 Gennady M. Verkhivker
Gennady M. Verkhivker Steve Agajanian
Steve Agajanian Guang Hu
Guang Hu Peng Tao
Peng Tao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Biosci. , 09 July 2020
Sec. Biological Modeling and Simulation
Volume 7 - 2020 | https://doi.org/10.3389/fmolb.2020.00136
This article is part of the Research Topic Understanding Protein Dynamics, Binding and Allostery for Drug Design View all 21 articles
Allosteric regulation is a common mechanism employed by complex biomolecular systems for regulation of activity and adaptability in the cellular environment, serving as an effective molecular tool for cellular communication. As an intrinsic but elusive property, allostery is a ubiquitous phenomenon where binding or disturbing of a distal site in a protein can functionally control its activity and is considered as the “second secret of life.” The fundamental biological importance and complexity of these processes require a multi-faceted platform of synergistically integrated approaches for prediction and characterization of allosteric functional states, atomistic reconstruction of allosteric regulatory mechanisms and discovery of allosteric modulators. The unifying theme and overarching goal of allosteric regulation studies in recent years have been integration between emerging experiment and computational approaches and technologies to advance quantitative characterization of allosteric mechanisms in proteins. Despite significant advances, the quantitative characterization and reliable prediction of functional allosteric states, interactions, and mechanisms continue to present highly challenging problems in the field. In this review, we discuss simulation-based multiscale approaches, experiment-informed Markovian models, and network modeling of allostery and information-theoretical approaches that can describe the thermodynamics and hierarchy allosteric states and the molecular basis of allosteric mechanisms. The wealth of structural and functional information along with diversity and complexity of allosteric mechanisms in therapeutically important protein families have provided a well-suited platform for development of data-driven research strategies. Data-centric integration of chemistry, biology and computer science using artificial intelligence technologies has gained a significant momentum and at the forefront of many cross-disciplinary efforts. We discuss new developments in the machine learning field and the emergence of deep learning and deep reinforcement learning applications in modeling of molecular mechanisms and allosteric proteins. The experiment-guided integrated approaches empowered by recent advances in multiscale modeling, network science, and machine learning can lead to more reliable prediction of allosteric regulatory mechanisms and discovery of allosteric modulators for therapeutically important protein targets.
Allosteric regulation is an efficient and robust mechanism for molecular communication and signaling in the cell employed by proteins for regulation of activity and adaptability during processes of signal transduction, catalysis, and gene regulation (Monod et al., 1965; Koshland, 1998; Changeux and Edelstein, 2005; Popovych et al., 2006; Changeux, 2012). The recent breakthroughs in nuclear magnetic resonance (NMR) technologies have enabled dynamic studies of large biomolecules at atomic resolution, and are now frequently employed as powerful diagnostic tools of allosteric communications in proteins (Boehr et al., 2006; Jarymowycz and Stone, 2006; Mittermaier and Kay, 2006, 2009; Sprangers et al., 2007; Korzhnev and Kay, 2008; Kalodimos, 2011; Kay, 2011, 2016; Rosenzweig and Kay, 2014; Lisi and Loria, 2016, 2017; Huang and Kalodimos, 2017; Jiang and Kalodimos, 2017). Allosteric molecular events can involve complex cascades of thermodynamic and rapid dynamic changes that occur on different spatial and temporal scales. The thermodynamic-centric energy landscape concepts and conformational selection models of allosteric regulation have gained a considerable prominence in recent years, rooted in the assumption that statistical ensembles of preexisting conformational states and communication pathways are intrinsic to a given protein system (Astl et al., 2019) and allow for modulation and redistribution induced by external perturbations, ligand binding, and mutations (Gunasekaran et al., 2004; Tsai et al., 2008, 2009; del Sol et al., 2009; Csermely et al., 2010, Zhuravlev and Papoian, 2010; Ma et al., 2011; Wrabl et al., 2011; Hilser et al., 2012; Nussinov, 2012; Motlagh et al., 2014; Tsai and Nussinov, 2014; Nussinov and Tsai, 2015; Guo and Zhou, 2016; Liu and Nussinov, 2016; Astl et al., 2019). Conformational dynamics redistributions in the absence of appreciable structural transformations are the hallmark of the “entropy-driven” allosteric mechanisms in which allosteric interactions can be mediated through alterations of functional motions and rebalancing of rigid and flexible protein regions (Cooper and Dryden, 1984; Stevens et al., 2001; Dam et al., 2002; Kern and Zuiderweg, 2003; Frederick et al., 2007; Tzeng and Kalodimos, 2009, Nesmelova et al., 2010; Kalodimos, 2011, 2012; McLeish et al., 2013; Li et al., 2014; Buchenberg et al., 2017; Stock and Hamm, 2018; Wodak et al., 2019). The quantitative elucidation of these highly dynamic and often elusive processes continues to present formidable technical and conceptual challenges. Despite significant advances, the quantitative characterization and prediction of functional allosteric states, interactions and mechanisms continue to present highly challenging problems in the field. The fundamental biological importance and complexity of these processes require innovative computational and experimental approaches that can advance current understanding of allosteric regulatory processes. A systematic interdisciplinary effort is needed to leverage the burgeoning knowledge about allosterically regulated proteins to develop robust experiment-informed computational tools for atomistic prediction of allosteric mechanisms. In this review we discuss and analyze how recent advances in biophysical simulations and network science can be integrated with NMR spectroscopy experiments and leverage the rising power of machine learning (ML) approaches to enable the reliable quantitative characterization of allosteric regulation mechanisms and facilitate allosteric drug discovery. We discuss in details computational strategies that leverage biophysical and network-based modeling with NMR experiments for characterization and probing of allosteric regulatory mechanisms. The review also critically discusses advantages and limitations of emerging approaches including Markovian modeling and the information-theoretical analysis of dynamic flows in allosteric networks in addressing present challenges and open questions of allosteric regulation mechanisms.
It has been recognized that allosteric regulation is a global property of protein systems that can be described by the residue interaction networks in which the effector binding initiates a cascade of coupled fluctuations propagating through the network and eliciting long-range functional responses at distal sites (Atilgan et al., 2004; Brinda and Vishveshwara, 2005, 2010; del Sol and O'Meara, 2005; Bode et al., 2007; Sethi et al., 2009; Vijayabaskar and Vishveshwara, 2010; Csermely et al., 2013; Di Paola and Giuliani, 2015; Dokholyan, 2016). The graph-based network approaches have offered a simple and effective formalism for describing allosteric interactions, where the dynamic fluctuations are mapped onto a graph with nodes representing residues and edges representing weights of the measured dynamic properties. The network-centric methods have represented a powerful complementary strategy to physics-based landscape models of protein dynamics by quantifying global functional changes (Vendruscolo et al., 2002; Atilgan et al., 2004; Brinda and Vishveshwara, 2005, 2010; Ghosh and Vishveshwara, 2007, 2008; Hansia et al., 2009; Bhattacharyya and Vishveshwara, 2011; Ghosh et al., 2011; Csermely et al., 2012; Gasper et al., 2012; Bhattacharya and Vaidehi, 2014; General et al., 2014; Dokholyan, 2016; Adhireksan et al., 2017), identifying key functional centers and allosteric communication pathways (Verkhivker et al., 2002; del Sol and O'Meara, 2005; del Sol et al., 2006; Sethi et al., 2009, 2013; Vijayabaskar and Vishveshwara, 2010; Rivalta et al., 2012; Vanwart et al., 2012; Farabella et al., 2014; Di Paola and Giuliani, 2015; Kalescky et al., 2015, 2016; Hertig et al., 2016; Ricci et al., 2016; Stolzenberg et al., 2016; Palermo et al., 2017; Zhou et al., 2017, 2019a,b; Liang et al., 2019; Li et al., 2019). Recent years have witnessed the proliferation of numerous computational tools for predicting allosteric pathways and communications in proteins (Ming and Wall, 2005, 2006; McClendon et al., 2009; Tehver et al., 2009; Mitternacht and Berezovsky, 2011; Bowman and Geissler, 2012; Panjkovich and Daura, 2012, 2014; Goncearenco et al., 2013; Kaya et al., 2013; Stetz and Verkhivker, 2017). The network studies have also suggested that rapid signal transmission of allosteric interactions through small-world networks encoded in protein folds may be a universal signature encoded in protein families (Tsai et al., 2009; Di Paola and Giuliani, 2015). Significant bodies of computational and experimental studies have shown that integration of network-based approaches with structural and biochemical studies can provide a robust platform for further exploration and atomistic characterization of allosteric states and regulatory mechanisms controlled by allostery.
Functional residues in residue networks are often connected via strong evolutionary relationships (Lockless and Ranganathan, 1999; Suel et al., 2003; Halabi et al., 2009; Aguilar et al., 2012; McLaughlin et al., 2012; Simonetti et al., 2013). Coevolution of protein residues can reflect correlated functional dynamics of these sites in mediating residue-residue contacts (Socolich et al., 2005), protein folding transitions (Morcos et al., 2011), and allosteric signaling in protein complexes (Wang et al., 2019). Coevolving residues could also form direct communication paths in the interaction networks with connections weighted according to dynamic couplings and coevolutionary interaction strengths between nodes (Chakrabarti and Panchenko, 2009, 2010; Nishi et al., 2011). Dynamic and coevolutionary residue correlations may also act as synchronizing forces that determine modular organization of allosteric interaction networks and enable efficient allosteric regulation (Stetz and Verkhivker, 2017). These results have motivated the development of novel community-based methods for modeling ensembles of allosteric communication pathways in protein structures (Tse and Verkhivker, 2015a,b; Verkhivker et al., 2016; Stetz and Verkhivker, 2017). Using this computational framework, it was found that efficient allosteric communications in various signaling proteins could be controlled by structurally stable functional centers that exploit dynamically coupled residues in their local communities to propagate cooperative structural changes. The important revelation of these studies was that dynamic and evolutionary residue correlations may act as synchronizing forces to enable efficient and robust allosteric regulation.
Examining proteins as dynamic regulatory machineries that fluctuate between functional allosteric states and modulated by ligand binding or mutations is critical to understanding the molecular principles of signaling in the cell. Computational studies of allosteric regulation in signaling proteins have led to important mechanistic insights, better atomistic understanding of complex regulatory processes and continuous integration with structural and functional experiments. A variety of computational approaches have been extensively explored in investigations of allosteric mechanisms in protein kinases. These studies included experiment-guided structural modeling and protein folding analysis (Levinson et al., 2006; Zhang et al., 2006; Kornev et al., 2008; Han et al., 2011; Jura et al., 2011; Shan et al., 2011, 2012, 2013; Taylor and Kornev, 2011; Tzeng and Kalodimos, 2011; Levinson and Boxer, 2012, 2014; Taylor et al., 2012a,b; Meharena et al., 2013; Shaw et al., 2014; Shukla et al., 2014; Kornev and Taylor, 2015; Schulze et al., 2016; Narayanan et al., 2017; Levinson, 2018; Ruff et al., 2018), molecular simulations and free energy computations (Yang and Roux, 2008; Dixit and Verkhivker, 2009, 2011a,b; Yang et al., 2009; Arkhipov et al., 2013; Lin and Roux, 2013; Lin et al., 2013, 2014; Dixit and Verkhivker, 2014; Meng and Roux, 2014; Fajer et al., 2017; Kim et al., 2017; Meng et al., 2017), and network modeling (James and Verkhivker, 2014; Tse and Verkhivker, 2015a,b,c; Czemeres et al., 2017; Stetz et al., 2017; Astl and Verkhivker, 2019a,b). By examining residue interaction networks in protein kinases a unifying mechanistic model of allosteric coupling between the ATP-binding and substrate binding sites conserved among kinases was proposed (Tse and Verkhivker, 2015a,b,c; Stetz et al., 2017). A theoretical framework for rationalizing binding preferences of the kinase inhibitors was developed establishing the relationships between ligand binding and modulation of the residue interaction networks (Tse and Verkhivker, 2015a,b,c). Atomistic modeling of the ABL kinase regulation using a combination molecular dynamics (MD) simulations, structural perturbation methods and network-centric analysis (Astl and Verkhivker, 2019a,b) has provided evidence of allosteric interactions and communication pathways in the ABL interaction networks that supported and explained the underlying mechanisms proposed in the pioneering NMR studies (Saleh et al., 2017).
Computational studies of allosteric regulation in molecular chaperones Hsp90 and Hsp70 have also been instrumental to the progress in the field by complementing biochemical experiments and providing a detailed dynamic view of the functional cycle and mechanisms (Colombo et al., 2008; Morra et al., 2009, Verkhivker et al., 2009; Morra et al., 2010, 2012; Matts et al., 2011a,b; Chiappori et al., 2012, 2016; Dixit and Verkhivker, 2012; Lawless et al., 2013; Verkhivker, 2014, 2018a,b; Paladino et al., 2015; Stetz and Verkhivker, 2015, 2016, 2017, 2018; Czemeres et al., 2017; Stetz et al., 2017). Using a network-based formalism of allostery, computational studies have captured NMR-observed direction-specific nature of signal propagation pathways in the Hsp70 chaperone (Stetz and Verkhivker, 2015, 2017).
Studies of allosteric mechanisms have indicated that integration of experiment-informed molecular simulations with network-based formalisms of allostery may provide a convenient and powerful platform for atomistic characterization of allosteric states and regulatory mechanisms. The lessons from studies of signaling proteins including protein kinases and molecular chaperones have suggested that allosteric regulation mechanisms can proceed via a non-trivial and often elusive combination of the three classical models of allostery: induced fit, conformational selection, and dynamic allostery. Computational modeling and atomistic simulations of protein systems and functional assemblies have shown that allosteric mechanisms may not necessarily imply a simple switching between the crystal structures of the inactive and active states, but often represent a complex regulatory machinery in which binding and external perturbations could give rise to a spectrum of functionally relevant and yet often hidden allosteric conformations exhibiting a range of activity levels.
The growing number of high-resolution crystal structures and wealth of structural information about protein systems have had an enormous impact on computational and simulation approaches, facilitating development of knowledge-based methods and advanced sampling techniques. However, allosteric functional states in proteins are often highly dynamic and short-lived representing low populated, high energy states that are rarely directly observed in X-ray crystallography experiment. A large amount of conformational sampling is typically needed to uncover and isolate high-energy functional states simulations. For instance, cryptic (or hidden) allosteric sites sporadically appear during conformational transitions of a protein in the presence of a bound ligand. These hidden allosteric sites are invisible in crystal structures and can be detected due to the stabilization of the low-populated, higher-energy conformation by certain compounds. Even with the advanced sampling techniques and enormous computer power that is now available, the experimental validation and confirmation of allosteric states represents the key component to ensure robust quantitative modeling and analysis of allosteric mechanisms.
NMR spectroscopy is a powerful method for studying protein dynamics and allosteric mechanisms by probing multiple time scales and detecting residue-specific conformational changes associated with ligand binding (Boehr et al., 2006; Jarymowycz and Stone, 2006; Mittermaier and Kay, 2006, 2009; Sprangers et al., 2007; Korzhnev and Kay, 2008; Kalodimos, 2011; Kay, 2011, 2016; Rosenzweig and Kay, 2014; Lisi and Loria, 2016, 2017; Huang and Kalodimos, 2017; Jiang and Kalodimos, 2017). The micro- to milli-second time scale protein motions measured in relaxation-dispersion experiments can provide information about the distribution of conformational states and thermodynamics and kinetics of allosteric protein changes. Protein dynamics can also be investigated by NMR methods other than traditional relaxation experiments. Residual dipolar couplings are sensitive to motions occurring across a vast time scale, ranging from seconds to faster than nanoseconds. Conformational changes in isotopically labeled proteins upon ligand binding can be detected by two-dimensional heteronuclear single quantum coherence (HSQC) spectroscopy for large protein systems (Sprangers et al., 2007; Korzhnev and Kay, 2008). Chemical shift mapping of protein residues upon ligand binding provides a specific and precise fingerprint of allosteric propagation effects that allows to detect site-specific binding responses, characterize pathways of allosteric communication and differentiate between competitive and allosteric inhibitor binding (Grutsch et al., 2016; Berjanskii and Wishart, 2017; Krivdin, 2017; Nerli et al., 2018). The NMR technologies have enabled structural studies of conformational dynamic processes at atomic resolution and are used to identify coupled networks and communication pathways in allosteric proteins (Swain and Gierasch, 2006; Smock and Gierasch, 2009; Shi and Kay, 2014; Grutsch et al., 2016). Relaxation dispersion NMR methods have enabled detection and characterization of rare and energetically excited conformational states that play significant role in dynamic activation of protein function and allosteric mechanisms (Vallurupalli et al., 2012; Kalbitzer et al., 2013; Munte et al., 2013; Sekhar and Kay, 2013, 2019; Williamson and Kitahara, 2019). Characterization of low-lying excited states of proteins by high-pressure NMR under equilibrium conditions can allow for detection of reversible transitions that are functionally relevant, providing means for probing dynamic energy landscapes of allosteric mechanisms (Kalbitzer et al., 2013; Williamson and Kitahara, 2019). High-pressure NMR can help to identify these conformations, including low populated functional states, and characterize their energies and kinetics of conformational changes (Williamson and Kitahara, 2019). By measuring redistributions in the conformational entropy, pressure-dependent chemical shifts can help to sequester low-populated functional states (Kalbitzer et al., 2013; Munte et al., 2013; Williamson and Kitahara, 2019).
Recent years have witnessed the development of various approaches that investigate NMR chemical shift perturbations to identify potential allosteric networks and structural dynamics in proteins (Selvaratnam et al., 2011, 2012; Robustelli et al., 2012; Cembran et al., 2014). NMR chemical exchange saturation transfer (CEST) experiments can provide adequate characterization of slower exchange processes, identify invisible states, and slow conformational exchange (Long et al., 2014; Anthis and Clore, 2015; Yuwen et al., 2017). NMR chemical shift covariance (CHESCA) and projection (CHESPA) analyses can identify blocks of dynamically coupled residues collectively forming allosteric interaction networks (Selvaratnam et al., 2011, 2012; Boulton et al., 2014, 2018; Boulton and Melacini, 2016). Allosteric proteins subjected to specific perturbations (ligand binding, mutations) cause residues that belong to the same effector-dependent allosteric network to exhibit a concerted response signal. CHESCA approach can detect patterns of correlated changes in the chemical shifts between apo and holo states due to perturbations and isolate allosterically coupled regions (Figure 1). This method is particularly effective in detecting allosteric networks within dynamic and partially unstructured regions (Boulton and Melacini, 2016; Boulton et al., 2018). NMR chemical shift perturbations have been recently used in combination with Markov model network analysis to reveal the dynamic flow of communication between allosteric communities in the protein kinases (Aoto et al., 2016). NMR-guided computational modeling can leverage CHESCA approach for computation of the chemical shift correlation matrices in the known allosteric states obtained using crystal structures of complexes with allosteric ligands. The experimental NMR chemical shifts can guide molecular simulations and network analysis by reporting on blocks of dynamically coupled residues forming allosteric interaction networks. Through integration of these experimental data into accelerated atomistic simulations, a more detailed mapping of the functional landscapes and relevant allosteric states can be achieved.
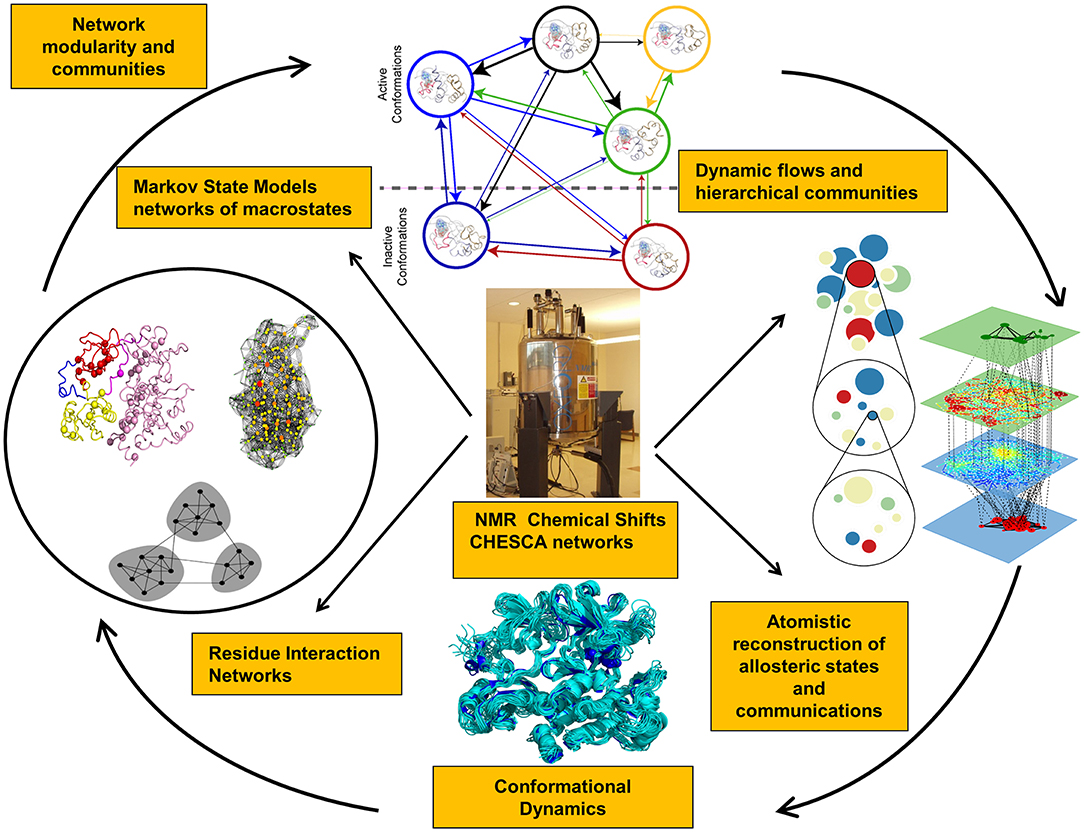
Figure 1. Integration of NMR experiments and computational approaches for experiment-guided analysis of allosteric states and mechanisms.
Protein systems can be efficiently simulated on longer time scales by accelerated meta-dynamics approaches (Limongelli et al., 2013; Palazzesi et al., 2013, 2017; Sutto and Gervasio, 2013; Bonomi and Camilloni, 2017; Kuzmanic et al., 2017; Yang et al., 2018; Brotzakis and Parrinello, 2019) where the experimental and computed NMR chemical shifts (Shen and Bax, 2010; Han et al., 2011) are often used to determine collective variables (Granata et al., 2013; Xia et al., 2013; Palazzesi et al., 2017). NMR chemical shifts can be also evaluated using structure-based CamShift approach (Kohlhoff et al., 2009) with collective variables defined as the difference between experimental and calculated chemical shifts. These NMR-guided simulation techniques have enabled adequate sampling of the conformational space and robust structure reconstruction using experimental constraints (Robustelli et al., 2010; Cavalli et al., 2011; Granata et al., 2013). NMR chemical shift observables can be also used in combination with other collective variables in meta-dynamics simulations to guide the efficient exploration of allosteric states and functional transitions (Kimanius et al., 2015; Ansari et al., 2016).
A combination of powerful and expensive NMR spectroscopy equipment, biophysical techniques and protein expression platforms is often required to obtain structures of allosteric states for protein systems and experimental validation of short-lived hidden functional conformations. Despite unique abilities of NMR spectroscopy to detect highly dynamic events and examine conformational landscapes of allosteric proteins, the NMR applications for high-resolution reconstruction of allosteric states are still fairly limited owing to complexity and cost of these experiments. Hence, development of novel research strategies based on innovative integration of NMR spectroscopy and experiment-guided simulation approaches become especially important and clearly represent the most promising avenue for further explorations going forward.
Given the complexity of thermodynamic and kinetic factors underlying allosteric regulatory events, the information-based theory of signal propagation (Chennubhotla and Bahar, 2006, 2007; Chennubhotla et al., 2008) and stochastic Markov state models (MSMs) (Prinz et al., 2011; McGibbon et al., 2014; Pande, 2014; Shukla et al., 2015, 2017; Wu et al., 2016; Husic and Pande, 2018) have become increasingly useful states-and-rates network models with the continuously developing open source software infrastructure (Cronkite-Ratcliff and Pande, 2013; Bowman, 2014; Bowman and Noe, 2014; Harrigan et al., 2017). The MSMs have been successfully adopted for describing the transitions between functional states during allosteric events (Bowman et al., 2015; Hart et al., 2016; Sengupta and Strodel, 2018). Combined with MD simulations, MSM approaches can provide connectivity maps of states on the free energy landscape, estimate the effect of allosteric perturbations on the conformational equilibrium, and rigorously describe kinetics of allosteric transitions. Recent advances in the field have highlighted how MSM tools can help to recognize structural and dynamic patterns of conformational ensembles, identify functional allosteric states hidden in the conformational ensembles and reconstruct allosteric mechanisms (Sengupta and Strodel, 2018). Markov models have been employed for understanding of the reaction mechanisms, thermodynamics and free-energy landscape population shifts, the hierarchy of timescales and the structural basis of allosteric events (Prinz et al., 2011; Pande, 2014; Shukla et al., 2015, 2017; Zhou et al., 2017, 2019a,b).
When combined with appropriate general coordinates, MSM could be a very powerful tool to reveal intrinsic states of the proteins (Malmstrom et al., 2015). The important component of the MSM approach in studies of allosteric systems is the employment of robust dimensionality reduction techniques to identify experimentally-informed collective variables that can enhance sampling and provide efficient detection and separation of functional allosteric states. Dimension reduction is often performed using time-lagged independent component analysis (TICA) (Schwantes and Pande, 2014; Perez-Hernandez and Noe, 2016; Noe and Clementi, 2017; Olsson et al., 2017). In these approaches, the simulation samples can be divided into substates assuming that conformations within each substate share kinetic similarity and could interconvert rapidly (Bowman et al., 2009; Zhou and Tao, 2018; Zhou et al., 2018a,b). t-SNE method was recently developed as a dimensionality reduction method with minimum structural information loss revealing that both one-dimensional (1D) and two-dimensional (2D) models of t-SNE method are superior to other tools in distinguishing functional states of allosteric proteins (Zhou et al., 2018a,b). MSMs and transition network models are widely applied to extract kinetic descriptors from equilibrium simulations. Directed Kinetic Transition Network (DKTN) which is a graph representation of a master equation was developed for describing non-equilibrium kinetics in allosteric proteins (Zhou et al., 2019a,b). Markov modeling studies have also examined the timescales and intra-molecular pathways implicated in allostery by introducing master equation-based approach for allostery by population shift (Long and Bruschweiler, 2011). Another study employed a graph-theoretic approach and Markov stability analysis for modeling of signaling pathways and characterization of allosteric sites (Amor et al., 2014).
Current allosteric models have suggested that conformational and dynamical distribution phase space accessible for allosteric interactions in proteins is much larger than the experimentally visible landscapes provided through crystallographic and NMR experiments. As a result, external perturbations, such as mutations and/or ligand binding that could significantly affect conformational space and dynamic distribution of allosteric proteins and can be employed as probes to explore functional consequences of allosteric phenomena. The recently developed Rigid Residue Scan (RRS) simulation method has been shown as effective tool to perturb protein dynamics and assess both conformational and dynamical redistributions in allosteric systems (Kalescky et al., 2015, 2016). Using the RRS method, the predictive models for light-oxygen-voltage-sensing (LOV) domains allostery have been developed that identified the experimentally verified mutants with distinctive allosteric regulatory effects. The results of this analysis have suggested how manipulating functional regions with light in LOV proteins could link chemistry and allostery, providing a path for rational engineering of LOV ontogenetic tools.
Multiscale simulations and MSM approaches have shown that allosteric mechanisms may not necessarily imply a simple two-state switch between the major inactive and active states, but often represent a dynamic multilayered regulatory machine in which binding and external perturbations could give rise to a discrete spectrum of functionally relevant and yet often hidden allosteric conformations exhibiting a range of activity levels. Experiment-informed Markovian modeling studies have shown a promise in adequately describing the hierarchy of allosteric states by recognizing structural and dynamic patterns of conformational ensembles and identifying functional allosteric states that are hidden in the crystal structures of allosteric proteins. Discovery of multiple hidden allosteric sites by combining Markov state models and experiments has been advanced and applied for antibiotic target TEM-1 β-lactamase (Bowman et al., 2015). Bowman et al. used MSM approach of a ligand-free protein to identify allosteric sites based on several signatures of collective dynamics, namely the presence of a pocket in a significant fraction of the population and the presence of correlated motions between the newly discovered pocket and the active site which provides means for allosteric communication between distant sites. The central to this pioneering work is a close integration with labeling experiments on TEM-1 β-lactamase that were performed to test the existence of hidden allosteric sites as feasible targets for allosteric drug design (Bowman et al., 2015). These illuminating studies have shown for the first time the power of integrated tools to identify, characterize and exploit hidden allosteric sites, highlighting the robust nature of Markov modeling tools in guiding the experiments. It has been argued that the wealth of thermodynamic, kinetic and structural data derived from MSMs can guide further development of experimental tools for discovery of hidden allosteric states and invisible cryptic allosteric binding sites.
The results suggest there are many undiscovered hidden allosteric sites that can be characterized and targeted with rational drug design (Cimermancic et al., 2016; Oleinikovas et al., 2016; Beglov et al., 2018; Kuzmanic et al., 2020). The hidden allosteric sites are invisible in crystal structures and cryptic sites can emerge as a result of stabilization of rare, high-energy states by small fragment probes. The allosteric mechanisms of cryptic site formation may involve a delicate interplay between induced-fit and conformational selection that can be modeled using elaborate replica-exchange sampling techniques (Oleinikovas et al., 2016). Collectively, experiment-informed multiscale simulation studies have shown that these tools can adequately describe complexity and stochasticity that underlies the thermodynamics and hierarchy of allosteric states and the molecular basis of allosteric mechanisms.
Recent advances in understanding allosteric regulation and activation mechanisms of therapeutic signaling proteins such as protein kinases have fueled unprecedented efforts to discover targeted allosteric inhibitors. Allosteric kinase inhibitors do not compete with ATP and could be more selective by binding to the regulatory sites outside of the ATP binding site (Dar and Shokat, 2011). Allosteric kinase inhibitors can improve target specificity and play an important role in the precision medicine initiative in oncology. NMR and X-ray crystallography studies have revealed a detailed atomistic picture of allosteric regulation in many protein kinases, showing how interacting signaling modules form a multilayered regulatory mechanism that exploits various allosteric switch points powered by binding or phosphorylation at different sites of the regulatory kinase complexes (Saleh et al., 2017). Recently discovered allosteric inhibitors of the ABL kinase GNF-2, GNF-5, and ABL001 (Asciminib) bind to the allosteric pocket near the C terminus of the ABL kinase domain stabilizes the inactive conformation of the kinase (Adrian et al., 2006; Zhang et al., 2010). Using solution NMR, X-ray crystallography, mutagenesis and hydrogen exchange mass spectrometry, it was shown that allosteric inhibitors can induce long-range structural and dynamic changes in the remote ATP-binding site (Adrian et al., 2006; Zhang et al., 2010; Wylie et al., 2017; Schoepfer et al., 2018). While the field of kinase inhibitors has enjoyed unprecedented success manifested in multiple FDA approved drugs, the development of allosteric kinase activators has been lagging behind. The mechanisms underpinning allosteric action of kinase activators can proceed by destabilization of the inactive state, stabilization of the active state, facilitating of the active state, and dynamic responses to phosphorylation in regulatory sites (Dar and Shokat, 2011; Fang et al., 2013; Hu et al., 2013; Cowan-Jacob et al., 2014). Structural and biochemical studies of allosteric inhibitors and activators of ABL kinase have indicated that structural environment near the allosteric pocket can serve as a sensor of ligand binding, triggering either stabilization of the inactive state or large conformational shift and activation. Furthermore, synergistic actions of allosteric and ATP competitive inhibitors have provided evidence that binding can perturb dynamics at distal regions and elicit ligand-specific communication between binding sites. Computational studies have detailed how allosteric inhibitors and activators may exert a differential control on allosteric signaling between binding sites (Astl and Verkhivker, 2019a). It was found that while inhibitor binding can strengthen the inhibitory ABL state and induce allosteric communications directed from the allosteric pocket to the ATP binding site, DPH activator may induce a more dynamic kinase state and preferentially activate allosteric couplings between the ATP and substrate binding sites (Astl and Verkhivker, 2019a).
By combining computational and experimental approaches a significant progress has been made in discovery of allosteric modulators of Hsp90 and Hsp70 chaperones. Recent studies have demonstrated that the C-terminal domain (CTD) of Hsp90 is important for dimerization of the chaperone and contains a second nucleotide binding site (Marcu et al., 2000a,b). The bacterial gyrase inhibitor novobiocin, a member of the coumeromycin family of antibiotics, is an Hsp90 antagonist that was found to inhibit a second ATP binding site at the C-terminus. Novobiocin binds the C-terminal nucleotide pocket and displaces both ATP and geldanamycin, and inhibits Hsp90's function (Marcu et al., 2000a,b). The principal advantage of C-terminal inhibition over N-terminal inhibition is the lack of a heat shock response upon ligand binding at the C-terminus of Hsp90. The first compounds that clearly differentiated between the C-terminus of Hsp90 and DNA gyrase, and converted a well-established gyrase inhibitor into a selective Hsp90 inhibitor were initially reported by Donnelly and Blagg (2008), Matts et al. (2011a), Matts et al. (2011b), Garg et al. (2016, 2017a,b), Hall et al. (2016), Khandelwal et al. (2016), and Kumar MV et al. (2018). The first experimental-guided computational prediction and mapping of hidden allosteric sites in Hsp90 combined NMR analysis, proteolytic fingerprinting and photoaffinity labeling with multiscale modeling of Hsp90 interactions and docking (Matts et al., 2011a,b). Computational predictions provided the first atomic resolution model of Hsp90 binding with the C-terminal modulator that fully satisfies the available experimental data and provide key insight for the structure-based design of allosteric Hsp90 inhibitors. In the subsequent study, a novel, computational approach for the discovery of allosteric inhibitors based on the physical characterization of signal propagation mechanisms was applied to the Hsp90 chaperone (Morra et al., 2010). By characterizing the allosteric hotspots of the inter-domain communication pathways, dynamic pharmacophore models to screen small molecules were developed. The computational predictions were combined with experimental validation showing that the selected molecules bind the allosteric sites of Hsp90, exhibit anti-proliferative activity in different tumor cell lines, and destabilize Hsp90 client proteins (Morra et al., 2010). The recent series of studies by Colombo and colleague have reported results of computer-aided design and synthesis of new allosteric ligands with micromolar to nanomolar anticancer activities, demonstrating the power of computational approaches in discovering allosteric modulators that can probe the relationships between structure dynamics and function of the Hsp90 proteins and regulatory complexes with client proteins (Sattin et al., 2015; D'Annessa et al., 2017; Masgras et al., 2017; Ferraro et al., 2019; Hu et al., 2020; Sanchez-Martin et al., 2020). Computational targeting of the Hsp90 client proteins based on the prediction of locally unstable substructures in proteins was used to develop potent probes and peptides blocking Hsp90-client interactions (Colombo et al., 2020). Recent efforts have also produced small molecules that can inhibit the inter-chaperone protein-protein interactions for Hsp70 chaperone (Gestwicki and Shao, 2019). These chemical probes have shown a considerable promise in interrogating chaperone networks in a range of models. Design, synthesis, and biological evaluation of small molecules that regulate the interaction between two Hsp70 and HOP chaperones reported the first class of compounds that specifically modulate these protein-protein interactions and inhibit protein folding events (Zaiter et al., 2019). An integrated computational and experimental approach probed allosteric mechanisms of Hsp70 binding, showing that symbiotic employment of different research tools in dissecting allosteric events in signaling proteins can be instrumental to discover selective allosteric modulators of protein functions (Rinaldi et al., 2018).
The emerging computational approaches that are now employed for studies of allosteric states and mechanisms include experiment-informed network approaches, Markovian modeling and also the information-theoretical methods that model dynamic flows and entropy transfer in complex systems. By describing protein dynamics as a dynamically evolving network of interconnected modules, the topological regularities of the network structure can be identified, while filtering out the relatively unimportant details. A modular description of a network can be viewed as a compression of that network topology, and the problem of community identification can be viewed as a problem of finding an efficient compression of the network structure and topology. Using this premise, the challenge of identifying the community structure of complex networks describing dynamic energy landscapes of allosteric proteins can be reformulated as an information-theoretic approach. Flow-based model methods operate through a stochastic walk on the dynamics of the network rather than on its topological structure, where communities consist of dynamically interconverting conformations among which the dynamic flow can persist for a long time and define functionally significant states (Rosvall and Bergstrom, 2007, 2008, 2010, 2011; Lancichinetti and Fortunato, 2009; Schaub et al., 2012; Rosvall et al., 2014; Kawamoto and Rosvall, 2015). This information-theoretical analysis can quantify the structure and dynamics of the proteins from a unified perspective in which short term dynamics is integrated into a long term behavior of the system through a modular description of dynamic flows occurring on a given network (Figure 2). In this approach, a random walk is used as a proxy for the dynamic flow on the network. The map equation method implemented by the Infomap algorithm (Edler et al., 2017) can find the optimal community partition of the dynamic conformational ensembles on different time scales (derived from MD simulations or MSM maps of macrostates) and identify dynamically persistent (as opposed to topology-derived) communities of functional macrostates. This dynamic flows method compresses the flows by aggregating nodes (states) with rapid stochastic movements, revealing network regularities as distinct dynamic modules in which flows are contained on a given time scale. The map equation has been also extended to the higher-order Markov dynamics (Lancichinetti and Fortunato, 2009; Lambiotte et al., 2011, 2019; Schaub et al., 2012; Rosvall et al., 2014; Delvenne et al., 2015; Salnikov et al., 2016). NMR chemical shift perturbations have been combined with Markov modeling and information-theoretical analysis to reveal the dynamic flow of communication between allosteric communities and identify critical residue nodes within the communication pathways in protein kinases (Aoto et al., 2016).
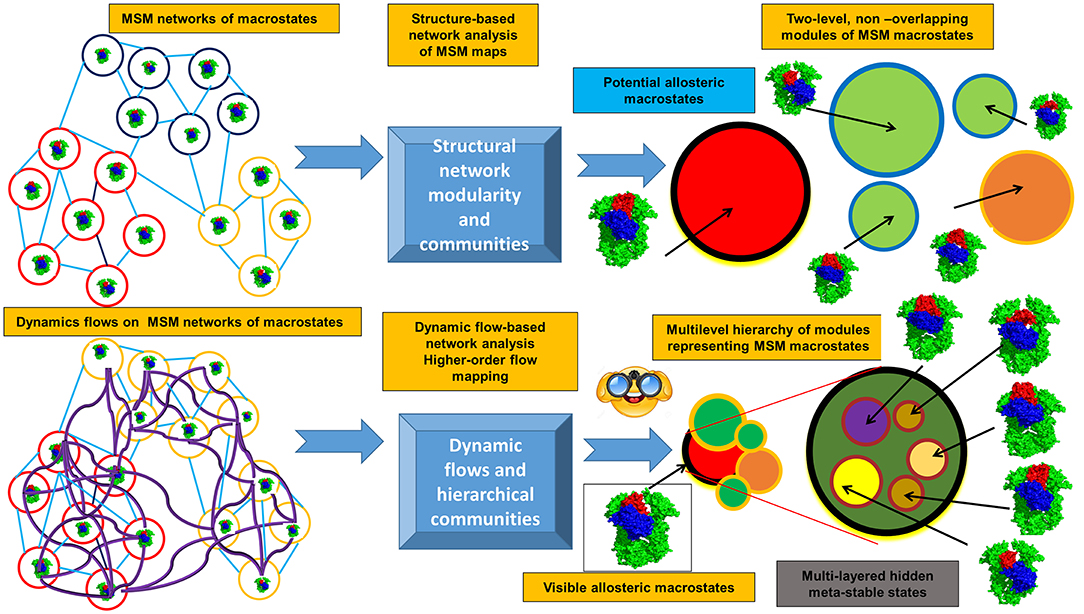
Figure 2. Overview of the information-theoretic framework for modeling of allosteric states and communications. The upper panel presents structure-based community detection. The lower panel illustrates modeling of the dynamical flows on the MSM maps of states and hierarchical dynamics-based detection of allosteric states and persistent communities.
This information-theoretical approach can also explore the dynamic evolution of the hierarchical multi-layered interaction networks and has a potential to uncover hidden allosteric states associated with the different dynamic time scales (Figure 2). Synchronization and causality are basic non-linear phenomenon observed in diverse complex systems, including allosterically regulated proteins. When studying allosteric mechanisms and communications in proteins, it is important not only to detect synchronized allosteric states, but also to identify causal relationships between them. The knowledge of information-theoretic measures is essential for the analysis of information flow between allosteric states and presents a challenging problem (Hlavácková-Schindler et al., 2007). The problem of finding a measure that is sensitive to the directionality of the flow of information has been explored using non-linear Granger causality of time series (Ancona et al., 2004). An asymmetric quantity termed Transfer Entropy (TE), has been proposed to estimate the directionality of the coupling and flow of information (Schreiber, 2000). The information-theoretic approaches measuring causal influences in multivariate time series (Hlavácková-Schindler et al., 2007; Ito, 2016; Darmon and Rapp, 2017) can be also applied to studies of allosteric protein states and mechanisms. The quantitative models of information flow between two correlated processes (Schreiber, 2000) were adopted to quantify time delayed correlations and entropy transfer between residue pairs as a measure of allosteric coupling in proteins (Hacisuleyman and Erman, 2017a,b). Through analysis of entropy transfer, one can determine residues that act as drivers of the fluctuations of other residues, thereby determining causality in the correlations and identifying residues that act as drivers of allosteric communication in proteins (Hacisuleyman and Erman, 2017a,b). The relative entropy concept from information theory was used as a quantitative metric to develop a method for measurement of the population shift during allosteric transitions (Zhou and Tao, 2019). The developed relative entropy-based dynamical allosteric network (REDAN) model was sucessfully applied to the second PDZ domain (PDZ2) in the human PTP1E protein, providing an accurate assessment of allosteic pathways (Zhou and Tao, 2019). A rigorous mathematical framework offered by the information-theoretical formalism of dynamic network flows combined with biophysical simulations may prove to be useful for finding modular patterns and dynamically persistent communities of macrostates. The integration of this methodology with NMR experiments can aid in the better identification of functional allosteric states by matching evolution of dynamic communities against the NMR chemical shift patterns and biophysical experiments.
Over the last few years, advances in the ML field have driven the design of new computational systems that improve with experience and are able to model increasingly complex chemical and biological phenomena (Goh et al., 2017; Korotcov et al., 2017; Chen et al., 2018a; Popova et al., 2018; Dimitrov et al., 2019; Mater and Coote, 2019). ML techniques have been successfully applied to various computational chemistry challenges (Husic and Pande, 2018), pharmaceutical data analysis (Burbidge et al., 2001), protein–ligand binding affinity prediction problems (Ballester and Mitchell, 2010, Decherchi et al., 2015), dissecting molecular determinants of protein mechanisms and biochemical reactions (Li et al., 2015, Cortina and Kasson, 2018, Shcherbinin et al., 2019). Data-intensive ML modeling can be also applied for detection and classification of allosteric protein states. The integration of Markov modeling, simulations and ML approaches into robust and reproducible computational pipelines with the experimental feedback can be explored for atomistic modeling and classification of allosteric states (Figure 3). Several ML algorithms including decision tree and artificial neural networks were employed in combination with MSM approaches to develop classification models of functionally relevant allosteric conformations that exhibit very similar tertiary structures (Zhou et al., 2018a,b). Despite the lack of significant conformational change between allosteric states of the second PDZ domain (PDZ2) in the human PTP1E protein, which is a prototypical example of dynamics-driven allostery, it was demonstrated that both algorithms could build effective prediction models and provide reliable quantitative evaluation of the contributions from individual residues to the difference between the two allosteric states (Zhou et al., 2018a,b). A high prediction accuracy and sensitivity of the ML models to small structural and dynamic changes have demonstrated the utility of these approaches in probing subtle allosteric changes. Deep neural networks were used in combination with MD simulations of the PDZ3 domain of PDS-95 revealing that allosteric effects can be quantified as residue-specific properties through two-dimensional property-residue maps (Hayatshahi et al., 2019). These residue response maps could accurately describe how different protein residues are affected by allosteric perturbations exerted on the protein system. Another ML-based analysis of protein dynamics was employed to compare the binding modes of TEM-1 β-lactamase in different catalytic states (Wang et al., 2019). While conventional analysis methods including principal components analysis (PCA) could not differentiate TEM-1 in different binding modes, neural network models resulted in an excellent classification scheme that differentiated between closely related functional states (Wang et al., 2019). This study has provided a unique insight into the role and specific function of individual residues, highlighting their contributions to the delicate thermodynamic balance between allosteric states.
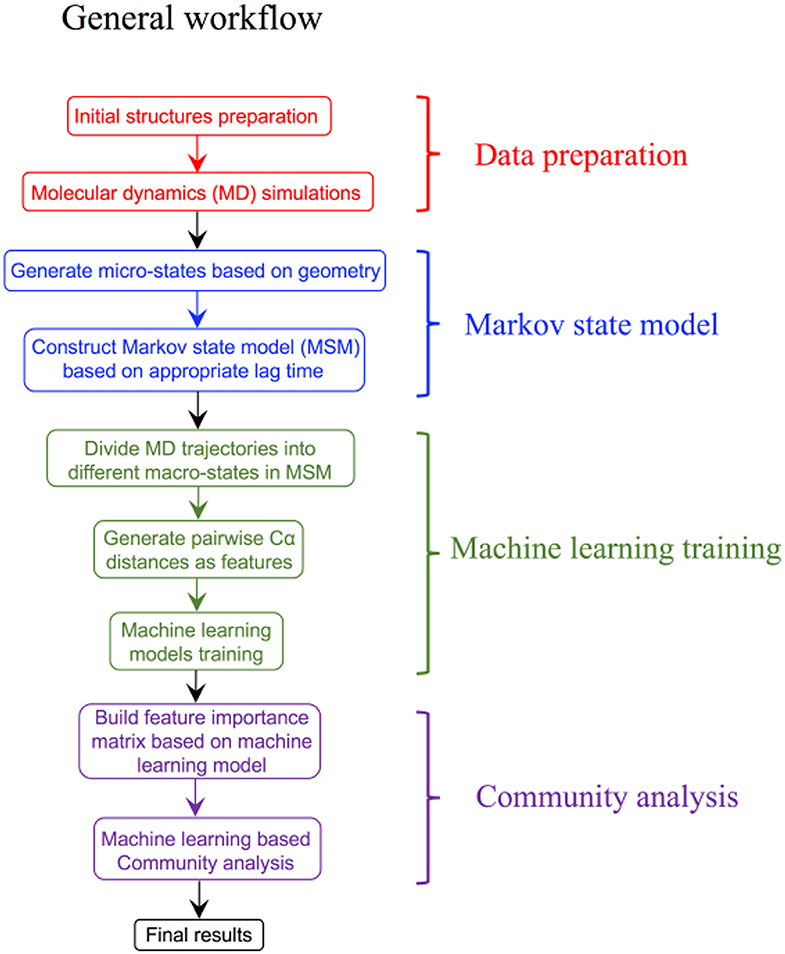
Figure 3. A general prototypical workflow of MSM approaches and ML modeling for detection and classification of functional allosteric states.
The remarkable rise of deep learning (DL) relying on the robust function approximations and representation properties of deep neural networks has provided us with new tools to automatically find compact low-dimensional representations (features) of high-dimensional data (LeCun et al., 2015). DL models have achieved outstanding predictive performance making dramatic breakthroughs in a wide range of applications, including automatic speech processing and image recognition (Toledano et al., 2018; Kim et al., 2019; Hey et al., 2020; Wu et al., 2020). In the words of Geoffrey Hinton who is the founder of DL technologies “Deep Learning is an algorithm which has no theoretical limitations on what it can learn; the more data you give and the more computational time you provide the better it is” (LeCun et al., 2015). Deep neural network methods were successfully applied to predict intrinsic molecular properties such as atomization energy based on simple molecular geometry and element types (Rupp et al., 2012). DL models were recently used for structure-functional prediction of cancer mutations and functional hotspots of ligand binding in cancer-associated genes (Agajanian et al., 2018). The developed models can capture ~90% of experimentally validated mutational hotspots and yield novel information about molecular signatures of driver mutations. In the recent studies, we have proposed novel DL architectures capable of predicting functional protein hotspots directly from raw nucleotide sequence information (Agajanian et al., 2019). These studies have shown that DL models can learn high importance features from raw genomic information and produce reliable recognition and classification of functionally significant cancer mutation hotspots. Moreover, these DL models can often outperform computational predictors of cancer mutations that are based on protein sequence and structure features (Agajanian et al., 2019). The success of DL tools in deciphering important functional phenotypes directly from primary sequence information is encouraging as these models can bypass the need for a large number of empirically-derived functional and structural features. However, ML methods often result in “black box” models with limited interpretability. There has been an explosion of interest in transparent and interpretable ML models to enable more efficient data mining and scientific knowledge discovery (Holzinger et al., 2014). Our investigations have also provided a roadmap how to combine DL predictions of functional sites with subsequent biophysical analysis to aid in the interpretability of ML models and facilitate their applications in biological problems (Agajanian et al., 2018, 2019).
One of the primary goals of artificial intelligence (AI) is to produce fully autonomous agents that interact with their environments to learn optimal behavior, improving over time through trial and error. An important mathematical framework for experience-driven autonomous learning through interactions with the environment is reinforcement learning (RL) (Sutton and Barto, 1981; Barto, 1994; Botvinick, 2012; Hassabis et al., 2017). While previous RL approaches lacked scalability and were limited to fairly low-dimensional problems, a marriage between deep neural networks and RL resulted in the new rapidly evolving field of deep reinforcement learning (DRL) that has achieved remarkable success in game-oriented and various scientific applications, attaining a wide popularity and celebrity-like following among researchers (Mnih et al., 2015; Silver et al., 2017; Botvinick et al., 2019; Jaderberg et al., 2019; Senior et al., 2019). DRL concepts leverage and symbiotically combine neural network modeling with reinforcement learning, in which optimization strategies are crafted based on the trade-offs and competition between rewards and punishments rather than conventional deterministic or stochastic exploration methods. After years of serving as a mere inspiration rather than a practical tool, DRL techniques have taken off overcoming scalability and data limitation issues, and exploding into one of the most intense areas of AI research. Recent years have witnessed the expansion of DRL applications into biomedical research including but not limited to biomedical informatics, drug discovery (Baskin, 2020; Grebner et al., 2020), and toxicology (Chary et al., 2020).
The rationale for employing DRL technologies in studies of allosteric regulation is to capitalize on conceptual and algorithmic similarity between Markov decisions processes (MDPs) which are at the core of RL methods and Markovian modeling of allosteric states in proteins. Several methods adopted RL-based conceptualization to develop MDP algorithms for conformational mapping of the protein landscapes and detection of functional allosteric states. REinforcement learning based Adaptive samPling (REAP) algorithm has shown a considerable promise by adopting RL principles in which an agent (or learning algorithm) takes actions in an environment (conformational protein landscape) to maximize a reward function (Shamsi et al., 2018). In this study, the action is associated with launching a pool of simulations along different collective variables (reaction coordinates), with the reward function proportional to the efficiency of a reaction coordinate to sample space and detect unknown states, and the agent selecting the directions which are most rewarding ultimately leading to the optimal adaptive strategy (Shamsi et al., 2018). Similar concepts were used to develop a goal-oriented sampling method, termed fluctuation amplification of specific traits (FAST) for rapid search of conformational space and identification of distinct functional states by balancing search near promising solutions (exploitation) and attempts to find novel solutions (exploration). Inspired by the RL ideas, this methods runs pools of simulations from starting points chosen based on the reward functions that encourages discovery of new conformations with selected physical properties (Zimmerman and Bowman, 2015; Zimmerman et al., 2018). Generative neural networks have been recently proposed as a tool for the discovery of efficient collective variables that are fundamental for adaptive exploration of the conformational landscapes and finding functional states and hidden allosteric states by guiding sampling toward poorly explored regions (Chiavazzo et al., 2017; Chen et al., 2018b; Hernandez et al., 2018; Mardt et al., 2018). MD simulations were combined with DL approach to train an autoencoder (Hinton and Salakhutdinov, 2006) in order to generate new protein conformations and mine conformational space of the bound state from an ensemble of unbound protein structures (Degiacomi, 2019). Another interesting study employed autoencoder-based detection algorithm to explore dynamic allostery induced by ligand binding based on the comparison of time fluctuations of distance matrices obtained from MD simulations in ligand-bound and unbound forms (Tsuchiya et al., 2019). In this method, the autoencoder neural network is first trained on the time fluctuations of protein motions in the apo form, and the trained autoencoder is then applied to analyze patterns of fluctuations in the holo form. Using this elegant implementation of RL approach, the authors mapped allosteric communication networks of the dynamically coupled residues and ligand pockets in the PDZ2 domain induced by binding (Tsuchiya et al., 2019). Allosteric pocket crosstalk defined as a temporal exchange of atoms between adjacent pockets in the MD trajectories can produce a fingerprint of hidden allosteric communication networks (La Sala et al., 2017). The recent RL-inspired studies of allosteric systems suggested that simulation-driven ML modeling and analysis of conformational landscapes may uncover rarely-populated functional states and shed the light on the key features of allosteric communications between visible and hidden binding pockets in proteins.
DRL is a continuous trial-and-error based sampling-learning process where the agent tries to apply different combination of actions on a state to find the highest cumulative reward. Although DRL methods can tackle a wide range of learning problems with a rigorous mathematical formulation, the challenges posed by the properly crafted interplay between rich data acquisition and delayed rewards remains a significant impediment to the widespread of RL methods in many application domains, including prediction of allosteric protein states and mechanisms. The challenges of DRL approaches often lie in the art of designing robust reward function. The hybrid reward functions with a weighted combination of topological, dynamic, and network-based rewards describing different characteristics of allosteric states may represent a potentially interesting strategy to mitigate the inherent deficiencies of RL and DRL methods. For this, the rewards may be treated as neural networks trained on the set of known allosteric states. A new saga in the rapidly evolving DRL field was the development of episodic-based DRL algorithms that estimate the value of actions and states using episodic memories where the agent stores each encountered state along with the sum of rewards obtained during the n time steps (Botvinick et al., 2019). The episodic memory-based models can be extended to develop curiosity reward bonus functions for efficient exploration of the environment and detecting states in the poorly accessible regions (Han et al., 2020). In this context, DRL framework that iterates episodes of DRL and community decomposition of the dynamic network flows on the conformational landscapes may enhance the interplay between sampling and learning, thus facilitating identification of hidden allosteric states. Different from traditional DRL approaches, this strategy can consider communities of states as intermediate stages in the learning process, rather than unique states, which could potentially lead to a more robust and versatile learning procedure (Figure 4).
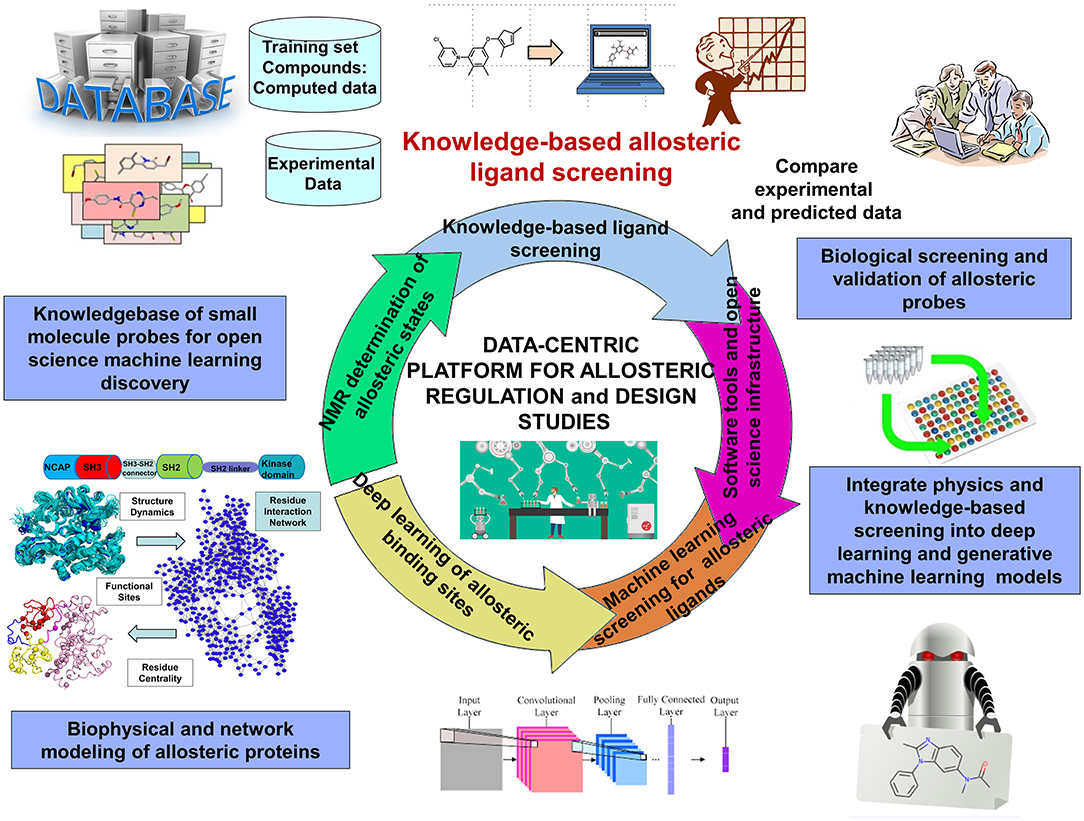
Figure 4. An overview of data-intensive ML platform for allosteric research and allosteric drug discovery.
Deep neural network (DNN) models, most notably variational autoencoder (VAE) (Gomez-Bombarelli et al., 2018) and generative adversarial networks (GANs) (Sorin et al., 2020; Zhong et al., 2020) have proven fruitful in chemical discovery and molecular design of novel synthesizable chemical probes. Automated chemical design approaches employed VAE neural networks for a data-driven continuous representation of molecules (Gomez-Bombarelli et al., 2018). GAN models are often considered as one of the most significant advances in the field of machine learning, and their success has generated a considerable momentum with growing number of applications including molecular design of novel chemical probes and materials (Guimaraes et al., 2017; Kadurin et al., 2017a,b; Olivecrona et al., 2017; Yu et al., 2017; Gupta et al., 2018; Polykovskiy et al., 2018; Putin et al., 2018a,b) (Figure 4). By leveraging sequence data generation (SeqGAN) approach (Yu et al., 2017); Objective-Reinforced Generative Adversarial Networks (ORGAN) (Guimaraes et al., 2017) combines GANs and RL to apply the GAN framework to molecular design with domain-specific rewards and feedback. Of particular importance is MolGAN, an implicit, generative model for small molecular graphs that circumvents the need for expensive graph matching procedures and adapts GAN approach to operate directly on graph-structured data (Cao and Kipf, 2018). CycleGAN provides unpaired image-to-image translation using Cycle-Consistent Adversarial Networks (Zhu et al., 2017). MolCycleGAN, which extended the CycleGAN framework with an added loss and extra encoding network, maps from distribution to distribution on unpaired samples, so it can amplify the size of our dataset in the process by taking all of the pairing combinations rather than relying on a training dataset of predefined molecule-inhibitor pairs (Maziarka et al., 2020). The advantage of MolCycleGAN is the ability to learn transformation rules from the sets of compounds with desired and undesired values of the considered property. The methodological and algorithmic progress in GAN applications to molecular discovery has been further catalyzed by the development of several comprehensive benchmarking sets embedded into a sophisticated cheminformatics infrastructure supporting open-source implementations of molecular features and learning algorithms (Olson et al., 2017; Polykovskiy et al., 2018; Racz et al., 2019). Despite recent developments in GANs models, the applicability of these tools for molecular design continues to present a promise rather than a validated strategy, lacking systematic and comprehensive tools to target specific protein families and interrogate molecular mechanisms. There is also growing interest in generative models which can incorporate both chemical and structural information about small molecules and their interactions with protein targets.
A systematic interdisciplinary effort is needed to leverage the burgeoning knowledge about allosterically regulated proteins in the development of experiment-informed data-oriented computational tools for prediction of allosteric mechanisms and allosteric drug discovery. The main advantage of experiment-informed Markovian modeling is the ability of this technique to adequately describe hierarchy of allosteric states and the molecular basis of allosteric mechanisms. Using a combination of NMR-guided simulations and MSM approach, we can determine structural and dynamic patterns of conformational ensembles and identify functional allosteric states that are hidden in the conformational ensembles. The critical challenges of these methodologies for modeling allosteric regulation phenomenon is selecting a set of experimentally-informed collective variables defined by the intrinsic dynamics to provide the optimal projection of the landscape into functional allosteric states. In this context, the newly emerging information-theoretical flow approaches and modeling of entropy transfer in proteins can represent viable complementary tools for adequate reconstruction of functional conformational landscapes in proteins. The proposed integration of biomolecular simulations and NMR experiments with machine learning into a comprehensive research platform is expected to produce a toolkit of approaches for prediction of allosteric states and mapping of allosteric mechanisms.
Network algorithms, information-theoretical approaches and DL models may be time-consuming and require a systematic exploration and engineering of features and neural net architectures with a constant and evolving feedback from NMR experiments to validate and confirm predictions. Several different ML architectures can be further explored to address potential efficiency and convergence problems including transfer learning, imitation learning, episodic control and dueling networks. To achieve synergies and robust integration of emerging technologies for predicting allosteric regulation mechanisms, a new open science infrastructure development is required which implies extensive sharing of experimental and computational data, software and knowledge across many discipline. Through integrative studies of allosteric mechanisms empowered by biophysical and data science approaches we can expand the toolkit of to dissect and interrogate allosteric mechanisms and functions in the therapeutically important protein families.
The growing body of computational and experimental studies has shown that integration of data-driven biophysical and ML approaches can bring about new drug discovery paradigms, opening up unexplored venues for further scientific innovation and unique biological insights. The integration of computational and NMR approaches into a novel research platform that explores experiment-informed physical simulations, Markov state modeling, information-theoretical formalism of dynamic allosteric networks under the unified umbrella of machine learning will key to dissect molecular rules of allosteric regulation. The innovative cross-disciplinary approaches that expand the knowledge, resources and tools for studies of allosteric regulation can promote a broader usage of new technologies to understand and exploit allosteric phenomenon through the lens of chemical biology, material science, synthetic biology and bioengineering. By developing an open science infrastructure for machine learning studies of allosteric regulation and validating computational approaches using integrative studies of allosteric mechanisms, the scientific community can expand the toolkit of approaches and chemical probes for dissecting and interrogation allosteric mechanisms in many therapeutically important proteins. The development of community-accessible tools that uniquely leverage the existing experimental and simulation knowledgebase to enable interrogation of the allosteric functions can provide much needed impetus to further experimental technologies and enable steady progress.
GV, PT, and GH conceived and designed the research, analyzed the results, and wrote the manuscript. GV, SA, PT, and GH performed the research. GV wrote the final version of the manuscript and supervised the project. All authors contributed to the article and approved the submitted version.
This work was partly supported by institutional funding from Chapman University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors acknowledge the technical assistance of Schmid College Grand Challenge Initiative Postdoctoral Fellow Dr. Anne Sonnenschein.
Adhireksan, Z., Palermo, G., Riedel, T., Ma, Z., Muhammad, R., Rothlisberger, U., et al. (2017). Allosteric cross-talk in chromatin can mediate drug-drug synergy. Nat. Commun. 8:14860. doi: 10.1038/ncomms14860
Adrian, F. J., Ding, Q., Sim, T., Velentza, A., Sloan, C., Liu, Y., et al. (2006). Allosteric inhibitors of Bcr-abl-dependent cell proliferation. Nat. Chem. Biol. 2, 95–102. doi: 10.1038/nchembio760
Agajanian, S., Odeyemi, O., Bischoff, N., Ratra, S., and Verkhivker, G. M. (2018). Machine learning classification and structure-functional analysis of cancer mutations reveal unique dynamic and network signatures of driver sites in oncogenes and tumor suppressor genes. J. Chem. Inf. Model. 58, 2131–2150. doi: 10.1021/acs.jcim.8b00414
Agajanian, S., Oluyemi, O., and Verkhivker, G. M. (2019). Integration of random forest classifiers and deep convolutional neural networks for classification and biomolecular modeling of cancer driver mutations. Front. Mol. Biosci. 6:44. doi: 10.3389/fmolb.2019.00044
Aguilar, D., Oliva, B., and Marino Buslje, C. (2012). Mapping the mutual information network of enzymatic families in the protein structure to unveil functional features. PLoS ONE 7:e41430. doi: 10.1371/journal.pone.0041430
Amor, B., Yaliraki, S. N., Woscholski, R., and Barahona, M. (2014). Uncovering allosteric pathways in caspase-1 using Markov transient analysis and multiscale community detection. Mol. Biosyst. 10, 2247–2258. doi: 10.1039/C4MB00088A
Ancona, N., Marinazzo, D., and Stramaglia, S. (2004). Radial basis function approach to nonlinear granger causality of time series. Phys. Rev. E Stat. Nonlin. Soft. Matter. Phys. 70(5 Pt 2):056221. doi: 10.1103/PhysRevE.70.056221
Ansari, S. M., Coletta, A., Kirkeby Skeby, K., Sorensen, J., Schiott, B., and Palmer, D. S. (2016). Allosteric-activation mechanism of bovine chymosin revealed by bias-exchange metadynamics and molecular dynamics simulations. J. Phys. Chem. B 120, 10453–10462. doi: 10.1021/acs.jpcb.6b07491
Anthis, N. J., and Clore, G. M. (2015). Visualizing transient dark states by NMR spectroscopy. Q Rev. Biophys. 48, 35–116. doi: 10.1017/S0033583514000122
Aoto, P. C., Martin, B. T., and Wright, P. E. (2016). NMR characterization of information flow and allosteric communities in the MAP kinase p38gamma. Sci. Rep. 6:28655. doi: 10.1038/srep28655
Arkhipov, A., Shan, Y., Das, R., Endres, N. F., Eastwood, M. P., Wemmer, D. E., et al. (2013). Architecture and membrane interactions of the EGF receptor. Cell 152, 557–569. doi: 10.1016/j.cell.2012.12.030
Astl, L., Tse, A., and Verkhivker, G. M. (2019). Interrogating regulatory mechanisms in signaling proteins by allosteric inhibitors and activators: a dynamic view through the lens of residue interaction networks. Adv. Exp. Med. Biol. 1163, 187–223. doi: 10.1007/978-981-13-8719-7_9
Astl, L., and Verkhivker, G. M. (2019a). Atomistic modeling of the abl kinase regulation by allosteric modulators using structural perturbation analysis and community-based network reconstruction of allosteric communications. J. Chem. Theory Comput. 15, 3362–3380. doi: 10.1021/acs.jctc.9b00119
Astl, L., and Verkhivker, G. M. (2019b). Data-driven computational analysis of allosteric proteins by exploring protein dynamics, residue coevolution and residue interaction networks. Biochim. Biophys. Acta Gen. Subj. doi: 10.1016/j.bbagen.2019.07.008. [Epub ahead of print].
Atilgan, A. R., Akan, P., and Baysal, C. (2004). Small-world communication of residues and significance for protein dynamics. Biophys. J. 86(Pt. 1), 85–91. doi: 10.1016/S0006-3495(04)74086-2
Ballester, P. J., and Mitchell, J. B. (2010). A machine learning approach to predicting protein-ligand binding affinity with applications to molecular docking. Bioinformatics 26, 1169–1175. doi: 10.1093/bioinformatics/btq112
Barto, A. G. (1994). Reinforcement learning control. Curr. Opin. Neurobiol. 4, 888–893. doi: 10.1016/0959-4388(94)90138-4
Baskin, I. I. (2020). The power of deep learning to ligand-based novel drug discovery. Expert. Opin. Drug Discov. doi: 10.1080/17460441.2020.1745183. [Epub ahead of print].
Beglov, D., Hall, D. R., Wakefield, A. E., Luo, L., Allen, K. N., Kozakov, D., et al. (2018). Exploring the structural origins of cryptic sites on proteins. Proc. Natl. Acad. Sci. U.S.A. 115, E3416–E3425. doi: 10.1073/pnas.1711490115
Berjanskii, M. V., and Wishart, D. S. (2017). Unraveling the meaning of chemical shifts in protein NMR. Biochim. Biophys. Acta Proteins Proteom. 1865(Pt. B), 1564–1576. doi: 10.1016/j.bbapap.2017.07.005
Bhattacharya, S., and Vaidehi, N. (2014). Differences in allosteric communication pipelines in the inactive and active states of a GPCR. Biophys. J. 107, 422–434. doi: 10.1016/j.bpj.2014.06.015
Bhattacharyya, M., and Vishveshwara, S. (2011). Probing the allosteric mechanism in pyrrolysyl-tRNA synthetase using energy-weighted network formalism. Biochemistry 50, 6225–6236. doi: 10.1021/bi200306u
Bode, C., Kovacs, I. A., Szalay, M. S., Palotai, R., Korcsmaros, T., and Csermely, P. (2007). Network analysis of protein dynamics. FEBS Lett. 581, 2776–2782. doi: 10.1016/j.febslet.2007.05.021
Boehr, D. D., Dyson, H. J., and Wright, P. E. (2006). An NMR perspective on enzyme dynamics. Chem. Rev. 106, 3055–3079. doi: 10.1021/cr050312q
Bonomi, M., and Camilloni, C. (2017). Integrative structural and dynamical biology with PLUMED-ISDB. Bioinformatics 33, 3999–4000. doi: 10.1093/bioinformatics/btx529
Botvinick, M., Ritter, S., Wang, J. X., Kurth-Nelson, Z., Blundell, C., and Hassabis, D. (2019). Reinforcement learning, fast and slow. Trends Cogn. Sci. 23, 408–422. doi: 10.1016/j.tics.2019.02.006
Botvinick, M. M. (2012). Hierarchical reinforcement learning and decision making. Curr. Opin. Neurobiol. 22, 956–962. doi: 10.1016/j.conb.2012.05.008
Boulton, S., Akimoto, M., Selvaratnam, R., Bashiri, A., and Melacini, G. (2014). A tool set to map allosteric networks through the NMR chemical shift covariance analysis. Sci. Rep. 4:7306. doi: 10.1038/srep07306
Boulton, S., and Melacini, G. (2016). Advances in NMR methods to map allosteric sites: from models to translation. Chem. Rev. 116, 6267–6304. doi: 10.1021/acs.chemrev.5b00718
Boulton, S., Selvaratnam, R., Ahmed, R., and Melacini, G. (2018). Implementation of the NMR CHEmical shift covariance analysis (CHESCA): a chemical biologist's approach to allostery. Methods Mol. Biol. 1688, 391–405. doi: 10.1007/978-1-4939-7386-6_18
Bowman, G. R. (2014). A tutorial on building markov state models with MSMBuilder and coarse-graining them with BACE. Methods Mol. Biol. 1084, 141–158. doi: 10.1007/978-1-62703-658-0_8
Bowman, G. R., Bolin, E. R., Hart, K. M., Maguire, B. C., and Marqusee, S. (2015). Discovery of multiple hidden allosteric sites by combining markov state models and experiments. Proc. Natl. Acad. Sci. U.S.A. 112, 2734–2739. doi: 10.1073/pnas.1417811112
Bowman, G. R., and Geissler, P. L. (2012). Equilibrium fluctuations of a single folded protein reveal a multitude of potential cryptic allosteric sites. Proc. Natl. Acad. Sci. U.S.A. 109, 11681–11686. doi: 10.1073/pnas.1209309109
Bowman, G. R., Huang, X., and Pande, V. S. (2009). Using generalized ensemble simulations and markov state models to identify conformational states. Methods 49, 197–201. doi: 10.1016/j.ymeth.2009.04.013
Bowman, G. R., and Noe, F. (2014). Software for building markov state models. Adv. Exp. Med. Biol. 797:139. doi: 10.1007/978-94-007-7606-7_11
Brinda, K. V., and Vishveshwara, S. (2005). A network representation of protein structures: implications for protein stability. Biophys. J. 89, 4159–4170. doi: 10.1529/biophysj.105.064485
Brinda, K. V., and Vishveshwara, S. (2010). Random network behaviour of protein structures. Mol. Biosyst. 6, 391–398. doi: 10.1039/B903019K
Brotzakis, Z. F., and Parrinello, M. (2019). Enhanced sampling of protein conformational transitions via dynamically optimized collective variables. J. Chem. Theory Comput. 15, 1393–1398. doi: 10.1021/acs.jctc.8b00827
Buchenberg, S., Sittel, F., and Stock, G. (2017). Time-resolved observation of protein allosteric communication. Proc. Natl. Acad. Sci. U.S.A. 114, E6804–E6811. doi: 10.1073/pnas.1707694114
Burbidge, R., Trotter, M., Buxton, B., and Holden, S. (2001). Drug design by machine learning: support vector machines for pharmaceutical data analysis. Comput. Chem. 26, 5–14. doi: 10.1016/S0097-8485(01)00094-8
Cao, N. D., and Kipf, T. (2018). MolGAN: An implicit generative model for small molecular graphs. arXiv [Preprint]. arXiv: 1805.11973.
Cavalli, A., Montalvao, R. W., and Vendruscolo, M. (2011). Using chemical shifts to determine structural changes in proteins upon complex formation. J. Phys. Chem. B 115, 9491–9494. doi: 10.1021/jp202647q
Cembran, A., Kim, J., Gao, J., and Veglia, G. (2014). NMR mapping of protein conformational landscapes using coordinated behavior of chemical shifts upon ligand binding. Phys. Chem. Chem. Phys. 16, 6508–6518. doi: 10.1039/C4CP00110A
Chakrabarti, S., and Panchenko, A. R. (2009). Coevolution in defining the functional specificity. Proteins 75, 231–240. doi: 10.1002/prot.22239
Chakrabarti, S., and Panchenko, A. R. (2010). Structural and functional roles of coevolved sites in proteins. PLoS ONE 5:e8591. doi: 10.1371/journal.pone.0008591
Changeux, J. P. (2012). Allostery and the monod-wyman-changeux model after 50 years. Annu. Rev. Biophys. 41, 103–133. doi: 10.1146/annurev-biophys-050511-102222
Changeux, J. P., and Edelstein, S. J. (2005). Allosteric mechanisms of signal transduction. Science 308, 1424–1428. doi: 10.1126/science.1108595
Chary, M. A., Manini, A. F., Boyer, E. W., and Burns, M. (2020). The role and promise of artificial intelligence in medical toxicology. J. Med. Toxicol. doi: 10.1007/s13181-020-00769-5. [Epub ahead of print].
Chen, H., Engkvist, O., Wang, Y., Olivecrona, M., and Blaschke, T. (2018a). The rise of deep learning in drug discovery. Drug Discov. Today 23, 1241–1250. doi: 10.1016/j.drudis.2018.01.039
Chen, W., Tan, A. R., and Ferguson, A. L. (2018b). Collective variable discovery and enhanced sampling using autoencoders: innovations in network architecture and error function design. J. Chem. Phys. 149:072312. doi: 10.1063/1.5023804
Chennubhotla, C., and Bahar, I. (2006). Markov propagation of allosteric effects in biomolecular systems: application to GroEL-GroES. Mol. Syst. Biol. 2:36. doi: 10.1038/msb4100075
Chennubhotla, C., and Bahar, I. (2007). Signal propagation in proteins and relation to equilibrium fluctuations. PLoS Comput. Biol. 3, 1716–1726. doi: 10.1371/journal.pcbi.0030172
Chennubhotla, C., Yang, Z., and Bahar, I. (2008). Coupling between global dynamics and signal transduction pathways: a mechanism of allostery for chaperonin GroEL. Mol. Biosyst. 4, 287–292. doi: 10.1039/b717819k
Chiappori, F., Merelli, I., Colombo, G., Milanesi, L., and Morra, G. (2012). Molecular mechanism of allosteric communication in Hsp70 revealed by molecular dynamics simulations. PLoS Comput. Biol. 8:e1002844. doi: 10.1371/journal.pcbi.1002844
Chiappori, F., Merelli, I., Milanesi, L., Colombo, G., and Morra, G. (2016). An atomistic view of Hsp70 allosteric crosstalk: from the nucleotide to the substrate binding domain and back. Sci. Rep. 6:23474. doi: 10.1038/srep23474
Chiavazzo, E., Covino, R., Coifman, R. R., Gear, C. W., Georgiou, A. S., Hummer, G., et al. (2017). Intrinsic map dynamics exploration for uncharted effective free-energy landscapes. Proc. Natl. Acad. Sci. U.S.A. 114, E5494–E5503. doi: 10.1073/pnas.1621481114
Cimermancic, P., Weinkam, P., Rettenmaier, T. J., Bichmann, L., Keedy, D. A., Woldeyes, R. A., et al. (2016). CryptoSite: expanding the druggable proteome by characterization and prediction of cryptic binding sites. J. Mol. Biol. 428, 709–719. doi: 10.1016/j.jmb.2016.01.029
Colombo, G., Morra, G., Meli, M., and Verkhivker, G. (2008). Understanding ligand-based modulation of the Hsp90 molecular chaperone dynamics at atomic resolution. Proc. Natl. Acad. Sci. U.S.A. 105, 7976–7981. doi: 10.1073/pnas.0802879105
Colombo, G., Paladino, A., Woodford, M. R., Backe, S. J., Sager, R. A., Kancherla, P., et al. (2020). Chemical perturbation of oncogenic protein folding: from the prediction of locally unstable structures to the design of disruptors of Hsp90-client interactions. Chemistry. doi: 10.1002/chem.202000615. [Epub ahead of print].
Cooper, A., and Dryden, D. T. (1984). Allostery without conformational change. A plausible model. Eur. Biophys. J. 11, 103–109. doi: 10.1007/BF00276625
Cortina, G. A., and Kasson, P. M. (2018). Predicting allostery and microbial drug resistance with molecular simulations. Curr. Opin. Struct. Biol. 52, 80–86. doi: 10.1016/j.sbi.2018.09.001
Cowan-Jacob, S. W., Jahnke, W., and Knapp, S. (2014). Novel approaches for targeting kinases: allosteric inhibition, allosteric activation and pseudokinases. Fut. Med. Chem. 6, 541–561. doi: 10.4155/fmc.13.216
Cronkite-Ratcliff, B., and Pande, V. (2013). MSMExplorer: visualizing markov state models for biomolecule folding simulations. Bioinformatics 29, 950–952. doi: 10.1093/bioinformatics/btt051
Csermely, P., Korcsmaros, T., Kiss, H. J., London, G., and Nussinov, R. (2013). Structure and dynamics of molecular networks: a novel paradigm of drug discovery: a comprehensive review. Pharmacol. Ther. 138, 333–408. doi: 10.1016/j.pharmthera.2013.01.016
Csermely, P., Palotai, R., and Nussinov, R. (2010). Induced fit, conformational selection and independent dynamic segments: an extended view of binding events. Trends Biochem. Sci. 35, 539–546. doi: 10.1016/j.tibs.2010.04.009
Csermely, P., Sandhu, K. S., Hazai, E., Hoksza, Z., Kiss, H. J., Miozzo, F., et al. (2012). Disordered proteins and network disorder in network descriptions of protein structure, dynamics and function: hypotheses and a comprehensive review. Curr. Protein. Pept. Sci. 13, 19–33. doi: 10.2174/138920312799277992
Czemeres, J., Buse, K., and Verkhivker, G. M. (2017). Atomistic simulations and network-based modeling of the Hsp90-Cdc37 chaperone binding with Cdk4 client protein: a mechanism of chaperoning kinase clients by exploiting weak spots of intrinsically dynamic kinase domains. PLoS ONE 12:34. doi: 10.1371/journal.pone.0190267
Dam, T. K., Roy, R., Page, D., and Brewer, C. F. (2002). Negative cooperativity associated with binding of multivalent carbohydrates to lectins. Thermodynamic analysis of the “multivalency effect”. Biochemistry 41, 1351–1358. doi: 10.1021/bi015830j
D'Annessa, I., Sattin, S., Tao, J., Pennati, M., Sanchez-Martin, C., Moroni, E., et al. (2017). Design of allosteric stimulators of the Hsp90 ATPase as new anticancer leads. Chemistry 23, 5188–5192. doi: 10.1002/chem.201700169
Dar, A. C., and Shokat, K. M. (2011). The evolution of protein kinase inhibitors from antagonists to agonists of cellular signaling. Annu. Rev. Biochem. 80, 769–795. doi: 10.1146/annurev-biochem-090308-173656
Darmon, D., and Rapp, P. E. (2017). Specific transfer entropy and other state-dependent transfer entropies for continuous-state input-output systems. Phys. Rev. E 96:022121. doi: 10.1103/PhysRevE.96.022121
Decherchi, S., Berteotti, A., Bottegoni, G., Rocchia, W., and Cavalli, A. (2015). The ligand binding mechanism to purine nucleoside phosphorylase elucidated via molecular dynamics and machine learning. Nat. Commun. 6:6155. doi: 10.1038/ncomms7155
Degiacomi, M. T. (2019). Coupling molecular dynamics and deep learning to mine protein conformational space. Structure 27, 1034–1040.e1033. doi: 10.1016/j.str.2019.03.018
del Sol, A., Fujihashi, H., Amoros, D., and Nussinov, R. (2006). Residues crucial for maintaining short paths in network communication mediate signaling in proteins. Mol. Syst. Biol. 2:2006.0019. doi: 10.1038/msb4100063
del Sol, A., and O'Meara, P. (2005). Small-world network approach to identify key residues in protein-protein interaction. Proteins 58, 672–682. doi: 10.1002/prot.20348
del Sol, A., Tsai, C. J., Ma, B., and Nussinov, R. (2009). The origin of allosteric functional modulation: multiple pre-existing pathways. Structure 17, 1042–1050. doi: 10.1016/j.str.2009.06.008
Delvenne, J. C., Lambiotte, R., and Rocha, L. E. (2015). Diffusion on networked systems is a question of time or structure. Nat. Commun. 6:7366. doi: 10.1038/ncomms8366
Di Paola, L., and Giuliani, A. (2015). Protein contact network topology: a natural language for allostery. Curr. Opin. Struct. Biol. 31, 43–48. doi: 10.1016/j.sbi.2015.03.001
Dimitrov, T., Kreisbeck, C., Becker, J. S., Aspuru-Guzik, A., and Saikin, S. K. (2019). Autonomous molecular design: then and now. ACS Appl. Mater. Interfaces 11, 24825–24836. doi: 10.1021/acsami.9b01226
Dixit, A., and Verkhivker, G. M. (2009). Hierarchical modeling of activation mechanisms in the ABL and EGFR kinase domains: thermodynamic and mechanistic catalysts of kinase activation by cancer mutations. PLoS Comput. Biol. 5:e1000487. doi: 10.1371/journal.pcbi.1000487
Dixit, A., and Verkhivker, G. M. (2011a). Computational modeling of allosteric communication reveals organizing principles of mutation-induced signaling in ABL and EGFR kinases. PLoS Comput. Biol. 7:e1002179. doi: 10.1371/journal.pcbi.1002179
Dixit, A., and Verkhivker, G. M. (2011b). The energy landscape analysis of cancer mutations in protein kinases. PLoS ONE 6:e26071. doi: 10.1371/journal.pone.0026071
Dixit, A., and Verkhivker, G. M. (2012). Probing molecular mechanisms of the Hsp90 chaperone: biophysical modeling identifies key regulators of functional dynamics. PLoS ONE 7:e37605. doi: 10.1371/journal.pone.0037605
Dixit, A., and Verkhivker, G. M. (2014). Structure-functional prediction and analysis of cancer mutation effects in protein kinases. Comput. Math. Methods Med. 2014:653487. doi: 10.1155/2014/653487
Dokholyan, N. V. (2016). Controlling allosteric networks in proteins. Chem. Rev. 116, 6463–6487. doi: 10.1021/acs.chemrev.5b00544
Donnelly, A., and Blagg, B. S. (2008). Novobiocin and additional inhibitors of the Hsp90 C-terminal nucleotide-binding pocket. Curr. Med. Chem. 15, 2702–2717. doi: 10.2174/092986708786242895
Edler, D., Guedes, T., Zizka, A., Rosvall, M., and Antonelli, A. (2017). Infomap bioregions: interactive mapping of biogeographical regions from species distributions. Syst. Biol. 66, 197–204. doi: 10.1093/sysbio/syw087
Fajer, M., Meng, Y., and Roux, B. (2017). The activation of c-Src tyrosine kinase: conformational transition pathway and free energy landscape. J. Phys. Chem. B 121, 3352–3363. doi: 10.1021/acs.jpcb.6b08409
Fang, Z., Grutter, C., and Rauh, D. (2013). Strategies for the selective regulation of kinases with allosteric modulators: exploiting exclusive structural features. ACS Chem. Biol. 8, 58–70. doi: 10.1021/cb300663j
Farabella, I., Pham, T., Henderson, N. S., Geibel, S., Phan, G., Thanassi, D. G., et al. (2014). Allosteric signalling in the outer membrane translocation domain of PapC usher. Elife 3:e0532. doi: 10.7554/eLife.03532.020
Ferraro, M., D'Annessa, I., Moroni, E., Morra, G., Paladino, A., Rinaldi, S., et al. (2019). Allosteric modulators of HSP90 and HSP70: dynamics meets function through structure-based drug design. J. Med. Chem. 62, 60–87. doi: 10.1021/acs.jmedchem.8b00825
Frederick, K. K., Marlow, M. S., Valentine, K. G., and Wand, A. J. (2007). Conformational entropy in molecular recognition by proteins. Nature 448, 325–329. doi: 10.1038/nature05959
Garg, G., Forsberg, L. K., Zhao, H., and Blagg, B. S. J. (2017a). Development of phenyl cyclohexylcarboxamides as a novel class of Hsp90 C-terminal inhibitors. Chemistry 23, 16574–16585. doi: 10.1002/chem.201703206
Garg, G., Khandelwal, A., and Blagg, B. S. (2016). Anticancer inhibitors of Hsp90 function: beyond the usual suspects. Adv. Cancer Res. 129, 51–88. doi: 10.1016/bs.acr.2015.12.001
Garg, G., Zhao, H., and Blagg, B. S. (2017b). Design, synthesis and biological evaluation of alkylamino biphenylamides as Hsp90 C-terminal inhibitors. Bioorg. Med. Chem. 25, 451–457. doi: 10.1016/j.bmc.2016.11.030
Gasper, P. M., Fuglestad, B., Komives, E. A., Markwick, P. R., and McCammon, J. A. (2012). Allosteric networks in thrombin distinguish procoagulant vs. anticoagulant activities. Proc. Natl. Acad. Sci. U.S.A. 109, 21216–21222. doi: 10.1073/pnas.1218414109
General, I. J., Liu, Y., Blackburn, M. E., Mao, W., Gierasch, L. M., and Bahar, I. (2014). ATPase subdomain IA is a mediator of interdomain allostery in Hsp70 molecular chaperones. PLoS Comput. Biol. 10:e1003624. doi: 10.1371/journal.pcbi.1003624
Gestwicki, J. E., and Shao, H. (2019). Inhibitors and chemical probes for molecular chaperone networks. J. Biol. Chem. 294, 2151–2161. doi: 10.1074/jbc.TM118.002813
Ghosh, A., Sakaguchi, R., Liu, C., Vishveshwara, S., and Hou, Y. M. (2011). Allosteric communication in cysteinyl tRNA synthetase: a network of direct and indirect readout. J. Biol. Chem. 286, 37721–37731. doi: 10.1074/jbc.M111.246702
Ghosh, A., and Vishveshwara, S. (2007). A study of communication pathways in methionyl- tRNA synthetase by molecular dynamics simulations and structure network analysis. Proc. Natl. Acad. Sci. U.S.A. 104, 15711–15716. doi: 10.1073/pnas.0704459104
Ghosh, A., and Vishveshwara, S. (2008). Variations in clique and community patterns in protein structures during allosteric communication: investigation of dynamically equilibrated structures of methionyl tRNA synthetase complexes. Biochemistry 47, 11398–11407. doi: 10.1021/bi8007559
Goh, G. B., Hodas, N. O., and Vishnu, A. (2017). Deep learning for computational chemistry. J. Comput. Chem. 38, 1291–1307. doi: 10.1002/jcc.24764
Gomez-Bombarelli, R., Wei, J. N., Duvenaud, D., Hernandez-Lobato, J. M., Sanchez-Lengeling, B., Sheberla, D., et al. (2018). Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent. Sci. 4, 268–276. doi: 10.1021/acscentsci.7b00572
Goncearenco, A., Mitternacht, S., Yong, T., Eisenhaber, B., Eisenhaber, F., and Berezovsky, I. N. (2013). SPACER: server for predicting allosteric communication and effects of regulation. Nucleic Acids Res. 41, W266–W272. doi: 10.1093/nar/gkt460
Granata, D., Camilloni, C., Vendruscolo, M., and Laio, A. (2013). Characterization of the free-energy landscapes of proteins by NMR-guided metadynamics. Proc. Natl. Acad. Sci. U.S.A. 110, 6817–6822. doi: 10.1073/pnas.1218350110
Grebner, C., Matter, H., Plowright, A. T., and Hessler, G. (2020). Automated de novo design in medicinal chemistry: which types of chemistry does a generative neural network learn? J. Med. Chem. doi: 10.1021/acs.jmedchem.9b02044. [Epub ahead of print].
Grutsch, S., Bruschweiler, S., and Tollinger, M. (2016). NMR methods to study dynamic allostery. PLoS Comput. Biol. 12:e1004620. doi: 10.1371/journal.pcbi.1004620
Guimaraes, G. L., Sanchez-Lengeling, B., Outeiral, C., Farias, P.L. C., and Aspuru-Guzik, A. (2017). Objective-reinforced generative adversarial networks (organ) for sequence generation models. arXiv [Preprint]. arXiv:1705.10843.
Gunasekaran, K., Ma, B., and Nussinov, R. (2004). Is allostery an intrinsic property of all dynamic proteins? Proteins 57, 433–443. doi: 10.1002/prot.20232
Guo, J., and Zhou, H. X. (2016). Protein allostery and conformational dynamics. Chem. Rev. 116, 6503–6515. doi: 10.1021/acs.chemrev.5b00590
Gupta, A., Muller, A. T., Huisman, B. J. H., Fuchs, J. A., Schneider, P., and Schneider, G. (2018). Generative recurrent networks for de novo drug design. Mol. Inform. 37:1700111. doi: 10.1002/minf.201700111
Hacisuleyman, A., and Erman, B. (2017a). Causality, transfer entropy, and allosteric communication landscapes in proteins with harmonic interactions. Proteins 85, 1056–1064. doi: 10.1002/prot.25272
Hacisuleyman, A., and Erman, B. (2017b). Entropy transfer between residue pairs and allostery in proteins: quantifying allosteric communication in ubiquitin. PLoS Comput. Biol. 13:e1005319. doi: 10.1371/journal.pcbi.1005319
Halabi, N., Rivoire, O., Leibler, S., and Ranganathan, R. (2009). Protein sectors: evolutionary units of three-dimensional structure. Cell 138, 774–786. doi: 10.1016/j.cell.2009.07.038
Hall, J. A., Seedarala, S., Zhao, H., Garg, G., Ghosh, S., and Blagg, B. S. (2016). Novobiocin analogues that inhibit the MAPK pathway. J. Med. Chem. 59, 925–933. doi: 10.1021/acs.jmedchem.5b01354
Han, B., Liu, Y., Ginzinger, S. W., and Wishart, D. S. (2011). SHIFTX2: significantly improved protein chemical shift prediction. J. Biomol. NMR 50, 43–57. doi: 10.1007/s10858-011-9478-4
Han, R., Chen, K., and Tan, C. (2020). Curiosity-driven recommendation strategy for adaptive learning via deep reinforcement learning. Br. J. Math. Stat. Psychol. doi: 10.1111/bmsp.12199. [Epub ahead of print].
Hansia, P., Ghosh, A., and Vishveshwara, S. (2009). Ligand dependent intra and inter subunit communication in human tryptophanyl tRNA synthetase as deduced from the dynamics of structure networks. Mol. Biosyst. 5, 1860–1872. doi: 10.1039/b903807h
Harrigan, M. P., Sultan, M. M., Hernandez, C. X., Husic, B. E., Eastman, P., Schwantes, C. R., et al. (2017). MSMBuilder: statistical models for biomolecular dynamics. Biophys. J. 112, 10–15. doi: 10.1016/j.bpj.2016.10.042
Hart, K. M., Ho, C. M., Dutta, S., Gross, M. L., and Bowman, G. R. (2016). Modelling proteins' hidden conformations to predict antibiotic resistance. Nat. Commun. 7:12965. doi: 10.1038/ncomms12965
Hassabis, D., Kumaran, D., Summerfield, C., and Botvinick, M. (2017). Neuroscience-inspired artificial intelligence. Neuron 95, 245–258. doi: 10.1016/j.neuron.2017.06.011
Hayatshahi, H. S., Ahuactzin, E., Tao, P., Wang, S., and Liu, J. (2019). Probing protein allostery as a residue-specific concept via residue response maps. J. Chem. Inf. Model. 59, 4691–4705. doi: 10.1021/acs.jcim.9b00447
Hernandez, C. X., Wayment-Steele, H. K., Sultan, M. M., Husic, B. E., and Pande, V. S. (2018). Variational encoding of complex dynamics. Phys. Rev. E 97:062412. doi: 10.1103/PhysRevE.97.062412
Hertig, S., Latorraca, N. R., and Dror, R. O. (2016). Revealing atomic-level mechanisms of protein allostery with molecular dynamics simulations. PLoS Comput. Biol. 12:e1004746. doi: 10.1371/journal.pcbi.1004746
Hey, T., Butler, K., Jackson, S., and Thiyagalingam, J. (2020). Machine learning and big scientific data. Philos. Trans. A Math. Phys. Eng. Sci. 378:20190054. doi: 10.1098/rsta.2019.0054
Hilser, V. J., Wrabl, J. O., and Motlagh, H. N. (2012). Structural and energetic basis of allostery. Annu. Rev. Biophys. 41, 585–609. doi: 10.1146/annurev-biophys-050511-102319
Hinton, G. E., and Salakhutdinov, R. R. (2006). Reducing the dimensionality of data with neural networks. Science 313, 504–507. doi: 10.1126/science.1127647
Hlavácková-Schindler, K., Paluš, M., Vejmelka, M., and Bhattacharya, J. (2007). Causality detection based on information-theoretic approaches in time series analysis. Phys. Rep. 441, 1–46. doi: 10.1016/j.physrep.2006.12.004
Holzinger, A., Dehmer, M., and Jurisica, I. (2014). Knowledge discovery and interactive data mining in bioinformatics–state-of-the-art, future challenges and research directions. BMC Bioinformatics 15(Suppl. 6):I1. doi: 10.1186/1471-2105-15-S6-I1
Hu, J., Stites, E. C., Yu, H., Germino, E. A., Meharena, H. S., Stork, P. J., et al. (2013). Allosteric activation of functionally asymmetric RAF kinase dimers. Cell 154, 1036–1046. doi: 10.1016/j.cell.2013.07.046
Hu, S., Ferraro, M., Thomas, A. P., Chung, J. M., Yoon, N. G., Seol, J. H., et al. (2020). Dual binding to orthosteric and allosteric sites enhances the anticancer activity of a TRAP1-targeting drug. J. Med. Chem. 63, 2930–2940. doi: 10.1021/acs.jmedchem.9b01420
Huang, C., and Kalodimos, C. G. (2017). Structures of large protein complexes determined by nuclear magnetic resonance spectroscopy. Annu. Rev. Biophys. 46, 317–336. doi: 10.1146/annurev-biophys-070816-033701
Husic, B. E., and Pande, V. S. (2018). Markov state models: from an art to a science. J. Am. Chem. Soc. 140, 2386–2396. doi: 10.1021/jacs.7b12191
Ito, S. (2016). Backward transfer entropy: informational measure for detecting hidden markov models and its interpretations in thermodynamics, gambling and causality. Sci. Rep. 6:36831. doi: 10.1038/srep36831
Jaderberg, M., Czarnecki, W. M., Dunning, I., Marris, L., Lever, G., Castaneda, A. G., et al. (2019). Human-level performance in 3D multiplayer games with population-based reinforcement learning. Science 364, 859–865. doi: 10.1126/science.aau6249
James, K. A., and Verkhivker, G. M. (2014). Structure-based network analysis of activation mechanisms in the ErbB family of receptor tyrosine kinases: the regulatory spine residues are global mediators of structural stability and allosteric interactions. PLoS ONE 9:e113488. doi: 10.1371/journal.pone.0113488
Jarymowycz, V. A., and Stone, M. J. (2006). Fast time scale dynamics of protein backbones: NMR relaxation methods, applications, and functional consequences. Chem. Rev. 106, 1624–1671. doi: 10.1021/cr040421p
Jiang, Y., and Kalodimos, C. G. (2017). NMR studies of large proteins. J. Mol. Biol. 429, 2667–2676. doi: 10.1016/j.jmb.2017.07.007
Jura, N., Zhang, X., Endres, N. F., Seeliger, M. A., Schindler, T., and Kuriyan, J. (2011). Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Mol. Cell. 42, 9–22. doi: 10.1016/j.molcel.2011.03.004
Kadurin, A., Aliper, A., Kazennov, A., Mamoshina, P., Vanhaelen, Q., Khrabrov, K., et al. (2017a). The cornucopia of meaningful leads: applying deep adversarial autoencoders for new molecule development in oncology. Oncotarget 8, 10883–10890. doi: 10.18632/oncotarget.14073
Kadurin, A., Nikolenko, S., Khrabrov, K., Aliper, A., and Zhavoronkov, A. (2017b). druGAN: an advanced generative adversarial autoencoder model for de novo generation of new molecules with desired molecular properties in silico. Mol. Pharm. 14, 3098–3104. doi: 10.1021/acs.molpharmaceut.7b00346
Kalbitzer, H. R., Rosnizeck, I. C., Munte, C. E., Narayanan, S. P., Kropf, V., and Spoerner, M. (2013). Intrinsic allosteric inhibition of signaling proteins by targeting rare interaction states detected by high-pressure NMR spectroscopy. Angew. Chem. Int. Ed. Engl. 52, 14242–14246. doi: 10.1002/anie.201305741
Kalescky, R., Liu, J., and Tao, P. (2015). Identifying key residues for protein allostery through rigid residue scan. J. Phys. Chem. A 119, 1689–1700. doi: 10.1021/jp5083455
Kalescky, R., Zhou, H., Liu, J., and Tao, P. (2016). Rigid residue scan simulations systematically reveal residue entropic roles in protein allostery. PLoS Comput. Biol. 12:e1004893. doi: 10.1371/journal.pcbi.1004893
Kalodimos, C. G. (2011). NMR reveals novel mechanisms of protein activity regulation. Protein Sci. 20, 773–782. doi: 10.1002/pro.614
Kalodimos, C. G. (2012). Protein function and allostery: a dynamic relationship. Ann. N Y Acad. Sci. 1260, 81–86. doi: 10.1111/j.1749-6632.2011.06319.x
Kawamoto, T., and Rosvall, M. (2015). Estimating the resolution limit of the map equation in community detection. Phys. Rev. E Stat. Nonlin. Soft. Matter. Phys. 91:012809. doi: 10.1103/PhysRevE.91.012809
Kay, L. E. (2011). NMR studies of protein structure and dynamics - a look backwards and forwards. J. Magn. Reson. 213, 492–494. doi: 10.1016/j.jmr.2011.08.010
Kay, L. E. (2016). New views of functionally dynamic proteins by solution NMR spectroscopy. J. Mol. Biol. 428(Pt A), 323–331. doi: 10.1016/j.jmb.2015.11.028
Kaya, C., Armutlulu, A., Ekesan, S., and Haliloglu, T. (2013). MCPath: monte carlo path generation approach to predict likely allosteric pathways and functional residues. Nucleic Acids Res. 41, W249–W255. doi: 10.1093/nar/gkt284
Kern, D., and Zuiderweg, E. R. (2003). The role of dynamics in allosteric regulation. Curr. Opin. Struct. Biol. 13, 748–757. doi: 10.1016/j.sbi.2003.10.008
Khandelwal, A., Crowley, V. M., and Blagg, B. S. J. (2016). Natural product inspired N-terminal Hsp90 inhibitors: from bench to bedside? Med. Res. Rev. 36, 92–118. doi: 10.1002/med.21351
Kim, J., Ahuja, L. G., Chao, F. A., Xia, Y., McClendon, C. L., Kornev, A. P., et al. (2017). A dynamic hydrophobic core orchestrates allostery in protein kinases. Sci. Adv. 3:e1600663. doi: 10.1126/sciadv.1600663
Kim, M., Yun, J., Cho, Y., Shin, K., Jang, R., Bae, H. J., et al. (2019). Deep learning in medical imaging. Neurospine 16, 657–668. doi: 10.14245/ns.1938396.198
Kimanius, D., Pettersson, I., Schluckebier, G., Lindahl, E., and Andersson, M. (2015). SAXS-guided metadynamics. J. Chem. Theory Comput. 11, 3491–3498. doi: 10.1021/acs.jctc.5b00299
Kohlhoff, K. J., Robustelli, P., Cavalli, A., Salvatella, X., and Vendruscolo, M. (2009). Fast and accurate predictions of protein NMR chemical shifts from interatomic distances. J. Am. Chem. Soc. 131, 13894–13895. doi: 10.1021/ja903772t
Kornev, A. P., and Taylor, S. S. (2015). Dynamics-driven allostery in protein kinases. Trends Biochem. Sci. 40, 628–647. doi: 10.1016/j.tibs.2015.09.002
Kornev, A. P., Taylor, S. S., and Ten Eyck, L. F. (2008). A helix scaffold for the assembly of active protein kinases. Proc. Natl. Acad. Sci. U.S.A. 105, 14377–14382. doi: 10.1073/pnas.0807988105
Korotcov, A., Tkachenko, V., Russo, D. P., and Ekins, S. (2017). Comparison of deep learning with multiple machine learning methods and metrics using diverse drug discovery data sets. Mol. Pharm. 14, 4462–4475. doi: 10.1021/acs.molpharmaceut.7b00578
Korzhnev, D. M., and Kay, L. E. (2008). Probing invisible, low-populated states of protein molecules by relaxation dispersion NMR spectroscopy: an application to protein folding. Acc. Chem. Res. 41, 442–451. doi: 10.1021/ar700189y
Koshland, D. E. Jr. (1998). Conformational changes: how small is big enough? Nat. Med. 4, 1112–1114. doi: 10.1038/2605
Krivdin, L. B. (2017). Calculation of (15)N NMR chemical shifts: recent advances and perspectives. Prog. Nucl. Magn. Reson. Spectrosc. 102–103, 98–119. doi: 10.1016/j.pnmrs.2017.08.001
Kumar MV, V., Ebna Noor, R., Davis, R. E., Zhang, Z., Sipavicius, E., Keramisanou, D., et al. (2018). Molecular insights into the interaction of Hsp90 with allosteric inhibitors targeting the C-terminal domain. MedChemComm 9, 1323–1331. doi: 10.1039/C8MD00151K
Kuzmanic, A., Bowman, G. R., Juarez-Jimenez, J., Michel, J., and Gervasio, F. L. (2020). Investigating cryptic binding sites by molecular dynamics simulations. Acc. Chem. Res. 53, 654–661. doi: 10.1021/acs.accounts.9b00613
Kuzmanic, A., Sutto, L., Saladino, G., Nebreda, A. R., Gervasio, F. L., and Orozco, M. (2017). Changes in the free-energy landscape of p38alpha MAP kinase through its canonical activation and binding events as studied by enhanced molecular dynamics simulations. Elife 6:e22175. doi: 10.7554/eLife.22175.024
La Sala, G., Decherchi, S., De Vivo, M., and Rocchia, W. (2017). Allosteric communication networks in proteins revealed through pocket crosstalk analysis. ACS Cent. Sci. 3, 949–960. doi: 10.1021/acscentsci.7b00211
Lambiotte, R., Rosvall, M., and Scholtes, I. (2019). From networks to optimal higher-order models of complex systems. Nat. Phys. 15, 313–320. doi: 10.1038/s41567-019-0459-y
Lambiotte, R., Sinatra, R., Delvenne, J. C., Evans, T. S., Barahona, M., and Latora, V. (2011). Flow graphs: interweaving dynamics and structure. Phys. Rev. E Stat. Nonlin. Soft. Matter. Phys. 84(Pt. 2):017102. doi: 10.1103/PhysRevE.84.017102
Lancichinetti, A., and Fortunato, S. (2009). Community detection algorithms: a comparative analysis. Phys. Rev. E Stat. Nonlin. Soft. Matter. Phys. 80(Pt. 2):056117. doi: 10.1103/PhysRevE.80.056117
Lawless, N., Blacklock, K., Berrigan, E., and Verkhivker, G. (2013). Structural bioinformatics and protein docking analysis of the molecular chaperone-kinase interactions: towards allosteric inhibition of protein kinases by targeting the hsp90-cdc37 chaperone machinery. Pharmaceuticals 6, 1407–1428. doi: 10.3390/ph6111407
LeCun, Y., Bengio, Y., and Hinton, G. (2015). Deep learning. Nature 521, 436–444. doi: 10.1038/nature14539
Levinson, N. M. (2018). The multifaceted allosteric regulation of aurora kinase A. Biochem. J. 475, 2025–2042. doi: 10.1042/BCJ20170771
Levinson, N. M., and Boxer, S. G. (2012). Structural and spectroscopic analysis of the kinase inhibitor bosutinib and an isomer of bosutinib binding to the Abl tyrosine kinase domain. PLoS ONE 7:e29828. doi: 10.1371/journal.pone.0029828
Levinson, N. M., and Boxer, S. G. (2014). A conserved water-mediated hydrogen bond network defines bosutinib's kinase selectivity. Nat. Chem. Biol. 10, 127–132. doi: 10.1038/nchembio.1404
Levinson, N. M., Kuchment, O., Shen, K., Young, M. A., Koldobskiy, M., Karplus, M., et al. (2006). A Src-like inactive conformation in the abl tyrosine kinase domain. PLoS Biol 4:e144. doi: 10.1371/journal.pbio.0040144
Li, C., Ma, N., Wang, Y., and Chen, G. (2014). Molecular dynamics simulation studies on the positive cooperativity of the Kemptide substrate with protein kinase A induced by the ATP ligand. J. Phys. Chem. B 118, 1273–1287. doi: 10.1021/jp411111g
Li, Q., Luo, R., and Chen, H. F. (2019). Dynamical important residue network (DIRN): network inference via conformational change. Bioinformatics 35, 4664–4670. doi: 10.1093/bioinformatics/btz298
Li, Z., Kermode, J. R., and De Vita, A. (2015). Molecular dynamics with on-the-fly machine learning of quantum-mechanical forces. Phys. Rev. Lett. 114:096405. doi: 10.1103/PhysRevLett.114.096405
Liang, Z., Verkhivker, G. M., and Hu, G. (2019). Integration of network models and evolutionary analysis into high-throughput modeling of protein dynamics and allosteric regulation: theory, tools and applications. Brief. Bioinform. 21, 815–835. doi: 10.1093/bib/bbz029
Limongelli, V., Bonomi, M., and Parrinello, M. (2013). Funnel metadynamics as accurate binding free-energy method. Proc. Natl. Acad. Sci. U.S.A. 110, 6358–6363. doi: 10.1073/pnas.1303186110
Lin, Y. L., Meng, Y., Huang, L., and Roux, B. (2014). Computational study of gleevec and G6G reveals molecular determinants of kinase inhibitor selectivity. J. Am. Chem. Soc. 136, 14753–14762. doi: 10.1021/ja504146x
Lin, Y. L., Meng, Y., Jiang, W., and Roux, B. (2013). Explaining why gleevec is a specific and potent inhibitor of Abl kinase. Proc. Natl. Acad. Sci. U.S.A. 110, 1664–1669. doi: 10.1073/pnas.1214330110
Lin, Y. L., and Roux, B. (2013). Computational analysis of the binding specificity of gleevec to Abl, c-Kit, Lck, and c-Src tyrosine kinases. J. Am. Chem. Soc. 135, 14741–14753. doi: 10.1021/ja405939x
Lisi, G. P., and Loria, J. P. (2016). Solution NMR spectroscopy for the study of enzyme allostery. Chem. Rev. 116, 6323–6369. doi: 10.1021/acs.chemrev.5b00541
Lisi, G. P., and Loria, J. P. (2017). Allostery in enzyme catalysis. Curr. Opin. Struct. Biol. 47, 123–130. doi: 10.1016/j.sbi.2017.08.002
Liu, J., and Nussinov, R. (2016). Allostery: an overview of its history, concepts, methods, and applications. PLoS Comput. Biol. 12:e1004966. doi: 10.1371/journal.pcbi.1004966
Lockless, S. W., and Ranganathan, R. (1999). Evolutionarily conserved pathways of energetic connectivity in protein families. Science 286, 295–299. doi: 10.1126/science.286.5438.295
Long, D., Bouvignies, G., and Kay, L. E. (2014). Measuring hydrogen exchange rates in invisible protein excited states. Proc. Natl. Acad. Sci. U.S.A. 111, 8820–8825. doi: 10.1073/pnas.1405011111
Long, D., and Bruschweiler, R. (2011). Atomistic kinetic model for population shift and allostery in biomolecules. J. Am. Chem. Soc. 133, 18999–19005. doi: 10.1021/ja208813t
Ma, B., Tsai, C. J., Haliloglu, T., and Nussinov, R. (2011). Dynamic allostery: linkers are not merely flexible. Structure 19, 907–917. doi: 10.1016/j.str.2011.06.002
Malmstrom, R. D., Kornev, A. P., Taylor, S. S., and Amaro, R. E. (2015). Allostery through the computational microscope: cAMP activation of a canonical signalling domain. Nat. Commun. 6:7588. doi: 10.1038/ncomms8588
Marcu, M. G., Chadli, A., Bouhouche, I., Catelli, M., and Neckers, L. M. (2000a). The heat shock protein 90 antagonist novobiocin interacts with a previously unrecognized ATP-binding domain in the carboxyl terminus of the chaperone. J. Biol. Chem. 275, 37181–37186. doi: 10.1074/jbc.M003701200
Marcu, M. G., Schulte, T. W., and Neckers, L. (2000b). Novobiocin and related coumarins and depletion of heat shock protein 90-dependent signaling proteins. J. Natl. Cancer Inst. 92, 242–248. doi: 10.1093/jnci/92.3.242
Mardt, A., Pasquali, L., Wu, H., and Noe, F. (2018). VAMPnets for deep learning of molecular kinetics. Nat. Commun. 9:5. doi: 10.1038/s41467-017-02388-1
Masgras, I., Sanchez-Martin, C., Colombo, G., and Rasola, A. (2017). The chaperone TRAP1 as a modulator of the mitochondrial adaptations in cancer cells. Front. Oncol. 7:58. doi: 10.3389/fonc.2017.00058
Mater, A. C., and Coote, M. L. (2019). Deep learning in chemistry. J. Chem. Inf. Model. 59, 2545–2559. doi: 10.1021/acs.jcim.9b00266
Matts, R. L., Brandt, G. E. L., Lu, Y. M., Dixit, A., Mollapour, M., Wang, S. Q., et al. (2011a). A systematic protocol for the characterization of Hsp90 modulators. Bioorg. Med. Chem. 19, 684–692. doi: 10.1016/j.bmc.2010.10.029
Matts, R. L., Dixit, A., Peterson, L. B., Sun, L., Voruganti, S., Kalyanaraman, P., et al. (2011b). Elucidation of the Hsp90 C-terminal inhibitor binding site. ACS Chem. Biol. 6, 800–807. doi: 10.1021/cb200052x
Maziarka, Ł., Pocha, A., Kaczmarczyk, J., Rataj, K., Danel, T., and Warchoł, M. (2020). Mol-CycleGAN: a generative model for molecular optimization. J. Cheminform. 12:2. doi: 10.1186/s13321-019-0404-1
McClendon, C. L., Friedland, G., Mobley, D. L., Amirkhani, H., and Jacobson, M. P. (2009). Quantifying correlations between allosteric sites in thermodynamic ensembles. J. Chem. Theory Comput. 5, 2486–2502. doi: 10.1021/ct9001812
McGibbon, R. T., Schwantes, C. R., and Pande, V. S. (2014). Statistical model selection for markov models of biomolecular dynamics. J. Phys. Chem. B 118, 6475–6481. doi: 10.1021/jp411822r
McLaughlin, R. N. Jr., Poelwijk, F. J., Raman, A., Gosal, W. S., and Ranganathan, R. (2012). The spatial architecture of protein function and adaptation. Nature 491, 138–142. doi: 10.1038/nature11500
McLeish, T. C., Rodgers, T. L., and Wilson, M. R. (2013). Allostery without conformation change: modelling protein dynamics at multiple scales. Phys. Biol. 10:056004. doi: 10.1088/1478-3975/10/5/056004
Meharena, H. S., Chang, P., Keshwani, M. M., Oruganty, K., Nene, A. K., Kannan, N., et al. (2013). Deciphering the structural basis of eukaryotic protein kinase regulation. PLoS Biol. 11:e1001680. doi: 10.1371/journal.pbio.1001680
Meng, Y., Pond, M. P., and Roux, B. (2017). Tyrosine kinase activation and conformational flexibility: lessons from src-family tyrosine kinases. Acc. Chem. Res. 50, 1193–1201. doi: 10.1021/acs.accounts.7b00012
Meng, Y., and Roux, B. (2014). Locking the active conformation of c-Src kinase through the phosphorylation of the activation loop. J. Mol. Biol. 426, 423–435. doi: 10.1016/j.jmb.2013.10.001
Ming, D., and Wall, M. E. (2005). Quantifying allosteric effects in proteins. Proteins 59, 697–707. doi: 10.1002/prot.20440
Ming, D., and Wall, M. E. (2006). Interactions in native binding sites cause a large change in protein dynamics. J. Mol. Biol. 358, 213–223. doi: 10.1016/j.jmb.2006.01.097
Mittermaier, A., and Kay, L. E. (2006). New tools provide new insights in NMR studies of protein dynamics. Science 312, 224–228. doi: 10.1126/science.1124964
Mittermaier, A. K., and Kay, L. E. (2009). Observing biological dynamics at atomic resolution using NMR. Trends Biochem. Sci. 34, 601–611. doi: 10.1016/j.tibs.2009.07.004
Mitternacht, S., and Berezovsky, I. N. (2011). Binding leverage as a molecular basis for allosteric regulation. PLoS Comput. Biol. 7:e1002148. doi: 10.1371/journal.pcbi.1002148
Mnih, V., Kavukcuoglu, K., Silver, D., Rusu, A. A., Veness, J., Bellemare, M. G., et al. (2015). Human-level control through deep reinforcement learning. Nature 518, 529–533. doi: 10.1038/nature14236
Monod, J., Wyman, J., and Changeux, J. P. (1965). On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 12, 88–118. doi: 10.1016/S0022-2836(65)80285-6
Morcos, F., Pagnani, A., Lunt, B., Bertolino, A., Marks, D. S., Sander, C., et al. (2011). Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc. Natl. Acad. Sci. U.S.A. 108, E1293–1301. doi: 10.1073/pnas.1111471108
Morra, G., Neves, M. A. C., Plescia, C. J., Tsustsumi, S., Neckers, L., Verkhivker, G., et al. (2010). Dynamics-based discovery of allosteric inhibitors: selection of new ligands for the C-terminal domain of Hsp90. J. Chem. Theory Comput. 6, 2978–2989. doi: 10.1021/ct100334n
Morra, G., Potestio, R., Micheletti, C., and Colombo, G. (2012). Corresponding functional dynamics across the Hsp90 chaperone family: insights from a multiscale analysis of MD simulations. PLoS Comput. Biol. 8:e1002433. doi: 10.1371/journal.pcbi.1002433
Morra, G., Verkhivker, G., and Colombo, G. (2009). Modeling signal propagation mechanisms and ligand-based conformational dynamics of the Hsp90 molecular chaperone full-length dimer. PLoS Comput. Biol. 5:e1000323. doi: 10.1371/journal.pcbi.1000323
Motlagh, H. N., Wrabl, J. O., Li, J., and Hilser, V. J. (2014). The ensemble nature of allostery. Nature 508, 331–339. doi: 10.1038/nature13001
Munte, C. E., Beck Erlach, M., Kremer, W., Koehler, J., and Kalbitzer, H. R. (2013). Distinct conformational states of the alzheimer beta-amyloid peptide can be detected by high-pressure NMR spectroscopy. Angew. Chem. Int. Ed. Engl. 52, 8943–8947. doi: 10.1002/anie.201301537
Narayanan, C., Bafna, K., Roux, L. D., Agarwal, P. K., and Doucet, N. (2017). Applications of NMR and computational methodologies to study protein dynamics. Arch. Biochem. Biophys. 628, 71–80. doi: 10.1016/j.abb.2017.05.002
Nerli, S., McShan, A. C., and Sgourakis, N. G. (2018). Chemical shift-based methods in NMR structure determination. Prog. Nucl Magn. Reson. Spectrosc. 106–107, 1–25. doi: 10.1016/j.pnmrs.2018.03.002
Nesmelova, I. V., Ermakova, E., Daragan, V. A., Pang, M., Menendez, M., Lagartera, L., et al. (2010). Lactose binding to galectin-1 modulates structural dynamics, increases conformational entropy, and occurs with apparent negative cooperativity. J. Mol. Biol. 397, 1209–1230. doi: 10.1016/j.jmb.2010.02.033
Nishi, H., Hashimoto, K., and Panchenko, A. R. (2011). Phosphorylation in protein-protein binding: effect on stability and function. Structure 19, 1807–1815. doi: 10.1016/j.str.2011.09.021
Noe, F., and Clementi, C. (2017). Collective variables for the study of long-time kinetics from molecular trajectories: theory and methods. Curr. Opin. Struct. Biol. 43, 141–147. doi: 10.1016/j.sbi.2017.02.006
Nussinov, R. (2012). How do dynamic cellular signals travel long distances? Mol. Biosyst. 8, 22–26. doi: 10.1039/C1MB05205E
Nussinov, R., and Tsai, C. J. (2015). Allostery without a conformational change? Revisiting the paradigm. Curr. Opin. Struct. Biol. 30, 17–24. doi: 10.1016/j.sbi.2014.11.005
Oleinikovas, V., Saladino, G., Cossins, B. P., and Gervasio, F. L. (2016). Understanding cryptic pocket formation in protein targets by enhanced sampling simulations. J. Am. Chem. Soc. 138, 14257–14263. doi: 10.1021/jacs.6b05425
Olivecrona, M., Blaschke, T., Engkvist, O., and Chen, H. (2017). Molecular de-novo design through deep reinforcement learning. J. Cheminform. 9:48. doi: 10.1186/s13321-017-0235-x
Olson, R. S., La Cava, W., Orzechowski, P., Urbanowicz, R. J., and Moore, J. H. (2017). PMLB: a large benchmark suite for machine learning evaluation and comparison. BioData Min. 10:36. doi: 10.1186/s13040-017-0154-4
Olsson, S., Wu, H., Paul, F., Clementi, C., and Noe, F. (2017). Combining experimental and simulation data of molecular processes via augmented markov models. Proc. Natl. Acad. Sci. U.S.A. 114, 8265–8270. doi: 10.1073/pnas.1704803114
Paladino, A., Morra, G., and Colombo, G. (2015). Structural stability and flexibility direct the selection of activating mutations in epidermal growth factor receptor kinase. J. Chem. Inf. Model. 55, 1377–1387. doi: 10.1021/acs.jcim.5b00270
Palazzesi, F., Barducci, A., Tollinger, M., and Parrinello, M. (2013). The allosteric communication pathways in KIX domain of CBP. Proc. Natl. Acad. Sci. U.S.A. 110, 14237–14242. doi: 10.1073/pnas.1313548110
Palazzesi, F., Valsson, O., and Parrinello, M. (2017). Conformational entropy as collective variable for proteins. J. Phys. Chem. Lett. 8, 4752–4756. doi: 10.1021/acs.jpclett.7b01770
Palermo, G., Ricci, C. G., Fernando, A., Basak, R., Jinek, M., Rivalta, I., et al. (2017). Protospacer adjacent motif-induced allostery activates CRISPR-Cas9. J. Am. Chem. Soc. 139, 16028–16031. doi: 10.1021/jacs.7b05313
Pande, V. S. (2014). Understanding protein folding using markov state models. Adv. Exp. Med. Biol. 797, 101–106. doi: 10.1007/978-94-007-7606-7_8
Panjkovich, A., and Daura, X. (2012). Exploiting protein flexibility to predict the location of allosteric sites. BMC Bioinformatics 13:273. doi: 10.1186/1471-2105-13-273
Panjkovich, A., and Daura, X. (2014). PARS: a web server for the prediction of protein allosteric and regulatory sites. Bioinformatics 30, 1314–1315. doi: 10.1093/bioinformatics/btu002
Perez-Hernandez, G., and Noe, F. (2016). Hierarchical time-lagged independent component analysis: computing slow modes and reaction coordinates for large molecular systems. J. Chem. Theory Comput. 12, 6118–6129. doi: 10.1021/acs.jctc.6b00738
Polykovskiy, D., Zhebrak, A., Vetrov, D., Ivanenkov, Y., Aladinskiy, V., Mamoshina, P., et al. (2018). Entangled conditional adversarial autoencoder for de novo drug discovery. Mol. Pharm. 15, 4398–4405. doi: 10.1021/acs.molpharmaceut.8b00839
Popova, M., Isayev, O., and Tropsha, A. (2018). Deep reinforcement learning for de novo drug design. Sci. Adv. 4:eaap7885. doi: 10.1126/sciadv.aap7885
Popovych, N., Sun, S., Ebright, R. H., and Kalodimos, C. G. (2006). Dynamically driven protein allostery. Nat. Struct. Mol. Biol. 13, 831–838. doi: 10.1038/nsmb1132
Prinz, J. H., Wu, H., Sarich, M., Keller, B., Senne, M., Held, M., et al. (2011). Markov models of molecular kinetics: generation and validation. J. Chem. Phys. 134:174105. doi: 10.1063/1.3565032
Putin, E., Asadulaev, A., Ivanenkov, Y., Aladinskiy, V., Sanchez-Lengeling, B., Aspuru-Guzik, A., et al. (2018a). Reinforced adversarial neural computer for de novo molecular design. J. Chem. Inf. Model. 58, 1194–1204. doi: 10.1021/acs.jcim.7b00690
Putin, E., Asadulaev, A., Vanhaelen, Q., Ivanenkov, Y., Aladinskaya, A. V., Aliper, A., et al. (2018b). Adversarial threshold neural computer for molecular de novo design. Mol. Pharm. 15, 4386–4397. doi: 10.1021/acs.molpharmaceut.7b01137
Racz, A., Bajusz, D., and Heberger, K. (2019). Multi-level comparison of machine learning classifiers and their performance metrics. Molecules 24:2811. doi: 10.3390/molecules24152811
Ricci, C. G., Silveira, R. L., Rivalta, I., Batista, V. S., and Skaf, M. S. (2016). Allosteric pathways in the PPARgamma-RXRalpha nuclear receptor complex. Sci. Rep. 6:19940. doi: 10.1038/srep19940
Rinaldi, S., Assimon, V. A., Young, Z. T., Morra, G., Shao, H., Taylor, I. R., et al. (2018). A local allosteric network in heat shock protein 70 (Hsp70) links inhibitor binding to enzyme activity and distal protein-protein interactions. ACS Chem. Biol. 13, 3142–3152. doi: 10.1021/acschembio.8b00712
Rivalta, I., Sultan, M. M., Lee, N. S., Manley, G. A., Loria, J. P., and Batista, V. S. (2012). Allosteric pathways in imidazole glycerol phosphate synthase. Proc. Natl. Acad. Sci. U.S.A. 109, E1428–1436. doi: 10.1073/pnas.1120536109
Robustelli, P., Kohlhoff, K., Cavalli, A., and Vendruscolo, M. (2010). Using NMR chemical shifts as structural restraints in molecular dynamics simulations of proteins. Structure 18, 923–933. doi: 10.1016/j.str.2010.04.016
Robustelli, P., Stafford, K. A., and Palmer, A. G. 3rd. (2012). Interpreting protein structural dynamics from NMR chemical shifts. J. Am. Chem. Soc. 134, 6365–6374. doi: 10.1021/ja300265w
Rosenzweig, R., and Kay, L. E. (2014). Bringing dynamic molecular machines into focus by methyl-TROSY NMR. Annu. Rev. Biochem. 83, 291–315. doi: 10.1146/annurev-biochem-060713-035829
Rosvall, M., and Bergstrom, C. T. (2007). An information-theoretic framework for resolving community structure in complex networks. Proc. Natl. Acad. Sci. U.S.A. 104, 7327–7331. doi: 10.1073/pnas.0611034104
Rosvall, M., and Bergstrom, C. T. (2008). Maps of random walks on complex networks reveal community structure. Proc. Natl. Acad. Sci. U.S.A. 105, 1118–1123. doi: 10.1073/pnas.0706851105
Rosvall, M., and Bergstrom, C. T. (2010). Mapping change in large networks. PLoS ONE 5:e8694. doi: 10.1371/journal.pone.0008694
Rosvall, M., and Bergstrom, C. T. (2011). Multilevel compression of random walks on networks reveals hierarchical organization in large integrated systems. PLoS ONE 6:e18209. doi: 10.1371/journal.pone.0018209
Rosvall, M., Esquivel, A. V., Lancichinetti, A., West, J. D., and Lambiotte, R. (2014). Memory in network flows and its effects on spreading dynamics and community detection. Nat. Commun. 5:4630. doi: 10.1038/ncomms5630
Ruff, E. F., Muretta, J. M., Thompson, A. R., Lake, E. W., Cyphers, S., Albanese, S. K., et al. (2018). A dynamic mechanism for allosteric activation of aurora kinase A by activation loop phosphorylation. Elife 7:e32766. doi: 10.7554/eLife.32766.019
Rupp, M., Tkatchenko, A., Muller, K. R., and von Lilienfeld, O. A. (2012). Fast and accurate modeling of molecular atomization energies with machine learning. Phys. Rev. Lett. 108:058301. doi: 10.1103/PhysRevLett.108.058301
Saleh, T., Rossi, P., and Kalodimos, C. G. (2017). Atomic view of the energy landscape in the allosteric regulation of Abl kinase. Nat. Struct. Mol. Biol. 24, 893–901. doi: 10.1038/nsmb.3470
Salnikov, V., Schaub, M. T., and Lambiotte, R. (2016). Using higher-order markov models to reveal flow-based communities in networks. Sci. Rep. 6:23194. doi: 10.1038/srep23194
Sanchez-Martin, C., Moroni, E., Ferraro, M., Laquatra, C., Cannino, G., Masgras, I., et al. (2020). Rational design of allosteric and selective inhibitors of the molecular chaperone TRAP1. Cell. Rep. 31:107531. doi: 10.1016/j.celrep.2020.107531
Sattin, S., Tao, J., Vettoretti, G., Moroni, E., Pennati, M., Lopergolo, A., et al. (2015). Activation of Hsp90 enzymatic activity and conformational dynamics through rationally designed allosteric ligands. Chemistry 21, 13598–13608. doi: 10.1002/chem.201502211
Schaub, M. T., Lambiotte, R., and Barahona, M. (2012). Encoding dynamics for multiscale community detection: markov time sweeping for the map equation. Phys. Rev. E Stat. Nonlin. Soft. Matter. Phys. 86(Pt. 2):026112. doi: 10.1103/PhysRevE.86.026112
Schoepfer, J., Jahnke, W., Berellini, G., Buonamici, S., Cotesta, S., Cowan-Jacob, S. W., et al. (2018). Discovery of asciminib (ABL001), an allosteric inhibitor of the tyrosine kinase activity of BCR-ABL1. J. Med. Chem. 61, 8120–8135. doi: 10.1021/acs.jmedchem.8b01040
Schreiber, T. (2000). Measuring information transfer. Phys. Rev. Lett. 85, 461–464. doi: 10.1103/PhysRevLett.85.461
Schulze, J. O., Saladino, G., Busschots, K., Neimanis, S., Suss, E., Odadzic, D., et al. (2016). Bidirectional allosteric communication between the ATP-binding site and the regulatory PIF pocket in PDK1 protein kinase. Cell. Chem. Biol. 23, 1193–1205. doi: 10.1016/j.chembiol.2016.06.017
Sekhar, A., and Kay, L. E. (2013). NMR paves the way for atomic level descriptions of sparsely populated, transiently formed biomolecular conformers. Proc. Natl. Acad. Sci. U.S.A. 110, 12867–12874. doi: 10.1073/pnas.1305688110
Sekhar, A., and Kay, L. E. (2019). An NMR view of protein dynamics in health and disease. Annu. Rev. Biophys. 48, 297–319. doi: 10.1146/annurev-biophys-052118-115647
Selvaratnam, R., Chowdhury, S., VanSchouwen, B., and Melacini, G. (2011). Mapping allostery through the covariance analysis of NMR chemical shifts. Proc. Natl. Acad. Sci. U.S.A. 108, 6133–6138. doi: 10.1073/pnas.1017311108
Selvaratnam, R., VanSchouwen, B., Fogolari, F., Mazhab-Jafari, M. T., Das, R., and Melacini, G. (2012). The projection analysis of NMR chemical shifts reveals extended EPAC autoinhibition determinants. Biophys. J. 102, 630–639. doi: 10.1016/j.bpj.2011.12.030
Sengupta, U., and Strodel, B. (2018). Markov models for the elucidation of allosteric regulation. Philos. Trans. R Soc. Lond. B Biol. Sci. 373:20170178. doi: 10.1098/rstb.2017.0178
Senior, A. W., Evans, R., Jumper, J., Kirkpatrick, J., Sifre, L., Green, T., et al. (2019). Protein structure prediction using multiple deep neural networks in the 13th critical assessment of protein structure prediction (CASP13). Proteins 87, 1141–1148. doi: 10.1002/prot.25834
Sethi, A., Eargle, J., Black, A. A., and Luthey-Schulten, Z. (2009). Dynamical networks in tRNA:protein complexes. Proc. Natl. Acad. Sci. U.S.A. 106, 6620–6625. doi: 10.1073/pnas.0810961106
Sethi, A., Tian, J., Derdeyn, C. A., Korber, B., and Gnanakaran, S. (2013). A mechanistic understanding of allosteric immune escape pathways in the HIV-1 envelope glycoprotein. PLoS Comput. Biol. 9:e1003046. doi: 10.1371/journal.pcbi.1003046
Shamsi, Z., Cheng, K. J., and Shukla, D. (2018). Reinforcement learning based adaptive sampling: REAPing rewards by exploring protein conformational landscapes. J. Phys. Chem. B 122, 8386–8395. doi: 10.1021/acs.jpcb.8b06521
Shan, Y., Arkhipov, A., Kim, E. T., Pan, A. C., and Shaw, D. E. (2013). Transitions to catalytically inactive conformations in EGFR kinase. Proc. Natl. Acad. Sci. U.S.A. 110, 7270–7275. doi: 10.1073/pnas.1220843110
Shan, Y., Eastwood, M. P., Zhang, X., Kim, E. T., Arkhipov, A., Dror, R. O., et al. (2012). Oncogenic mutations counteract intrinsic disorder in the EGFR kinase and promote receptor dimerization. Cell 149, 860–870. doi: 10.1016/j.cell.2012.02.063
Shan, Y., Kim, E. T., Eastwood, M. P., Dror, R. O., Seeliger, M. A., and Shaw, D. E. (2011). How does a drug molecule find its target binding site? J. Am. Chem. Soc. 133, 9181–9183. doi: 10.1021/ja202726y
Shaw, A. S., Kornev, A. P., Hu, J., Ahuja, L. G., and Taylor, S. S. (2014). Kinases and pseudokinases: lessons from RAF. Mol. Cell. Biol. 34, 1538–1546. doi: 10.1128/MCB.00057-14
Shcherbinin, D., Veselovsky, A., Rubtsova, M., Grigorenko, V., and Egorov, A. (2019). The impact of long-distance mutations on the omega-loop conformation in TEM type beta-lactamases. J. Biomol. Struct. Dyn. 38, 2369–2376. doi: 10.1080/07391102.2019.1634642
Shen, Y., and Bax, A. (2010). SPARTA+: a modest improvement in empirical NMR chemical shift prediction by means of an artificial neural network. J. Biomol. NMR 48, 13–22. doi: 10.1007/s10858-010-9433-9
Shi, L., and Kay, L. E. (2014). Tracing an allosteric pathway regulating the activity of the HslV protease. Proc. Natl. Acad. Sci. U.S.A. 111, 2140–2145. doi: 10.1073/pnas.1318476111
Shukla, D., Hernandez, C. X., Weber, J. K., and Pande, V. S. (2015). Markov state models provide insights into dynamic modulation of protein function. Acc. Chem. Res. 48, 414–422. doi: 10.1021/ar5002999
Shukla, D., Meng, Y., Roux, B., and Pande, V. S. (2014). Activation pathway of Src kinase reveals intermediate states as targets for drug design. Nat. Commun. 5:3397. doi: 10.1038/ncomms4397
Shukla, S., Shamsi, Z., Moffett, A. S., Selvam, B., and Shukla, D. (2017). Application of hidden markov models in biomolecular simulations. Methods Mol. Biol. 1552, 29–41. doi: 10.1007/978-1-4939-6753-7_3
Silver, D., Schrittwieser, J., Simonyan, K., Antonoglou, I., Huang, A., Guez, A., et al. (2017). Mastering the game of go without human knowledge. Nature 550, 354–359. doi: 10.1038/nature24270
Simonetti, F. L., Teppa, E., Chernomoretz, A., Nielsen, M., and Marino Buslje, C. (2013). MISTIC: mutual information server to infer coevolution. Nucleic Acids Res. 41, W8–14. doi: 10.1093/nar/gkt427
Smock, R. G., and Gierasch, L. M. (2009). Sending signals dynamically. Science 324, 198–203. doi: 10.1126/science.1169377
Socolich, M., Lockless, S. W., Russ, W. P., Lee, H., Gardner, K. H., and Ranganathan, R. (2005). Evolutionary information for specifying a protein fold. Nature 437, 512–518. doi: 10.1038/nature03991
Sorin, V., Barash, Y., Konen, E., and Klang, E. (2020). Creating artificial images for radiology applications using generative adversarial networks (GANs) – a systematic review. Acad. Radiol. doi: 10.1016/j.acra.2019.12.024. [Epub ahead of print].
Sprangers, R., Velyvis, A., and Kay, L. E. (2007). Solution NMR of supramolecular complexes: providing new insights into function. Nat. Methods 4, 697–703. doi: 10.1038/nmeth1080
Stetz, G., Tse, A., and Verkhivker, G. M. (2017). Ensemble-based modeling and rigidity decomposition of allosteric interaction networks and communication pathways in cyclin-dependent kinases: differentiating kinase clients of the Hsp90-Cdc37 chaperone. PLoS ONE 12:e0186089. doi: 10.1371/journal.pone.0186089
Stetz, G., and Verkhivker, G. M. (2015). Dancing through life: molecular dynamics simulations and network-centric modeling of allosteric mechanisms in Hsp70 and Hsp110 chaperone proteins. PLoS ONE 10:e0143752. doi: 10.1371/journal.pone.0143752
Stetz, G., and Verkhivker, G. M. (2016). Probing allosteric inhibition mechanisms of the Hsp70 chaperone proteins using molecular dynamics simulations and analysis of the residue interaction networks. J. Chem. Inf. Model. 56, 1490–1517. doi: 10.1021/acs.jcim.5b00755
Stetz, G., and Verkhivker, G. M. (2017). Computational analysis of residue interaction networks and coevolutionary relationships in the Hsp70 chaperones: a community-hopping model of allosteric regulation and communication. PLoS Comput. Biol. 13:e1005299. doi: 10.1371/journal.pcbi.1005299
Stetz, G., and Verkhivker, G. M. (2018). Functional role and hierarchy of the intermolecular interactions in binding of protein kinase clients to the Hsp90-Cdc37 chaperone: structure-based network modeling of allosteric regulation. J. Chem. Inf. Model. 58, 405–421. doi: 10.1021/acs.jcim.7b00638
Stevens, S. Y., Sanker, S., Kent, C., and Zuiderweg, E. R. (2001). Delineation of the allosteric mechanism of a cytidylyltransferase exhibiting negative cooperativity. Nat. Struct. Biol. 8, 947–952. doi: 10.1038/nsb1101-947
Stock, G., and Hamm, P. (2018). A non-equilibrium approach to allosteric communication. Philos. Trans. R Soc. Lond. B Biol. Sci. 373:20170187. doi: 10.1098/rstb.2017.0187
Stolzenberg, S., Michino, M., LeVine, M. V., Weinstein, H., and Shi, L. (2016). Computational approaches to detect allosteric pathways in transmembrane molecular machines. Biochim. Biophys. Acta 1858(Pt. B), 1652–1662. doi: 10.1016/j.bbamem.2016.01.010
Suel, G. M., Lockless, S. W., Wall, M. A., and Ranganathan, R. (2003). Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nat. Struct. Biol. 10, 59–69. doi: 10.1038/nsb881
Sutto, L., and Gervasio, F. L. (2013). Effects of oncogenic mutations on the conformational free-energy landscape of EGFR kinase. Proc. Natl. Acad. Sci. U.S.A. 110, 10616–10621. doi: 10.1073/pnas.1221953110
Sutton, R. S., and Barto, A. G. (1981). Toward a modern theory of adaptive networks: expectation and prediction. Psychol. Rev. 88, 135–170. doi: 10.1037/0033-295X.88.2.135
Swain, J. F., and Gierasch, L. M. (2006). The changing landscape of protein allostery. Curr. Opin. Struct. Biol. 16, 102–108. doi: 10.1016/j.sbi.2006.01.003
Taylor, S. S., Ilouz, R., Zhang, P., and Kornev, A. P. (2012a). Assembly of allosteric macromolecular switches: lessons from PKA. Nat. Rev. Mol. Cell. Biol. 13, 646–658. doi: 10.1038/nrm3432
Taylor, S. S., Keshwani, M. M., Steichen, J. M., and Kornev, A. P. (2012b). Evolution of the eukaryotic protein kinases as dynamic molecular switches. Philos. Trans. R Soc. Lond. B Biol. Sci. 367, 2517–2528. doi: 10.1098/rstb.2012.0054
Taylor, S. S., and Kornev, A. P. (2011). Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem. Sci. 36, 65–77. doi: 10.1016/j.tibs.2010.09.006
Tehver, R., Chen, J., and Thirumalai, D. (2009). Allostery wiring diagrams in the transitions that drive the GroEL reaction cycle. J. Mol. Biol. 387, 390–406. doi: 10.1016/j.jmb.2008.12.032
Toledano, D. T., Fernandez-Gallego, M. P., and Lozano-Diez, A. (2018). Multi-resolution speech analysis for automatic speech recognition using deep neural networks: experiments on TIMIT. PLoS ONE 13:e0205355. doi: 10.1371/journal.pone.0205355
Tsai, C. J., del Sol, A., and Nussinov, R. (2008). Allostery: absence of a change in shape does not imply that allostery is not at play. J. Mol. Biol. 378, 1–11. doi: 10.1016/j.jmb.2008.02.034
Tsai, C. J., Del Sol, A., and Nussinov, R. (2009). Protein allostery, signal transmission and dynamics: a classification scheme of allosteric mechanisms. Mol. Biosyst. 5, 207–216. doi: 10.1039/b819720b
Tsai, C. J., and Nussinov, R. (2014). A unified view of “how allostery works”. PLoS Comput. Biol. 10:e1003394. doi: 10.1371/journal.pcbi.1003394
Tse, A., and Verkhivker, G. M. (2015a). Molecular determinants underlying binding specificities of the ABL kinase inhibitors: combining alanine scanning of binding hot spots with network analysis of residue interactions and coevolution. PLoS ONE 10:e0130203. doi: 10.1371/journal.pone.0130203
Tse, A., and Verkhivker, G. M. (2015b). Molecular dynamics simulations and structural network analysis of c-Abl and c-Src kinase core proteins: capturing allosteric mechanisms and communication pathways from residue centrality. J. Chem. Inf. Model. 55, 1645–1662. doi: 10.1021/acs.jcim.5b00240
Tse, A., and Verkhivker, G. M. (2015c). Small-world networks of residue interactions in the Abl kinase complexes with cancer drugs: topology of allosteric communication pathways can determine drug resistance effects. Mol. Biosyst. 11, 2082–2095. doi: 10.1039/C5MB00246J
Tsuchiya, Y., Taneishi, K., and Yonezawa, Y. (2019). Autoencoder-based detection of dynamic allostery triggered by ligand binding based on molecular dynamics. J. Chem. Inf. Model. 59, 4043–4051. doi: 10.1021/acs.jcim.9b00426
Tzeng, S. R., and Kalodimos, C. G. (2009). Dynamic activation of an allosteric regulatory protein. Nature 462, 368–372. doi: 10.1038/nature08560
Tzeng, S. R., and Kalodimos, C. G. (2011). Protein dynamics and allostery: an NMR view. Curr. Opin. Struct. Biol. 21, 62–67. doi: 10.1016/j.sbi.2010.10.007
Vallurupalli, P., Bouvignies, G., and Kay, L. E. (2012). Studying “invisible” excited protein states in slow exchange with a major state conformation. J. Am. Chem. Soc. 134, 8148–8161. doi: 10.1021/ja3001419
Vanwart, A. T., Eargle, J., Luthey-Schulten, Z., and Amaro, R. E. (2012). Exploring residue component contributions to dynamical network models of allostery. J. Chem. Theory Comput. 8, 2949–2961. doi: 10.1021/ct300377a
Vendruscolo, M., Dokholyan, N. V., Paci, E., and Karplus, M. (2002). Small-world view of the amino acids that play a key role in protein folding. Phys. Rev. E Stat. Nonlin. Soft. Matter. Phys. 65(Pt. 1):061910. doi: 10.1103/PhysRevE.65.061910
Verkhivker, G., Blacklock, K., and Buchner, J. (2016). Dissecting allosteric regulatory mechanisms of the Hsp90 chaperone interactions with the protein kinase clients: Integrating structural bioinformatics with multiscale atomistic simulations and biophysical experiments. Abst. Pap. Am. Chem. Soc. 251:2.
Verkhivker, G. M. (2014). Computational studies of allosteric regulation in the hsp90 molecular chaperone: from functional dynamics and protein structure networks to allosteric communications and targeted anti-cancer modulators. Israel J. Chem. 54, 1052–1064. doi: 10.1002/ijch.201300143
Verkhivker, G. M. (2018a). Computational modeling of the Hsp90 interactions with cochaperones and small-molecule inhibitors. Methods Mol. Biol. 1709, 253–273. doi: 10.1007/978-1-4939-7477-1_19
Verkhivker, G. M. (2018b). Dynamics-based community analysis and perturbation response scanning of allosteric interaction networks in the TRAP1 chaperone structures dissect molecular linkage between conformational asymmetry and sequential ATP hydrolysis. Biochim. Biophys. Acta Proteins Proteom. 1866, 899–912. doi: 10.1016/j.bbapap.2018.04.008
Verkhivker, G. M., Bouzida, D., Gehlhaar, D. K., Rejto, P. A., Freer, S. T., and Rose, P. W. (2002). Complexity and simplicity of ligand-macromolecule interactions: the energy landscape perspective. Curr. Opin. Struct. Biol. 12, 197–203. doi: 10.1016/S0959-440X(02)00310-X
Verkhivker, G. M., Dixit, A., Morra, G., and Colombo, G. (2009). Structural and computational biology of the molecular chaperone Hsp90: from understanding molecular mechanisms to computer-based inhibitor design. Curr. Top. Med. Chem. 9, 1369–1385. doi: 10.2174/156802609789895700
Vijayabaskar, M. S., and Vishveshwara, S. (2010). Interaction energy based protein structure networks. Biophys. J. 99, 3704–3715. doi: 10.1016/j.bpj.2010.08.079
Wang, F., Shen, L., Zhou, H., Wang, S., Wang, X., and Tao, P. (2019). Machine learning classification model for functional binding modes of TEM-1 beta-lactamase. Front. Mol. Biosci. 6:47. doi: 10.3389/fmolb.2019.00047
Williamson, M. P., and Kitahara, R. (2019). Characterization of low-lying excited states of proteins by high-pressure NMR. Biochim. Biophys. Acta Proteins Proteom. 1867, 350–358. doi: 10.1016/j.bbapap.2018.10.014
Wodak, S. J., Paci, E., Dokholyan, N. V., Berezovsky, I. N., Horovitz, A., Li, J., et al. (2019). Allostery in its many disguises: from theory to applications. Structure 27, 566–578. doi: 10.1016/j.str.2019.01.003
Wrabl, J. O., Gu, J., Liu, T., Schrank, T. P., Whitten, S. T., and Hilser, V. J. (2011). The role of protein conformational fluctuations in allostery, function, and evolution. Biophys. Chem. 159, 129–141. doi: 10.1016/j.bpc.2011.05.020
Wu, H., Paul, F., Wehmeyer, C., and Noe, F. (2016). Multiensemble markov models of molecular thermodynamics and kinetics. Proc. Natl. Acad. Sci. U.S.A. 113, E3221–E3230. doi: 10.1073/pnas.1525092113
Wu, J., Yilmaz, E., Zhang, M., Li, H., and Tan, K. C. (2020). Deep spiking neural networks for large vocabulary automatic speech recognition. Front. Neurosci. 14:199. doi: 10.3389/fnins.2020.00199
Wylie, A. A., Schoepfer, J., Jahnke, W., Cowan-Jacob, S. W., Loo, A., Furet, P., et al. (2017). The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature 543, 733–737. doi: 10.1038/nature21702
Xia, J., Deng, N. J., and Levy, R. M. (2013). NMR relaxation in proteins with fast internal motions and slow conformational exchange: model-free framework and markov state simulations. J. Phys. Chem. B 117, 6625–6634. doi: 10.1021/jp400797y
Yang, S., Banavali, N. K., and Roux, B. (2009). Mapping the conformational transition in Src activation by cumulating the information from multiple molecular dynamics trajectories. Proc. Natl. Acad. Sci. U.S.A. 106, 3776–3781. doi: 10.1073/pnas.0808261106
Yang, S., and Roux, B. (2008). Src kinase conformational activation: thermodynamics, pathways, and mechanisms. PLoS Comput. Biol. 4:e1000047. doi: 10.1371/journal.pcbi.1000047
Yang, Y. I., Niu, H., and Parrinello, M. (2018). Combining metadynamics and integrated tempering sampling. J. Phys. Chem. Lett. 9, 6426–6430. doi: 10.1021/acs.jpclett.8b03005
Yu, L., Zhang, W., Wang, J., and Yu, Y. (2017). SeqGAN: sequence generative adversarial nets with policy gradient. arXiv [Preprint]. arXiv:1609.05473.
Yuwen, T., Sekhar, A., and Kay, L. E. (2017). Separating dipolar and chemical exchange magnetization transfer processes in (1) H-CEST. Angew. Chem. Int. Ed. Engl. 56, 6122–6125. doi: 10.1002/anie.201610759
Zaiter, S. S., Huo, Y., Tiew, F. Y., Gestwicki, J. E., and McAlpine, S. R. (2019). Designing de novo small molecules that control heat shock protein 70 (Hsp70) and heat shock organizing protein (HOP) within the chaperone protein-folding machinery. J. Med. Chem. 62, 742–761. doi: 10.1021/acs.jmedchem.8b01436
Zhang, J., Adrian, F. J., Jahnke, W., Cowan-Jacob, S. W., Li, A. G., Iacob, R. E., et al. (2010). Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature 463, 501–506. doi: 10.1038/nature08675
Zhang, X., Gureasko, J., Shen, K., Cole, P. A., and Kuriyan, J. (2006). An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 125, 1137–1149. doi: 10.1016/j.cell.2006.05.013
Zhong, G., Gao, W., Liu, Y., Yang, Y., Wang, D. H., and Huang, K. (2020). Generative adversarial networks with decoder-encoder output noises. Neural Netw. 127, 19–28. doi: 10.1016/j.neunet.2020.04.005
Zhou, H., Dong, Z., and Tao, P. (2018a). Recognition of protein allosteric states and residues: machine learning approaches. J. Comput. Chem. 39, 1481–1490. doi: 10.1002/jcc.25218
Zhou, H., Dong, Z., Verkhivker, G., Zoltowski, B. D., and Tao, P. (2019a). Allosteric mechanism of the circadian protein vivid resolved through markov state model and machine learning analysis. PLoS Comput. Biol. 15:e1006801. doi: 10.1371/journal.pcbi.1006801
Zhou, H., and Tao, P. (2018). Dynamics sampling in transition pathway space. J. Chem. Theory Comput. 14, 14–29. doi: 10.1021/acs.jctc.7b00606
Zhou, H., and Tao, P. (2019). REDAN: Relative entropy-based dynamical allosteric network model. Mol. Phys. 117, 1334–1343. doi: 10.1080/00268976.2018.1543904
Zhou, H., Wang, F., Bennett, D. I. G., and Tao, P. (2019b). Directed kinetic transition network model. J. Chem. Phys. 151:144112. doi: 10.1063/1.5110896
Zhou, H., Wang, F., and Tao, P. (2018b). t-Distributed stochastic neighbor embedding method with the least information loss for macromolecular simulations. J. Chem. Theory Comput. 14, 5499–5510. doi: 10.1021/acs.jctc.8b00652
Zhou, H., Zoltowski, B. D., and Tao, P. (2017). Revealing hidden conformational space of LOV protein VIVID through rigid residue scan simulations. Sci. Rep. 7:46626. doi: 10.1038/srep46626
Zhu, J.-Y., Park, T., Isola, P., and Efros, A. A. (2017). “Unpaired image-to-image translation using cycle-consistent adversarial networks,” in 2017 IEEE International Conference on Computer Vision (ICCV) (Venice: IEEE), 2242–2251. doi: 10.1109/ICCV.2017.244
Zhuravlev, P. I., and Papoian, G. A. (2010). Protein functional landscapes, dynamics, allostery: a tortuous path towards a universal theoretical framework. Q Rev. Biophys. 43, 295–332. doi: 10.1017/S0033583510000119
Zimmerman, M. I., and Bowman, G. R. (2015). FAST conformational searches by balancing exploration/exploitation trade-offs. J. Chem. Theory Comput. 11, 5747–5757. doi: 10.1021/acs.jctc.5b00737
Keywords: allosteric regulation, multiscale modeling, Markov state models, network analysis, deep learning, reinforcement learning, drug discovery
Citation: Verkhivker GM, Agajanian S, Hu G and Tao P (2020) Allosteric Regulation at the Crossroads of New Technologies: Multiscale Modeling, Networks, and Machine Learning. Front. Mol. Biosci. 7:136. doi: 10.3389/fmolb.2020.00136
Received: 01 May 2020; Accepted: 08 June 2020;
Published: 09 July 2020.
Edited by:
Shozeb Haider, UCL School of Pharmacy, United KingdomReviewed by:
Sarath Chandra Dantu, Brunel University London, United KingdomCopyright © 2020 Verkhivker, Agajanian, Hu and Tao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gennady M. Verkhivker, dmVya2hpdmtAY2hhcG1hbi5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.