 Mark R. Dalman1*
Mark R. Dalman1* W. Brian Simison2
W. Brian Simison2 Danny Nielsen3Sabana Bhatta4Noor Ramahi4Clair Yee4Dipendra Thapaliya4Jhalka Kadariya4Shanice Cheatham4Hailee Olson4
Danny Nielsen3Sabana Bhatta4Noor Ramahi4Clair Yee4Dipendra Thapaliya4Jhalka Kadariya4Shanice Cheatham4Hailee Olson4 Tara C. Smith4
Tara C. Smith4- 1College of Podiatric Medicine, Kent State University, Kent, OH, United States
- 2Center for Comparative Genomics, California Academy of Sciences, San Francisco, CA, United States
- 3Department of Natural Resources and Environmental Science, University of Nevada, Reno, Reno, NV, United States
- 4College of Public Health, Kent State University, Kent, OH, United States
Background: Staphylococcus aureus is a gram-positive bacterium commonly found in the nares and oropharynx of one in three individuals and has the potential to cause significant health problems. With antibiotic-resistant strains causing 11,000 deaths yearly and ~2% of the population nasally colonized with methicillin-resistant S. aureus, a search for predictive markers and associative relationships between carriage have been long-sought goals. Within our study, we leveraged monozygotic twin participants in concert with multi-site microbiome analyses to characterize the impacts of S. aureus on composition.
Results: We recruited 147 monozygotic twin pairs and characterized three sites, i.e., the nares, oropharynx, and hand microbiomes, using 16S rRNA v3-v4 sequencing in addition to S. aureus carriage status. The prevalence of S. aureus was highest in the oropharynx followed by nares and hand with concordance between twin pairs highest in the nares, followed by oropharynx. The detection of S. aureus was statistically correlated with differences in microbiome composition across sites, as indicated by beta diversity and DESeq2 analyses. Microbiome composition was most similar in twins’ nares that were S. aureus culture-positive concordant, whereas twins that were culture-negative concordant had the most similarity in the oropharynx. Of significance, Moraxella nonliquefacians and Capnocytophaga were inversely associated with S. aureus in the nares and oropharynx, respectively.
Conclusions: This improved understanding of S. aureus colonization in nares, oropharynx, and hand microbiomes in monozygotic twin pairs is a further step towards unraveling the degree to which the microbiome is influenced by host genetics and S. aureus carriage.
Introduction
Symbiotic relationships between Eukaryotes and Prokaryotes have a long history (Baldo and Werren, 2021). Recent research has deepened our understanding of microbial colonization dynamics in relation to human health and disease (Campbell and Koch, 2017). The human microbiome, a community of over 1014 microbial cells (Sender et al., 2016), is passed from generation to generation (Rosenberg and Zilber-Rosenberg, 2016). It plays a critical role in priming the immune system (Round et al., 2010) and even shaping metabolism (Dabke et al., 2019). To better understand perturbations from normal healthy microbiomes (Kong and Segre, 2017; Oh et al., 2016), in 2008, the Human Microbiome Project (HMP1) sought to establish microbial compositional characteristics across core body sites (i.e., nasal, oropharyngeal, and skin). In doing so, studies have noted that certain microbiome perturbations, such as inverse relationships involving Staphylococcus epidermidis interfering with colonization of microbes such as Leishmania major (Naik et al., 2012) and methicillin-resistant Staphylococcus aureus (MRSA) (Johnson et al., 2015; Park et al., 2011) can impact composition.
S. aureus is a major nosocomial pathogen, contaminating surfaces (Dalman et al., 2019) and is even asymptomatically carried by over a third of the population (Vanegas et al., 2021). In the U.S., it causes over 11,000 deaths and 80,000 invasive infections annually (Dantes et al., 2013), commonly colonizing the nares and oropharynx of individuals and posing a risk for active infections. S. aureus is known to inhibit colonization by other bacterial species through antimicrobial peptides and toxins, potentially causing dysbiosis (Garrett and Palmer, 2024; Park et al., 2011). Our group has identified reservoirs of methicillin-sensitive and resistant strains (MSSA and MRSA) in human and environmental samples (Kates et al., 2019; Thapaliya et al., 2017); however, the associated impact on composition remains underexplored. Twin studies offer a controlled framework to explore these associations, providing insights into how host genetics influence microbial communities and S. aureus carriage.
Twin studies provide a valuable framework for investigating the relative contributions of genetic and environmental factors to complex traits, such as disease susceptibility and physiological characteristics (Galton, 1876; Ganna et al., 2013). By eliminating genetic variability, monozygotic (MZ) twins are particularly well-suited for studying microbiome composition and exploring associations with S. aureus carriage. Studies focused exclusively on monozygotic twins have advanced our understanding of the oral microbiota, revealing minimal differences in S. aureus presence in saliva (Bockmann et al., 2011; Wu et al., 2018). In contrast, traditional twin studies incorporating dizygotic (DZ) twins have shown mixed results, with some studies suggesting a modest genetic influence on S. aureus nasal carriage (Andersen et al., 2012), while other studies in closed genetic pools in Amish communities suggest that environmental factors play a larger role (Roghmann et al., 2011). Additionally, genetic studies have further identified variants associated with S. aureus susceptibility, such as single nucleotide variants (SNVs) in the KAT2B (linked to intermittent carriage), PDE4B, VRK1, and BCL11B genes (Brown et al., 2015; Ye et al., 2014), suggesting that genetic factors may be inherited and influence both S. aureus carriage and microbial composition.
The aim of this study was to characterize the microbial community of the nares, oropharynx, and hands in monozygotic twins, and to quantify any potential effect of S. aureus colonization on microbiome composition. We predicted that microbiome composition and complexity would differ in S. aureus culture-positive participants, potentially due to competitive interactions. Additionally, we predicted that twin pairs would show similar microbiome compositions in the nares and oropharynx, irrespective of S. aureus carriage status. This observational study, rather than testing causative mechanisms such as competitive exclusion, provides a controlled framework to assess associations between S. aureus presence and microbiome characteristics across multiple different body sites in only monozygotic twins. By focusing on genetically identical pairs, we minimize confounding genetic variability to better explore microbial composition and its host associations.
Materials and methods
Ethical statement, sample collection, and processing
All study protocols were approved by the Kent State University Institutional Review Board (#16-293). We recruited 147 monozygotic twin pairs (n=294) at the Twins Day Festival in Twinsburg, OH, in August 2016. Eligibility criteria included: 8 to 80 years of age, English-speaking, no antibiotic or inhaled corticosteroid use within 90 days, no nasal influenza vaccine in the past month, no active hand or upper respiratory infections, and no hospitalization exceeding 24 hours within 90 days. Participants provided written consent or assent (for minors), along with a parent’s consent.
Participants completed questionnaires on medical history, housing, and hygiene practices (Supplementary Data Sheet 1). They were asked to refrain from eating, drinking, chewing gum, brushing teeth (30 minutes), or washing hands (1 hour) prior to sample collection. Anterior nares and oropharynx swabs were collected in duplicate, along with dominant hand samples using the glove juice technique (Leyden et al., 1989; Kampf et al., 2013). All the samples were processed at the Kent State University (KSU) Smith Emerging Infections Laboratory on the day of their collection. Anterior nares and oropharynx swabs were collected with sterile nylon flocked swabs (Copan Diagnostics, Murrieta, CA), and hand microbes were collected using sterile gloves and sterile peptone. All samples were subjected to a bifurcated protocol in the lab. Of the duplicate swabs taken from the anterior nares and oropharynx, one swab was subjected to bacterial DNA isolation for 16S rRNA V3-V4 sequencing, and the other duplicate swab was subjected to a culture-based technique for S. aureus isolation. S. aureus was phenotypically assessed and confirmed using a combination of agglutination and biochemical assays as recently described by Dalman et al. (2019).
Microbiome extraction
Microbial DNA from anterior nares, oropharynx, and hand samples was isolated using the MO BIO PowerSoil DNA isolation kit (Mo BIO Laboratories Inc, Carlsbad, CA). For swabs, the swab head was vortexed in DNA extraction buffer for 10 minutes, followed by centrifugation and resuspension before proceeding with the kit protocol. DNA was quantified using the Qubit BR dsDNA kit (Invitrogen, Waltham, MA, USA) and sent to Case Western Reserve Genomics Core for 16S rRNA V3-V4 amplification, library preparation, and sequencing using 2x250 paired-end reads (Illumina MiSeq protocol). Data was uploaded to the Illumina Basespace cloud for bioinformatics analysis following the standard Illumina MiSeq protocol. Data was uploaded to the Illumina Basespace cloud server and retrieved for bioinformatics.
Microbiome analyses and comparisons
Raw sequence reads were processed and analyzed with QIIME2 v2021.8 (Bolyen et al., 2019) using modified scripts from Amplicon SOP v2 of Microbiome Helper (Comeau et al., 2017). Raw read quality was evaluated with FASTQC v0.11.8 (Andrews, 2010) and MultiQC v1.12 (Ewels et al., 2016). Raw FASTQ reads were imported as QIIME2 artifacts (QZA format) using the QIIME2 import tool. Primers were trimmed with Cutadapt v3.4 (Martin, 2011). The reads were denoised after joining with VSEARCH v2.13.3 (Rognes et al., 2016). All sequences were trimmed to the same length prior to using Deblur v1.1.0 (Amir et al., 2017) to correct reads and identify amplicon sequence variants (ASVs). We used the Naive-Bayes approach in the scikit-learn v1.1 Python library and the SILVA database (silva-138-99-nb-classifier.qza) to assign the ASVs to taxonomy. We removed rare ASVs that had frequencies of less than 0.1% of the mean sample depth using the QIIME2 filter-features tool. This was followed by filtering out contaminants, including chloroplast, mitochondria, unclassified ASVs, and low-coverage samples using the QIIME2 filter-table tool. To confirm that the sequence depths were sufficient, we generated rarefaction curves to identify appropriate depth cutoffs with the QIIME2 alpha-rarefaction tool. Bar charts of the taxonomic abundance across samples were generated using the QIIME2 barplot tool. Differential abundances between groups were identified using ANCOM 2.0 (Mandal et al., 2015). Finally, we generated and exported a BIOM file with taxonomy as metadata using a Microbiome helper script using sed and the QIIME2 export tool. All statistical analyses were conducted in R version 4.2.0 (R Core Team, 2022).
The QIIME2 artifacts including the feature count table, phylogenetic tree, taxonomy table, and sample metadata were converted to a phyloseq (McMurdie and Holmes, 2013) object in R using the qza_to_phyloseq function of the qiime2R package (Bisanz, 2018). Using the rarefy_even_depth function of phyloseq we rarefied microbial reads to the smallest sequencing depth of all samples. Using the plot_richness function of phyloseq, we compared the Shannon, Chao1, and Simpson diversity estimates, and used Wilcox tests to determine significant differences. To detect differences between tissue locations, we compared alpha diversity across the hand, nares, and oropharynx samples and between S. aureus-positive and -negative samples. To determine if S. aureus carriage influenced microbial diversity within sample locations, we also compared diversity between S. aureus positive and negative samples and correspondingly across tissue locations.
We visualized the relative read abundances (RRA) of microbial lineages at the level of phylum, class, order, family, genus, and species across location and S. aureus carriage status. We first agglomerated ASV reads to the desired taxonomic level using the tax_glom function of phyloseq before merging sample types using the merge_samples function. We then transformed the read count results to RRA using the transform_sample_counts function of phyloseq. We then plotted RRA using ggplot (Wickham, 2016).
Monozygotic twin’s compositional differences were tested using a G-test with Yates’ continuity correction with applied Fishers exact test when contingency table <20 (Agresti, 1992; Rivals et al., 2007; Yates, 1934). Benjamini–Hochberg FDR multiple testing correction was used to control for false discovery rate along with the Newcombe–Wilson method to test confidence intervals (Newcombe, 1998). A scatter plot and corresponding R2 was calculated in STAMP (Parks et al., 2014; Parks and Beiko, 2010) for all monozygotic twin pairs based on their culture-dependent status [concordant (CC), discordant (DC), and not colonized] with corresponding R2 values cumulatively plotted for each twin pair based on site location. An R2 of 1.00 indicates the identical composition of features at the hierarchical level of comparison (all analyses were done at the genus level).
We used the ordinate function to perform Principal Coordinates Analysis (PCoA) using the Bray–Curtis method, and both weighted and unweighted UniFrac distances. We then plotted the PCoAs using the plot_ordination function of phyloseq. We visualized PCoAs with combinations of the first three PCoA axes. To quantify the amount of variation in microbiome composition explained by location and S. aureus carriage, we performed permutation analysis of variance (PERMANOVA) using 999 permutations with the adonis2 function of the vegan package (Oksanen et al., 2020). We performed PERMANOVA using the Bray–Curtis method, and both weighted and unweighted UniFrac distance matrices.
Differentially abundant microbiota
To detect differentially abundant microbial lineages, we used the DESeq2 package (Love et al., 2014). We performed the analysis in multiple pairwise contrasts: 1) between the nares and oropharynx in S. aureus-positive samples; 2) between the nares and oropharynx in S. aureus-negative samples; 3) between S. aureus-negative and -positive samples in only the nares; and 4) between S. aureus-negative and -positive samples in only the oropharynx. Within the DESeq function, we used the Wald test, parametric fitType, and poscounts sfType parameters. Microbial ASVs were considered significantly differentially abundant with adjusted P values < 0.05 (corrected using the Benjamini–Hochberg method). Differentially abundant microbial taxa were plotted for each of the above-described analyses using ggplot2 (Wickham, 2016).
Data access
Sequences for the project can be found under BioProject ID: PRJNA1026518. Code for analysis can be found at: https://github.com/calacademy-research/Dalman_Twin_Microbiome_Scripts.
Results
Participant demographics and carriage rates
Full demographic data can be found in Table 1. A total of 294 twins (147 twin pairs) were convenience sampled at an International Twins Festival in Twinsburg, Ohio. The average cohort age was 32.5 years (95% CI: 30.4-34.6), with S. aureus-positive participants averaging 30.7 years (95% CI: 27.8-33.6) and culture-negative participants 34.1 years (95% CI: 31.1-37.0). A total of 220 monozygotic twins were female (74.8%) and overall, 87.8% of participants identified as white, 158 twins (54%) lived with their twin sibling, and 53% of these cohabiting twins (84/158) shared a room.
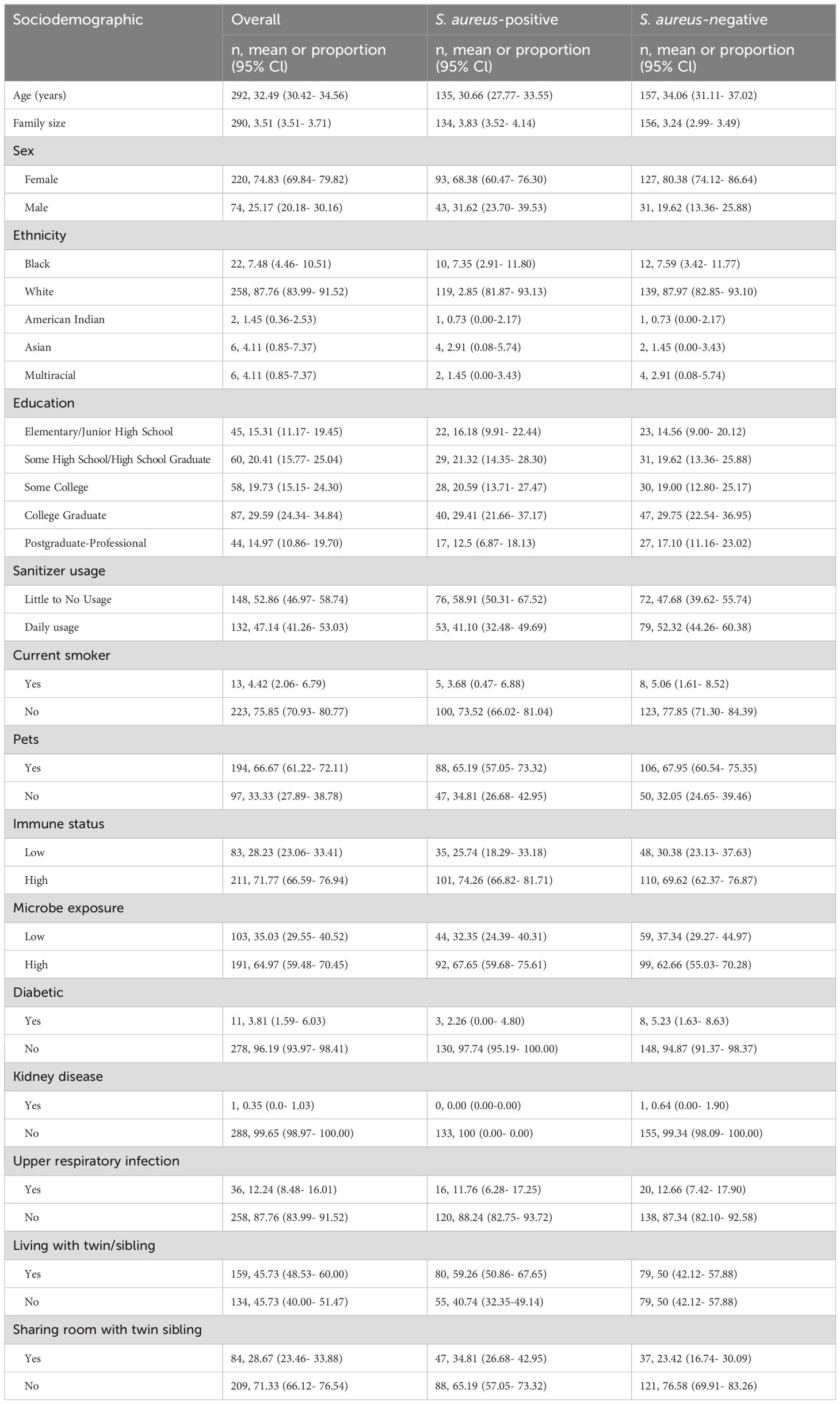
Table 1. Sociodemographic characteristics of monozygotic twin participants by Staphylococcus aureus culture-dependent status.
From 294 twin participant samples, 178 sites (178/882, 20.2%) were positive for S. aureus via culture-dependent techniques. Overall, carriage for the nares, oropharynx, and hand were 26.2%, 29.9%, and 4.4%, respectively. For 43 participants, S. aureus was restricted to the nares, 52 were restricted to the oropharynx, and 4 were restricted to the hand. Twenty-eight participants were co-colonized in the nares and oropharynx, three co-colonized in the oropharynx and on the hand, and one participant was co-colonized in the nares and on the hand. Five participants were colonized at all three sites (nares, oropharynx, and hand).
Concordance
We calculated concordance as the number of twin pairs that were concordant positive for S. aureus divided by the cumulative number of twin pairs that were either concordant or discordant positive. Among monozygotic twin pairs, a total of 25 (25/54; 46.3%) twin pairs were concordant culture-positive for S. aureus and 29 (29/54; 53.7%) were discordant within the anterior nares (Table 2). Within oropharynx culture-positive samples, a total of 19 (19/69; 27.5%) twin pairs were concordant culture-positive for S. aureus whereas 50 (50/69; 72.5%) were discordant. Only 1 (1/12; 8.3%) set of twins were concordant for S. aureus carriage in the hand samples with 11 (11/12; 91.7%) twin pairs being discordant. A Chi-square analysis revealed significant variation in concordance patterns across the three sites. In the nares, concordance showed a strong and statistically significant association (χ2 = 34.33, p<0.001). The oropharynx showed a moderate but statistically significant association (χ2 = 4.751, p= 0.029), while no significant concordance was observed in the hand samples (χ2 = 0.176, p=0.675).
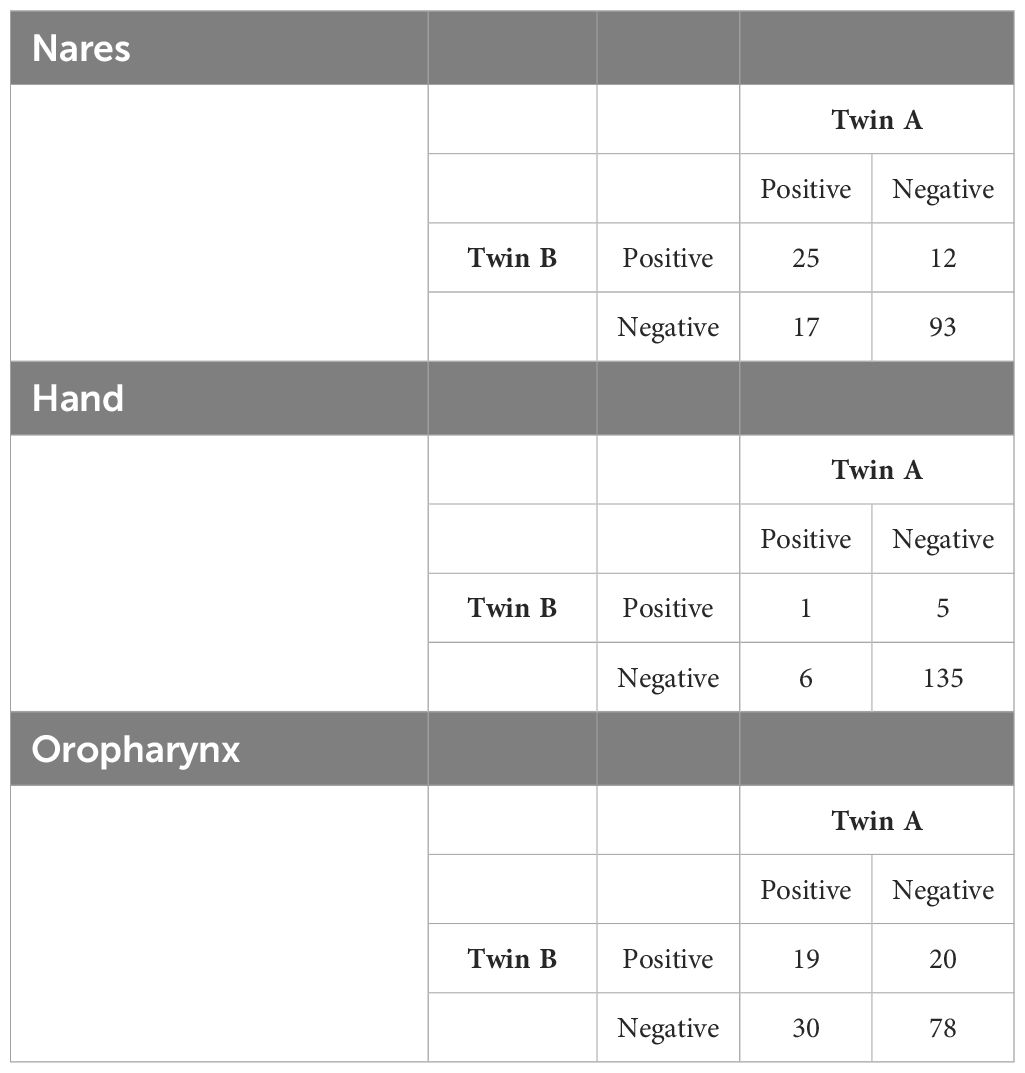
Table 2. Concordance of Staphylococcus aureus carriage in monozygotic twin pairs in the nares, oropharynx, and on hands. Each number represents a twin pair comparison.
Alpha and beta diversity
We obtained 4,680,299 high-quality filtered reads across 1,351 ASVs. Sample sequencing depth ranged from 2,527 to 46,685 reads with an average of 10,589 reads. We rarefied all samples to an even sampling depth of 2,527 reads. Alpha diversity was calculated to measure the richness of community microbial species within each sample. The Chao1 diversity (Figure 1) was greatest in the hand samples, regardless of S. aureus culture status (p <0.001) when compared to other sites (nares and oropharynx) and their corresponding culture status (positive/negative). There was no significant difference in the Chao1 diversity between hand culture-positive and culture-negative samples (p>0.05). The oropharynx exhibited the second-highest alpha diversity of the three sites sampled. The nares had the lowest alpha diversity of all sites sampled, with no significant difference in alpha diversity between culture-positive nares and culture-negative nares (p> 0.05). The Chao1 alpha diversity was the only index tested of three (Shannon and Simpson index figures can be found in Supplementary Data Sheet 2) to find a significant difference between culture-positive and culture-negative oropharynx samples (p=0.05). Diversity did not differ between culture statuses within the nares nor hand sites (p >0.05). There were, however, significant differences in alpha diversity when comparing collective nares to oropharynx, oropharynx to hand, and nares to hand samples (Figure 1; p<0.001).
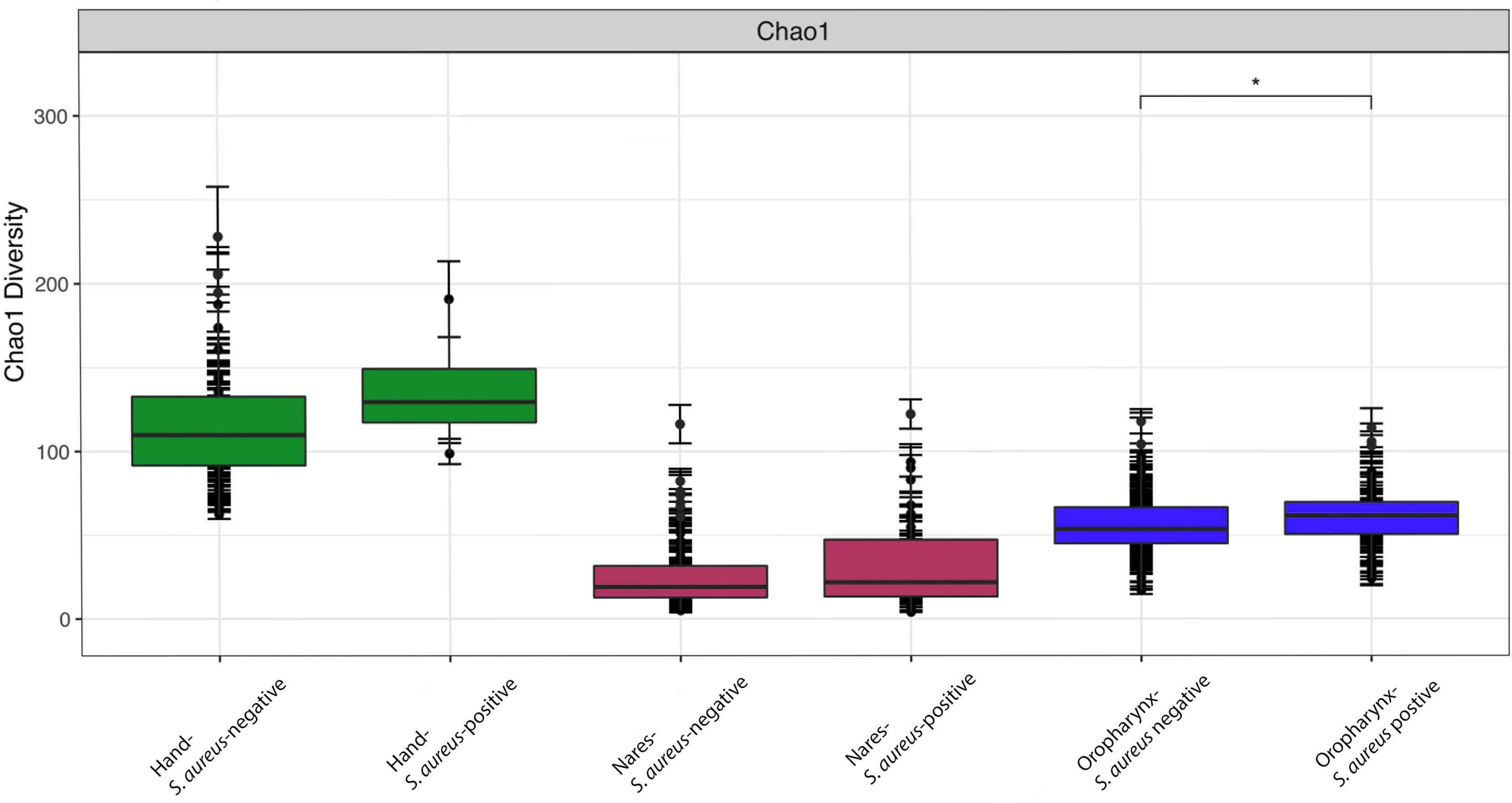
Figure 1. Chao1 alpha diversity indices indicating community species richness for the nares, oropharynx, and hand samples, separated by S. aureus culture status. Data are presented as mean ± SD with median and interquartile ranges shown. All site comparisons were significant at p ≤ 0.05. Within the oropharynx samples, only culture-positive versus culture-negative sites was significantly different (*p = 0.05).
Beta diversity was calculated to measure the diversity across all samples using the Bray–Curtis method (both weighted and unweighted UniFrac were also calculated and can be found in Supplementary Data Sheet 3). The ordination plot of the Bray–Curtis distances for all samples is shown in Figure 2. The samples primarily clustered by sample location (nares, oropharynx, and hand; a PERMANOVA was conducted to test for differences in mean and variance for the groups’ centroids, p= 0.001) but also by S. aureus culture status (p= 0.001). Additionally, the interaction between variables, location, and culture was also highly significant (p= 0.001). To test what was driving the differences in beta diversity, a PERMDISP function was run to test differences in dispersion (variance) between any of the groups. As shown in Figure 3, the beta dispersion of the locations was highly significant (p=0.001) whilst the dispersion of the S. aureus culture status of samples was not significant (p=0.151) and was not driving the observed beta diversity.
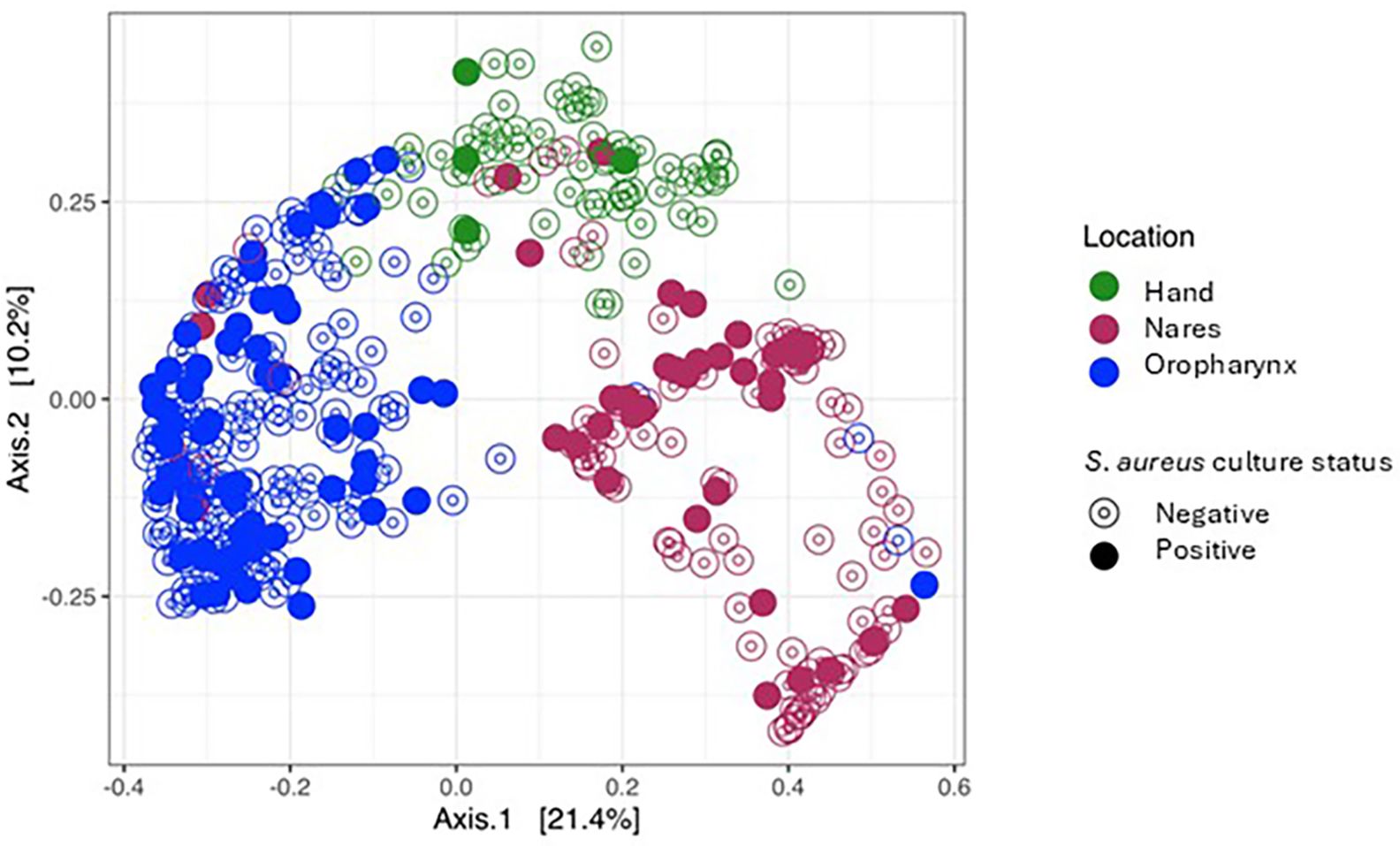
Figure 2. Principal Coordinates Analysis (PCoA) of monozygotic twin participants by location and Staphylococcus aureus culture-dependent status (Bray–Curtis).
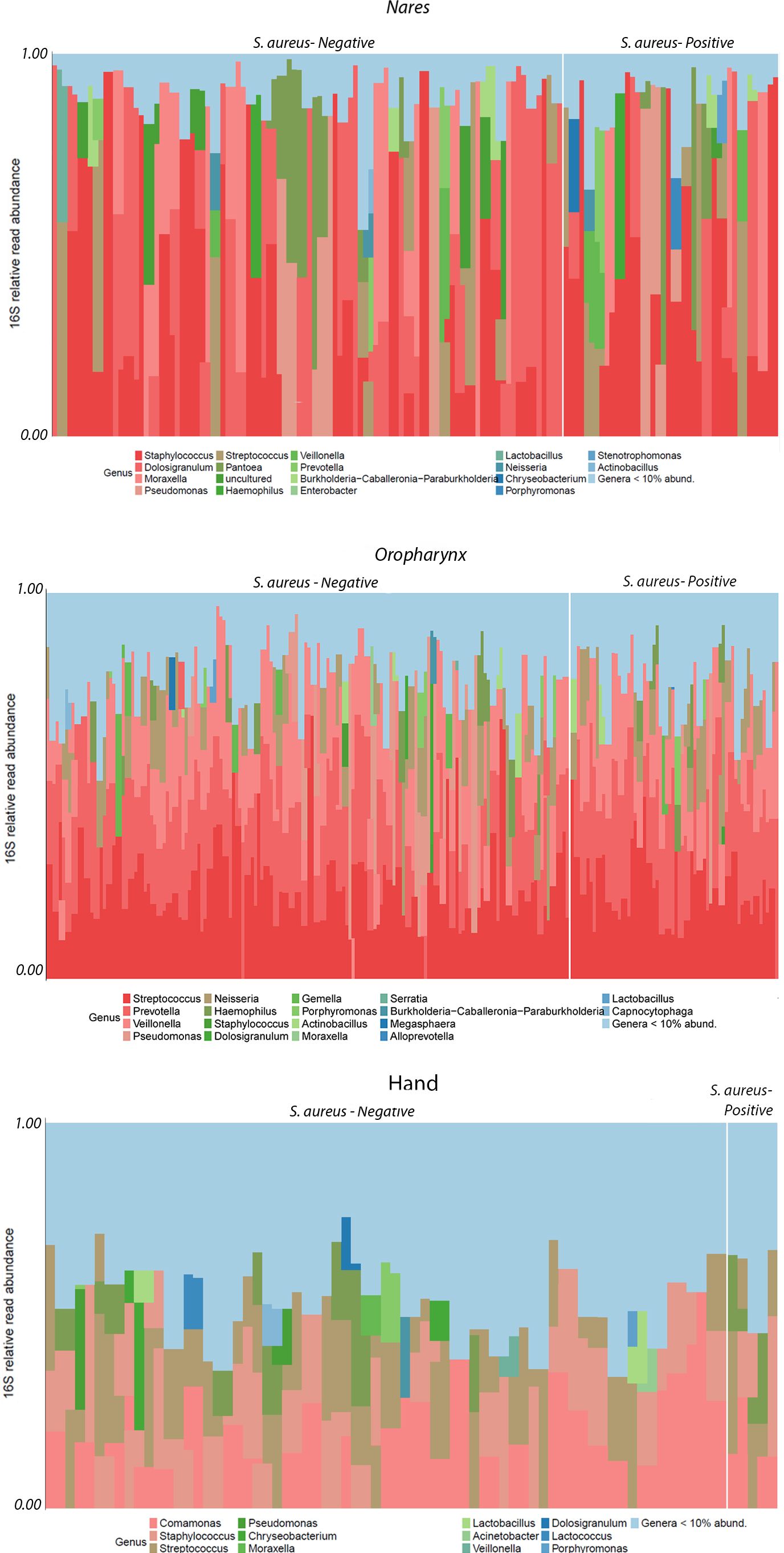
Figure 3. 16S Relative Read Abundance (RRA) of monozygotic twin samples as a function of location (nares, oropharynx, and hand) and culture-dependent status (S. aureus positive/negative) plotted at the genus level.
Relative read abundance (%)
The RRA of taxa at the phylum and class levels showed no observable differences between culture statuses across location sites (data not shown). However, descriptive differences in RRA were noted between locations (hand, nares, and oropharynx) at the order and genus levels (Figure 3). The most pronounced variations in RRA between culture-positive and culture-negative samples within locations were observed at the order and genus levels, with more taxa identified in culture-negative samples from the nares, oropharynx, and hand. A total of six bacterial orders were found in the nares, with Burkholderiales being enriched in the S. aureus-negative samples. In both hand and oropharynx culture-negative samples, there was enrichment of Pseudomonales compared to their respective culture-positive samples at the order level. Additionally, the oropharynx was the only site (compared to nares and hand) to have increased RRA of Veillonellales (Selenomonadales) and Bacteroidales in their samples. At the genus level, the nares had the highest RRA of all sites with almost 50% (0.48 16S RRA) of Staphylococcus in culture-positive samples. The second most abundant in the nares was the genus Dolosigranulum. The most dominant genus on the hands was a collective number of genera with <5% abundance making up more than 30% of the composition in both culture-positive and -negative hand samples. The next most abundant in the hand samples were Prevotella, Comamonas, and Staphylococcus in that order. In the oropharynx, Streptococcus was the most abundant genera followed closely by Prevotella and Veillonella.
Querying shared microbiota composition between monozygotic twins
In the nares, concordant culture-positive twins exhibited greater similarity in bacterial genera composition (R² = 0.73 ± 0.10) than concordant culture-negative twins (R² = 0.53 ± 0.65), with discordant twins showing the lowest similarity (R² = 0.42 ± 0.09) (Figure 4). In the oropharynx microbiome, concordant culture-positive twins (R² = 0.73 ± 0.09) and discordant culture-positive twins (R² = 0.72 ± 0.04) displayed comparable levels of bacterial genera similarity. Notably, concordant culture-negative twins had the highest similarity in the oropharynx microbiome (R² = 0.85 ± 0.02). Although data for the hand microbiome was limited, culture-negative twin pairs showed a distinct similarity in bacterial composition (R² = 0.69 ± 0.06).
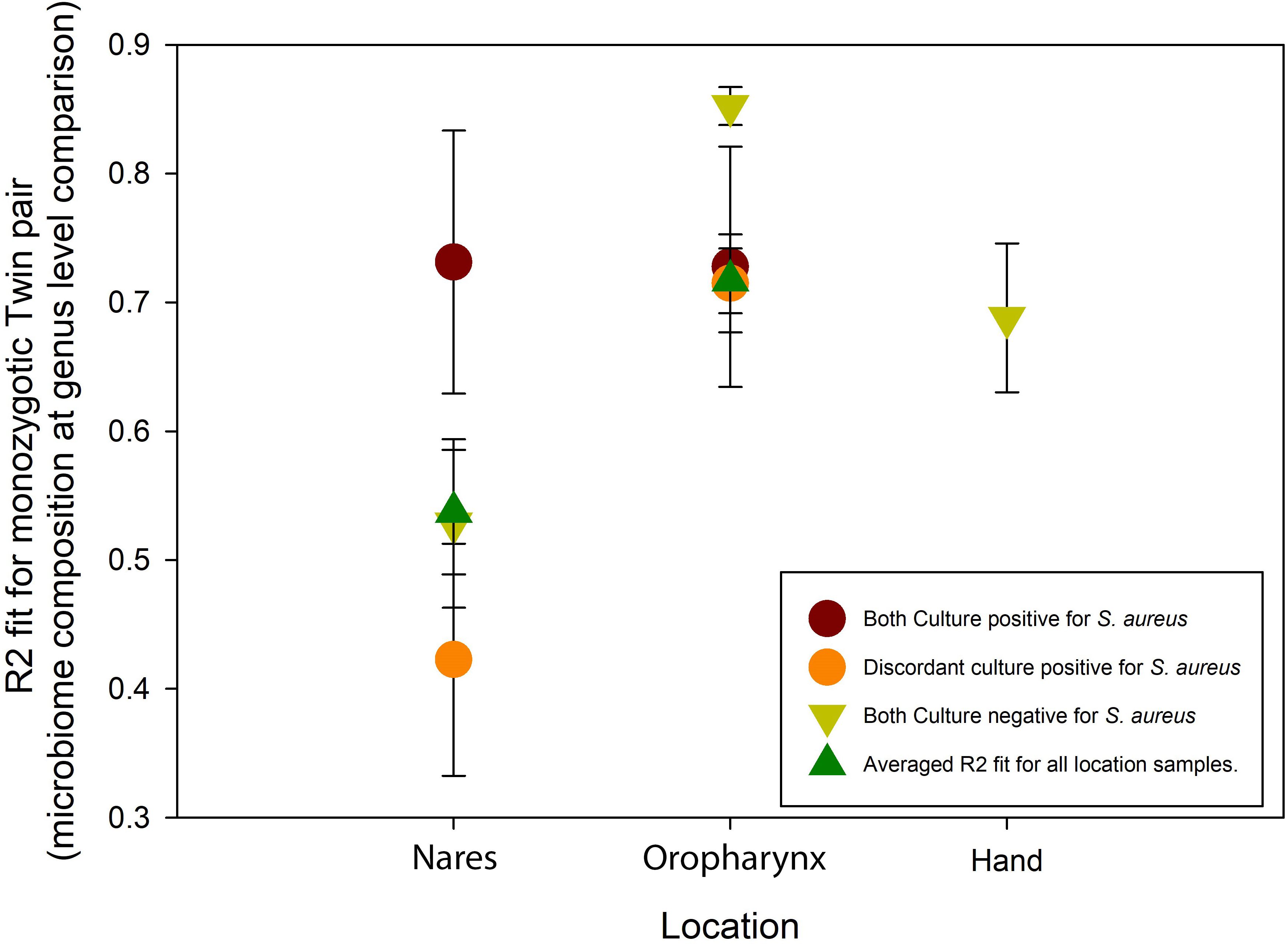
Figure 4. R2 fit of microbiome composition results between monozygotic twin pairs (analyzed at the genus level). Data are plotted as mean± SEM.
Differentially abundant taxa
The DESeq2 method (log(Count +1) transformed data) was used to identify differentially abundant taxa across locations (nares, oropharynx, hand) and S. aureus culture status (positive vs. negative) (Figure 5). In nares samples, Moraxella nonliquefaciens (Proteobacteria) was enriched in culture-negative samples (padj = 0.0065), while Capnocytophaga (Bacteroidota) was enriched in culture-negative oropharynx samples (padj = 0.0032). No significant differences were observed in hand samples between culture statuses.
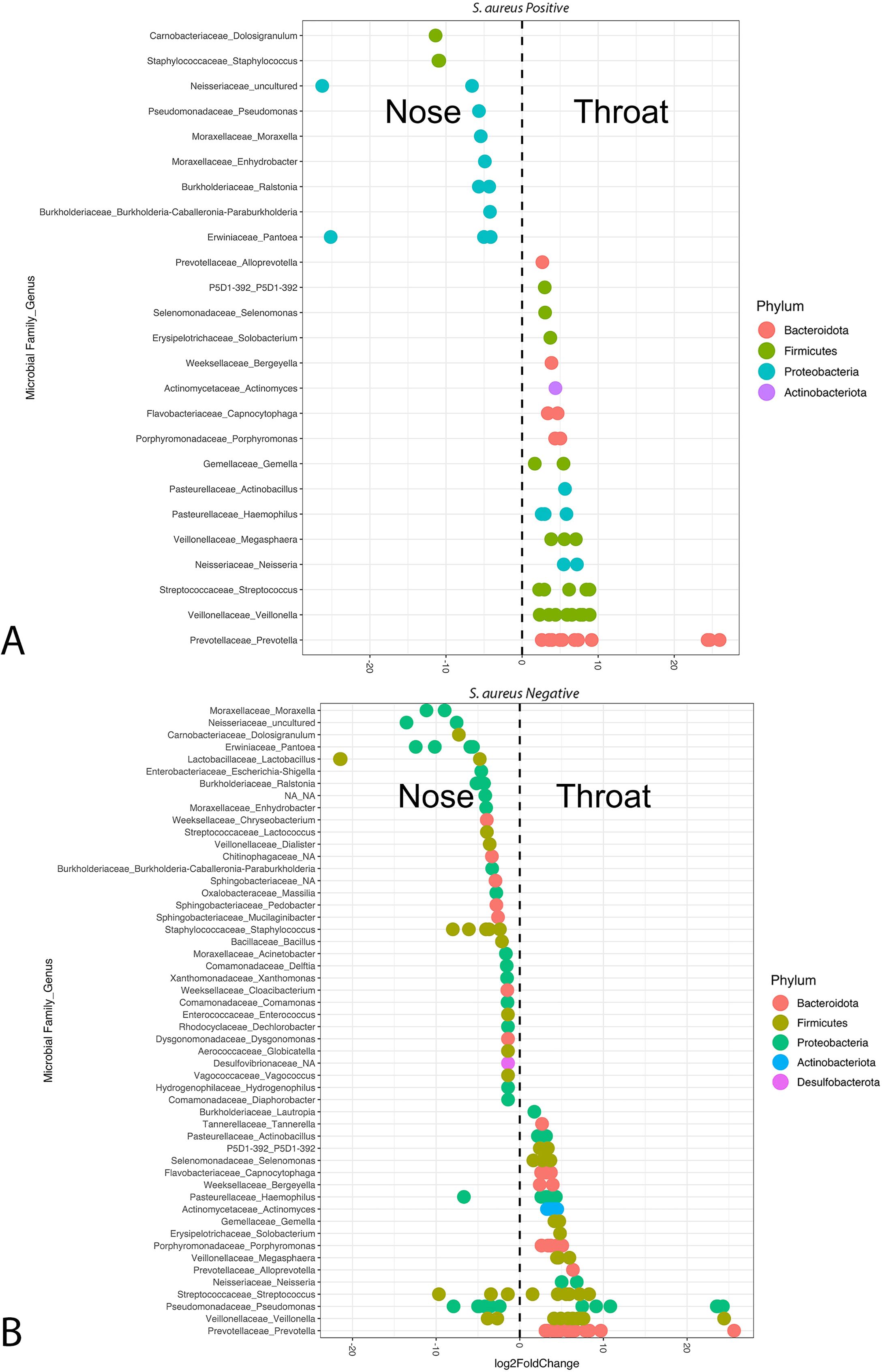
Figure 5. Differential abundance of bacteria taxa using DeSeq2 from (A) S. aureus culture-positive and (B) S. aureus culture-negative nares and oropharynx samples. These plots show the microbial taxonomy at the family and genus level (y-axis). Multiple points along a single y-axis row indicate that multiple microbial ASVs were differentially abundant within that family and genus level, representing different species or variants within the genus.
A total of 61 genera were significantly enriched between culture-positive samples from the nares and oropharynx (Figure 5A). In the nares, nine unique genera, all within Proteobacteria or Firmicutes, were significantly enriched compared to the oropharynx, with uncultured Neisseriaceae, Pantoea, Dolosigranulum, and Staphylococcus as the most abundant (padj < 0.001). In contrast, 16 unique genera were enriched in the oropharynx compared to the nares, with Prevotella as the most significantly enriched genus, followed by Veillonella and Streptococcus (padj < 0.001 each). Notably, the oropharynx microbiome showed diversity across four phyla—Bacteroidota, Firmicutes, Proteobacteria, and Actinobacteria—while the nares were primarily enriched with Firmicutes and Proteobacteria.
Among culture-negative samples from the nares and oropharynx, 145 genera were significantly enriched, with 52 unique genera shown in Figure 5B. In the nares, four phyla (Bacteroidota, Firmicutes, Proteobacteria, and Desulfobacterota) were enriched, with 64 genera (33 unique) showing significance; Lactobacillus was the most enriched (padj < 0.001), followed by uncultured Neisseriaceae, Staphylococcus (-6.08 ± 1.29 log2FoldChange; padj < 0.001), and Escherichia-shigella (padj < 0.001). Conversely, oropharynx samples had 81 significantly enriched genera (19 unique), with Prevotella, Veillonella, Pseudomonas, Streptococcus, and Actinomyces as the most abundant (padj < 0.001 each).
Discussion
S. aureus is a common commensal of the human microbiome, and its carriage is a significant risk factor for severe invasive infections with high mortality. Understanding factors that are associated with S. aureus carriage may lead to strategies for preventing these infections. Previous traditional twin studies have suggested that host genetics play a role in S. aureus carriage within the microbiome, though the degree of genetic influence remains debated (Andersen et al., 2012; Cyr et al., 2017; Peacock et al., 2001). Studies on the gastrointestinal microbiome have similarly identified a genetic contribution to microbiome composition (Goodrich et al., 2016; Turnbaugh et al., 2009; Vilchez-Vargas et al., 2022) though these effects seem to be in microbiomes less impacted by the environment. Using an observational approach in monozygotic twins, we investigated S. aureus concordance and microbiome composition across the nares, oropharynx, and hand. Our analysis revealed that location was the most significant modifier of microbiome composition while S. aureus carriage was significantly associated with microbial diversity and composition in monozygotic twins, albeit not similarly across all sampled locations.
Concordance
We characterized the microbiome composition of the nares, oropharynx, and hand of 147 monozygotic twin pairs, and described the compositional association of S. aureus culture status. Our study demonstrates that S. aureus culture status does associate with distinct microbiome composition in genetically identical twin pairs and that despite S. aureus concordance rates between MZ pairs ranging from 8%-46% at the three sites sampled, microbiome similarity was highest for concordant pairs. Microbiome composition was more similar between CC S. aureus pairs than DC S. aureus pairs (at the genus level) in both the nares and oropharynx [R² = Nares 0.73 (CC) vs 0.42 (DC); Oropharynx 0.85 (CC) vs 0.72 (DC)]. A recent study on Korean monozygotic twins also found that MZ twins’ microbiomes are more similar than DZ twins (Si et al., 2015) and our study reaffirms this finding while also noting that discordance for S. aureus carriage resulted in the least similar microbiome composition in monozygotic twin pairs at the genus level.
Our twin concordance rate for S. aureus positivity in the nares (46.3%) was notably higher than concordance rates for autoimmune conditions in Finnish twins, such as type 2 (34%) and type 1 diabetes (23%) (Kaprio et al., 1992), as well as S. aureus concordance rates reported in previous nasal microbiome studies by Andersen et al. (37.5%, 2012) and Liu et al. (2015) (26.1%, 2015). However, it remains lower than rates associated with conditions influenced by microbial presence, such as Crohn’s disease, inflammatory bowel disease, and ulcerative colitis (approximately 50%; Jess et al., 2005). Concordance in the oropharynx was 27.5%, while the hand exhibited a lower concordance of 8.2%, consistent with findings that suggest the hand microbiome is more variable and likely environmentally driven (Edmonds-Wilson et al., 2015). Notably, the oropharynx microbiome showed the highest genus-level similarity among concordant twins, with similarity strongest among twin pairs who were both non-colonized by S. aureus (R² = 0.85), suggesting that S. aureus colonization may increase microbiome variability in the oropharynx more than in the nares. Similar microbiome shifts impacting alpha diversity have been observed in other vertebrates (Barbagelata et al., 2011; Khamash et al., 2018; Park et al., 2011). Overall, our S. aureus concordance data from strictly monozygotic twins suggest an intermediate position, aligning with traditional twin studies that indicate minimal genetic influence (Andersen et al., 2012; Roghmann et al., 2011) but also supporting findings of modest genetic contributions to concordance rates (Aly et al., 1974; Hoeksma and Winkler, 1963; Si et al., 2015).
Prevalence
The overall prevalence of S. aureus we identified in the nares, oropharynx, and hand (26.2%, 29.9%, and 4.4%, respectively) aligns with prior studies showing approximately 30% of the general population are carriers (Gorwitz et al., 2008; Sollid et al., 2014). However, our rates were lower than those reported for medical students (39.3%; López-Aguilera et al., 2013), healthy Midwestern adults and children (44.1% and 36.1%, respectively; Hanson et al., 2018), and young women not using hormonal contraceptives (34%; Stensen et al., 2019). Although this study did not include longitudinal sampling, studies from Mexico reported higher S. aureus carriage (59.3%) across the nares and oropharynx (Hamdan-Partida et al., 2010). When comparing oropharynx to nares prevalence, our study observed a higher carriage rate in the oropharynx, similar to findings in prison populations (42.7% vs. 35.0%; Lee et al., 2011), healthy Iowans (37.9% vs. 25.4%; Hanson et al., 2018), and orthopedic patients (40% vs. 31%; (Nilsson and Ripa, 2006), but opposite to young children, where nares prevalence was higher (25.9% vs. 39.2%; (Esposito et al., 2014). Interestingly, S. aureus prevalence on the hand was markedly lower than in studies of food handlers (28.7%; (Beyene et al., 2019) and closer to healthcare worker rates (10.5%; Tammelin et al., 2003). Although we did not control for the time since the last handwashing (which could influence hand carriage rates), previous studies have shown that post-decolonization time can affect S. aureus prevalence and may explain the lower rates observed on hands in our study (Beyene et al., 2019).
Differential biodiversity
When comparing biodiversity within (alpha) and between (beta) sampled sites differentially colonized by S. aureus, we found that only the oropharynx site showed a significant difference in alpha diversity between S. aureus culture-positive and culture-negative samples (Chao1 index, p=0.05), using an index suited for low-abundance studies (Kers and Saccenti, 2022). No significant differences were observed using the Shannon and Simpson diversity indices (p>0.05; Supplementary Data Sheet 2). In contrast, other studies have reported that oropharynx alpha diversity can vary significantly over time (Bach et al., 2021), and although we sampled only healthy participants, Zhao et al. (2021) found that low alpha diversity is linked with increased S. aureus bacteremia in ICU neonates, suggesting that S. aureus presence may impact microbiome composition. Comparing alpha diversity across sites (nares, oropharynx, and hand), significant differences emerged (p<0.01; Figure 2), supporting prior findings that skin microbiome environments differ by site (Perez et al., 2016). Overall, the hand exhibited the highest alpha diversity, while the nares had the lowest (Figure 2), consistent with studies showing variability in alpha diversity across human ecological niches, including nares, oropharynx, sputum, feet, and skin (De Boeck et al., 2017; Han et al., 2020; Kates et al., 2019). Given that hand microbiome composition is known to vary widely between individuals and is influenced by factors such as sex (Basic-Cicak and Hasic Telalovic, 2023; Edmonds-Wilson et al., 2015), our findings of higher alpha diversity on the hand relative to the nares and oropharynx were not unexpected.
Biodiversity across sampling sites was measured using Bray–Curtis distance to assess taxon-based compositional differences in the nares, oropharynx, and hand samples based on S. aureus culture status. PCoA plots supported the hypothesis that S. aureus presence modifies beta diversity (p=0.001) and interacts with sampling sites (p=0.001). Previous twin studies found no significant differences in alpha or beta diversity in conditions such as rosacea (Zaidi et al., 2018) or the skin microbiome of Korean twins (Si et al., 2015). However, other studies have reported inverse relationships between S. aureus colonization and other microbiota (Bessesen et al., 2015) or even increased biodiversity due to S. aureus colonization (Reyes et al., 2020). To further clarify, while S. aureus status contributes to statistical differences in beta diversity, these findings may not represent biologically meaningful shifts in composition across all body sites. Despite this, our data do support the hypothesis that S. aureus carriage influences beta diversity, reflected in increased RRA at the family level: Neisseriaceae and Staphylococcaceae on the hands; increased Staphylococcaceae and low-abundance families in the nares for S. aureus culture-positive individuals; increased Carnobacteriaceae in culture-negative participants; increased Prevotellaceae and low-abundance families in S. aureus culture-positive oropharynx samples; and increased Pseudomonadaceae in culture-negative oropharynx samples. Increased Neisseriaceae abundance has been linked to chronic rhinosinusitis and S. aureus carriage (Wagner Mackenzie et al., 2021). Carnobacteriaceae, found in low abundance in other nasal studies (5.7%; (Lu et al., 2018), shows an inverse relationship with S. aureus (Flores Ramos et al., 2021), supporting our findings in the culture-negative samples. Additionally, our study found increased Prevotellaceae abundance correlated with S. aureus, consistent with findings in MRSA skin and soft tissue infections (SSTI) lesions (Misic et al., 2015), though our study did not assess MRSA SSTIs. Interestingly, the increased Pseudomonadaceae abundance in culture-negative participants aligns with other research suggesting that P. aeruginosa can outcompete S. aureus in wounds (Yung et al., 2021), despite reports of synergistic effects in other contexts (Alves et al., 2018; DeLeon et al., 2014).
To identify differentially abundant taxa, we used DESeq2 (Calgaro et al., 2020). In the nares, only one species, M. nonliquefaciens, was significantly more abundant in samples negative for S. aureus. Other studies have similarly reported Moraxella species in healthy controls (van den Munckhof et al., 2020), noting that Moraxella acts independently of S. aureus in the upper respiratory tract (Claassen-Weitz et al., 2021). However, M. nonliquefaciens can also cause infections similar to S. aureus, such as botryomycosis, a chronic granulomatous skin disease (Feldman and Petersen, 1989). In the oropharynx, only the genus Capnocytophaga was significantly more abundant in S. aureus culture-negative monozygotic twins. Known as a common oral commensal in humans and pets, Capnocytophaga supports oral health (Aas et al., 2008; Gross et al., 2010; Sixou et al., 2006) but has been linked to severe endocarditis in humans (Hayani et al., 2009; Sakai et al., 2019), similarly to S. aureus (Baddour et al., 2015). No differentially abundant taxa were observed between culture-positive and culture-negative hand samples.
When comparing differentially expressed taxa between the nares and oropharynx in S. aureus culture-positive samples, the most abundant taxa in the nares included Dolosigranulum, Staphylococcus, and Pantoea, while Prevotella, Streptococcus, Neisseria, and Alloprevotella were more notable in the oropharynx. Studies have shown that Dolosigranulum is commonly found in the nares (De Boeck et al., 2017) and is inversely associated with S. aureus carriage (Brugger et al., 2020). This inverse relationship is reflected in our data, as Dolosigranulum was significantly more abundant in the nares than Staphylococcus. Additionally, the presence of Neisseriaceae in the oropharynx aligns with studies showing their frequent co-occurrence (Drayß et al., 2019; Weyand, 2017), while Pantoea has also been identified in the nasal-oropharyngeal region (Walterson and Stavrinides, 2015). Similarly, co-presence of S. aureus and Prevotella has been observed in foot and hand paronychia (Grande-Del-Arco et al., 2020) and murine MRSA pneumonia models (Yamashita et al., 2020).
When evaluating differentially abundant taxa between S. aureus culture-negative and culture-positive nares and oropharynx samples, more abundant taxa were observed in S. aureus culture-negative samples. For instance, the most prevalent taxa in culture-negative nares were Lactobacillus, Moraxella, and Ralstonia, all typically found in the oropharyngeal microbiome. In the absence of S. aureus, these taxa may be more prominent in the nares. Lactobacillus, particularly strains with catalase function, is known to inhabit healthy nasal microbiomes (De Boeck et al., 2017), while Moraxella is frequently observed in healthy elderly individuals (van den Munckhof et al., 2020). Ralstonia, though commonly a soil microbe, appears in patients under mechanical ventilation (Waugh et al., 2010) and has a symbiotic relationship with Rhizopus, enhancing virulence and resistance to phagocytosis (Itabangi et al., 2022). Conversely, in the S. aureus culture-negative oropharynx samples, Haemophilus, Prevotella, and Streptococcus were among the most abundant taxa. Studies have shown Haemophilus can support S. aureus attachment on surfaces (Esin et al., 2015), potentially aiding carriage, though both taxa are sometimes found together (Nair et al., 2014). As noted previously, Prevotella was common in both S. aureus culture-positive and -negative oropharynx samples, suggesting that its abundance is influenced by location rather than S. aureus presence. The increased abundance of Streptococcus in S. aureus culture-negative oropharynx samples aligns with studies showing Streptococcus can inhibit S. aureus through hydrogen peroxide activity (Bogaert et al., 2004; Regev-Yochay et al., 2006), though S. aureus also modifies streptococcal biofilms (Schnurr et al., 2021), with antioxidants potentially affecting both taxa in biofilms (Sempere et al., 2022).
Limitations
This study has some limitations that could be addressed in future research. First, this study only investigated monozygotic twins. Although this minimized genetic variability, disentangling genetics from environment would require the addition of dizygotic twins to the study design and even investigating twins reared apart. Second, we did not account for the time twins spent together, which may influence S. aureus transmission via shared surfaces. Next, molecular typing was not performed, meaning concordant twins could carry two distinctly different S. aureus strains, potentially driving microbiome differences beyond simple presence or absence. Fourth, as a point-prevalence study, we lack longitudinal data. Although the International Twin Festival setting restricted repeated sampling, longitudinal data would provide clearer insights into the dynamics of S. aureus carriage over time. Additionally, we did not investigate the mechanistic impact of S. aureus carriage on microbiome composition or manipulate its composition. Despite these limitations, the study’s strengths include a large sample size and extensive sampling per participant, which enhances our understanding of S. aureus carriage associations within monozygotic twin pairs.
Conclusions
Our findings support our first prediction that S. aureus carriage is associated with reduced microbial diversity, as indicated by RRA and DESeq2 analyses showing fewer abundant taxa, potentially due to competitive exclusion by S. aureus. Specifically, in the S. aureus-negative samples, M. nonliquefaciens and Capnocytophaga were enriched in the nares and oropharynx, respectively, suggesting that these taxa may be outcompeted in S. aureus-positive environments.
Chao1 diversity narrowly achieved significance in only the oropharynx samples, pointing to a subtle yet detectable deviation from our hypothesis. Consistent with our second hypothesis, monozygotic twin pairs showed the highest concordance for S. aureus colonization in the nares, followed by the oropharynx and hand. STAMP analyses further revealed that microbiome composition was most similar in the S. aureus-positive nares samples, while the S. aureus-negative oropharynx samples showed an inverse pattern, likely reflecting influences of sampling location and/or timing. Bray–Curtis beta diversity analysis confirmed that location was the main factor shaping microbiome composition, with S. aureus status introducing additional, site-specific variation that acted as a modifier to compositional differences.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The studies involving humans were approved by Kent State University Institutional Review Board. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
MD: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. WS: Data curation, Investigation, Methodology, Software, Writing – review & editing. DN: Data curation, Investigation, Methodology, Software, Visualization, Writing – review & editing. SB: Data curation, Methodology, Writing – review & editing. NR: Data curation, Methodology, Writing – review & editing. CY: Data curation, Methodology, Writing – review & editing. DT: Data curation, Methodology, Writing – review & editing. JK: Data curation, Methodology, Writing – review & editing. SC: Data curation, Methodology, Writing – review & editing. HO: Methodology, Writing – review & editing, Data curation. TS: Funding acquisition, Investigation, Methodology, Resources, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Funding was provided to TS through startup funds from Kent State University.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/frmbi.2024.1457940/full#supplementary-material
Abbreviations
DNA, Deoxyribonucleic acid; MSSA, methicillin-sensitive S. aureus; MRSA, methicillin-resistant S. aureus; RNA, ribosomal RNA; SNVs, Single nucleotide variants; PCoA, Principal Coordinates Analysis; ASV, Amplicon sequence variant; PERMANOVA, permutational multivariate ANOVA; CI, Confidence interval; SEM, standard error of mean; RRA, relative read abundance.
References
Aas J. A., Griffen A. L., Dardis S. R., Lee A. M., Olsen I., Dewhirst F. E., et al. (2008). Bacteria of dental caries in primary and permanent teeth in children and young adults. J. Clin. Microbiol. 46, 1407–1417. doi: 10.1128/JCM.01410-07
Agresti A. (1992). A survey of exact inference for contingency tables. Stat. Sci. 7, 131–153. doi: 10.1214/ss/1177011454
Alves P. M., Al-Badi E., Withycombe C., Jones P. M., Purdy K. J., Maddocks S. E. (2018). Interaction between Staphylococcus aureus and Pseudomonas aeruginosa is beneficial for colonisation and pathogenicity in a mixed biofilm. Pathog. Dis. 76, 1–10. doi: 10.1093/femspd/fty003
Aly R., Maibach H. I., Shinefield H. R., Mandel A. D. (1974). Staphylococcus aureus carriage in twins. Am. J. Dis. Children 127, 486–488. doi: 10.1001/archpedi.1974.02110230032004
Amir A., McDonald D., Navas-Molina J. A., Kopylova E., Morton J. T., Zech Xu Z., et al. (2017). Deblur rapidly resolves single-nucleotide community sequence patterns. mSystems 2, e00191–e00116. doi: 10.1128/mSystems.00191-16
Andersen P. S., Pedersen J. K., Fode P., Skov R. L., Fowler V. G. Jr., Stegger M., et al. (2012). Influence of host genetics and environment on nasal carriage of Staphylococcus aureus in Danish middle-aged and elderly twins. J. Infect. Dis. 206, 1178–1184. doi: 10.1093/infdis/jis491
Andrews S. (2010). FastQC: a quality control tool for high throughput sequence data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (Accessed August 1, 2023).
Bach L. L., Ram A., Ijaz U. Z., Evans T. J., Lindström J. (2021). A longitudinal study of the human oropharynx microbiota over time reveals a common core and significant variations with self-reported disease. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.573969
Baddour L. M., Wilson W. R., Bayer A. S., Fowler V. G. Jr., Tleyjeh I. M., Rybak M. J., et al. (2015). Infective endocarditis in adults: Diagnosis, antimicrobial therapy, and management of complications: a scientific statement for healthcare professionals from the American Heart Association. Circulation 132, 1435–1486. doi: 10.1161/CIR.0000000000000296
Baldo L., Werren J. H. (2021). Evolutionary genetics of microbial symbiosis. Genes 12, 327. doi: 10.3390/genes12030327
Barbagelata M. S., Alvarez L., Gordiola M., Tuchscherr L., von Eiff C., Becker K., et al. (2011). Auxotrophic mutant of Staphylococcus aureus interferes with nasal colonization by the wild type. Microbes Infection 13, 1081–1090. doi: 10.1016/j.micinf.2011.06.010
Basic-Cicak D., Hasic Telalovic J. (2023). “Data science of microbiome: does gender matter,” in Advanced technologies, systems, and applications VII. Eds. Ademović N., Mujčić E., Mulić M., Kevrić J., Akšamija Z. (Springer, Cham: Springer International Publishing), 650–663. doi: 10.1007/978-3-031-17697-5_49
Bessesen M. T., Kotter C. V., Wagner B. D., Adams J. C., Kingery S., Benoit J. B., et al. (2015). MRSA colonization and the nasal microbiome in adults at high risk of colonization and infection. J. Infection 71, 649–657. doi: 10.1016/j.jinf.2015.08.008
Beyene G., Mamo G., Kassa T., Tasew G., Mereta S. T. (2019). Nasal and Hand Carriage Rate of Staphylococcus aureus among Food Handlers Working in Jimma Town, Southwest Ethiopia. Ethiopian J. Health Sci. 29, 5. doi: 10.4314/ejhs.v29i5
Bisanz J. E. (2018). qiime2R: Importing QIIME2 artifacts and associated data into R sessions. Version 0.99. Available at: https://scholar.google.com/citations?view_op=view_citation&hl=en&user=zRK8-hgAAAAJ&citation_for_view=zRK8-hgAAAAJ:NMxIlDl6LWMC.
Bockmann M. R., Harris A. V., Bennett C. N., Odeh R., Hughes T. E., Townsend G. C. (2011). Timing of colonization of caries-producing bacteria: an approach based on studying monozygotic twin pairs. Int. J. Dentistry 2011, 571573. doi: 10.1155/2011/571573
Bogaert D., van Belkum A., Sluijter M., Luijendijk A., de Groot R., Rümke H. C., et al. (2004). Colonisation by Streptococcus pneumoniae and Staphylococcus aureus in healthy children. Lancet 363, 1871–1872. doi: 10.1016/S0140-6736(04)16357-5
Bolyen E., Rideout J. R., Dillon M. R., Bokulich N. A., Abnet C. C., Al-Ghalith G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 8. doi: 10.1038/s41587-019-0209-9
Brown E. L., Below J. E., Fischer R. S. B., Essigmann H. T., Hu H., Huff C., et al. (2015). Genome-wide association study of Staphylococcus aureus carriage in a community-based sample of mexican-americans in starr county, texas. PloS One 10, e0142130. doi: 10.1371/journal.pone.0142130
Brugger S. D., Eslami S. M., Pettigrew M. M., Escapa I. F., Henke M. T., Kong Y., et al. (2020). Dolosigranulum pigrum cooperation and competition in human nasal microbiota. Msphere 5, e00852–e00820. doi: 10.1128/mSphere.00852-20
Calgaro M., Romualdi C., Waldron L., Risso D., Vitulo N. (2020). Assessment of statistical methods from single cell, bulk RNA-seq, and metagenomics applied to microbiome data. Genome Biol. 21, 1–31. doi: 10.1186/s13059-020-02104-1
Campbell D. J., Koch M. A. (2017). Living in peace: host-microbiota mutualism in the skin. Cell Host Microbe 21, 419–420. doi: 10.1016/j.chom.2017.03.012
Claassen-Weitz S., Lim K. Y., Mullally C., Zar H. J., Nicol M. P. (2021). The association between bacteria colonizing the upper respiratory tract and lower respiratory tract infection in young children: A systematic review and meta-analysis. Clin. Microbiol. Infection 27, 1262–1270. doi: 10.1016/j.cmi.2021.05.034
Comeau A. M., Douglas G. M., Langille M. G. I. (2017). Microbiome helper: A custom and streamlined workflow for microbiome research. mSystems 2, e00127–e00116. doi: 10.1128/mSystems.00127-16
Cyr D. D., Allen A. S., Du G.-J., Ruffin F., Adams C., Thaden J. T., et al. (2017). Evaluating genetic susceptibility to Staphylococcus aureus bacteremia in African Americans using admixture mapping. Genes Immun. 18, 2. doi: 10.1038/gene.2017.6
Dabke K., Hendrick G., Devkota S. (2019). The gut microbiome and metabolic syndrome. J. Clin. Invest. 129, 4050–4057. doi: 10.1172/JCI129194
Dalman M., Bhatta S., Nagajothi N., Thapaliya D., Olson H., Naimi H. M., et al. (2019). Characterizing the molecular epidemiology of Staphylococcus aureus across and within fitness facility types. BMC Infect. Dis. 19, 69. doi: 10.1186/s12879-019-3699-7
Dantes R., Mu Y., Belflower R., Aragon D., Dumyati G., Harrison L. H., et al. (2013). National burden of invasive methicillin-resistant Staphylococcus aureus infections, United States 2011. JAMA Internal Med. 173, 1970–1978. doi: 10.1001/jamainternmed.2013.10423
De Boeck I., Wittouck S., Wuyts S., Oerlemans E. F. M., van den Broek M. F. L., Vandenheuvel D., et al. (2017). Comparing the healthy nose and nasopharynx microbiota reveals continuity as well as niche-specificity. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.02372
DeLeon S., Clinton A., Fowler H., Everett J., Horswill A. R., Rumbaugh K. P. (2014). Synergistic interactions of Pseudomonas aeruginosa and Staphylococcus aureus in an in vitro Wound Model. Infection Immun. 82, 4718–4728. doi: 10.1128/IAI.02198-14
Drayß M., Claus H., Hubert K., Thiel K., Berger A., Sing A., et al. (2019). Asymptomatic carriage of Neisseria meningitidis, Haemophilus influenzae, Streptococcus pneumoniae, Group A Streptococcus and Staphylococcus aureus among adults aged 65 years and older. PloS One 14, e0212052. doi: 10.1371/journal.pone.0212052
Edmonds-Wilson S. L., Nurinova N. I., Zapka C. A., Fierer N., Wilson M. (2015). Review of human hand microbiome research. J. Dermatol. Sci. 80, 3–12. doi: 10.1016/j.jdermsci.2015.07.006
Esin L., Antonelli P. J., Ojano-Dirain C. (2015). Effect of Haemophilus influenzae exposure on Staphylococcus aureus tympanostomy tube attachment and biofilm formation. JAMA Otolaryngology–Head Neck Surg. 141, 148–153. doi: 10.1001/jamaoto.2014.3208
Esposito S., Terranova L., Zampiero A., Ierardi V., Rios W. P., Pelucchi C., et al. (2014). Oropharyngeal and nasal Staphylococcus aureus carriage by healthy children. BMC Infect. Dis. 14, 723. doi: 10.1186/s12879-014-0723-9
Ewels P., Magnusson M., Lundin S., Käller M. (2016). MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048. doi: 10.1093/bioinformatics/btw354
Feldman S. R., Petersen M. J. (1989). Botryomycosis caused by Moraxella nonliquefaciens. Cutis 43, 140–142.
Flores Ramos S., Brugger S. D., Escapa I. F., Skeete C. A., Cotton S. L., Eslami S. M., et al. (2021). Genomic stability and genetic defense systems in dolosigranulum pigrum, a candidate beneficial bacterium from the human microbiome. mSystems 6, e00425–e00421. doi: 10.1128/mSystems.00425-21
Galton F. (1876). The history of twins, as a criterion of the relative powers of nature and nurture. J. Anthropological Institute Great Britain Ireland 5, 391–406. doi: 10.2307/2840900
Ganna A., Ortega-Alonso A., Havulinna A., Salomaa V., Kaprio J., Pedersen N. L., et al. (2013). Utilizing twins as controls for non-twin case-materials in genome wide association studies. PloS One 8, e83101. doi: 10.1371/journal.pone.0083101
Garrett S. R., Palmer T. (2024). The role of proteinaceous toxins secreted by Staphylococcus aureus in interbacterial competition. FEMS Microbes 5, xtae006. doi: 10.1093/femsmc/xtae006
Goodrich J. K., Davenport E. R., Beaumont M., Jackson M. A., Knight R., Ober C., et al. (2016). Genetic determinants of the gut microbiome in UK twins. Cell Host Microbe 19, 731–743. doi: 10.1016/j.chom.2016.04.017
Gorwitz R. J., Kruszon-Moran D., McAllister S. K., McQuillan G., McDougal L. K., Fosheim G. E., et al. (2008). Changes in the prevalence of nasal colonization with Staphylococcus aureus in the United States 2001–2004. J. Infect. Dis. 197, 1226–1234. doi: 10.1086/533494
Grande-Del-Arco J., Jimenez-Cristino M. D., García-de-la-Peña R., Fernández-Espejo E., Córdoba-Fernández A. (2020). A Rare Paronychia with Superinfection with Prevotella bivia and Staphylococcus haemolyticus: The Importance of Early Microbiological Diagnosis. Pathogens 9, 12. doi: 10.3390/pathogens9120999
Gross E. L., Leys E. J., Gasparovich S. R., Firestone N. D., Schwartzbaum J. A., Janies D. A., et al. (2010). Bacterial 16S sequence analysis of severe caries in young permanent teeth. J. Clin. Microbiol. 48, 4121–4128. doi: 10.1128/JCM.01232-10
Hamdan-Partida A., Sainz-Espuñes T., Bustos-Martínez J. (2010). Characterization and persistence of Staphylococcus aureus strains isolated from the anterior nares and throats of healthy carriers in a mexican community. J. Clin. Microbiol. 48, 1701–1705. doi: 10.1128/JCM.01929-09
Han S. H., Lee J. S., Song K.-H., Choe Y. B., Ahn K. J., Lee Y. W. (2020). Differences in foot skin microbiomes between patients with type 2 diabetes and healthy individuals. Mycoses 63, 314–322. doi: 10.1111/myc.13046
Hanson B. M., Kates A. E., O’Malley S. M., Mills E., Herwaldt L. A., Torner J. C., et al. (2018). Staphylococcus aureus in the nose and throat of Iowan families. Epidemiol. Infection 146, 1777–1784. doi: 10.1017/S0950268818001644
Hayani O., Higginson L. A., Toye B., Burwash I. G. (2009). Man’s best friend? Infective endocarditis due to Capnocytophaga canimorsus. Can. J. Cardiol. 25, e130–e132. doi: 10.1016/S0828-282X(09)70076-5
Hoeksma A., Winkler K. C. (1963). The normal flora of the nose in twins. Acta Leidensia 32, 123–133.
Itabangi H., Sephton-Clark P. C., Tamayo D. P., Zhou X., Starling G. P., Mahamoud Z., et al. (2022). A bacterial endosymbiont of the fungus Rhizopus microsporus drives phagocyte evasion and opportunistic virulence. Curr. Biol. 32, 1115–1130. doi: 10.1016/j.cub.2022.01.028
Jess T., Riis L., Jespersgaard C., Hougs L., Andersen P. S., Orholm M. K., et al. (2005). Disease concordance, zygosity, and NOD2/CARD15 status: follow-up of a population-based cohort of danish twins with inflammatory bowel disease. Off. J. Am. Coll. Gastroenterol. | ACG 100, 2486–2492. doi: 10.1111/j.1572-0241.2005.00224.x
Johnson R. C., Ellis M. W., Lanier J. B., Schlett C. D., Cui T., Merrell D. S. (2015). Correlation between nasal microbiome composition and remote purulent skin and soft tissue infections. Infection Immun. 83, 802–811. doi: 10.1128/IAI.02664-14
Kampf G., Ruselack S., Eggerstedt S., Nowak N., Bashir M. (2013). Less and less–influence of volume on hand coverage and bactericidal efficacy in hand disinfection. BMC Infect Dis 13, 472. doi: 10.1186/1471-2334-13-472
Kaprio J., Tuomilehto J., Koskenvuo M., Romanov K., Reunanen A., Eriksson J., et al. (1992). Concordance for Type 1 (insulin-dependent) and Type 2 (non-insulin-dependent) diabetes mellitus in a population-based cohort of twins in Finland. Diabetologia 35, 1060–1067. doi: 10.1007/BF02221682
Kates A. E., Dalman M., Torner J. C., Smith T. C. (2019). The nasal and oropharyngeal microbiomes of healthy livestock workers. PloS One 14, e0212949. doi: 10.1371/journal.pone.0212949
Kers J. G., Saccenti E. (2022). The power of microbiome studies: some considerations on which alpha and beta metrics to use and how to report results. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.796025
Khamash D. F., Voskertchian A., Milstone A. M. (2018). Manipulating the microbiome: Evolution of a strategy to prevent S. aureus disease in children. J. Perinatology 38, 105–109. doi: 10.1038/jp.2017.155
Kong H. H., Segre J. A. (2017). The molecular revolution in cutaneous biology: investigating the skin microbiome. J. Invest. Dermatol. 137, e119–e122. doi: 10.1016/j.jid.2016.07.045
Lee C. J., Sankaran S., Mukherjee D. V., Apa Z. L., Hafer C. A., Wright L., et al. (2011). Staphylococcus aureus oropharyngeal carriage in a prison population. Clin. Infect. Dis. 52, 775–778. doi: 10.1093/cid/cir026
Leyden J. J., McGinley K. J., Kates S. G., Myung K. B. (1989). Subungual bacteria of the hand contribution to the glove juice test: efficacy of antimicrobial detergents. Infection Control & Hospital Epidemiology 10, 451–454. doi: 10.1086/645920
Liu C. M., Price L. B., Hungate B. A., Abraham A. G., Larsen L. A., Christensen K., et al. (2015). Staphylococcus aureus and the ecology of the nasal microbiome. Sci. Adv. 1, e1400216. doi: 10.1126/sciadv.1400216
López-Aguilera S., Goñi-Yeste M. D. M., Barrado L., González-Rodríguez-Salinas M. C., Otero J. R., Chaves F. (2013). Staphylococcus aureus nasal colonization in medical students: Importance in nosocomial transmission. Enfermedades infecciosas y microbiologia clinica 31, 500–505. doi: 10.1016/j.eimc.2012.12.005
Love M. I., Huber W., Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. doi: 10.1186/s13059-014-0550-8
Lu Y. J., Sasaki T., Kuwahara-Arai K., Uehara Y., Hiramatsu K. (2018). Development of a new application for comprehensive viability analysis based on microbiome analysis by next-generation sequencing: insights into staphylococcal carriage in human nasal cavities. Appl. Environ. Microbiol. 84, e00517–e00518. doi: 10.1128/AEM.00517-18
Mandal S., Van Treuren W., White R. A., Eggesbø M., Knight R., Peddada S. D. (2015). Analysis of composition of microbiomes: A novel method for studying microbial composition. Microbial Ecol. Health Dis. 26, 27663. doi: 10.3402/mehd.v26.27663
Martin M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.Journal 17, 1. doi: 10.14806/ej.17.1.200
McMurdie P. J., Holmes S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PloS One 8, e61217. doi: 10.1371/journal.pone.0061217
Misic A. M., Davis M. F., Tyldsley A. S., Hodkinson B. P., Tolomeo P., Hu B., et al. (2015). The shared microbiota of humans and companion animals as evaluated from Staphylococcus carriage sites. Microbiome 3, 2. doi: 10.1186/s40168-014-0052-7
Naik S., Bouladoux N., Wilhelm C., Molloy M. J., Salcedo R., Kastenmuller W., et al. (2012). Compartmentalized control of skin immunity by resident commensals. Science 337, 1115–1119. doi: 10.1126/science.1225152
Nair N., Biswas R., Götz F., Biswas L. (2014). Impact of Staphylococcus aureus on pathogenesis in polymicrobial infections. Infection Immun. 82, 2162–2169. doi: 10.1128/IAI.00059-14
Newcombe R. G. (1998). Two-sided confidence intervals for the single proportion: Comparison of seven methods. Stat Med. 17, 857–872. doi: 10.1002/(sici)1097-0258(19980430)17:8<857::aid-sim777>3.0.co;2-e
Nilsson P., Ripa T. (2006). Staphylococcus aureus Throat Colonization Is More Frequent than Colonization in the Anterior Nares. J. Clin. Microbiol. 44, 3334–3339. doi: 10.1128/JCM.00880-06
Oh J., Byrd A. L., Park M., Kong H. H., Segre J. A., Program N. C. S. (2016). Temporal stability of the human skin microbiome. Cell 165, 854–866. doi: 10.1016/j.cell.2016.04.008
Oksanen J., Blanchet F., Friendly M., Kindt R., Legendre P., McGlinn D., et al. (2020). vegan: community ecology package (2.5-7). Available online at: https://CRAN.R-project.org/package=vegan (Accessed August 1, 2023).
Park B., Iwase T., Liu G. Y. (2011). Intranasal application of S. epidermidis prevents colonization by methicillin-resistant Staphylococcus aureus in mice. PloS One 6, e25880. doi: 10.1371/journal.pone.0025880
Parks D. H., Beiko R. G. (2010). Identifying biologically relevant differences between metagenomic communities. Bioinformatics 26, 715–721. doi: 10.1093/bioinformatics/btq041
Parks D. H., Tyson G. W., Hugenholtz P., Beiko R. G. (2014). STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 30, 3123–3124. doi: 10.1093/bioinformatics/btu494
Peacock S. J., de Silva I., Lowy F. D. (2001). What determines nasal carriage of Staphylococcus aureus? Trends Microbiol. 9, 605–610. doi: 10.1016/S0966-842X(01)02254-5
Perez G. I. P., Gao Z., Jourdain R., Ramirez J., Gany F., Clavaud C., et al. (2016). Body site is a more determinant factor than human population diversity in the healthy skin microbiome. PloS One 11, e0151990. doi: 10.1371/journal.pone.0151990
R Core Team. (2022). R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. Available at: https://www.R-project.org/ (Accessed February 1, 2022).
Regev-Yochay G., Carmeli Y., Raz M., Pinco E., Etienne J., Leavitt A., et al. (2006). Prevalence and genetic relatedness of community-acquired methicillin-resistant Staphylococcus aureus in Israel. Eur. J. Clin. Microbiol. Infect. Dis. 25, 719–722. doi: 10.1007/s10096-006-0210-3
Reyes N., Montes O., Figueroa S., Tiwari R., Sollecito C. C., Emmerich R., et al. (2020). Staphylococcus aureus nasal carriage and microbiome composition among medical students from Colombia: A cross-sectional study. F1000Research 9, 78. doi: 10.12688/f1000research.22035.2
Rivals I., Personnaz L., Taing L., Potier M.-C. (2007). Enrichment or depletion of a GO category within a class of genes: Which test? Bioinformatics 23, 401–407. doi: 10.1093/bioinformatics/btl633
Roghmann M.-C., Johnson J. K., Stine O. C., Lydecker A. D., Ryan K. A., Mitchell B. D., et al. (2011). Persistent Staphylococcus aureus colonization is not a strongly heritable trait in Amish families. PloS One 6, e17368. doi: 10.1371/journal.pone.0017368
Rognes T., Flouri T., Nichols B., Quince C., Mahé F. (2016). VSEARCH: A versatile open source tool for metagenomics. PeerJ 4, e2584. doi: 10.7717/peerj.2584
Rosenberg E., Zilber-Rosenberg I. (2016). Microbes drive evolution of animals and plants: the hologenome concept. mBio 7, e01395–e01315. doi: 10.1128/mBio.01395-15
Round J. L., O’Connell R. M., Mazmanian S. K. (2010). Coordination of tolerogenic immune responses by the commensal microbiota. J. Autoimmun. 34, J220–J225. doi: 10.1016/j.jaut.2009.11.007
Sakai J., Imanaka K., Kodana M., Ohgane K., Sekine S., Yamamoto K., et al. (2019). Infective endocarditis caused by Capnocytophaga canimorsus; a case report. BMC Infect. Dis. 19, 1–5. doi: 10.1186/s12879-019-4492-3
Schnurr E., Paqué P. N., Attin T., Nanni P., Grossmann J., Holtfreter S., et al. (2021). Staphylococcus aureus Interferes with Streptococci Spatial Distribution and with Protein Expression of Species within a Polymicrobial Oral Biofilm. Antibiotics 10, 116. doi: 10.3390/antibiotics10020116
Sempere J., Llamosí M., Román F., Lago D., González-Camacho F., Pérez-García C., et al. (2022). Clearance of mixed biofilms of Streptococcus pneumoniae and methicillin-susceptible/resistant Staphylococcus aureus by antioxidants N-acetyl-l-cysteine and cysteamine. Sci. Rep. 12, 1–12. doi: 10.1038/s41598-022-10609-x
Sender R., Fuchs S., Milo R. (2016). Are we really vastly outnumbered? Revisiting the ratio of bacterial to host cells in humans. Cell 164, 337–340. doi: 10.1016/j.cell.2016.01.013
Si J., Lee S., Park J. M., Sung J., Ko G. (2015). Genetic associations and shared environmental effects on the skin microbiome of Korean twins. BMC Genomics 16, 992–992. doi: 10.1186/s12864-015-2131-y
Sixou J. L., Aubry-Leuliette A., De Medeiros-Battista O., Lejeune S., Jolivet-Gougeon A., Solhi-Pinsard H., et al. (2006). Capnocytophaga in the dental plaque of immunocompromised children with cancer. Int. J. Paediatric Dentistry 16, 75–80. doi: 10.1111/j.1365-263X.2006.00697.x
Sollid J. U. E., Furberg A. S., Hanssen A. M., Johannessen M. (2014). Staphylococcus aureus: Determinants of human carriage. Infection Genet. Evol. 21, 531–541. doi: 10.1016/j.meegid.2013.03.020
Stensen D. B., Småbrekke L., Olsen K., Grimnes G., Nielsen C. S., Simonsen G. S., et al. (2019). Hormonal contraceptive use and Staphylococcus aureus nasal and throat carriage in a Norwegian youth population. PloS One 14, e0218511. doi: 10.1371/journal.pone.0218511
Tammelin A., Klötz F., Hambræus A., Ståhle E., Ransjö U. (2003). Nasal and Hand Carriage of Staphylococcus aureus in Staff at a Department for Thoracic and Cardiovascular Surgery: Endogenous or Exogenous Source? Infection Control Hosp. Epidemiol. 24, 686–689. doi: 10.1086/502277
Thapaliya D., Taha M., Dalman M. R., Kadariya J., Smith T. C. (2017). Environmental contamination with Staphylococcus aureus at a large, Midwestern university campus. Sci. Total Environ. 599, 1363–1368. doi: 10.1016/j.scitotenv.2017.05.080
Turnbaugh P. J., Hamady M., Yatsunenko T., Cantarel B. L., Duncan A., Ley R. E., et al. (2009). A core gut microbiome in obese and lean twins. Nature 457, 7228. doi: 10.1038/nature07540
van den Munckhof E. H., Hafkamp H. C., de Kluijver J., Kuijper E. J., de Koning M. N., Quint W. G., et al. (2020). Nasal microbiota dominated by Moraxella spp. is associated with respiratory health in the elderly population: A case control study. Respir. Res. 21, 1–9. doi: 10.1186/s12931-020-01443-8
Vanegas J. M., Salazar-Ospina L., Gallego M. A., Jiménez J. N. (2021). A longitudinal study shows intermittent colonization by Staphylococcus aureus with a high genetic diversity in hemodialysis patients. Int. J. Med. Microbiol. 311, 151471. doi: 10.1016/j.ijmm.2020.151471
Vilchez-Vargas R., Skieceviciene J., Lehr K., Varkalaite G., Thon C., Urba M., et al. (2022). Gut microbial similarity in twins is driven by shared environment and aging. eBioMedicine 79, 104011. doi: 10.1016/j.ebiom.2022.104011
Wagner Mackenzie B., Zoing M., Clow F., Waite D. W., Radcliff F. J., Taylor M. W., et al. (2021). Characterising clinical Staphylococcus aureus isolates from the sinuses of patients with chronic rhinosinusitis. Sci. Rep. 11, 1. doi: 10.1038/s41598-021-01297-0
Walterson A. M., Stavrinides J. (2015). Pantoea: Insights into a highly versatile and diverse genus within the Enterobacteriaceae. FEMS Microbiol. Rev. 39, 968–984. doi: 10.1093/femsre/fuv027
Waugh J. B., Granger W. M., Gaggar A. (2010). Incidence, relevance and response for Ralstonia respiratory infections. Clin. Lab. Science: J. Am. Soc. Med. Technol. 23, 99–106.
Weyand N. J. (2017). Neisseria models of infection and persistence in the upper respiratory tract. Pathog. Dis. 75, ftx031. doi: 10.1093/femspd/ftx031
Wickham H. (2016). “Data analysis,” in Ggplot2: elegant graphics for data analysis. Ed. Wickham H. (Springer, Cham: Springer International Publishing), 189–201. doi: 10.1007/978-3-319-24277-4_9
Wu H., Zeng B., Li B., Ren B., Zhao J., Li M., et al. (2018). Research on oral microbiota of monozygotic twins with discordant caries experience—In vitro and in vivo study. Sci. Rep. 8, 7267. doi: 10.1038/s41598-018-25636-w
Yamashita Y., Nagaoka K., Kimura H., Suzuki M., Fukumoto T., Hayasaka K., et al. (2020). Pathogenic Effect of Prevotella intermedia on a Mouse Pneumonia Model Due to Methicillin-Resistant Staphylococcus aureus With Up-Regulated α-Hemolysin Expression. Front. Microbiol. 11, 587235. doi: 10.3389/fmicb.2020.587235
Yates F. (1934). Contingency tables involving small numbers and the χ2 test. Supplement to J. R. Stat. Soc. 1, 217–235. doi: 10.2307/2983604
Ye Z., Vasco D. A., Carter T. C., Brilliant M. H., Schrodi S. J., Shukla S. K. (2014). Genome wide association study of SNP-, gene-, and pathway-based approaches to identify genes influencing susceptibility to Staphylococcus aureus infections. Front. Genet. 5. doi: 10.3389/fgene.2014.00125
Yung D. B. Y., Sircombe K. J., Pletzer D. (2021). Friends or enemies? The complicated relationship between Pseudomonas aeruginosa and Staphylococcus aureus. Mol. Microbiol. 116, 1–15. doi: 10.1111/mmi.v116.1
Zaidi A. K., Spaunhurst K., Sprockett D., Thomason Y., Mann M. W., Fu P., et al. (2018). Characterization of the facial microbiome in twins discordant for rosacea. Exp. Dermatol. 27, 295–298. doi: 10.1111/exd.13491
Keywords: microbiome, 16S rRNA, Staphylococcus aureus, monozygotic twins, nares, oropharynx, hand, carriage
Citation: Dalman MR, Simison WB, Nielsen D, Bhatta S, Ramahi N, Yee C, Thapaliya D, Kadariya J, Cheatham S, Olson H and Smith TC (2025) Staphylococcus aureus carriage is associated with microbiome composition in the nares and oropharynx, not the hand, of monozygotic twins. Front. Microbiomes 3:1457940. doi: 10.3389/frmbi.2024.1457940
Received: 01 July 2024; Accepted: 06 December 2024;
Published: 20 January 2025.
Edited by:
Tim Dumonceaux, Agriculture and Agri-Food Canada (AAFC), CanadaReviewed by:
Sean Mathias Hemmingsen, National Research Council Canada (NRC), CanadaScott Jorge Dos Santos, Western University, Canada
Copyright © 2025 Dalman, Simison, Nielsen, Bhatta, Ramahi, Yee, Thapaliya, Kadariya, Cheatham, Olson and Smith. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark R. Dalman, bWRhbG1hbkBrZW50LmVkdQ==