 Niokhor Dione
Niokhor Dione Kodjovi D. Mlaga
Kodjovi D. Mlaga Siyi Liang
Siyi Liang Guillaume Jospin
Guillaume Jospin Zara Marfori
Zara Marfori Nancy Alvarado
Nancy Alvarado Elisa Scarsella
Elisa Scarsella Ruchita Uttarwar
Ruchita Uttarwar Holly H. Ganz*
Holly H. Ganz*- AnimalBiome, Oakland, CA, United States
Introduction: Escherichia ruysiae is a newly identified species within the Escherichia genus, yet its presence in domestic animals remains largely unexamined. This study characterizes four isolates detected for the first time in the domestic dog (Canis lupus familiaris), focusing on their phenotypic and genomic features.
Methods: We used culturomic methods to isolate four E. ruysiae isolates that were initially identified as Escherichia coli using MALDI-TOF mass spectrometry. Whole-genome sequencing confirmed that the isolates belonged to E. ruysiae, not E. coli. Phenotypic characterization included enzymatic activity assays and antimicrobial susceptibility testing. Comparative genomic analyses were performed on these four isolates, along with 14 additional E. ruysiae and representative genomes from the five other Escherichia species in order to assess genetic diversity and functional gene distribution.
Results and discussion: All strains exhibited similar enzymatic activities and resistance to clindamycin, erythromycin, and metronidazole. The pangenome analysis revealed that most missing gene orthologs are related to motility followed by metabolism, including synthetases, reductases, phosphatases, permeases, transferases, and epimerases, as well as structural genes like efflux pumps and transporters. Phylogroup typing using the ClermonTyping method identified two main groups within the E. ruysiae species, Clade III and IV. Typical virulence genes associated with E. coli are absent in these strains. The multiple approaches used in this study expand our understanding of the diverse aspects of the recently described species, E. ruysiae.
Introduction
The gut microbiota of humans and animals comprises a diverse array of microbes that play critical roles in digestion, immune system function, and overall health. Advances in sequencing technologies, particularly metagenomics, have greatly enhanced our understanding of microbiome composition, enabling the discovery and characterization of new microorganisms and elucidating their roles in host biology. However, the identification of novel microorganisms remains largely dependent on existing databases of known species, limiting the scope of metagenomics. Only culture-based methods can recover live and previously unknown microorganisms (Ju et al., 2022).
Culturomics has emerged in the last decade as a powerful tool to culture and identify unknown bacteria in humans, animals, and the environment. The method consists of diversifying the culture media and conditions for the same sample by modifying parameters such as incubation time, temperature, pH, and atmosphere, or by adding inhibitory or growth factors such as antibiotics, promoters such as blood, and rumen fluid to culture low abundant and or fastidious populations, followed by the rapid identification by MALDI-TOF mass spectrometry. It enables the culture of enormous numbers of new microorganisms, including new species and genera associated with animal and human health and diseases, providing new perspectives on host–microbe interactions (Lagier et al., 2018, 2024).
We are conducting a major effort to identify and characterize the microbiome of domestic dogs. As part of this initiative we have identified over 400 E. coli isolates, through different steps: MALDI- TOF for initial screening, qPCR for species confirmation, multiplex PCR for phylogrouping (Dogan et al., 2020) and Whole Genome Sequencing (WGS) to classify serotype and pathogenicity groups. Following WGS we discovered a subset of isolates with a distinct core genome compared to the rest. Further analysis using the Average Nucleotide Identity (ANI) confirmed that these isolates represent a different species within the Escherichia genus: E. ruysiae.
Non-Escherichia coli encompasses cryptic clades I, II, III, and IV, and E. ruysiae is the latest species described within the Escherichia genus. This genus also includes four other species: Escherichia coli, Escherichia albertii, Escherichia fergusonii, and Escherichia marmotae. The first E. ruysiae strain was isolated from a fecal sample collected in the Netherlands from a human (Homo sapiens) who may have acquired the strain while traveling internationally for a month in Asia and developed traveler’s diarrhea (van der Putten et al., 2021). So far, its dissemination and potential role in human and animal health is not well characterized. Escherichia ruysiae was subsequently isolated from a fecal sample collected from a Lohmann Brown layer hen (Gallus gallus domesticus) on a farm in England (Thomson et al., 2022) and from a fecal sample collected from an urban dwelling large-billed crow (Corvus macrorhynchos) in Japan (Sakaguchi et al., 2023).
In this context of wide dissemination, it is important to understand the genomic features that underline their spread, mechanisms of acquisition of antimicrobial and virulence genes, and the genomic characteristics that support their evolution. Here we describe the phenotypic and genomic features of these new isolates from the domestic dog and perform a comparative genomics analysis with representative species of other Escherichia available in the public repository NCBI.
Materials and methods
Sample information, culture, and identification
The stool sample used in this study was collected from a healthy spayed female 9-year-old Shepherd-Husky mix living in Oakland, California, United States. After collection, the samples were transported to the laboratory and homogenized using BIOME-Preserve AS-930 (Anaerobe Systems, Morgan Hill, CA) in a one-to-one ratio. This sample was processed as part of our culturomics efforts to characterize the microbiome of companion pets by culturing, isolating, identifying, and characterizing bacteria. The E. ruysiae strains that are the focus of this study were isolated following culturomics protocols as previously described by Lagier et al. (2012). Briefly, 100 μL of the aliquoted sample was serially diluted (10−1–10−10) in Dilution Blank AS-9183 (Anaerobe Systems, Morgan Hill, CA) then 70 μL of each dilution was inoculated into a MacConkey agar plate (Becton Dickinson, Franklin Lakes, NJ 07417), and incubated aerobically at 37°C for 24 h. Following standard procedures, grown colonies were first identified using the MALDI–TOF mass spectrometer (MS). A scrapped colony is deposited on a disposable MALDI-TOF MS target plate (Bruker Daltonics, Bremen, Germany) and covered first with 2 μL of 70% formic acid solution and dried before being covered by 2 μL of a MALDI matrix solution (saturated α-cyano acid-4- hydroxycinnamic in 50% acetonitrile, 47.5% HPLC water, and 2.5% trifluoroacetic acid). After drying, analysis was performed using MALDI Biotyper smart System RUO microflex LT/SH smart with the BIDAL database v12 (Bruker Daltonics, Bremen, Germany). A triplex PCR was carried out using the Clermont typing method (Clermont et al., 2000) followed by whole genome sequencing using the Pacific Biosciences Sequel IIe.
Phenotypic characterization
The newly isolated bacterial strains were identified first using the MALDI-TOF MS instrument (Bruker Daltonics, Bremen, Germany) as per a previous study (Asare et al., 2023). Additionally, bacterial characterization was conducted utilizing the Gram staining technique (Sigma Aldrich, MA, United States), while motility was observed using the hanging drop method under a light microscope (Accu-Scope, NY, United States), as described in earlier studies (Jain et al., 2020). The catalase, oxidase (bioMérieux, Marcy-l’Étoile, France), and biochemical tests API ZYM (bioMérieux, Marcy-l’Étoile, France) were conducted to study the bacterial phenotypic characteristics. Furthermore, these strains were also visualized to characterize their morphology and structure. Fixed bacteria were placed on a 400 mesh Formvar-coated copper grid that had been previously subjected to glow discharge (Electron Microscopy Sciences, PA, United States). Following a settlement period of 2 min for the bacteria, the grids were thoroughly rinsed with UranyLess EM Stain (Electron Microscopy Sciences, PA, United States) and the remaining UranyLess EM Stain was removed. Once dried, the grids were imaged at a voltage of 120 kV using a Tecnai 12 (Thermo Fisher Scientific, Waltham, MA, United States) Transmission Electron Microscope (TEM), located in the Electron Microscope Lab at the University of California, Berkeley.
Antimicrobial susceptibility testing
We used the disk diffusion method to screen the antimicrobial susceptibility of all bacteria isolates. We screened the E. ruysiae isolates against 11 antibiotic drugs, sourced from Thermo Fisher Scientific (Waltham, MA, United States), using the following disks: Metronidazole 50 μg, Cefpodoxime 10 μg, Enrofloxacin 5 μg, Erythromycin 15 μg, Amoxicillin/Clavulanic acid 30 μg, Tetracycline 30 μg, Sulfamethoxazole/Trimethoprim 25 μg, Doxycycline 30 μg, Clindamycin 2 μg, Gentamicin 10 μg, and Chloramphenicol 30 μg. We inoculated E. ruysiae isolates onto Columbia agar plates and incubated them at 37°C for 24 h. Pure colonies were introduced into Dilution Blank AS-9183 (Anaerobe Systems, Morgan Hill, CA) to prepare a 0.5 McFarland bacterial suspension. This suspension was then used to inoculate Mueller Hinton agar plates Becton Dickinson BBL™ (Franklin Lakes, NJ, United States) using a wet swab following the SLCI guidelines. Antibiotic disks were then placed on the surface of the plates with the aid of a Sensi-disk self-tamping 8-place dispenser Becton Dickinson BBL™ (Franklin Lakes, NJ, United States). The plates were incubated at 37°C for 24 h and inhibition zones were read by BIOMIC V3 (Giles Scientific in Santa Barbara, CA, United States). Each isolate was tested in triplicate.
DNA extraction and whole genome sequencing
We extracted DNA using the ZymoBIOMICS™ DNA Miniprep Kit for bacterial cells (Zymo Research, Irvine, CA, United States). The samples were first prepared by inoculating a bacterial colony into a 2 mL microcentrifuge tube containing 1,000 μL phosphate buffered saline (PBS) (Thermo Fisher Scientific, Waltham, MA, United States) and centrifuging at 5,000 RPM for 10 min. The PBS was removed without dislodging the pellet. Twenty μl of 20 mg/mL lysozyme & 750 μL ZymoBIOMICS lysis solution was then added and incubated at 37°C for 40 min. The samples were then transferred to ZR BashingBead™ Lysis tubes and secured in a Vortex-Genie® 2 mixer fitted with a 2 mL tube holder adapter and vortexed at full speed for 40 min. DNA concentration was measured using a Qubit 4 Fluorometer (Thermo Fisher Scientific, Waltham, MA, United States). Genomic DNA was sheared to a 7–12 kb fragment length on a Megaruptor 3 (Diagenode LLC., Denville, NJ, United States). The quality of the DNA was examined on a 2% agarose gel. The sequencing library was prepared following the manufacturer’s protocol using the SMRTbell prep kit 3.0 and and the isolates were barcoded using the SMRTbell barcoded adapter plate 3.0 (Pacific Biosciences, Menlo Park, CA, United States). We loaded 115 μL of the pooled library onto a 96-well sample plate and prepared it for a sequencing run on a Sequel IIe sequencer using Binding kit 3.2, Sequencing kit 2.0, and SMRT Cell 8 M (Pacific Biosciences, Menlo Park, CA, United States). The library was sequenced using a 15-h movie. HiFi data was obtained from the raw subreads using the circular consensus sequencing process.
Comparative genomics
Genome assembly and annotation
All sequencing reads were screened for quality control using Fastqc v0.12.1 (Andrews, 2010). The sequencing reads were filtered with a Phred score > 30, renamed by sample ID, and assembled using Canu assembler v2.3 (Koren, et al. 2017) with default parameters. The final scaffold files produced were screened for level of contamination and purity using CheckM2 v1.0.2 (Chklovski et al., 2023) and taxonomically classified using GTDB-Tk v2.4.0 (RefDB version r220) (Chaumeil et al., 2022). We annotated the four genomes using Prokka v1.14.6 (Seemann, 2014) with an e-value set to 0.00001, a minimum coverage of 95%, using the specific database of the genus Escherichia (Supplementary Table S1). In addition, 14 genomes of E. ruysiae (Supplementary Table S2) and one representative genome of each other species in the Escherichia genus (Supplementary Table S3) were also retrieved from the NCBI Refseq database for comparative analysis purposes and re-annotated using the same methods.
Isolate sequence typing, serogrouping and phylogrouping
We performed an in silico Multi-Locus Sequence Type (MLST) analysis using MLST package, version 2.23.0,1 adapted from https://pubmlst.org (Jolley and Maiden, 2010) using these seven housekeeping genes: adk, fumC, gyrB, icd, mdh, purA, and recA. Escherichia ruysiae species are part of the cryptic clades I, II, III, and IV (Walk, 2015). We used the software ClermonTyping version 1.4.0 (Beghain et al., 2018) to determine the phylogroups of all the genomes.
Pangenome analysis
We performed a pangenome analysis, searching for clusters of ortholog genes using get_homologues v22082022 (Contreras-Moreira and Vinuesa, 2013; Vinuesa and Contreras-Moreira, 2015). We used two approaches to search for ortholog genes, namely OrthoMCL v1.4 (OMCL) (Li et al., 2003) and Clusters of Orthologous Genes (COG) triangles v2.1 (Kurtz et al., 2004; Kristensen et al., 2010) clustering algorithm. Sequence similarity requires an equal Pfam domain, and all clusters detected were reported. The processes were performed locally using default parameters. First we calculated the Average Nucleotide Identity (ANI) and the Percentage of Conserved Proteins (POCPs) of the Escherichia genus in order to determine the level of delineation and to determine the relatedness of the newly isolated four strains to publicly available E. ruysiae genomes. Then in order to support the ANI-based species delineation, we constructed a core genome midpoint-rooted Maximum Likelihood phylogenetic tree of 23 species, including 18 E. ruysiae strains and five representative genomes from other Escherichia species, using 1,000 bootstrap replicates. We computed the core and pan-genomes, including their evolution, composition, and the parsimony tree of E. ruysiae strains using the intersection of both OMCL and COG approaches. In addition, we queried the pangenome to determine accessory (shell) and specific (cloud) ortholog genes of our strains versus others.
Phylogenetic reconstruction, genomics markers, resistome and virulome
We performed a whole and a core genome alignment of all 18 genomes of E. ruysiae using scapper,2 a simple and fast SNP alignment tool that uses mummer v4.0.0 (Kurtz et al., 2004) and Trimal v1.4.rev22 (Capella-Gutiérrez et al., 2009). We reconstructed the phylogenetic evolution of the core genome using FastTree v2.1 (Price et al., 2009). We screened the resistome profile by mapping the full genome of E. ruysiae to various resistance gene databases including NCBI (Feldgarden et al., 2019), and ARGANNOT (Gupta et al., 2014), ResFinder (Zankari et al., 2012), CARD (McArthur et al., 2013) and MEGARes 2.0 (Bonin et al., 2023) to determine genes associated to antimicrobial resistance using ABRicate software.3
Results
Culture, isolation, and phenotypic characterization
Strains AB134, AB135, AB136, and AB137 were isolated in July 2022 after 24 h of incubation on a MacConkey Agar plate (BD) following the culturomics process. A pure colony of each isolate was tentatively identified using MALDI-TOF mass spectrometry (MBT Sirius One RUO; Bruker Daltonics, Bremen, Germany). The four strains were identified as E. coli with a score of 1.8 which is the identification threshold at the genus level. This result was due to the absence of a reference spectrum of E. ruysiae in the Bruker BIDAL database v12 used in the analysis. The triplex PCR also did not allow clear identification of E. ruysiae We performed whole genome sequencing using PacBio sequencing technology and identified the strains AB134, AB135, AB136, AB137 as E. ruysiae with Gtdbtk based on the Average Nucleic Identity (ANI), which were 98.94, 98.93, 98.92, and 97.1%, respectively. Gram staining performed using the Sigma Aldrich Gram Staining Kit (St. Louis MI, United States) showed Gram-negative bacilli rod-shaped (Figure 1A). After identification, we performed a protein extraction as recommended by Bruker to generate a reference spectrum (Figure 1B) using 12 individual colonies of strains AB134 grown on in-house LB agar (Waltham, MA, United States) made from a dehydrated medium.
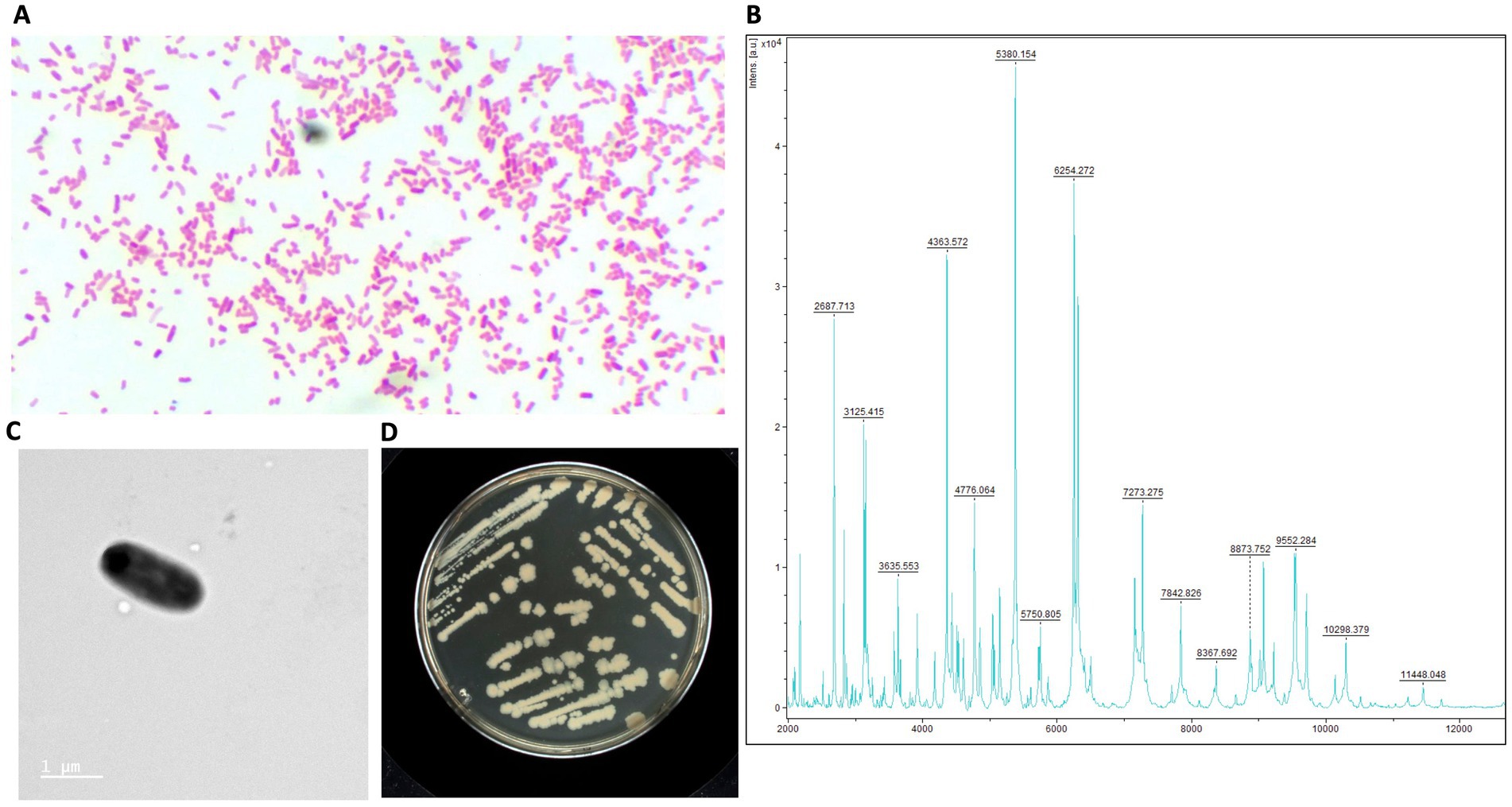
Figure 1. Structural and phenotypic characterization of E. ruysiae (strain AB135). (A) Gram stain of colonies under 100X light microscopy, (B) MALDI-TOF spectrum, (C) Electronic microscopy with rod-shape bacterium, (D) Colony morphology and color on LB agar.
The rod-shaped bacilli form was confirmed by electron microscopy, where strain AB134 exhibited a mean length of 1.8 μm and width of 0.6 μm (Figure 1C). When grown on MacConkey agar plates, E. ruysiae exhibit irregular dark white colonies with a diameter of about 0.3–0.8 mm (Figure 1D). Evaluation of enzymatic activity of E. ruysiae strains AB134, AB135, AB136, and AB137 using API ZYM revealed that all strains exhibited similar enzymatic activities with positive reactions observed with alkaline phosphatase, leucine arylamidase, acid phosphatase, naphthol-AS-BI-phosphohydrolase α-galactosidase, ß-galactosidase, and α-glucosidase. All E. ruysiae show negative activity on the other hand with the following enzymes: esterase (C4), esterase lipase (C8), lipase (C14), valine arylamidase, cystine arylamidase, trypsin, α-chymotrypsin, α-galactosidase, ß-glucuronidase, ß-glucosidase, N-acetyl-ß-glucosaminidase, α-mannosidase, and α-fucosidase (Table 1). The API 20E profile evaluating amino acids, salts, activity, and carbon source shows Positive activity for 2-nitrophenyl-ȕD galactopyranoside, L-ornithine, sodium pyruvate, D-glucose, D-mannitol, D-sorbitol, L-rhamnose, and L-arabiose, and negative activity for L-arginine, L-lysine, trisodium citrate, sodium thiosulfate, urea, L-tryptophan, gelatin (bovine origin), D-saccharose, D-melibiose, and amygdalin (Table 1).
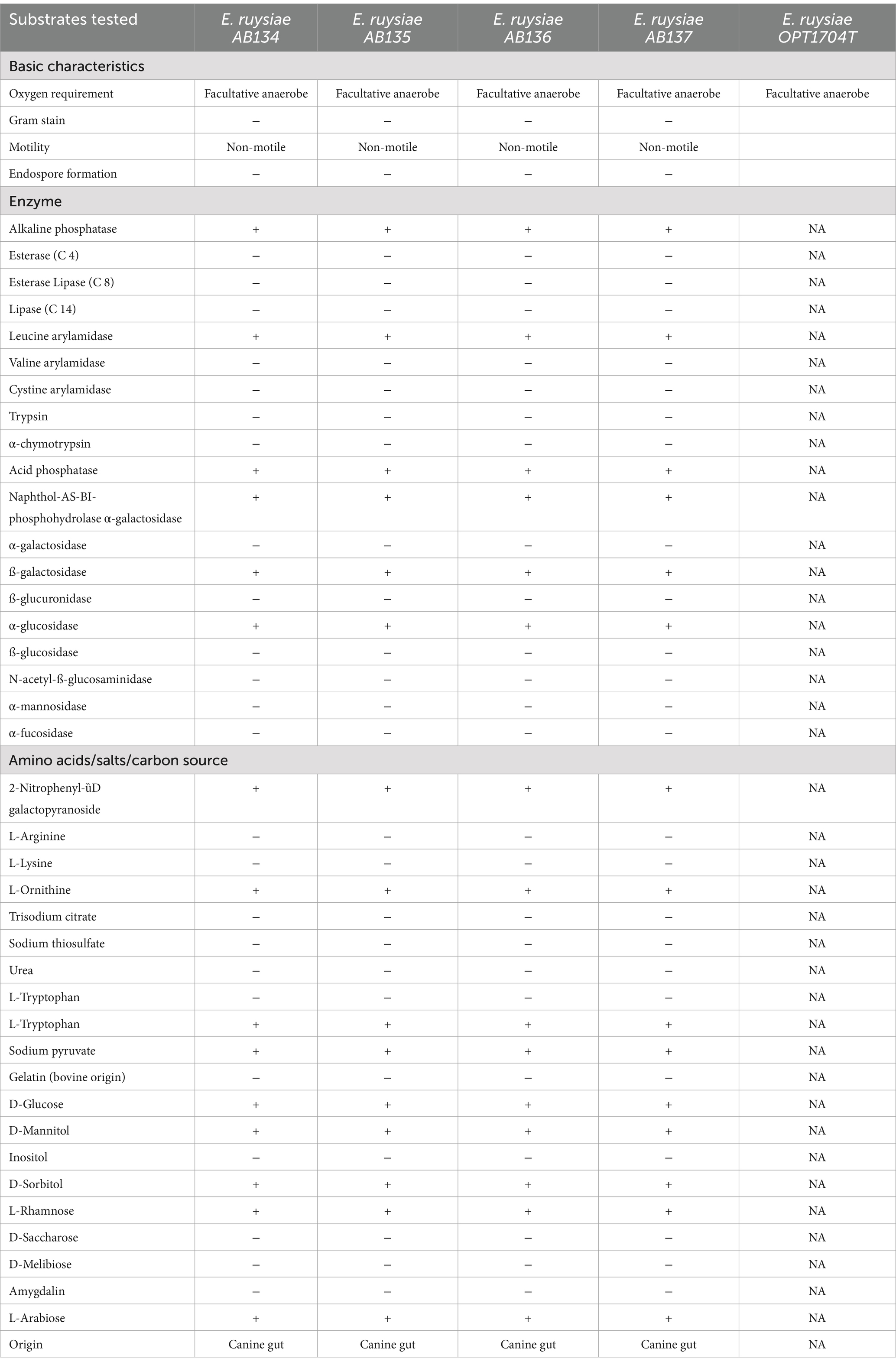
Table 1. Structure, morphology, and phenotypic characteristics of E. ruysiae, strains AB134, AB135, AB136, and AB137. The type strain OPT1704T is listed here to indicate that it was not characterized phenotypically.
From the disk diffusion method, these E. ruysiae strains exhibited susceptibility to cefpodoxime, enrofloxacin, chloramphenicol, tetracycline, doxycycline, trimethoprim-sulfamethoxazole, and gentamicin. The strains exhibited resistance to clindamycin, erythromycin, and metronidazole, while showing an intermediate response for amoxicillin-clavulanate (Table 2; Figure 2).
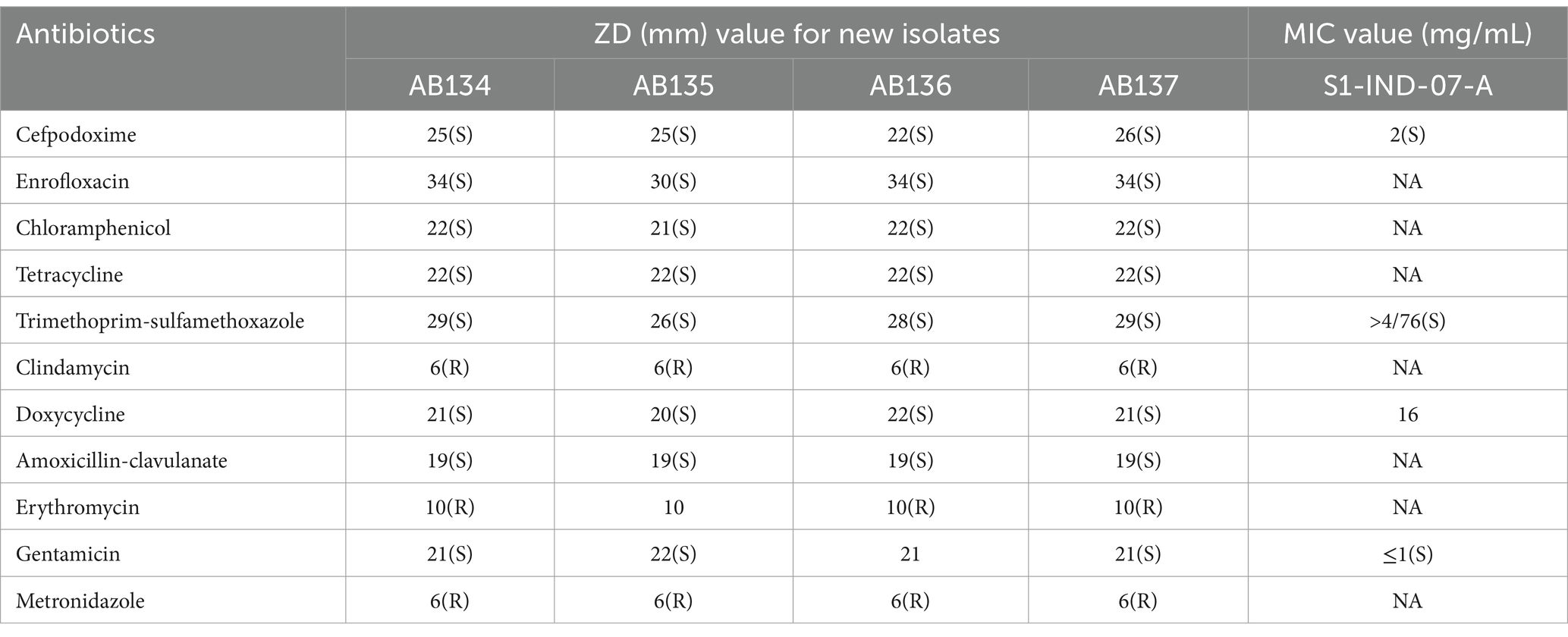
Table 2. Antimicrobial Susceptibility testing results of E. rusyiae strains AB134, AB135, AB135, AB136 compared to E. ruysiae strains S1-IND-07-A (Campos-Madueno et al., 2023).
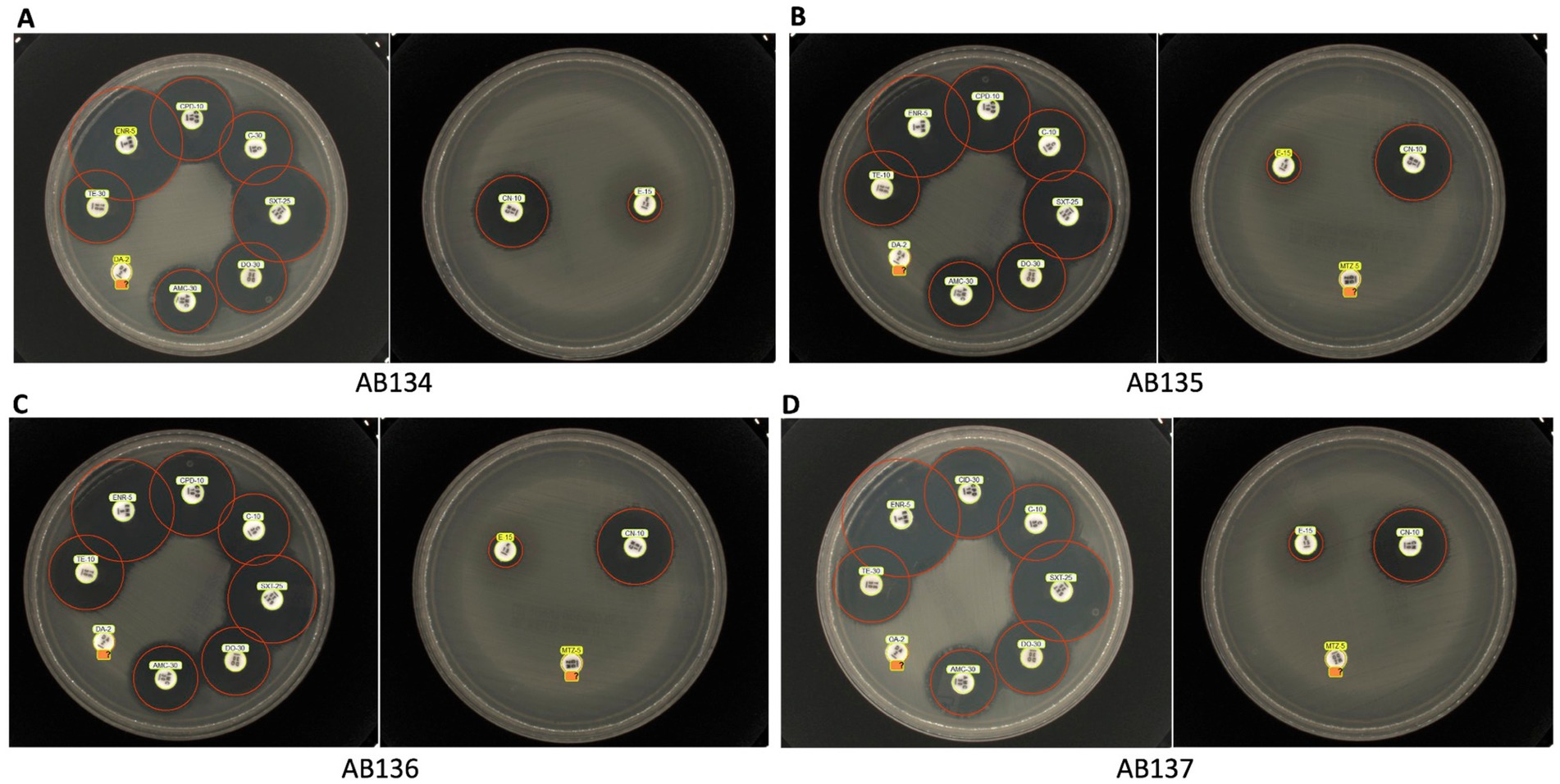
Figure 2. Antimicrobial susceptibility testing of E. ruysiae isolates. The figure shows results for multiple isolates AB134 (A), AB135 (B), AB136 (C), and AB137 (D) against trimethoprim-sulfamethoxazole, cefpodoxime, enrofloxacin, chloramphenicol, tetracycline, doxycycline, amoxicillin-clavulanate, gentamicin, clindamycin, erythromycin, and metronidazole (with the exception that metronidazole susceptibility was not tested for AB134).
Genome features, taxono-genomics, and genomic description
Genomes of the four strains were submitted and referenced in the NCBI database with the respective accession numbers: E. ruysiae strain AB134 (JAVIWR000000000), E. ruysiae strain AB135 (JAVIWS000000000), E. ruysiae strain AB136 (JAVIWT000000000), and E. ruysiae strain AB137 (JAVIWU000000000). The genomes were assembled, annotated, and genome features described (Table 3). All genomes are circularized in one contig of 4.5 Mb average size and 95 kb of circularized plasmid. We checked the quality of the assemblies with CheckM which indicated 100% completeness with an average GC% of 50.6%. Genomic accession numbers, bioproject number, and links for the four new E. ruysiae genomes are provided in Supplementary material (Supplementary Table S1). The genomes were taxonomically classified using Gtdbtk and the results indicated that all four genomes are identified as E. ruysiae with a minimum of 97.1% of Average Nucleotide Identity (ANI) and 0.95 of Alignment Fraction (AF) with E. ruysiae GCF_902498915.1 (Supplementary material). We computed the overall ANI of all the available genomes of E. ruysiae including reference genomes of E. coli, E. marmotae, E albertii, E. fergusonii, and E. whittamii using the OMCL ortholog finding algorithm in get_homologes with default parameters. The heatmap (Figure 3A) indicates that all the E. ruysiae (red square) forms a distinct cluster, separate from other Escherichia species (white to blue). Additionally, E. ruysiae are clustered in two distinct groups, with a minimum of 97.47% and a maximum of 98.6% ANI. The four strains sequenced in this study AB134, AB135, AB136, and AB137 belong to the same group with ANI ranging from 98.94 to 97.6%. The closest related species is E. coli NC000913 with 95% on average and the most distant species is E. fergusonii strain FDAARGOS 1499 (ANI: 92.41–92.51%). The core genome midpoint-rooted Maximum Likelihood tree indicates that all E. ruysiae strains are closely related, forming two distinct clusters separate from the other species (Figure 3B). In contrast, E. coli, E. marmotae, E. whittamii, E. albertii, and E. fergusonii exhibit more distant relationships.
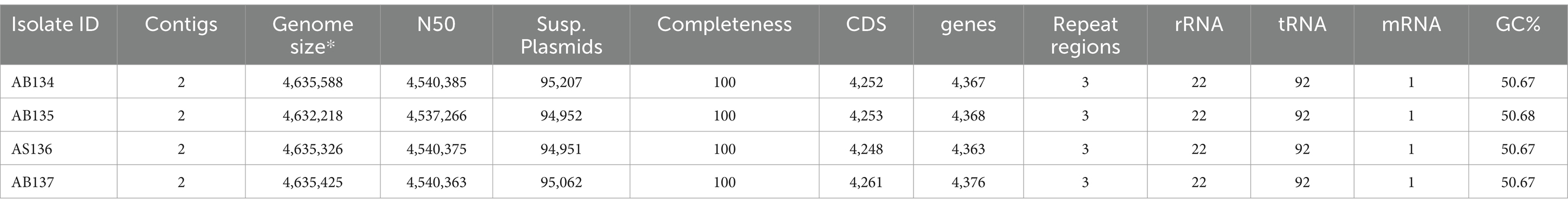
Table 3. E. ruysiae genomes features from genome assembly and annotation.
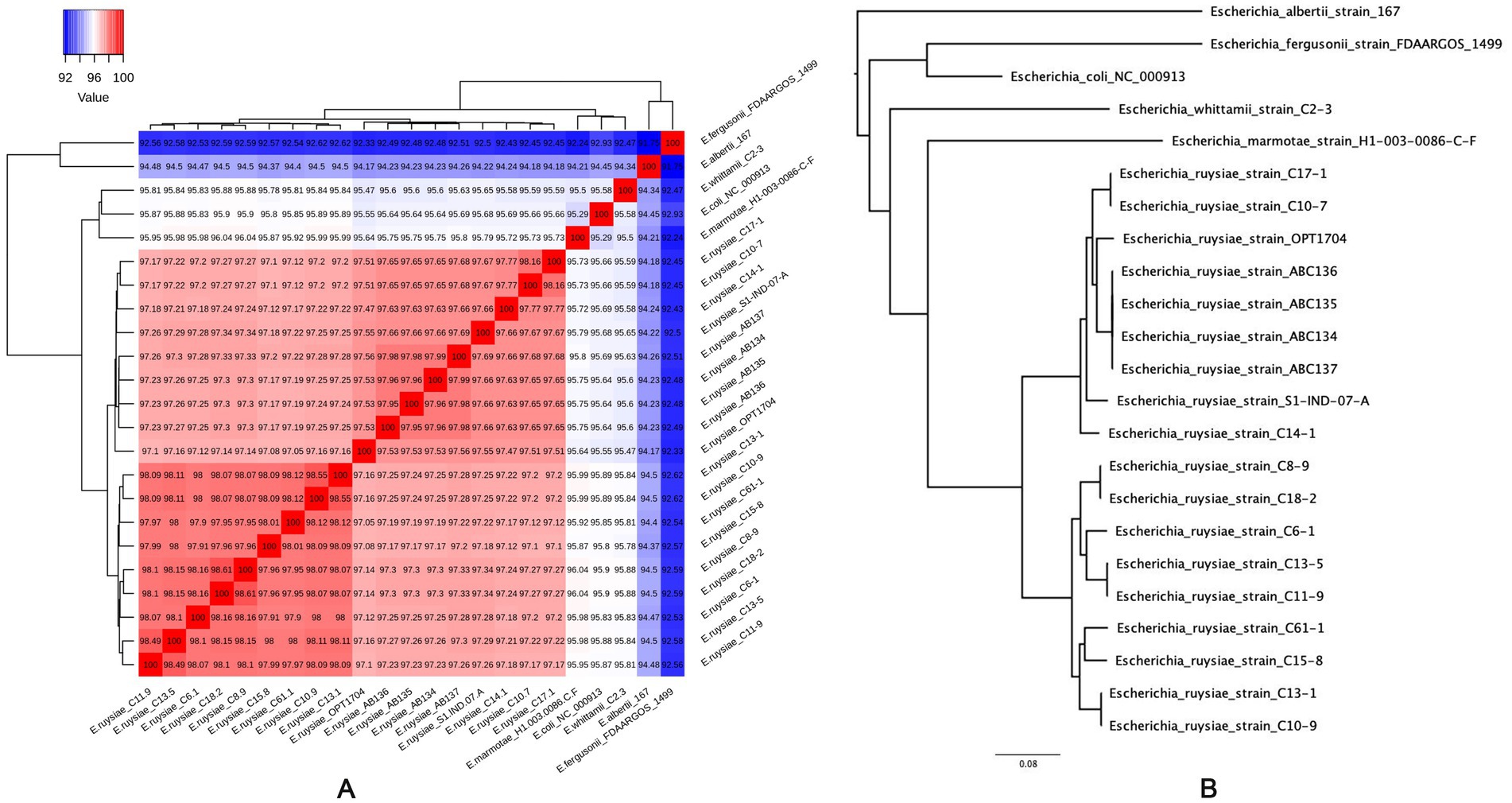
Figure 3. Average Nucleotide Identity (ANI) of E. ruysiae, E. coli, E. marmotae, E. whittami, E. albertii, and E. fergusonii. (A) Heatmap with overall ANI of all the available genomes of E. ruysiae. (B) Midpoint rooting maximum likelihood tree based on core genome alignment (RAxML best tree).
Pangenome description of E. ruysiae species
We clustered the genes from all genomes into ortholog genes using get homolog. From the 18 publicly available genomes (Supplementary Table S2), we identified 6,714 total genes from both OMCL (173 specifics) and COG (186 specifics) with 3,231 genes included in the hard core genes (present in all 18 genomes) and 3,330 genes in the soft core (at least 17 genomes possess these genes). One thousand four hundred eighty-five genes were shared across 10 to 95% of the genomes (accessory or shell genes) and 1899 genes are specific to each genome (cloud genes). The hard core genome estimation using the Tettelin fit converged with residual standard error = 100.76 and fitting value from 4,105 genes to 3,284 following the equation:
Using the Willenbrock method, the residual error was estimated to be 95.08 with the fitting value from 4,142 to 3,256 following the equation (Figure 4A):
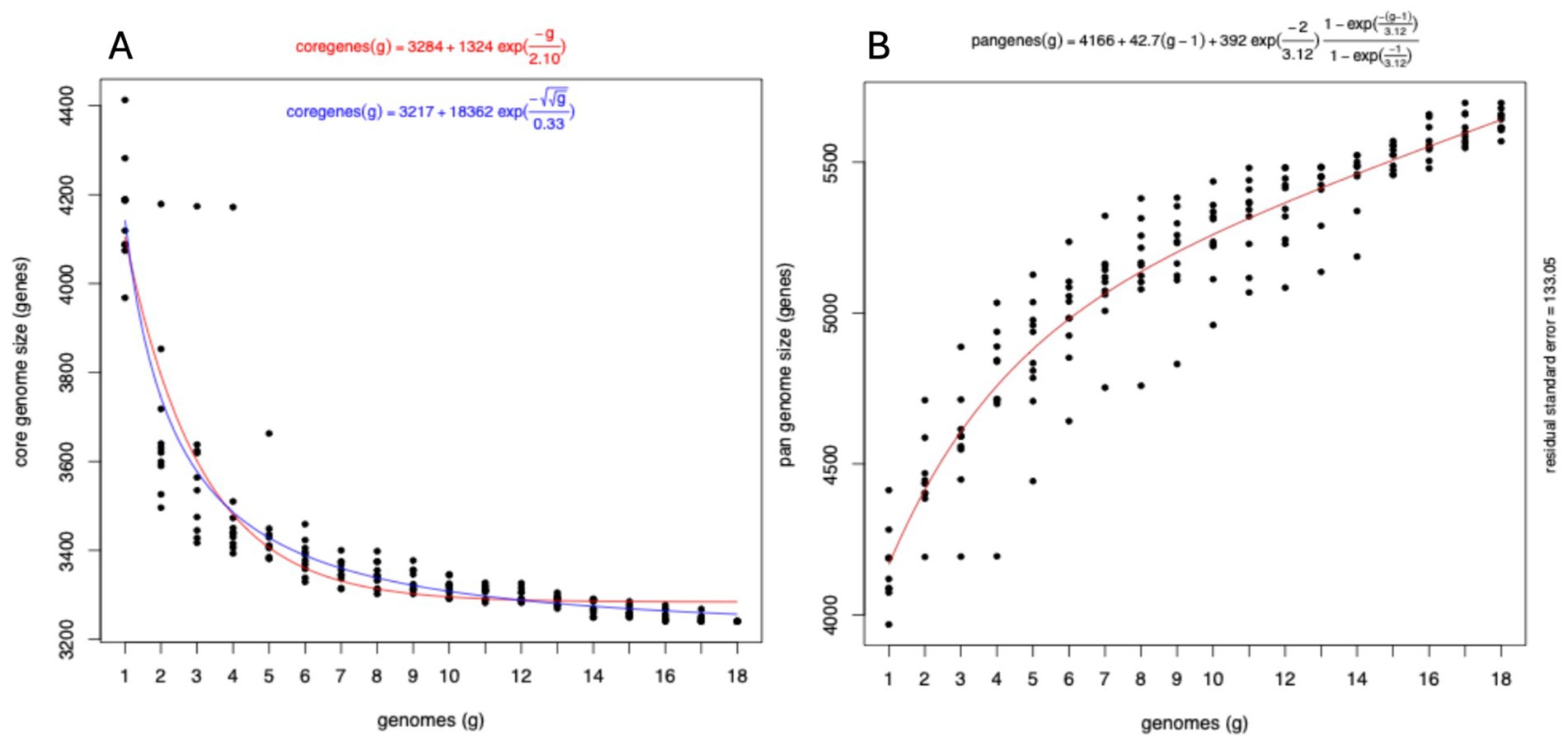
Figure 4. Evolution of the pangenome of E. ruysiae. (A) Hard core genome estimation using the Tettelin fit with residual error estimated using the Willenbrock method. (B) Pangenome fit is consistent with an open pangenome.
Tettelin fitting methods revealed that the variation in gene numbers from the 16th genome onwards is negligible, with only four additional genes identified after the 11th genome. This suggests that the hard core genome has reached a stable state and undergoes minimal variation. The pan-genome also fit converged with the estimated residual error of 133.05 following the curve equation of
This equation shows the pangenome size keeps increasing indefinitely when adding a new genome, hence is interpreted as representing an open pangenome (Figure 4B). The four genomes sequenced for this study: AB134, AB135, AB136, and AB137 have altogether 147 unique groups of genes including accessory and unique genes, and are missing 923 groups of genes. The unique group of genes includes genes associated with virulence such as invasin (group 1,338), type II secretion system protein J, G, F, and E (xcpW, epsG, epsF, and epsE respectively), flagella-associated system genes (flgL, flgH, flgG, flgH, flgE, flgC, tagD, fliI, fliE, vnfA, fliP, flihB), flagellin, and adhesin BmaC. we also identified groups of genes associated with evolution mechanisms that include transposon and phage transfer and acquisition [Insertion sequence family transposase (ISSen1 and ISEc48 among many others), endonuclease and endoribonuclease, recombinase, prophage integrase (IntS)]. The majority of missing groups of genes are motility-associated genes and metabolism-associated genes including synthetases, reductases, phosphatases, permeases, transferases, and epimerases, as well as structural-related genes such as efflux pumps and transporters (Supplementary Figure S1).
Phylogenetic analysis and evolution of E. ruysiae species
Phylogroup typing using the ClermonTyping method showed that there are two main groups within the E. ruysiea species, namely Clade III characterized by genes trpA, trpBA, and chuIII and Clade IV characterized by the genes trpBA, chuIV, and facultatively trpAgpC. All typical genes for E. coli typing were absent (arpA, chuA, yjaA, and TspE4.C2). All genomes sequenced for this study belong to Clade IV. The topology of the estimated phylogenetic tree, based on the core genome, shows two distinct clades positioned on separate branches with a midpoint rooting layout (Figure 5A). The phylogenetic distribution and the clade type are neither geographically- nor host-dependent. Strains isolated from chicken occur in both Clades III and IV while the human and dog strains isolated to date only occur in Clade IV (Figure 5B). All Clade III strains including the sequencing type (ST): ST6540, ST3568, ST9287, ST2371, and ST11513 are located on the top branch, and the Clade IV with ST: ST6467, ST11516, ST4103, ST5792, ST9858 on the bottom branch (Figure 5B). Analysis of the 18 E. ruysiae isolates suggests that phylogenetic distribution and clade type are not strongly associated with geography or host species. However, this pattern likely reflects the limited number of isolates identified to date, given that E. ruysiae is a recently described cryptic species. Expanding efforts to isolate E. ruysiae from diverse hosts across a broad geographical range may provide deeper insights into the evolution of these clades and their adaptations to different environments.
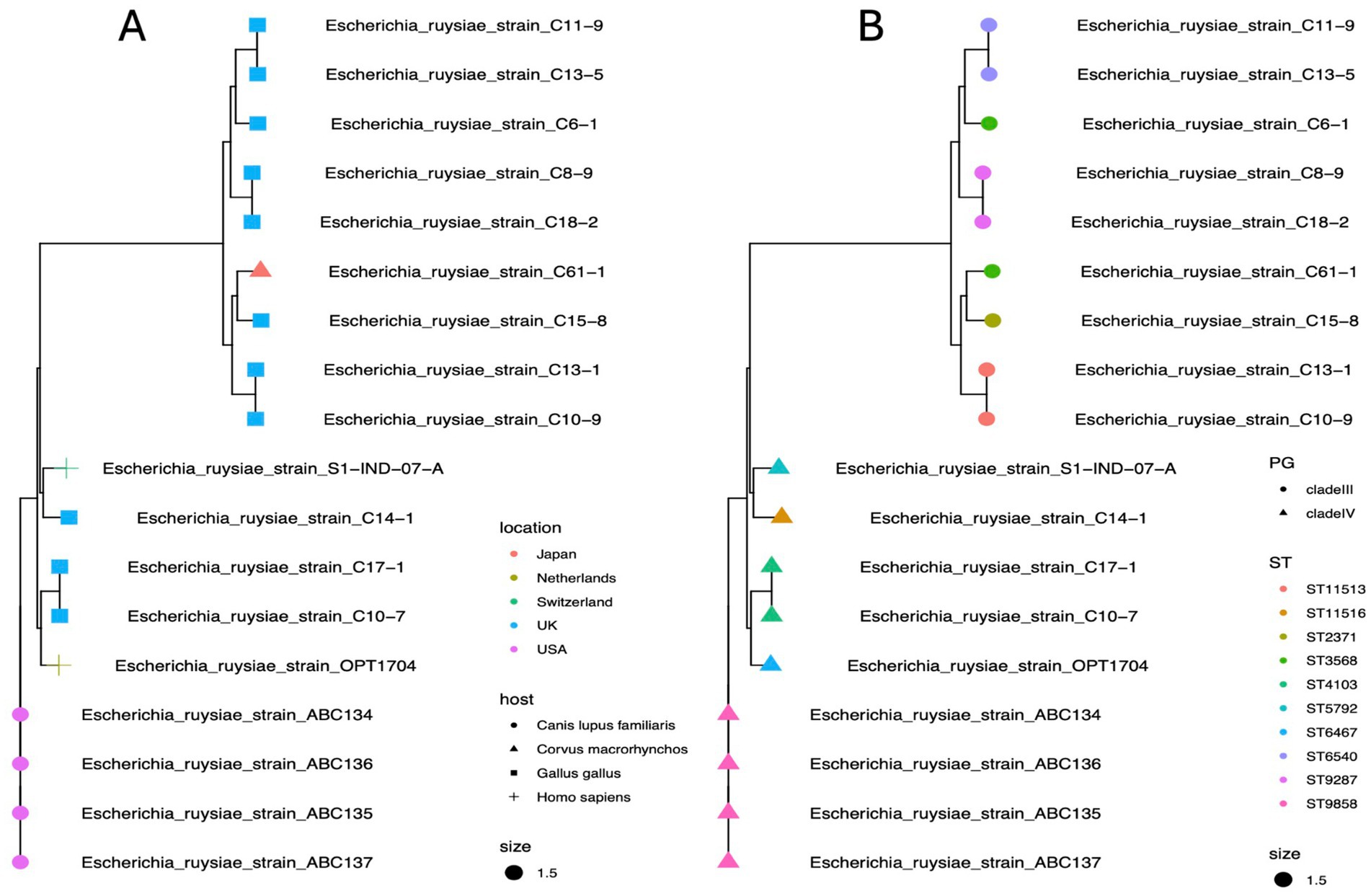
Figure 5. Maximum likelihood phylogenetic reconstruction of E. ruysiae using core genome alignment. (A) Tree showing diversity by geographical location (tip colors) and host (tip shapes). (B) Tree showing diversity by phylogroups (tip shapes) and sequence types (ST, tip colors).
Resistome and virulome description
We identified genes associated with antimicrobial resistance in all genomes. Aminoglycoside resistance genes Agly-aadA (aminoglycoside-3-adenyltransferase) and penicillin-binding protein (Bla-Pbinding protein) occurred in all strains. Beta-lactamase BlaCTX-14, Bla-CTX-8, and Bla-CTX-15 can be identified among both clades. The strain C6-1 isolated from chicken feces possesses some virulence genes but lacks common resistance genes, suggesting potential for infection but possibly treatable with a wider range of antibiotics. Human strains (OPT1704, S1-IND-07-A) harbor both virulence genes and multiple resistance genes, indicating a potential risk for antibiotic-resistant infections (Figure 6).
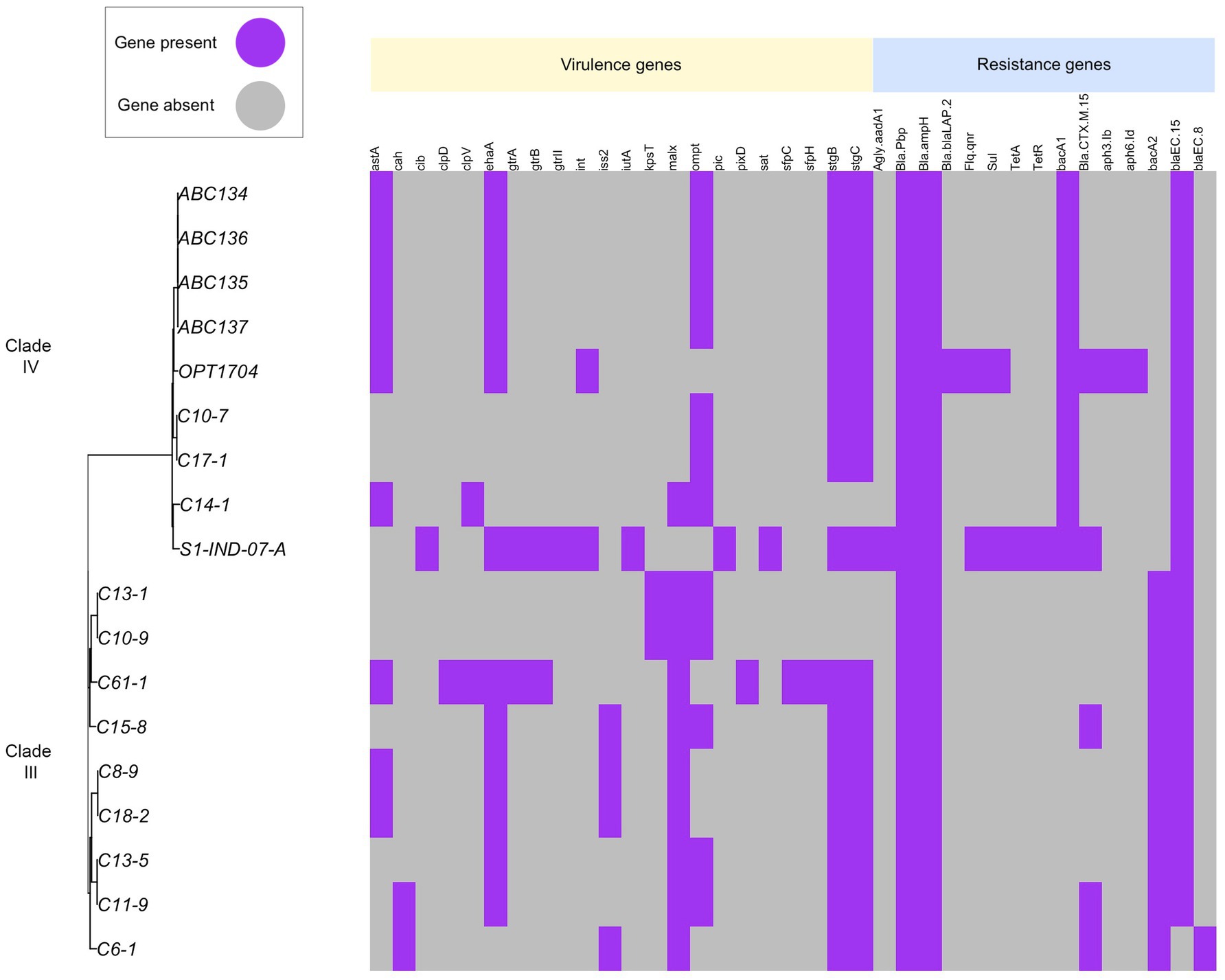
Figure 6. Distribution of virulence genes and antimicrobial resistance genes across E. ruysiae. Strain C6-1 isolated from chicken feces possesses some virulence genes but lacks common resistance genes, suggesting potential for infection but possibly treatable with a relatively wide range of antibiotics. Strains from humans (OPT1704, S1-IND-07-A) harbor both virulence genes and multiple resistance genes, indicating a potentially high risk for antibiotic-resistant infections. Aminoglycoside resistance genes Agly-aadA (aminoglycoside-3-adenyltransferase) and penicillin-binding protein (Bla-Pbinding protein) were found in all strains. Beta-lactamase BlaCTX-14, Bla-CTX-8, and Bla-CTX-15 were identified in both clades.
Discussion
The first successful isolation and culturing of E. ruysiae from the fecal microbiome of a healthy domestic dog in western United States expands our understanding of its host range and geographic distribution. The reported isolation of E. ruysiae from a diversity of animal hosts (a human traveler (van der Putten et al., 2021), an urban wild crow (Sakaguchi et al., 2023), a domestic chicken (Thomson et al., 2022), and now a domestic dog) on three different continents (Europe, Asia, North America) underscores the adaptability of this species and suggests a global distribution. Using culturomic and genomic techniques, we gained new insights into the previously elusive growth requirements and metabolic capabilities of E. ruysiae, findings that may help facilitate future isolation efforts.
All strains isolated in this study grew on MacConkey Agar plates, forming colonies with distinct size and color after 18–24 h aerobic incubation at 37°C, following the culturomics process (Lagier et al., 2018). Previous studies reported that E. ruysiae forms circular, gray-white colonies on a Columbia sheep blood agar plates after overnight incubation at 37°C (Van Der Putten et al., 2021). MALDI-TOF was unable to distinguish between E. coli and E. ruysiae because the spectral profile for E. ruysiae had not yet been included in the databases of both the MALDI-TOF (Bruker Daltonics, Bremen, Germany) and VITEK2 (BioMérieux, Marcy-l’Étoile, France) systems (van der Putten et al., 2021). The differences between these species became apparent only when we compared the genomes of the isolates with those of E. coli. Whole genome sequencing, which confirmed the identification of E. ruysiae, remains the gold standard for accurate taxonomic identification, and highlights the limitations of MALDI-TOF when using an incomplete database. Nonetheless the MALDI-TOF remains an excellent identification tool, particularly for well characterized species and is invaluable for culturing-based microbiome analysis (Demir and Hazırolan, 2024).
A detailed phenotypic characterization of E. ruysiae strains revealed distinctive features that may contribute to the bacterium’s ecological niche. Analysis of biochemical properties and antibiotic susceptibility profiles highlighted the species’ phenotypic diversity. Notably, reduced motility and probable loss of flagella were consistent with the initial description of E. ruysiae (van der Putten et al., 2021) and may represent an adaptation to a commensal lifestyle. Similar loss of flagellar motility was observed in an experimental evolution study where non-symbiotic E. coli evolved into an insect mutualist (Koga et al., 2022). Additional virulence-associated traits warrant further investigation to better understand the ecological relevance and potential pathogenicity of E. ruysiae across different environments.
Comparative analysis of the antimicrobial susceptibility of our strains and E. ruysiea strain S1-IND-07-A from Campos-Madueno et al. (2023), revealed similar susceptibility to gentamicin, cefpodoxime, and trimethoprim-sulfamethoxazole., Importantly, the antimicrobial resistance findings in our study are not too concerning from a public health perspective because only the Bla-CTX-15 gene was detected in the genomes of the four isolates. However, all four strains exhibited resistance to clindamycin (lincosamide), erythromycin (macrolide), and metronidazole (nitroimidazole) in the in vitro disk diffusion AST profiles.
Pangenome analysis provided further insights into the genomic diversity and adaptive potential of E. ruysiae. The expansive accessory genome and the exponential pangenome evolution underscore the species’ genomic plasticity and its ability to acquire and integrate genetic materials from diverse sources, thereby shaping its evolutionary trajectory. This plasticity likely supports the organism’s capacity to live and adapt to various environments and niches. Additionally, phylogrouping based on core genome analysis allowed us to categorize E. ruysiae strains into two distinct phylogenetic groups, providing a useful framework for understanding their evolutionary relationships and tracking the geographical and ecological distribution of the species.
In conclusion, our study integrates culturomic, phenotypic, pangenomic, and phylogenetic analyses to provide a robust foundation for further exploration of the ecological, evolutionary, and clinical significance of the recently described species E. ruysiae.
Data availability statement
All four genomes sequenced for the study were submitted and referenced in the NCBI database (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1010808/) with the respective accession numbers E. ruysiae strain AB134 (JAVIWR000000000), E. ruysiae strain AB135 (JAVIWS000000000), E. ruysiae strain AB136 (JAVIWT000000000), and E. ruysiae strain AB137 (JAVIWU000000000).
Ethics statement
The stool sample used to isolate E. ruysiae was obtained non-invasively with the informed consent of the pet owner.
Author contributions
ND: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Supervision, Validation, Writing – original draft, Writing – review & editing. KM: Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. SL: Investigation, Writing – review & editing. GJ: Data curation, Software, Writing – review & editing. ZM: Investigation, Writing – review & editing. NA: Investigation, Writing – review & editing. ES: Investigation, Methodology, Writing – review & editing. RU: Investigation, Writing – review & editing. HG: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by AnimalBiome.
Acknowledgments
The authors thank the pet parents for their consent and contributions to our research on the microbiota in companion animals, including Escherichia spp. In particular, we thank Cindy S. for providing Rosie’s donations, which yielded these interesting findings.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1558802/full#supplementary-material
Footnotes
References
Andrews, S. (2010). FastQC: a quality control tool for high throughput sequence data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
Asare, P. T., Lee, C.-H., Hürlimann, V., Teo, Y., Cuénod, A., Akduman, N., et al. (2023). Corrigendum: a MALDI-TOF MS library for rapid identification of human commensal gut bacteria from the class Clostridia. Front. Microbiol. 14:1208177. doi: 10.3389/fmicb.2023.1208177
Beghain, J., Bridier-Nahmias, A., Le Nagard, H., Denamur, E., and Clermont, O. (2018). ClermonTyping: an easy-to-use and accurate in silico method for Escherichia genus strain phylotyping. Microb. Genom. 4:e000192. doi: 10.1099/mgen.0.000192
Bonin, N., Doster, E., Worley, H., Pinnell, L. J., Bravo, J. E., Ferm, P., et al. (2023). MEGARes and AMR++, v3.0: an updated comprehensive database of antimicrobial resistance determinants and an improved software pipeline for classification using high-throughput sequencing. Nucleic Acids Res. 51, D744–D752. doi: 10.1093/nar/gkac1047
Chklovski, A., Parks, D. H., Woodcroft, B. J., and Tyson, G. W. (2023). CheckM2: a rapid, scalable and accurate tool for assessing microbial genome quality using machine learning. Nature methods, 20, 1203–1212.
Clermont, O., Bonacorsi, S., and Bingen, E. (2000). Rapid and simple determination of the Escherichia coli phylogenetic group. AEM, 66, 4555–4558.
Campos-Madueno, E. I., Aldeia, C., Sendi, P., and Endimiani, A. (2023). Escherichia ruysiae may serve as a reservoir of antibiotic resistance genes across multiple settings and regions. Microbiol. Spectr. 11:e0175323. doi: 10.1128/spectrum.01753-23
Capella-Gutiérrez, S., Silla-Martínez, J. M., and Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P., and Parks, D. H. (2022). GTDB-Tk v2: memory friendly classification with the genome taxonomy database. bioRxiv. doi: 10.1101/2022.07.11.499641
Contreras-Moreira, B., and Vinuesa, P. (2013). GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Am. Soc. Microbiol. 79, 7696–7701. doi: 10.1128/AEM.02411-13
Demir, M., and Hazırolan, G. (2024). Rapid bacterial identification from positive blood cultures by MALDI-TOF MS following short-term incubation on solid media. Infect. Dis. Clin. Microbiol. 6, 141–146. doi: 10.36519/idcm.2024.319
Dogan, B., Zhang, S., Kalla, S. E., Dogan, E. I., Guo, C., Ang, C. R., et al. (2020). Molecular and phenotypic characterization of Escherichia coli associated with granulomatous colitis of boxer dogs. Antibiotics, 9, 540.
Feldgarden, M., Brover, V., Haft, D. H., Prasad, A. B., Slotta, D. J., Tolstoy, I., et al. (2019). Validating the AMRFinder tool and resistance gene database by using antimicrobial resistance genotype-phenotype correlations in a collection of isolates. Antimicrob. Agents Chemother. 63:e00483-19. doi: 10.1128/AAC.00483-19
Gupta, S. K., Padmanabhan, B. R., Diene, S. M., Lopez-Rojas, R., Kempf, M., Landraud, L., et al. (2014). ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob. Agents Chemother. 58, 212–220. doi: 10.1128/AAC.01310-13
Jain, A., Jain, R., and Jain, S., (2020). Basic techniques in biochemistry, microbiology and molecular biology (pp. 181–183). New York, NY, USA: Springer.
Jolley, K. A., and Maiden, M. C. J. (2010). BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11:595. doi: 10.1186/1471-2105-11-595
Ju, Y., Wang, X., Wang, Y., Li, C., Yue, L., and Chen, F. (2022). Application of metagenomic and culturomic technologies in fecal microbiota transplantation: a review. Sheng Wu Gong Cheng Xue Bao 38, 3594–3605. doi: 10.13345/j.cjb.220573
Kristensen, D. M., Kannan, L., Coleman, M. K., Wolf, Y. I., Sorokin, A., Koonin, E. V., et al. (2010). A low-polynomial algorithm for assembling clusters of orthologous groups from intergenomic symmetric best matches. Bioinformatics 26, 1481–1487. doi: 10.1093/bioinformatics/btq229
Koga, R., Moriyama, M., Onodera-Tanifuji, N., Ishii, Y., Takai, H., Mizutani, M., et al. (2022). Single mutation makes Escherichia coli an insect mutualist. Nature Microbiology, 7, 1141–1150.
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M., (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome research, 27, 722–736.
Kurtz, S., Phillippy, A., Delcher, A. L., Smoot, M., Shumway, M., Antonescu, C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5:R12. doi: 10.1186/gb-2004-5-2-r12
Lagier, J. C., Armougom, F., Million, M., Hugon, P., Pagnier, I., Robert, C., et al. (2012). Microbial culturomics: paradigm shift in the human gut microbiome study. Clin. Microbiol. Infect, 18, 1185–1193.
Lagier, J.-C., Dubourg, G., Million, M., Cadoret, F., Bilen, M., Fenollar, F., et al. (2018). Culturing the human microbiota and culturomics. Nat. Rev. Microbiol. 16, 540–550. doi: 10.1038/s41579-018-0041-0
Lagier, J.-C., Khelaifia, S., Alou, M. T., Ndongo, S., Dione, N., Hugon, P., et al. (2024). Retraction note: culture of previously uncultured members of the human gut microbiota by culturomics. Nat. Microbiol. 10:601. doi: 10.1038/s41564-024-01894-5
Li, L., Stoeckert, C. J. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
McArthur, A. G., Waglechner, N., Nizam, F., Yan, A., Azad, M. A., Baylay, A. J., et al. (2013). The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 57, 3348–3357. doi: 10.1128/AAC.00419-13
Price, M. N., Dehal, P. S., and Arkin, A. P. (2009). FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650. doi: 10.1093/molbev/msp077
Sakaguchi, K., Tanabe, M., Takizawa, S., Kasahara, S., Denda, T., Koide, S., et al. (2023). Zoonotic potential and antimicrobial resistance of Escherichia spp. in urban crows in Japan-first detection of E. Marmotae and E. Ruysiae. Comp. Immunol. Microbiol. Infect. Dis. 100:102040. doi: 10.1016/j.cimid.2023.102040
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Thomson, N. M., Gilroy, R., Getino, M., Foster-Nyarko, E., van Vliet, A. H. M., La Ragione, R. M., et al. (2022). Remarkable genomic diversity among Escherichia isolates recovered from healthy chickens. PeerJ 10:e12935. doi: 10.7717/peerj.12935
Van Der Putten, B. C. L., Matamoros, S., Mende, D. R., Scholl, E. R., Consortium, C., and Schultsz, C. (2021). Escherichia ruysiae sp. nov., a novel gram-stain-negative bacterium, isolated from a faecal sample of an international traveller. Int. J. Syst. Evol. Microbiol. 71:004609. doi: 10.1099/ijsem.0.004609
Vinuesa, P., and Contreras-Moreira, B. (2015). Robust identification of orthologues and paralogues for microbial pan-genomics using GET_HOMOLOGUES: a case study of pIncA/C plasmids. Methods Mol. Biol. 1231, 203–232. doi: 10.1007/978-1-4939-1720-4_14
Walk, S. T. (2015). The “cryptic” Escherichia. EcoSal Plus 6, 10–1128. doi: 10.1128/ecosalplus.ESP-0002-2015
Keywords: Escherichia ruysiae , canine microbiome, culturomics, whole genome sequencing, taxono-genomic
Citation: Dione N, Mlaga KD, Liang S, Jospin G, Marfori Z, Alvarado N, Scarsella E, Uttarwar R and Ganz HH (2025) Comparative genomic and phenotypic description of Escherichia ruysiae: a newly identified member of the gut microbiome of the domestic dog. Front. Microbiol. 16:1558802. doi: 10.3389/fmicb.2025.1558802
Edited by:
Qinghong Li, Nestle Purina Research, United StatesReviewed by:
Yanfen Cheng, Nanjing Agricultural University, ChinaTales Fernando da Silva, Institute of Biological Sciences, Brazil
Copyright © 2025 Dione, Mlaga, Liang, Jospin, Marfori, Alvarado, Scarsella, Uttarwar and Ganz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Holly H. Ganz, aG9sbHlAYW5pbWFsYmlvbWUuY29t