 Christine M. Bassis1
Christine M. Bassis1 Kaylie A. Bullock1
Kaylie A. Bullock1 Daniel E. Sack1
Daniel E. Sack1 Katie Saund2Ali Pirani2Evan S. Snitkin1,2Veronica I. Alaniz3Elisabeth H. Quint3Jason D. Bell3†
Katie Saund2Ali Pirani2Evan S. Snitkin1,2Veronica I. Alaniz3Elisabeth H. Quint3Jason D. Bell3† Vincent B. Young1,2*†
Vincent B. Young1,2*†- 1Department of Internal Medicine/Division of Infectious Diseases, University of Michigan Medical School, Ann Arbor, MI, United States
- 2Department of Microbiology & Immunology, University of Michigan Medical School, Ann Arbor, MI, United States
- 3Department of Obstetrics and Gynecology, University of Michigan Medical School, Ann Arbor, MI, United States
Introduction: Factors that influence vaginal microbiota composition, including its source, are not well understood.
Methods: To determine if vaginal microbiota transmission from mother to daughter at birth could possibly influence the human vaginal microbiota composition in adolescence, we conducted a preliminary study to investigate the relationship between the vaginal microbiota of 13 adolescents (ages 15-21) and their mothers and the daughter’s birth mode.
Results and discussion: Based on analysis of bacterial 16S rRNA gene sequences, the vaginal microbiotas of mother/daughter pairs were more similar to each other if the daughter was born by vaginal delivery rather than by C-section. Specifically, within pair β-diversity, measured by the Yue and Clayton θ (θYC) distance metric, was significantly lower if the daughter was born by vaginal delivery. Additionally, genome sequences from an important member of the vaginal microbiota, Lactobacillus crispatus, isolated from one mother/daughter pair in which the daughter was born by vaginal delivery, were highly similar based on recombination-filtered single nucleotide polymorphisms (SNPs). Both community-level analysis and isolate genome sequence analysis are consistent with birth-mode dependent transmission and persistence of at least some members of the vaginal microbiota.
1 Introduction
The vaginal microbiota plays an important role in human health. The community structure of the vaginal microbiota is linked to infection susceptibility and preterm birth (Callahan et al., 2017; Klatt et al., 2017; McClelland et al., 2018; Bayigga et al., 2019; Tamarelle et al., 2019; Sun et al., 2022). The composition of the vaginal microbiota is distinct from other body sites and contains types of bacteria that seem specific to this anatomic location (Human Microbiome Project Consortium, H.M.P, 2012). For example, the vaginal microbiota is often dominated by specific types of Lactobacillus, most commonly L. crispatus and L. iners (Ravel et al., 2011; Bassis et al., 2017). Vaginal Lactobacillus sp. are thought to maintain dominance and inhibit colonization of other microbes through lactic acid production (O’Hanlon et al., 2013; Tachedjian et al., 2018).
Despite strong evidence that the vaginal microbiota can have significant impacts on health, the factors that influence the composition of the vaginal microbiota are not well understood. It is not known how this vagina-specific community is maintained from generation to generation. One possibility is that at least some members of the vaginal microbiota are transmitted from mother to daughter at birth and maintained in daughters through adolescence.
In healthy babies, the first large, direct exposure to microbes occurs at birth. Birth mode has been shown to influence the composition of the newborn microbiota (gut, skin, mouth), likely due to different bacterial exposure in vaginal delivery and C-sections (Dominguez-Bello et al., 2010; Madan et al., 2016). However, the effect of birth mode on the composition of the vaginal microbiota has not been investigated. In this study, we compared the vaginal microbiotas of 13 mother/daughter pairs and investigated the effect of birth mode on mother/daughter microbiota similarity. We also compared the genome sequences from Lactobacillus crispatus isolates from one mother/daughter pair. In this study, we took the first steps toward addressing our hypothesis that the vaginal microbiota of mothers and daughters would be more similar if the daughter was born by vaginal delivery than by C-section.
2 Materials and methods
2.1 Subject recruitment and sample collection
Mother/daughter pairs were recruited from the Pediatric and Adolescent Gynecology Clinic at the University of Michigan Health System in 2014 and 2015. Exclusions were pregnancy and age of less than 15 years. Written, informed consent was obtained and participants completed a baseline Qualtrics survey on their demographics and pertinent gynecologic and medical history. Vaginal samples were self-collected using a dual-headed swab (Starplex Scientific, S09D) at baseline and then weekly for 4 weeks. The baseline swab was obtained in the clinic, with immediate storage on ice and transfer to -80°C within a few hours. The subsequent swabs were returned via mail at ambient temperature. For each swab, subjects were asked to record the sampling date and if they were menstruating at the time of sampling. After the fifth swab was received and a completion incentive was mailed to the subject, the link between samples and subject names was destroyed, irreversibly de-identifying all samples. The study was approved by the University of Michigan IRB (HUM00086661).
2.2 DNA isolation and 16S rRNA gene sequencing
One of the swab heads from each sample was clipped directly into the bead plate of a PowerMag Microbiome RNA/DNA Isolation Kit (Mo Bio Laboratories, Inc.). DNA isolation was performed according to the manufacturer’s instructions using an epMotion 5075 liquid handling system. The V4 region of the 16S rRNA gene was amplified from 1 or 7μl DNA and sequenced with a MiSeq (Illumina, San Diego, CA) using the 500-cycle MiSeq Reagent Kit, v. 2 (Illumina, catalog No. MS-102–2003) by the University of Michigan Microbial Systems Molecular Biology Laboratory as described previously (Seekatz et al., 2015). The other swab head was used for cultivation or stored at -80°C.
2.3 Bacterial community analysis
The 16S rRNA gene sequences were processed using mothur v.1.36.1 and v.1.39.5 following the mothur MiSeq SOP (Schloss et al., 2009; Kozich et al., 2013). Details of the processing steps are available in mother.daughter_mothur.batch (https://github.com/cbassis/MotherDaughter_Vaginal_Microbiota.study). After sequence processing and alignment to the SILVA reference alignment (Release 102) (Schloss, 2009), sequences were binned into operational taxonomic units (OTUs) based on 97% sequence similarity using the average neighbor method (Schloss and Westcott, 2011; Westcott and Schloss, 2015). Samples with fewer than 1000 sequences were excluded from the analysis. OTUs were classified to the genus level within mothur using a modified version of the Ribosomal Database Project (RDP) training set (version 9) (Wang et al., 2007; Cole et al., 2014). To further classify the Lactobacillus OTUs, representative sequences were analyzed using standard nucleotide BLAST for highly similar sequences (megablast) on the National Center for Biotechnology Information (NCBI) BLAST web page (https://blast.ncbi.nlm.nih.gov/Blast.cgi) (Morgulis et al., 2008). OTU relative abundances were calculated and plotted in a heatmap. To compare bacterial communities between pairs, within pairs and within subjects, we calculated θYC distances (a metric that takes relative abundances of both shared and non-shared OTUs into account) (Yue and Clayton, 2005). A Kruskal-Wallis test with a Dunn’s posttest or a Wilcoxon (Mann-Whitney) test were used to determine if differences in θYC distances were statistically significant. Principal coordinates analysis (PCoA) was used to visualize the θYC distances between samples. R Studio (Version 1.1.456) with R (Version 3.5.1) was used for the statistical tests and plotting the heat map, box and whisker plots, and the ordination using the code available: https://github.com/cbassis/MotherDaughter_Vaginal_Microbiota.study/tree/master/R_code. Adobe Illustrator (CS6) was used for labeling and formatting figures.
2.4 Lactobacillus crispatus isolation
For pair I, the second swab head from the freshly collected baseline vaginal sample was swabbed onto an MRS agar plate and incubated in an anaerobic chamber (Coy Laboratory Products) at 37°C. DNA from individual isolates was amplified with primers targeting the V1-V5 region of the 16S rRNA gene (“B” adapter oligo sequence +27F (5’-CCTATCCCCTGTGTGCCTTGGCAGTCTCAGAGAGTTTGATCCTGGCTCAG-3’) and XLR_926R_v2bBar8L (5’-CCATCTCATCCCTGCGTGTCTCCGACTCAGCACGCCCGTCAATTCMTTTRAGT-3’)) (Jumpstart Consortium Human Microbiome Project Data Generation Working Group, 2010) under the following PCR conditions: 2 minutes @ 95°C; 30x(20 seconds @ 95°C, 30 seconds @ 50°C, 30 sseconds @ 72°C); 5 minutes @ 72°C. Amplicon sequences obtained by via Sanger sequencing were used for isolate identification.
2.5 DNA isolation and genome sequencing
Three Lactobacillus crispatus isolates from pair I, 2 from the mother and 1 from the daughter, were grown overnight in 1 ml liquid MRS in an anaerobic chamber (Coy Laboratory Products) at 37°C. Genomic DNA was isolated from the liquid cultures using the PowerMicrobiome™ RNA Isolation Kit (Mo Bio Laboratories, Inc.) without the DNase treatment. Genome sequencing was performed by the Microbial Systems Molecular Biology Laboratory at the University of Michigan using a Nextera™ XT DNA Library Preparation kit (Illumina, San Diego, CA) and a 600-cycle MiSeq Reagent Kit, v. 3 (Illumina, San Diego, CA) on a MiSeq (Illumina, San Diego, CA).
2.6 Genome sequence analysis
Phylogenetic relationships between L. crispatus isolates from mother/daughter pair I and all L. crispatus strains with genome sequences available as fastq files from NCBI on December 27th, 2018 were determined based on recombination-filtered single nucleotide polymorphisms (SNPs). Quality of reads was assessed with FastQC v0.11.3 (Andrews), and Trimmomatic 0.36 (Bolger et al., 2014) was used for trimming adapter sequences and low-quality bases. Variants were identified by (i) mapping filtered reads to reference genome sequence L. crispatus ST1 (SAMEA2272191) using the Burrows-Wheeler short-read aligner (bwa-0.7.17) (Li and Durbin, 2009), (ii) discarding polymerase chain reaction duplicates with Picard (picard-tools-2.5.0), and (iii) calling variants with SAMtools (samtools-1.2) and bcftools. Variants were filtered from raw results using GATK ‘s (GenomeAnalysisTK-3.3-0) VariantFiltration (QUAL, >100; MQ, >50; >=10 reads supporting variant; and FQ, <0.025) (McKenna et al., 2010). In addition, a custom python script was used to filter out single-nucleotide variants that were (i) <5 base pairs (bp) in proximity to indels, (ii) fell under Phage and Repeat region of the reference genome (identified using Phaster (Pon et al., 2016) and Nucmer (MUMmer3.23) (Kurtz et al., 2004)), (iii) not present in the core genome, or (iv) in a recombinant region identified by Gubbins 2.3.1 (Croucher et al., 2015). A maximum likelihood tree was constructed in RAxML 8.2.8 (Stamatakis, 2014) using a general-time reversible model of sequence evolution. Bootstrap analysis was performed with the number of bootstrap replicates determined using the bootstrap convergence test and the autoMRE convergence criteria (-N autoMRE). Bootstrap support values were overlaid on the best scoring tree identified during rapid bootstrap analysis (-f a). The final maximum likelihood tree was plotted and pairwise SNP distances were calculated in R Studio (Version 1.1.463) with R (Version 3.5.3): https://github.com/cbassis/MotherDaughter_Vaginal_Microbiota.study/blob/master/R_code/Mother_Daughter_Figure_3_Genome_Tree_and_Genome_Analysis.Rmd. Adobe Illustrator (CS6) was used for labeling and formatting the figure.
2.7 Calculation of doubling time estimate for vaginal L. crispatus in vivo
We used the number of SNPs between the pair I mother and daughter L. crispatus isolates to estimate the doubling time of vaginal L. crispatus in vivo if all SNPs in the recombination-filtered core genome were due to mutations acquired since the daughter’s birth:
The mutation rate of L. crispatus is unknown, so for this estimate we used the published mutation rate of another Lactobacillus, L. casei Zhang, in vitro, without antibiotics (1.0x10-9 bp/generation) (Wang et al., 2017). The pair I daughter’s age in hours was:
The average length of the recombination-filtered core genome (940,943 bp) was used for genome length. We assumed that the isolates arose from a common ancestor and that all mutations were non-convergent, so the number of mutations acquired by each isolate would equal the number of SNPs between the mother’s isolate and the daughter’s isolate divided by 2. We also estimated the number of mutations acquired per isolate core genome per year as:
3 Results
3.1 Subject characteristics and sequencing results
A total of 107 self-collected, vaginal swab samples were obtained from 26 subjects (13 mother/daughter pairs) (Table 1 and Supplemental Table 1). Each subject returned 1-5 weekly samples (median=5 samples/subject, IQR=1). After sequence processing and exclusion of samples with fewer than 1000 sequences, a total of 2,336,437 high quality bacterial 16S rRNA gene sequences from 101 samples were analyzed with an average of 23,133 +/- 10,212 sequences per sample. Sequence counts and α-diversity metrics for each sample included in our analysis are listed in Supplemental Table 2.

Table 1 Subject Characteristics.
3.2 An individual’s vaginal microbiota is relatively stable over 4 weeks
During the sampling period, the vaginal microbiota of each subject was relatively stable. The high stability of the vaginal microbiota is apparent from the consistent within subject community composition (Figure 1 and Supplemental Table 3). For example, OTU1 (L. crispatus) and/or OTU2 (L. iners) persisted from week to week in many subjects. Additionally, average θYC distances were significantly lower within subjects than between subjects (Kruskal-Wallis p-value= 8.154e-10) (Figure 2A) and samples clustered by subject in a PCoA based on θYC distances (Supplemental Figure 1).

Figure 1 Vaginal bacterial community compositions of mother/daughter pairs. Relative abundances of OTUs in weekly vaginal swab samples from 13 mother/daughter pairs. Mother/daughter pairs were ordered by average within pair θYC distances, with the most similar pair (I) on top and the least similar pair (XIII) on the bottom. OTUs with a minimum of 200 sequences in the dataset overall and present at a relative abundance greater than 2% in at least 1 sample were included in the heat map. Taxonomic classifications to genus level of these OTUs are listed in Supplemental Table 3.
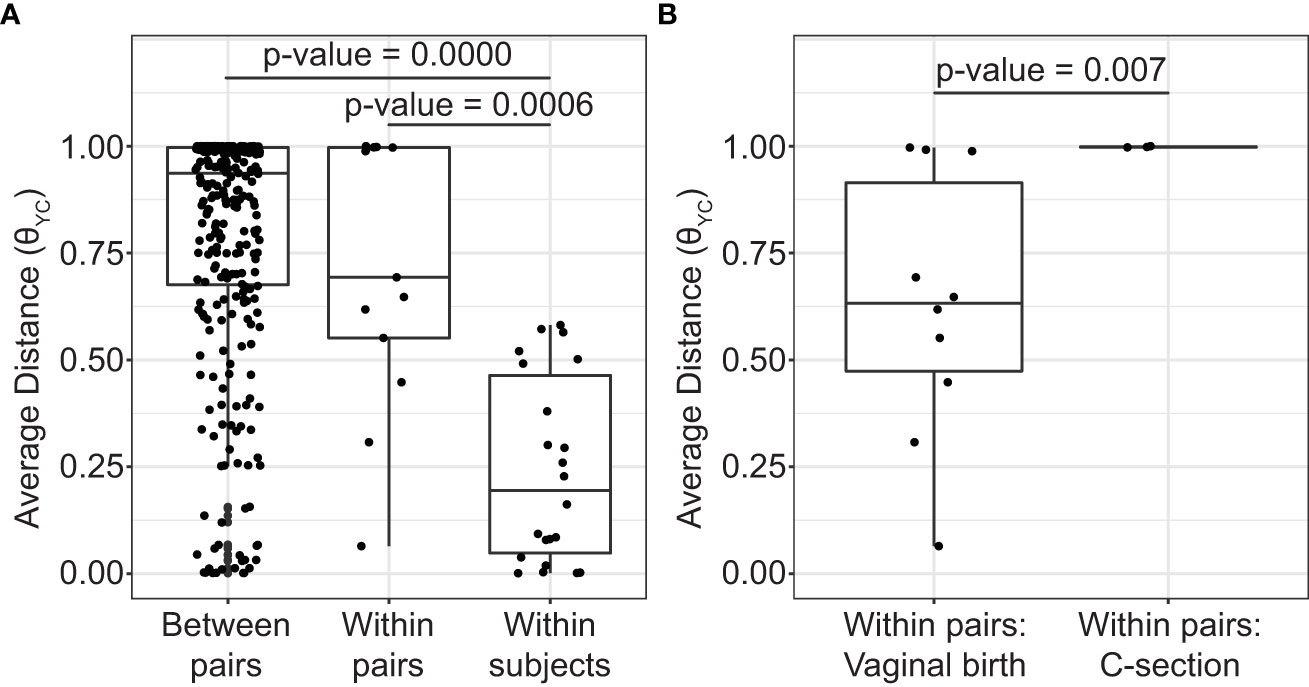
Figure 2 Average distances between vaginal bacterial communities. (A) Mean θYC distances between subjects from different mother/daughter pairs (between pairs), between subjects within a mother/daughter pair (within pair) and between samples from the same subject (within subject). P-values for comparisons that were significantly different by Dunn’s posttest are shown (Kruskal-Wallis p-value= 8.154e-10). (B) Mean θYC distances between subjects within a mother/daughter pair for daughters born by vaginal birth and by C-section. Wilcoxon (Mann-Whitney) test p-value is shown. In the box and whiskers plots, the median θYC distance is indicated by a line, values within the first to the third quartiles are inside the box and the whiskers extend to the smallest and largest values within 1.5x the interquartile range.
3.3 Factors associated with similarity between vaginal microbiotas of mothers and their daughters
To determine if mothers and their daughters had more similar vaginal microbiotas than unrelated subjects, we compared the average θYC distances between all unrelated subjects (between pairs) and the average θYC distances between mothers and their own daughters (within pairs) (Figure 2A). There was a trend toward greater similarity (lower θYC distances) within all mother/daughter pairs than between subjects in different mother/daughter pairs. To determine if birth mode was related to vaginal microbiota similarity within mother/daughter pairs, we compared the average within pair θYC distances for pairs in which the daughter was born by vaginal delivery and by C-section (Figure 2B). The average within pair θYC distances were significantly lower for pairs in which the daughter was born by vaginal delivery compared to C-section (Wilcoxon test p-value=0.007) (Figure 2B). Additional factors, including reproductive status, are also likely to contribute to mother/daughter vaginal microbiota similarity. Indeed, we also found a strong trend toward greater mother-daughter vaginal microbiota similarity (lower within pair θYC distances) if the mother was in her reproductive stage rather than postmenopausal (Wilcoxon test p-value=0.07) (Supplemental Figure 2).
3.4 Lactobacillus crispatus isolates from mother/daughter pair I have highly similar genome sequences
The birth mode-dependent similarity of the vaginal microbiotas of mothers and their daughters suggested that vaginal bacteria could be transmitted between generations at birth and persist into adolescence. However, it is possible that genetic or environmental factors shared by a mother and her daughter lead to acquisition of similar bacteria later, resulting in the de novo establishment of similar vaginal communities. To investigate the possibility of direct transmission and persistence of one member of the vaginal microbiota, we generated draft genome sequences of Lactobacillus crispatus strains isolated from the freshly collected second swab head of mother/daughter pair I. The draft genome sequences of these isolates were compared with publicly available L. crispatus genome sequences by constructing a maximum likelihood phylogenetic tree based on a recombination-filtered core genome alignment. Interestingly, the three strains of L. crispatus from mother/daughter pair I, UMP1M1, UMP1M2 and UMP1D1, were more similar to each other than to any of the other strains, including others isolated from the female reproductive tract (Figure 3).
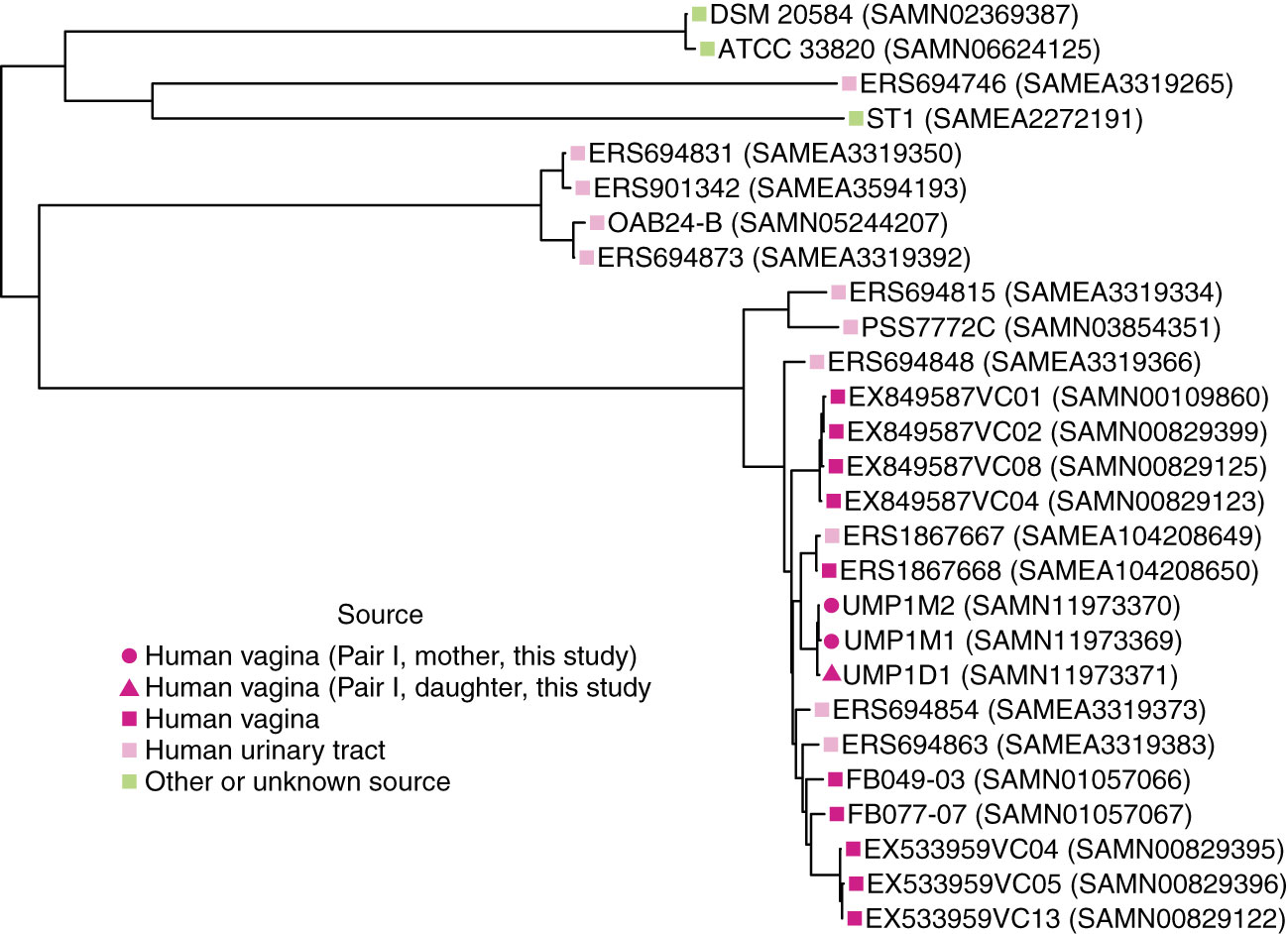
Figure 3 Phylogenetic relationships between L. crispatus strains. Maximum likelihood tree based on recombination-filtered SNP distances between L. crispatus genome sequences of isolates from mother/daughter pair I and other L. crispatus strains with publicly available genomes. Tip labels indicate L. crispatus strain names and NCBI BioSample identifiers. Bootstrap values were greater than or equal to 0.65.
We also calculated the number of SNPs between our isolates using the recombination-filtered core genome alignment. There were 11 recombination-filtered SNPs between the 2 isolates from the mother (UMP1M1 and UMP1M2) and 25 and 16 recombination-filtered SNPs between the daughter’s isolate (UMP1D1) and the 2 isolates from the mother (UMP1M1 and UMP1M2, respectively).
3.5 Estimate of in vivo doubling time and mutation rate for vaginal L. crispatus
To further investigate the plausibility that the L. crispatus strain isolated from daughter I descended from a strain transmitted from her mother at birth, we estimated the doubling time that would allow our isolates to acquire the observed number of SNPs over 20 years. Based on the 25 SNPs between UMP1M1 and UMP1D1, the estimated doubling time for L. crispatus in vivo would be 13.2 hours. Based on the 16 SNPs between UMP1M2 and UMP1D1, the estimated doubling time would be 20.6 hours. We also estimated the in vivo mutation rate of the core genome of the L. crispatus isolates to be 0.4-0.6 mutations per year.
4 Discussion
Our study provides preliminary evidence that the vaginal microbiota may be vertically transmitted from mother to daughter at birth via vaginal delivery and persists into adolescence. Because the daughters in our study were 15-21 years old, both transmission and persistence were required to observe evidence of vertical transmission. The first piece of evidence supporting vertical transmission is that the vaginal microbiotas of mothers and their adolescent daughters were more similar if their daughter was born by vaginal delivery rather than C-section. The second piece of evidence supporting vertical transmission and persistence is that an important member of the vaginal microbiota, L. crispatus, isolated from a vaginally-born, 20-year-old daughter and her mother (pair I) had highly similar genome sequences.
Lactobacillus dominance may not be typical in young children but very few vaginal microbiota studies include samples prior to menarche (France et al., 2022a). By ages 10-12, prior to menarche, one study found Lactobacillus dominance to be common and the vaginal microbiotas resemble reproductive-stage subjects (Hickey et al., 2015). If Lactobacillus persists from birth to adolescence, it may require persistence at low levels for many years. A recent study provides evidence of persistence of several vaginal bacterial strains in reproductive age women for more than a year (France et al., 2022b).
Other studies have compared the vaginal microbiotas of mothers and daughters without analyzing the effect of birth mode (Lee et al., 2013; Hickey et al., 2015; Si et al., 2016; France et al., 2022c). One study found greater similarity between the vaginal microbiotas of mothers and daughters than between unrelated subjects (Lee et al., 2013). This was similar to the trend we observed toward greater community similarity within mother/daughter pairs, regardless of birth mode, than between unrelated subjects in different mother/daughter pairs (Figure 2A). Greater similarity between the vaginal microbiota of mothers and daughters was not detected in the other studies (Hickey et al., 2015; Si et al., 2016; France et al., 2022c). If many of the daughters in the other studies were born by C-section then high similarity between mothers and daughters would not be expected. With C- section rates of ~30% in the United States (study site for (Hickey et al., 2015; France et al., 2022c)) and ~36% in South Korea (study site for (Si et al., 2016)) this is a possibility (Caughey et al., 2014; Kim et al., 2017). Additionally, our study focused on adolescent daughters (age 15-21) while the Hickey et al. (2015) study focused on younger daughters and the Si et al. (2016) study focused on older daughters. Reproductive stage has been shown to influence the structure of the vaginal microbiota (Brotman et al., 2018) and we see a similar effect in this study (Supplemental Figure 2). Therefore differences in reproductive stage may also contribute to differences in vaginal community composition between mothers and daughters.
Although an overall community similarity was not observed in these studies, specific community members (Lactobacillus and Prevotella) were identified as most heritable in one study (Si et al., 2016).
Based on the number of SNPs observed between the mother and daughter L. crispatus isolates and published mutations rates for L. casei Zhang (Wang et al., 2017), we estimated that L. crispatus would have an in vivo doubling time of 13.2-20.6 hours, depending on the specific isolates compared. The doubling time estimates of 13.2 hours and 20.6 hours for L. crispatus in vivo are within the range estimated for other bacteria in their natural environments, including Escherichia coli (15 hours) and Salmonella enterica (25 hours) (Gibson et al., 2018). These doubling times are faster than the 4.1-5.6 days doubling times measured for L. casei Shirota in mouse intestines, where its growth rate was insufficient to maintain colonization (Lee et al., 2004). Although the actual growth and mutation rates of L. crispatus in the human vagina have not been measured, we estimated reasonable in vivo doubling times for vaginal L. crispatus based on the observed number of SNPs between L. crispatus isolates from mother/daughter pair I, the age of daughter I and L. casei Zhang mutation rates. Considering the uncertainty in the estimates, transmission of L. crispatus from mother to daughter at birth followed by the accumulation of independent mutations during 20 years of persistence in the mother and daughter is a plausible explanation for the observed recombination-filtered SNPs. Future studies comparing genomes of L. crispatus isolates from more mother/daughter pairs with a variety of daughter ages are needed. Future metagenomic studies could also be used to infer in vivo growth rates (Korem et al., 2015).
The 2 L. crispatus isolates from the mother had highly similar genomes, differing by only 11 recombination-filtered SNPs. A previous study also observed high similarity between the genomes of multiple vaginal L. crispatus isolates from one individual, noting that they were indistinguishable (Abdelmaksoud et al., 2016). Future investigations of L. crispatus genomic variation within an individual may yield further insight on colonization and dynamics of the vaginal microbiota.
Consistent with a previous study, L. crispatus isolates from the human vagina were phylogenetically intermixed with isolates from the human urinary tract, including highly similar vaginal (ERS1867668 (SAMEA104208650)) and bladder (ERS1867667 (SAMEA104208649)) isolates from the same subject (Figure 3) (Thomas-White et al., 2018).
The health implications of vertical transmission of the vaginal microbiota are unknown and were not addressed in this study. However, if vertical transmission is an important factor in determining the composition of the vaginal microbiota there may be important consequences. Vertical transmission of the vaginal microbiota may be one mechanism for maintaining human microbiota over generations via a consistent and specific seeding of the newborn microbiota. Delivery mode is an important factor in determining the early composition of the gut microbiota (Rutayisire et al., 2016; Wampach et al., 2018) and is a risk factor for development of immune-related disorders later in life (Tamburini et al., 2016). This suggests an important role for the mother’s vaginal microbiota in seeding the infant and setting the stage for development of the gut microbiota. Therefore, maintenance of the vaginal microbiota between generations may be critical for gut microbiota development in each generation.
Additionally, the vaginal microbiota plays an important if not well understood role in reproductive health, with associations between vaginal microbiota composition and infection susceptibility, bacterial vaginosis (BV) and preterm birth (Callahan et al., 2017; Klatt et al., 2017; McClelland et al., 2018; Bayigga et al., 2019; Tamarelle et al., 2019; Sun et al., 2022). Evidence from this study suggests that transmission of microbes from mother to daughter at birth may influence the composition of the daughter’s microbiota later in life and may contribute to the maintenance of specific members of the human vaginal microbiota over generations.
This study provides evidence consistent with vertical transmission contributing to the vaginal microbiota. As this was a preliminary study, there are some key limitations. This was a small study with only 13 mother/daughter pairs (92% white) and 3/13 daughters born by C-section. Mothers with daughters born by C-section were on average older than mothers with daughters born by vaginal delivery (Table 1 and Supplemental Table 2) and two of the three mothers with daughters born by C-section were post-menopausal which could also contribute to a greater difference in mother/daughter vaginal microbiotas (Brotman et al., 2018). Beyond birth mode and reproductive status, other factors including genetics and shared environment could contribute to mother/daughter vaginal microbiota similarity. Of the eleven pairs asked about cohabitation, only one pair (IV) reported that they didn’t currently live together full or part-time (Supplemental Table 2). Therefore, the influence of cohabitation on vaginal microbiota similarity could not be addressed in our study. Genomic analysis of isolates was limited to one member of the vaginal microbiota from 1 mother/daughter pair. Despite these limitations, we feel that these results are intriguing and provide impetus for future studies in larger populations, including more racially diverse subjects, more daughters born by C-section and analysis of more isolate genome sequences or metagenomes to validate these findings.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA547595; https://www.ncbi.nlm.nih.gov/bioproject/PRJNA547620; https://github.com/cbassis/MotherDaughter_Vaginal_Microbiota.study.
Ethics statement
The studies involving human participants were reviewed and approved by University of Michigan IRB. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
CB was involved in study design and planning, data analysis, figure preparation and manuscript writing. KB was involved in subject recruitment, sample processing, isolation of L. crispatus genomic DNA for sequencing, data analysis and manuscript editing. DS was involved in subject recruitment, sample processing and manuscript editing. KS was involved in genomic data analysis, interpretation of genomic data, phylogenetic tree construction and manuscript editing. AP was involved in genomic data analysis and manuscript editing. ES was involved in genomic data analysis and interpretation and manuscript review. VA was involved in subject recruitment and planning. EQ was involved in study design and planning, subject recruitment and manuscript editing. VY was involved in study design and manuscript editing. JB was involved with study design and planning, subject recruitment and manuscript editing. All authors contributed to the article and approved the submitted version.
Acknowledgments
We’d like to thank the participants who contributed samples. We’d also like to thank Megan Lemieur for help with study organization and Qualtrics survey implementation and the University of Michigan Microbial Systems Molecular Biology Laboratory for swab DNA isolation, bacterial community and genomic library preparation and sequencing. We’d also like to thank Ingrid Bergin and members of the Young lab for many discussions related to this project over the years. This research was also supported in part through computational resources and services provided by Advanced Research Computing at the University of Michigan, Ann Arbor.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/frmbi.2023.1129394/full#supplementary-material
Supplementary Figure 1 | Principal coordinates analysis (PCoA) of vaginal microbiota from 13 mother/daughter pairs. The θYC distances between 101 vaginal microbiota samples are represented by PCoA. Samples from daughters are represented by triangles and samples from mothers by circles. Each mother/daughter pair is represented by a unique color. Biplot arrows represent the 3 OTUs most correlated with position on the PCoA plot.
Supplementary Figure 2 | Mean θYC distances between subjects within a mother/daughter pair by the mother’s reproductive stage. Wilcoxon (Mann-Whitney) test p-value is shown. In the box and whiskers plots, the median thetaYC distance is indicated by a line, values within the first to the third quartiles are inside the box and the whiskers extend to the smallest and largest values within 1.5x the interquartile range.
References
Abdelmaksoud A. A., Koparde V. N., Sheth N. U., Serrano M. G., Glascock A. L., Fettweis J. M., et al. (2016). Comparison of Lactobacillus crispatus isolates from Lactobacillus-dominated vaginal microbiomes with isolates from microbiomes containing bacterial vaginosis-associated bacteria. Microbiology 162 (3), 466–475. doi: 10.1099/mic.0.000238
Andrews S., FastQC. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
Bassis C. M., Allsworth J. E., Wahl H. N., Sack D. E., Young V. B., Bell J. D. (2017). Effects of intrauterine contraception on the vaginal microbiota. Contraception 96 (3), 189–195. doi: 10.1016/j.contraception.2017.05.017
Bayigga L., Kateete D. P., Anderson D. J., Sekikubo M., Nakanjako D. (2019). Diversity of vaginal microbiota in sub-Saharan Africa and its effects on HIV transmission and prevention. Am. J. Obstet Gynecol 220 (2), 155–166. doi: 10.1016/j.ajog.2018.10.014
Bolger A. M., Lohse M., Usadel B. (2014). Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 30 (15), 2114–2120. doi: 10.1093/bioinformatics/btu170
Brotman R. M., Shardell M. D., Gajer P., Fadrosh D., Chang K., Silver M. I., et al. (2018). Association between the vaginal microbiota, menopause status, and signs of vulvovaginal atrophy. Menopause 25 (11), 1321–1330. doi: 10.1097/GME.0000000000001236
BWA. Available at: http://bio-bwa.sourceforge.net.
Callahan B. J., DiGiulio D. B., Goltsman D. S. A., Sun C. L., Costello E. K., Jeganathan P., et al. (2017). Replication and refinement of a vaginal microbial signature of preterm birth in two racially distinct cohorts of US women. Proc. Natl. Acad. Sci. 114(37), 9966–71. doi: 10.1073/pnas.1705899114
Calling SNPs/INDELs with SAMtools/BCFtools. Available at: http://samtools.sourceforge.net/mpileup.shtml.
Caughey A. B., Cahill A. G., Guise J.-M., Rouse D. J. (2014). Safe prevention of the primary cesarean delivery. Am. J. Obstetrics Gynecology 210 (3), 179–193. doi: 10.1016/j.ajog.2014.01.026
Cole J. R., Wang Q., Fish J. A., Chai B., McGarrell D. M., Sun Y., et al. (2014). Ribosomal database project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. (Online) 42 (Database issue), D633–D642. doi: 10.1093/nar/gkt1244
Croucher N. J., Page A. J., Connor T. R., Delaney A. J., Keane J. A., Bentley S. D., et al. (2015). Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using gubbins. Nucleic Acids Res. 43 (3), e15. doi: 10.1093/nar/gku1196
Dominguez-Bello M. G., Costello E. K., Contreras M., Magris M., Hidalgo G., Fierer N., et al. (2010). Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. 107 (26), 11971–11975. doi: 10.1073/pnas.1002601107
France M., Alizadeh M., Brown S., Ma B., Ravel J. (2022a). Towards a deeper understanding of the vaginal microbiota. Nat. Microbiol. 7 (3), 367–378. doi: 10.1038/s41564-022-01083-2
France M. T., Brown S. E., Rompalo A. M., Brotman R. M., Ravel J. (2022c). Identification of shared bacterial strains in the vaginal microbiota of related and unrelated reproductive-age mothers and daughters using genome-resolved metagenomics. PloS One 17 (10), e0275908. doi: 10.1371/journal.pone.0275908
France M., Ma B., Ravel J. (2022b). Persistence and In vivo evolution of vaginal bacterial strains over a multiyear time period. mSystems 7 (6), e0089322. doi: 10.1128/msystems.00893-22
Gibson B., Wilson Daniel J., Feil E., Eyre-Walker A. (2018). The distribution of bacterial doubling times in the wild. Proc. R. Soc. B: Biol. Sci. 285 (1880), 20180789. doi: 10.1098/rspb.2018.0789
Group J.C.H.M.P.D.G.W. (2010) 16S 454 sequencing protocol HMP consortium. Available at: https://www.hmpdacc.org/hmp/doc/16S_Sequencing_SOP_4.2.2.pdf.
Hickey R. J., Zhou X., Settles M. L., Erb J., Malone K., Hansmann M. A., et al. (2015). Vaginal microbiota of adolescent girls prior to the onset of menarche resemble those of reproductive-age women. mBio 6 (2), e00097–e00015. doi: 10.1128/mBio.00097-15
Human Microbiome Project Consortium, H.M.P. (2012). Structure, function and diversity of the healthy human microbiome. Nature 486 (7402), 207–214. doi: 10.1038/nature11234
Kim S. J., Kim S. J., Han K.-T., Park E.-C. (2017). Medical costs, cesarean delivery rates, and length of stay in specialty hospitals vs. non-specialty hospitals in south Korea. PloS One 12 (11), e0188612. doi: 10.1371/journal.pone.0188612
Klatt N. R., Cheu R., Birse K., Zevin A. S., Perner M., Noël-Romas L., et al. (2017). Vaginal bacteria modify HIV tenofovir microbicide efficacy in African women. Science 356 (6341), 938. doi: 10.1126/science.aai9383
Korem T., Zeevi D., Suez J., Weinberger A., Avnit-Sagi T., Pompan-Lotan M., et al. (2015). Growth dynamics of gut microbiota in health and disease inferred from single metagenomic samples. Science 349 (6252), 1101–1106. doi: 10.1126/science.aac4812
Kozich J. J., Westcott S. L., Baxter N. T., Highlander S. K., Schloss P. D. (2013). Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq illumina sequencing platform. Appl. Environ. Microbiol. 79 (17), 5112–5120. doi: 10.1128/AEM.01043-13
Kurtz S., Phillippy A., Delcher A. L., Smoot M., Shumway M., Antonescu C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5 (2), R12. doi: 10.1186/gb-2004-5-2-r12
Lee Y. K., Ho P. S., Low C. S., Arvilommi H., Salminen S. (2004). Permanent colonization by Lactobacillus casei is hindered by the low rate of cell division in mouse gut. Appl. Environ. Microbiol. 70 (2), 670. doi: 10.1128/AEM.70.2.670-674.2004
Lee J. E., Lee S., Lee H., Song Y. M., Lee K., Han M. J., et al. (2013). Association of the vaginal microbiota with human papillomavirus infection in a Korean twin cohort. PloS One 8 (5), e63514. doi: 10.1371/journal.pone.0063514
Li H., Durbin R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25 (14), 1754–1760. doi: 10.1093/bioinformatics/btp324
Madan J. C., Hoen A. G., Lundgren S. N., Farzan S. F., Cottingham K. L., Morrison H. G., et al. (2016). Association of cesarean delivery and formula supplementation with the intestinal microbiome of 6-week-old infants. JAMA Pediatr. 170 (3), 212–219. doi: 10.1001/jamapediatrics.2015.3732
McClelland R. S., Lingappa J. R., Srinivasan S., Kinuthia J., John-Stewart G. C., Jaoko W., et al. (2018). Evaluation of the association between the concentrations of key vaginal bacteria and the increased risk of HIV acquisition in African women from five cohorts: a nested case-control study. Lancet Infect. Dis. 18 (5), 554–564. doi: 10.1016/S1473-3099(18)30058-6
McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., et al. (2010). The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20 (9), 1297–1303. doi: 10.1101/gr.107524.110
Morgulis A., Coulouris G., Raytselis Y., Madden T. L., Agarwala R., Schäffer A. A. (2008). Database indexing for production MegaBLAST searches. Bioinformatics 24 (16), 1757–1764. doi: 10.1093/bioinformatics/btn322
O’Hanlon D. E., Moench T. R., Cone R. A. (2013). Vaginal pH and microbicidal lactic acid when lactobacilli dominate the microbiota. PloS One 8 (11), e80074. doi: 10.1371/journal.pone.0080074
Picard. Available at: https://broadinstitute.github.io/picard/.
Pon A., Marcu A., Arndt D., Grant J. R., Sajed T., Liang Y., et al. (2016). PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44 (W1), W16–W21. doi: 10.1093/nar/gkw387
Ravel J., Gajer P., Abdo Z., Schneider G. M., Koenig S. S. K., McCulle S. L., et al. (2011). Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. 108 (Supplement 1), 4680–4687. doi: 10.1073/pnas.1002611107
Rutayisire E., Huang K., Liu Y., Tao F. (2016). The mode of delivery affects the diversity and colonization pattern of the gut microbiota during the first year of infants' life: a systematic review. BMC Gastroenterol. 16 (1), 86. doi: 10.1186/s12876-016-0498-0
Schloss P. D. (2009). A high-throughput DNA sequence aligner for microbial ecology studies. PloS One 4 (12), e8230. doi: AEM.01541-09
Schloss P. D., Westcott S. L. (2011). Assessing and improving methods used in operational taxonomic unit-based approaches for 16S rRNA gene sequence analysis. Appl. Environ. Microbiol. 77 (10), 3219–3226. doi: 10.1128/AEM.02810-10
Schloss P. D., Westcott S. L., Ryabin T., Hall J. R., Hartmann M., Hollister E. B., et al. (2009). Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75 (23), 7537–7541. doi: 10.1128/aem.01541-09
Seekatz A. M., Theriot C. M., Molloy C. T., Wozniak K. L., Bergin I. L., Young V. B. (2015). Fecal microbiota transplantation eliminates Clostridium difficile in a murine model of relapsing disease. Infection Immun. 83 (10), 3838–3846. doi: 10.1128/iai.00459-15
Si J., You H. J., Yu J., Sung J., Ko G. (2016). Prevotella as a hub for vaginal microbiota under the influence of host genetics and their association with obesity. Cell Host Microbe 21 (1), 97–105. doi: 10.1016/j.chom.2016.11.010
Stamatakis A. (2014). RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinf. (Oxford England) 30 (9), 1312–1313. doi: 10.1093/bioinformatics/btu033
Sun S., Serrano M. G., Fettweis J. M., Basta P., Rosen E., Ludwig K., et al. (2022). Race, the vaginal microbiome, and spontaneous preterm birth. mSystems 7 (3), e0001722. doi: 10.1128/msystems.00017-22
Tachedjian G., O’Hanlon D. E., Ravel J. (2018). The implausible “in vivo” role of hydrogen peroxide as an antimicrobial factor produced by vaginal microbiota. Microbiome 6 (1), 29. doi: 10.1186/s40168-018-0418-3
Tamarelle J., Thiébaut A. C. M., de Barbeyrac B., Bébéar C., Ravel J., Delarocque-Astagneau E. (2019). The vaginal microbiota and its association with human papillomavirus, Chlamydia trachomatis, Neisseria gonorrhoeae and Mycoplasma genitalium infections: a systematic review and meta-analysis. Clin. Microbiol. Infection 25 (1), 35–47. doi: 10.1016/j.cmi.2018.04.019
Tamburini S., Shen N., Wu H. C., Clemente J. C. (2016). The microbiome in early life: implications for health outcomes. Nat. Med. 22, 713. doi: 10.1038/nm.4142
Thomas-White K., Forster S. C., Kumar N., Van Kuiken M., Putonti C., Stares M. D., et al. (2018). Culturing of female bladder bacteria reveals an interconnected urogenital microbiota. Nat. Commun. 9 (1), 1557. doi: 10.1038/s41467-018-03968-5
Wampach L., Heintz-Buschart A., Fritz J. V., Ramiro-Garcia J., Habier J., Herold M., et al. (2018). Birth mode is associated with earliest strain-conferred gut microbiome functions and immunostimulatory potential. Nat. Commun. 9 (1), 5091. doi: 10.1038/s41467-018-07631-x
Wang J., Dong X., Shao Y., Guo H., Pan L., Hui W., et al. (2017). Genome adaptive evolution of Lactobacillus casei under long-term antibiotic selection pressures. BMC Genomics 18 (1), 320. doi: 10.1186/s12864-017-3710-x
Wang Q., Garrity G. M., Tiedje J. M., Cole J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73 (16), 5261–5267. doi: 10.1128/aem.00062-07
Westcott S. L., Schloss P. D. (2015). De novo clustering methods outperform reference-based methods for assigning 16S rRNA gene sequences to operational taxonomic units. PeerJ 3, e1487. doi: 10.7717/peerj.1487
Keywords: vaginal microbiota, vertical transmission, microbiome, 16S rRNA gene sequences, Lactobacillus crispatus genomics, mother-daughter vaginal microbiota
Citation: Bassis CM, Bullock KA, Sack DE, Saund K, Pirani A, Snitkin ES, Alaniz VI, Quint EH, Bell JD and Young VB (2023) Vaginal microbiota of adolescents and their mothers: A preliminary study of vertical transmission and persistence. Front. Microbiomes 2:1129394. doi: 10.3389/frmbi.2023.1129394
Received: 21 December 2022; Accepted: 07 March 2023;
Published: 24 March 2023.
Edited by:
Marius Vital, Hannover Medical School, GermanyReviewed by:
Cornelia Gottschick, Martin Luther University of Halle-Wittenberg, GermanyMichael France, University of Maryland, United States
Copyright © 2023 Bassis, Bullock, Sack, Saund, Pirani, Snitkin, Alaniz, Quint, Bell and Young. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vincent B. Young, eW91bmd2aUB1bWljaC5lZHU=
†These authors share senior authorship