 Erik Henke
Erik Henke Rajender Nandigama
Rajender Nandigama Süleyman Ergün
Süleyman Ergün
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Biosci. , 31 January 2020
Sec. Molecular Diagnostics and Therapeutics
Volume 6 - 2019 | https://doi.org/10.3389/fmolb.2019.00160
Solid tumors are complex organ-like structures that consist not only of tumor cells but also of vasculature, extracellular matrix (ECM), stromal, and immune cells. Often, this tumor microenvironment (TME) comprises the larger part of the overall tumor mass. Like the other components of the TME, the ECM in solid tumors differs significantly from that in normal organs. Intratumoral signaling, transport mechanisms, metabolisms, oxygenation, and immunogenicity are strongly affected if not controlled by the ECM. Exerting this regulatory control, the ECM does not only influence malignancy and growth of the tumor but also its response toward therapy. Understanding the particularities of the ECM in solid tumor is necessary to develop approaches to interfere with its negative effect. In this review, we will also highlight the current understanding of the physical, cellular, and molecular mechanisms by which the pathological tumor ECM affects the efficiency of radio-, chemo-, and immunotherapy. Finally, we will discuss the various strategies to target and modify the tumor ECM and how they could be utilized to improve response to therapy.
The last 25 years saw a massive shift in our approach to understand the biology of solid tumors. While research centered for a long time nearly exclusively on the individual tumor cells, the processes leading to their transformation, or conveying their malignancy, the tumor in its entirety and full complexity moved more and more the focus of cancer research. Starting from the concept of viewing the tumor as a complex organ, we meanwhile use the term tumor microenvironment (TME) to describe the entirety of the tumor components that are not malignant by themselves. Thus, the TME consists of the tumor's vasculature, connective tissue, infiltrating immune cells, and the extracellular matrix (ECM), and increasingly, all these individual components of the TME became the focus of new research communities within the fast-growing cancer field. These research efforts already resulted in the development and successful clinical implementation of TME-targeted drugs, starting with antiangiogenic agents and the potentially game-changing introduction of immunotherapeutics (Hurwitz et al., 2004; Robert et al., 2015)1,2. Although the ECM is probably the component of the TME that initially received the least attention, this also has changed considerably over the last decade, and numerous articles have bit by bit complemented our understanding of the tumor ECM and its role in malignancy and response to therapy.
In this article, we will focus on the impact of the ECM on the various forms of cancer therapy and on recent efforts to modulate the ECM for improved therapeutic efficacy. Of course, the ECM not only affects our efforts to treat cancer but also is directly involved in tumor establishment and disease progression. Effects of ECM-targeted approach on the efficacy of antitumor therapy often cannot be distinguished from direct effects on tumor behavior or progression. Therefore, it will be necessary to also discuss to a certain degree the immediate involvement of the ECM in malignancy and how its modification affects the course of the disease. However, to establish a base for the following review of our current efforts to include ECM targeting in the management of malignant diseases, we will first recapitulate the particularities of the ECM in solid tumors.
Many solid tumors express high levels of various ECM molecules like fibrillar collagens, fibronectin, elastin, and laminins (Provenzano et al., 2008; Mammoto et al., 2013). In addition, some cancers, that is pancreatic ductal adenocarcinomas (PDACs), are particularly rich in hyaluronan (Provenzano and Hingorani, 2013). In many tumors, the ECM compromises up to 60% of the tumor mass. Source of these ECM molecules are the tumor cells themselves, but to an even larger degree cancer-associated fibroblasts (CAFs) (Casey et al., 2009; Naba et al., 2012). Indeed, the infiltration of fibroblasts/myofibroblasts and the subsequent accumulation of significant amounts of collagenous ECM is observed in many solid tumors. This process, called desmoplasia, is strongly linked to poor prognosis and resistance to systemic therapy (Conti et al., 2008; Schober et al., 2014). By supporting tumor cells via paracrine stromal cell-derived factor-1 (SDF1) and transforming growth factor beta (TGFβ) signals, CAFs further contribute not only to a more malignant tumor phenotype by driving epithelial-to-mesenchymal transition (EMT) but also induce production of collagen and other ECM molecules (Zode et al., 2009; Porsch et al., 2013; Garcia et al., 2016). All components of the TME vary significantly from their respective counterparts in non-malignant tissues. Consequently, also the tumor ECM diverges strongly not only in amount of deposition but also in composition, organization, and post-translational modification from the ECM in surrounding normal tissue. In invasive ductal carcinomas, collagen production is shifted toward Col I and Col III compared to benign mammary lesions (Deak et al., 1991; Kauppila et al., 1998). The increased expression is mainly observed in the stromal part of the tumor. As the benign lesions also consist mainly of cells with fibroblastic characteristics, this demonstrated the tendency and capacity of breast tumor cells to shift the secreted matrisome of its stroma. In the desmoplastic stroma of breast carcinomas, up to 15% of the collagenous matrix consists of Col V, a collagen isoform of low abundance in normal and fibrocystic (<0.1%) breast tissue (Barsky et al., 1982). Experiments like second harmonics observation of fiber formation indicate that increasing the Col V/Col I ratio reduces length and organization of collagen fibers (Ajeti et al., 2011). At higher ratios, fiber formation can be completely inhibited, resulting in a gel-like ECM (Pucci-Minafra and Luparello, 1991). Col IV, on the other hand, is downregulated in ovarian carcinomas compared to benign tissue, and the expression is inversely correlated with stage and markers of malignancy (Bar et al., 2004). The same pattern of enhanced Col I/Col III expression and reduced Col IV expression is observed in human lung tumors (Fang et al., 2019). In melanoma, increased Col I expression is observed and correlated with invasiveness, angiogenesis, and reduced survival (van Kempen et al., 2008; Miskolczi et al., 2018). High collagen messenger RNA (mRNA) expression, the aberrant form of fibrous collagen spindles, and the increased expression of matrix proteases are signs of an fast turnover of the collagen in tumors (Kauppila et al., 1998).
The synthesis and maturation of collagens is a complex process. The increased collagen production in the TME requires not only increased transcription and translation of collagen encoding genes but also the upregulation of enzymes that are necessary for proper post-translational processing and secretion of collagen molecules. Indeed, collagen-processing enzymes like lysyl hydroxylases and prolyl hydrolases are often strongly expressed in the TME (Erler et al., 2006; Gilkes et al., 2013; Xiong et al., 2014). Lysyl oxidases are also significantly upregulated in many tumors, especially in desmoplastic cancers (Peyrol et al., 1997; Erler et al., 2006; Barry-Hamilton et al., 2010). This leads to an increased cross-linking of collagens and elastin, rendering the tumor more rigid and contributing—in combination with the already increased ECM deposition—to its palpability. The highly cross-linked collagenous matrix increases mechanical stress and focal adhesion kinase (FAK)-mediated signaling and reduces overall supply with oxygen and in the TME (Levental et al., 2009; Taylor et al., 2011; Baker et al., 2012; Rossow et al., 2018). Prolyl-4-hydroxylases (P4HAs) are ascorbic-acid-dependent enzymes that modify collagens intracellularly (reviewed in Gorres and Raines, 2010). The formation of hydroxyproline in this process is necessary to increase stability of helical collagen under physiological conditions. High P4HA expression increases intratumoral collagen deposition (Xiong et al., 2014). It is also strongly correlated with resistance to chemotherapy and reduced survival in triple-negative breast cancer patients (Xiong et al., 2018).
Similar to collagens, proteoglycans (PGs), also constituting an important part of the ECM, require enzymes for correct production and assembly. PGs consist of a core protein, which is heavily glycosylated with longer shorter chains of glycosaminoglycans (GAGs). The attachment of the GAGs happens in the Golgi apparatus, where the core protein is translocated. Glycosyltransferases first attach a tetra saccharide to a serine of the core protein that functions as linker. Other glycosyltransferases then conjugate monosaccharides to the growing GAG chain. In addition to various glycosyltransferases, sulfotransferases and epimerases are needed for proper chain modification. The GAGs consist of repetitive disaccharide patterns. Depending on the composition of these disaccharides, GAGs are distinguished in chondroitin sulfate, dermatan sulfate, heparan sulfate, and keratan sulfate. PGs can differ significantly in molecular weight. Together with collagens—mainly Col II—PGs are the constituents of cartilage, underlining their capacity to contribute to tissue stiffness and sturdiness. In many tumors, increased levels of PGs are observed, but the story seems to be more complex as the changes in PG content between normal and malignant tissue lie mainly in shifts between various PGs often between low- and high-molecular weight PGs. Comparing prostate cancer to normal prostate tissue, Suhovskih et al. found increased expression of aggrecan and CSPG4 but reduced decorin levels (Suhovskih et al., 2013), while gastric cancers showed a doubling of overall GAG content and an increase in versican and decorin expression (Theocharis et al., 2003). Similarly, in squamous cell laryngeal carcinoma, versican and decorin are stronger represented than in normal tissue, while aggrecan is completely downregulated (Vynios et al., 2008). Brown et al. found breast carcinoma stroma to be strongly enriched in both versican and decorin expression along with Col I and fibronectin (Brown et al., 1999). In prostate cancer, decorin, lumican, and versican were strongly overexpressed, although this study relied on mRNA expression data of the core proteins; thus, conclusions about the actual PG content are difficult (Koninger et al., 2004). In peritumoral stroma of melanoma, versican is enriched compared to benign nevi (Gambichler et al., 2008). Sulfation patterns of GAGs also seem to change in cancer. Interestingly, it seems that, in colorectal cancer (CRC) and gastric cancer, a shift from 4- to 6-sulfation occurs, while in laryngeal carcinomas, the shift is reversed with more 4-sulfattion vs. 6-sulfation in normal tissue (Theocharis, 2002; Theocharis et al., 2003; Vynios et al., 2008).
Hyaluronic acid (HA) is a GAG that is not conjugated to peptides, and in contrast to the three others, it is not synthesized in the Golgi apparatus. Synthesis is performed by a family of three transmembrane glycosyltransferases, hyaluronan synthetase 1–3 (HAS1-3), which alternately conjugate gluconic acid and N-acetylglucosamine (reviewed in Weigel and DeAngelis, 2007; Passi et al., 2019). HASs are plasma-membrane-bound proteins, which probably are not only responsible for the synthesis but also for the extracellular export of the synthesized HA macromolecule via direct extrusion through an enzymatic pore. Some controversy exists about whether HA is exported via ABC transporters like MRP5 or MDR1 (Schulz et al., 2007). While treatment with ABC-transporter inhibitors like S-decylglutathione or trequinsin reduced HA production in human fibroblasts (Prehm and Schumacher, 2004), this approach using various inhibitors failed to block HA release in breast cancer cell lines that express the targeted ABC transporters (Thomas and Brown, 2010). However, while the involvement of ABC transporters in the production of HA is still debated, 4-methylumbelliferone (4-MU) inhibits HAS1–3 with some specificity and is effective in blocking HA synthesis (Nakamura et al., 1995; Kakizaki et al., 2004; Urakawa et al., 2012; Ikuta et al., 2017; Karalis et al., 2018). HA production is increased in many cancers, most notably in pancreatic carcinomas (Theocharis et al., 2000; Cheng et al., 2013) but also in breast cancers (Bertrand et al., 1992; Auvinen et al., 1997), CRC (Wang et al., 1996), prostate cancer (Lipponen et al., 2001), and even in brain tumors (Jadin et al., 2015). Stromal cells, i.e., fibroblasts, are often identified histologically as the main source of HA in the tumor, and tumor cells can increase HA synthesis in cocultured fibroblasts (Knudson et al., 1984). HA levels, and expression of HAS1–3, are correlated with poor prognosis (Bertrand et al., 1992; Ropponen et al., 1998; Auvinen et al., 2000; Zhang H. et al., 2016). HA acts as a ligand for CD44 and might thereby play a role in EMT, resulting in increased invasiveness and metastasis (Ghatak et al., 2010; Heldin et al., 2014). Expression of proteins that are thought to be cancer stem cell markers (CD90, CD133, EpCAM) are also reduced after inhibition of HA synthesis (Sukowati et al., 2019).
Laminins are also often stronger expressed in malignant tissue. In normal tissue, laminins are components exclusive of the basement membrane, resulting in a continuous well-delineated linear staining by immunohistology. This appearance is often distorted in tumors, or laminin appears to be ubiquitously distributed in the stromal parts of the tumors (Gusterson et al., 1982; Hand et al., 1985; Alon et al., 1986; Qiu et al., 2018). The loss of adherence to a defined basement membrane and the disruption of this basement membrane is of course a characteristic of invasive behavior. Conclusively, the increased laminin expression and aberrant distribution is correlated with poor prognosis and invasiveness, a fact that is documented since the early 1980s (Albrechtsen et al., 1981; Siegal et al., 1981). In breast cancer, laminin overexpression is generally observed (Alon et al., 1986).
Using a sophisticated and elaborated proteomics approach, Naba et al. analyzed the matrisome of human melanoma xenografts grown in severe combined immunodeficiency mice. As the differences in amino acid sequence between murine and human orthologs is sufficiently high for most proteins, the approach allowed them to distinguish the contribution of the (human) tumor cells and the (murine) stromal cells (Naba et al., 2012). The experiments revealed that some proteins are expressed exclusively by the tumor cells (e.g., Col7a1, Lama4, Lamb1) and others by the stromal cells (e.g., Col5a3, Lama2, Eln). Moreover, the ECM composition changes significantly during progression and metastasis.
Many cancers from the same tissue of origin can be subdivided according to the molecular expression profile. These molecular subtypes yield a lot of information about the tumor's metabolism, misregulation of survival and apoptotic pathways, presence of oncogenic drivers, and therefore the sensitivity and resistance toward different treatment modalities. Not surprisingly, ECM expression and deposition also differs significantly between molecular tumor subtypes. Breast cancers can be subdivided according to their expression status for estrogen receptor, progesterone receptor, and Her2 into luminal A (ER/PR+, Her2−), luminal B (ER/PR+, Her2+), Her2-positive (ER/PR−, Her2+), and basal-like/triple negative (triple-negative breast cancer, ER/PR−, Her2−) (Perou et al., 2000). Triple-negative breast cancer and, to a lesser extent, Her2 tumors show not only increased deposition of collagen but also enhanced invasion with CAFs (Acerbi et al., 2015; Takai et al., 2016).
The expression profile of ECM-related genes is also in many cancers a valuable prognostic factor. Bergamaschi et al. were able to divide breast cancer samples in four subgroups (ECM1–4) according to their ECM expression profile that correlated significantly with prognosis (Bergamaschi et al., 2008). Interestingly, the different expression subgroups correlated only slightly with results of histological evaluation that grouped the tumors according to the density of their appearance. A strong correlation of elevated ECM expression and poor prognosis was also found in luminal BCa (Riaz et al., 2012). In pediatric osteosarcoma, a signature of high ECM turnover was found to be highly prognostic for chemoresistance (Mintz et al., 2005) and, in gastric cancer expression of a signature containing Col1a1, Fn1, and Muc5a, was highly predictive of overall and disease-free survival (Jiang et al., 2019). Besides indicators for immune suppression, high expression of collagens Col3a1, Col4a1, and Col5a2 is correlated with poor prognosis in glioblastoma (Chen et al., 2018).
As we have seen, the ECM in tumors significantly differs in composition and architecture from that in normal tissue. Considering its physical properties, the tumor ECM is more abundant, denser, and stiffer. These altered characteristics can negatively affect response to therapy in multiple ways (Figure 1). Most obviously, an excessive accumulation of dense and rigid ECM, which histologically often encapsulates clusters of tumor cells, can act as a barrier, shielding the cells from therapeutic agents. This effect is directly linked to a reduced overall supply, as this barrier also impairs diffusion of oxygen, nutrients, and metabolites. Increased hypoxia and metabolic stress lead to activation of antiapoptotic and drug resistance pathways. Finally, cell–ECM contacts and increased tissue stiffness can directly contribute to chemoresistance of tumor via integrin and FAK-signaling.
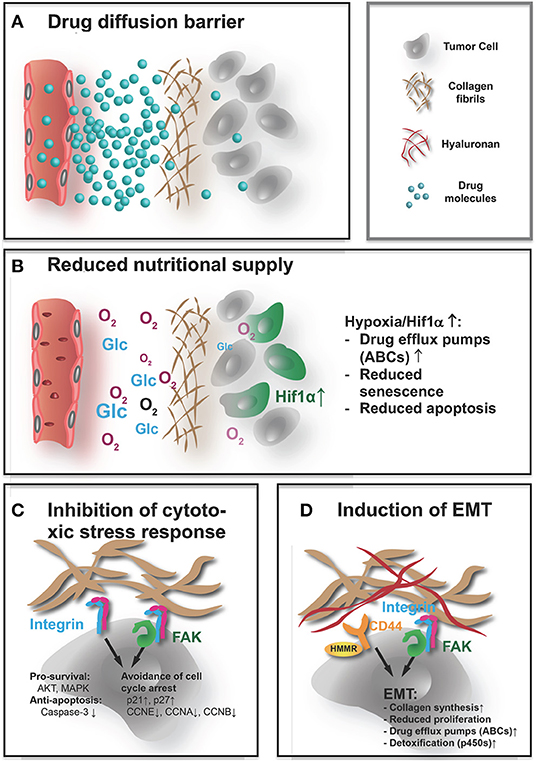
Figure 1. How the ECM affects the efficacy of systemic treatment. Systemically applied drugs, independently of their nature being small molecules or larger biomolecules, e.g., antibodies, peptides, or nucleic acids, have to reach their target cells and cause a therapeutic response. The abundant, highly cross-linked ECM interferes with the efficacy in both direct and indirect ways. (A) The rigid dense ECM acts as a diffusion barrier that impedes access of the drugs to the tumor cells, thereby acting as a shield protecting the tumor from therapeutically effective doses. (B) The reduced diffusion through the ECM also impairs supply with nutrients and oxygen. Pathological signaling in response to metabolic stress and hypoxia increase expression of drug efflux pumps and impair apoptosis and senescence, rendering drugs that reach the undersupplied cells less effective. (C) Direct contact with the ECM also affects these pathways that lead to a muted response to cytotoxic stress. Integrin and FAK activation increase prosurvival signaling, reduce apoptotic response, and help the cells to avoid cell cycle arrest when confronted with chemotherapy-induced damage. (D) Similarly, not only integrin and FAK but also hyaluronan induced CD44/HMMR signals can lead to EMT. The mesenchymal state is characterized by stem-like, chemoresistant traits. This includes again not only upregulation of ABC transporters and reduced proliferation but also activation of cell metabolism (cytochrome p450) that improves detoxification. That EMT also seems to increase collagen synthesis, and production of cross-linking enzymes in tumor cells might lead to a vicious cycle where the dense ECM induces EMT that again drives ECM build-up.
An aspect that is necessary to mention is meanwhile understood effect of fibrosis and CAF infiltration, and therefore also alterations in ECM composition and accumulation, on metastasis and EMT. Occurrence of metastasis imminently affects treatment options and therapeutic outcome. EMT is linked not only to increased metastasis (Yang et al., 2004; Mani et al., 2007; Ocana et al., 2012; Stankic et al., 2013) but also to chemoresistance (Singh and Settleman, 2010; Haslehurst et al., 2012; Ren et al., 2013; Fischer et al., 2015; Zheng et al., 2015; Li H. et al., 2019; Li N. et al., 2019). EMT in cancer is associated with the acquisition of a more stem-cell-like character. Accordingly, Fisher et al. found that the cyclophosphamide resistance of tumor cells that acquired a mesenchymal character could be attributed to reduced proliferation and increased expression of drug efflux pumps (ABCC1, ABCB1) and of enzymes involved in drug metabolism, like cytochrome p450s (Fischer et al., 2015), traits that are characteristic for stem and progenitor populations.
Different aspects of the ECM contribute to EMT. One critical factor is tissue stiffness. Increasing stiffness of the surrounding ECM drives EMT in breast cancer cells by promoting TWIST1 translocalization into the nucleus (Wei et al., 2015). Rice et al. found, by plating PDAC cells on substrates of different rigidity, that stiffness increases nuclear localization of the transcription factors YAP and TAZ, which can drive EMT (Rice et al., 2017). As a result, mesenchymal markers (vimentin) were upregulated, epithelial markers (E-cadherin) were reduced, and the cells demonstrated increased resistance to paclitaxel. Gemcitabine sensitivity was not affected, which was attributed to the fact that gemcitabine reversed the mesenchymal character on the stiff matrices as measured by normalized vimentin expression. Different components of the ECM might also trigger or increase EMT-like processes. The interaction of HA with hyaluronan-mediated motility receptor (HMMR) drives epicardial EMT during injury-induced heart regeneration in zebrafish (Missinato et al., 2015). In gastric cancer, the HA/HMMR axis can also be linked to EMT and 5-FU resistance (Zhang et al., 2019). In vitro Col I secreted by hepatic stellate cells induced EMT in hepatocarcinoma cells (Yang et al., 2014). A hallmark of EMT is the loss of epithelial polarization, which by itself is linked to anchorage of epithelial layers on a basement membrane (BM). Walter et al. found that defects in the BM and of Col IV deposition in particular can trigger EMT (Walter et al., 2018). In proximal tubular epithelial cells, Col IV helps to maintain an epithelial phenotype, while Col I promotes EMT (Zeisberg et al., 2001). Reduced Col IV synthesis or incorrect assembly and increased Col I synthesis thereby contributed to renal fibrosis. In general, the examination of the effect of collagen deposition on tumor EMT is complicated by the question of which comes first: is collagen build-up inducing EMT or are cells producing more collagen as a result of undergoing EMT. EMT is observed under pathological fibrosis in normal organs, and fibrotic collagen accumulation is often considered a result of the more mesenchymal character of the affected cells (Higgins et al., 2007; Hosper et al., 2013). This might be true for cancer, too. It has been shown that TWIST1, one of the earliest described transcription factors inducing EMT, is a potentially direct regulator of Col1a5 transcription (Garcia-Palmero et al., 2016). Similarly, the transcription factor ZEB1 positively regulates Col1 transcription and, in addition, promotes LOXL2 expression that contributes to collagen stabilization (Ponticos et al., 2004; Peng et al., 2017).
As the ECM composition within tumors itself is heterogeneous, these effects of the ECM on cell behavior and cell fate contribute strongly to tumor cell heterogeneity. In addition, there is evidence that ECM components can influence genetic instability. Deletion of the paired Col4A5 and Col4A6 genes contributes to the development of leiomyomatosis (Zhou et al., 1993). Elevated expression of MMP3 can transform cells in vitro, cause genomic instability in murine mammary glands, and promote carcinogenesis in these mice (Sternlicht et al., 1999; Radisky et al., 2005). Finally, blocking integrin β1 signaling attenuated tumorigenesis in an inducible human model for epidermal neoplasia (Reuter et al., 2009). Acquisition of genetic instability is an early event in tumorigenesis. As in these examples, ECM alterations drive tumorigenesis or directly genetic instability, it is likely that also later changes in ECM accumulation or composition influence genetic instability, thereby contributing to tumor progression and genetic heterogeneity.
Distribution of drugs in the tumor occurs mainly by diffusion. Owing to lack of lymphatic drainage, convection is in most solid tumors of less importance (Welter and Rieger, 2013; Dewhirst and Secomb, 2017). An abundant and highly condensed ECM therefore can significantly reduce drug transport, resulting in only a small volume of the surrounding tissue being supplied by the individual vessels. Solid tumors are already characterized by low microvessel density (Offersen et al., 1998; da Silva et al., 2009). The often observed abundant, highly cross-linked ECM further aggravates the supply situation, leading to hypoxia and metabolic stress. Hypoxia again is directly linked to resistance vs. various forms of cytotoxic therapy and radiotherapy (Moeller and Dewhirst, 2004; Doublier et al., 2012; Jain, 2014; Horsman and Overgaard, 2016; Graham and Unger, 2018).
The family of the five lysyl oxidase isoenzymes (LOX and LOXL1–4) catalyzes the formation of cross-links between collagen molecules and in elastin networks (reviewed in Smith-Mungo and Kagan, 1998; Lucero and Kagan, 2006; Siddikuzzaman et al., 2011). They are essential for stabilization of collagen networks in a last extracellular maturation step. Thus, increased lysyl oxidase levels affect drug distribution in two ways: through better collagen stabilization toward degradation, they enhance collagen accumulation and the increasing quantity of cross-links turns the collagen network denser, further decreasing diffusivity (Rohrig et al., 2017; Rossow et al., 2018). Stromal fibroblasts contribute strongly to the elevated lysyl oxidase levels observed in many cancers (Peyrol et al., 1997). Using real-time confocal imaging of multicellular tumor spheroids, Schutze et al. demonstrated that diffusion of chemotherapeutics like doxorubicin is significantly hampered by LOX or LOXL2 overexpression (Schutze et al., 2015). Inhibition of lysyl oxidases with 2-aminopropionitril reversed the effect. Lysyl oxidases can directly confer intrinsic chemoresistance in human tumors (Rossow et al., 2018). The initially findings of this study, that lysyl oxidases are upregulated in a defined subgroup of intrinsically resistant tumors of breast, ovarian, and CRC, were substantiated by the observation that overexpression of lysyl oxidases rendered previously sensitive and lysyl oxidase–low murine tumors completely resistant to chemotherapeutic treatment. Inhibition of lysyl oxidases generally improves oxygenation (Erler et al., 2006; Rossow et al., 2018). In response, vascular endothelial growth factor A (VEGF-A) expression, the main angiogenic factor, that is a Hif-1α-regulated protein, is reduced with positive effects on tumor vascular maturation and patency (Maxwell et al., 1997; Rossow et al., 2018). Lysyl oxidases might also have a direct effect on VEGF-A expression via oxidation of the platelet-derived growth factor receptor extracellular domain (Baker et al., 2013). In addition, the increased tissue rigidity in LOXL2-high tumors facilitates endothelial invasion of the tumor tissue, a critical step in neo-vessel formation, presumably by increasing motility via FAK signaling (Zaffryar-Eilot et al., 2013).
Treatment with hyaluronidases in vivo reduces HA content and improves gemcitabine and DOX uptake in murine pancreatic ductal adenocarcinoma (PDAC) models (Provenzano et al., 2012; Jacobetz et al., 2013). In osteosarcoma, xenografts uptake of liposomal DOX could be improved with hyaluronidase treatment (Eikenes et al., 2005). Especially, PDACs display high hyaluronan content and can bind large amounts of water in the ECM leading to increase in interstitial fluid pressure (PIF). Some studies indicate that transcapillary transport and diffusion within the tumor might be hindered by high PIF resulting from high HA contend and/or vessel leakage. It has to be shown if also tumors with lower hyaluronan content respond to this treatment with better drug distribution. In two of these studies, also improved vascular perfusion and reduced vessel collapse were observed after hyaluronidase treatment (Eikenes et al., 2005; Jacobetz et al., 2013). This might indicate that the high PIF in hyaluronan-rich tumors restricts drug transport mainly by compressing the supplying vessels and less by interfering with interstitial drug diffusion. This would be in line with mathematical models that indicate that PIF has only a minor effect on diffusion (Eikenberry, 2009).
In conclusion, it remains to be stated that a close connection exists between the signaling pathways that regulate ECM formation and angiogenesis. Especially the shared regulation via the hypoxia-response axis results in the fact that interventions that alter either the tumor ECM or the vasculature will likely also affect the other. Effects on drug response and delivery are therefore often difficult to pinpoint on a clear ECM or vascular mechanism.
As carcinoma- or tumor-associated fibroblasts (CAFs) are the main source of the ECM in tumors, it is necessary to have a closer look at the particularities of these cells (Bagordakis et al., 2016; Pankova et al., 2016; Pasanen et al., 2016). CAFs are found in all solid tumors (Puram et al., 2017; Zhao et al., 2018). They differ substantially from the quiescent, metabolically inactive fibroblasts found in normal connective tissue, as they are migratory, growth and immune response promoting, and synthetically active (reviewed in Kalluri, 2016). The source of CAFs varies strongly and often according to tumor type. Stellate cells, bone-marrow-derived mesenchymal stem cells, mesenchymal stem cells from adipose tissue, and resident quiescent fibroblasts have been identified as cells of origin for CAFs (McDonald et al., 2015; Barcellos-de-Souza et al., 2016; Borriello et al., 2017; Ohlund et al., 2017). Not surprisingly, given their varying origin, CAFs are a heterogeneous cell population that can have strong differences in morphology, cell–cell interaction, and expression profile. However, they share common characteristics, as they are synthetically active, mobile, and invasive, and promote proliferation and immune response. All this is in stark contrast to the quiescent, metabolically inactive fibroblasts found in normal connective tissue (for a review on CAFs, see Kalluri, 2016).
CAFs already contribute to chemoresistance by themselves via various mechanisms: PAI-1, a cytokine produced by CAFs, activates Erk/Akt signaling and suppresses caspase-3 activation, which is necessary for tumor apoptosis following chemotherapeutic stress (Che et al., 2018). Similarly, interleukin 6 (IL6) is also produced predominantly by CAFs and induces expression of resistance-mediating CXCR7 in tumor cells (Qiao et al., 2018; Xu et al., 2018). Especially under hypoxic conditions, CAFs produce high levels of TGFβ, which induces stem cell-like properties in tumor cells including increased resistance to chemotherapy (Tang et al., 2018). Finally, the complex ECM produced by CAFs interferes with therapeutic response by forming a shielding barrier and providing prompts for protective signaling, e.g., via interaction with integrins and cadherins (Eke et al., 2010; Naci et al., 2012; McGrail et al., 2015; Jakubzig et al., 2018; Naik et al., 2018). Based on these observations, it should be possible to significantly improve drug distribution and response to therapy by depleting CAFs in the tumor stroma. However, CAFs seem to have an ambivalent role: depletion of αSMA+ CAFs model induced immunosuppression in a murine PDAC model and being strongly detrimental to the survival of the animals (Ozdemir et al., 2014). Thus, targeting CAFs might not be a straightforward solution. As outlined above, CAFs are a heterogeneous cell population, with in all likelihood different functions in the TME. Understanding these functions might help to separate the divers CAF populations and to develop strategies aimed at specific subgroups. Recently, it has been shown that chemoresistance is mainly driven by a population of CD10+GPR77+ CAFs (Su et al., 2018). Depletion of these CAFs with an anti-GPR77 antibody increased sensitivity to cytotoxic treatment. However, the authors pinpointed the mechanism on IL6 and IL8 that is produced specifically by CD10+GPR77+ CAFs and induces a stem-like phenotype in the cancer cells including increased chemoresistance. Whether CD10+GPR77+ CAFs also contribute disproportionally to ECM formation needs to be seen. Another idea is to reprogram CAFs back to a quiescent phenotype by inhibiting activating pathways, e.g., nuclear factor kappa B (NFκb) signaling that is increased in activated fibroblasts. Inhibition of NFκb reversed the typical CAF phenotype and improved response to cisplatin in ovarian cancer xenografts (Xu et al., 2018). The authors also showed that NFκb inhibition strongly reduced the deposition of collagen in the tumor matrix observed after cisplatin treatment. Activated CAFs also express vitamin D receptor (Sherman et al., 2014). Treatment with calcipotriol, a vitamin D analog, reversed CAFs back to stellate cells, normalized the TME, and increased gemcitabine concentration in the tumor resulting in improved response in PDAC models (Sherman et al., 2014). CAFs display an invasive and migratory phenotype that is also reflected in ROCK-pathway activation compared to quiescent fibroblasts. The ROCK inhibitor, fasudil, reduced collagen deposition, enhanced gemcitabine uptake, and improved treatment response in a transgenic PDAC model (Whatcott et al., 2017). Interestingly, the authors were able to attribute the improved treatment response to improved drug delivery after reduced collagen deposition that resulted from a reversal of the CAFs to a stellate cell phenotype.
In summary, the effects of the ECM and of CAFs on response to chemotherapy are multifaceted: forming a protective barrier that impedes drug diffusion, increased antiapoptotic effects through integrin and FAK signaling, and activation of drug resistance pathways by hypoxia and metabolic stress caused by reduced supply. In addition, ECM-targeting drugs can have toxic effects on the tumor cells themselves. Thus, an improved response to therapy after ECM targeting by itself cannot be attributed to a clear mechanism. In general, additional experiments have to be planned into the studies to prove that the ECM also directly blocks access of antineoplastic drugs to the tumor cells. The studies that were able to demonstrate a direct effect of the ECM on drug delivery and distribution within the tumor are summarized in Table 1.

Table 1. Effect of ECM components on tumor drug delivery.
Cells sense their direct environment via various cell surface receptors, most notably integrins (reviewed in Kechagia et al., 2019). These surface-derived signals enable complex cellular responses to changes in ECM composition and stiffness, which may include responses that alter the cells sensitivity to therapeutics.
Already 20 years ago, it was shown that adherence to ECM via integrin β1 can protect SCLC cells from etoposide-induced apoptosis by blocking proteolytic caspase-3 activation (Sethi et al., 1999). In addition to this PI3K-mediated antiapoptotic effect, ECM–integrin β1 interaction prevents G2/M arrest in response to radiation or chemotherapy by upregulation of p21 and p27 and downregulation of cyclins A, B, and E (Hodkinson et al., 2006). The authors showed that both fibronectin as well as laminin can interact with integrin β1 and cause the protective effect. A third effect of fibronectin–integrin β1 interaction might be the upregulation of survival signals via ILK/Akt/NF-κB, as proposed by another research team that demonstrated protection vs. 5-FU-induced apoptosis in oral squamous carcinoma cells (Nakagawa et al., 2014). Cells sense tissue stiffness through signals from FAK that again cooperates with integrins. FAK signals increase pro-survival pathways like AKT and MAPK. This has been shown to confer resistance to rapamycin an mTOR inhibitor (Yoon et al., 2017). Silencing of FAK with small-interfering RNA (siRNA)/short hairpin RNA (shRNA) reinstated sensitivity to docetaxel in OvCa cells and to 5-FU in CRC cells (Halder et al., 2005; Chen et al., 2010).
Recently, Xiao et al. were able to demonstrate that resistance to therapy can have even more complex causations, involving a combination of various ECM components and their respective receptors (Xiao et al., 2019). The authors used a biomatrix model that enabled them to expose glioblastoma cells to differently modified matrixes in a 3D setting. Thereby, they found that cooperation of HA/CD44 interaction and RGD-triggered integrin αv signaling increases resistance through coactivation of SRC and reduction in proapoptotic BCL-2 expression. Similarly, Nguyen et al. found that increased stiffness on a hydrogel screening platform only in cooperation with collagen-integrin β1 triggered JNK expression-mediated resistance toward sorafenib (Nguyen et al., 2014).
Radiotherapy is considered as one of the potentially curative modalities for cancer. However, preclinical studies suggested that tumor ECM might play a pivotal role in resistance and recurrences to radiotherapy in different cancers (Cordes and Meineke, 2003; Sandfort et al., 2007; Ou et al., 2012). Tumor hypoxia or increased inflammation in TME modifies tumor ECM components and increases collagen deposition, ECM density, and stiffness (Hui and Chen, 2015; Willumsen et al., 2018). In addition, it is known that adhesion to the dense ECM modifies radiation sensitivity of cancer cells (Onoda et al., 1992).
Not only cell adhesion to the ECM but also ECM-induced signaling is mediated largely by integrins, a family of heterodimeric cell surface receptors that directly interact with the ECM (reviewed in Desgrosellier and Cheresh, 2010; Hamidi and Ivaska, 2018). Integrins play a crucial role in cell survival, proliferation, morphogenesis, tumorigenesis, and angiogenesis (Aumailley and Gayraud, 1998; Hapke et al., 2003; Reginato et al., 2003; Demircioglu and Hodivala-Dilke, 2016). Cell survival by integrins is mediated by several signaling pathways such as FAK, Src kinases, PI3K/Akt, MAP kinase signaling, NFκB signaling, p130cas/paxillin, and upregulation of Bcl2-family antiapoptotic proteins (Uhm et al., 1999; Damiano et al., 2001; Cordes et al., 2006; Hehlgans et al., 2007; Serebriiskii et al., 2008). It has also been shown that by altering integrin expression and increasing secretion of survival-promoting ECM molecules, tumor cells develop resistance to anoikis and improve survival in inappropriate ECM environments (Khwaja et al., 1997; Gilmore, 2005). Given their role in cancer cell survival, it is not surprising that integrins, and therefore the ECM-molecules they interact with, are also strongly involved in regulating resistance to radiotherapy: recent studies reported that β1 integrins are upregulated after radiotherapy and play a role in mediating resistance to radiotherapy (Onoda et al., 1992). Experiments in melanoma and sarcoma cells showed that downregulation of the p53 tumor suppressor gene resulted in increased survival following cell detachment and resistance to apoptosis (Lewis et al., 2002; Hazlehurst et al., 2003). Blocking ECM-induced signaling by targeting either β1 integrins or downstream PI3/AKT signaling enhanced the efficacy of radiotherapy in cultured breast cancer cells and implanted BCa xenografts (Liang et al., 2003; Park et al., 2006, 2008). Another study performed on A549 lung cancer cells demonstrated that cell adhesion to the ECM protein fibronectin promotes resistance to radiotherapy (Cordes and Beinke, 2004). The protective function of fibronectin is mediated by α5β1 integrin, as shown in A549 and H1299 lung cancer cells, where the increased fibronectin synthesis after cetuximab treatment attenuated cytotoxic and radio sensitivity (Eke et al., 2013). Similarly, in matrigel-embedded 3D cultures of human malignant human breast cancer cells targeting the interaction between fibronectin and α5β1 integrin enhanced radioresponse by promoting apoptosis (Nam et al., 2010). Further studies also revealed that integrin α5β3 is upregulated after radiotherapy and that treatment with an α5β3 antagonist enhanced radiosensitivity (Abdollahi et al., 2005).
Matrix metalloproteinases (MMPs) are involved in turnover and modulation of ECM components. The MMP-caused fragmentation of ECM components observed in many cancers in TME is generally associated with poor prognosis (Noel et al., 2012). In vitro studies have demonstrated that increased MMP activity and ECM proteolysis leads to enhanced migration, angiogenesis, and metastasis after radiotherapy, and breast cancer cells showed increased invasion capacity with increased expression of MMP2-activating molecules MT1-MMP and TIMP-2 (Paquette et al., 2007; Artacho-Cordon et al., 2012). MMPs also promote angiogenesis by degradation of ECM components and basement membrane, which furthermore can affect outcome of radiotherapy in particular by enhancing escape from stress after the treatment (Nambiar et al., 2015). Furthermore, in preclinical studies, pretreatment with MMP2 inhibitors enhanced sensitivity to radiotherapy (Qian et al., 2002; Kaliski et al., 2005; Badiga et al., 2011). Similarly, MT1-MMP blockade in murine breast carcinomas with a neutralizing antibody enhanced radiosensitivity via increased tumor perfusion (Ager et al., 2015). Interestingly, this was likely caused by a shift from proangiogenic M2 to a phagocytic M1 macrophage phenotype. Other studies also reported that NF-κB causes resistance to radiotherapy by inducing MMP2/9, which promote tumor metastasis and invasion. These studies also showed that treatment of CRC cells with nafamostat mesilate (FUT175), a synthetic serine protease inhibitor, results in downregulation of NF-κB and enhances sensitivity to radiotherapy by inhibiting MMP2/9 (Sugano et al., 2018).
TGFβ signaling modulates the TME by stimulation of myofibroblasts and other stromal cells and by increasing collagen cross-linking enzymes, particularly lysyl oxidases (Egeblad et al., 2010). Both TGFβ and lysyl oxidases can be targeted to improve response to radiotherapy: inhibition of TGFβ signaling enhances radiation sensitivity of non-small-cell lung cancer (NSCLC) cells in vitro and in a Lewis lung carcinoma mouse model (Kirshner et al., 2006; Du et al., 2015; Zhao et al., 2016). While radiotherapy increases LOX secretion in several tumor cell types and in in vivo lung adenocarcinoma xenograft models (Shen et al., 2014), knockdown of LOX2 in DU145 prostate cancer cells using a siRNA approach enhanced their radiosensitivity not only in vitro but also in a xenograft model (Xie et al., 2019). P4HA is another enzyme necessary for correct collagen deposition and directly responsible for increased collagen deposition in tumors (Xiong et al., 2014). Its expression is strongly correlated with response to radiotherapy in breast cancer patients (Toss et al., 2018).
The effect of ionizing radiation on cells is also strongly dependent on their oxygenation status. Hypoxia significantly impairs the effectiveness of radiotherapy (reviewed in Griffioen et al., 2001; Horsman and Overgaard, 2016; Graham and Unger, 2018). Consequently, strategies to improve oxygenation status, in tumors before radiotherapy—e.g., by pretreatment with antiangiogenic drugs—have been devised and tested. Although antiangiogenic drugs were initially designed to reduce tumor growth by starving it, there might be a period after administration when antiangiogenics briefly improve supply by impairing non-functional vessel formation (Claes and Leenders, 2008). Dings et al. found that treatment with Avastin or the antiangiogenic peptide anginex improved oxygenation in various murine tumor models (Griffioen et al., 2001; Dings et al., 2007). Scheduling radiation to line up with this improved oxygenation window enhanced the efficacy of radiotherapy in these models. Correspondingly, response to radiotherapy increased in murine tumors during the improved oxygenation observed 2 days after treatment with the VEGF-R2-inhibitor sunitinib (Matsumoto et al., 2011). However, some evidence suggests that the protective effect of hypoxia on radiation damage is at least in part mediated by Hif1α controlled release of proangiogenic and endothelial protective cytokines that restrict radiation damage on the tumor vessels (Moeller and Dewhirst, 2004). Thus, the beneficial effect of antiangiogenic therapy on response to radiotherapy might not stem from reduced hypoxia but from increased sensitivity of the growth factor signaling-deprived endothelium. Moreover, antiangiogenic treatment leads, in most settings, not to improved supply but to increased hypoxia, both in murine models and in patients (Henke et al., 2007; Keunen et al., 2011; Van der Veldt et al., 2012; Miyazaki et al., 2014; Rohrig et al., 2017). Interestingly, Riesterer et al. showed that fractionated radiation reversed the increased hypoxia after Vatalinib treatment (Riesterer et al., 2006).
Cancer immunotherapy is a promising concept that yielded impressive breakthroughs in recent years. The term immunotherapy is used for a variety of therapeutic approaches that all aim to engage the patient's immune system against cancer. Adoptive transfer methods are based on patient-derived lymphocytes that are expanded, genetically modified, or activated ex vivo before being reinfused (Robbins et al., 2015; Lu et al., 2017). In addition, the application of antitumor vaccination approaches have made considerable progress in recent years (reviewed in Rammensee and Singh-Jasuja, 2013; Accolla et al., 2019; Peng et al., 2019). The method most widely established in the clinic is the treatment with checkpoint inhibitors. Many tumor cells express ligands to T-cell receptors that, upon engagement, block immune surveillance. These ligand/receptor interactions act as inhibitory checkpoints for the adaptive immune system to prevent indiscriminate attacks on the hosts own cells. Currently, the interactions of the ligands CD80/CD86 with the receptor CTLA4 and the PD-L1 with its receptor programmed cell death receptor (PD-1) are of the most importance in cancer immunotherapy. The therapeutic success that draw so much attention in recent years can be mainly attributed to the clinical introduction of inhibitory antibodies against CTLA4 and PD-L1/PD-1 (Eggermont et al., 2018; Gandhi et al., 2018; Paz-Ares et al., 2018)3 As a detailed discussion of the different immunotherapeutic approaches, the regulatory pathways, and various T-lymphocyte populations involved in cancer immune surveillance is beyond the scope of this article, we want to refer the reader to the many excellent review articles that focus on these topics (e.g., Lim et al., 2018; Sharpe and Pauken, 2018; Ganesh et al., 2019).
While some cancers, e.g., melanoma or NSCLC, respond well to immunotherapy and checkpoint inhibition in particular (Robert et al., 2015; Gandhi et al., 2018; Paz-Ares et al., 2018), results in other cancers, e.g., breast carcinomas or PDACs, are less striking (McArthur et al., 2016; Parra et al., 2017; Adams et al., 2018; Rugo et al., 2018). Immunotherapeutic approaches need to get both the drug and T lymphocytes deep into the tumor and in contact with the tumor cells to be effective. A major obstacle for the successful application of the immune therapeutics in some cancer patients seem to be the low infiltration with T lymphocytes, as infiltration rate is highly predictive of response (Issa-Nummer et al., 2013). The infiltration rate is not only determined by the degree the malignant cells are able to provoke an immune response (hence the tumor's immunogenicity) but also by the ECM that can act as a protective shield. Immune cells that are first attracted to side of tumor growth by cytokine gradients (chemotaxis) are often diverted from this direction when confronted with the rigid, ECM-rich encapsulation around the tumor cell clusters. The immune cells migrate then along the gradient of increasing rigidity and ECM-provided adhesion sites (haptotaxis), being diverted from the tumor cells (see Figure 2 for a detailed representation of various ways the ECM might affect immunotherapy). Thereby, the high density of the tumor ECM strongly determines not only distribution of immunomodulatory drugs but also infiltration of immune cells into the tumor (Hallmann et al., 2015; Raave et al., 2018). Increased hypoxia and metabolic stress that are in part a result of the poor diffusion in ECM-rich tumors lead to an upregulation of immunosuppressive factors like IL-10, CCL18, CCL22, TGFβ, and prostaglandin E2 and also VEGF-A (Wei et al., 2011; Xue and Shah, 2013; Schaaf et al., 2018). Especially TGFβ acts in the TME as a suppressor of infiltrating CD8+-cytotoxic lymphocytes (CTLs) and natural killer (NK) cells. TGFβ exercises this effect by attracting regulatory T cells (Tregs) and by working as a M2-polarizing agent for macrophages (Φ) (Ostroukhova et al., 2006; Zhang F. et al., 2016; Schaaf et al., 2018). Both M2-Φ and Tregs negatively regulate infiltration and activity of CD8+-CTLs (Ruella et al., 2017). VEGF-A also is able to recruit Tregs that express NRP1, a VEGF coreceptor, and can directly suppresses activation of T cells (Gavalas et al., 2012; Powell et al., 2018). Consequently, tumor ECM remodeling, stabilization, and accumulation are increasingly recognized as crucial factors controlling infiltration and also differentiation, activation, and polarization of immune cells in the TME (Mushtaq et al., 2018). For the distribution of immunomodulatory drugs, the same consideration apply as for other antineoplastic drugs. Many immunomodulatory drugs, meanwhile, in the clinic are therapeutic antibodies, e.g., the CTLA-4 directed ipilimumab and PD-1 directed pembrolizumab (Robert et al., 2015; Adams et al., 2018; Hodi et al., 2018). Because of their large hydrodynamic diameter, diffusion of these macromolecular drugs is even more affected by a dense, strongly cross-linked ECM.
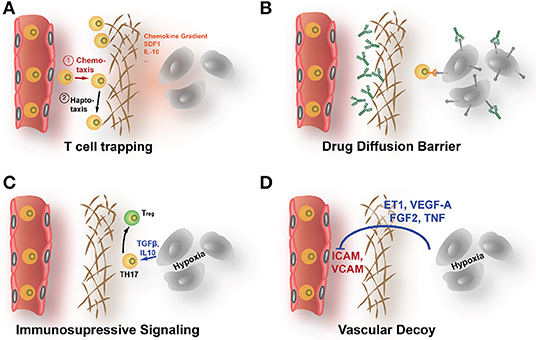
Figure 2. How the ECM affects the efficacy of immunotherapy. (A) The dense ECM can prevent immune cells to reach the tumor cells even in highly immunogenic cancers. Upon contact with areas of increased stiffness, lymphocytes are prone to follow less a chemoattractive gradient but to migrate along the fields of elevated rigidity (haptotaxis). (B) The shielding diffusion barrier that the ECM forms prevents also immunotherapeutic drugs, like checkpoint inhibitory ABs, to reach the tumor. (C) The increased hypoxia that results from poor supply behind the diffusion barrier can directly enhance immune escape by upregulation of immunomodulatory factors like IL-10 or TGF-β. (D) Hypoxia also increases angiogenic signals. Activated blood vessels show reduced ICAM1 expression, impeding attachment and extravasation of immune cells.
Neither T cells nor dendritic cells are able to penetrate dense fibrils in tumor ECM. Salmon et al. showed in human lung tumor specimen that the migration and finally distribution of T cells is dictated by the aligned collagen fibers surrounding tumor islets and perivascular regions in the tumor stroma (Salmon and Donnadieu, 2012; Salmon et al., 2012). This resulted in an accumulation of T cells in the stroma, where most of these immune cells got trapped without being able to reach the targeted tumor cells for destruction. Collagenase treatment ameliorated the trapping effect and increased infiltration into the areas of malignant cells. Another study in PDAC suggested that hyaluronan, by forming dense architecture, impedes infiltration of effector immune cells and drugs in a similar manner as collagen (Jacobetz et al., 2013). The clinical relevance of this shielding function of the stromal ECM that keeps immune cells at distance from the tumor cells was most strikingly demonstrated by Mariathasan et al. that showed in a urothelial cancer patient cohort that non-response to PD-L1 checkpoint inhibition correlated with CTL entrapment in the stromal ECM (Mariathasan et al., 2018).
Hypoxia in the TME, which is partially caused by the dense ECM, leads to an upregulation of angiogenic factors like VEGF. This also impairs infiltration with CTLs as endothelial cells downregulate in response to VEGF cell surface glycoproteins, such as selectins and cell adhesion molecules like ICAM1, ICAM2, and VCAM1 (Griffioen et al., 1996; Achen et al., 2005; Castermans and Griffioen, 2007; Lund and Swartz, 2010). This results in a masking of the supplying blood vessels, as the CTLs cannot longer attach to the endothelium void of the necessary adhesion proteins. Other studies found that the constantly activated tumor endothelium selectively promote transmigration of immunosuppressive Tregs by upregulating adhesion molecules like MadCAM1, CD62-E, CD166, and stabilin1 (Nummer et al., 2007; Shetty et al., 2011). Voron et al. reported that VEGF-A promotes immune escape by inducing Tregs and causing PD-1 expression on VEGF-R2 expressing CD8+ T cells (Voron et al., 2015). CAFs, which regulate stromal matrix and serve as a primary source for matrix-associated proteins, also play an essential role in the infiltration of leukocytes into the tumor (reviewed in Turley et al., 2015).
ECM proteins collagens, laminin, and fibronectin regulate polarization and activation of immune cells in TME (Vaday and Lider, 2000; Simon and Bromberg, 2017; Mushtaq et al., 2018). Transmembrane collagens like Col XVII can trigger immune inhibitory signaling in NK cells via leukocyte associated Ig-like receptor-1 as has be shown in multiple cell lines (Rygiel et al., 2011). In cell culture, high molecular weight HA (in contrast to low molecular weight HA) suppress the immune system by increasing activity of Tregs, presumably acting as an TLR ligand (Bollyky et al., 2009).
MMP and ADAM metalloproteinases also modulate immune and inflammatory responses by degradation of the ECM. The ECM acts as a reservoir of immunomodulatory cytokines and growth factors that are released upon its proteolytic degradation. In addition, the cleavage products of the ECM (e.g., matrikines) can, by themselves, affect immune surveillance. Lastly, metalloproteinases are involved in release of immunoactive factors from the cell surface. ADAM10, ADAM17, and MMP9, for example, are responsible for shedding of major histocompatibility complex class I chain-related molecule A (MICA) from tumor cells (Waldhauer et al., 2008; Chitadze et al., 2013). MICA is a surface ligand and activates the immunoreceptor NKG2D, effectively marking MICA-expressing cells for elimination. Although many cancers express MICA, the upregulation of metalloproteinases enables the tumors cells to escape immune surveillance as has been shown in human prostate cancer, breast cancer, and osteosarcoma cells (Barsoum et al., 2011; Sun et al., 2011). A further link to the ECM exists in the form that ADAM10 is hypoxia regulated, that itself is increased by ECM accumulation (Barsoum et al., 2011). Enzymatic proteolysis of versican results in the release of the matrikine versikine that triggers the generation of conventional dendritic cell in CRC, which subsequently promotes T-cell infiltration (Hope et al., 2017). An immune-enhancing quality of versikine was also detected in myeloma (Hope et al., 2016). Here, however, versikine induced production of inflammatory IL1β and IL6 from myeloma-associated macrophages that increased infiltration of CD8+ T cells.
The ECM also directly regulates escape mechanisms, e.g., the expression of checkpoint molecules. Inhibition of HA synthesis by 4-MU in a mesothelioma xenograft lead to a significant increase in both PD-1 and PD-L1 expression (Cho et al., 2017).
Tumor-associated macrophages are the most frequent immune cells found in the TME (Lewis and Pollard, 2006). TAMs play an important role in mediating adaptive immune response in cancer. Interestingly, macrophages can have both anti- and proinflammatory roles, which is often linked to their M1 or M2 polarization status (reviewed in Tariq et al., 2017). Various ECM components are involved in TAM polarization: At least in cell culture, HA alone is able to strongly drive macrophage polarization toward a protumorigenic and anti-inflammatory M2 phenotype (Kim et al., 2019). This effect of ECM components on macrophage polarization was reported already much earlier for Col I that also drives M2 polarization (Kaplan, 1983; Wesley et al., 1998). Conversely, fibronectin-rich ECM strongly enhances the cytotoxic activity of macrophages toward tumor cells, consistent with M1 polarization (Perri et al., 1982). Although it has not been explicitly shown that ECM targeting affects immune surveillance and the response to immunotherapy by changing TAM polarization, the clear connection between the ECM and macrophage polarization, on the one hand, and macrophage polarization and immune escape, on the other, strongly imply the possibility of such an effect.
Considering the wide and negative effects of the abundant and pathologically altered tumor ECM on various treatment modalities, the interest in targeting the ECM to improve therapeutic efficacy is evident. Of course, removing the treatment-impeding ECM in situ is a major challenge. Nevertheless, in preclinical models, the ECM degradation has been shown to improve drug uptake and response (Table 2). For example, Eikenes et al. studied the possibility of enzymatic hydrolysis of collagen in vivo: Treatment of osteosarcoma xenografts with systemically injected collagenase increased uptake of an antibody specific for the implanted tumor (Eikenes et al., 2004). The collagenase treatment improved overall accumulation of the therapeutic antibody and resulted in a more homogeneous distribution. Wang et al. immobilized collagenase on nanogels and used it to treat a hepatocellular mouse model, in which it improved distribution and response to doxorubicin (Wang et al., 2018). Similarly, hyaluronidase was used to treat hyaluronan-rich tumors and to increase uptake and efficacy of gemcitabine and DOX in experimental PDAC (Provenzano et al., 2012; Jacobetz et al., 2013) and of liposomal DOX in osteosarcoma xenografts (Eikenes et al., 2005). Currently, several clinical trials with pegylated hyaluronidase (PEGPH20) are ongoing (see below).
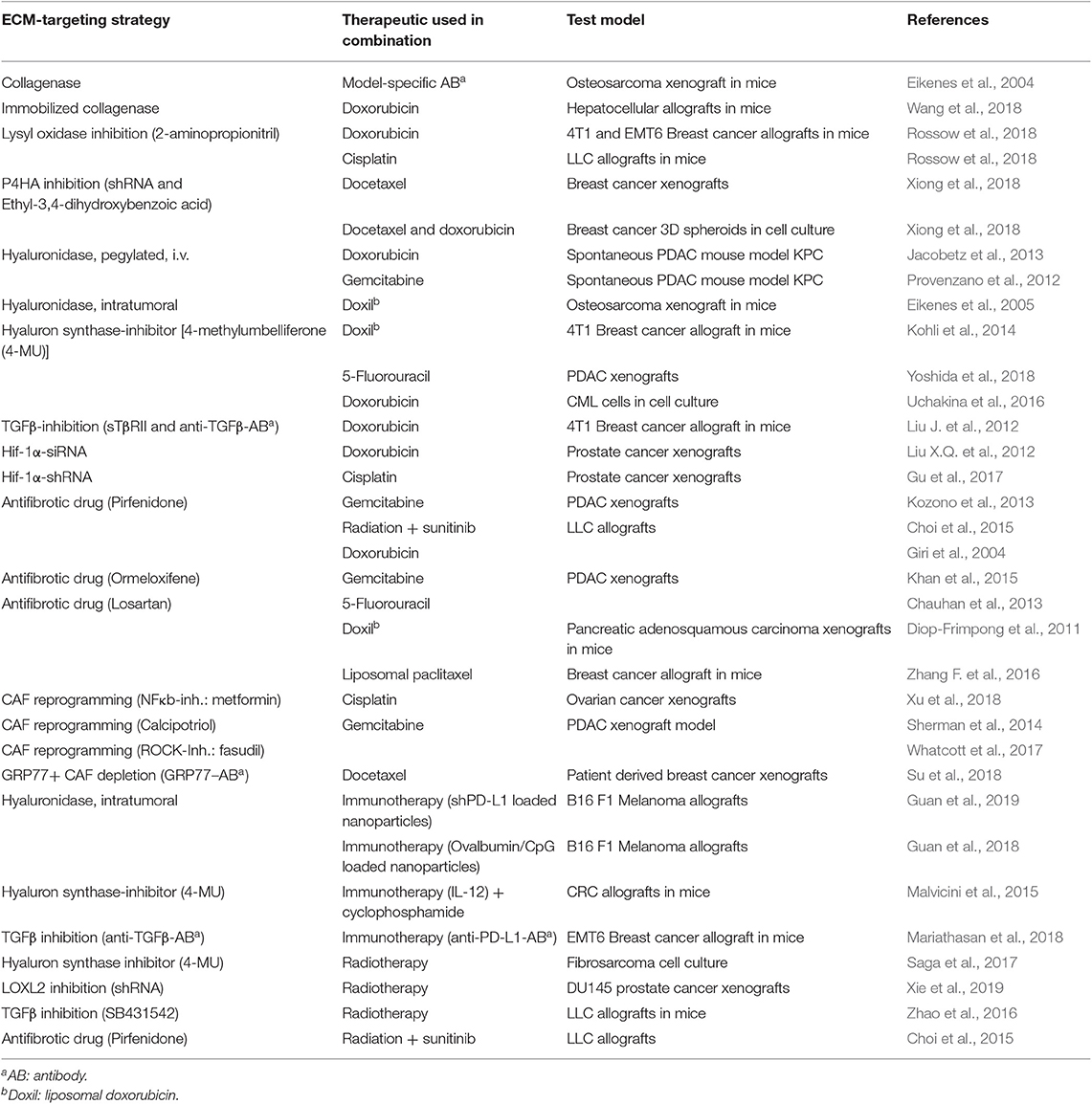
Table 2. Overview on preclinical and cell culture approaches to improve response to therapy by targeting the ECM.
However, as the ECM in tumors is constantly remodeled, it might not be necessary to remove already existent ECM. The perpetual turnover is signified by concomitantly high-level synthesis of ECM macromolecules and degradation of the ECM by tumor-secreted hydrolytic enzymes like MMPs and cathepsins, on the other hand. This opens the possibility to shift the turnover toward net degradation by blocking de novo synthesis. This can be accomplished by either reducing cues that lead to increased expression of ECM molecules, like TGFβ signaling or hypoxia-response pathways, or by inhibiting the various modifying enzymes necessary for proper production, secretion, and maturation of these ECM molecules.
The complex post-translational modifications that collagens have to be subjected to for proper assembly, secretion, and extracellular maturation offer multiple possibilities to interfere with their accumulation in the TME. These modifications are catalyzed by enzymes that are preferred subjects for targeting with small molecule drugs.
Inhibition of lysyl oxidases reduces tissue stiffness on overall collagen deposition, as the maturation process in the form of LOX-induced cross-links further stabilizes collagens and protects them from degradation. Treatment with 2-aminopropionitrile reduced significantly collagen deposition and drug accumulation in various murine allograft models (Rossow et al., 2018). In breast cancer allografts, this leads to increased sensitivity toward DOX and improved efficacy against metastatic disease. In Lewis lung carcinomas, it improved response to cisplatin. Post-translational hydroxylation by collagen P4HA is necessary for correct intracellular processing of collagen molecules. Xiong et al. showed that inhibition of P4HA1 significantly reduced collagen deposition in breast cancer xenografts (Xiong et al., 2018). While inhibition of P4HA1 in these xenografts either with shRNAs or ethyl-3,4-dihydroxybenzoic acid had no antitumor effect on itself, it significantly improved response to docetaxel in reducing tumor growth and pulmonal metastasis. Latest clinical data might underline the necessity to conduct further studies. Recently, phase II trials combining treatment with a LOXL2-directed antibody (simtuzumab) with chemotherapy—gemcitabine in PDAC and FOLFIRI regiment in CRC—ended with unsatisfactory results (Benson et al., 2017; Hecht et al., 2017). Drug scheduling in these trials were not designed to synergistically profit from improved drug delivery after ECM modification, but on the assumption of an additive effect of the cytotoxic drugs with the antitumoral effect that has been demonstrated preclinically for LOXL2 blockade by itself (Barry-Hamilton et al., 2010; Rodriguez et al., 2010). If, as indicated in preclinical tests, the improved drug distribution after collagen destabilization is indeed a deciding mechanism by which LOX targeting can improve treatment outcome, this has to be reflected in the trial design. In patients, the changes of the ECM would take much longer to manifest, than in the generally much faster growing murine test models. Another problem in targeting lysyl oxidases is the shared substrate spectrum and the completely redundant biological activity of the five family members (Molnar et al., 2003). Targeting LOXL2 with an inhibiting antibody might be too specific, and using a small molecule drug that equally inhibits all five lysyl oxidases might yield better results. Incidentally, simtuzumab did not fare well as a stand-alone therapeutic in the treatment for various fibrotic diseases either (Raghu et al., 2017; Verstovsek et al., 2017; Harrison et al., 2018).
As discussed above, hyaluronan synthases (HAS1-3) can be inhibited by 4-MU. Treatment with 4-MU significantly reduces HA accumulation in various murine tumor models. This already confers antiproliferative, proapoptotic, and antimetastatic effects in cultured tumor cells and implanted tumors (Yates et al., 2015; Nagase et al., 2017). The antitumor effect results from reduced CD44 activation that lowers PI3K signaling and AKT and ERK phosphorylation (Kundu et al., 2013; Lompardia et al., 2017). Others have shown that 4-MU increases p38 activation and caspase-3, caspase-9, and PARP cleavage, explaining not only the apoptotic effect but might also indicate an increased sensitivity toward cytotoxic stress (Lokeshwar et al., 2010; Uchakina et al., 2016). Importantly, these proapoptotic effects can be rescued by HA addition to cultured cells, demonstrating that these are direct results of reduction in HAS activity and not off-target effects. The intrinsic antitumor properties of 4-MU complicate concluding whether HAS inhibition improves therapeutic response in cotreatment studies with chemotherapeutics and other tumor-cell-targeted drugs. However, several studies also looked on drug distribution and accumulation. Kohli et al. used liposome-encapsulated 4-MU to treat 4T1 allografts, resulting not only in HA reduction but also in a more heterogeneous distribution of Doxil (liposomal DOX) and reduced tumor growth in the combination treatment group (Kohli et al., 2014). 4-MU increased intracellular accumulation of fluorouracil, indicating an influence on either cell permeability or drug efflux and improved response to 5-fluorouracil in PDAC xenografts (Yoshida et al., 2018).
HA targeting also can improve immunotherapy. 4-MU treatment in syngeneic C26 CRC allografts improved infiltration of T lymphocytes and subsequent therapy with a combination of cyclophosphamide and adenovirally delivered IL12 (Malvicini et al., 2015). Intratumoral application of hyaluronidase improved response to PD-L1-directed shRNA-based immunotherapy and vaccination with ovalbumin/CpG in a melanoma mouse model (Guan et al., 2018, 2019). The authors attributed the improved response to both immunotherapy modalities to an increased T-cell infiltration rate after the hyaluronidase treatment. A positive effect of 4-MU treatment was also reported on the sensitivity of fibrosarcoma cells to radiotherapy (Saga et al., 2017). Under the trade name Hychromone, 4-MU is already marketed in Asia and Europe as a choleretic agent (reviewed in Kudo et al., 2017). As clinical experience with this drug already exists, application in a different cancer treatment setting could be probably enhanced.
A further alternative to targeting either ECM molecules or the enzymes necessary for their production can be to interfere with the signaling pathways that lead to their upregulation in the first place. This might be an interesting strategy, as at least some of these pathways seem to be master regulators of many pathological alterations in the TME. TGFβ might be one of these key factors. TGFβ signaling has been shown to induce production of collagen, lysyl oxidases, and hyaluronan (Zode et al., 2009; Voloshenyuk et al., 2011; Porsch et al., 2013; Xie et al., 2013; Garcia et al., 2016). Treatment of murine syngeneic 4T1 breast carcinomas with a TGFβ inhibitor reduced Col I content and improved accumulation and distribution of DOX (Liu J. et al., 2012). After, establishing that TGFβ expression was a prognostic factor for the response of urothelial cancer patients to PD-L1 directed immunotherapy and that TGFβ correlated with entrapment of CD8+ CTLs in the stroma of the non-responsive tumors, Mariathasan et al. showed that concomitant TGFβ inhibition strongly improved CTL infiltration and outcome of checkpoint therapy in mouse models (Mariathasan et al., 2018). Another signaling pathway that affects ECM synthesis is the hypoxia response via Hif-1α-stabilization. Hif-1 signaling induces collagen expression and lysyl oxidases (Erler et al., 2009; Eisinger-Mathason et al., 2013; Schutze et al., 2015). Thus, targeting Hif-1α could improve response to therapy, and several approaches to inhibit Hif-1α signaling are at various stages of development (reviewed in Hu et al., 2013). As a transcription factor acting by protein–protein and protein–DNA interaction, Hif-1α is notoriously difficult to target. Antisense siRNA and shRNA strategies are mainly used to target Hif-1α in preclinical models: in prostate xenografts, a nanocarrier approach to deliver Hif-1α siRNA improved the efficacy of DOX treatment in rats (Liu X.Q. et al., 2012). In addition, in prostate xenografts, the effective downregulation of Hif-1α using salmonella to deliver shRNA-expressing plasmids resulted in increased sensitivity to cisplatin (CDDP) (Gu et al., 2017). However, for the improved drug distribution and response after either TGFβ- or Hif-1α targeting, alternative mechanisms cannot be excluded: TGFβ does not only induce increased ECM build-up and stabilization, but it is also an important regulator of tumor angiogenesis (reviewed in Goumans et al., 2009; van Meeteren et al., 2011). In the treatment study of 4T1 breast carcinomas with TGFβ inhibitors, Liu et al. observed not only a reduction in Col I deposition but also increased vessel maturation and improved vascular perfusion (Liu J. et al., 2012). Similarly, Hif-1α induces expression of the angiogenic factors VEGF-A, FGF-2, and SDF1 (Enholm et al., 1997; Tang et al., 2004; Du et al., 2008). Moreover, as outlined above, hypoxia and subsequent Hif-1α stabilization by itself upregulates various pathways mediating tumor cell resistance to chemotherapy and radiation (Sullivan et al., 2008; Kolenda et al., 2011). However, killing two birds with one stone is far from an undesirable, and Hif-1α targeting has the potential to simultaneously reduce metastatic and invasive behavior, sensitize tumor cells to therapy, and ameliorate the detrimental effects of a pathologically altered ECM and dysfunctional vasculature on drug supply, distribution, and resistance.
As mentioned before, stromal cells are a major source of the vast amounts of ECM that, in many tumors, obstruct efficient and homogeneous drug delivery. The infiltration and often encapsulation of tumors with CAFs parallels in many respects fibrosis under other pathological conditions. Therefore, it is of course evident to test antifibrotic drugs that are in clinical use for other ailments for their potential to reduce the malignant tumor ECM and to increase drug transport. Konzono et al. found that the efficacy of gemcitabine in pancreas cancer xenografts was increased after treatment with the antifibrotic drug pirfenidone (Kozono et al., 2013). However, pirfenidone had a significant antitumor effect by itself. Thus, it is possible that the observed increased efficacy of the combination only reflects an additive effect. In a trimodal combination approach, pirfenidone improved response to radiation and sunitinib in a murine Lewis lung carcinoma model (Choi et al., 2015). Histological analysis indicated that the increased collagen production caused by radiation reduced efficacy of sunitinib. This desmoplastic response was prevented by pirfenidone, restoring sunitinib sensitivity. Pirfenidone might have additional beneficial effects in the treatment of cancer: DOX has significant renal and cardiac toxicity limiting its lifetime doses in cancer patients. Pirfenidone ameliorated these toxic effects in rats, presumably by reducing the fibrotic reaction in the affected organs (Giri et al., 2004). Pirfenidone has also some sensitizing effect in 2D coculture experiments, indicating that it is not only improving therapy response by interfering with ECM production: by treating CAF-NSCLC cell cocultures with pirfenidone, the sensitivity of both cells was increased (Mediavilla-Varela et al., 2016). Ormeloxifene, an estrogen receptor modulator, in contrast to pirfenidone, had no substantial antitumor effect in the PDAC xenografts it was tested in Khan et al. (2015). Still, it improved the efficacy of gemcitabine treatment. Ormeloxifene also inhibits sonic hedgehog signaling, a pathway involved in desmoplasia in PDAC and reduced fibroblast infiltration and Col I deposition. Losartan, an angiotensin receptor antagonist, increased perfused vessel density and reduced Col I and hyaluronan synthesis in various murine tumors. This resulted in improved delivery and efficacy of 5-FU and DOX (Diop-Frimpong et al., 2011; Chauhan et al., 2013). Losartan also improved response to liposomal paclitaxel in a murine model for stage IV metastatic breast cancer (Zhang L. et al., 2016). This study also showed that losartan reduced TGFβ1 expression and subsequently the production of Col I and LOX.
Considering the interesting results already obtained in preclinical tests, it is not surprising that several of the approaches to alter the ECM to improve response to concomitant therapy are already in at least early clinical test phases (see Table 3 for details).
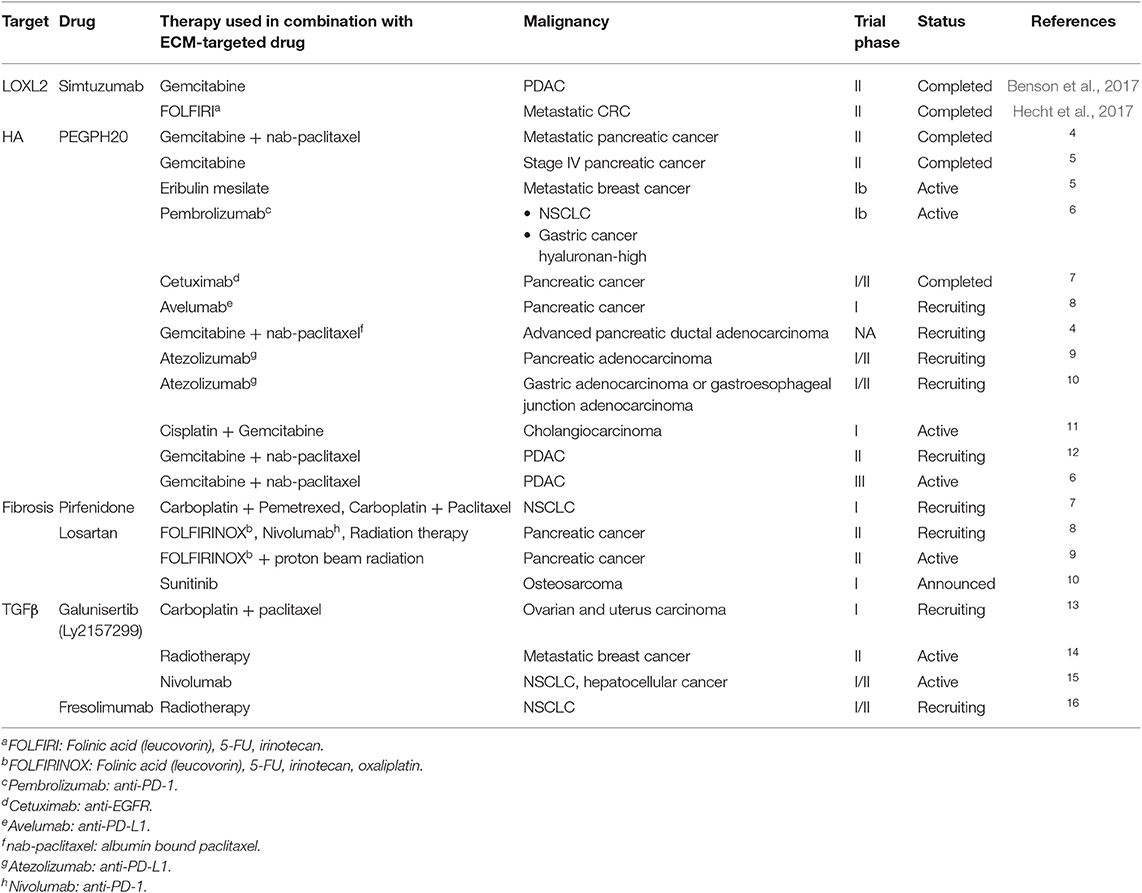
Table 3. ECM-targeted drugs in combination with tumor-directed therapy in clinical trials.
We already mentioned the completed phase II trials using LOXL2 antibody in combination with gemcitabine and FOLFIRI in PDAC and CRC patients, respectively (Benson et al., 2017; Hecht et al., 2017). The furthest progress was probably the clinical evaluation of hyaluronidase as an auxiliary treatment. Pegylated hyaluronidase (PEGPH20) is tested in PDAC in combination with gemcitabine, nab-paclitaxel, avelumab, and cetuximab (Hingorani et al., 2016; Doherty et al., 2018; Gourd, 2018; Infante et al., 2018)17, in breast cancer with erlotinib18 and a range of other drugs in various different cancers. As far as the studies are completed and evaluated, the results appear more encouraging than those from the LOXL2 trials, and a phase III trial combining PEGPH20 with nab-paclitaxel plus gemcitabine in PDAC started already in early 201619. Pirfenidone plus standard of care chemotherapy is undergoing phase I evaluation in NSCLC20. In addition, pirfenidone is tested for its ability to ameliorate fibrosis in cancer patients resulting from radiotherapy. As mentioned above, the angiotensin-II inhibitor losartan might, in addition to its vascular effects, considerably affect the ECM. It is under clinical investigation in combination with immuno-, radio-, and chemotherapy21, 22,23.
The ECM in solid tumors is strongly involved in determining the course of the disease and also the results of our efforts to treat malignancies. Interfering with the synthesis and accumulation of ECM or related processes has the potential to significantly improve the outcome of concomitant therapeutic approaches, whether these are conventional cytotoxic treatments, radiotherapy, or targeted therapy including immunotherapy. We have already begun to explore the potential of ECM targeting to improve response to antineoplastic therapy. These research efforts are still mainly in the realms of basic and early translational research and often far from reaching the clinic. However, there are major challenges we will face over the next decades. First, from the view of basic scientist interested in understanding the fundamental mechanisms behind how the ECM influences cancer treatment, there is the problem that targeting ECM synthesis almost always affects other processes in the TME. Likewise, targeting other components of the TME, e.g., angiogenesis or immune cells also affects the ECM. This mutual interdependence of various processes in the tumor often complicates the interpretation of experiments. A meticulous understanding of the underlying mechanisms, however, is often necessary for successful translation in the clinic. Second, the ECM is a complex mixture of numerous macromolecules, with divers characteristics, that often rely on complicated multistep processes for synthesis. This enables interfering with tumoral ECM accumulation in various ways and opens the possibility for tailor-made ECM-targeting strategies for various malignancies. However, it is also a challenge to select from this multitude of possible points of attack, the right ones, that promise the strongest effects and that are least likely cause problems with the functions of the ECM in physiological processes. Finally, although ECM destabilization can already positively affect the course of neoplastic diseases, by reducing invasiveness, progression, and metastasis, the most likely application for ECM-targeted approaches is not a stand-alone therapy but modalities where they are used to cause synergistic improvements in combination with other tumor-directed forms of therapy. Combining various forms that are supposed to synergistically improve each other's performance asks for successively more complicated forms of clinical evaluation, with more extensive planning, more detailed follow-up procedures, more surrogate endpoints, and possibly more trial arms. If we master these challenges, ECM-targeting approaches have the potential to significantly add to our repertoire of cancer-fighting strategies and moreover strongly improve the performance of the therapies already at our hand.
EH, RN, and SE wrote the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thankfully acknowledge funding by the Wilhelm Sander-Stiftung (2018.001.1) and the Interdisciplinary Centre for Clinical Research (IZKF) of the Universitätsklinikum Würzburg (B-369).
1. ^FDA Approves Keytruda (pembrolizumab) for Advanced Melanoma. Available online at: https://www.drugs.com/newdrugs/fda-approves-keytruda-pembrolizumab-advanced-melanoma-4079.html (accessed December 22, 2018).
2. ^FDA Approves YERVOY™ (ipilimumab) for the Treatment of Patients with Newly Diagnosed or Previously-Treated Unresectable or Metastatic Melanoma, the Deadliest form of Skin Cancer. Available online at: https://news.bms.com/press-release/rd-news/fda-approves-yervoy-ipilimumab-treatment-patients-newly-diagnosed-or-previousl.
3. ^FDA Grants Accelerated Approval to Ipilimumab for MSI-H or dMMR Metastatic Colorectal Cance. Available online at: https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm613227.htm.
4. ^PEGPH20 Plus Nab-Paclitaxel Plus Gemcitabine Compared with Nab-Paclitaxel Plus Gemcitabine in Subjects with Stage IV Untreated Pancreatic Cancer (HALO-109-202). Available online at: https://clinicaltrials.gov/ct2/show/record/NCT01839487 (accessed May 2, 2019).
5. ^Study of Gemcitabine + PEGPH20 vs Gemcitabine Alone in Stage IV Previously Untreated Pancreatic Cancer. Available online at: https://clinicaltrials.gov/ct2/show/NCT01453153 (accessed May 2, 2019).
6. ^Phase 1b Open-Label Study of PEGylated Recombinant Human Hyaluronidase (PEGPH20) with Pembrolizumab. Available online at: https://clinicaltrials.gov/ct2/show/NCT02563548 (accessed May 2, 2019).
7. ^Two Stage Study of Single Dose PEGPH20 and Cetuximab in Patients with Pancreatic Adenocarcinoma Prior to Surgical Resection. Available online at: https://clinicaltrials.gov/ct2/show/NCT02241187 (accessed May 2, 2019).
8. ^A Trial of PEGPH20 in Combination with Avelumab in Chemotherapy Resistant Pancreatic Cancer. Available online at: https://clinicaltrials.gov/ct2/show/NCT03481920 (accessed May 2, 2019).
9. ^A Study of Multiple Immunotherapy-Based Treatment Combinations in Participants with Metastatic Pancreatic Ductal Adenocarcinoma (Morpheus-Pancreatic Cancer). Available online at: https://clinicaltrials.gov/ct2/show/NCT03193190 (accessed May 2, 2019).
10. ^A Study of Multiple Immunotherapy-Based Treatment Combinations in Patients with Locally Advanced Unresectable or Metastatic Gastric or Gastroesophageal Junction Cancer (G/GEJ) (Morpheus-Gastric Cancer). Available online at: https://clinicaltrials.gov/ct2/show/NCT03281369 (accessed May 2, 2019).
11. ^Study of PEGPH20 with Cisplatin (CIS) and Gemcitabine (GEM); PEGPH20 with Atezolizumab, CIS, and GEM; and CIS and GEM Alone in Participants with Previously Untreated, Unresectable, Locally Advanced, or Metastatic Intrahepatic and Extrahepatic Cholangiocarcinoma and Gallbladder Adenocarcinoma. Available online at: https://clinicaltrials.gov/ct2/show/NCT03267940 (accessed May 2, 2019).
12. ^PEGPH20, Gemicitabine and Nab-Paclitaxel for Pancreatic Ductal Adenocarcinoma. Available online at: https://clinicaltrials.gov/ct2/show/NCT02487277 (accessed May 2, 2019).
13. ^Paclitaxel/Carboplatin + Galunisertib for Patients with Carcinosarcoma of the Uterus or Ovary. Available online at: https://clinicaltrials.gov/ct2/show/NCT03206177 (accessed May 2, 2019).
14. ^LY2157299 Monohydrate (LY2157299) and Radiotherapy in Metastatic Breast Cancer. Available online at: https://clinicaltrials.gov/ct2/show/NCT02538471 (accessed May 2, 2019).
15. ^A Study of Galunisertib (LY2157299) in Combination with Nivolumab in Advanced Refractory Solid Tumors and in Recurrent or Refractory NSCLC, or Hepatocellular Carcinoma. Available online at: https://clinicaltrials.gov/ct2/show/NCT02423343 (accessed May 2, 2019).
16. ^SABR-ATAC: A Trial of TGF-beta Inhibition and Stereotactic Ablative Radiotherapy for Early Stage Non-small Cell Lung Cancer. Available online at: https://clinicaltrials.gov/ct2/show/NCT02581787 (accessed May 2, 2019).
17. ^Study of Gemcitabine, Nab-paclitaxel, PEGPH20 and Rivaroxaban for Advanced Pancreatic Adenocarcinoma. Available online at: https://clinicaltrials.gov/ct2/show/NCT02921022 (accessed May 2, 2019).
18. ^Study of Eribulin Mesylate in Combination with PEGylated Recombinant Human Hyaluronidase (PEGPH20) Versus Eribulin Mesylate Alone in Subjects with Human Epidermal Growth Factor Receptor 2 (HER2)-Negative, High-Hyaluronan (HA) Metastatic Breast Cancer (MBC). Available online at: https://clinicaltrials.gov/ct2/show/NCT02753595 (accessed May 2, 2019).
19. ^A Study of PEGylated Recombinant Human Hyaluronidase in Combination with Nab-Paclitaxel Plus Gemcitabine Compared with Placebo Plus Nab-Paclitaxel and Gemcitabine in Participants with Hyaluronan-High Stage IV Previously Untreated Pancreatic Ductal Adenocarcinoma. Available online at: https://clinicaltrials.gov/ct2/show/NCT02715804 (accessed May 2, 2019).
20. ^Pirfenidone Combined with Standard First-Line Chemotherapy in Advanced-Stage Lung NSCLC. Available online at: https://clinicaltrials.gov/ct2/show/NCT03177291 (accessed May 2, 2019).
21. ^Losartan and Nivolumab in Combination with FOLFIRINOX and SBRT in Localized Pancreatic Cancer. Available online at: https://clinicaltrials.gov/ct2/show/NCT03563248 (accessed May 2, 2019).
22. ^Proton w/FOLFIRINOX-Losartan for Pancreatic Cancer. Available online at: https://clinicaltrials.gov/ct2/show/NCT01821729 (accessed May 2, 2019).
23. ^Losartan + Sunitinib in Treatment of Osteosarcoma. Available online at: https://clinicaltrials.gov/ct2/show/NCT03900793 (accessed May 2, 2019).
Abdollahi, A., Griggs, D. W., Zieher, H., Roth, A., Lipson, K. E., Saffrich, R., et al. (2005). Inhibition of alpha(v)beta3 integrin survival signaling enhances antiangiogenic and antitumor effects of radiotherapy. Clin. Cancer Res. 11, 6270–6279. doi: 10.1158/1078-0432.CCR-04-1223
Accolla, R. S., Ramia, E., Tedeschi, A., and Forlani, G. (2019). CIITA-driven MHC class II expressing tumor cells as antigen presenting cell performers: toward the construction of an optimal anti-tumor vaccine. Front. Immunol. 10:1806. doi: 10.3389/fimmu.2019.01806
Acerbi, I., Cassereau, L., Dean, I., Shi, Q., Au, A., Park, C., et al. (2015). Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr. Biol. 7, 1120–1134. doi: 10.1039/c5ib00040h
Achen, M. G., McColl, B. K., and Stacker, S. A. (2005). Focus on lymphangiogenesis in tumor metastasis. Cancer Cell 7, 121–127. doi: 10.1016/j.ccr.2005.01.017
Adams, S., Schmid, P., Rugo, H. S., Winer, E. P., Loirat, D., Awada, A., et al. (2018). Pembrolizumab monotherapy for previously treated metastatic triple-negative breast cancer: cohort a of the phase 2 KEYNOTE-086 study. Ann. Oncol. 30, 405–411. doi: 10.1093/annonc/mdy517
Ager, E. I., Kozin, S. V., Kirkpatrick, N. D., Seano, G., Kodack, D. P., Askoxylakis, V., et al. (2015). Blockade of MMP14 activity in murine breast carcinomas: implications for macrophages, vessels, and radiotherapy. J. Natl. Cancer Inst. 107:djv017. doi: 10.1093/jnci/djv017
Ajeti, V., Nadiarnykh, O., Ponik, S. M., Keely, P. J., Eliceiri, K. W., and Campagnola, P. J. (2011). Structural changes in mixed Col I/Col V collagen gels probed by SHG microscopy: implications for probing stromal alterations in human breast cancer. Biomed. Opt. Express 2, 2307–2316. doi: 10.1364/BOE.2.002307
Albrechtsen, R., Nielsen, M., Wewer, U., Engvall, E., and Ruoslahti, E. (1981). Basement membrane changes in breast cancer detected by immunohistochemical staining for laminin. Cancer Res. 41, 5076–5081.
Alon, Y., Horowitz, A. T., Biran, S., Weiss, D. W., and Doljanski, F. (1986). Immunofluorescent characterization of fibronectin, laminin, and keratin in normal and neoplastic human mammary epithelial cells in culture and in breast tissue sections. Int. J. Tissue React. 8, 401–410.
Artacho-Cordon, F., Rios-Arrabal, S., Lara, P. C., Artacho-Cordon, A., Calvente, I., and Nunez, M. I. (2012). Matrix metalloproteinases: potential therapy to prevent the development of second malignancies after breast radiotherapy. Surg. Oncol. 21, e143–e151. doi: 10.1016/j.suronc.2012.06.001
Aumailley, M., and Gayraud, B. (1998). Structure and biological activity of the extracellular matrix. J. Mol. Med. 76, 253–265. doi: 10.1007/s001090050215
Auvinen, P., Tammi, R., Parkkinen, J., Tammi, M., Agren, U., Johansson, R., et al. (2000). Hyaluronan in peritumoral stroma and malignant cells associates with breast cancer spreading and predicts survival. Am. J. Pathol. 156, 529–536. doi: 10.1016/S0002-9440(10)64757-8
Auvinen, P. K., Parkkinen, J. J., Johansson, R. T., Agren, U. M., Tammi, R. H., Eskelinen, M. J., et al. (1997). Expression of hyaluronan in benign and malignant breast lesions. Int. J. Cancer 74, 477–481. doi: 10.1002/(sici)1097-0215(19971021)74:5<477::aid-ijc1>3.0.co;2-0
Badiga, A. V., Chetty, C., Kesanakurti, D., Are, D., Gujrati, M., Klopfenstein, J. D., et al. (2011). MMP-2 siRNA inhibits radiation-enhanced invasiveness in glioma cells. PLoS ONE 6:e20614. doi: 10.1371/journal.pone.0020614
Bagordakis, E., Sawazaki-Calone, I., Macedo, C. C., Carnielli, C. M., de Oliveira, C. E., Rodrigues, P. C., et al. (2016). Secretome profiling of oral squamous cell carcinoma-associated fibroblasts reveals organization and disassembly of extracellular matrix and collagen metabolic process signatures. Tumour Biol. 37, 9045–9057. doi: 10.1007/s13277-015-4629-y
Baker, A. M., Bird, D., Lang, G., Cox, T. R., and Erler, J. T. (2012). Lysyl oxidase enzymatic function increases stiffness to drive colorectal cancer progression through FAK. Oncogene 32, 1863–1868. doi: 10.1038/onc.2012.202
Baker, A. M., Bird, D., Welti, J. C., Gourlaouen, M., Lang, G., Murray, G. I., et al. (2013). Lysyl oxidase plays a critical role in endothelial cell stimulation to drive tumor angiogenesis. Cancer Res. 73, 583–594. doi: 10.1158/0008-5472.CAN-12-2447
Bar, J. K., Grelewski, P., Popiela, A., Noga, L., and Rabczynski, J. (2004). Type IV collagen and CD44v6 expression in benign, malignant primary and metastatic ovarian tumors: correlation with Ki-67 and p53 immunoreactivity. Gynecol. Oncol. 95, 23–31. doi: 10.1016/j.ygyno.2004.06.046
Barcellos-de-Souza, P., Comito, G., Pons-Segura, C., Taddei, M. L., Gori, V., Becherucci, V., et al. (2016). Mesenchymal stem cells are recruited and activated into carcinoma-associated fibroblasts by prostate cancer microenvironment-derived TGF-beta1. Stem Cells 34, 2536–2547. doi: 10.1002/stem.2412
Barry-Hamilton, V., Spangler, R., Marshall, D., McCauley, S., Rodriguez, H. M., Oyasu, M., et al. (2010). Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat. Med. 16, 1009–1017. doi: 10.1038/nm.2208
Barsky, S. H., Rao, C. N., Grotendorst, G. R., and Liotta, L. A. (1982). Increased content of type V collagen in desmoplasia of human breast carcinoma. Am. J. Pathol. 108, 276–283.
Barsoum, I. B., Hamilton, T. K., Li, X., Cotechini, T., Miles, E. A., Siemens, D. R., et al. (2011). Hypoxia induces escape from innate immunity in cancer cells via increased expression of ADAM10: role of nitric oxide. Cancer Res. 71, 7433–7441. doi: 10.1158/0008-5472.CAN-11-2104
Benson, A. B. III., Wainberg, Z. A., Hecht, J. R., Vyushkov, D., Dong, H., Bendell, J., et al. (2017). A phase II randomized, double-blind, placebo-controlled study of simtuzumab or placebo in combination with gemcitabine for the first-line treatment of pancreatic adenocarcinoma. Oncologist 22, 241–e15. doi: 10.1634/theoncologist.2017-0024
Bergamaschi, A., Tagliabue, E., Sorlie, T., Naume, B., Triulzi, T., Orlandi, R., et al. (2008). Extracellular matrix signature identifies breast cancer subgroups with different clinical outcome. J. Pathol. 214, 357–367. doi: 10.1002/path.2278
Bertrand, P., Girard, N., Delpech, B., Duval, C., d'Anjou, J., and Dauce, J. P. (1992). Hyaluronan (hyaluronic acid) and hyaluronectin in the extracellular matrix of human breast carcinomas: comparison between invasive and non-invasive areas. Int. J. Cancer 52, 1–6. doi: 10.1002/ijc.2910520102
Bollyky, P. L., Falk, B. A., Wu, R. P., Buckner, J. H., Wight, T. N., and Nepom, G. T. (2009). Intact extracellular matrix and the maintenance of immune tolerance: high molecular weight hyaluronan promotes persistence of induced CD4+CD25+ regulatory T cells. J. Leukoc. Biol. 86, 567–572. doi: 10.1189/jlb.0109001
Borriello, L., Nakata, R., Sheard, M. A., Fernandez, G. E., Sposto, R., Malvar, J., et al. (2017). Cancer-associated fibroblasts share characteristics and protumorigenic activity with mesenchymal stromal cells. Cancer Res. 77, 5142–5157. doi: 10.1158/0008-5472.CAN-16-2586
Brown, L. F., Guidi, A. J., Schnitt, S. J., Van De Water, L., Iruela-Arispe, M. L., Yeo, T. K., et al. (1999). Vascular stroma formation in carcinoma in situ, invasive carcinoma, and metastatic carcinoma of the breast. Clin. Cancer Res. 5, 1041–1056.
Casey, T., Bond, J., Tighe, S., Hunter, T., Lintault, L., Patel, O., et al. (2009). Molecular signatures suggest a major role for stromal cells in development of invasive breast cancer. Breast Cancer Res. Treat. 114, 47–62. doi: 10.1007/s10549-008-9982-8
Castermans, K., and Griffioen, A. W. (2007). Tumor blood vessels, a difficult hurdle for infiltrating leukocytes. Biochim. Biophys. Acta 1776, 160–174. doi: 10.1016/j.bbcan.2007.07.005
Chauhan, V. P., Martin, J. D., Liu, H., Lacorre, D. A., Jain, S. R., Kozin, S. V., et al. (2013). Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun. 4:2516. doi: 10.1038/ncomms3516
Che, Y., Wang, J., Li, Y., Lu, Z., Huang, J., Sun, S., et al. (2018). Cisplatin-activated PAI-1 secretion in the cancer-associated fibroblasts with paracrine effects promoting esophageal squamous cell carcinoma progression and causing chemoresistance. Cell Death Dis. 9:759. doi: 10.1038/s41419-018-0808-2
Chen, D., Chen, D., Cao, D., Hu, J., and Yao, Y. (2018). A signature based on survival-related genes identifies high-risk glioblastomas harboring immunosuppressive and aggressive ECM characteristics. Zhong Nan Da Xue Xue Bao Yi Xue Ban 43, 368–382. doi: 10.11817/j.issn.1672-7347.2018.04.006
Chen, Y., Wang, Z., Chang, P., Xiang, L., Pan, F., Li, J., et al. (2010). The effect of focal adhesion kinase gene silencing on 5-fluorouracil chemosensitivity involves an Akt/NF-kappaB signaling pathway in colorectal carcinomas. Int. J. Cancer 127, 195–206. doi: 10.1002/ijc.25025
Cheng, X. B., Sato, N., Kohi, S., and Yamaguchi, K. (2013). Prognostic impact of hyaluronan and its regulators in pancreatic ductal adenocarcinoma. PLoS ONE 8:e80765. doi: 10.1371/journal.pone.0080765
Chitadze, G., Lettau, M., Bhat, J., Wesch, D., Steinle, A., Furst, D., et al. (2013). Shedding of endogenous MHC class I-related chain molecules A and B from different human tumor entities: heterogeneous involvement of the “a disintegrin and metalloproteases” 10 and 17. Int. J. Cancer 133, 1557–1566. doi: 10.1002/ijc.28174
Cho, H., Matsumoto, S., Fujita, Y., Kuroda, A., Menju, T., Sonobe, M., et al. (2017). Trametinib plus 4-methylumbelliferone exhibits antitumor effects by ERK blockade and CD44 downregulation and affects PD-1 and PD-L1 in malignant pleural mesothelioma. J. Thorac. Oncol. 12, 477–490. doi: 10.1016/j.jtho.2016.10.023
Choi, S. H., Nam, J. K., Jang, J., Lee, H. J., and Lee, Y. J. (2015). Pirfenidone enhances the efficacy of combined radiation and sunitinib therapy. Biochem. Biophys. Res. Commun. 462, 138–143. doi: 10.1016/j.bbrc.2015.04.107
Claes, A., and Leenders, W. (2008). Vessel normalization by VEGF inhibition. A complex story. Cancer Biol. Ther. 7, 1014–1016. doi: 10.4161/cbt.7.7.6474
Conti, J. A., Kendall, T. J., Bateman, A., Armstrong, T. A., Papa-Adams, A., Xu, Q., et al. (2008). The desmoplastic reaction surrounding hepatic colorectal adenocarcinoma metastases aids tumor growth and survival via alphav integrin ligation. Clin. Cancer Res. 14, 6405–6413. doi: 10.1158/1078-0432.CCR-08-0816
Cordes, N., and Beinke, C. (2004). Fibronectin alters cell survival and intracellular signaling of confluent A549 cultures after irradiation. Cancer Biol. Ther. 3, 47–53. doi: 10.4161/cbt.3.1.570
Cordes, N., and Meineke, V. (2003). Cell adhesion-mediated radioresistance (CAM-RR). Extracellular matrix-dependent improvement of cell survival in human tumor and normal cells in vitro. Strahlenther. Onkol. 179, 337–344. doi: 10.1007/s00066-003-1074-4
Cordes, N., Seidler, J., Durzok, R., Geinitz, H., and Brakebusch, C. (2006). β1-integrin-mediated signaling essentially contributes to cell survival after radiation-induced genotoxic injury. Oncogene 25, 1378–1390. doi: 10.1038/sj.onc.1209164
da Silva, B. B., Lopes-Costa, P. V., dos Santos, A. R., de Sousa-Junior, E. C., Alencar, A. P., Pires, C. G., et al. (2009). Comparison of three vascular endothelial markers in the evaluation of microvessel density in breast cancer. Eur. J. Gynaecol. Oncol. 30, 285–288.
Damiano, J. S., Hazlehurst, L. A., and Dalton, W. S. (2001). Cell adhesion-mediated drug resistance (CAM-DR) protects the K562 chronic myelogenous leukemia cell line from apoptosis induced by BCR/ABL inhibition, cytotoxic drugs, and gamma-irradiation. Leukemia 15, 1232–1239. doi: 10.1038/sj.leu.2402179
Deak, S. B., Glaug, M. R., Pierce, R. A., Bancila, E., Amenta, P., Mackenzie, J. W., et al. (1991). Desmoplasia in benign and malignant breast disease is characterized by alterations in level of mRNAs coding for types I and III procollagen. Matrix 11, 252–258. doi: 10.1016/S0934-8832(11)80232-5
Demircioglu, F., and Hodivala-Dilke, K. (2016). alphavbeta3 integrin and tumour blood vessels-learning from the past to shape the future. Curr. Opin. Cell Biol. 42, 121–127. doi: 10.1016/j.ceb.2016.07.008
Desgrosellier, J. S., and Cheresh, D. A. (2010). Integrins in cancer: biological implications and therapeutic opportunities. Nat. Rev. Cancer 10, 9–22. doi: 10.1038/nrc2748
Dewhirst, M. W., and Secomb, T. W. (2017). Transport of drugs from blood vessels to tumour tissue. Nat. Rev. Cancer 17, 738–750. doi: 10.1038/nrc.2017.93
Dings, R. P., Loren, M., Heun, H., McNiel, E., Griffioen, A. W., Mayo, K. H., et al. (2007). Scheduling of radiation with angiogenesis inhibitors anginex and Avastin improves therapeutic outcome via vessel normalization. Clin. Cancer Res. 13, 3395–3402. doi: 10.1158/1078-0432.CCR-06-2441
Diop-Frimpong, B., Chauhan, V. P., Krane, S., Boucher, Y., and Jain, R. K. (2011). Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc. Natl. Acad. Sci. U.S.A. 108, 2909–2914. doi: 10.1073/pnas.1018892108
Doherty, G. J., Tempero, M., and Corrie, P. G. (2018). HALO-109-301: a phase III trial of PEGPH20 (with gemcitabine and nab-paclitaxel) in hyaluronic acid-high stage IV pancreatic cancer. Future Oncol. 14, 13–22. doi: 10.2217/fon-2017-0338
Doublier, S., Belisario, D. C., Polimeni, M., Annaratone, L., Riganti, C., Allia, E., et al. (2012). HIF-1 activation induces doxorubicin resistance in MCF7 3-D spheroids via P-glycoprotein expression: a potential model of the chemo-resistance of invasive micropapillary carcinoma of the breast. BMC Cancer 12, 4. doi: 10.1186/1471-2407-12-4
Du, R., Lu, K. V., Petritsch, C., Liu, P., Ganss, R., Passegue, E., et al. (2008). HIF1α induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 13, 206–220. doi: 10.1016/j.ccr.2008.01.034
Du, S., Bouquet, S., Lo, C. H., Pellicciotta, I., Bolourchi, S., Parry, R., et al. (2015). Attenuation of the DNA damage response by transforming growth factor-beta inhibitors enhances radiation sensitivity of non-small-cell lung cancer cells in vitro and in vivo. Int. J. Radiat. Oncol. Biol. Phys. 91, 91–99. doi: 10.1016/j.ijrobp.2014.09.026
Egeblad, M., Rasch, M. G., and Weaver, V. M. (2010). Dynamic interplay between the collagen scaffold and tumor evolution. Curr. Opin. Cell Biol. 22, 697–706. doi: 10.1016/j.ceb.2010.08.015
Eggermont, A. M. M., Blank, C. U., Mandala, M., Long, G. V., Atkinson, V., Dalle, S., et al. (2018). Adjuvant pembrolizumab versus placebo in resected stage III melanoma. N. Engl. J. Med. 378, 1789–1801. doi: 10.1056/NEJMoa1802357
Eikenberry, S. (2009). A tumor cord model for doxorubicin delivery and dose optimization in solid tumors. Theor. Biol. Med. Model. 6:16. doi: 10.1186/1742-4682-6-16
Eikenes, L., Bruland, O. S., Brekken, C., and Davies Cde, L. (2004). Collagenase increases the transcapillary pressure gradient and improves the uptake and distribution of monoclonal antibodies in human osteosarcoma xenografts. Cancer Res. 64, 4768–4773. doi: 10.1158/0008-5472.CAN-03-1472
Eikenes, L., Tari, M., Tufto, I., Bruland, O. S., and de Lange Davies, C. (2005). Hyaluronidase induces a transcapillary pressure gradient and improves the distribution and uptake of liposomal doxorubicin (Caelyx) in human osteosarcoma xenografts. Br. J. Cancer 93, 81–88. doi: 10.1038/sj.bjc.6602626
Eisinger-Mathason, T. S., Zhang, M., Qiu, Q., Skuli, N., Nakazawa, M. S., Karakasheva, T., et al. (2013). Hypoxia-dependent modification of collagen networks promotes sarcoma metastasis. Cancer Discov. 3, 1190–1205. doi: 10.1158/2159-8290.CD-13-0118
Eke, I., Koch, U., Hehlgans, S., Sandfort, V., Stanchi, F., Zips, D., et al. (2010). PINCH1 regulates Akt1 activation and enhances radioresistance by inhibiting PP1alpha. J. Clin. Invest. 120, 2516–2527. doi: 10.1172/JCI41078
Eke, I., Storch, K., Krause, M., and Cordes, N. (2013). Cetuximab attenuates its cytotoxic and radiosensitizing potential by inducing fibronectin biosynthesis. Cancer Res. 73, 5869–5879. doi: 10.1158/0008-5472.CAN-13-0344
Enholm, B., Paavonen, K., Ristimaki, A., Kumar, V., Gunji, Y., Klefstrom, J., et al. (1997). Comparison of VEGF, VEGF-B, VEGF-C and Ang-1 mRNA regulation by serum, growth factors, oncoproteins and hypoxia. Oncogene 14, 2475–2483. doi: 10.1038/sj.onc.1201090
Erler, J. T., Bennewith, K. L., Cox, T. R., Lang, G., Bird, D., Koong, A., et al. (2009). Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 15, 35–44. doi: 10.1016/j.ccr.2008.11.012
Erler, J. T., Bennewith, K. L., Nicolau, M., Dornhofer, N., Kong, C., Le, Q. T., et al. (2006). Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 440, 1222–1226. doi: 10.1038/nature04695
Fang, S., Dai, Y., Mei, Y., Yang, M., Hu, L., Yang, H., et al. (2019). Clinical significance and biological role of cancer-derived type I collagen in lung and esophageal cancers. Thorac Cancer 10, 277–288. doi: 10.1111/1759-7714.12947
Fischer, K. R., Durrans, A., Lee, S., Sheng, J., Li, F., Wong, S. T., et al. (2015). Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 527, 472–476. doi: 10.1038/nature15748
Gambichler, T., Kreuter, A., Grothe, S., Altmeyer, P., Brockmeyer, N. H., and Rotterdam, S. (2008). Versican overexpression in cutaneous malignant melanoma. Eur. J. Med. Res. 13, 500–504.
Gandhi, L., Rodriguez-Abreu, D., Gadgeel, S., Esteban, E., Felip, E., De Angelis, F., et al. (2018). Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N. Engl. J. Med. 378, 2078–2092. doi: 10.1056/NEJMoa1801005
Ganesh, K., Stadler, Z. K., Cercek, A., Mendelsohn, R. B., Shia, J., Segal, N. H., et al. (2019). Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 16, 361–375. doi: 10.1038/s41575-019-0126-x
Garcia, R., Merino, D., Gomez, J. M., Nistal, J. F., Hurle, M. A., Cortajarena, A. L., et al. (2016). Extracellular heat shock protein 90 binding to TGFbeta receptor I participates in TGFbeta-mediated collagen production in myocardial fibroblasts. Cell. Signal. 28, 1563–1579. doi: 10.1016/j.cellsig.2016.07.003
Garcia-Palmero, I., Torres, S., Bartolome, R. A., Pelaez-Garcia, A., Larriba, M. J., Lopez-Lucendo, M., et al. (2016). Twist1-induced activation of human fibroblasts promotes matrix stiffness by upregulating palladin and collagen alpha1(VI). Oncogene 35, 5224–5236. doi: 10.1038/onc.2016.57
Gavalas, N. G., Tsiatas, M., Tsitsilonis, O., Politi, E., Ioannou, K., Ziogas, A. C., et al. (2012). VEGF directly suppresses activation of T cells from ascites secondary to ovarian cancer via VEGF receptor type 2. Br. J. Cancer 107, 1869–1875. doi: 10.1038/bjc.2012.468
Ghatak, S., Hascall, V. C., Markwald, R. R., and Misra, S. (2010). Stromal hyaluronan interaction with epithelial CD44 variants promotes prostate cancer invasiveness by augmenting expression and function of hepatocyte growth factor and androgen receptor. J. Biol. Chem. 285, 19821–19832. doi: 10.1074/jbc.M110.104273
Gilkes, D. M., Chaturvedi, P., Bajpai, S., Wong, C. C., Wei, H., Pitcairn, S., et al. (2013). Collagen prolyl hydroxylases are essential for breast cancer metastasis. Cancer Res. 73, 3285–3296. doi: 10.1158/0008-5472.CAN-12-3963
Gilmore, A. P. (2005). Anoikis. Cell Death Differ. 12(Suppl. 2), 1473–1477. doi: 10.1038/sj.cdd.4401723
Giri, S. N., Al-Bayati, M. A., Du, X., Schelegle, E., Mohr, F. C., and Margolin, S. B. (2004). Amelioration of doxorubicin-induced cardiac and renal toxicity by pirfenidone in rats. Cancer Chemother. Pharmacol. 53, 141–150. doi: 10.1007/s00280-003-0703-z
Gorres, K. L., and Raines, R. T. (2010). Prolyl 4-hydroxylase. Crit. Rev. Biochem. Mol. Biol. 45, 106–124. doi: 10.3109/10409231003627991
Goumans, M. J., Liu, Z., and ten Dijke, P. (2009). TGF-beta signaling in vascular biology and dysfunction. Cell Res. 19, 116–127. doi: 10.1038/cr.2008.326
Gourd, E. (2018). PEGPH20 for metastatic pancreatic ductal adenocarcinoma. Lancet Oncol. 19:e81. doi: 10.1016/S1470-2045(17)30953-1
Graham, K., and Unger, E. (2018). Overcoming tumor hypoxia as a barrier to radiotherapy, chemotherapy and immunotherapy in cancer treatment. Int. J. Nanomedicine 13, 6049–6058. doi: 10.2147/IJN.S140462
Griffioen, A. W., Damen, C. A., Blijham, G. H., and Groenewegen, G. (1996). Tumor angiogenesis is accompanied by a decreased inflammatory response of tumor-associated endothelium. Blood 88, 667–673. doi: 10.1182/blood.V88.2.667.bloodjournal882667
Griffioen, A. W., van der Schaft, D. W., Barendsz-Janson, A. F., Cox, A., Struijker Boudier, H. A., Hillen, H. F., et al. (2001). Anginex, a designed peptide that inhibits angiogenesis. Biochem. J. 354, 233–242. doi: 10.1042/bj3540233
Gu, J., Li, Y., Zeng, J., Wang, B., Ji, K., Tang, Y., et al. (2017). Knockdown of HIF-1alpha by siRNA-expressing plasmid delivered by attenuated Salmonella enhances the antitumor effects of cisplatin on prostate cancer. Sci. Rep. 7:7546. doi: 10.1038/s41598-017-07973-4
Guan, X., Chen, J., Hu, Y., Lin, L., Sun, P., Tian, H., et al. (2018). Highly enhanced cancer immunotherapy by combining nanovaccine with hyaluronidase. Biomaterials 171, 198–206. doi: 10.1016/j.biomaterials.2018.04.039
Guan, X., Lin, L., Chen, J., Hu, Y., Sun, P., Tian, H., et al. (2019). Efficient PD-L1 gene silence promoted by hyaluronidase for cancer immunotherapy. J. Control. Release 293, 104–112. doi: 10.1016/j.jconrel.2018.11.022
Gusterson, B. A., Warburton, M. J., Mitchell, D., Ellison, M., Neville, A. M., and Rudland, P. S. (1982). Distribution of myoepithelial cells and basement membrane proteins in the normal breast and in benign and malignant breast diseases. Cancer Res. 42, 4763–4770.
Halder, J., Landen, C. N. Jr., Lutgendorf, S. K., Li, Y., Jennings, N. B., Fan, D., et al. (2005). Focal adhesion kinase silencing augments docetaxel-mediated apoptosis in ovarian cancer cells. Clin. Cancer Res. 11, 8829–8836. doi: 10.1158/1078-0432.CCR-05-1728
Hallmann, R., Zhang, X., Di Russo, J., Li, L., Song, J., Hannocks, M. J., et al. (2015). The regulation of immune cell trafficking by the extracellular matrix. Curr. Opin. Cell Biol. 36, 54–61. doi: 10.1016/j.ceb.2015.06.006
Hamidi, H., and Ivaska, J. (2018). Every step of the way: integrins in cancer progression and metastasis. Nat. Rev. Cancer 18, 533–548. doi: 10.1038/s41568-018-0038-z
Hand, P. H., Thor, A., Schlom, J., Rao, C. N., and Liotta, L. (1985). Expression of laminin receptor in normal and carcinomatous human tissues as defined by a monoclonal antibody. Cancer Res. 45, 2713–2719.
Hapke, S., Kessler, H., Luber, B., Benge, A., Hutzler, P., Hofler, H., et al. (2003). Ovarian cancer cell proliferation and motility is induced by engagement of integrin alpha(v)beta3/Vitronectin interaction. Biol. Chem. 384, 1073–1083. doi: 10.1515/BC.2003.120
Harrison, S. A., Abdelmalek, M. F., Caldwell, S., Shiffman, M. L., Diehl, A. M., Ghalib, R., et al. (2018). Simtuzumab is ineffective for patients with bridging fibrosis or compensated cirrhosis caused by nonalcoholic steatohepatitis. Gastroenterology 155, 1140–1153. doi: 10.1053/j.gastro.2018.07.006
Haslehurst, A. M., Koti, M., Dharsee, M., Nuin, P., Evans, K., Geraci, J., et al. (2012). EMT transcription factors snail and slug directly contribute to cisplatin resistance in ovarian cancer. BMC Cancer 12:91. doi: 10.1186/1471-2407-12-91
Hazlehurst, L. A., Landowski, T. H., and Dalton, W. S. (2003). Role of the tumor microenvironment in mediating de novo resistance to drugs and physiological mediators of cell death. Oncogene 22, 7396–7402. doi: 10.1038/sj.onc.1206943
Hecht, J. R., Benson, A. B. 3rd, Vyushkov, D., Yang, Y., Bendell, J., and Verma, U. (2017). A phase II, randomized, double-blind, placebo-controlled study of simtuzumab in combination with FOLFIRI for the second-line treatment of metastatic KRAS mutant colorectal adenocarcinoma. Oncologist 22, 243–e23. doi: 10.1634/theoncologist.2016-0479
Hehlgans, S., Haase, M., and Cordes, N. (2007). Signalling via integrins: implications for cell survival and anticancer strategies. Biochim. Biophys. Acta 1775, 163–180. doi: 10.1016/j.bbcan.2006.09.001
Heldin, P., Basu, K., Kozlova, I., and Porsch, H. (2014). HAS2 and CD44 in breast tumorigenesis. Adv. Cancer Res. 123, 211–229. doi: 10.1016/B978-0-12-800092-2.00008-3
Henke, E., Perk, J., Vider, J., de Candia, P., Chin, Y., Solit, D., et al. (2007). “Targeted inhibition of Id1 in the tumor endothelium,” in Gordon Research Conference: Angiogenesis and Microcirculation (Newport, RI).
Higgins, D. F., Kimura, K., Bernhardt, W. M., Shrimanker, N., Akai, Y., Hohenstein, B., et al. (2007). Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J. Clin. Invest. 117, 3810–3820. doi: 10.1172/JCI30487
Hingorani, S. R., Harris, W. P., Beck, J. T., Berdov, B. A., Wagner, S. A., Pshevlotsky, E. M., et al. (2016). Phase Ib study of pegylated recombinant human hyaluronidase and gemcitabine in patients with advanced pancreatic cancer. Clin. Cancer Res. 22, 2848–2854. doi: 10.1158/1078-0432.CCR-15-2010
Hodi, F. S., Chiarion-Sileni, V., Gonzalez, R., Grob, J. J., Rutkowski, P., Cowey, C. L., et al. (2018). Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 19, 1480–1492. doi: 10.1016/S1470-2045(18)30700-9
Hodkinson, P. S., Elliott, T., Wong, W. S., Rintoul, R. C., Mackinnon, A. C., Haslett, C., et al. (2006). ECM overrides DNA damage-induced cell cycle arrest and apoptosis in small-cell lung cancer cells through beta1 integrin-dependent activation of PI3-kinase. Cell Death Differ. 13, 1776–1788. doi: 10.1038/sj.cdd.4401849
Hope, C., Emmerich, P. B., Papadas, A., Pagenkopf, A., Matkowskyj, K. A., Van De Hey, D. R., et al. (2017). Versican-derived matrikines regulate Batf3-dendritic cell differentiation and promote T cell infiltration in colorectal cancer. J. Immunol. 199, 1933–1941. doi: 10.4049/jimmunol.1700529
Hope, C., Foulcer, S., Jagodinsky, J., Chen, S. X., Jensen, J. L., Patel, S., et al. (2016). Immunoregulatory roles of versican proteolysis in the myeloma microenvironment. Blood 128, 680–685. doi: 10.1182/blood-2016-03-705780
Horsman, M. R., and Overgaard, J. (2016). The impact of hypoxia and its modification of the outcome of radiotherapy. J. Radiat. Res. 57(Suppl. 1), i90–i98. doi: 10.1093/jrr/rrw007
Hosper, N. A., van den Berg, P. P., de Rond, S., Popa, E. R., Wilmer, M. J., Masereeuw, R., et al. (2013). Epithelial-to-mesenchymal transition in fibrosis: collagen type I expression is highly upregulated after EMT, but does not contribute to collagen deposition. Exp. Cell Res. 319, 3000–3009. doi: 10.1016/j.yexcr.2013.07.014
Hu, Y., Liu, J., and Huang, H. (2013). Recent agents targeting HIF-1alpha for cancer therapy. J. Cell. Biochem. 114, 498–509. doi: 10.1002/jcb.24390
Hui, L., and Chen, Y. (2015). Tumor microenvironment: sanctuary of the devil. Cancer Lett. 368, 7–13. doi: 10.1016/j.canlet.2015.07.039
Hurwitz, H., Fehrenbacher, L., Novotny, W., Cartwright, T., Hainsworth, J., Heim, W., et al. (2004). Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 350, 2335–2342. doi: 10.1056/NEJMoa032691
Ikuta, K., Ota, T., Zhuo, L., Urakawa, H., Kozawa, E., Hamada, S., et al. (2017). Antitumor effects of 4-methylumbelliferone, a hyaluronan synthesis inhibitor, on malignant peripheral nerve sheath tumor. Int. J. Cancer 140, 469–479. doi: 10.1002/ijc.30460
Infante, J. R., Korn, R. L., Rosen, L. S., LoRusso, P., Dychter, S. S., Zhu, J., et al. (2018). Phase 1 trials of PEGylated recombinant human hyaluronidase PH20 in patients with advanced solid tumours. Br. J. Cancer 118, 153–161. doi: 10.1038/bjc.2017.327
Issa-Nummer, Y., Darb-Esfahani, S., Loibl, S., Kunz, G., Nekljudova, V., Schrader, I., et al. (2013). Prospective validation of immunological infiltrate for prediction of response to neoadjuvant chemotherapy in HER2-negative breast cancer–a substudy of the neoadjuvant GeparQuinto trial. PLoS ONE 8:e79775. doi: 10.1371/journal.pone.0079775
Jacobetz, M. A., Chan, D. S., Neesse, A., Bapiro, T. E., Cook, N., Frese, K. K., et al. (2013). Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 62, 112–120. doi: 10.1136/gutjnl-2012-302529
Jadin, L., Pastorino, S., Symons, R., Nomura, N., Jiang, P., Juarez, T., et al. (2015). Hyaluronan expression in primary and secondary brain tumors. Ann. Transl. Med. 3:80. doi: 10.3978/j.issn.2305-5839.2015.04.07
Jain, R. K. (2014). Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell 26, 605–622. doi: 10.1016/j.ccell.2014.10.006
Jakubzig, B., Baltes, F., Henze, S., Schlesinger, M., and Bendas, G. (2018). Mechanisms of matrix-induced chemoresistance of breast cancer cells-deciphering novel potential targets for a cell sensitization. Cancers 10:E495. doi: 10.3390/cancers10120495
Jiang, K., Liu, H., Xie, D., and Xiao, Q. (2019). Differentially expressed genes ASPN, COL1A1, FN1, VCAN and MUC5AC are potential prognostic biomarkers for gastric cancer. Oncol. Lett. 17, 3191–3202. doi: 10.3892/ol.2019.9952
Kakizaki, I., Kojima, K., Takagaki, K., Endo, M., Kannagi, R., Ito, M., et al. (2004). A novel mechanism for the inhibition of hyaluronan biosynthesis by 4-methylumbelliferone. J. Biol. Chem. 279, 33281–33289. doi: 10.1074/jbc.M405918200
Kaliski, A., Maggiorella, L., Cengel, K. A., Mathe, D., Rouffiac, V., Opolon, P., et al. (2005). Angiogenesis and tumor growth inhibition by a matrix metalloproteinase inhibitor targeting radiation-induced invasion. Mol. Cancer Ther. 4, 1717–1728. doi: 10.1158/1535-7163.MCT-05-0179
Kalluri, R. (2016). The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 16, 582–598. doi: 10.1038/nrc.2016.73
Kaplan, G. (1983). In vitro differentiation of human monocytes. Monocytes cultured on glass are cytotoxic to tumor cells but monocytes cultured on collagen are not. J. Exp. Med. 157, 2061–2072. doi: 10.1084/jem.157.6.2061
Karalis, T. T., Heldin, P., Vynios, D. H., Neill, T., Buraschi, S., Iozzo, R. V., et al. (2018). Tumor-suppressive functions of 4-MU on breast cancer cells of different ER status: regulation of hyaluronan/HAS2/CD44 and specific matrix effectors. Matrix Biol. 78–79, 118–138. doi: 10.1016/j.matbio.2018.04.007
Kauppila, S., Stenback, F., Risteli, J., Jukkola, A., and Risteli, L. (1998). Aberrant type I and type III collagen gene expression in human breast cancer in vivo. J. Pathol. 186, 262–268. doi: 10.1002/(SICI)1096-9896(1998110)186:3<262::AID-PATH191>3.0.CO;2-3
Kechagia, J. Z., Ivaska, J., and Roca-Cusachs, P. (2019). Integrins as biomechanical sensors of the microenvironment. Nat. Rev. Mol. Cell Biol. 20, 457–473. doi: 10.1038/s41580-019-0134-2
Keunen, O., Johansson, M., Oudin, A., Sanzey, M., Rahim, S. A., Fack, F., et al. (2011). Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc. Natl. Acad. Sci. U.S.A. 108, 3749–3754. doi: 10.1073/pnas.1014480108
Khan, S., Ebeling, M. C., Chauhan, N., Thompson, P. A., Gara, R. K., Ganju, A., et al. (2015). Ormeloxifene suppresses desmoplasia and enhances sensitivity of gemcitabine in pancreatic cancer. Cancer Res. 75, 2292–2304. doi: 10.1158/0008-5472.CAN-14-2397
Khwaja, A., Rodriguez-Viciana, P., Wennstrom, S., Warne, P. H., and Downward, J. (1997). Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. EMBO J. 16, 2783–2793. doi: 10.1093/emboj/16.10.2783
Kim, H., Cha, J., Jang, M., and Kim, P. (2019). Hyaluronic acid-based extracellular matrix triggers spontaneous M2-like polarity of monocyte/macrophage. Biomater. Sci. 7, 2264–2271. doi: 10.1039/C9BM00155G
Kirshner, J., Jobling, M. F., Pajares, M. J., Ravani, S. A., Glick, A. B., Lavin, M. J., et al. (2006). Inhibition of transforming growth factor-beta1 signaling attenuates ataxia telangiectasia mutated activity in response to genotoxic stress. Cancer Res. 66, 10861–10869. doi: 10.1158/0008-5472.CAN-06-2565
Knudson, W., Biswas, C., and Toole, B. P. (1984). Interactions between human tumor cells and fibroblasts stimulate hyaluronate synthesis. Proc. Natl. Acad. Sci. U.S.A. 81, 6767–6771. doi: 10.1073/pnas.81.21.6767
Kohli, A. G., Kivimae, S., Tiffany, M. R., and Szoka, F. C. (2014). Improving the distribution of Doxil(R) in the tumor matrix by depletion of tumor hyaluronan. J. Control. Release 191, 105–114. doi: 10.1016/j.jconrel.2014.05.019
Kolenda, J., Jensen, S. S., Aaberg-Jessen, C., Christensen, K., Andersen, C., Brunner, N., et al. (2011). Effects of hypoxia on expression of a panel of stem cell and chemoresistance markers in glioblastoma-derived spheroids. J. Neurooncol. 103, 43–58. doi: 10.1007/s11060-010-0357-8
Koninger, J., Giese, T., di Mola, F. F., Wente, M. N., Esposito, I., Bachem, M. G., et al. (2004). Pancreatic tumor cells influence the composition of the extracellular matrix. Biochem. Biophys. Res. Commun. 322, 943–949. doi: 10.1016/j.bbrc.2004.08.008
Kozono, S., Ohuchida, K., Eguchi, D., Ikenaga, N., Fujiwara, K., Cui, L., et al. (2013). Pirfenidone inhibits pancreatic cancer desmoplasia by regulating stellate cells. Cancer Res. 73, 2345–2356. doi: 10.1158/0008-5472.CAN-12-3180
Kudo, D., Suto, A., and Hakamada, K. (2017). The development of a novel therapeutic strategy to target hyaluronan in the extracellular matrix of pancreatic ductal adenocarcinoma. Int. J. Mol. Sci. 18:E600. doi: 10.3390/ijms18030600
Kundu, B., Saha, P., Datta, K., and Kundu, S. C. (2013). A silk fibroin based hepatocarcinoma model and the assessment of the drug response in hyaluronan-binding protein 1 overexpressed HepG2 cells. Biomaterials 34, 9462–9474. doi: 10.1016/j.biomaterials.2013.08.047
Levental, K. R., Yu, H., Kass, L., Lakins, J. N., Egeblad, M., Erler, J. T., et al. (2009). Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 139, 891–906. doi: 10.1016/j.cell.2009.10.027
Lewis, C. E., and Pollard, J. W. (2006). Distinct role of macrophages in different tumor microenvironments. Cancer Res. 66, 605–612. doi: 10.1158/0008-5472.CAN-05-4005
Lewis, J. M., Truong, T. N., and Schwartz, M. A. (2002). Integrins regulate the apoptotic response to DNA damage through modulation of p53. Proc. Natl. Acad. Sci. U.S.A. 99, 3627–3632. doi: 10.1073/pnas.062698499
Li, H., Li, J., Chen, L., Qi, S., Yu, S., Weng, Z., et al. (2019). HERC3-mediated SMAD7 ubiquitination degradation promotes autophagy-induced EMT and chemoresistance in glioblastoma. Clin. Cancer Res. 25, 3602–3616. doi: 10.1158/1078-0432.CCR-18-3791
Li, N., Babaei-Jadidi, R., Lorenzi, F., Spencer-Dene, B., Clarke, P., Domingo, E., et al. (2019). An FBXW7-ZEB2 axis links EMT and tumour microenvironment to promote colorectal cancer stem cells and chemoresistance. Oncogenesis 8:13. doi: 10.1038/s41389-019-0125-3
Liang, K., Lu, Y., Jin, W., Ang, K. K., Milas, L., and Fan, Z. (2003). Sensitization of breast cancer cells to radiation by trastuzumab. Mol. Cancer Ther. 2, 1113–1120.
Lim, M., Xia, Y., Bettegowda, C., and Weller, M. (2018). Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 15, 422–442. doi: 10.1038/s41571-018-0003-5
Lipponen, P., Aaltomaa, S., Tammi, R., Tammi, M., Agren, U., and Kosma, V. M. (2001). High stromal hyaluronan level is associated with poor differentiation and metastasis in prostate cancer. Eur. J. Cancer 37, 849–856. doi: 10.1016/S0959-8049(00)00448-2
Liu, J., Liao, S., Diop-Frimpong, B., Chen, W., Goel, S., Naxerova, K., et al. (2012). TGF-beta blockade improves the distribution and efficacy of therapeutics in breast carcinoma by normalizing the tumor stroma. Proc. Natl. Acad. Sci. U.S.A. 109, 16618–16623. doi: 10.1073/pnas.1117610109
Liu, X. Q., Xiong, M. H., Shu, X. T., Tang, R. Z., and Wang, J. (2012). Therapeutic delivery of siRNA silencing HIF-1 alpha with micellar nanoparticles inhibits hypoxic tumor growth. Mol. Pharm. 9, 2863–2874. doi: 10.1021/mp300193f
Lokeshwar, V. B., Lopez, L. E., Munoz, D., Chi, A., Shirodkar, S. P., Lokeshwar, S. D., et al. (2010). Antitumor activity of hyaluronic acid synthesis inhibitor 4-methylumbelliferone in prostate cancer cells. Cancer Res. 70, 2613–2623. doi: 10.1158/0008-5472.CAN-09-3185
Lompardia, S. L., Diaz, M., Papademetrio, D. L., Pibuel, M., Alvarez, E., and Hajos, S. E. (2017). 4-methylumbelliferone and imatinib combination enhances senescence induction in chronic myeloid leukemia cell lines. Invest. New Drugs 35, 1–10. doi: 10.1007/s10637-016-0397-9
Lu, Y. C., Parker, L. L., Lu, T., Zheng, Z., Toomey, M. A., White, D. E., et al. (2017). Treatment of patients with metastatic cancer using a major histocompatibility complex class II-restricted T-cell receptor targeting the cancer germline antigen MAGE-A3. J. Clin. Oncol. 35, 3322–3329. doi: 10.1200/JCO.2017.74.5463
Lucero, H. A., and Kagan, H. M. (2006). Lysyl oxidase: an oxidative enzyme and effector of cell function. Cell. Mol. Life Sci. 63, 2304–2316. doi: 10.1007/s00018-006-6149-9
Lund, A. W., and Swartz, M. A. (2010). Role of lymphatic vessels in tumor immunity: passive conduits or active participants? J. Mammary Gland Biol. Neoplasia 15, 341–352. doi: 10.1007/s10911-010-9193-x
Malvicini, M., Fiore, E., Ghiaccio, V., Piccioni, F., Rizzo, M., Olmedo Bonadeo, L., et al. (2015). Tumor microenvironment remodeling by 4-methylumbelliferone boosts the antitumor effect of combined immunotherapy in murine colorectal carcinoma. Mol. Ther. 23, 1444–1455. doi: 10.1038/mt.2015.112
Mammoto, T., Jiang, A., Jiang, E., Panigrahy, D., Kieran, M. W., and Mammoto, A. (2013). Role of collagen matrix in tumor angiogenesis and glioblastoma multiforme progression. Am. J. Pathol. 183, 1293–1305. doi: 10.1016/j.ajpath.2013.06.026
Mani, S. A., Yang, J., Brooks, M., Schwaninger, G., Zhou, A., Miura, N., et al. (2007). Mesenchyme forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal-like breast cancers. Proc. Natl. Acad. Sci. U.S.A. 104, 10069–10074. doi: 10.1073/pnas.0703900104
Mariathasan, S., Turley, S. J., Nickles, D., Castiglioni, A., Yuen, K., Wang, Y., et al. (2018). TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548. doi: 10.1038/nature25501
Matsumoto, S., Batra, S., Saito, K., Yasui, H., Choudhuri, R., Gadisetti, C., et al. (2011). Antiangiogenic agent sunitinib transiently increases tumor oxygenation and suppresses cycling hypoxia. Cancer Res. 71, 6350–6359. doi: 10.1158/0008-5472.CAN-11-2025
Maxwell, P. H., Dachs, G. U., Gleadle, J. M., Nicholls, L. G., Harris, A. L., Stratford, I. J., et al. (1997). Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc. Natl. Acad. Sci. U.S.A. 94, 8104–8109. doi: 10.1073/pnas.94.15.8104
McArthur, H. L., Diab, A., Page, D. B., Yuan, J., Solomon, S. B., Sacchini, V., et al. (2016). A pilot study of preoperative single-dose ipilimumab and/or cryoablation in women with early-stage breast cancer with comprehensive immune profiling. Clin. Cancer Res. 22, 5729–5737. doi: 10.1158/1078-0432.CCR-16-0190
McDonald, L. T., Russell, D. L., Kelly, R. R., Xiong, Y., Motamarry, A., Patel, R. K., et al. (2015). Hematopoietic stem cell-derived cancer-associated fibroblasts are novel contributors to the pro-tumorigenic microenvironment. Neoplasia 17, 434–448. doi: 10.1016/j.neo.2015.04.004
McGrail, D. J., Khambhati, N. N., Qi, M. X., Patel, K. S., Ravikumar, N., Brandenburg, C. P., et al. (2015). Alterations in ovarian cancer cell adhesion drive taxol resistance by increasing microtubule dynamics in a FAK-dependent manner. Sci. Rep. 5:9529. doi: 10.1038/srep09529
Mediavilla-Varela, M., Boateng, K., Noyes, D., and Antonia, S. J. (2016). The anti-fibrotic agent pirfenidone synergizes with cisplatin in killing tumor cells and cancer-associated fibroblasts. BMC Cancer 16:176. doi: 10.1186/s12885-016-2162-z
Mintz, M. B., Sowers, R., Brown, K. M., Hilmer, S. C., Mazza, B., Huvos, A. G., et al. (2005). An expression signature classifies chemotherapy-resistant pediatric osteosarcoma. Cancer Res. 65, 1748–1754. doi: 10.1158/0008-5472.CAN-04-2463
Miskolczi, Z., Smith, M. P., Rowling, E. J., Ferguson, J., Barriuso, J., and Wellbrock, C. (2018). Collagen abundance controls melanoma phenotypes through lineage-specific microenvironment sensing. Oncogene 37, 3166–3182. doi: 10.1038/s41388-018-0209-0
Missinato, M. A., Tobita, K., Romano, N., Carroll, J. A., and Tsang, M. (2015). Extracellular component hyaluronic acid and its receptor Hmmr are required for epicardial EMT during heart regeneration. Cardiovasc. Res. 107, 487–498. doi: 10.1093/cvr/cvv190
Miyazaki, S., Kikuchi, H., Iino, I., Uehara, T., Setoguchi, T., Fujita, T., et al. (2014). Anti-VEGF antibody therapy induces tumor hypoxia and stanniocalcin 2 expression and potentiates growth of human colon cancer xenografts. Int. J. Cancer 135, 295–307. doi: 10.1002/ijc.28686
Moeller, B. J., and Dewhirst, M. W. (2004). Raising the bar: how HIF-1 helps determine tumor radiosensitivity. Cell Cycle 3, 1107–1110. doi: 10.4161/cc.3.9.1099
Molnar, J., Fong, K. S., He, Q. P., Hayashi, K., Kim, Y., Fong, S. F., et al. (2003). Structural and functional diversity of lysyl oxidase and the LOX-like proteins. Biochim. Biophys. Acta 1647, 220–224. doi: 10.1016/S1570-9639(03)00053-0
Mushtaq, M. U., Papadas, A., Pagenkopf, A., Flietner, E., Morrow, Z., Chaudhary, S. G., et al. (2018). Tumor matrix remodeling and novel immunotherapies: the promise of matrix-derived immune biomarkers. J. Immunother. Cancer 6:65. doi: 10.1186/s40425-018-0376-0
Naba, A., Clauser, K. R., Hoersch, S., Liu, H., Carr, S. A., and Hynes, R. O. (2012). The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell. Proteomics 11:M111.014647. doi: 10.1074/mcp.M111.014647
Naci, D., El Azreq, M. A., Chetoui, N., Lauden, L., Sigaux, F., Charron, D., et al. (2012). alpha2beta1 integrin promotes chemoresistance against doxorubicin in cancer cells through extracellular signal-regulated kinase (ERK). J. Biol. Chem. 287, 17065–17076. doi: 10.1074/jbc.M112.349365
Nagase, H., Kudo, D., Suto, A., Yoshida, E., Suto, S., Negishi, M., et al. (2017). 4-methylumbelliferone suppresses hyaluronan synthesis and tumor progression in SCID mice intra-abdominally inoculated with pancreatic cancer cells. Pancreas 46, 190–197. doi: 10.1097/MPA.0000000000000741
Naik, A., Al-Yahyaee, A., Abdullah, N., Sam, J. E., Al-Zeheimi, N., Yaish, M. W., et al. (2018). Neuropilin-1 promotes the oncogenic Tenascin-C/integrin beta3 pathway and modulates chemoresistance in breast cancer cells. BMC Cancer 18:533. doi: 10.1186/s12885-018-4446-y
Nakagawa, Y., Nakayama, H., Nagata, M., Yoshida, R., Kawahara, K., Hirosue, A., et al. (2014). Overexpression of fibronectin confers cell adhesion-mediated drug resistance (CAM-DR) against 5-FU in oral squamous cell carcinoma cells. Int. J. Oncol. 44, 1376–1384. doi: 10.3892/ijo.2014.2265
Nakamura, T., Takagaki, K., Shibata, S., Tanaka, K., Higuchi, T., and Endo, M. (1995). Hyaluronic-acid-deficient extracellular matrix induced by addition of 4-methylumbelliferone to the medium of cultured human skin fibroblasts. Biochem. Biophys. Res. Commun. 208, 470–475. doi: 10.1006/bbrc.1995.1362
Nam, J. M., Onodera, Y., Bissell, M. J., and Park, C. C. (2010). Breast cancer cells in three-dimensional culture display an enhanced radioresponse after coordinate targeting of integrin alpha5beta1 and fibronectin. Cancer Res. 70, 5238–5248. doi: 10.1158/0008-5472.CAN-09-2319
Nambiar, D. K., Rajamani, P., and Singh, R. P. (2015). Silibinin attenuates ionizing radiation-induced pro-angiogenic response and EMT in prostate cancer cells. Biochem. Biophys. Res. Commun. 456, 262–268. doi: 10.1016/j.bbrc.2014.11.069
Nguyen, T. V., Sleiman, M., Moriarty, T., Herrick, W. G., and Peyton, S. R. (2014). Sorafenib resistance and JNK signaling in carcinoma during extracellular matrix stiffening. Biomaterials 35, 5749–5759. doi: 10.1016/j.biomaterials.2014.03.058
Noel, A., Gutierrez-Fernandez, A., Sounni, N. E., Behrendt, N., Maquoi, E., Lund, I. K., et al. (2012). New and paradoxical roles of matrix metalloproteinases in the tumor microenvironment. Front. Pharmacol. 3:140. doi: 10.3389/fphar.2012.00140
Nummer, D., Suri-Payer, E., Schmitz-Winnenthal, H., Bonertz, A., Galindo, L., Antolovich, D., et al. (2007). Role of tumor endothelium in CD4+ CD25+ regulatory T cell infiltration of human pancreatic carcinoma. J. Natl. Cancer Inst. 99, 1188–1199. doi: 10.1093/jnci/djm064
Ocana, O. H., Corcoles, R., Fabra, A., Moreno-Bueno, G., Acloque, H., Vega, S., et al. (2012). Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell 22, 709–724. doi: 10.1016/j.ccr.2012.10.012
Offersen, B. V., Borre, M., and Overgaard, J. (1998). Immunohistochemical determination of tumor angiogenesis measured by the maximal microvessel density in human prostate cancer. APMIS 106, 463–469. doi: 10.1111/j.1699-0463.1998.tb01372.x
Ohlund, D., Handly-Santana, A., Biffi, G., Elyada, E., Almeida, A. S., Ponz-Sarvise, M., et al. (2017). Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 214, 579–596. doi: 10.1084/jem.20162024
Onoda, J. M., Piechocki, M. P., and Honn, K. V. (1992). Radiation-induced increase in expression of the alpha IIb beta 3 integrin in melanoma cells: effects on metastatic potential. Radiat. Res. 130, 281–288. doi: 10.2307/3578372
Ostroukhova, M., Qi, Z., Oriss, T. B., Dixon-McCarthy, B., Ray, P., and Ray, A. (2006). Treg-mediated immunosuppression involves activation of the Notch-HES1 axis by membrane-bound TGF-beta. J. Clin. Invest. 116, 996–1004. doi: 10.1172/JCI26490
Ou, J., Luan, W., Deng, J., Sa, R., and Liang, H. (2012). αV integrin induces multicellular radioresistance in human nasopharyngeal carcinoma via activating SAPK/JNK pathway. PLoS ONE 7:e38737. doi: 10.1371/journal.pone.0038737
Ozdemir, B. C., Pentcheva-Hoang, T., Carstens, J. L., Zheng, X., Wu, C. C., Simpson, T. R., et al. (2014). Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25, 719–734. doi: 10.1016/j.ccr.2014.04.005
Pankova, D., Chen, Y., Terajima, M., Schliekelman, M. J., Baird, B. N., Fahrenholtz, M., et al. (2016). Cancer-associated fibroblasts induce a collagen cross-link switch in tumor stroma. Mol. Cancer Res. 14, 287–295. doi: 10.1158/1541-7786.MCR-15-0307
Paquette, B., Baptiste, C., Therriault, H., Arguin, G., Plouffe, B., and Lemay, R. (2007). In vitro irradiation of basement membrane enhances the invasiveness of breast cancer cells. Br. J. Cancer 97, 1505–1512. doi: 10.1038/sj.bjc.6604072
Park, C. C., Zhang, H., Pallavicini, M., Gray, J. W., Baehner, F., Park, C. J., et al. (2006). β1 integrin inhibitory antibody induces apoptosis of breast cancer cells, inhibits growth, and distinguishes malignant from normal phenotype in three dimensional cultures and in vivo. Cancer Res. 66, 1526–1535. doi: 10.1158/0008-5472.CAN-05-3071
Park, C. C., Zhang, H. J., Yao, E. S., Park, C. J., and Bissell, M. J. (2008). β1 integrin inhibition dramatically enhances radiotherapy efficacy in human breast cancer xenografts. Cancer Res. 68, 4398–4405. doi: 10.1158/0008-5472.CAN-07-6390
Parra, K., Valenzuela, P., Lerma, N., Gallegos, A., Reza, L. C., Rodriguez, G., et al. (2017). Impact of CTLA-4 blockade in conjunction with metronomic chemotherapy on preclinical breast cancer growth. Br. J. Cancer 116, 324–334. doi: 10.1038/bjc.2016.429
Pasanen, I., Lehtonen, S., Sormunen, R., Skarp, S., Lehtilahti, E., Pietila, M., et al. (2016). Breast cancer carcinoma-associated fibroblasts differ from breast fibroblasts in immunological and extracellular matrix regulating pathways. Exp. Cell Res. 344, 53–66. doi: 10.1016/j.yexcr.2016.04.016
Passi, A., Vigetti, D., Buraschi, S., and Iozzo, R. V. (2019). Dissecting the role of hyaluronan synthases in the tumor microenvironment. FEBS J. 286, 2937–2949. doi: 10.1111/febs.14847
Paz-Ares, L., Luft, A., Vicente, D., Tafreshi, A., Gumus, M., Mazieres, J., et al. (2018). Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N. Engl. J. Med. 379, 2040–2051. doi: 10.1056/NEJMoa1810865
Peng, D. H., Ungewiss, C., Tong, P., Byers, L. A., Wang, J., Canales, J. R., et al. (2017). ZEB1 induces LOXL2-mediated collagen stabilization and deposition in the extracellular matrix to drive lung cancer invasion and metastasis. Oncogene 36, 1925–1938. doi: 10.1038/onc.2016.358
Peng, M., Mo, Y., Wang, Y., Wu, P., Zhang, Y., Xiong, F., et al. (2019). Neoantigen vaccine: an emerging tumor immunotherapy. Mol. Cancer 18:128. doi: 10.1186/s12943-019-1055-6
Perou, C. M., Sorlie, T., Eisen, M. B., van de Rijn, M., Jeffrey, S. S., Rees, C. A., et al. (2000). Molecular portraits of human breast tumours. Nature 406, 747–752. doi: 10.1038/35021093
Perri, R. T., Kay, N. E., McCarthy, J., Vessella, R. L., Jacob, H. S., and Furcht, L. T. (1982). Fibronectin enhances in vitro monocyte-macrophage-mediated tumoricidal activity. Blood 60, 430–435. doi: 10.1182/blood.V60.2.430.430
Peyrol, S., Raccurt, M., Gerard, F., Gleyzal, C., Grimaud, J. A., and Sommer, P. (1997). Lysyl oxidase gene expression in the stromal reaction to in situ and invasive ductal breast carcinoma. Am. J. Pathol. 150, 497–507.
Ponticos, M., Partridge, T., Black, C. M., Abraham, D. J., and Bou-Gharios, G. (2004). Regulation of collagen type I in vascular smooth muscle cells by competition between Nkx2.5 and deltaEF1/ZEB1. Mol. Cell. Biol. 24, 6151–6161. doi: 10.1128/MCB.24.14.6151-6161.2004
Porsch, H., Bernert, B., Mehic, M., Theocharis, A. D., Heldin, C. H., and Heldin, P. (2013). Efficient TGFbeta-induced epithelial-mesenchymal transition depends on hyaluronan synthase HAS2. Oncogene 32, 4355–4365. doi: 10.1038/onc.2012.475
Powell, J., Mota, F., Steadman, D., Soudy, C., Miyauchi, J. T., Crosby, S., et al. (2018). Small molecule neuropilin-1 antagonists combine antiangiogenic and antitumor activity with immune modulation through reduction of transforming growth factor beta (TGFβ) production in regulatory T-cells. J. Med. Chem. 61, 4135–4154. doi: 10.1021/acs.jmedchem.8b00210
Prehm, P., and Schumacher, U. (2004). Inhibition of hyaluronan export from human fibroblasts by inhibitors of multidrug resistance transporters. Biochem. Pharmacol. 68, 1401–1410. doi: 10.1016/j.bcp.2004.06.017
Provenzano, P. P., Cuevas, C., Chang, A. E., Goel, V. K., Von Hoff, D. D., and Hingorani, S. R. (2012). Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 21, 418–429. doi: 10.1016/j.ccr.2012.01.007
Provenzano, P. P., and Hingorani, S. R. (2013). Hyaluronan, fluid pressure, and stromal resistance in pancreas cancer. Br. J. Cancer 108, 1–8. doi: 10.1038/bjc.2012.569
Provenzano, P. P., Inman, D. R., Eliceiri, K. W., Knittel, J. G., Yan, L., Rueden, C. T., et al. (2008). Collagen density promotes mammary tumor initiation and progression. BMC Med. 6:11. doi: 10.1186/1741-7015-6-11
Pucci-Minafra, I., and Luparello, C. (1991). Type V/type I collagen interactions in vitro and growth-inhibitory effect of hybrid substrates on 8701-BC carcinoma cells. J. Submicrosc. Cytol. Pathol. 23, 67–74.
Puram, S. V., Tirosh, I., Parikh, A. S., Patel, A. P., Yizhak, K., Gillespie, S., et al. (2017). Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 171, 1611–1624.e24. doi: 10.1016/j.cell.2017.10.044
Qian, L. W., Mizumoto, K., Urashima, T., Nagai, E., Maehara, N., Sato, N., et al. (2002). Radiation-induced increase in invasive potential of human pancreatic cancer cells and its blockade by a matrix metalloproteinase inhibitor, CGS27023. Clin. Cancer Res. 8, 1223–1227.
Qiao, Y., Zhang, C., Li, A., Wang, D., Luo, Z., Ping, Y., et al. (2018). IL6 derived from cancer-associated fibroblasts promotes chemoresistance via CXCR7 in esophageal squamous cell carcinoma. Oncogene 37, 873–883. doi: 10.1038/onc.2017.387
Qiu, X., Tan, H., Fu, D., Zhu, Y., and Zhang, J. (2018). Laminin is over expressed in breast cancer and facilitate cancer cell metastasis. J. Cancer Res. Ther. 14, S1170–S1172. doi: 10.4103/0973-1482.191035
Raave, R., van Kuppevelt, T. H., and Daamen, W. F. (2018). Chemotherapeutic drug delivery by tumoral extracellular matrix targeting. J. Control. Release 274, 1–8. doi: 10.1016/j.jconrel.2018.01.029
Radisky, D. C., Levy, D. D., Littlepage, L. E., Liu, H., Nelson, C. M., Fata, J. E., et al. (2005). Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 436, 123–127. doi: 10.1038/nature03688
Raghu, G., Brown, K. K., Collard, H. R., Cottin, V., Gibson, K. F., Kaner, R. J., et al. (2017). Efficacy of simtuzumab versus placebo in patients with idiopathic pulmonary fibrosis: a randomised, double-blind, controlled, phase 2 trial. Lancet Respir Med. 5, 22–32. doi: 10.1016/S2213-2600(16)30421-0
Rammensee, H. G., and Singh-Jasuja, H. (2013). HLA ligandome tumor antigen discovery for personalized vaccine approach. Expert Rev. Vaccines 12, 1211–1217. doi: 10.1586/14760584.2013.836911
Reginato, M. J., Mills, K. R., Paulus, J. K., Lynch, D. K., Sgroi, D. C., Debnath, J., et al. (2003). Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nat. Cell Biol. 5, 733–740. doi: 10.1038/ncb1026
Ren, J., Chen, Y., Song, H., Chen, L., and Wang, R. (2013). Inhibition of ZEB1 reverses EMT and chemoresistance in docetaxel-resistant human lung adenocarcinoma cell line. J. Cell. Biochem. 114, 1395–1403. doi: 10.1002/jcb.24481
Reuter, J. A., Ortiz-Urda, S., Kretz, M., Garcia, J., Scholl, F. A., Pasmooij, A. M., et al. (2009). Modeling inducible human tissue neoplasia identifies an extracellular matrix interaction network involved in cancer progression. Cancer Cell 15, 477–488. doi: 10.1016/j.ccr.2009.04.002
Riaz, M., Sieuwerts, A. M., Look, M. P., Timmermans, M. A., Smid, M., Foekens, J. A., et al. (2012). High TWIST1 mRNA expression is associated with poor prognosis in lymph node-negative and estrogen receptor-positive human breast cancer and is co-expressed with stromal as well as ECM related genes. Breast Cancer Res. 14:R123. doi: 10.1186/bcr3317
Rice, A. J., Cortes, E., Lachowski, D., Cheung, B. C. H., Karim, S. A., Morton, J. P., et al. (2017). Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis 6:e352. doi: 10.1038/oncsis.2017.54
Riesterer, O., Honer, M., Jochum, W., Oehler, C., Ametamey, S., and Pruschy, M. (2006). Ionizing radiation antagonizes tumor hypoxia induced by antiangiogenic treatment. Clin. Cancer Res. 12, 3518–3524. doi: 10.1158/1078-0432.CCR-05-2816
Robbins, P. F., Kassim, S. H., Tran, T. L., Crystal, J. S., Morgan, R. A., Feldman, S. A., et al. (2015). A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin. Cancer Res. 21, 1019–1027. doi: 10.1158/1078-0432.CCR-14-2708
Robert, C., Schachter, J., Long, G. V., Arance, A., Grob, J. J., Mortier, L., et al. (2015). Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 372, 2521–2532. doi: 10.1056/NEJMoa1503093
Rodriguez, H. M., Vaysberg, M., Mikels, A., McCauley, S., Velayo, A. C., Garcia, C., et al. (2010). Modulation of lysyl oxidase-like 2 enzymatic activity by an allosteric antibody inhibitor. J. Biol. Chem. 285, 20964–20974. doi: 10.1074/jbc.M109.094136
Rohrig, F., Vorlova, S., Hoffmann, H., Wartenberg, M., Escorcia, F. E., Keller, S., et al. (2017). VEGF-ablation therapy reduces drug delivery and therapeutic response in ECM-dense tumors. Oncogene 36, 1–12. doi: 10.1038/onc.2016.182
Ropponen, K., Tammi, M., Parkkinen, J., Eskelinen, M., Tammi, R., Lipponen, P., et al. (1998). Tumor cell-associated hyaluronan as an unfavorable prognostic factor in colorectal cancer. Cancer Res. 58, 342–347.
Rossow, L., Veitl, S., Vorlova, S., Wax, J. K., Kuhn, A. E., Maltzahn, V., et al. (2018). LOX-catalyzed collagen stabilization is a proximal cause for intrinsic resistance to chemotherapy. Oncogene 37, 4921–4940. doi: 10.1038/s41388-018-0320-2
Ruella, M., Klichinsky, M., Kenderian, S. S., Shestova, O., Ziober, A., Kraft, D. O., et al. (2017). Overcoming the immunosuppressive tumor microenvironment of hodgkin lymphoma using chimeric antigen receptor T cells. Cancer Discov. 7, 1154–1167. doi: 10.1158/2159-8290.CD-16-0850
Rugo, H. S., Delord, J. P., Im, S. A., Ott, P. A., Piha-Paul, S. A., Bedard, P. L., et al. (2018). Safety and antitumor activity of pembrolizumab in patients with estrogen receptor-positive/human epidermal growth factor receptor 2-negative advanced breast cancer. Clin. Cancer Res. 24, 2804–2811. doi: 10.1158/1078-0432.CCR-17-3452
Rygiel, T. P., Stolte, E. H., de Ruiter, T., van de Weijer, M. L., and Meyaard, L. (2011). Tumor-expressed collagens can modulate immune cell function through the inhibitory collagen receptor LAIR-1. Mol. Immunol. 49, 402–406. doi: 10.1016/j.molimm.2011.09.006
Saga, R., Monzen, S., Chiba, M., Yoshino, H., Nakamura, T., and Hosokawa, Y. (2017). Anti-tumor and anti-invasion effects of a combination of 4-methylumbelliferone and ionizing radiation in human fibrosarcoma cells. Oncol. Lett. 13, 410–416. doi: 10.3892/ol.2016.5385
Salmon, H., and Donnadieu, E. (2012). Within tumors, interactions between T cells and tumor cells are impeded by the extracellular matrix. Oncoimmunology 1, 992–994. doi: 10.4161/onci.20239
Salmon, H., Franciszkiewicz, K., Damotte, D., Dieu-Nosjean, M. C., Validire, P., Trautmann, A., et al. (2012). Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Invest. 122, 899–910. doi: 10.1172/JCI45817
Sandfort, V., Koch, U., and Cordes, N. (2007). Cell adhesion-mediated radioresistance revisited. Int. J. Radiat. Biol. 83, 727–732. doi: 10.1080/09553000701694335
Schaaf, M. B., Garg, A. D., and Agostinis, P. (2018). Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis. 9:115. doi: 10.1038/s41419-017-0061-0
Schober, M., Jesenofsky, R., Faissner, R., Weidenauer, C., Hagmann, W., Michl, P., et al. (2014). Desmoplasia and chemoresistance in pancreatic cancer. Cancers 6, 2137–2154. doi: 10.3390/cancers6042137
Schulz, T., Schumacher, U., and Prehm, P. (2007). Hyaluronan export by the ABC transporter MRP5 and its modulation by intracellular cGMP. J. Biol. Chem. 282, 20999–21004. doi: 10.1074/jbc.M700915200
Schutze, F., Rohrig, F., Vorlova, S., Gatzner, S., Kuhn, A., Ergun, S., et al. (2015). Inhibition of lysyl oxidases improves drug diffusion and increases efficacy of cytotoxic treatment in 3D tumor models. Sci. Rep. 5:17576. doi: 10.1038/srep17576
Serebriiskii, I., Castello-Cros, R., Lamb, A., Golemis, E. A., and Cukierman, E. (2008). Fibroblast-derived 3D matrix differentially regulates the growth and drug-responsiveness of human cancer cells. Matrix Biol. 27, 573–585. doi: 10.1016/j.matbio.2008.02.008
Sethi, T., Rintoul, R. C., Moore, S. M., MacKinnon, A. C., Salter, D., Choo, C., et al. (1999). Extracellular matrix proteins protect small cell lung cancer cells against apoptosis: a mechanism for small cell lung cancer growth and drug resistance in vivo. Nat. Med. 5, 662–668. doi: 10.1038/9511
Sharpe, A. H., and Pauken, K. E. (2018). The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 18, 153–167. doi: 10.1038/nri.2017.108
Shen, C. J., Sharma, A., Vuong, D. V., Erler, J. T., Pruschy, M., and Broggini-Tenzer, A. (2014). Ionizing radiation induces tumor cell lysyl oxidase secretion. BMC Cancer 14:532. doi: 10.1186/1471-2407-14-532
Sherman, M. H., Yu, R. T., Engle, D. D., Ding, N., Atkins, A. R., Tiriac, H., et al. (2014). Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 159, 80–93. doi: 10.1016/j.cell.2014.08.007
Shetty, S., Weston, C. J., Oo, Y. H., Westerlund, N., Stamataki, Z., Youster, J., et al. (2011). Common lymphatic endothelial and vascular endothelial receptor-1 mediates the transmigration of regulatory T cells across human hepatic sinusoidal endothelium. J. Immunol. 186, 4147–4155. doi: 10.4049/jimmunol.1002961
Siddikuzzaman Grac, V. M., and Guruvayoorappan, C. (2011). Lysyl oxidase: a potential target for cancer therapy. Inflammopharmacology 19, 117–129. doi: 10.1007/s10787-010-0073-1
Siegal, G. P., Barsky, S. H., Terranova, V. P., and Liotta, L. A. (1981). Stages of neoplastic transformation of human breast tissue as monitored by dissolution of basement membrane components. An immunoperoxidase study. Invasion Metastasis 1, 54–70.
Simon, T., and Bromberg, J. S. (2017). Regulation of the immune system by laminins. Trends Immunol. 38, 858–871. doi: 10.1016/j.it.2017.06.002
Singh, A., and Settleman, J. (2010). EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 29, 4741–4751. doi: 10.1038/onc.2010.215
Smith-Mungo, L. I., and Kagan, H. M. (1998). Lysyl oxidase: properties, regulation and multiple functions in biology. Matrix Biol. 16, 387–398. doi: 10.1016/S0945-053X(98)90012-9
Stankic, M., Pavlovic, S., Chin, Y., Brogi, E., Padua, D., Norton, L., et al. (2013). TGF-beta-Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition. Cell Rep. 5, 1228–1242. doi: 10.1016/j.celrep.2013.11.014
Sternlicht, M. D., Lochter, A., Sympson, C. J., Huey, B., Rougier, J. P., Gray, J. W., et al. (1999). The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell 98, 137–146. doi: 10.1016/S0092-8674(00)81009-0
Su, S., Chen, J., Yao, H., Liu, J., Yu, S., Lao, L., et al. (2018). CD10(+)GPR77(+) cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell 172, 841–856.e16. doi: 10.1016/j.cell.2018.01.009
Sugano, H., Shirai, Y., Horiuchi, T., Saito, N., Shimada, Y., Eto, K., et al. (2018). Nafamostat mesilate enhances the radiosensitivity and reduces the radiation-induced invasive ability of colorectal cancer cells. Cancers 10:335. doi: 10.3390/cancers10100386
Suhovskih, A. V., Mostovich, L. A., Kunin, I. S., Boboev, M. M., Nepomnyashchikh, G. I., Aidagulova, S. V., et al. (2013). Proteoglycan expression in normal human prostate tissue and prostate cancer. ISRN Oncol. 2013:680136. doi: 10.1155/2013/680136
Sukowati, C. H. C., Anfuso, B., Fiore, E., Ie, S. I., Raseni, A., Vascotto, F., et al. (2019). Hyaluronic acid inhibition by 4-methylumbelliferone reduces the expression of cancer stem cells markers during hepatocarcinogenesis. Sci. Rep. 9:4026. doi: 10.1038/s41598-019-40436-6
Sullivan, R., Pare, G. C., Frederiksen, L. J., Semenza, G. L., and Graham, C. H. (2008). Hypoxia-induced resistance to anticancer drugs is associated with decreased senescence and requires hypoxia-inducible factor-1 activity. Mol. Cancer Ther. 7, 1961–1973. doi: 10.1158/1535-7163.MCT-08-0198
Sun, D., Wang, X., Zhang, H., Deng, L., and Zhang, Y. (2011). MMP9 mediates MICA shedding in human osteosarcomas. Cell Biol. Int. 35, 569–574. doi: 10.1042/CBI20100431
Takai, K., Le, A., Weaver, V. M., and Werb, Z. (2016). Targeting the cancer-associated fibroblasts as a treatment in triple-negative breast cancer. Oncotarget 7, 82889–82901. doi: 10.18632/oncotarget.12658
Tang, N., Wang, L., Esko, J., Giordano, F. J., Huang, Y., Gerber, H. P., et al. (2004). Loss of HIF-1α in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell 6, 485–495. doi: 10.1016/j.ccr.2004.09.026
Tang, Y. A., Chen, Y. F., Bao, Y., Mahara, S., Yatim, S., Oguz, G., et al. (2018). Hypoxic tumor microenvironment activates GLI2 via HIF-1alpha and TGF-beta2 to promote chemoresistance in colorectal cancer. Proc. Natl. Acad. Sci. U.S.A. 115, E5990–E5999. doi: 10.1073/pnas.1801348115
Tariq, M., Zhang, J., Liang, G., Ding, L., He, Q., and Yang, B. (2017). Macrophage polarization: anti-cancer strategies to target tumor-associated macrophage in breast cancer. J. Cell. Biochem. 118, 2484–2501. doi: 10.1002/jcb.25895
Taylor, M. A., Amin, J. D., Kirschmann, D. A., and Schiemann, W. P. (2011). Lysyl oxidase contributes to mechanotransduction-mediated regulation of transforming growth factor-beta signaling in breast cancer cells. Neoplasia 13, 406–418. doi: 10.1593/neo.101086
Theocharis, A. D. (2002). Human colon adenocarcinoma is associated with specific post-translational modifications of versican and decorin. Biochim. Biophys. Acta 1588, 165–172. doi: 10.1016/S0925-4439(02)00161-8
Theocharis, A. D., Tsara, M. E., Papageorgacopoulou, N., Karavias, D. D., and Theocharis, D. A. (2000). Pancreatic carcinoma is characterized by elevated content of hyaluronan and chondroitin sulfate with altered disaccharide composition. Biochim. Biophys. Acta 1502, 201–206. doi: 10.1016/S0925-4439(00)00051-X
Theocharis, A. D., Vynios, D. H., Papageorgakopoulou, N., Skandalis, S. S., and Theocharis, D. A. (2003). Altered content composition and structure of glycosaminoglycans and proteoglycans in gastric carcinoma. Int. J. Biochem. Cell Biol. 35, 376–390. doi: 10.1016/S1357-2725(02)00264-9
Thomas, N. K., and Brown, T. J. (2010). ABC transporters do not contribute to extracellular translocation of hyaluronan in human breast cancer in vitro. Exp. Cell Res. 316, 1241–1253. doi: 10.1016/j.yexcr.2010.01.004
Toss, M. S., Miligy, I. M., Gorringe, K. L., AlKawaz, A., Khout, H., Ellis, I. O., et al. (2018). Prolyl-4-hydroxylase A subunit 2 (P4HA2) expression is a predictor of poor outcome in breast ductal carcinoma in situ (DCIS). Br. J. Cancer 119, 1518–1526. doi: 10.1038/s41416-018-0337-x
Turley, S. J., Cremasco, V., and Astarita, J. L. (2015). Immunological hallmarks of stromal cells in the tumour microenvironment. Nat. Rev. Immunol. 15, 669–682. doi: 10.1038/nri3902
Uchakina, O. N., Ban, H., Hostetler, B. J., and McKallip, R. J. (2016). Inhibition of hyaluronic acid formation sensitizes chronic myelogenous leukemia to treatment with doxorubicin. Glycobiology 26, 1171–1179. doi: 10.1093/glycob/cww064
Uhm, J. H., Gladson, C. L., and Rao, J. S. (1999). The role of integrins in the malignant phenotype of gliomas. Front. Biosci. 4, D188–D199. doi: 10.2741/Uhm
Urakawa, H., Nishida, Y., Wasa, J., Arai, E., Zhuo, L., Kimata, K., et al. (2012). Inhibition of hyaluronan synthesis in breast cancer cells by 4-methylumbelliferone suppresses tumorigenicity in vitro and metastatic lesions of bone in vivo. Int. J. Cancer 130, 454–466. doi: 10.1002/ijc.26014
Vaday, G. G., and Lider, O. (2000). Extracellular matrix moieties, cytokines, and enzymes: dynamic effects on immune cell behavior and inflammation. J. Leukoc. Biol. 67, 149–159. doi: 10.1002/jlb.67.2.149
Van der Veldt, A. A., Lubberink, M., Bahce, I., Walraven, M., de Boer, M. P., Greuter, H. N., et al. (2012). Rapid decrease in delivery of chemotherapy to tumors after anti-VEGF therapy: implications for scheduling of anti-angiogenic drugs. Cancer Cell 21, 82–91. doi: 10.1016/j.ccr.2011.11.023
van Kempen, L. C., Rijntjes, J., Mamor-Cornelissen, I., Vincent-Naulleau, S., Gerritsen, M. J., Ruiter, D. J., et al. (2008). Type I collagen expression contributes to angiogenesis and the development of deeply invasive cutaneous melanoma. Int. J. Cancer 122, 1019–1029. doi: 10.1002/ijc.23147
van Meeteren, L. A., Goumans, M. J., and ten Dijke, P. (2011). TGF-β receptor signaling pathways in angiogenesis; emerging targets for anti-angiogenesis therapy. Curr. Pharm. Biotechnol. 12, 2108–2120. doi: 10.2174/138920111798808338
Verstovsek, S., Savona, M. R., Mesa, R. A., Dong, H., Maltzman, J. D., Sharma, S., et al. (2017). A phase 2 study of simtuzumab in patients with primary, post-polycythaemia vera or post-essential thrombocythaemia myelofibrosis. Br. J. Haematol. 176, 939–949. doi: 10.1111/bjh.14501
Voloshenyuk, T. G., Landesman, E. S., Khoutorova, E., Hart, A. D., and Gardner, J. D. (2011). Induction of cardiac fibroblast lysyl oxidase by TGF-beta1 requires PI3K/Akt, Smad3, and MAPK signaling. Cytokine 55, 90–97. doi: 10.1016/j.cyto.2011.03.024
Voron, T., Colussi, O., Marcheteau, E., Pernot, S., Nizard, M., Pointet, A. L., et al. (2015). VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 212, 139–148. doi: 10.1084/jem.20140559
Vynios, D. H., Theocharis, D. A., Papageorgakopoulou, N., Papadas, T. A., Mastronikolis, N. S., Goumas, P. D., et al. (2008). Biochemical changes of extracellular proteoglycans in squamous cell laryngeal carcinoma. Connect. Tissue Res. 49, 239–243. doi: 10.1080/03008200802147662
Waldhauer, I., Goehlsdorf, D., Gieseke, F., Weinschenk, T., Wittenbrink, M., Ludwig, A., et al. (2008). Tumor-associated MICA is shed by ADAM proteases. Cancer Res. 68, 6368–6376. doi: 10.1158/0008-5472.CAN-07-6768
Walter, C., Davis, J. T., Mathur, J., and Pathak, A. (2018). Physical defects in basement membrane-mimicking collagen-IV matrices trigger cellular EMT and invasion. Integr. Biol. 10, 342–355. doi: 10.1039/c8ib00034d
Wang, C., Tammi, M., Guo, H., and Tammi, R. (1996). Hyaluronan distribution in the normal epithelium of esophagus, stomach, and colon and their cancers. Am. J. Pathol. 148, 1861–1869.
Wang, X., Luo, J., He, L., Cheng, X., Yan, G., Wang, J., et al. (2018). Hybrid pH-sensitive nanogels surface-functionalized with collagenase for enhanced tumor penetration. J. Colloid Interface Sci. 525, 269–281. doi: 10.1016/j.jcis.2018.04.084
Wei, J., Wu, A., Kong, L. Y., Wang, Y., Fuller, G., Fokt, I., et al. (2011). Hypoxia potentiates glioma-mediated immunosuppression. PLoS ONE 6:e16195. doi: 10.1371/journal.pone.0016195
Wei, S. C., Fattet, L., Tsai, J. H., Guo, Y., Pai, V. H., Majeski, H. E., et al. (2015). Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat. Cell Biol. 17, 678–688. doi: 10.1038/ncb3157
Weigel, P. H., and DeAngelis, P. L. (2007). Hyaluronan synthases: a decade-plus of novel glycosyltransferases. J. Biol. Chem. 282, 36777–36781. doi: 10.1074/jbc.R700036200
Welter, M., and Rieger, H. (2013). Interstitial fluid flow and drug delivery in vascularized tumors: a computational model. PLoS ONE 8:e70395. doi: 10.1371/journal.pone.0070395
Wesley, R. B. II., Meng, X., Godin, D., and Galis, Z. S. (1998). Extracellular matrix modulates macrophage functions characteristic to atheroma: collagen type I enhances acquisition of resident macrophage traits by human peripheral blood monocytes in vitro. Arterioscler. Thromb. Vasc. Biol. 18, 432–440. doi: 10.1161/01.ATV.18.3.432
Whatcott, C. J., Ng, S., Barrett, M. T., Hostetter, G., Von Hoff, D. D., and Han, H. (2017). Inhibition of ROCK1 kinase modulates both tumor cells and stromal fibroblasts in pancreatic cancer. PLoS ONE 12:e0183871. doi: 10.1371/journal.pone.0183871
Willumsen, N., Thomsen, L. B., Bager, C. L., Jensen, C., and Karsdal, M. A. (2018). Quantification of altered tissue turnover in a liquid biopsy: a proposed precision medicine tool to assess chronic inflammation and desmoplasia associated with a pro-cancerous niche and response to immuno-therapeutic anti-tumor modalities. Cancer Immunol. Immunother. 67, 1–12. doi: 10.1007/s00262-017-2074-z
Xiao, W., Wang, S., Zhang, R., Sohrabi, A., Yu, Q., Liu, S., et al. (2019). Bioengineered scaffolds for 3D culture demonstrate extracellular matrix-mediated mechanisms of chemotherapy resistance in glioblastoma. Matrix Biol. doi: 10.1016/j.matbio.2019.04.003
Xie, J., Wang, C., Huang, D. Y., Zhang, Y., Xu, J., Kolesnikov, S. S., et al. (2013). TGF-beta1 induces the different expressions of lysyl oxidases and matrix metalloproteinases in anterior cruciate ligament and medial collateral ligament fibroblasts after mechanical injury. J. Biomech. 46, 890–898. doi: 10.1016/j.jbiomech.2012.12.019
Xie, P., Yu, H., Wang, F., Yan, F., and He, X. (2019). Inhibition of LOXL2 enhances the radiosensitivity of castration-resistant prostate cancer cells associated with the reversal of the EMT process. Biomed Res. Int. 2019:4012590. doi: 10.1155/2019/4012590
Xiong, G., Deng, L., Zhu, J., Rychahou, P. G., and Xu, R. (2014). Prolyl-4-hydroxylase alpha subunit 2 promotes breast cancer progression and metastasis by regulating collagen deposition. BMC Cancer 14:1. doi: 10.1186/1471-2407-14-1
Xiong, G., Stewart, R. L., Chen, J., Gao, T., Scott, T. L., Samayoa, L. M., et al. (2018). Collagen prolyl 4-hydroxylase 1 is essential for HIF-1alpha stabilization and TNBC chemoresistance. Nat. Commun. 9:4456. doi: 10.1038/s41467-018-06893-9
Xu, S., Yang, Z., Jin, P., Yang, X., Li, X., Wei, X., et al. (2018). Metformin suppresses tumor progression by inactivating stromal fibroblasts in ovarian cancer. Mol. Cancer Ther. 17, 1291–1302. doi: 10.1158/1535-7163.MCT-17-0927
Xue, X., and Shah, Y. M. (2013). Hypoxia-inducible factor-2alpha is essential in activating the COX2/mPGES-1/PGE2 signaling axis in colon cancer. Carcinogenesis 34, 163–169. doi: 10.1093/carcin/bgs313
Yang, J., Mani, S. A., Donaher, J. L., Ramaswamy, S., Itzykson, R. A., Come, C., et al. (2004). Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117, 927–939. doi: 10.1016/j.cell.2004.06.006
Yang, M. C., Wang, C. J., Liao, P. C., Yen, C. J., and Shan, Y. S. (2014). Hepatic stellate cells secretes type I collagen to trigger epithelial mesenchymal transition of hepatoma cells. Am. J. Cancer Res. 4, 751–763.
Yates, T. J., Lopez, L. E., Lokeshwar, S. D., Ortiz, N., Kallifatidis, G., Jordan, A., et al. (2015). Dietary supplement 4-methylumbelliferone: an effective chemopreventive and therapeutic agent for prostate cancer. J. Natl. Cancer Inst. 107:djv085. doi: 10.1093/jnci/djv085
Yoon, S. O., Shin, S., Karreth, F. A., Buel, G. R., Jedrychowski, M. P., Plas, D. R., et al. (2017). Focal adhesion- and IGF1R-dependent survival and migratory pathways mediate tumor resistance to mTORC1/2 inhibition. Mol Cell 67, 512–527.e4. doi: 10.1016/j.molcel.2017.06.033
Yoshida, E., Kudo, D., Nagase, H., Suto, A., Shimoda, H., Suto, S., et al. (2018). 4-methylumbelliferone decreases the hyaluronan-rich extracellular matrix and increases the effectiveness of 5-fluorouracil. Anticancer Res. 38, 5799–5804. doi: 10.21873/anticanres.12919
Zaffryar-Eilot, S., Marshall, D., Voloshin, T., Bar-Zion, A., Spangler, R., Kessler, O., et al. (2013). Lysyl oxidase-like-2 promotes tumour angiogenesis and is a potential therapeutic target in angiogenic tumours. Carcinogenesis 34, 2370–2379. doi: 10.1093/carcin/bgt241
Zeisberg, M., Bonner, G., Maeshima, Y., Colorado, P., Muller, G. A., Strutz, F., et al. (2001). Renal fibrosis: collagen composition and assembly regulates epithelial-mesenchymal transdifferentiation. Am. J. Pathol. 159, 1313–1321. doi: 10.1016/S0002-9440(10)62518-7
Zhang, F., Wang, H., Wang, X., Jiang, G., Liu, H., Zhang, G., et al. (2016). TGF-β induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget 7, 52294–52306. doi: 10.18632/oncotarget.10561
Zhang, H., Ren, L., Ding, Y., Li, F., Chen, X., Ouyang, Y., et al. (2019). Hyaluronan-mediated motility receptor confers resistance to chemotherapy via TGFbeta/Smad2-induced epithelial-mesenchymal transition in gastric cancer. FASEB J. 33, 6365–6377. doi: 10.1096/fj.201802186R
Zhang, H., Tsang, J. Y., Ni, Y. B., Chan, S. K., Chan, K. F., Cheung, S. Y., et al. (2016). Hyaluronan synthase 2 is an adverse prognostic marker in androgen receptor-negative breast cancer. J. Clin. Pathol. 69, 1055–1062. doi: 10.1136/jclinpath-2016-203617
Zhang, L., Wang, Y., Xia, T., Yu, Q., Zhang, Q., Yang, Y., et al. (2016). Suppression for lung metastasis by depletion of collagen I and lysyl oxidase via losartan assisted with paclitaxel-loaded pH-sensitive liposomes in breast cancer. Drug Deliv. 23, 2970–2979. doi: 10.3109/10717544.2015.1132798
Zhao, Q., Eichten, A., Parveen, A., Adler, C., Huang, Y., Wang, W., et al. (2018). Single-cell transcriptome analyses reveal endothelial cell heterogeneity in tumors and changes following antiangiogenic treatment. Cancer Res. 78, 2370–2382. doi: 10.1158/0008-5472.CAN-17-2728
Zhao, Y., Wang, L., Huang, Q., Jiang, Y., Wang, J., Zhang, L., et al. (2016). Radiosensitization of non-small cell lung cancer cells by inhibition of TGF-beta1 signaling with SB431542 is dependent on p53 status. Oncol. Res. 24, 1–7. doi: 10.3727/096504016X14570992647087
Zheng, X., Carstens, J. L., Kim, J., Scheible, M., Kaye, J., Sugimoto, H., et al. (2015). Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 527, 525–530. doi: 10.1038/nature16064
Zhou, J., Mochizuki, T., Smeets, H., Antignac, C., Laurila, P., de Paepe, A., et al. (1993). Deletion of the paired alpha 5(IV) and alpha 6(IV) collagen genes in inherited smooth muscle tumors. Science 261, 1167–1169. doi: 10.1126/science.8356449
Keywords: extracellular matrix, cancer therapy, drug transport, immunotherapy, chemotherapy (CH), radiotherapy, tumor microenvironment, ECM
Citation: Henke E, Nandigama R and Ergün S (2020) Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front. Mol. Biosci. 6:160. doi: 10.3389/fmolb.2019.00160
Received: 04 May 2019; Accepted: 20 December 2019;
Published: 31 January 2020.
Edited by:
William Cho, Queen Elizabeth Hospital (QEH), Hong KongReviewed by:
Haiyan Xiao, Augusta University, United StatesCopyright © 2020 Henke, Nandigama and Ergün. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Erik Henke, ZXJpay5oZW5rZUB1bmktd3VlcnpidXJnLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.