 Susanne C. S. Bantele
Susanne C. S. Bantele Boris Pfander
Boris Pfander- Max Planck Institute of Biochemistry, DNA Replication and Genome Integrity, Martinsried, Germany
Many cellular pathways are dedicated to maintain the integrity of the genome. In eukaryotes, the underlying DNA transactions occur in the context of chromatin. Cells utilize chromatin and its dynamic nature to regulate those genome integrity pathways. Accordingly, chromatin becomes restructured and modified around DNA damage sites. Here, we review the current knowledge of a chromatin remodeler Fun30SMARCAD1, which plays a key role in genome maintenance. Fun30SMARCAD1 promotes DNA end resection and the repair of DNA double-stranded breaks (DSBs). Notably, however, Fun30SMARCAD1 plays additional roles in maintaining heterochromatin and promoting transcription. Overall, Fun30SMARCAD1 is involved in distinct processes and the specific roles of Fun30SMARCAD1 at DSBs, replication forks and sites of transcription appear discordant at first view. Nonetheless, a picture emerges in which commonalities within these context-dependent roles of Fun30SMARCAD1 exist, which may help to gain a more global understanding of chromatin alterations induced by Fun30SMARCAD1.
Fun30 (function unknown now, budding yeast) and its homologs Fft3 (fission yeast) and SMARCAD1 (human; Etl1 in mouse) are non-essential Snf2-like Etl1-subfamily nucleosome remodelers which function in DNA replication, heterochromatin stability, transcription, meiotic hotspot activity, and regulation of DNA repair (Flaus, 2006; Okazaki et al., 2008; Neves-Costa et al., 2009; Rowbotham et al., 2011; Strålfors et al., 2011; Yu et al., 2011; Chen et al., 2012; Costelloe et al., 2012; Eapen et al., 2012; Byeon et al., 2013; Steglich et al., 2015; Densham et al., 2016; Doiguchi et al., 2016; Bantele et al., 2017; Lee et al., 2017; Xiao et al., 2017; Chakraborty et al., 2018; Ding et al., 2018; Jahn et al., 2018; Storey et al., 2018; Terui et al., 2018; Sachs et al., 2019). Notably, during DNA double-strand break (DSB) repair a major function of Fun30 orthologs appears to be in DNA end resection, a process that requires the mobilization and likely eviction of nucleosomes (Chen et al., 2012; Costelloe et al., 2012; Eapen et al., 2012; Bantele et al., 2017). In apparent contrast, during DNA replication of heterochromatin, Fun30 orthologs in fission yeast and human cells seem to rather provide stability of nucleosomes and to prevent loss of heterochromatic histone marks (Rowbotham et al., 2011; Taneja et al., 2017).
With this review, we aim to summarize current data in order to show commonalities and highlight regulatory mechanisms controlling Fun30SMARCAD1 remodelers with a special focus on the DNA damage response. The different Fun30SMARCAD1 functions appear discrepant at first view, but in this review we will also attempt to point toward commonalities.
Domain Structure of Fun30 and its Orthologs
Fun30 and its orthologs are ~1,000 amino acids large, single-subunit nucleosome remodelers, which appear to act in homodimeric form (Awad et al., 2010). A bioinformatic analysis showed that Fun30 shares the highest degree of homology with Swr1 and Ino80 of the Snf2 remodeler family (Flaus, 2006). It comprises the catalytic Snf2 nucleosome remodeling domain, but with a Fun30-specific yet uncharacterized insert at the C-terminus (Liu and Jiang, 2017). The N-terminal half of the protein appears to be regulatory and harbors specific regions with the ability to engage in protein-protein interactions (Flaus, 2006; Neves-Costa et al., 2009; Bantele et al., 2017). At the N-terminus, conserved Cyclin-dependent kinase (CDK) phosphorylation sites in yeast Fun30 and human SMARCAD1 (Chen X. et al., 2016; Bantele et al., 2017) are followed by ubiquitin-binding CUE (Coupling of Ubiquitin conjugation to ER degradation) domains, which exist in one or more copies in almost all Fun30 orthologs. In human SMARCAD1, further regulatory ATM phosphorylation sites and phosphorylation-dependent RING1 ubiquitylation sites are targeted after DNA damage and located at the C-terminus (Matsuoka et al., 2007; Densham et al., 2016; Chakraborty et al., 2018). In the following, we will view Fun30SMARCAD1 from N to C and summarize the molecular role of the additional regulatory elements.
CDK Phosphorylation at the N-Terminus of Fun30 and SMARCAD1
Several studies have established Fun30 as CDK substrate in vitro and in vivo (Ubersax et al., 2003; Chen et al., 2012; Chen X. et al., 2016; Bantele et al., 2017). Specifically, Fun30 is targeted by CDK on S20, S28, and S34 (Chen X. et al., 2016; Bantele et al., 2017). Similarly, SMARCAD1 can be phosphorylated by CDK on T71 (Bantele et al., 2017). Once phosphorylated, S20 and S28 in Fun30 and T71 in SMARCAD1 mediate a direct protein-protein interaction with the N-terminal BRCT repeats of the scaffold protein Dpb11 (in yeast) and TOPBP1 (in human) (Bantele et al., 2017). In yeast, this interaction leads to formation of a ternary complex with the 9-1-1 checkpoint clamp and contributes to targeting Fun30 to sites of DNA damage (Bantele et al., 2017, Figure 1). These data suggest that phosphorylation is a means to localize Fun30, but additionally it is possible that phosphorylation and the associated protein-protein interactions are involved in activating the remodeller toward its substrate.
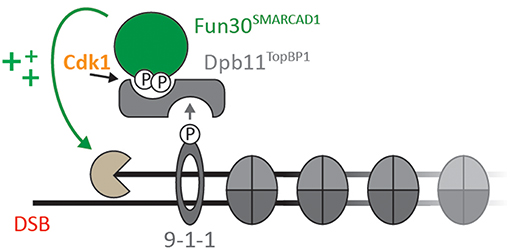
Figure 1. Cell cycle- and DNA damage-activated kinases lead to formation of a ternary complex formed by Fun30SMARCAD1, Dpb11TOPBP1, and the 9-1-1 complex (adapted from Bantele et al., 2017; Bantele, 2018). Upon CDK-dependent phosphorylation of Fun30 S20/S28 or SMARCAD1 T71, respectively, Fun30 and SMARCAD1 associate with BRCT1+2 of Dpb11 or BRCT0/1/2 of TOPBP1. In yeast, binding to the 9-1-1 complex (in a DNA damage-induced manner) contributes to localization of Fun30-Dpb11 to sites of DNA end resection, where it stimulates long-range resection (Chen et al., 2012; Costelloe et al., 2012; Eapen et al., 2012; Bantele et al., 2017).
CUE Domains
CUE domains are known for their ability to bind ubiquitin (Donaldson et al., 2003; Kang et al., 2003; Shih et al., 2003), and the N-terminal CUE domain in SMARCAD1 was shown to mediate interactions with the chromatin regulator KAP1 (Rowbotham et al., 2011; Ding et al., 2018; Lim et al., 2019). Furthermore, a CUE domain-dependent interaction of SMARCAD1 with ubiquitylated histone H2A has been identified recently (Densham et al., 2016). Whether binding of ubiquitylated histones is a conserved property of Fun30 orthologs remains to be shown. While a CUE domain has been bioinformatically predicted for Fun30 as well (Neves-Costa et al., 2009), so far no binding partner of the Fun30 CUE domain could be identified. In vitro experiments also failed to provide evidence for Fun30 binding to ubiquitylated histones (Awad et al., 2010). Nonetheless, the CUE-dependent protein-protein interactions seem to contribute to context-dependent chromatin localization in the human protein (Densham et al., 2016; Ding et al., 2018). Interestingly, the SMARCAD1-KAP1 interaction has very recently been shown to occur between the SMARCAD1 N-terminal CUE domain and a specific patch in KAP1 that does structurally not resemble ubiquitin (Lim et al., 2019). This finding suggests alternative and still-to-be explored interaction modes of Fun30SMARCAD1.
DNA Damage-Dependent Phosphorylation at the C-Terminus of SMARCAD1
SMARCAD1 is a substrate of the ATM kinase and gets phosphorylated on T906 upon DNA damage (Matsuoka et al., 2007; Densham et al., 2016; Chakraborty et al., 2018). This modification is a prerequisite for the subsequent ubiquitylation on K905 in a RING1-dependent manner (Chakraborty et al., 2018). Both, DNA damage-dependent phosphorylation and ubiquitylation of SMARCAD1 were connected to functions in the DNA damage response, but do not seem to be conserved in the yeast protein. Interestingly, Fun30 was suggested to interact with other proteins of the DNA damage response, such as DNA end resection enzymes Exo1 and Dna2, as well as RPA (Chen et al., 2012). Where the specific interaction sites are located, whether all interactions are direct and in how far they are regulated by post-translational modification remains to be determined.
Biochemical Activities of Fun30SMARCAD1
Nucleosome remodelers use ATP to remodel histone-DNA contacts in order to move or position nucleosomes, evict them or change their composition (Clapier and Cairns, 2009; Hargreaves and Crabtree, 2011). These molecular activities can be studied well in vitro and analogous experiments have been performed for Fun30 (Awad et al., 2010; Adkins et al., 2013, 2017).
Fun30 has the general ability to directly bind DNA in vitro. Interestingly, both single-stranded (ss) and double-stranded (ds) DNA as well as nucleosome-associated DNA was bound (Awad et al., 2010; Adkins et al., 2017). In line with these findings, presence of ssDNA, dsDNA, or chromatin stimulated the ATPase activity of Fun30 as was observed for other remodelers (Awad et al., 2010; reviewed in Zhou et al., 2016; Clapier et al., 2017). In vitro, nucleosomes were seen to be repositioned in the presence of Fun30 and H2A-H2B dimers were found to be liberated from chromatin templates, suggesting that Fun30 has nucleosome sliding and histone dimer exchange activity (Awad et al., 2010; Byeon et al., 2013). In vivo, evidence for a dimer exchange activity of Fun30 is currently lacking, but fun30Δ cells showed alterations in the nucleosome-free region at the 5′ end of gene bodies, as well as altered occupancy of −1, +2, and +3 nucleosomes (Byeon et al., 2013). Consistently, also fission yeast Fft3 was found to be required for chromatin architecture (Durand-Dubief et al., 2012). Overall, these data are in good agreement with a role of Fun30 and its orthologs in nucleosome sliding and perhaps positioning, but at this point indirect effects on cellular chromatin architecture cannot be ruled out.
In the context of DNA damage, it is unknown whether Fun30 is involved in nucleosome sliding and/or positioning or whether it plays other roles. While a previous study did not find evidence for Fun30 mediating changes in nucleosome positioning in the proximity of a DSB (Costelloe et al., 2012), it might be technically challenging to visualize such changes during dynamic repair.
A possible role in H2A-H2B dimer exchange may manifest in changes in occupancy of the H2A variant H2A.Z or perhaps also of post-translationally modified forms of H2A (such as γH2A). Distribution of H2A.Z was indeed influenced by Fft3 and Fun30, both genome-wide and particularly in centromeric, pericentromeric, and subtelomeric chromatin (Strålfors et al., 2011; Durand-Dubief et al., 2012). Given that H2A.Z is a well-known regulator of DSB repair (van Attikum et al., 2007; Kalocsay et al., 2009; Xu et al., 2012; Adkins et al., 2013; Lademann et al., 2017), it is tempting to hypothesize that Fun30 regulates H2A.Z at DSBs as well. So far, it remains to be determined whether the changes in H2A.Z distribution induced by Fun30 occur at DSBs and might potentially contribute to resection regulation.
Notably, Fun30 has particular binding preferences when it comes to nucleosome structure and modification, for example it seems to be repelled by S129-phosphorylated H2A (γH2A, induced by DNA damage) (Eapen et al., 2012; Adkins et al., 2017). One could therefore speculate that Fun30 might antagonize γH2A via an H2A/H2B dimer exchange activity. Experimental data however argue against such a model, as no changes in γH2A phosphorylation after DNA damage could be observed in mutants lacking Fun30 (Eapen et al., 2012).
Lastly, during maintenance of heterochromatin/ transcriptionally silent chromatin, Fun30/Fft3/SMARCAD1 appear to function as stabilizers of chromatin marks (Durand-Dubief et al., 2012; Byeon et al., 2013; Taneja et al., 2017), but whether this can be explained by sliding/dimer exchange activities or whether this function involves an additional activity is not known.
Overall impressive progress has been made toward understanding the catalytic activities of Fun30SMARCAD1, but nonetheless we currently do not understand the specific nature of the substrate toward which the remodeling activity is directed to, nucleosomes or modified nucleosomes are a possibility, but the function in DSB repair (see below) suggests that it might also be nucleosomes in complex with an additional protein(s) or maybe even a nucleosome-bound protein.
Biological Functions of Fun30SMARCAD1
At first glance, the biological functions of Fun30 and its orthologs appear at least as diverse as its biochemical activities (sliding, positioning, dimer exchange). In the following, we therefore aim to not only summarize the known functions, but also to highlight commonalities, since a common model describing Fun30 function would help discriminate direct from indirect consequences of a loss of Fun30 function and facilitate future research.
Gene Expression Control
Orthologs of Fun30 promote gene expression. Fission yeast Fft3 facilitates the progression of RNA Polymerase II through actively transcribed genes by mediating nucleosome dissociation (Lee et al., 2017). Also, SMARCAD1 was found to act as transcriptional activator and enhances CBP-mediated histone acetylation (Doiguchi et al., 2016). The overall importance of the contribution of Fun30 orthologs to transcription regulation remains however to be elucidated. At least in budding yeast, absence of Fun30 caused only minor changes in the expression of few proteins (Chen et al., 2012), possibly reflecting redundancy with other nucleosome remodelers (Barbaric et al., 2007; Smolle et al., 2012).
Maintenance of Silent Chromatin
All Fun30 orthologs were shown to localize to heterochromatic or repressed genomic loci and contribute to their establishment and preservation. Fission yeast Fft3 and budding yeast Fun30 localize to insulator elements and are involved in silencing at subtelomeres, centromeres, rDNA repeats, and mating type loci (Neves-Costa et al., 2009; Strålfors et al., 2011; Durand-Dubief et al., 2012; Steglich et al., 2015; Taneja et al., 2017; Jahn et al., 2018). In absence of Fft3, the composition and nuclear localization of heterochromatin is altered and accumulates euchromatic histone modifications such as H4K12Ac and H3K9Ac, as well as histone variants like H2A.Z (Strålfors et al., 2011; Steglich et al., 2015). Fun30 contributes to transcriptional repression of genes and across centromeres in order to ensure unhampered chromosome segregation (Strålfors et al., 2011; Durand-Dubief et al., 2012; Byeon et al., 2013). In vivo, Fft3, Fun30, and SMARCAD1 thus seem to ensure maintenance and inheritance of boundaries between chromatin states by stabilizing nucleosomes and preserving heterochromatic histone marks (Durand-Dubief et al., 2012; Byeon et al., 2013; Taneja et al., 2017; Xiao et al., 2017; Ding et al., 2018; Sachs et al., 2019).
Notably, Fun30 and SMARCAD1 are not only involved in maintenance of heterochromatin or silent chromatin, but are also involved in generating repressed chromatin de novo, where an interaction with HDAC1/2 mediating H3/H4 deacetylation might be involved (Okazaki et al., 2008; Rowbotham et al., 2011; Yu et al., 2011).
Inheritance of nucleosomes is crucial for heterochromatin maintenance and therefore is tightly linked to DNA replication (Saredi et al., 2016; Yadav and Whitehouse, 2016; Yang et al., 2016; reviewed in Serra-Cardona and Zhang, 2018). In line with this, it was not only shown that SMARCAD1 is required for heterochromatin maintenance in proliferating cells, but SMARCAD1 was also shown to bind to the replication factor PCNA (Rowbotham et al., 2011) suggesting a possible mechanism for how it could be targeted to sites of DNA replication. Also, the CUE domains of SMARCAD1 are specifically required and could play a role in targeting (Rowbotham et al., 2011; Ding et al., 2018). The first CUE domain of SMARCAD1 binds to KAP1 (Ding et al., 2018), but a universal function of the CUE domains as well as the link between CUE-dependent interactors and PCNA has not been established. A putative role of Fun30SMARCAD1 in chromatin inheritance during DNA replication is also interesting, since DNA replication involves formation of ssDNA and nucleosome eviction and therefore features mechanistic similarities to DNA resection, another process where Fun30 is crucially involved in (see below).
DNA Damage Response and DSB Repair
First connections of Fun30 to the DNA damage response were made by several genetic screens in budding yeast—a screen identifying factors involved in chromosome stability and segregation (Ouspenski et al., 1999), several genetic interaction screens with DNA repair mutants (Krogan et al., 2003, 2006; Collins et al., 2007; Beltrao et al., 2009; Costanzo et al., 2010), a screen for mutants affecting gene targeting (Chen et al., 2012), and a screen for mutants affecting break-induced replication (Costelloe et al., 2012). Fun30 and SMARCAD1 were furthermore connected to the DNA mismatch repair pathway (MMR; Chen Z. et al., 2016; Goellner et al., 2018; Terui et al., 2018) and shown to be required for the resistance to irradiation and camptothecin (CPT) (Neves-Costa et al., 2009; Costelloe et al., 2012; Chakraborty et al., 2018). In 2012, a series of pioneering publications established a key role of Fun30 and SMARCAD1 during the repair of DNA DSBs by homologous recombination (Chen et al., 2012; Costelloe et al., 2012; Eapen et al., 2012). Together with more recent work (Chen X. et al., 2016; Densham et al., 2016; Bantele et al., 2017) these publications convincingly demonstrate a molecular function in promoting DNA end resection, the nucleolytic digestion of dsDNA at DSBs that leads to the formation of 3′ssDNA overhangs.
Enhancement of DSB Resection
DNA DSBs can be repaired by non-homologous end-joining (NHEJ) or homologous recombination (HR) pathways (Symington and Gautier, 2011). The choice between these two repair regimes depends strongly on the cell cycle state and is determined at the step of DNA end resection, where DSB ends are nucleolytically digested so that 3′ overhangs are formed. These overhangs constitute crucial intermediates of repair by homologous recombination and moreover have a central signaling function at DSBs.
It is reasonable to assume that nucleosomes constitute a barrier to DNA end resection into undamaged chromatin, and indeed chromatinized DNA is resected less efficiently with increasing nucleosome density in vitro (Adkins et al., 2013). Notably, two nuclease complexes are mainly responsible for spreading of resection (long range resection) (Zhu et al., 2008; Mimitou and Symington, 2009). These nucleases—Exo1 alone and Dna2 in conjunction with the Sgs1-Top3-Rmi1 complex—bypass nucleosomes with distinct mechanisms, suggesting that they might require different forms of nucleosome remodeling (Adkins et al., 2013). Furthermore, different chromatin states such as heterochromatin might require additional means to promote resection (Baldeyron et al., 2011; Eapen et al., 2012; Soria and Almouzni, 2013; Batté et al., 2017).
Notably, while Fun30 and SMARCAD1 are required for efficient long-range resection through chromatin in vivo (Chen et al., 2012; Costelloe et al., 2012; Eapen et al., 2012; Bantele et al., 2017), initial studies could not demonstrate an effect of Fun30 on resection through chromatinized DNA in vitro, at least in case of Exo1 (Adkins et al., 2013). Most likely, the in vitro system therefore fails to recapitulate the in vivo situation. This allows to speculate that the specific substrate of Fun30 remodeling during DNA end resection might have been missing from the in vitro reaction.
In this regard, it is interesting to note that genetics have revealed a major function of Fun30 and SMARCAD1 in counteracting a resection inhibitor—Rad9 in yeast, 53BP1 in humans (Chen et al., 2012; Densham et al., 2016; Bantele et al., 2017). The role of Rad9 and 53BP1 as inhibitors of DNA end resection is clearly established. However, it is not clear whether the specific mechanism of resection inhibition is conserved through evolution. Notably, both Rad9 and 53BP1, as well as fission yeast Crb2 are nucleosome binders and appear to recognize several (modified) histones, suggesting multivalency (Huyen et al., 2004; Nakamura et al., 2004; Sanders et al., 2004; Wysocki et al., 2005; Botuyan et al., 2006; Toh et al., 2006; Grenon et al., 2007; Hammet et al., 2007; Fradet-Turcotte et al., 2013; Wilson et al., 2016). Again, the specific nature of binding sites does not appear to be conserved, but the multivalent interaction with nucleosomes is shared by Rad9 orthologs. In the absence of Rad9 or 53BP1, the remodeling activity of Fun30 or SMARCAD1 seems to be at least partly dispensable and phenotypes such as CPT sensitivity are suppressed (Chen et al., 2012; Densham et al., 2016; Bantele et al., 2017). Collectively, these data establish Rad953BP1-bound nucleosomes as excellent candidate substrate for Fun30 activity. Consistently, Rad953BP1 was shown to accumulate around DSBs when Fun30SMARCAD1 was absent (Chen et al., 2012; Costelloe et al., 2012; Densham et al., 2016). In both yeast and human cells, the ATPase activity of the remodeler is required to facilitate resection, implying active remodeling as part of the resection-promoting mechanism (Bantele et al., 2017; Chakraborty et al., 2018). One can therefore conclude that the opposition to Rad953BP1 is a central task of Fun30SMARCAD1-dependent remodeling.
The molecular nature of the Rad953BP1-Fun30SMARCAD1 antagonism is currently elusive. Figure 2 highlights plausible models for the yeast proteins, where Rad9 could act as specific inhibitor of resection, directly or indirectly inhibiting the resection nucleases via chromatin, for example by inhibiting Fun30 (Figures 2A,B). Conversely, Fun30 might overcome resection-inhibition by Rad9 and in this instance either directly remove Rad9 from chromatin, block its association in an indirect manner or counteract its downstream effects (Figures 2C,D). Since Rad9 is a nucleosome-binder, an indirect effect on Rad9 association could depend on the histone marks recognized by Rad9. For example, removal of the γH2A phosphorylation mark would lead to a defect in Rad9 chromatin association (Javaheri et al., 2006; Hammet et al., 2007; Eapen et al., 2012; Clerici et al., 2014). Such a model would be consistent with the described H2A-H2B dimer exchange ability of Fun30 (Awad et al., 2010), but would be inconsistent with experimental data, where cells lacking the γH2A modification still partially require Fun30 for efficient resection (Eapen et al., 2012). We anticipate that biochemical reconstitution will identify the remodeling substrate of Fun30SMARCAD1 and allow to reveal the mechanism by which Rad953BP1 and Fun30SMARCAD1 antagonize each other.
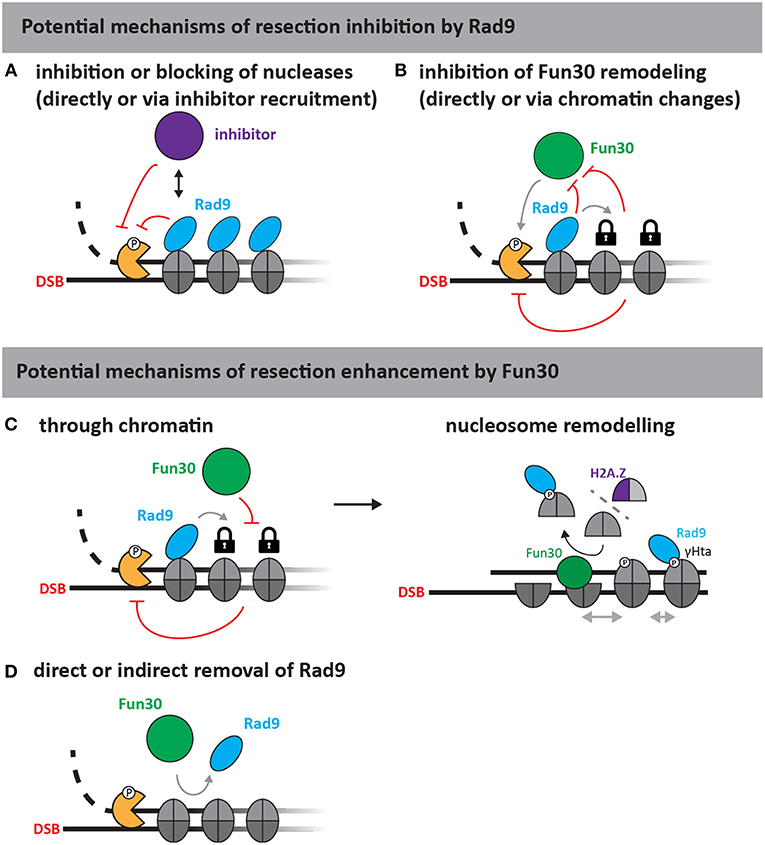
Figure 2. Putative mechanisms of resection regulation by Fun30 and Rad9 (adapted from Bantele, 2018). As Rad9 is a chromatin-binding protein without apparent catalytic activity, at least two mechanisms of resection inhibition can be envisioned (upper part). First, Rad9 could directly block or slow down the progression of nucleases either by inhibiting the nucleases (A) or by stabilizing chromatin in a configuration that is non-permissive to resection (B) for example by inhibiting Fun30, if the latter was required to help overcome resection-inhibition by nucleosomes. Fun30 could also promote resection by several different mechanisms (lower part). As a nucleosome remodeler, Fun30 could either act through chromatin (C), or by removing Rad9 from chromatin (D). The action through chromatin could involve its putative remodeling activities and potentially H2A/H2B dimer exchange, which might affect γH2A and H2A.Z dynamics or repositioning of nucleosomes by nucleosome sliding (C, right side).
It is also interesting to note that several of the binding partners of Fun30 are shared by its antagonist Rad9. These include histones, but also the BRCT repeat protein Dpb11 (Javaheri et al., 2006; Hammet et al., 2007; Pfander and Diffley, 2011; Bantele et al., 2017). Notably, in case of Dpb11, Fun30 and Rad9 even share the binding site, suggesting direct competition (Bantele et al., 2017). However, while competition might contribute to the antagonistic relationship, it is certainly not the exclusive source of this antagonism, as resection depends on the catalytic activity of Fun30 even in a context of a Fun30-Dpb11 fusion protein (Bantele et al., 2017).
Also in human cells, SMARCAD1 antagonizes 53BP1, as depletion of SMARCAD1 leads to a stabilization of 53BP1 at DSBs (Densham et al., 2016). Compared to the budding yeast system, the situation in human cells appears to be more complex. First, human cells have a second, well-established pro-resection factor and 53BP1 antagonist—BRCA1 (Cao et al., 2009; Bothmer et al., 2010; Bouwman et al., 2010; Bunting et al., 2010, 2012; Escribano-Díaz et al., 2013). BRCA1 forms a ubiquitin ligase complex together with BARD1 and BRCA1-BARD1 were shown to mediate ubiquitylation of histone H2A (Kalb et al., 2014; Densham et al., 2016). Notably, this might be a point where the two pro-resection pathways converge, since ubiquitylated H2A appears to stabilize SMARCAD1 at the DSB site, likely via direct CUE domain-dependent binding of SMARCAD1 to ubiquitylated H2A (Densham et al., 2016). A second layer of complexity comes in the form of 53BP1 effectors, such as RIF1, REV3, and the Shieldin complex (Xu et al., 2015; Dev et al., 2018; Findlay et al., 2018; Gupta et al., 2018; Mirman et al., 2018; Noordermeer et al., 2018). These effectors may inhibit resection by changing PTMs on damaged chromatin (RIF1 is a PP1 phosphatase-associated factor) or even promoting fill-in DNA synthesis (Hiraga et al., 2014; Mirman et al., 2018; Bhowmick et al., 2019; Garzón et al., 2019). Similar mechanisms have not yet been described in yeast and might represent metazoan-specific additions to an evolutionary conserved chromatin-dependent control of DNA end resection.
Cell Cycle Control and DSB Repair Pathway Choice
DSB repair by homologous recombination is coupled to the presence of a sister chromatid and therefore DSB repair pathway choice is cell cycle-regulated. This cell cycle-regulation impinges on the control of resection by Fun30, as Fun30 is phosphorylated by CDK (Chen X. et al., 2016; Bantele et al., 2017). Mechanistically, CDK-phosphorylation generates a binding site for the scaffold protein Dpb11, which in turn binds to the 9-1-1 checkpoint clamp thus leading to a ternary complex between Fun30, Dpb11, and 9-1-1 (Figure 1; Bantele et al., 2017). Formation of this complex mediates targeting to and likely activation of Fun30 at sites of DNA end resection (Bantele et al., 2017). A similar mechanism is likely occurring also in mammalian cells as SMARCAD1 can be phosphorylated by CDK as well, leading to interaction with the Dpb11 ortholog TOPBP1 (Bantele et al., 2017).
Budding yeast cells arrested in M phase show DNA end resection of DSBs that strongly depends on Fun30 and the Fun30 targeting complex (Bantele et al., 2017). Nonetheless, additional factors are clearly involved in the cell cycle control of DNA end resection. Overall, these findings raise the question of which specific DNA end resection pathway and pathway decision Fun30 is actually involved in. In this regard it has been shown that repair by homologous recombination requires resection of only a few 100 base pairs (Jinks-Robertson et al., 1993; Ira and Haber, 2002; Zhu et al., 2008), while for alternative recombination pathways and repair by single-strand annealing (SSA) in particular longer stretches of resected DNA are required (Zhu et al., 2008). One can therefore reason that the switch between NHEJ and HR is already done once resection initiates and that activation of long-range resection by Fun30 would rather further shift repair to an SSA-type mechanism. Indeed, a mild decrease of DNA end resection efficiency for example in an exo1Δ or fun30Δ mutant strain seems beneficial for HR efficiency (Lee et al., 2016), while it impedes SSA repair (Chen et al., 2012; Eapen et al., 2012; Bantele et al., 2017). Already now, one can however conclude that the Fun30-Rad9 switch and its effect on the DSB surrounding chromatin adds a further layer to DSB repair pathway choice and its changing nature during the cell cycle. It will be exciting to explore, in how far genetic tools such as a fusion of Fun30 to the 9-1-1 complex (Bantele et al., 2017) can be used to bypass these controls and whether they can be utilized for HR-dependent genome editing reactions.
Role During Chromatin Disruption and Regulation by DNA Clamps
Past research has given us very valuable insights into the individual functions of Fun30SMARCAD1, but is there commonality or can we link them to a specific enzymatic activity? The most obvious commonality at least between the function during DNA end resection and the function in the maintenance of silent chromatin regions during DNA replication is that both DNA end resection and DNA replication involve the formation of ssDNA and eviction of nucleosomes. While neither Fun30 nor any of its orthologs have been directly tested for an “evictase” function, the presence of Fun30 did not overcome the barrier function of nucleosomes toward the Exo1 exonuclease, arguing that Fun30 is not an “evictase” (Adkins et al., 2013). Moreover, the function of Fun30SMARCAD1 at DSBs and replication forks seems generally discordant, since during DNA end resection and most likely also transcription Fun30SMARCAD1 seems to open-up chromatin, while during replication it rather seems to be involved in stabilizing nucleosomes (Rowbotham et al., 2011; Chen et al., 2012; Costelloe et al., 2012; Eapen et al., 2012; Lee et al., 2018).
Nonetheless, there is more commonality—in particular seen in the regulation of Fun30SMARCAD1 by DNA clamps. During resection, Fun30 acts in complex with the 9-1-1 clamp (connected by the Dpb11 bridge) and this complex is likely conserved in humans as well (Takeishi et al., 2010; Ohashi et al., 2014; Bantele et al., 2017). Strikingly, SMARCAD1 was also found to bind to PCNA (Rowbotham et al., 2011), a processivity factor during DNA replication and key platform for protein recruitment at replication forks (Moldovan et al., 2007). While this similarity is striking, the connection to PCNA and 9-1-1 can in fact rather offer an explanation for the discrepant roles of Fun30SMARCAD1 during replication and DSB resection. PCNA and 9-1-1 were shown to be loaded onto double-stranded DNA in in vitro experiments and our unpublished data suggest that the same is true in vivo (Gomes and Burgers, 2001; Gomes et al., 2001; Majka et al., 2006; reviewed in Majka and Burgers, 2004; Peritore and Pfander, unpublished). Interestingly, the DNA clamps are located at very different positions, if one compares a replication fork to sites of DSB resection. At sites of resection, dsDNA is present upstream of the ss-dsDNA junction. The 9-1-1 complex and associated factors are therefore loaded “in front” of the resecting nucleases (see Figure 1). By associating with the 9-1-1 complex, Fun30SMARCAD1 is therefore in an ideal position to remove potential obstacles ahead of the resecting nucleases. PCNA in turn is loaded at the primer-template junction and as such will travel behind the replicative polymerase and helicase (Moldovan et al., 2007). As such, binding to PCNA will allow SMARCAD1 not only to associate with the replisome, but exactly to the place where chromatin is restored (Rowbotham et al., 2011). Thus, while similar mechanisms are used to control Fun30SMARCAD1 in different processes, the combination of localization and activity leads to different or discordant outcomes.
Concluding Remarks and Future Research
Fun30 has a key role in promoting DNA end resection and is differentially regulated at different cell cycle stages. Such regulation appears to be of high importance for the maintenance of genomic integrity, and accordingly deregulation of the human ortholog of Fun30, SMARCAD1, was found to play a crucial role during the progression of triple-negative breast cancer, which is specifically characterized by an HR-defect (Kubaisy et al., 2016; Arafat et al., 2018).
It will therefore be highly exciting for future research to further unravel the different functions and molecular mechanisms that Fun30SMARCAD1 employ to promote genome integrity. Notably, Fun30 is involved in several aspects of DNA metabolism. The major task here will be to elucidate commonalities and differences between the underlying mechanisms in order to achieve an overarching understanding of Fun30 remodeling activity.
Author Contributions
BP and SB wrote the review.
Funding
The Pfander lab work on Fun30SMARCAD1 was supported by the Max-Planck-Gesellschaft, by a grants within the Collaborative Research Cluster SFB1064 Chromatin Dynamics (project A23) and a grant from the Deutsche Forschungsgemeinschaft (DFG, German Research Council, PF794 3/1).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Sigurd Braun, Sebastian Eustermann, and members of the Pfander lab for stimulating discussion and critical reading of the manuscript.
References
Adkins, N. L., Niu, H., Sung, P., and Peterson, C. L. (2013). Nucleosome dynamics regulates DNA processing. Nat. Struct. Mol. Biol. 20, 836–842. doi: 10.1038/nsmb.2585
Adkins, N. L., Swygert, S. G., Kaur, P., Niu, H., Grigoryev, S. A., Sung, P., et al. (2017). Nucleosome-like, single-stranded DNA (ssDNA)-histone octamer complexes and the implication for DNA double strand break repair. J. Biol. Chem. 292, 5271–5281. doi: 10.1074/jbc.M117.776369
Arafat, K., Al Kubaisy, E., Sulaiman, S., Karam, S. M., Al Natour, Z., Hassan, A. H., et al. (2018). SMARCAD1 in breast cancer progression. Cell. Physiol. Biochem. 50, 489–500. doi: 10.1159/000494163
Awad, S., Ryan, D., Prochasson, P., Owen-Hughes, T., and Hassan, A. H. (2010). The Snf2 homolog Fun30 acts as a homodimeric ATP-dependent chromatin-remodeling enzyme. J. Biol. Chem. 285, 9477–9484. doi: 10.1074/jbc.M109.082149
Baldeyron, C., Soria, G., Roche, D., Cook, A. J. L., and Almouzni, G. (2011). HP1alpha recruitment to DNA damage by p150CAF-1 promotes homologous recombination repair. J. Cell Biol. 193, 81–95. doi: 10.1083/jcb.201101030
Bantele, S., Ferreira, P., Gritenaite, D., Boos, D., and Pfander, B. (2017). Targeting of the Fun30 nucleosome remodeller by the Dpb11 scaffold facilitates cell cycle-regulated DNA end resection. Elife 6:e21687. doi: 10.7554/eLife.21687
Bantele, S. C. S. (2018). Cell Cycle Regulated Signaling and Remodeling at DNA Double-Strand Breaks (Dissertation). Munich: LMU.
Barbaric, S., Luckenbach, T., Schmid, A., Blaschke, D., Hörz, W., and Korber, P. (2007). Redundancy of chromatin remodeling pathways for the induction of the yeast PHO5 promoter in vivo. J. Biol. Chem. 282, 27610–27621. doi: 10.1074/jbc.M700623200
Batté, A., Brocas, C., Bordelet, H., Hocher, A., Ruault, M., Adjiri, A., et al. (2017). Recombination at subtelomeres is regulated by physical distance, double-strand break resection and chromatin status. EMBO J. 36, 2609–2625. doi: 10.15252/embj.201796631
Beltrao, P., Trinidad, J. C., Fiedler, D., Roguev, A., Lim, W. A., Shokat, K. M., et al. (2009). Evolution of phosphoregulation: comparison of phosphorylation patterns across yeast species. PLoS Biol. 7:e1000134. doi: 10.1371/journal.pbio.1000134
Bhowmick, R., Thakur, R. S., Venegas, A. B., Liu, Y., Nilsson, J., Barisic, M., et al. (2019). The RIF1-PP1 axis controls abscission timing in human cells. Curr. Biol. 29, 1232–1242.e5. doi: 10.1016/j.cub.2019.02.037
Bothmer, A., Robbiani, D. F., Feldhahn, N., Gazumyan, A., Nussenzweig, A., and Nussenzweig, M. C. (2010). 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J. Exp. Med. 207, 855–865. doi: 10.1084/jem.20100244
Botuyan, M. V., Lee, J., Ward, I. M., Kim, J.-E., Thompson, J. R., Chen, J., et al. (2006). Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell 127, 1361–1373. doi: 10.1016/j.cell.2006.10.043
Bouwman, P., Aly, A., Escandell, J. M., Pieterse, M., Bartkova, J., van der Gulden, H., et al. (2010). 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 17, 688–695. doi: 10.1038/nsmb.1831
Bunting, S. F., Callen, E., Kozak, M. L., Kim, J. M., Wong, N., López-Contreras, A. J., et al. (2012). BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol. Cell. 46, 125–135. doi: 10.1016/j.molcel.2012.02.015
Bunting, S. F., Callen, E., Wong, N., Chen, H.-T., Polato, F., Gunn, A., et al. (2010). 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141, 243–254. doi: 10.1016/j.cell.2010.03.012
Byeon, B., Wang, W., Barski, A., Ranallo, R. T., Bao, K., Schones, D. E., et al. (2013). The ATP-dependent chromatin remodeling enzyme Fun30 represses transcription by sliding promoter-proximal nucleosomes. J. Biol. Chem. 288, 23182–23193. doi: 10.1074/jbc.M113.471979
Cao, L., Xu, X., Bunting, S. F., Liu, J., Wang, R.-H., Cao, L. L., et al. (2009). A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol. Cell. 35, 534–541. doi: 10.1016/j.molcel.2009.06.037
Chakraborty, S., Pandita, R. K., Hambarde, S., Mattoo, A. R., Charaka, V., Ahmed, K. M., et al. (2018). SMARCAD1 phosphorylation and ubiquitination are required for resection during DNA double-strand break repair. iScience 2, 123–135. doi: 10.1016/j.isci.2018.03.016
Chen, X., Cui, D., Papusha, A., Zhang, X., Chu, C.-D., Tang, J., et al. (2012). The Fun30 nucleosome remodeller promotes resection of DNA double-strand break ends. Nature 489, 576–580. doi: 10.1038/nature11355
Chen, X., Niu, H., Yu, Y., Wang, J., Zhu, S., Zhou, J., et al. (2016). Enrichment of Cdk1-cyclins at DNA double-strand breaks stimulates Fun30 phosphorylation and DNA end resection. Nucleic Acids Res. 44, 2742–2753. doi: 10.1093/nar/gkv1544
Chen, Z., Tran, M., Tang, M., Wang, W., Gong, Z., and Chen, J. (2016). Proteomic analysis reveals a novel Mutator S (MutS) partner involved in mismatch repair pathway. Mol. Cell. Proteom. 15, 1299–1308. doi: 10.1074/mcp.M115.056093
Clapier, C. R., and Cairns, B. R. (2009). The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 78, 273–304. doi: 10.1146/annurev.biochem.77.062706.153223
Clapier, C. R., Iwasa, J., Cairns, B. R., and Peterson, C. L. (2017). Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol. 18, 407–422. doi: 10.1038/nrm.2017.26
Clerici, M., Trovesi, C., Galbiati, A., Lucchini, G., and Longhese, M. P. (2014). Mec1/ATR regulates the generation of single-stranded DNA that attenuates Tel1/ATM signaling at DNA ends. EMBO J. 33, 198–216. doi: 10.1002/embj.201386041
Collins, S. R., Miller, K. M., Maas, N. L., Roguev, A., Fillingham, J., Chu, C. S., et al. (2007). Functional dissection of protein complexes involved in yeast chromosome biology using a genetic interaction map. Nature 446, 806–810. doi: 10.1038/nature05649
Costanzo, M., Baryshnikova, A., Bellay, J., Kim, Y., Spear, E. D., Sevier, C. S., et al. (2010). The genetic landscape of a cell. Science 327, 425–431. doi: 10.1126/science.1180823
Costelloe, T., Louge, R., Tomimatsu, N., Mukherjee, B., Martini, E., Khadaroo, B., et al. (2012). The yeast Fun30 and human SMARCAD1 chromatin remodellers promote DNA end resection. Nature 489, 581–510. doi: 10.1038/nature11353
Densham, R. M., Garvin, A. J., Stone, H. R., Strachan, J., Baldock, R. A., Daza-Martin, M., et al. (2016). Human BRCA1–BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat. Struct. Mol. Biol. 23, 647–655. doi: 10.1038/nsmb.3236
Dev, H., Chiang, T.-W. W., Lescale, C., Krijger, I., Martin, A. G., Pilger, D., et al. (2018). Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol. 20, 1–19. doi: 10.1038/s41556-018-0140-1
Ding, D., Bergmaier, P., Sachs, P., Klangwart, M., Rückert, T., Bartels, N., et al. (2018). The CUE1 domain of the SNF2-like chromatin remodeler SMARCAD1 mediates its association with KRAB-associated protein 1 (KAP1) and KAP1 target genes. J. Biol. Chem. 293, 2711–2724. doi: 10.1074/jbc.RA117.000959
Doiguchi, M., Nakagawa, T., Imamura, Y., Yoneda, M., Higashi, M., Kubota, K., et al. (2016). SMARCAD1 is an ATP-dependent stimulator of nucleosomal H2A acetylation via CBP, resulting in transcriptional regulation. Sci. Rep. 6:20179. doi: 10.1038/srep20179
Donaldson, K. M., Yin, H. W., Gekakis, N., Supek, F., and Joazeiro, C. (2003). Ubiquitin signals protein trafficking via interaction with a novel ubiquitin binding domain in the membrane fusion regulator, Vps9p. Curr. Biol. 13, 258–262. doi: 10.1016/S0960-9822(03)00043-5
Durand-Dubief, M., Will, W. R., Petrini, E., Theodorou, D., Harris, R. R., Crawford, M. R., et al. (2012). SWI/SNF-like chromatin remodeling factor Fun30 supports point centromere function in S. cerevisiae. PLoS Genet. 8:e1002974. doi: 10.1371/journal.pgen.1002974
Eapen, V. V., Sugawara, N., Tsabar, M., Wu, W.-H., and Haber, J. E. (2012). The Saccharomyces cerevisiae chromatin remodeler Fun30 regulates DNA end resection and checkpoint deactivation. Mol. Cell. Biol. 32, 4727–4740. doi: 10.1128/MCB.00566-12
Escribano-Díaz, C., Orthwein, A., Fradet-Turcotte, A., Xing, M., Young, J. T. F., Tkáč, J., et al. (2013). A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell. 49, 872–883. doi: 10.1016/j.molcel.2013.01.001
Findlay, S., Heath, J., Luo, V. M., Malina, A., Morin, T., Coulombe, Y., et al. (2018). SHLD2/FAM35A co-operates with REV7 to coordinate DNA double-strand break repair pathway choice. EMBO J. 37:e100158. doi: 10.15252/embj.2018100158
Flaus, A. (2006). Identification of multiple distinct Snf2 subfamilies with conserved structural motifs. Nucleic Acids Res. 34, 2887–2905. doi: 10.1093/nar/gkl295
Fradet-Turcotte, A., Canny, M. D., Escribano-Díaz, C., Orthwein, A., Leung, C. C. Y., Huang, H., et al. (2013). 53BP1 is a reader of the DNA-damage- induced H2A Lys 15 ubiquitin mark. Nature 498, 50–54. doi: 10.1038/nature12318
Garzón, J., Ursich, S., Lopes, M., Hiraga, S.-I., and Donaldson, A. D. (2019). Human RIF1-protein phosphatase 1 prevents degradation and breakage of nascent DNA on replication stalling. Cell Rep. 27, 2558–2566.e4. doi: 10.1016/j.celrep.2019.05.002
Goellner, E. M., Putnam, C. D., Graham, W. J., Rahal, C. M., Li, B.-Z., and Kolodner, R. D. (2018). Identification of Exo1-Msh2 interaction motifs in DNA mismatch repair and new Msh2-binding partners. Nat. Struct. Mol. Biol. 25, 650–659. doi: 10.1038/s41594-018-0092-y
Gomes, X. V., and Burgers, P. M. (2001). ATP utilization by yeast replication factor C. I. ATP-mediated interaction with DNA and with proliferating cell nuclear antigen. J. Biol. Chem. 276, 34768–34775. doi: 10.1074/jbc.M011631200
Gomes, X. V., Schmidt, S. L., and Burgers, P. M. (2001). ATP utilization by yeast replication factor C. II. Multiple stepwise ATP binding events are required to load proliferating cell nuclear antigen onto primed DNA. J. Biol. Chem. 276, 34776–34783. doi: 10.1074/jbc.M011743200
Grenon, M., Costelloe, T., Jimeno, S., O'Shaughnessy, A., FitzGerald, J., Zgheib, O., et al. (2007). Docking onto chromatin via theSaccharomyces cerevisiae Rad9 Tudor domain. Yeast 24, 105–119. doi: 10.1002/yea.1441
Gupta, R., Somyajit, K., Narita, T., Maskey, E., Stanlie, A., Kremer, M., et al. (2018). DNA repair network analysis reveals shieldin as a key regulator of NHEJ and PARP inhibitor sensitivity. Cell 173, 972–974.e23. doi: 10.1016/j.cell.2018.03.050
Hammet, A., Magill, C., Heierhorst, J., and Jackson, S. P. (2007). Rad9 BRCT domain interaction with phosphorylated H2AX regulates the G1 checkpoint in budding yeast. EMBO Rep. 8, 851–857. doi: 10.1038/sj.embor.7401036
Hargreaves, D. C., and Crabtree, G. R. (2011). ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res. 21, 396–420. doi: 10.1038/cr.2011.32
Hiraga, S. I., Alvino, G. M., Chang, F., Lian, H. Y., Sridhar, A., Kubota, T., et al. (2014). Rif1 controls DNA replication by directing Protein Phosphatase 1 to reverse Cdc7-mediated phosphorylation of the MCM complex. Genes Dev. 28, 372–383. doi: 10.1101/gad.231258.113
Huyen, Y., Zgheib, O., Ditullio, R. A., Gorgoulis, V. G., Zacharatos, P., Petty, T. J., et al. (2004). Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature 432, 406–411. doi: 10.1038/nature03114
Ira, G., and Haber, J. E. (2002). Characterization of RAD51-independent break-induced replication that acts preferentially with short homologous sequences. Am. Soc. Microbiol. 22, 6384–6392. doi: 10.1128/MCB.22.18.6384-6392.2002
Jahn, L. J., Mason, B., Brøgger, P., Toteva, T., Nielsen, D. K., and Thon, G. (2018). Dependency of heterochromatin domains on replication factors. G3. 8, 477–489. doi: 10.1534/g3.117.300341
Javaheri, A., Wysocki, R., Jobin-Robitaille, O., Altaf, M., Côté, J., and Kron, S. J. (2006). Yeast G1 DNA damage checkpoint regulation by H2A phosphorylation is independent of chromatin remodeling. Proc. Natl. Acad. Sci. U.S.A. 103, 13771–13776. doi: 10.1073/pnas.0511192103
Jinks-Robertson, S., Michelitch, M., and Ramcharan, S. (1993). Substrate length requirements for efficient mitotic recombination in Saccharomyces cerevisiae. Mol. Cell. Biol. 13, 3937–3950. doi: 10.1128/MCB.13.7.3937
Kalb, R., Mallery, D. L., Larkin, C., Huang, J. T. J., and Hiom, K. (2014). BRCA1 is a histone-H2A-specific ubiquitin ligase. Cell Rep. 8, 999–1005. doi: 10.1016/j.celrep.2014.07.025
Kalocsay, M., Hiller, N. J., and Jentsch, S. (2009). Chromosome-wide Rad51 spreading and SUMO-H2A.Z-dependent chromosome fixation in response to a persistent DNA double-strand break. Mol. Cell. 33, 335–343. doi: 10.1016/j.molcel.2009.01.016
Kang, R. S., Daniels, C. M., Francis, S. A., Shih, S. C., Salerno, W. J., Hicke, L., et al. (2003). Solution structure of a CUE-ubiquitin complex reveals a conserved mode of ubiquitin binding. Cell 113, 621–630. doi: 10.1016/S0092-8674(03)00362-3
Krogan, N. J., Cagney, G., Yu, H., Zhong, G., Guo, X., Ignatchenko, A., et al. (2006). Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440, 637–643. doi: 10.1038/nature04670
Krogan, N. J., Keogh, M.-C., Datta, N., Sawa, C., Ryan, O. W., Ding, H., et al. (2003). A Snf2 family ATPase complex required for recruitment of the histone H2A variant Htz1. Mol. Cell. 12, 1565–1576. doi: 10.1016/S1097-2765(03)00497-0
Kubaisy, A.l, E., Arafat, K., De Wever, O., Hassan, A. H., and Attoub, S. (2016). SMARCAD1 knockdown uncovers its role in breast cancer cell migration, invasion, and metastasis. Expert Opin. Ther. Targets 20, 1035–1043. doi: 10.1080/14728222.2016.1195059
Lademann, C. A., Renkawitz, J., Pfander, B., and Jentsch, S. (2017). The INO80 complex removes H2A.Z to promote presynaptic filament formation during homologous recombination. Cell Rep. 19, 1294–1303. doi: 10.1016/j.celrep.2017.04.051
Lee, C.-S., Wang, R. W., Chang, H.-H., Capurso, D., Segal, M. R., and Haber, J. E. (2016). Chromosome position determines the success of double-strand break repair. Proc. Natl. Acad. Sci. U.S.A. 113, E146–E154. doi: 10.1073/pnas.1523660113
Lee, J., Choi, E. S., and Lee, D. (2018). It's fun to transcribe with Fun30: a model for nucleosome dynamics during RNA polymerase II-mediated elongation. Transcription 9, 108–116. doi: 10.1080/21541264.2017.1356434
Lee, J., Choi, E. S., Seo, H. D., Kang, K., Gilmore, J. M., Florens, L., et al. (2017). Chromatin remodeller Fun30Fft3 induces nucleosome disassembly to facilitate RNA polymerase II elongation. Nat. Commun. 8:14527. doi: 10.1038/ncomms14527
Lim, M., Newman, J. A., Williams, H. L., Masino, L., Aitkenhead, H., Gravard, A. E., et al. (2019). A ubiquitin-binding domain that binds a structural fold distinct from that of ubiquitin. Structure 27, 1316–1325.e6. doi: 10.1016/j.str.2019.05.003
Liu, L., and Jiang, T. (2017). Crystal structure of the ATPase-C domain of the chromatin remodeller Fun30 from Saccharomyces cerevisiae. Acta Crystallographica. Acta Crystallogr. F Struct. Biol. Commun. 73(Pt 1), 9–15. doi: 10.1107/S2053230X16019269
Majka, J., Binz, S. K., Wold, M. S., and Burgers, P. M. J. (2006). Replication protein A directs loading of the DNA damage checkpoint clamp to 5'-DNA junctions. J. Biol. Chem. 281, 27855–27861. doi: 10.1074/jbc.M605176200
Majka, J., and Burgers, P. (2004). The PCNA-RFC families of DNA clamps and clamp loaders. Prog. Nucleic Acid Res. Mol. Biol. 78, 227–260. doi: 10.1016/S0079-6603(04)78006-X
Matsuoka, S., Ballif, B. A., Smogorzewska, A., McDonald, E. R. I., Hurov, K. E., Luo, J., et al. (2007). ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160–1166. doi: 10.1126/science.1140321
Mimitou, E. P., and Symington, L. S. (2009). DNA end resection: many nucleases make light work. DNA Repair. 8, 983–995. doi: 10.1016/j.dnarep.2009.04.017
Mirman, Z., Lottersberger, F., Takai, H., Kibe, T., Gong, Y., Takai, K., et al. (2018). 53BP1–RIF1–shieldin counteracts DSB resection through CST- and Polα-dependent fill-in. Nature 560, 112–116. doi: 10.1038/s41586-018-0324-7
Moldovan, G.-L., Pfander, B., and Jentsch, S. (2007). PCNA, the maestro of the replication fork. Cell 129, 665–679. doi: 10.1016/j.cell.2007.05.003
Nakamura, T. M., Du, L.-L., Redon, C., and Russell, P. (2004). Histone H2A phosphorylation controls Crb2 recruitment at DNA breaks, maintains checkpoint arrest, and influences DNA repair in fission yeast. Mol. Cell. Biol. 24, 6215–6230. doi: 10.1128/MCB.24.14.6215-6230.2004
Neves-Costa, A., Will, W. R., Vetter, A. T., Miller, J. R., and Varga-Weisz, P. (2009). The SNF2-family member Fun30 promotes gene silencing in heterochromatic loci. PLoS ONE 4:e8111. doi: 10.1371/journal.pone.0008111
Noordermeer, S. M., Adam, S., Setiaputra, D., Barazas, M., Pettitt, S. J., Ling, A. K., et al. (2018). The shieldin complex mediates 53BP1-dependent DNA repair. Nature 560, 117–121. doi: 10.1038/s41586-018-0340-7
Ohashi, E., Takeishi, Y., Ueda, S., and Tsurimoto, T. (2014). Interaction between Rad9–Hus1–Rad1 and TopBP1 activates ATR–ATRIP and promotes TopBP1 recruitment to sites of UV-damage. DNA Repair 21, 1–11. doi: 10.1016/j.dnarep.2014.05.001
Okazaki, N., Ikeda, S., Ohara, R., Shimada, K., Yanagawa, T., Nagase, T., et al. (2008). The novel protein complex with SMARCAD1/KIAA1122 binds to the vicinity of TSS. J. Mol. Biol. 382, 257–265. doi: 10.1016/j.jmb.2008.07.031
Ouspenski, I. I., Elledge, S. J., and Brinkley, B. R. (1999). New yeast genes important for chromosome integrity and segregation identified by dosage effects on genome stability. Nucleic Acids Res. 27, 3001–3008. doi: 10.1093/nar/27.15.3001
Pfander, B., and Diffley, J. F. X. (2011). Dpb11 coordinates Mec1 kinase activation with cell cycle-regulated Rad9 recruitment. EMBO J. 30, 4897–4907. doi: 10.1038/emboj.2011.345
Rowbotham, S. P., Barki, L., Neves-Costa, A., Santos, F., Dean, W., Hawkes, N., et al. (2011). Maintenance of silent chromatin through replication requires SWI/SNF-like chromatin remodeler SMARCAD1. Mol. Cell. 42, 285–296. doi: 10.1016/j.molcel.2011.02.036
Sachs, P., Ding, D., Bergmaier, P., Lamp, B., Schlagheck, C., Finkernagel, F., et al. (2019). SMARCAD1 ATPase activity is required to silence endogenous retroviruses in embryonic stem cells. Nat. Commun. 10:1335. doi: 10.1038/s41467-019-09078-0
Sanders, S. L., Portoso, M., Mata, J., Bähler, J., Allshire, R. C., and Kouzarides, T. (2004). Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell 119, 603–614. doi: 10.1016/j.cell.2004.11.009
Saredi, G., Huang, H., Hammond, C. M., Alabert, C., Bekker-Jensen, S., Forne, I., et al. (2016). H4K20me0 marks post-replicative chromatin and recruits the TONSL–MMS22L DNA repair complex. Nature 534, 714–718. doi: 10.1038/nature18312
Serra-Cardona, A., and Zhang, Z. (2018). Replication-coupled nucleosome assembly in the passage of epigenetic information and cell identity. Trends Biochem. Sci. 43, 136–148. doi: 10.1016/j.tibs.2017.12.003
Shih, S. C., Prag, G., Francis, S. A., Sutanto, M. A., Hurley, J. H., and Hicke, L. (2003). A ubiquitin-binding motif required for intramolecular monoubiquitylation, the CUE domain. EMBO J. 22, 1273–1281. doi: 10.1093/emboj/cdg140
Smolle, M., Venkatesh, S., Gogol, M. M., Li, H., Zhang, Y., Florens, L., et al. (2012). Chromatin remodelers Isw1 and Chd1 maintain chromatin structure during transcription by preventing histone exchange. Nat. Struct. Mol. Biol. 19, 884–892. doi: 10.1038/nsmb.2312
Soria, G., and Almouzni, G. (2013). Differential contribution of HP1 proteins to DNA end resection and homology-directed repair. Cell Cycle 12, 422–429. doi: 10.4161/cc.23215
Steglich, B., Strålfors, A., Khorosjutina, O., Persson, J., Smialowska, A., Javerzat, J.-P., et al. (2015). The Fun30 chromatin remodeler Fft3 controls nuclear organization and chromatin structure of insulators and subtelomeres in fission yeast. PLoS Genet. 11:e1005101. doi: 10.1371/journal.pgen.1005101
Storey, A. J., Wang, H.-P., Protacio, R. U., Davidson, M. K., Tackett, A. J., and Wahls, W. P. (2018). Chromatin-mediated regulators of meiotic recombination revealed by proteomics of a recombination hotspot. Epigenet. Chromatin 11:64. doi: 10.1186/s13072-018-0233-x
Strålfors, A., Walfridsson, J., Bhuiyan, H., and Ekwall, K. (2011). The FUN30 chromatin remodeler, Fft3, protects centromeric and subtelomeric domains from euchromatin formation. PLoS Genet. 7:e1001334. doi: 10.1371/journal.pgen.1001334
Symington, L. S., and Gautier, J. (2011). Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 45, 247–271. doi: 10.1146/annurev-genet-110410-132435
Takeishi, Y., Ohashi, E., Ogawa, K., Masai, H., Obuse, C., and Tsurimoto, T. (2010). Casein kinase 2-dependent phosphorylation of human Rad9 mediates the interaction between human Rad9-Hus1-Rad1 complex and TopBP1. Genes Cells 15, 761–771. doi: 10.1111/j.1365-2443.2010.01418.x
Taneja, N., Zofall, M., Balachandran, V., Thillainadesan, G., Sugiyama, T., Wheeler, D., et al. (2017). SNF2 family protein Fft3 suppresses nucleosome turnover to promote epigenetic inheritance and proper replication. Mol. Cell. 66, 50–62.e6. doi: 10.1016/j.molcel.2017.02.006
Terui, R., Nagao, K., Kawasoe, Y., Taki, K., Higashi, T. L., Tanaka, S., et al. (2018). Nucleosomes around a mismatched base pair are excluded via an Msh2-dependent reaction with the aid of SNF2 family ATPase Smarcad1. Genes Dev. 32, 806–821. doi: 10.1101/gad.310995.117
Toh, G. W. L., O'Shaughnessy, A. M., Jimeno, S., Dobbie, I. M., Grenon, M., Maffini, S., et al. (2006). Histone H2A phosphorylation and H3 methylation are required for a novel Rad9 DSB repair function following checkpoint activation. DNA Repair 5, 693–703. doi: 10.1016/j.dnarep.2006.03.005
Ubersax, J. A., Woodbury, E. L., Quang, P. N., Paraz, M., Blethrow, J. D., Shah, K., et al. (2003). Targets of the cyclin-dependent kinase Cdk1. Nature 425, 859–864. doi: 10.1038/nature02062
van Attikum, H., Fritsch, O., and Gasser, S. M. (2007). Distinct roles for SWR1 and INO80 chromatin remodeling complexes at chromosomal double-strand breaks. EMBO J. 26, 4113–4125. doi: 10.1038/sj.emboj.7601835
Wilson, M. D., Benlekbir, S., Fradet-Turcotte, A., Sherker, A., Julien, J.-P., McEwan, A., et al. (2016). The structural basis of modified nucleosome recognition by 53BP1. Nature 536, 100–103. doi: 10.1038/nature18951
Wysocki, R., Javaheri, A., Allard, S., Sha, F., Cote, J., and Kron, S. J. (2005). Role of Dot1-dependent histone H3 methylation in G1 and S phase DNA damage checkpoint functions of Rad9. Mol. Cell. Biol. 25, 8430–8443. doi: 10.1128/MCB.25.19.8430-8443.2005
Xiao, S., Lu, J., Sridhar, B., Cao, X., Yu, P., Zhao, T., et al. (2017). SMARCAD1 contributes to the regulation of naive pluripotency by interacting with histone citrullination. Cell Rep. 18, 3117–3128. doi: 10.1016/j.celrep.2017.02.070
Xu, G., Chapman, J. R., Brandsma, I., Yuan, J., Mistrik, M., Bouwman, P., et al. (2015). REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 521, 541–544. doi: 10.1038/nature14328
Xu, Y., Ayrapetov, M. K., Xu, C., Gursoy-Yuzugullu, O., Hu, Y., and Price, B. D. (2012). Histone H2A.Z controls a critical chromatin remodeling step required for DNA double-strand break repair. Mol. Cell. 48, 723–733. doi: 10.1016/j.molcel.2012.09.026
Yadav, T., and Whitehouse, I. (2016). Replication-coupled nucleosome assembly and positioning by ATP-dependent chromatin- remodeling enzymes. Cell Rep. 15, 715–723. doi: 10.1016/j.celrep.2016.03.059
Yang, J., Zhang, X., Feng, J., Leng, H., Li, S., Xiao, J., et al. (2016). The histone chaperone FACT contributes to DNA replication-coupled nucleosome assembly. Cell Rep. 14, 1128–1141. doi: 10.1016/j.celrep.2015.12.096
Yu, Q., Zhang, X., and Bi, X. (2011). Roles of chromatin remodeling factors in the formation and maintenance of heterochromatin structure. J. Biol. Chem. 286, 14659–14669. doi: 10.1074/jbc.M110.183269
Zhou, C. Y., Johnson, S. L., Gamarra, N. I., and Narlikar, G. J. (2016). Mechanisms of ATP-dependent chromatin remodeling motors. Annu. Rev. Biophys. 45, 153–181. doi: 10.1146/annurev-biophys-051013-022819
Keywords: Fun30/SMARCAD1, nucleosome remodeling, DNA double-stranded break, DNA end resection, cell cycle, post-translational modification, genome stability
Citation: Bantele SCS and Pfander B (2019) Nucleosome Remodeling by Fun30SMARCAD1 in the DNA Damage Response. Front. Mol. Biosci. 6:78. doi: 10.3389/fmolb.2019.00078
Received: 23 July 2019; Accepted: 19 August 2019;
Published: 04 September 2019.
Edited by:
Sergio Fernandes De Almeida, University of Lisbon, PortugalReviewed by:
Jo R. Morris, University of Birmingham, United KingdomXin Bi, University of Rochester, United States
Copyright © 2019 Bantele and Pfander. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Boris Pfander, YnBmYW5kZXJAYmlvY2hlbS5tcGcuZGU=
†Present address: Susanne C. S. Bantele, Faculty of Health and Medical Sciences, Novo Nordisk Foundation Center for Protein Research, University of Copenhagen, Copenhagen, Denmark