 1998; Sukhodolets et al., 2001; Deaconescu et al., 2006; Shaw et al., 2008; Guy et al., 2009; Boubakri et al., 2010; Jin et al., 2011; Epshtein et al., 2014; Wiedermannova et al., 2014; Liu et al., 2015; Sanders et al., 2017; Le et al., 2018). These enzymes have been also implicated: (i) in preventing or mitigating the impact of protein-DNA complexes or spontaneous non-bulky DNA lesions of oxidative nature that halt transcription or replication, (ii) in avoiding the conflicts generated by the collision between the replication and transcription machineries, and (iii) in promoting RNAP recycling, sliding backward along the template (backtracking or retreating) or RNAP removal, or when replication forks are arrested by the formation of R-loops (Ayora et al., 1996; Komissarova and Kashlev, 1997; Sukhodolets et al., 2001; Trautinger et al., 2005; Deaconescu et al., 2006; Guy et al., 2009; Yawn et al., 2009; Boubakri et al., 2010; Gupta et al., 2013; Bruning et al., 2014; Wiedermannova et al., 2014; Merrikh et al., 2015; Sanders et al., 2017; Ho et al., 2018; Le et al., 2018).
To study whether the lack of PcrA destabilization of transcription complexes is the primary cause of inviability, the ΔhelD pcrAT, ΔrecD2 pcrAT, ΔhepA pcrAT, ΔywqA pcrAT, and Δmfd pcrAT strains were constructed as described (Materials and methods). PcrA depletion, which decreases cell survival in the ΔrecD2 context (Torres et al., 2017), was used as control. Except in the Δmfd strain, the H2O2 or MMS concentrations used were not sufficient to reveal a reduced viability phenotype (Figure 3).
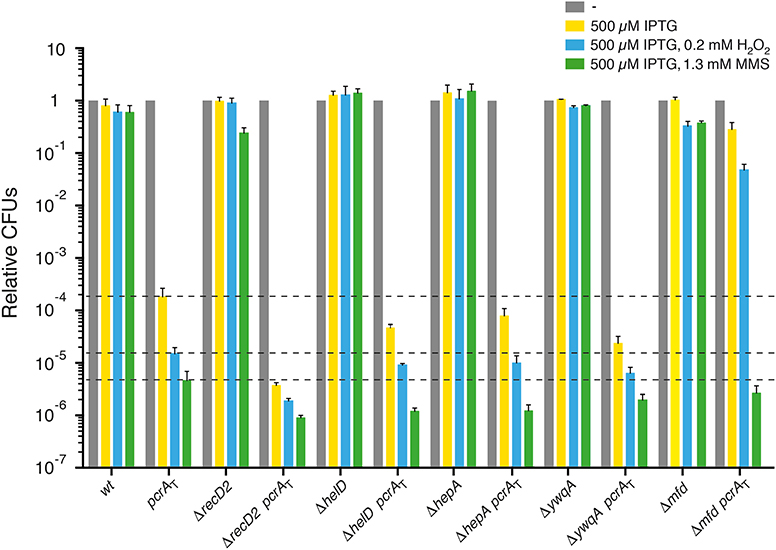
Figure 3. PcrA lethality is suppressed by mfd inactivation, but not by recD2, helD, hepA, ywqA inactivation. Log phase cultures of wt, single or double mutant strains were diluted and plated on LB agar containing 500 μM IPTG (yellow bars) or lacking it (gray bars). Lethality assays showing cell viability upon PcrA depletion in recD2, helD, hepA, ywqA, or mfd cells. Log phase cultures of indicated strains were diluted and plated on LB agar containing IPTG and 0.2 mM H2O2 (blue bars) or IPTG and 1.3 mM MMS (green bars). Experiments were performed at least four times. The dotted lines mark the survival rate upon PcrA depletion. Data are shown as mean fractional survival ± SEM.
Upon addition of IPTG, the lethality observed following PcrA depletion was not suppressed by recD2 inactivation (Figure 3). In the presence of IPTG, the viability was significantly decreased (by ~25-fold) in ΔrecD2 pcrAT when compared to the pcrAT strain. Addition of both IPTG and H2O2 or MMS significantly reduced cell survival (by ~8- and ~5-fold, respectively) in the ΔrecD2 pcrAT cells when compared to the pcrAT strain (Figure 3), suggesting that pcrA is not epistatic to recD2 in response to H2O2- or MMS-induced DNA damage.
Inactivation of the helD helicase partially suppresses the DNA repair defect of recF15, ΔrecO, and ΔrecR cells (Carrasco et al., 2001). The absence of HelD did not suppress cell inviabilily upon PcrA depletion; in this background, cell viability was not significantly decreased when compared to the pcrAT control upon addition of IPTG (Figure 3, yellow bar [+ IPTG]). Upon addition of IPTG and MMS, cell survival was significantly decreased (by ~4-fold), when compared to the pcrAT strain (Figure 3, green bar [+ IPTG and MMS]). In contrast, in the absence of E. coli HelD and UvrD cells remain recombination proficient and apparently are as capable of repairing MMS-induced DNA damage as the wt control (Mendonca et al., 1993). Addition of IPTG and H2O2 marginally decreased cell survival (by ~2-fold) (Figure 3, blue bar [+ IPTG and H2O2]).
The absence of HepA or YwqA did not suppress cell inviability upon PcrA depletion (Figure 3). Cell viability was significantly decreased (by ~8-fold) in the ΔywqA pcrAT when compared to the pcrAT control, but not in the ΔhepA pcrAT (decreased by ~3-fold) when compared to the pcrAT control (Figure 3).
Addition of IPTG and H2O2 did not significantly reduce survival in ΔhepA pcrAT or ΔywqA pcrAT cells when compared to the pcrAT strain (Figure 3). The presence of IPTG and MMS significantly reduced cell survival in the ΔhepA pcrAT, but marginally reduced cell survival in ΔywqA pcrAT cells when compared to the pcrAT strain (Figure 3). These data altogether suggest that decreasing the probability of backtracking events contributes to maintaining genome stability, but not to suppress the lethality of PcrA depleted cells. Unlike in E. coli cells (Shaw et al., 2008; Jin et al., 2011; Liu et al., 2015), we have little information about how the B. subtilis HepA or YwqA ATPase propels backward translocation of the RNAP along the DNA template or release a sequestered RNAP.
E. coli cells lacking Mfd show a weak sensitivity to UV irradiation (Witkin, 1969). E. coli Mfd recruits UvrA to the site of a roadblock that stalls RNAP. This activity is crucial for the recognition and removal of a stalled RNAP, but Mfd is subsequently displaced by UvrB to initiate transcription coupled repair (Selby and Sancar, 1993, 1994; Ayora et al., 1996; Manelyte et al., 2010; Epshtein, 2015; Ho et al., 2018, 2020; Le et al., 2018). In contrast, inactivation of B. subtilis mfd renders cells significantly sensitive to the UV mimetic 4-nitroquinoline-1-oxide and also to oxidative non-bulky lesions as those generated by exposure to MMS (Ayora et al., 1996). Our results suggested that Mfd is also required in B. subtilis cells to repair non-bulky lesions (Figure 3). It is likely that in B. subtilis: (i) bulky and non-bulky DNA lesions stall RNAP; and (ii) Mfd interacts with and dislodges RNAP from the damaged DNA template.
Upon addition of IPTG, inactivation of mfd significantly suppressed (P < 0.001) the lethality induced by PcrA depletion (Figure 3), but the colonies were minute and with an area ~19-fold smaller than in the absence of IPTG, suggesting that Mfd and PcrA play a crucial role in response to a replicative stress. The Δmfd mutation significantly suppressed the sensitivity of Δmfd pcrAT cells to H2O2 (addition of both IPTG and H2O2), but Δmfd pcrAT cells showed a non-significant decrease when compared to the pcrAT strain in the presence of both IPTG and MMS (Figure 3). It is likely that the pcrA gene is epistatic to mfd in response to MMS-induced DNA lesions. Inactivation of E. coli mfd partially suppresses the sensitivity to UV irradiation in the uvrD context (Epshtein et al., 2014).
Conclusions
We show that PcrA depletion lethality is suppressed by recJ, recO16, or recA inactivation, but not by addAB, recQ, rarA, recX, or recU inactivation when cells are grown in rich medium (Figures 1A,B). These data suggest that PcrA depleted cells primarily die due to their inability to resuscitate replisomes blocked by a RecA-ssDNA complex. Indeed, RecO loads RecA onto SsbA-coated ssDNA and a RecA nucleoprotein filament downregulates initiation of PriA-dependent DNA replication in vitro (Vlasic et al., 2014), and PcrA depletion inviability requires RecA for replication re-start (Million-Weaver et al., 2015). This is consistent with the observation that PcrA depletion inviability also requires RecO, which loads RecA onto ssDNA (Carrasco et al., 2015), but not AddAB. In a minimal synthetic medium, the pcrA inactivation lethality is also suppressed by inactivation of the recO or recR positive mediators or a leaky mutation in the positive recF17 modulator (Petit and Ehrlich, 2002).
In E. coli cells, the synthetic lethality of uvrD and rep mutations is partially suppressed by recJ or recQ inactivation in minimal medium (Lestini and Michel, 2008). We can envision that the discrepancies observed between E. coli and B. subtilis cells are related to genetic differences between these genetically distant bacteria. First, B. subtilis cells have two RecQ-like helicases, RecQ and RecS, with the latter potentially masking the outcome, whereas E. coli has only RecQ. Second, E. coli cells have two proteins (UvrD and RecX) to actively dismantle a RecA nucleoprotein filament (Petrova et al., 2015; Le et al., 2017), whereas B. subtilis cells have four different proteins (PcrA, RecX, RecU, and RecD2) to do this job (Anand et al., 2007; Le et al., 2017; Torres et al., 2017; Serrano et al., 2018). PcrA was also found to be necessary to survive DNA damage. The pcrA gene is not epistatic to genes involved in end resection (addAB, recJ, recQ), RecA mediators (recO16), or negative RecA modulators (recX, recU) in response to MMS- or H2O2-induced DNA damage, but it is epistatic to the recA gene, suggesting that PcrA also contributes to repair-by-recombination via poorly understood mechanisms. The role of the positive RecA modulator RarA upon PcrA depletion requires further studies.
As it has been seen previously for Srs2 (Marini and Krejci, 2010), PcrA might play a putative role in promoting SDSA, which does not entail the generation of COs. Depletion of PcrA leads to additive, in recJ, recO and recA, and to synergic accumulation of unsegregated chromosomes in recQ, rarA recU backgrounds. The dual activities of these proteins (see above) mask the interpretation of our results.
Finally, we show that PcrA depletion lethality is suppressed by mfd inactivation, but not by recD2, helD, hepA, or ywqA inactivation (Figure 3). We show that pcrA is not epistatic to recD2, helD, or hepA in response to non-bulky DNA damage, but it is epistatic to mfd. The role of the poorly characterized YwqA ATPase upon PcrA depletion requires further studies.
PcrA and Mfd might act on RTCs, both dependent (Ayora et al., 1996) and independently of the nucleotide excision repair pathway (Figure 3). The PcrA and Mfd translocases physically interact with stalled RNAPs at lesions on the DNA template. PcrA is a pro-backtracking factor by promoting forward RNAP translocation, and Mfd might be an anti-backtracking that dislodges a stalled RNAP, as previously postulated for the isolated protein in vitro (Selby and Sancar, 1993; Ayora et al., 1996; Park et al., 2002; Deaconescu et al., 2006; Epshtein et al., 2014; Sanders et al., 2017; Ho et al., 2018; Le et al., 2018). It is likely that when damaged template bases interfere with RNAP progression, it backtracks and becomes transiently arrested. Under this condition, Mfd and PcrA, which interact with UvrA and UvrB, respectively, are crucial factors involved in mitigating RTCs in the presence of DNA lesions that are or not targeted by transcription coupled repair. Thus, we propose that PcrA is crucial to remove a stalled RNAP that would otherwise hinder DNA replication even in the presence of DNA lesions that are not targeted by transcription coupled repair. In other words, in the absence of PcrA, RNAP may not be evicted from the damage site by Mfd, leading to a harmful genotoxic stress that induces lethality. PcrA allows genome duplication to occur concurrently with other essential DNA transactions (replication, transcription, repair, segregation). Partial PcrA depletion sensitizes cells to severe DNA transactions and its major role is to work in concert with recombination and repair proteins at stalled DNAP/RNAP complexes to facilitate replication progression beyond the conflict point.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
MM, RT, and JA designed the experiments and drafted the manuscript. MM, RT, CM, JR-M, and GS performed the experiments. JA coordinated the research and wrote the manuscript. MM, RT, CM, JR-M, GS, and JA interpreted the data. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Ministerio de Ciencia e Innovación, Agencia Estatal de Investigación (MCIU/AEI)/FEDER PGC2018-097054-B-I00 to JA.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Marie A. Petit for the ΔyqhH (ΔhepA) and ΔywqA strains, to Houra and Christopher Merrikh for the pcrA-ssrA sspB degron strain, and to Marielle C. Gruenig and Michael M. Cox for the early steps of recL16 mapping. We thank C. Marchisone for technical assistance and S. Ayora for comments on the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2020.00140/full#supplementary-material
References
Aboussekhra, A., Chanet, R., Adjiri, A., and Fabre, F. (1992). Semidominant suppressors of Srs2 helicase mutations of Saccharomyces cerevisiae map in the RAD51 gene, whose sequence predicts a protein with similarities to procaryotic RecA proteins. Mol. Cell. Biol. 12, 3224–3234. doi: 10.1128/MCB.12.7.3224
Albert, T. J., Dailidiene, D., Dailide, G., Norton, J. E., Kalia, A., Richmond, T. A., et al. (2005). Mutation discovery in bacterial genomes: metronidazole resistance in Helicobacter pylori. Nat. Methods 2, 951–953. doi: 10.1038/nmeth805
Alonso, J. C., Tailor, R. H., and Luder, G. (1988). Characterization of recombination-deficient mutants of Bacillus subtilis. J. Bacteriol. 170, 3001–3007. doi: 10.1128/JB.170.7.3001-3007.1988
Alonso, J. C., Viret, J. F., and Tailor, H. R. (1987). Plasmid maintenance in Bacillus subtilis recombination-deficient mutants. Mol. Gen. Genet. 208, 349–352. doi: 10.1007/BF00330464
Anand, S. P., Zheng, H., Bianco, P. R., Leuba, S. H., and Khan, A. S. (2007). DNA helicase activity of PcrA is not required for the displacement of RecA protein from DNA or inhibition of RecA-mediated strand exchange. J. Bacteriol. 189, 4502–4509. doi: 10.1128/JB.00376-07
Antony, E., Tomko, E. J., Xiao, Q., Krejci, L., Lohman, T. M., and Ellenberger, T. (2009). Srs2 disassembles Rad51 filaments by a protein-protein interaction triggering ATP turnover and dissociation of Rad51 from DNA. Mol. Cell. 35, 105–115. doi: 10.1016/j.molcel.2009.05.026
Ayora, S., Carrasco, B., Doncel, E., Lurz, R., and Alonso, C. J. (2004). Bacillus subtilis RecU protein cleaves Holliday junctions and anneals single-stranded DNA. Proc. Natl. Acad. Sci. U.S.A. 101, 452–457. doi: 10.1073/pnas.2533829100
Ayora, S., Rojo, F., Ogasawara, N., Nakai, S., and Alonso, C. J. (1996). The Mfd protein of Bacillus subtilis 168 is involved in both transcription-coupled DNA repair and DNA recombination. J. Mol. Biol. 256, 301–318. doi: 10.1006/jmbi.1996.0087
Baharoglu, Z., Lestini, R., Duigou, S., and Michel, B. (2010). RNA polymerase mutations that facilitate replication progression in the rep uvrD recF mutant lacking two accessory replicative helicases. Mol. Microbiol. 77, 324–336. doi: 10.1111/j.1365-2958.2010.07208.x
Bernstein, D. A., Zittel, M. C., and Keck, L. J. (2003). High-resolution structure of the E. coli RecQ helicase catalytic core. EMBO 22, 4910–4921. doi: 10.1093/emboj/cdg500
Boubakri, H., de Septenville, A. L., Viguera, E., and Michel, B. (2010). The helicases DinG, Rep and UvrD cooperate to promote replication across transcription units in vivo. EMBO J. 29, 145–157. doi: 10.1038/emboj.2009.308
Bruning, J. G., Howard, J. L., and McGlynn, P. (2014). Accessory replicative helicases and the replication of protein-bound DNA. J. Mol. Biol. 426, 3917–3928. doi: 10.1016/j.jmb.2014.10.001
Cañas, C., Suzuki, Y., Marchisone, C., Carrasco, B., Freire-Beneitez, V., Takeyasu, K., et al. (2014). Interaction of branch migration translocases with the Holliday junction-resolving enzyme and their implications in Holliday junction resolution. J. Biol. Chem. 289, 17634–17646. doi: 10.1074/jbc.M114.552794
Canosi, U., Iglesias, A., and Trautner, A. T. (1981). Plasmid transformation in Bacillus subtilis: effects of insertion of Bacillus subtilis DNA into plasmid pC194. Mol. Gen. Genet. 181, 434–440. doi: 10.1007/BF00428732
Cárdenas, P. P., Carrasco, B., Defeu Soufo, C., César, C. E., Herr, K., Kaufenstein, M., et al. (2012). RecX facilitates homologous recombination by modulating RecA activities. PLoS Genet. 8:e1003126. doi: 10.1371/journal.pgen.1003126
Carrasco, B., Ayora, S., Lurz, R., and Alonso, C. J. (2005). Bacillus subtilis RecU Holliday-junction resolvase modulates RecA activities. Nucleic Acids Res. 33, 3942–3952. doi: 10.1093/nar/gki713
Carrasco, B., Cozar, M. C., Lurz, R., Alonso, J. C., and Ayora, S. (2004). Genetic recombination in Bacillus subtilis 168: contribution of Holliday junction processing functions in chromosome segregation. J. Bacteriol. 186, 5557–5566. doi: 10.1128/JB.186.17.5557-5566.2004
Carrasco, B., Fernández, S., Petit, M. A., and Alonso, C. J. (2001). Genetic recombination in Bacillus subtilis 168: effect of DeltahelD on DNA repair and homologous recombination. Bacteriol. J. 183, 5772–5777. doi: 10.1128/JB.183.19.5772-5777.2001
Carrasco, B., Seco, E. M., López-Sanz, M., Alonso, J. C., and Ayora, S. (2018). Bacillus subtilis RarA modulates replication restart. Nucleic Acids Res. 46, 7206–7220. doi: 10.1093/nar/gky541
Carrasco, B., Yadav, T., Serrano, E., and Alonso, C. J. (2015). Bacillus subtilis RecO and SsbA are crucial for RecA-mediated recombinational DNA repair. Nucleic Acids Res. 43, 5984–5997. doi: 10.1093/nar/gkv545
de la Hoz, B., Ayora, S., Sitkiewicz, I., Fernández, S., Pankiewicz, R., Alonso, J. C., et al. (2000). Plasmid copy-number control and better-than-random segregation genes of pSM19035 share a common regulator. Proc. Natl. Acad. Sci. U.S.A. 97, 728–733. doi: 10.1073/pnas.97.2.728
Deaconescu, A. M., Chambers, A. L., Smith, A. J., Nickels, B. E., Hochschild, A., Savery, N. J., et al. (2006). Structural basis for bacterial transcription-coupled DNA repair. Cell 124, 507–520. doi: 10.1016/j.cell.2005.11.045
Dillingham, M. S. (2011). Superfamily I helicases as modular components of DNA-processing machines. Biochem. Soc. Trans. 39, 413–423. doi: 10.1042/BST0390413
Dillingham, M. S., Soultanas, P., and Wigley, B. D. (1999). Site-directed mutagenesis of motif III in PcrA helicase reveals a role in coupling ATP hydrolysis to strand separation. Nucleic Acids Res. 27, 3310–3317. doi: 10.1093/nar/27.16.3310
Epshtein, V. (2015). UvrD helicase: an old dog with a new trick: how one step backward leads to many steps forward. Bioessays 37, 12–19. doi: 10.1002/bies.201400106
Epshtein, V., Kamarthapu, V., McGary, K., Svetlov, V., Ueberheide, B., Proshkin, S., et al. (2014). UvrD facilitates DNA repair by pulling RNA polymerase backwards. Nature 505, 372–377. doi: 10.1038/nature12928
Fagerburg, M. V., Schauer, G. D., Thickman, K. R., Bianco, P. R., Khan, S. A., Leuba, S. H., et al. (2012). PcrA-mediated disruption of RecA nucleoprotein filaments–essential role of the ATPase activity of RecA. Nucleic Acids Res. 40, 8416–8424. doi: 10.1093/nar/gks641
Fernández, S., Sorokin, A., and Alonso, C. J. (1998). Genetic recombination in Bacillus subtilis 168: effects of recU and recS mutations on DNA repair and homologous recombination. J. Bacteriol. 180, 3405–3409. doi: 10.1128/JB.180.13.3405-3409.1998
Fu, D., Calvo, J. A., D., and Samson, L. (2012). Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer 12, 104–120. doi: 10.1038/nrc3185
Gassel, M., and Alonso, C. J. (1989). Expression of the recE gene during induction of the SOS response in Bacillus subtilis recombination-deficient strains. Mol. Microbiol. 3, 1269–1276. doi: 10.1111/j.1365-2958.1989.tb00277.x
Griffith, K. L., and Grossman, D. A. (2008). Inducible protein degradation in Bacillus subtilis using heterologous peptide tags and adaptor proteins to target substrates to the protease ClpXP. Mol. Microbiol. 70, 1012–1025. doi: 10.1111/j.1365-2958.2008.06467.x
Gupta, M. K., Guy, C. P., Yeeles, J. T., Atkinson, J., Bell, H., Lloyd, R. G., et al. (2013). Protein-DNA complexes are the primary sources of replication fork pausing in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 110, 7252–7257. doi: 10.1073/pnas.1303890110
Guy, C. P., Atkinson, J., Gupta, M. K., Mahdi, A. A., Gwynn, E. J., Rudolph, C. J., et al. (2009). Rep provides a second motor at the replisome to promote duplication of protein-bound DNA. Mol. Cell. 36, 654–666. doi: 10.1016/j.molcel.2009.11.009
Ho, H. N., van Oijen, A. M., and Ghodke, H. (2018). The transcription-repair coupling factor Mfd associates with RNA polymerase in the absence of exogenous damage. Nat. Commun. 9:1570. doi: 10.1038/s41467-018-03790-z
Ho, H. N., van Oijen, A. M., and Ghodke, H. (2020). Single-molecule imaging reveals molecular coupling between transcription and DNA repair machinery in live cells. Nat. Commun. 11:1478. doi: 10.1038/s41467-020-15182-3
Jin, D. J., Zhou, Y. N., Shaw, G., and Ji, X. (2011). Structure and function of RapA: a bacterial Swi2/Snf2 protein required for RNA polymerase recycling in transcription. Biochim. Biophys. Acta 1809, 470–475. doi: 10.1016/j.bbagrm.2011.03.003
Kamarthapu, V., Epshtein, V., Benjamin, B., Proshkin, S., Mironov, A., Cashel, M., et al. (2016). ppGpp couples transcription to DNA repair in E. coli. Science 352, 993–996. doi: 10.1126/science.aad6945
Kaniecki, K., De Tullio, L., Gibb, B., Kwon, Y., Sung, P. C., and Greene, E. C. (2017). Dissociation of Rad51 presynaptic complexes and heteroduplex DNA joints by tandem assemblies of Srs2. Cell Rep. 21, 3166–3177. doi: 10.1016/j.celrep.2017.11.047
Keiler, K. C., Waller, P. R., and Sauer, T. R. (1996). Role of a peptide tagging system in degradation of proteins synthesized from damaged messenger RN. Science 271, 990–993. doi: 10.1126/science.271.5251.990
Kidane, D., Sánchez, H., Alonso, J. C., and Graumann, L. P. (2004). Visualization of DNA double-strand break repair in live bacteria reveals dynamic recruitment of Bacillus subtilis RecF, RecO and RecN proteins to distinct sites on the nucleoids. Mol. Microbiol. 52, 1627–1639. doi: 10.1111/j.1365-2958.2004.04102.x
Komissarova, N., and Kashlev, M. (1997). Transcriptional arrest: Escherichia coli RNA polymerase translocates backward, leaving the 3' end of the RNA intact and extruded. Proc. Natl. Acad. Sci. U.S.A. 94, 1755–1760. doi: 10.1073/pnas.94.5.1755
Kowalczykowski, S. C. (2015). An overview of the molecular mechanisms of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 7:a016410. doi: 10.1101/cshperspect.a016410
Krejci, L., Van Komen, S., Li, Y., Villemain, J., Reddy, M. S., Klein, H., et al. (2003). DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature 423, 305–309. doi: 10.1038/nature01577
Le, S, Serrano, E., Kawamura, R., Carrasco, B., Yan, J., and Alonso, C. J. (2017). Bacillus subtilis RecA with DprA-SsbA antagonizes RecX function during natural transformation. Nucleic Acids Res. 45, 8873–8885. doi: 10.1093/nar/gkx583
Le, T. T., Yang, Y., Tan, C., Suhanovsky, M. M., Fulbright, R. M. Jr., Inman, J. T., et al. (2018). Mfd Dynamically regulates transcription via a release and catch-up mechanism. Cell 172, 344–357 e15. doi: 10.1016/j.cell.2017.11.017
Lenhart, J. S., Brandes, E. R., Schroeder, J. W., Sorenson, R. J., Showalter, H. D., and Simmons, A. L. (2014). RecO and RecR Are Necessary for RecA loading in response to DNA damage and replication fork stress. J. Bacteriol. 196, 2851–2860. doi: 10.1128/JB.01494-14
Lenhart, J. S., Pillon, M. C., Guarne, A., Biteen, J. S. A., and Simmons, L. (2016). Mismatch repair in Gram-positive bacteria. Res. Microbiol. 167, 4–12. doi: 10.1016/j.resmic.2015.08.006
Lestini, R., and Michel, B. (2007). UvrD controls the access of recombination proteins to blocked replication forks. EMBO J. 26, 3804–3814. doi: 10.1038/sj.emboj.7601804
Lestini, R., and Michel, B. (2008). UvrD and UvrD252 counteract RecQ, RecJ, and RecFOR in a rep mutant of Escherichia coli. J. Bacteriol. 190, 5995–6001. doi: 10.1128/JB.00620-08
Liu, B., Zuo, Y., and Steitz, A. T. (2015). Structural basis for transcription reactivation by RapA. Proc. Natl. Acad. Sci. U.S.A. 112, 2006–2010. doi: 10.1073/pnas.1417152112
Magner, D. B., Blankschien, M. D., Lee, J. A., Pennington, J. M., Lupski, J. R., and Rosenberg, M. S. (2007). RecQ promotes toxic recombination in cells lacking recombination intermediate-removal proteins. Mol. Cell. 26, 273–286. doi: 10.1016/j.molcel.2007.03.012
Manelyte, L., Kim, Y. I., Smith, A. J., Smith, R. M., and Savery, J. N. (2010). Regulation and rate enhancement during transcription-coupled DNA repair. Mol. Cell. 40, 714–724. doi: 10.1016/j.molcel.2010.11.012
Manfredi, C., Carrasco, B., Ayora, S., and Alonso, C. J. (2008). Bacillus subtilis RecO nucleates RecA onto SsbA-coated single-stranded DNA. J. Biol. Chem. 283, 24837–24847. doi: 10.1074/jbc.M802002200
Manfredi, C., Suzuki, Y., Yadav, T., Takeyasu, K., and Alonso, C. J. (2010). RecO-mediated DNA homology search and annealing is facilitated by SsbA. Nucleic Acids Res. 38, 6920–6929. doi: 10.1093/nar/gkq533
Marini, V., and Krejci, L. (2010). Srs2: the “Odd-Job Man” in DNA repair. DNA Repair 9, 268–275. doi: 10.1016/j.dnarep.2010.01.007
Mendonca, V. M., Kaiser-Rogers, K., and Matson, W. S. (1993). Double helicase II (uvrD)-helicase IV (helD) deletion mutants are defective in the recombination pathways of Escherichia coli. J. Bacteriol. 175, 4641–4651. doi: 10.1128/JB.175.15.4641-4651.1993
Merrikh, C. N., Brewer, B. J., and Merrikh, H. (2015). The B. subtilis accessory helicase PcrA facilitates DNA replication through transcription units. PLoS Genet. 11:e1005289. doi: 10.1371/journal.pgen.1005289
Million-Weaver, S., Samadpour, A. N., and Merrikh, H. (2015). Replication restart after replication-transcription conflicts requires RecA in Bacillus subtilis. J. Bacteriol. 197, 2374–2382. doi: 10.1128/JB.00237-15
Muzzin, O., Campbell, E. A., Xia, L., Severinova, E., Darst, S. A., and Severinov, K. (1998). Disruption of Escherichia coli hepA, an RNA polymerase-associated protein, causes UV sensitivity. J. Biol. Chem. 273, 15157–15161. doi: 10.1074/jbc.273.24.15157
Myka, K. K., Hawkins, M., Syeda, A. H., Gupta, M. K., Meharg, C., Dillingham, M. S., et al. (2017). Inhibiting translation elongation can aid genome duplication in Escherichia coli. Nucleic Acids Res. 45, 2571–2584. doi: 10.1093/nar/gkw1254
Park, J., Myong, S., Niedziela-Majka, A., Lee, K. S., Yu, J., Lohman, T. M., et al. (2010). PcrA helicase dismantles RecA filaments by reeling in DNA in uniform steps. Cell 142, 544–555. doi: 10.1016/j.cell.2010.07.016
Park, J. S., Marr, M. T., and Roberts, W. J. (2002). E. coli transcription repair coupling factor (Mfd protein) rescues arrested complexes by promoting forward translocation. Cell 109, 757–767. doi: 10.1016/S0092-8674(02)00769-9
Petit, M. A., Dervyn, E., Rose, M., Entian, K. D., McGovern, S., Ehrlich, S. D., et al. (1998). PcrA is an essential DNA helicase of Bacillus subtilis fulfilling functions both in repair and rolling-circle replication. Mol. Microbiol. 29, 261–273. doi: 10.1046/j.1365-2958.1998.00927.x
Petit, M. A., and Ehrlich, D. (2002). Essential bacterial helicases that counteract the toxicity of recombination proteins. EMBO J. 21, 3137–3147. doi: 10.1093/emboj/cdf317
Petrova, V., Chen, S. H., Molzberger, E. T., Tomko, E., Chitteni-Pattu, S., Jia, H., et al. (2015). Active displacement of RecA filaments by UvrD translocase activity. Nucleic Acids Res. 43, 4133–4149. doi: 10.1093/nar/gkv186
Quail, M. A., Kozarewa, I., Smith, F., Scally, A., Stephens, P. J., Durbin, R., et al. (2008). A large genome center's improvements to the Illumina sequencing system. Nat. Methods 5, 1005–1010. doi: 10.1038/nmeth.1270
Raguse, M., Torres, R., Seco, E. M., Gándara, C., Ayora, S., Moeller, R., et al. (2017). Bacillus subtilis DisA helps to circumvent replicative stress during spore revival. DNA Repair. 59, 57–68. doi: 10.1016/j.dnarep.2017.09.006
Rojo, F., and Alonso, C. J. (1995). The β recombinase of plasmid pSM19035 binds to two adjacent sites, making different contacts at each of them. Nucleic Acids Res. 23, 3181–3188. doi: 10.1093/nar/23.16.3181
Rojo, F. C., and Alonso, J. (1994). The β recombinase from the Streptococcal plasmid pSM 19035 represses its own transcription by holding the RNA polymerase at the promoter region. Nucleic Acids Res. 22, 1855–1860. doi: 10.1093/nar/22.10.1855
Romero, H., Rosch, T. C., Hernández-Tamayo, R., Lucena, D., Ayora, S., Alonso, J. C., et al. (2019b). Single molecule tracking reveals functions for RarA at replication forks but also independently from replication during DNA repair in Bacillus subtilis. Sci. Rep. 9:1997. doi: 10.1038/s41598-018-38289-6
Romero, H., Serrano, E., Hernández-Tamayo, R., Carrasco, B., Cárdenas, P. P., Ayora, S., et al. (2020). Bacillus subtilis RarA acts as a positive RecA accessory protein. Front. Microbiol. 11:92. doi: 10.3389/fmicb.2020.00092
Romero, H., Torres, R., Hernández-Tamayo, R., Carrasco, B., Ayora, S., Graumann, P. L., et al. (2019a). Bacillus subtilis RarA acts at the interplay between replication and repair-by-recombination. DNA Repair 78, 27–36. doi: 10.1016/j.dnarep.2019.03.010
Sánchez, H., Carrasco, B., Cozar, M. C., and Alonso, C. J. (2007). Bacillus subtilis RecG branch migration translocase is required for DNA repair and chromosomal segregation. Mol. Microbiol. 65, 920–935. doi: 10.1111/j.1365-2958.2007.05835.x
Sanchez, H., Kidane, D., Cozar, M. C., Graumann, P. L., and Alonso, C. J. (2006). Recruitment of Bacillus subtilis RecN to DNA double-strand breaks in the absence of DNA end processing. J. Bacteriol. 188, 353–360. doi: 10.1128/JB.188.2.353-360.2006
Sanchez, H., Kidane, D., Reed, P., Curtis, F. A., Cozar, M. C., Graumann, P. L., et al. (2005). The RuvAB branch migration translocase and RecU Holliday junction resolvase are required for double-stranded DNA break repair in Bacillus subtilis. Genetics 171, 873–883. doi: 10.1534/genetics.105.045906
Sanders, K., Lin, C. L., Smith, A. J., Cronin, N., Fisher, G., Eftychidis, V., et al. (2017). The structure and function of an RNA polymerase interaction domain in the PcrA/UvrD helicase. Nucleic Acids Res. 45, 3875–3887. doi: 10.1093/nar/gkx074
Sedgwick, B. (2004). Repairing DNA-methylation damage. Nat. Rev. Mol. Cell Biol. 5, 148–157. doi: 10.1038/nrm1312
Selby, C. P., and Sancar, A. (1993). Molecular mechanism of transcription-repair coupling. Science 260, 53–58. doi: 10.1126/science.8465200
Selby, C. P., and Sancar, A. (1994). Mechanisms of transcription-repair coupling and mutation frequency decline. Microbiol. Rev. 58, 317–329. doi: 10.1128/MMBR.58.3.317-329.1994
Serrano, E., Carrasco, B., Gilmore, J. L., Takeyasu, K., and Alonso, C. J. (2018). RecA regulation by RecU and DprA during Bacillus subtilis natural plasmid transformation. Front. Microbiol. 9:1514. doi: 10.3389/fmicb.2018.01514
Shaw, G., Gan, J., Zhou, Y. N., Zhi, H., Subburaman, P., Zhang, R., et al. (2008). Structure of RapA, a Swi2/Snf2 protein that recycles RNA polymerase during transcription. Structure 16, 1417–1427. doi: 10.1016/j.str.2008.06.012
Singleton, M. R., Dillingham, M. S., and Wigley, B. D. (2007). Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 76, 23–50. doi: 10.1146/annurev.biochem.76.052305.115300
Subramanya, H. S., Bird, L. E., Brannigan, J. A., and Wigley, B. D. (1996). Crystal structure of a DExx box DNA helicase. Nature 384, 379–383. doi: 10.1038/384379a0
Sukhodolets, M. V., Cabrera, J. E., Zhi, H., and Jin, J. D. (2001). RapA, a bacterial homolog of SWI2/SNF2, stimulates RNA polymerase recycling in transcription. Genes Dev. 15, 3330–3341. doi: 10.1101/gad.936701
Taucher-Scholtz, G., Abdel-Monem, M., and Hoffmann-Berling, H. (1983). “Functions of helicases in E. coli,” in Mechanisms of DNA Replication and Recombination, ed N. R. Cozzarelli (New York, NY: Alan R. Liss Inc.), 65–76.
Torres, R., Carrasco, B., Gandara, C., Baidya, A. K., Ben-Yehuda, S., and Alonso, C. J. (2019). Bacillus subtilis DisA regulates RecA-mediated DNA strand exchange. Nucleic Acids Res. 47, 5141–5154. doi: 10.1093/nar/gkz219
Torres, R., Romero, H., Rodríguez-Cerrato, V., C., and Alonso, J. (2017). Interplay between Bacillus subtilis RecD2 and the RecG or RuvAB helicase in recombinational repair. DNA Repair 55, 40–46. doi: 10.1016/j.dnarep.2017.05.004
Trautinger, B. W., Jaktaji, R. P., Rusakova, E., and Lloyd, G. R. (2005). RNA polymerase modulators and DNA repair activities resolve conflicts between DNA replication and transcription. Mol. Cell. 19, 247–258. doi: 10.1016/j.molcel.2005.06.004
Valero-Rello, A., Lopez-Sanz, M., Quevedo-Olmos, A., Sorokin, A., and Ayora, S. (2017). Molecular mechanisms that contribute to horizontal transfer of plasmids by the bacteriophage SPP1. Front. Microbiol. 8:1816. doi: 10.3389/fmicb.2017.01816
Veaute, X., Delmas, S., Selva, M., Jeusset, J., Le Cam, E., Matic, I., et al. (2005). UvrD helicase, unlike Rep helicase, dismantles RecA nucleoprotein filaments in Escherichia coli. EMBO J. 24, 180–189. doi: 10.1038/sj.emboj.7600485
Veaute, X., Jeusset, J., Soustelle, C., Kowalczykowski, S. C., Le Cam, E., and Fabre, F. (2003). The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature 423, 309–312. doi: 10.1038/nature01585
Vlasic, I., Mertens, R., Seco, E. M., Carrasco, B., Ayora, S., Reitz, G., et al. (2014). Bacillus subtilis RecA and its accessory factors, RecF, RecO, RecR and RecX, are required for spore resistance to DNA double-strand break. Nucleic Acids Res. 42, 2295–2307. doi: 10.1093/nar/gkt1194
Wiedermannova, J., Sudzinova, P., Koval, T., Rabatinova, A., Sanderova, H., Ramaniuk, O., et al. (2014). Characterization of HelD, an interacting partner of RNA polymerase from Bacillus subtilis. Nucleic Acids Res. 42, 5151–5163. doi: 10.1093/nar/gku113
Witkin, E. M. (1969). Ultraviolet-induced mutation and DNA repair. Annu. Rev. Microbiol. 23, 487–514. doi: 10.1146/annurev.mi.23.100169.002415
Wu, L. D., and Hickson, I. (2006). DNA helicases required for homologous recombination and repair of damaged replication forks. Annu. Rev. Genet. 40, 279–306. doi: 10.1146/annurev.genet.40.110405.090636
Keywords: replication fork stalling, RNA polymerase backtracking, replication-transcription conflict, RecL16, Rep, UvrD
Citation: Moreno-del Alamo M, Torres R, Manfredi C, Ruiz-Masó JA, del Solar G and Alonso JC (2020) Bacillus subtilis PcrA Couples DNA Replication, Transcription, Recombination and Segregation. Front. Mol. Biosci. 7:140. doi: 10.3389/fmolb.2020.00140
Received: 28 April 2020; Accepted: 10 June 2020;
Published: 21 July 2020.
Edited by:
Chew Chieng Yeo, Sultan Zainal Abidin University, MalaysiaReviewed by:
Arijit Dutta, The University of Texas Health Science Center at San Antonio, United StatesHarshad Ghodke, University of Wollongong, Australia
Copyright © 2020 Moreno-del Alamo, Torres, Manfredi, Ruiz-Masó, del Solar and Alonso. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan Carlos Alonso, amNhbG9uc29AY25iLmNzaWMuZXM=
†Present address: Candela Manfredi, Dept of Pediatrics, Emory University School of Medicine, Atlanta, GA, United States