
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Mol. Biosci. , 07 July 2020
Sec. Biophysics
Volume 7 - 2020 | https://doi.org/10.3389/fmolb.2020.00131
This article is part of the Research Topic Mechanisms of Fluorescent Proteins View all 7 articles
 Breland G. Oscar1Liangdong Zhu1Hayati Wolfendeen2Nikita D. Rozanov3Alvin Chang3Kenneth T. Stout1,3
Breland G. Oscar1Liangdong Zhu1Hayati Wolfendeen2Nikita D. Rozanov3Alvin Chang3Kenneth T. Stout1,3 Jason W. Sandwisch1Joseph J. Porter2Ryan A. Mehl2
Jason W. Sandwisch1Joseph J. Porter2Ryan A. Mehl2 Chong Fang1*
Chong Fang1*Tracking the structural dynamics of fluorescent protein chromophores holds the key to unlocking the fluorescence mechanisms in real time and enabling rational design principles of these powerful and versatile bioimaging probes. By combining recent chemical biology and ultrafast spectroscopy advances, we prepared the superfolder green fluorescent protein (sfGFP) and its non-canonical amino acid (ncAA) derivatives with a single chlorine, bromine, and nitro substituent at the ortho site to the phenolate oxygen of the embedded chromophore, and characterized them using an integrated toolset of femtosecond transient absorption and tunable femtosecond stimulated Raman spectroscopy (FSRS), aided by quantum calculations of the vibrational normal modes. A dominant vibrational cooling time constant of ~4 and 11 ps is revealed in Cl-GFP and Br-GFP, respectively, facilitating a ~30 and 12% increase of the fluorescent quantum yield vs. the parent sfGFP. Similar time constants were also retrieved from the transient absorption spectra, substantiating the correlated electronic and vibrational motions on the intrinsic molecular timescales. Key carbon-halogen stretching motions coupled with phenolate ring motions of the deprotonated chromophores at ca. 908 and 890 cm−1 in Cl-GFP and Br-GFP exhibit enhanced activities in the electronic excited state and blue-shift during a distinct vibrational cooling process on the ps timescale. The retrieved structural dynamics change due to targeted site-specific halogenation of the chromophore thus provides an effective means to design new GFP derivatives and enrich the bioimaging probe toolset for life and medical sciences.
Since its discovery several decades ago, green fluorescent protein (GFP) has been widely used for biolabeling and bioimaging due to its characteristic bright green emission, high fluorescence quantum yield (FQY), and stability (Shimomura et al., 1962; Chalfie et al., 1994; Tsien, 1998; Patterson and Lippincott-Schwartz, 2002; Zimmer, 2002; Betzig et al., 2006; Fang et al., 2009; Jung, 2012b; Dedecker et al., 2013). GFP is amenable to structural alterations like circular permutation, leading to the development of biosensors such as the GFP-calmodulin chimera, wherein the fluorescence response is modulated by varying concentrations of free calcium ion (Ca2+) (Baird et al., 1999; Zhao et al., 2011; Oscar et al., 2014). Due to the protein utility across broad science and engineering fields, spectroscopic studies have been performed on wild-type (wt)GFP and its derivatives to elucidate the underlying fluorescence mechanisms and predict how further structural changes may alter the optical response including unwanted events such as blinking and photobleaching. On the molecular level, excitation of neutral (A, 395 nm), or anionic (B, 475 nm) absorption bands of the wtGFP autocyclized Ser65-Tyr66-Gly67 (SYG) chromophore results in green fluorescence. After photoexcitation, the neutral chromophore A* undergoes a picosecond (ps) excited-state proton transfer (ESPT) reaction to reach a deprotonated intermediate state (I*) within an unrelaxed protein environment preceding green emission (Chattoraj et al., 1996; Lossau et al., 1996; Brejc et al., 1997; Fang et al., 2009).
The optical properties of GFP can be tuned by modifying either the surrounding protein pocket in the β-barrel or the three-residue chromophore (Table S1). For example, point mutation Thr203Tyr of the enhanced yellow fluorescent protein, EYFP, leads to red-shifted absorption and emission due to a π-π interaction between spatially close tyrosine rings (Ormö et al., 1996; Wachter et al., 1998). Since the Tyr sidechain is not mechanistically required for chromophore formation, Tyr66 can be replaced: mutation to His or Trp eliminates the ESPT pathway and shifts the absorption and emission bands to generate blue and cyan fluorescent proteins, respectively (Wachter et al., 1997; Kummer et al., 2002; Ai et al., 2007; Tomosugi et al., 2009). The red fluorescent proteins are typically formed by an extended conjugation along the chromophore N-acylimine carbonyl (Gross et al., 2000; Shaner et al., 2004; Piatkevich et al., 2010; Subach and Verkhusha, 2012). Because these strategies in tuning GFP spectral properties conventionally involve only 20 standard amino acids, they pose certain limitations in achieving desired properties. Notably, the site-specific modification of proteins with non-canonical amino acids (ncAAs) provides an appealing way to engineer spectral properties and encode new functionalities (Link et al., 2003; Wang et al., 2003; Peeler and Mehl, 2012). The GFP chromophore with a p-azido-L-phenylalanine mutation, for example, exhibits photoactivatable behavior originating from phenyl azide photolysis in the unnatural chromophore (Reddington et al., 2013). The ncAAs can further act as site-specific vibrational probes or spin labels, making them ideal for structural dynamics techniques such as electron paramagnetic resonance (EPR) spectroscopy, NMR, and time-resolved vibrational spectroscopy (Fleissner et al., 2009; Sripakdeevong et al., 2014; Hall et al., 2019).
In this work, we characterized a series of superfolder GFP (sfGFP) (Pédelacq et al., 2006) mutants that contain a single ncAA point mutation at the chromophore tyrosine residue and compared their attributes to model chromophores using both ultrafast electronic and vibrational spectroscopic signatures (Fang et al., 2018). The halogenated derivatives of sfGFP, 3-chlorotyrosine (Cl-GFP) and 3-bromotyrosine (Br-GFP), contain an electron-withdrawing substituent at the ortho site to the phenolic hydroxyl which introduces steric effects in the protein pocket and increases the polarizability of the aromatic bonds over the chromophore ring system. Meanwhile, the 3-nitrotyrosine (nitro-GFP) mutant contains a strong electron-withdrawing group capable of forming additional hydrogen bonds in addition to an ~30 Å3 increase in residue volume (De Filippis et al., 2006). Spectral properties such as absorption, emission, and excited-state dynamics are characterized by steady-state and time-resolved electronic spectroscopy; in addition, the chromophore structure and local environment can be revealed by femtosecond stimulated Raman spectroscopy (FSRS) (Dietze and Mathies, 2016; Fang et al., 2019). This integrated experimental platform resolving the coupled electronic and atomic motions in highly fluorescent systems allows us to better understand the effect of a ncAA mutation at the active site, which elucidates the conformational preference of a chromophore inside the protein matrix and the underlying photophysics/photochemistry of fluorescent proteins in the electronic excited states.
The incorporation of a nitrotyrosine (Cooley et al., 2014; Rauch et al., 2016) or halotyrosine (Jang et al., 2020) at a selected position of sfGFP was performed as previously described. Briefly, the codon codifying each of the tyrosine residues in the sfGFP sequence optimized for bacterial expression was replaced by an amber stop codon (TAG), which was recognized by the orthogonal nitro or halotyrosine-bearing suppressor tRNA and engineered tRNA synthetase. The modified proteins were then expressed, purified, and confirmed by mass spectrometry (Jang et al., 2020). The protein concentrations for our ultrafast spectroscopic characterization were 10 mg/mL at pH = 8.1 (10 mM Tris, 50 mM Na2HPO4, 100 mM NaCl) and 5.5 (100 mM citric acid, 200 mM Na2HPO4, 100 mM NaCl). As control samples, the monohalogenated 4-hydroxybenzylidene-1,2-dimethylimidazolinone (HBDI) chromophores were synthesized according to literature by combining an iminoglycine methyl ester with a Schiff base (Baldridge et al., 2010), as detailed in the Supplementary Text.
The UV/Visible and emission spectra of all proteins and small molecules were collected on a ThermoScientific Evolution 201 and Hitachi F-2500 fluorescence spectrophotometer, respectively. Quantum yield was measured relative to fluorescein in 0.1 M NaOH according to the reported method (Patterson et al., 1997). The tunable picosecond (ps) Raman pump, white light probe, and femtosecond (fs) actinic pump (see Supplementary Text) (Zhu et al., 2014; Liu et al., 2016) enable the acquisition of time-resolved FSRS data in the electronic excited state with simultaneously high spectral and time resolutions (Dietze and Mathies, 2016; Fang et al., 2018). Transient absorption (TA) spectra were collected before each experiment with the Raman pump blocked so that only the fs pulses interact with the sample. The UV/Visible spectra were recorded before and after time-resolved experiments to check sample integrity (<5% change commonly detected). A full description of the methods can be found in our earlier reports on fluorescent proteins (Tang et al., 2016, 2018a; Fang et al., 2018).
The absorption and emission spectra of sfGFP, Cl-GFP, Br-GFP, and nitro-GFP in pH = 5.5 buffer solution (Figure 1) are tabulated in Table S2. The pKa of sfGFP is ~6 so a distinct neutral chromophore population (λmax = 395 nm) is observed (Figure 1A), whereas in more alkaline conditions the deprotonated chromophore population (λmax = 488 nm) dominates (Figure S1A). We observed a lowered pKa of ~4.5 upon tyrosine halogenation inside sfGFP, so the neutral chromophore absorption around 400 nm is largely absent at pH = 5.5 while the anionic chromophore absorption bands red-shift (Figures S1B,C). These results are corroborated by the pKa of 3Cl-Tyr EGFP at ~4.7 and red-shifted spectral peaks vs. EGFP (Ayyadurai et al., 2012; Zhang et al., 2012). The emission profiles of Cl-GFP and Br-GFP are also red-shifted vs. sfGFP, and the shift magnitude increases with the mass of the halogen substituent (Figures 1A–C), the trend matching 3-iodotyrosine-GFP with a red-shifted emission beyond 520 nm (Young et al., 2011). Notably, changes in absorption and emission for chromophores in a protein matrix are more pronounced than those in small molecules. The absorption bands of the sfGFP model chromophore, neutral, and anionic p-HBDI with capped methyl groups, are found at 370 and 425 nm, respectively (Vengris et al., 2004; Taylor et al., 2019). The corresponding Cl (Br)-HBDI bands appear at 369 (368) nm and 424 (425) nm in pH = 3 and 7.6 aqueous solutions, respectively.
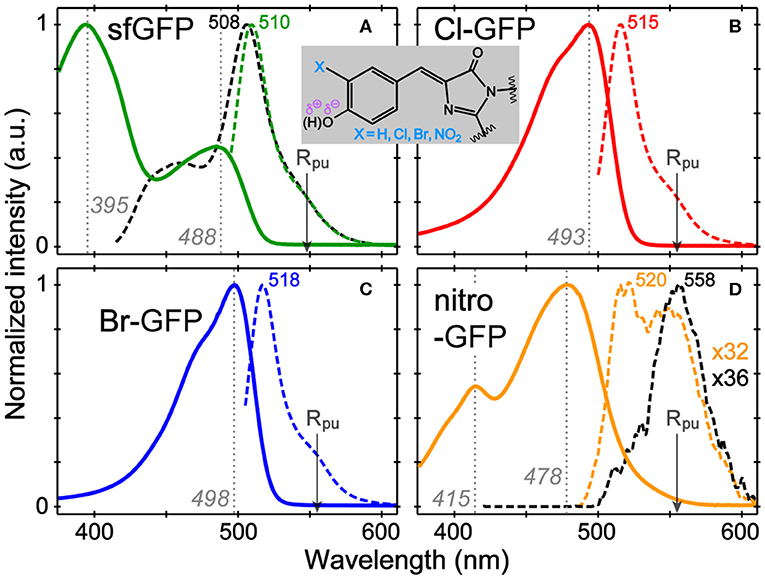
Figure 1. Normalized absorption (solid) and emission (dashed lines) spectra of (A) sfGFP, (B) Cl-GFP, (C) Br-GFP, and (D) nitro-GFP in pH = 5.5 aqueous buffer solution. The excitation wavelengths are marked by gray dotted lines. For sfGFP and nitro-GFP, emission after the bluer excitation is shown as black dashed lines. The nitro-GFP emission normalization factor is denoted to manifest weak emission. The Raman pump is indicated by the arrow. The chromophore chemical structure is shown in the inset.
In contrast, the nitrated GFP chromophore results in a non-fluorescent protein, which is not surprising given the photochemistry of nitrated aromatics (see Supplementary Text for the mechanism; De Filippis et al., 2006; Tang and Fang, 2019). The absorption spectrum shows two bands at 415 and 478 nm that exhibit a pH-dependent ratio change with the 415 nm peak becoming stronger under acidic conditions (Figure S1D). The 415 nm band likely corresponds to neutral chromophore in nitro-GFP and is red-shifted from the 395 nm absorption band in sfGFP, which could be due to an intramolecularly H-bonded form in nitro-GFP. Excitation of either absorption band produces negligible fluorescence (Φ < 0.0005, see Table S2) and the detectable emission is significantly red-shifted to ~550 nm. This behavior is reminiscent of free 3-nitrotyrosine in solution, which shows the pH-dependent changes of its visible absorption bands, a pKa near neutral pH, and an FQY below 0.0001 (Tang and Fang, 2019), leading to the effective use of 3-nitrotyrosine as a FRET quencher in peptides and proteins when a nearby fluorescent residue (e.g., Tyr, Trp) acts as the donor (Duus et al., 1998; De Filippis et al., 2006).
To verify the chromophore's ionization state and uncover local interactions within the protein pocket, we implemented the wavelength-tunable FSRS technique at different resonance conditions (Liu et al., 2016; Fang et al., 2018), wherein a narrowband Raman pump and broadband Raman probe induce the stimulated Raman scattering signal with desirable enhancement to achieve high signal-to-noise ratio. The ncAA chromophores exhibit unique spectral signatures when compared to sfGFP; for example, the halogenated chromophores contain highly polarizable groups that affect Raman peak frequencies, and the nitrated chromophore consists of the spectrally isolated –NO2 vibrational modes that act as sensitive probes for the local environment (De Filippis et al., 2006; Ayyadurai et al., 2012).
In the ground-state FSRS of the protein series in pH = 5.5 buffer (Figure 2), the Raman pump is energetically close to the absorption band of the anionic chromophore (Figure 1). In sfGFP, most of the Thr-Tyr-Gly (TYG) chromophore population is neutral, but the pre-resonance condition favors the anionic subpopulation (see Figure S1A) and amplifies its Raman features. The protonation state is confirmed by the 1,547 cm−1 marker band, attributed to the C=N, C=C, and C=O stretching motions in the anionic chromophore (see Table S3 for vibrational normal mode assignments) based on literature and our calculations (Bell et al., 2000; Schellenberg et al., 2001; Tozzini and Nifosı̀, 2001). For proteins with primarily neutral chromophores, this marker band shifts to ~1,566 cm−1, also observed in wtGFP (Fang et al., 2009), a series of GFP-based Ca2+ biosensors (Oscar et al., 2014; Tang et al., 2016), and in sfGFP at an off-resonance condition (Figure S2A). The ncAA-mutant proteins (Figures 2A–C, also see Figure S2B for the off-resonance FSRS data of Br-GFP) all exhibit strong peaks near 1,542 cm−1, corroborating the anionic chromophore as determined by the electronic absorption spectra (Figure 1). The ~1,576 cm−1 shoulder peak in mutant proteins is assigned to additional phenolate ring C=O and C=C stretch contributions in the anionic chromophore (Tables S4, S5), but this mode may indicate an H-bonded population of halogenated chromophores while the H-bond partner could be an adjacent water or protein residue in forming the O–H···X (X = Cl, Br) bond (Pal et al., 2005). In the anionic chromophores outside the protein matrix, strong C=C and C=O stretching modes appear at 1,560 cm−1 (Cl-HBDI) and 1,558 cm−1 (Br-HBDI) in Figure S3, slightly blue-shifted from the reported 1,556 cm−1 mode of HBDI in basic solution (Bell et al., 2000; Schellenberg et al., 2001; Taylor et al., 2019).
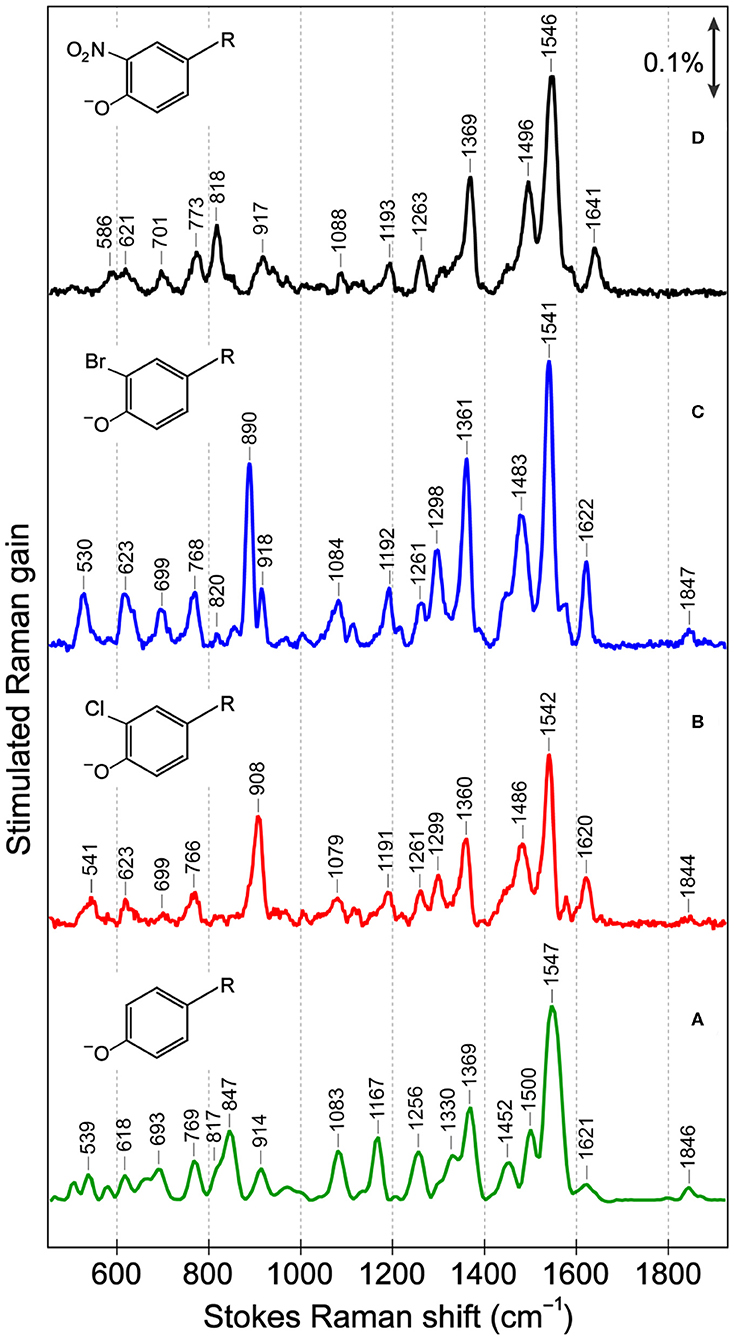
Figure 2. Ground-state Stokes FSRS data of (A) sfGFP, (B) Cl-GFP, (C) Br-GFP, and (D) nitro-GFP in pH = 5.5 aqueous buffer solution. In (B–D) the Raman pump was set at 555 nm while in (A) the Raman pump wavelength was 548 nm. The stimulated Raman gain is indicated by the double-headed arrow. The anionic chromophore chemical structures with various substitutions are depicted in the inset. R represents the remaining conjugated framework of the chromophore that connects to protein backbone.
The cluster of modes between ~1,200 and 1,400 cm−1 also probe the chromophore's protonation state: the 1,256 cm−1 mode in sfGFP involves the phenolate ring H-rock and CO stretch, which typically exhibits a frequency blueshift in the deprotonated state (Bell et al., 2000; Fang et al., 2009; Oscar et al., 2014). This mode blue-shifts to 1,261 cm−1 in Cl-GFP and Br-GFP, consistent with the incorporation of an electron-withdrawing group adjacent to the phenolate oxygen site and the increased acidity as well as photoacidity of the chromophore (Chen et al., 2019). In contrast, for the phenolate ring H-rocking and imidazolinone ring C–N stretching mode at 1,369 cm−1 (Table S3) that was also observed for the TYG chromophore inside a protein Ca2+-biosensor (Tang et al., 2016), due to steric hindrance this mode red-shifts to ~1,360 cm−1 upon chromophore halogenation (Figure 2). Moreover, the 1,167 cm−1 phenolate ring H-scissoring motion exhibits a notable blueshift to ~1,192 cm−1 in Cl-GFP and Br-GFP since the pertinent normal modes of the halogenated chromophores involve less imidazolinone ring contributions (Tables S3–S5). These in-plane vibrational motions thus serve as sensitive probes to elucidate the effect of ortho-halogenation of the largely planar chromophore inside a protein pocket.
Notably, the chromophore autocyclization during protein maturation is primarily a function of the protein backbone. A majority of GFP chromophores are observed in the Z (cis) stereoisomer, but the halogen substituent on tyrosine can occupy two distinct atropisomeric positions with the probability of each determined by the properties of the substituent itself as well as the local environment supplied by the protein interior (Bae et al., 2004; Pal et al., 2005; Jung, 2012a; Chang et al., 2019). For example, the crystal structure and electron density mapping of 3-fluorotyrosyl-EGFP revealed two conformations of the TYG chromophore with a major conformer wherein fluorine interacts with Thr203, equivalent to Configuration 1 in Figure 3 (Bae et al., 2004). Small-molecule analogs of the chromophore were reported with this conformation as well as the Trp-containing chromophores and the 3,4-dihydroxy-L-phenylalanine GFP chromophore (Hyun Bae et al., 2003; Hasegawa et al., 2007; Ayyadurai et al., 2011). However, 3-chlorotyrosine chromophores in the short H-bond (His148Asp) GFP system exhibit only one crystallographic occupancy corresponding to Configuration 2 in Figure 3 partly due to specific electrostatic interactions introduced by the nearby His148Asp mutation (Oltrogge and Boxer, 2015). Can FSRS provide evidence for the protein chromophore configuration?
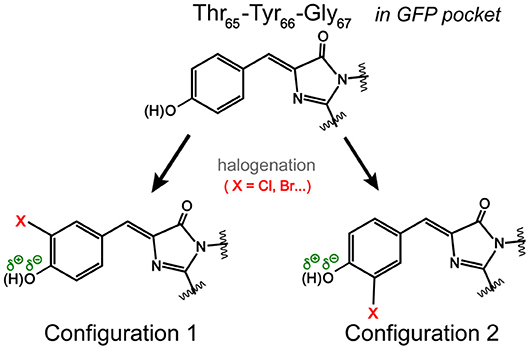
Figure 3. Possible orientations of the halogen atom (X) on the GFP chromophore. In Configuration 1, the halogen atom is oriented toward Thr203 in a typical GFP protein matrix. In Configuration 2, the halogen atom could face Ser205 (Pal et al., 2005). The dissociable hydroxyl group is highlighted.
Several low-frequency motions below 1,000 cm−1 are resolved for sfGFP and the mutated series (Figure 2), and these modes are sensitive to the chromophore conformation. The C–Cl stretch (550–800 cm−1), C–Br stretch (500–700 cm−1), and NO2 bend (~820 cm−1) are all expected to occur in this region (Kovács et al., 1998; Chen et al., 2020). Mode assignment of the anionic TYG chromophore and the halogenated derivatives in both conformations (Figure 3) was carried out by comparing the experimental Raman spectra to literature values and density functional theory (DFT) RB3LYP-level calculations with the 6-311G+(d, p) basis set performed in vacuo and in water. Strong vibrational modes at 908 cm−1 in Cl-GFP and 890 cm−1 in Br-GFP are assigned to phenolate ring breathing with a prominent C–X stretching component. The ring breathing mode has been previously assigned at ~820 cm−1 (Oscar et al., 2014) that is also listed in Table S3, but the C–X stretching contribution blue-shifts the calculated normal mode frequency (see Tables S4, S5). Similarly, the benzene ring breathing mode at 992 cm−1 was experimentally recorded at 1,001 cm−1 in chlorobenzene and 998 cm−1 in bromobenzene, so this mode is a useful reporter on halogen substitution (Meneely et al., 1971).
Resonance Raman spectra with a Raman pump close to the red edge of electronic absorption bands (Figure 1, Figure S1) were used to further assign the low-frequency vibrational bands (Figures 4A,B, Figure S4), supported by the anti-Stokes FSRS with a 580 nm Raman pump (Figure S5) (Tang et al., 2018b). With identical protein concentration and Raman pump power, the signal strength is increased by an order of magnitude with 507 nm pump. Resonance Raman peaks include excited-state contributions (Quick et al., 2015), which can be seen by strong agreement between the resonance Raman spectra and the excited-state spectra at 50 fs following 480 nm photoexcitation. Since halogen atoms are expected to increase chromophore polarizability, the modes strongly influenced by halogenation are enhanced (see Supplementary Text). The lineshape of high-frequency modes is largely preserved in all proteins, though the peaks in S1 are generally broader than those in S0 due to the shorter lifetime of the excited-state species, while the frequency shift is due to an interplay between anharmonicity and the intrinsic frequency change from S0→S1 due to electron redistribution (Chen et al., 2018; Fang et al., 2018).
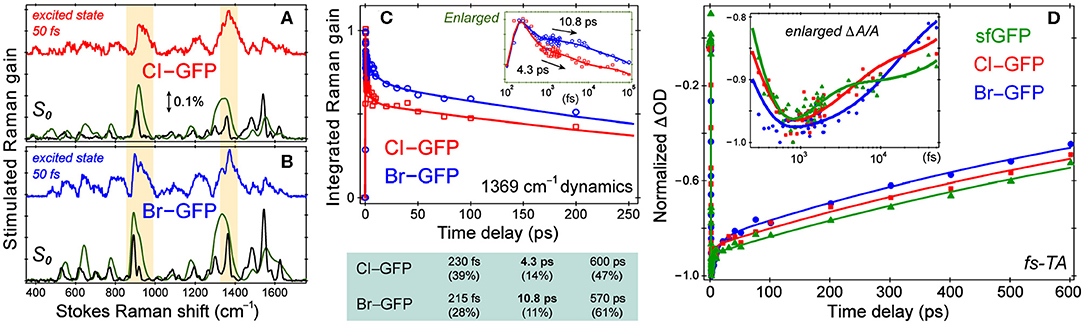
Figure 4. Spectroscopic characterization of ncAA-GFPs. Ground and excited-state Stokes FSRS of (A) Cl-GFP and (B) Br-GFP. The spectra with the 555 and 507 nm Raman pump are depicted in black and green (×0.05), respectively. The excited-state Raman spectrum at 50 fs after 480 nm photoexcitation is shown in red (Cl-GFP) and blue (Br-GFP). The stimulated Raman gain of 0.1% is indicated by the double-headed arrow. (C) Normalized Raman intensity dynamics of the 1,369 cm−1 band of Cl-GFP (red) and Br-GFP (blue) with the least-squares fit in solid lines. Early-time dynamics are highlighted in the inset on a semilogarithmic scale. The triexponential-fit components are listed below with the amplitude weight percentages of decay in parenthesis. (D) Fs-TA dynamics of the SE band (550−570 nm) of sfGFP (green), Cl-GFP (red), and Br-GFP (blue) in pH = 5.5 aqueous buffer solution following 480 nm excitation. The data are normalized at the maximal SE peak magnitude point for comparison. The least-squares fit for each data trace is shown as the color-coded solid curve. The inset shows the early-time dynamics on a semilogarithmic scale to highlight the multiple timescales involved.
Based on the ground-state FSRS and calculations, we tentatively assign Br-GFP to Configuration 1 in Figure 3 such that bromine interacts with the nearby Thr203 residue. Though DFT calculations of the model chromophore in vacuo cannot capture the myriad of interactions between the protein pocket and the chromophore, there is better agreement between the experimental and calculated vibrational modes of Configuration 1 of Br-GFP, especially in the low-frequency region (Merrick et al., 2007; Wang et al., 2015). In particular, the observed 890 and 918 cm−1 modes (Figure 2C) both have significant Br contributions and the calculated frequencies of 852 and 881 cm−1 in Configuration 1 (Table S5) better match the experimental energy gap of 28 cm−1 between the modes, instead of the exact mode frequencies that are highly subjective to the frequency scaling factor (Merrick et al., 2007). A crystallographic analysis is required to confirm if a minor population exists; however, we expect the bromine to experience more repulsive interactions with the sidechains of Ser205 and Glu222 in Configuration 2 based on the sfGFP crystal structure (PDB ID: 2B3P) (Pédelacq et al., 2006). Our preliminary molecular dynamics simulations based on free energy perturbation methods (Seeliger and de Groot, 2010) with a calculated chromophore force field (Malde et al., 2011), however, seem to suggest that while both configurations are stable in the protein pocket, Configuration 2 of Cl-GFP is thermodynamically more favorable. Further investigation is thus needed to better determine which configuration is dominant surrounded by dynamic protein residues in the system of interest.
While the small molecule analog HBDI undergoes a non-radiative cis-to-trans isomerization after photoexcitation (Mandal et al., 2004; Taylor et al., 2019), confinement in the protein pocket typically inhibits this pathway in favor of other energy dissipation routes. To rule out photoisomerization in the excited state and explore the photodynamics affected by halogenation, we implemented time-resolved electronic and vibrational spectroscopies (Liu et al., 2016; Fang et al., 2019). Using an fs photoexcitation pulse at 480 nm and a white light probe, TA spectra were collected to reveal dynamics in the first singlet excited state. The sfGFP, Cl-GFP, and Br-GFP all have a broad stimulated emission (SE) feature below 600 nm (in correlation with the fluorescence band in Figure 1, Figure S1) and a weak excited-state absorption band beyond 600 nm (Tang et al., 2015). We focus on TA dynamics by plotting the red-edge integrated signal of SE band from 550–570 nm, which rapidly reaches the maximum magnitude before a biexponential decay (see Figure 4D, Table S6). For sfGFP in pH = 5.5 buffer, only a small portion of the chromophore population is excited at 480 nm, and a ~1.2 ps component accounts for 20% of the SE dynamics while a longer 1.2 nanosecond (ns) component is dominant. A similar time constant of 1.9 ps for the initial rise of fluorescence signal was reported in the deprotonated chromophore of GFP after 478 nm excitation (Chattoraj et al., 1996), likely arising from an ultrafast process (e.g., intramolecular vibrational relaxation; Felker and Zewail, 1985) that populates the fluorescent state of the deprotonated species. The long time component is less accurately determined due to the 600 ps detection window (see Section 2.2), but generally approaches the fluorescence lifetime (~3 ns) of the photoexcited deprotonated chromophore (Chattoraj et al., 1996; Striker et al., 1999; Zimmer, 2002; Tang et al., 2018c).
Notably, the first recovery component of the SE band of halogenated proteins is significantly longer than the parent protein (see Figure 4D inset and Table S6), but the ns process is largely unchanged. The 4.1 ps component in Cl-GFP lengthens to 12.4 ps in Br-GFP, and these time constants are attributable to the excited state (S1) relaxation dynamics other than fluorescence (vide infra), especially with the excess energy provided by the 480 nm pump. The excellent match between these TA time constants and the aforementioned Raman mode intensity decay time constants (Figure 4C, lower panel) supports a unified picture for energy relaxation on molecular timescales of a photoexcited deprotonated chromophore inside the protein pocket (Tang et al., 2018c; Fang et al., 2019). Previous photoelectron spectroscopic results of the UV-irradiated anionic HBDI chromophore (Mooney et al., 2013) and that with chemical modifications (e.g., difluoro-substituents) (Bochenkova et al., 2017) corroborate the changes in the excited-state energy surfaces and variation of the 1.4 ps lifetime (in gas phase and solution) following an initial ~330 fs component. Recently with time-resolved action spectroscopy, the HBDI anion after 480 nm pump shows ca. 1–11 ps lifetimes at 300 K (Svendsen et al., 2017).
To verify that the observed dynamics arise from a vibrational progression, we performed the time-resolved FSRS experiments in S1 to directly track atomic motions (Fang et al., 2018). In previous FSRS reports on GFP derivatives, the modes with strong intensities are typically the phenol C–H bending motions, phenolic CO stretch, and the imidazolinone C=N stretch with frequencies at ~1,180, 1,265, and 1,565 cm−1 in S1 (Fang et al., 2009; Oscar et al., 2014; Tang et al., 2015, 2016). These high-frequency marker bands are also prominent in the pre-resonance ground-state Raman spectrum of sfGFP (Figure S4). Interestingly, halogenation changes this pattern by exhibiting several enhanced low-frequency modes. In the excited state, modes with major C–X contributions exhibit the strongest intensities, and the mode frequencies are blue-shifted from those in S0 (Figures 4A,B). Following 480 nm photoexcitation, the S1 vibrational modes of Cl-GFP and Br-GFP decay in time without the appearance of new peaks, while only small mode frequency blueshift occurs (Figure S6). The ~1,369 cm−1 mode is present in S0 and S1 spectra of halogenated proteins and could be the vibronically coupled mode based on the energy gap observed in the electronic spectra (see Supplementary Text and Figure 1). This marker band dynamics are fit with a triexponential function (Figure 4C), largely matching the fs-TA dynamics (Figure 4D). Notably, Cl-GFP exhibits a ~4 ps decay time constant that is much shorter than the ~11 ps counterpart of Br-GFP, in correlation with the noticeable mode frequency blueshift in Figure S6 that indicates vibrational cooling in S1 (Fang et al., 2019).
Interestingly, the amplitude weights of the initial ps components (i.e., 20, 37, and 27% in Table S6) correlate with the FQYs (i.e., 0.68, 0.88, and 0.76 in Table S2) of sfGFP, Cl-GFP, and Br-GFP, respectively. We surmise that the 4–12 ps components in Cl-GFP and Br-GFP (longer than 1.2 ps in sfGFP) involve certain nuclear motions associated with the phenolate ring as its size/weight increases, which allow effective vibrational cooling that promotes radiative emission from the lower portion of the potential energy surface of the deprotonated protein chromophore (Fang et al., 2018; Tang et al., 2018c). This mechanism is corroborated by a recent report on the introduction of asymmetric electronic structures and vibronic features to fluorophores, which can facilitate strong internal conversion with redder emission (Ren et al., 2018). Notably, photoisomerization typically leads to characteristic Raman mode frequency redshift due to the chromophore conformational change (Fang et al., 2019), which was not observed here (Figure S6). Moreover, the ring-twisting-induced non-radiative transition contradicts the high FQYs of halogenated sfGFP (Table S2), whereas the essentially non-fluorescent nitro-GFP likely involves an ultrafast nitroaromatic twisting motion leading to an S1/S0 conical intersection (Tang and Fang, 2019). One challenge that needs to be tackled before future FSRS experiments on nitro-GFP is the low signal-to-noise ratio without a prominent SE band (see Supplementary Text for details) like that in the halogenated sfGFP achieving resonance Raman enhancement in S1 (Figure S6).
In summary, we prepared and characterized a series of superfolder GFP mutants with ncAA chromophores using a combination of fs-TA spectroscopy, wavelength-tunable ground and excited-state FSRS (with ncAA chromophores in solution as control samples), and DFT calculations of normal mode frequencies. In particular, the single-site halogenated proteins display improved properties that include the red-shifted absorption and emission, increased concentration of deprotonated emissive species, and an increased fluorescence quantum yield. Such desirable application properties of the halogenated GFP mutants stem from a solid biophysical chemistry foundation in that they are a direct consequence of the engineerable molecular structure and dynamics of the photosensitive unit inside a protein matrix. The nitro-GFP provides a useful contrasting sample that will be further investigated.
We focused on the structural aspects of single-site halogenation at the protein active site to examine key conformational preference and elucidate the excited-state energy dissipation pathways in Cl-GFP and Br-GFP. Such a targeted analysis using a well-known series of electron-withdrawing groups with sufficient temporal and spectral resolution paints a more complete picture of chemically modified chromophores reacting to the incoming photons, thus enabling future rational design of functional molecular machines (Fang et al., 2019). The strong vibronic coupling that influences the SE dynamics may provide a useful direction to engineer probes for SE-depletion spectroscopy and imaging (Hell, 2009; Silva et al., 2016). Furthermore, these brighter protein mutants show that ncAA incorporation within the chromophore is a versatile and effective way to engineer photochemistry and protein functionality.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
RM and CF conceived and designed the research and acquired funding. BO and LZ performed spectroscopic experiments. BO, NR, AC, and JS performed data curation including calculations. HW, JP, and KS contributed new protein and chromophore samples. LZ and CF contributed advanced non-linear spectroscopic tools. BO and CF wrote the manuscript. All the authors have edited the final manuscript and approved it for publication.
CF was supported by the US National Science Foundation through a Career Grant (CHE-1455353). We thank the Betty Wang Discovery Fund (2019 to CF) at the OSU College of Science for the acquisition of a new Shimadzu RF-6000 spectrofluorophotometer. RM was supported by a National Institutes of Health Grant (RGM114653A) and National Science Foundation Grant (MCB-1518265).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Dr. Longteng Tang, Cheng Chen, Taylor Krueger, Sean Tachibana, and Sean Boulanger for helpful discussions.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2020.00131/full#supplementary-material
Ai, H. W., Shaner, N. C., Cheng, Z., Tsien, R. Y., and Campbell, R. E. (2007). Exploration of new chromophore structures leads to the identification of improved blue fluorescent proteins. Biochemistry 46, 5904–5910. doi: 10.1021/bi700199g
Ayyadurai, N., Deepankumar, K., Prabhu, N. S., Budisa, N., and Yun, H. (2012). Evaluation and biosynthetic incorporation of chlorotyrosine into recombinant proteins. Biotechnol. Bioproc. Eng. 17, 679–686. doi: 10.1007/s12257-012-0066-6
Ayyadurai, N., Saravanan Prabhu, N., Deepankumar, K., Lee, S.-G., Jeong, H.-H., Lee, C.-S., et al. (2011). Development of a selective, sensitive, and reversible biosensor by the genetic incorporation of a metal-binding site into green fluorescent protein. Angew. Chem. Int. Ed. 50, 6534–6537. doi: 10.1002/anie.201008289
Bae, J. H., Paramita Pal, P., Moroder, L., Huber, R., and Budisa, N. (2004). Crystallographic evidence for isomeric chromophores in 3-fluorotyrosyl-green fluorescent protein. ChemBioChem 5, 720–722. doi: 10.1002/cbic.200300818
Baird, G. S., Zacharias, D. A., and Tsien, R. Y. (1999). Circular permutation and receptor insertion within green fluorescent proteins. Proc. Natl. Acad. Sci. U. S. A. 96, 11241–11246. doi: 10.1073/pnas.96.20.11241
Baldridge, A., Kowalik, J., and Tolbert, L. M. (2010). Efficient synthesis of new 4-arylideneimidazolin-5-ones related to the GFP chromophore by 2+3 cyclocondensation of arylideneimines with imidate ylides. Synthesis 2010, 2424–2436. doi: 10.1055/s-0029-1218796
Bell, A. F., He, X., Wachter, R. M., and Tonge, P. J. (2000). Probing the ground state structure of the green fluorescent protein chromophore using Raman spectroscopy. Biochemistry 39, 4423–4431. doi: 10.1021/bi992675o
Betzig, E., Patterson, G. H., Sougrat, R., Lindwasser, O. W., Olenych, S., Bonifacino, J. S., et al. (2006). Imaging intracellular fluorescent proteins at nanometer resolution. Science 313, 1642–1645. doi: 10.1126/science.1127344
Bochenkova, A. V., Mooney, C. R. S., Parkes, M. A., Woodhouse, J. L., Zhang, L., Lewin, R., et al. (2017). Mechanism of resonant electron emission from the deprotonated GFP chromophore and its biomimetics. Chem. Sci. 8, 3154–3163. doi: 10.1039/C6SC05529J
Brejc, K., Sixma, T. K., Kitts, P. A., Kain, S. R., Tsien, R. Y., Orm,ö, M., et al. (1997). Structural basis for dual excitation and photoisomerization of the Aequorea victoria green fluorescent protein. Proc. Natl. Acad. Sci. U. S. A. 94, 2306–2311. doi: 10.1073/pnas.94.6.2306
Chalfie, M., Tu, Y., Euskirchen, G., Ward, W. W., and Prasher, D. C. (1994). Green fluorescent protein as a marker for gene expression. Science 263, 802–805. doi: 10.1126/science.8303295
Chang, J., Romei, M. G., and Boxer, S. G. (2019). Structural evidence of photoisomerization pathways in fluorescent proteins. J. Am. Chem. Soc. 141, 15504–15508. doi: 10.1021/jacs.9b08356
Chattoraj, M., King, B. A., Bublitz, G. U., and Boxer, S. G. (1996). Ultra-fast excited state dynamics in green fluorescent protein: multiple states and proton transfer. Proc. Natl. Acad. Sci. U. S. A. 93, 8362–8367. doi: 10.1073/pnas.93.16.8362
Chen, C., Zhu, L., Baranov, M. S., Tang, L., Baleeva, N. S., Smirnov, A. Y., et al. (2019). Photoinduced proton transfer of GFP-inspired fluorescent superphotoacids: principles and design. J. Phys. Chem. B 123, 3804–3821. doi: 10.1021/acs.jpcb.9b03201
Chen, C., Zhu, L., Boulanger, S. A., Baleeva, N. S., Myasnyanko, I. N., Baranov, M. S., et al. (2020). Ultrafast excited-state proton transfer dynamics in dihalogenated non-fluorescent and fluorescent GFP chromophores. J. Chem. Phys. 152:021101. doi: 10.1063/1.5138666
Chen, C., Zhu, L., and Fang, C. (2018). Femtosecond stimulated Raman line shapes: dependence on resonance conditions of pump and probe pulses. Chin. J. Chem. Phys. 31, 492–502. doi: 10.1063/1674-0068/31/cjcp1805125
Cooley, R. B., Feldman, J. L., Driggers, C. M., Bundy, T. A., Stokes, A. L., Karplus, P. A., et al. (2014). Structural basis of improved second-generation 3-nitro-tyrosine tRNA synthetases. Biochemistry 53, 1916–1924. doi: 10.1021/bi5001239
De Filippis, V., Frasson, R., and Fontana, A. (2006). 3-Nitrotyrosine as a spectroscopic probe for investigating protein–protein interactions. Protein Sci. 15, 976–986. doi: 10.1110/ps.051957006
Dedecker, P., De Schryver, F. C., and Hofkens, J. (2013). Fluorescent proteins: shine on, you crazy diamond. J. Am. Chem. Soc. 135, 2387–2402. doi: 10.1021/ja309768d
Dietze, D. R., and Mathies, R. A. (2016). Femtosecond stimulated Raman spectroscopy. ChemPhysChem 17, 1224–1251. doi: 10.1002/cphc.201600104
Duus, J. Ø., Meldal, M., and Winkler, J. R. (1998). Fluorescence energy-transfer probes of conformation in peptides: the 2-aminobenzamide/nitrotyrosine pair. J. Phys. Chem. B 102, 6413–6418. doi: 10.1021/jp973352+
Fang, C., Frontiera, R. R., Tran, R., and Mathies, R. A. (2009). Mapping GFP structure evolution during proton transfer with femtosecond Raman spectroscopy. Nature 462, 200–204. doi: 10.1038/nature08527
Fang, C., Tang, L., and Chen, C. (2019). Unveiling coupled electronic and vibrational motions of chromophores in condensed phases. J. Chem. Phys. 151:200901. doi: 10.1063/1.5128388
Fang, C., Tang, L., Oscar, B. G., and Chen, C. (2018). Capturing structural snapshots during photochemical reactions with ultrafast Raman spectroscopy: from materials transformation to biosensor responses. J. Phys. Chem. Lett. 9, 3253–3263. doi: 10.1021/acs.jpclett.8b00373
Felker, P. M., and Zewail, A. H. (1985). Dynamics of intramolecular vibrational-energy redistribution (IVR). I. Coherence effects. J. Chem. Phys. 82, 2961–2974. doi: 10.1063/1.448246
Fleissner, M. R., Brustad, E. M., Kálai, T., Altenbach, C., Cascio, D., Peters, F. B., et al. (2009). Site-directed spin labeling of a genetically encoded unnatural amino acid. Proc. Natl. Acad. Sci. U. S. A. 106, 21637–21642. doi: 10.1073/pnas.0912009106
Gross, L. A., Baird, G. S., Hoffman, R. C., Baldridge, K. K., and Tsien, R. Y. (2000). The structure of the chromophore within DsRed, a red fluorescent protein from coral. Proc. Natl. Acad. Sci. U. S. A. 97, 11990–11995. doi: 10.1073/pnas.97.22.11990
Hall, C. R., Tolentino Collado, J., Iuliano, J. N., Gil, A. A., Adamczyk, K., Lukacs, A., et al. (2019). Site-specific protein dynamics probed by ultrafast infrared spectroscopy of a non-canonical amino acid. J. Phys. Chem. B 123, 9592–9597. doi: 10.1021/acs.jpcb.9b09425
Hasegawa, J. Y., Fujimoto, K., Swerts, B., Miyahara, T., and Nakatsuji, H. (2007). Excited states of GFP chromophore and active site studied by the SAC-CI method: effect of protein-environment and mutations. J. Comput. Chem. 28, 2443–2452. doi: 10.1002/jcc.20667
Hyun Bae, J., Rubini, M., Jung, G., Wiegand, G., Seifert, M. H. J., Azim, M. K., et al. (2003). Expansion of the genetic code enables design of a novel “gold” class of green fluorescent proteins. J. Mol. Biol. 328, 1071–1081. doi: 10.1016/S0022-2836(03)00364-4
Jang, H. S., Gu, X., Cooley, R. B., Porter, J. J., Henson, R. L., Willi, T., et al. (2020). Efficient site-specific prokaryotic and eukaryotic incorporation of halotyrosine amino acids into proteins. ACS Chem. Biol. 15, 562–574. doi: 10.1021/acschembio.9b01026
Jung, G. (2012a). Fluorescent Proteins I: From Understanding to Design. (Berlin, Heidelberg: Springer-Verlag).
Jung, G. (2012b). Fluorescent Proteins II: Application of Fluorescent Protein Technology. (Berlin, Heidelberg: Springer-Verlag).
Kovács, A., Izvekov, V., Keresztury, G., and Pongor, G. (1998). Vibrational analysis of 2-nitrophenol. A joint FT-IR, FT-Raman and scaled quantum mechanical study. Chem. Phys. 238, 231–243. doi: 10.1016/S0301-0104(98)00307-3
Kummer, A. D., Wiehler, J., Schüttrigkeit, T. A., Berger, B. W., Steipe, B., and Michel-Beyerle, M. E. (2002). Picosecond time-resolved fluorescence from blue-emitting chromophore variants Y66F and Y66H of the green fluorescent protein. ChemBioChem 3, 659–663. doi: 10.1002/1439-7633(20020703)3:7<659::AID-CBIC659>3.0.CO;2-U Available online at: https://chemistry-europe.onlinelibrary.wiley.com/doi/abs/10.1002/1439-7633%2820020703%293%3A7%3C659%3A%3AAID-CBIC659%3E3.0.CO%3B2-U
Link, A. J., Mock, M. L., and Tirrell, D. A. (2003). Non-canonical amino acids in protein engineering. Curr. Opin. Biotechnol. 14, 603–609. doi: 10.1016/j.copbio.2003.10.011
Liu, W., Wang, Y., Tang, L., Oscar, B. G., Zhu, L., and Fang, C. (2016). Panoramic portrait of primary molecular events preceding excited state proton transfer in water. Chem. Sci. 7, 5484–5494. doi: 10.1039/C6SC00672H
Lossau, H., Kummer, A., Heinecke, R., Pollinger-Dammer, F., Kompa, C., Bieser, G., et al. (1996). Time-resolved spectroscopy of wild-type and mutant green fluorescent proteins reveals excited state deprotonation consistent with fluorophore-protein interactions. Chem. Phys. 213, 1–16. doi: 10.1016/S0301-0104(96)00340-0
Malde, A. K., Zuo, L., Breeze, M., Stroet, M., Poger, D., Nair, P. C., et al. (2011). An automated force field topology builder (ATB) and repository: version 1.0. J. Chem. Theory Comput. 7, 4026–4037. doi: 10.1021/ct200196m
Mandal, D., Tahara, T., and Meech, S. R. (2004). Excited-state dynamics in the green fluorescent protein chromophore. J. Phys. Chem. B 108, 1102–1108. doi: 10.1021/jp035816b
Meneely, C. T., She, C. Y., and Edwards, D. F. (1971). Line-shape parameters of the Raman lines of chloro-, bromo-, and iodobenzene. J. Mol. Spectrosc. 39, 73–78. doi: 10.1016/0022-2852(71)90277-3
Merrick, J. P., Moran, D., and Radom, L. (2007). An evaluation of harmonic vibrational frequency scale factors. J. Phys. Chem. A 111, 11683–11700. doi: 10.1021/jp073974n
Mooney, C. R. S., Horke, D. A., Chatterley, A. S., Simperler, A., Fielding, H. H., and Verlet, J. R. R. (2013). Taking the green fluorescence out of the protein: dynamics of the isolated GFP chromophore anion. Chem. Sci. 4, 921–927. doi: 10.1039/C2SC21737F
Oltrogge, L. M., and Boxer, S. G. (2015). Short hydrogen bonds and proton delocalization in green fluorescent protein (GFP). ACS Cent. Sci. 1, 148–156. doi: 10.1021/acscentsci.5b00160
Ormö, M., Cubitt, A. B., Kallio, K., Gross, L. A., Tsien, R. Y., and Remington, S. J. (1996). Crystal structure of the Aequorea victoria green fluorescent protein. Science 273, 1392–1395. doi: 10.1126/science.273.5280.1392
Oscar, B. G., Liu, W., Zhao, Y., Tang, L., Wang, Y., Campbell, R. E., et al. (2014). Excited-state structural dynamics of a dual-emission calmodulin-green fluorescent protein sensor for calcium ion imaging. Proc. Natl. Acad. Sci. U. S. A. 111, 10191–10196. doi: 10.1073/pnas.1403712111
Pal, P. P., Bae, J. H., Azim, M. K., Hess, P., Friedrich, R., Huber, R., et al. (2005). Structural and spectral response of Aequorea victoria green fluorescent proteins to chromophore fluorination. Biochemistry 44, 3663–3672. doi: 10.1021/bi0484825
Patterson, G. H., Knobel, S. M., Sharif, W. D., Kain, S. R., and Piston, D. W. (1997). Use of the green fluorescent protein and its mutants in quantitative fluorescence microscopy. Biophys. J. 73, 2782–2790. doi: 10.1016/S0006-3495(97)78307-3
Patterson, G. H., and Lippincott-Schwartz, J. (2002). A photoactivatable GFP for selective photolabeling of proteins and cells. Science 297, 1873–1877. doi: 10.1126/science.1074952
Pédelacq, J.-D., Cabantous, S., Tran, T., Terwilliger, T. C., and Waldo, G. S. (2006). Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 24, 79–88. doi: 10.1038/nbt1172
Peeler, J. C., and Mehl, R. A. (2012). “Site-specific incorporation of unnatural amino acids as probes for protein conformational changes,” in Unnatural Amino Acids: Methods and Protocols, eds L. Pollegioni, and S. Servi (New York, NY: Humana Press), 125–134.
Piatkevich, K. D., Malashkevich, V. N., Almo, S. C., and Verkhusha, V. V. (2010). Engineering ESPT pathways based on structural analysis of LSSmKate red fluorescent proteins with large Stokes shift. J. Am. Chem. Soc. 132, 10762–10770. doi: 10.1021/ja101974k
Quick, M., Dobryakov, A. L., Kovalenko, S. A., and Ernsting, N. P. (2015). Resonance femtosecond-stimulated Raman spectroscopy without actinic excitation showing low-frequency vibrational activity in the S2 state of all-trans β-carotene. J. Phys. Chem. Lett. 6, 1216–1220. doi: 10.1021/acs.jpclett.5b00243
Rauch, B. J., Porter, J. J., Mehl, R. A., and Perona, J. J. (2016). Improved incorporation of non-canonical amino acids by an engineered tRNATyr suppressor. Biochemistry 55, 618–628. doi: 10.1021/acs.biochem.5b01185
Reddington, S. C., Rizkallah, P. J., Watson, P. D., Pearson, R., Tippmann, E. M., and Jones, D. D. (2013). Different photochemical events of a genetically encoded phenyl azide define and modulate GFP fluorescence. Angew. Chem. Int. Ed. 52, 5974–5977. doi: 10.1002/anie.201301490
Ren, T.-B., Xu, W., Zhang, W., Zhang, X.-X., Wang, Z.-Y., Xiang, Z., et al. (2018). A general method to increase Stokes shift by introducing alternating vibronic structures. J. Am. Chem. Soc. 140, 7716–7722. doi: 10.1021/jacs.8b04404
Schellenberg, P., Johnson, E., Esposito, A. P., Reid, P. J., and Parson, W. W. (2001). Resonance Raman scattering by the green fluorescent protein and an analogue of its chromophore. J. Phys. Chem. B 105, 5316–5322. doi: 10.1021/jp0046243
Seeliger, D., and de Groot, B. L. (2010). Protein thermostability calculations using alchemical free energy simulations. Biophys. J. 98, 2309–2316. doi: 10.1016/j.bpj.2010.01.051
Shaner, N. C., Campbell, R. E., Steinbach, P. A., Giepmans, B. N. G., Palmer, A. E., and Tsien, R. Y. (2004). Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22, 1567–1572. doi: 10.1038/nbt1037
Shimomura, O., Johnson, F. H., and Saiga, Y. (1962). Extraction, purification and properties of Aequorin, a bioluminescent protein from the luminous Hydromedusan, Aequorea. J. Cell. Comp. Physiol. 59, 223–239. doi: 10.1002/jcp.1030590302
Silva, W. R., Graefe, C. T., and Frontiera, R. R. (2016). Toward label-free super-resolution microscopy. ACS Photonics 3, 79–86. doi: 10.1021/acsphotonics.5b00467
Sripakdeevong, P., Cevec, M., Chang, A. T., Erat, M. C., Ziegeler, M., Zhao, Q., et al. (2014). Structure determination of non-canonical RNA motifs guided by 1H NMR chemical shifts. Nat. Methods 11, 413–416. doi: 10.1038/nmeth.2876
Striker, G., Subramaniam, V., Seidel, C. A. M., and Volkmer, A. (1999). Photochromicity and fluorescence lifetimes of green fluorescent protein. J. Phys. Chem. B 103, 8612–8617. doi: 10.1021/jp991425e
Subach, F. V., and Verkhusha, V. V. (2012). Chromophore transformations in red fluorescent proteins. Chem. Rev. 112, 4308–4327. doi: 10.1021/cr2001965
Svendsen, A., Kiefer, H. V., Pedersen, H. B., Bochenkova, A. V., and Andersen, L. H. (2017). Origin of the intrinsic fluorescence of the green fluorescent protein. J. Am. Chem. Soc. 139, 8766–8771. doi: 10.1021/jacs.7b04987
Tang, L., and Fang, C. (2019). Nitration of tyrosine channels photoenergy through a conical intersection in water. J. Phys. Chem. B 123, 4915–4928. doi: 10.1021/acs.jpcb.9b03464
Tang, L., Liu, W., Wang, Y., Zhao, Y., Oscar, B. G., Campbell, R. E., et al. (2015). Unraveling ultrafast photoinduced proton transfer dynamics in a fluorescent protein biosensor for Ca2+ imaging. Chem. Eur. J. 21, 6481–6490. doi: 10.1002/chem.201500491
Tang, L., Liu, W., Wang, Y., Zhu, L., Han, F., and Fang, C. (2016). Ultrafast structural evolution and chromophore inhomogeneity inside a green-fluorescent-protein-based Ca2+ biosensor. J. Phys. Chem. Lett. 7, 1225–1230. doi: 10.1021/acs.jpclett.6b00236
Tang, L., Wang, Y., Zhu, L., Kallio, K., Remington, S. J., and Fang, C. (2018a). Photoinduced proton transfer inside an engineered green fluorescent protein: a stepwise-concerted-hybrid reaction. Phys. Chem. Chem. Phys. 20, 12517–12526. doi: 10.1039/C8CP01907J
Tang, L., Zhu, L., Taylor, M. A., Wang, Y., Remington, S. J., and Fang, C. (2018b). Excited state structural evolution of a GFP single-site mutant tracked by tunable femtosecond-stimulated Raman spectroscopy. Molecules 23:2226. doi: 10.3390/molecules23092226
Tang, L., Zhu, L., Wang, Y., and Fang, C. (2018c). Uncovering the hidden excited state toward fluorescence of an intracellular pH indicator. J. Phys. Chem. Lett. 9, 4969–4975. doi: 10.1021/acs.jpclett.8b02281
Taylor, M. A., Zhu, L., Rozanov, N. D., Stout, K. T., Chen, C., and Fang, C. (2019). Delayed vibrational modulation of the solvated GFP chromophore into a conical intersection. Phys. Chem. Chem. Phys. 21, 9728–9739. doi: 10.1039/C9CP01077G
Tomosugi, W., Matsuda, T., Tani, T., Nemoto, T., Kotera, I., Saito, K., et al. (2009). An ultramarine fluorescent protein with increased photostability and pH insensitivity. Nat. Methods 6, 351–353. doi: 10.1038/nmeth.1317
Tozzini, V., and Nifosì, R. (2001). Ab initio molecular dynamics of the green fluorescent protein (GFP) chromophore: an insight into the photoinduced dynamics of green fluorescent proteins. J. Phys. Chem. B 105, 5797–5803. doi: 10.1021/jp010052q
Tsien, R. Y. (1998). The green fluorescent protein. Annu. Rev. Biochem. 67, 509–544. doi: 10.1146/annurev.biochem.67.1.509
Vengris, M., van Stokkum, I. H. M., He, X., Bell, A. F., Tonge, P. J., van Grondelle, R., et al. (2004). Ultrafast excited and ground-state dynamics of the green fluorescent protein chromophore in solution. J. Phys. Chem. A 108, 4587–4598. doi: 10.1021/jp037902h
Wachter, R. M., Elsliger, M.-A., Kallio, K., Hanson, G. T., and Remington, S. J. (1998). Structural basis of spectral shifts in the yellow-emission variants of green fluorescent protein. Structure 6, 1267–1277. doi: 10.1016/S0969-2126(98)00127-0
Wachter, R. M., King, B. A., Heim, R., Kallio, K., Tsien, R. Y., Boxer, S. G., et al. (1997). Crystal structure and photodynamic behavior of the blue emission variant Y66H/Y145F of green fluorescent protein. Biochemistry 36, 9759–9765. doi: 10.1021/bi970563w
Wang, L., Xie, J. M., Deniz, A. A., and Schultz, P. G. (2003). Unnatural amino acid mutagenesis of green fluorescent protein. J. Org. Chem. 68, 174–176. doi: 10.1021/jo026570u
Wang, Y., Tang, L., Liu, W., Zhao, Y., Oscar, B. G., Campbell, R. E., et al. (2015). Excited state structural events of a dual-emission fluorescent protein biosensor for Ca2+ imaging studied by femtosecond stimulated Raman spectroscopy. J. Phys. Chem. B 119, 2204–2218. doi: 10.1021/jp505698z
Young, D. D., Jockush, S., Turro, N. J., and Schultz, P. G. (2011). Synthetase polyspecificity as a tool to modulate protein function. Bioorg. Med. Chem. Lett. 21, 7502–7504. doi: 10.1016/j.bmcl.2011.09.108
Zhang, W., Wang, Y. Q., and Wang, J. Y. (2012). Fluorescent protein engineering through genetic incorporation of 3-chlorotyrosine. Prog. Biochem. Biophys. 39, 378–387. doi: 10.3724/SP.J.1206.2011.00299
Zhao, Y., Araki, S., Wu, J., Teramoto, T., Chang, Y.-F., Nakano, M., et al. (2011). An expanded palette of genetically encoded Ca2+ indicators. Science 333, 1888–1891. doi: 10.1126/science.1208592
Zhu, L., Liu, W., and Fang, C. (2014). A versatile femtosecond stimulated Raman spectroscopy setup with tunable pulses in the visible to near infrared. Appl. Phys. Lett. 105, 041106. doi: 10.1063/1.4891766
Keywords: fluorescent proteins, ultrafast spectroscopy, structural dynamics, vibrational cooling, non-canonical amino acid, femtosecond stimulated Raman
Citation: Oscar BG, Zhu L, Wolfendeen H, Rozanov ND, Chang A, Stout KT, Sandwisch JW, Porter JJ, Mehl RA and Fang C (2020) Dissecting Optical Response and Molecular Structure of Fluorescent Proteins With Non-canonical Chromophores. Front. Mol. Biosci. 7:131. doi: 10.3389/fmolb.2020.00131
Received: 29 March 2020; Accepted: 02 June 2020;
Published: 07 July 2020.
Edited by:
Aljaz Godec, Max Planck Institute for Biophysical Chemistry, GermanyReviewed by:
Lars Henrik Andersen, Aarhus University, DenmarkCopyright © 2020 Oscar, Zhu, Wolfendeen, Rozanov, Chang, Stout, Sandwisch, Porter, Mehl and Fang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chong Fang, Y2hvbmcuZmFuZ0BvcmVnb25zdGF0ZS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.