
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 19 March 2025
Sec. Systems Microbiology
Volume 16 - 2025 | https://doi.org/10.3389/fmicb.2025.1544934
This article is part of the Research Topic Methods for Imaging and Omics Data Science: Advances, Applications, and Spatiotemporal Innovations View all 9 articles
 Eduarda Guimarães Sousa1*†
Eduarda Guimarães Sousa1*† Gabriela Munis Campos1†
Gabriela Munis Campos1† Marcus Vinícius Canário Viana1†
Marcus Vinícius Canário Viana1† Gabriel Camargos Gomes1†
Gabriel Camargos Gomes1† Diego Lucas Neres Rodrigues2†
Diego Lucas Neres Rodrigues2† Flavia Figueira Aburjaile2†
Flavia Figueira Aburjaile2† Belchiolina Beatriz Fonseca3†
Belchiolina Beatriz Fonseca3† Max Roberto Batista de Araújo1,4†
Max Roberto Batista de Araújo1,4† Mateus Matiuzzi da Costa5†
Mateus Matiuzzi da Costa5† Eric Guedon6†
Eric Guedon6† Bertram Brenig7†
Bertram Brenig7† Siomar Soares8†
Siomar Soares8† Vasco Azevedo1*†
Vasco Azevedo1*†Introduction: Next-generation sequencing (NGS) has played a pivotal role in the advancement of taxonomics, allowing for the accurate identification, differentiation, and reclassification of several bacteria species. Bacillus velezensis is a Gram-positive, facultatively aerobic, spore-forming bacterium known for its antimicrobial and antifungal properties. Strains of this species are highly relevant in agriculture, biotechnology, the food industry, and biomedicine.
Methods: In this study, we characterized the genomes of nine Bacillus strains isolated from soil in the state of Bahia (Brazil) using NGS with Illumina platform. Identification was performed by Average Nucleotide Identity (ANI) and digital DNA-DNA hybridization (dDDH) analyses, which revealed a match between the genomic information of the isolates and B. velezensis NRRL B-41580, with a variation of 89.3% to 91.8% by dDDH in TYGS and 95% to 98.04% by ANI in GTDBtk.
Results and discussion: Two strains, BAC144 and BAC1273, exhibited high similarity to B. amyloliquefaciens subsp. plantarum FZB42. However, the latter strain was subsequently reclassified as B. velezensis. The division pattern observed during identification was confirmed in the phylogenomic analysis, where BAC144 and BAC1273 clustered with Bacillus amyloliquefaciens subsp. plantarum, while the other strains clustered with B. velezensis NRRL B-41580, forming a clade with high genetic similarity, with a bootstrap value of 100%. Furthermore, a synteny analysis demonstrated greater conservation among the strains from this study compared to the reference strain, with the formation of distinct collinear groups. The pangenome analysis revealed an open pangenome, highlighting the genetic diversity within the species. Based on this analysis, a functional annotation was performed to compare exclusive gene repertoires across groups, uncovering distinct adaptations and functional profiles. The identification of bacterial strains belonging to this species is of great importance due to their high applicability. The strains identified in this study underscore the need for more robust taxonomic technologies to accurately classify prokaryotes, which are subject to constant evolutionary changes, requiring the reclassification of several species within the genus Bacillus, many of which are heterotypic synonyms of B. velezensis like Bacillus oryzicola, B. amyloliquefaciens subsp. plantarum and Bacillus methylotrophicus.
The genus Bacillus contains species of Gram-positive, rod-shaped, spore-forming, and facultative aerobic bacteria that occur in diverse natural and human-created environments, with environmental, biotechnological, and medical relevance. Some strains have agriculture, bioremediation, and pharmaceutical production applications, while others are human pathogens (Logan and Vos, 2015). After comparative phylogenomic analysis, most of the species were reclassified to other genera, with the remaining ones classified as “Bacillus subtilis group” (27 species), or “Bacillus cereus group” (19 species). The taxonomy of the B. subtilis group underwent various changes, including novel species and many reclassifications, and much work has to be done to benefit the research and the applications (Xu and Kovács, 2024).
Bacillus velezensis has agricultural, biotechnological, and environmental applications, due to the promotion of plant growth, inhibition of plant pathogens, probiotic effects in animal feed, production of biopolymers, antimicrobials, anticancer drugs, biosurfactants, and degradation of agroindustrial byproducts (Adeniji et al., 2019; Khalid et al., 2021; Keshmirshekan et al., 2024). The correct identification of B. velezensis strains is relevant due to their potential applications.
Taxono-genomics incorporates genome sequencing in taxonomic studies, offering reliable and reproducible data (Ramasamy et al., 2014). The taxonomic identification of bacteria from DNA sequences can be achieved using one of a few loci, such as 16S rRNA, rpoB (Christensen and Olsen, 2018), many loci, such as in the method Ribosomal Multilocus Sequence Typing (rMLST) (Jolley et al., 2012), or the whole genome for a more excellent resolution, in methods such as Average Nucleotide Identity (ANI), digital DNA–DNA hybridization (dDDH) (Christensen and Olsen, 2018), and Tetra-nucleotide Signature Correlation Index (TETRA) (Richter and Rosselló-Móra, 2009). Taxono-genomics have been used to describe new species (Ramasamy et al., 2014), and solve taxonomic classifications (Xu and Kovács, 2024).
The methods using whole genome, perform a pairwise comparison of the query sequence to a database of reference genomes, followed by a minimum cutoff value to consider them as from the same species, such as ≥95% for ANI (Jain et al., 2018). ANI facilitates taxonomic identification by leveraging the mean identity of all orthologous genes across a minimum of two genomes. This calculation enables the delineation of lineages within a single species. dDDH simulates traditional DNA–DNA hybridization techniques in silico, providing a similarity score between two genomes. A threshold of >70% similarity indicates that the compared genomes belong to the same species (Meier-Kolthoff et al., 2014). For TETRA a > 0.99 correlation coefficient (z-score) (Richter and Rosselló-Móra, 2009). Difference in G + C content of not greater than 1% is also expected within genomes from the same species (Meier-Kolthoff et al., 2014).
Previous studies have reported the importance of B. velezensis strains as potential probiotics, mainly applied in aquaculture and agriculture (Khalid et al., 2021; Dong et al., 2022). However, it has also been applied to humans, as shown in the work of Brutscher et al. (2024), where strain BV379 was evaluated for biosafety as a potential candidate probiotic for oral use. One of the most beneficial effects of B. velezensis for these applications is the production of bioactive compounds such as surfactin, bacilysin and fengycin, which are associated with antagonistic, biosurfactant, antioxidant and anti-inflammatory activities (Han X. et al., 2021; Barale et al., 2022; Medeot et al., 2023).
Given the high biotechnological applicability of this species, this work identified and characterized the genomes of nine strains isolated from soil in Bahia, Brazil, where the discovery of new genetic profiles will reinforce the importance of studies on its diversity and evolution, showing its value for future industrial and therapeutic applications, and the need to characterize strains for biological control or use as probiotics.
Nine Bacillus strains, BAC39, BAC118, BAC124, BAC137, BAC144, BAC156, BAC207, BAC238, and BAC1273 (Supplementary Table 1), isolated from the soil in Bahia, Brazil, were used. They were cultivated in Lysogeny Broth (LB) supplemented with 0.1% Tween 80 for 24 h at 37°C.
Furthermore, a Flex Control Microflex LT mass spectrometer (Bruker Daltonics) with a 60 Hz nitrogen laser was used to process the spectra of each sample using Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry (MALDI-TOF MS). This technique enables rapid bacterial identification by analyzing protein mass spectra. The MALDI Biotyper software, version 3 (Bruker Daltonics), was used to create a Master Spectral Library (MSP) for the strains, utilizing the BioTyper MSP breeding standard. The bacteria were prepared in accordance with the manufacturer’s instructions (Dos Santos et al., 2022).
DNA extraction was performed through the Wizard® Genomic DNA Purification Kit (Promega), following the instructions recommended by the manufacturer. Next-generation sequencing was performed using the Hi-Seq 2,500 platform (2x150bp) (Illumina®, United States), with the ThruPLEX DNA-Seq Kit (Takara) used for paired-end library construction.
Two different datasets were used for phylogenomic analyses. One dataset contains all Bacillus genomes from RefSeq, deposited in the National Center for Biotechnology Information (NCBI) database (accessed on 10 May 2024), and also present in the Genome Taxonomy Database (GTDB), as listed in Supplementary Table 2, with Pseudomonas aeruginosa DSM 50071T as an outgroup (Gupta et al., 2023), the B. velezensis NRRL B-41580 and Bacillus amyloliquefaciens subsp. plantarum FZB42 strains were also included in the dataset, identified by analyzing their Average Nucleotide Identity (ANI), which showed a similarity of over 95% to the samples in the collection, organisms belonging to the same species or closely related represent a cutoff value of ≥95% (Jain et al., 2018).
For the second dataset, strains that showed alignment with the isolated samples predicted by the Type Strain Genome Server1 were used for phylogenomic analyses, as listed in Table 1 and Supplementary Data 1. All genomes were downloaded from the RefSeq NCBI (National Center for Biotechnology Information) databases.
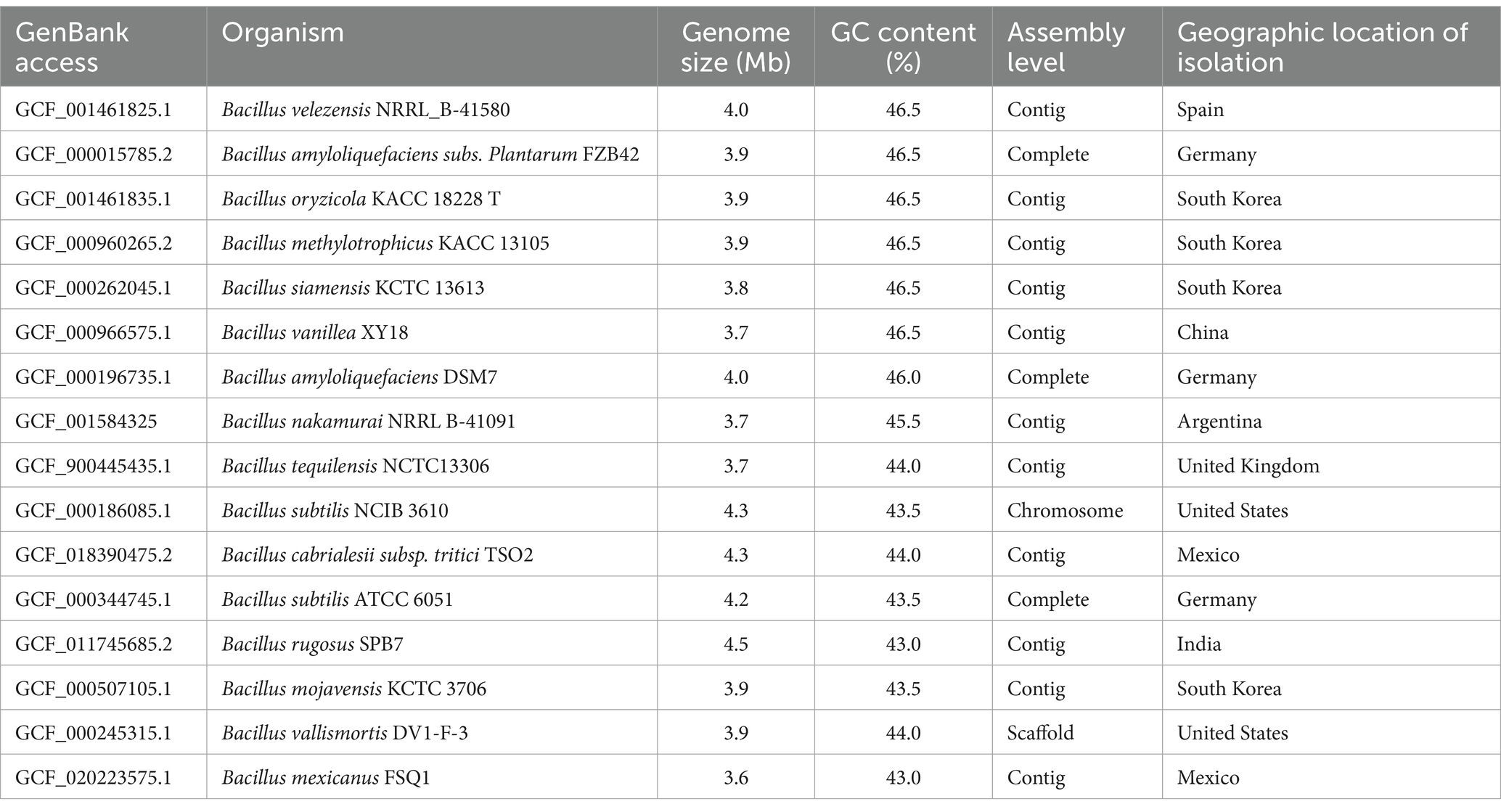
Table 1. Genome information for the dataset based on TYGS analysis.
The trimming and quality of the sequencing were analyzed using the Fastp v0.23.4 tool,2 an Ultrafast one-pass FASTQ data preprocessing, to assemble and annotate the genomes a posteriori, as indicated in the reports generated by the sequencing software (Babraham Bioinformatics – FastQC A Quality Control tool for High Throughput Sequence Data, n.d.; Chen, 2023). The de novo assembly software for prokaryotes, Unicycler v0.5.1 (Wick et al., 2017), was used to assemble the sequenced isolates. The Quast software3 (Gurevich et al., 2013) was used to evaluate the quality of the assembly.
The isolated, assembled, and annotated genomes were analyzed to assess their quality and identify potential errors, including contamination, completeness, and genome integrity. This was performed using the CheckM2 v1.0.2 software (Chklovski et al., 2023),4 which use universal machine learning models that performed analyses based on the amount of GC content. The presence of chimeric contigs was analyzed using GUNC v1.0.6 (Orakov et al., 2021).5
The annotation was performed using the Prokka software, designed for rapid prokaryotic genome annotation6 (Seemann, 2014). Prokka uses a variety of databases to identify the functional elements (features) of bacterial and archaeal genomes, including coding DNA sequences (CDSs), tRNAs, rRNAs, and protein functional annotations. The output is compatible with files that are suitable for use in subsequent analyses.
The taxonomic identification of the strains was determined using two distinct approaches. The first approach was the digital DNA–DNA Hybridization (dDDH) analysis implemented in TYGS7 (Meier-Kolthoff et al., 2013; Meier-Kolthoff and Göker, 2019). This method calculates the DNA similarity between the aligned fragments of a query genome to a Type Strain database, with a similarity cutoff above 70% for the same species. As a second approach to identification, ANI was analyzed using the GTDB-Tk v2.4.0,8 which performs taxonomic classification based on the Genome Database Taxonomy (GTDB) (Chaumeil et al., 2020). For this study, similarities more significant than 95% were considered to be indicative of a close genetic relationship (Ciufo et al., 2018).
The analogous genomes generated by TYGS were used as a dataset for subsequent analyses, including the heatmap construction to corroborate the identification of each lineage by the pyANI program v3.0 (Pritchard et al., 2019). This program generates the map by calculating the ANI through the MUMmer alignment method, with a 95% similarity threshold for distinct genomes of the same species.
Following the MLST scheme defined in PubMLST, the sequence type (ST) was determined in silico using the FastMLST script,9 taking into account the alleles of seven housekeeping genes (Guerrero-Araya et al., 2021). In addition, PhyloPhlAn v 3.1.68,10 an integrated pipeline for large-scale phylogenetic profiling (Asnicar et al., 2020), was used to construct a tree based on maximum likelihood strategies of the core genome generated by the PPanGGOLiN program v 2.2.0 and bootstrap values with 1,000 replicates. Moreover, a phylogenetic tree based on the 16S rRNA gene was constructed. The sequences were obtained from genomic annotation and the GenBank database at NCBI. The phylogenetic analysis was performed using MEGA version 12, using the maximum likelihood method with bootstrap values calculated from 1,000 replicates (Kumar et al., 2024). The both trees were visualized using iTOL v.611 (Letunic and Bork, 2024).
The Partitioned PanGenome Graph of Linked Neighbors (PPanGGoLin) program, a tool for analyzing the pangenome of prokaryotes (Gautreau et al., 2020), was used to analyze the shared gene repertoire of the strains isolated in this study with the species from the NCBI public database comparing with B. velezensis (370 complete genomes, accessed 07 August 2024). Additionally, we included strains from the same phylogenetic clade as highlighted in the gray square in Figure 1, ensuring a more refined comparative analysis. In order to check the proportion of genes shared by the bacteria in this study with those available in the database. The program uses a combination of graphical representation and machine learning to categorize genes based on their frequency and distribution, resulting in the following classification: Genes that are persistent and conserved, and which are present in almost all genomes, are associated with essential functions. These are followed by shell genes of intermediate occurrence, which are linked to ecological adaptation or resistance. Finally, there are cloud-specific genes, which are often acquired by horizontal transfer, and which indicate genomic plasticity.
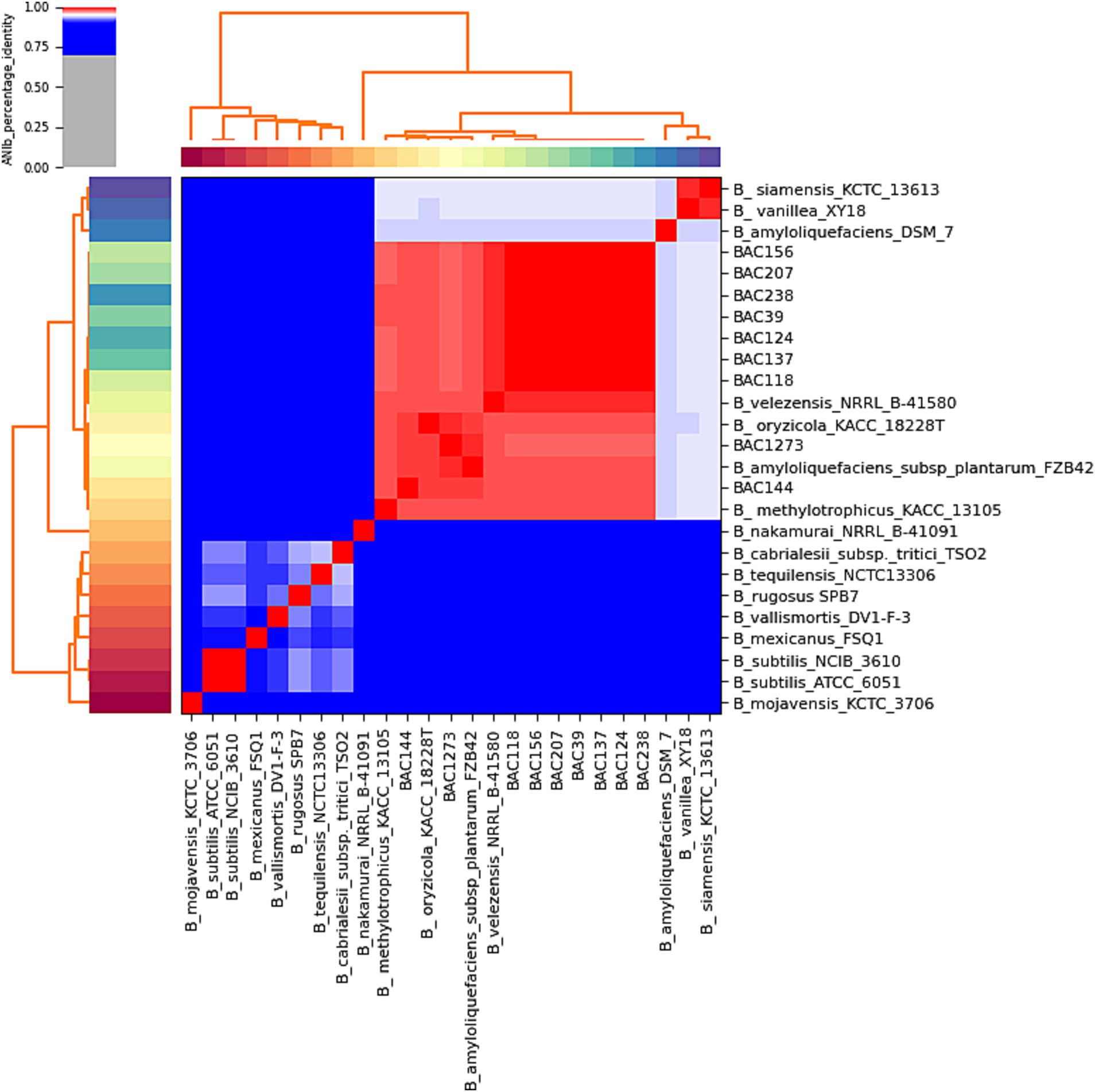
Figure 1. A heatmap of the analysis was carried out to show the similarity percentage, demonstrating very high similarity between the samples isolated in this study and B. velezensis strains from the database.
To assess possible gene rearrangements and to analyze collinearity between genes when comparing genomes, the Mauve program v.20150226 (Darling et al., 2004) was used, together with the progressive Mauve algorithm, to analyze gene synteny between the samples isolated and sequenced in this work and the genome identified by GTDB-Tk, B. velezensis NRRL B-41580 (GCF_001461825.1), which is also closely related by phylogenetic tree analysis.
In addition, Gegenees v3.1 software was used to obtain a distance matrix related to the species similarity to each other and to understand the differences between the lineages (Ågren et al., 2012). This tool uses BLASTn to perform an alignment of genome fragments of pre-defined sizes of 500 nucleotides and, in the end, produces a heat map showing the similarity between each strain in a range from 0 to 100 percent.
The program eggNOG-mapper v2 (Cantalapiedra et al., 2021) was performed for functional annotation, orthology assignments, and domain prediction. The PFAM, KEGG, and COG databases were used to describe the exclusive proteins of the groups determined based on the obtained results. To achieve this, the genomic repertoire matrix generated by PPanGGOLiN was used, and the annotation was performed using an in-house script in R. Comparative analysis of functional annotation was based on the results of taxonomic and phylogenomic analyses. A presence/absence-based approach was applied to identify genes exclusively present in each group while being absent in the remaining ones.
In this study, the utilization of MALDI-TOF MS technology led to the identification of two isolates, designated as genus Bacillus (BAC 118 and BAC 207), within the range of 1,717 to 1,819, as specified by the manufacturer. Four strains (BAC 39, BAC124, BAC156 and BAC 238) were unable to be identified, classified as having no peaks found (score < 0), and three (BAC137, BAC144 and BAC1273) exhibited a very low score and were therefore deemed unreliable for identification purposes, the color red is indicative of a low score, with minimal identifying power. Conversely, yellow signifies an intermediate score, yet still not conclusive (Table 2). This method is based on comparing the mass spectrometry profile of the isolate with a reference database. The score reflects the degree of similarity between the spectrum generated by the sample and the profiles present in the database. It was not possible to identify the species of the isolates from these scores.
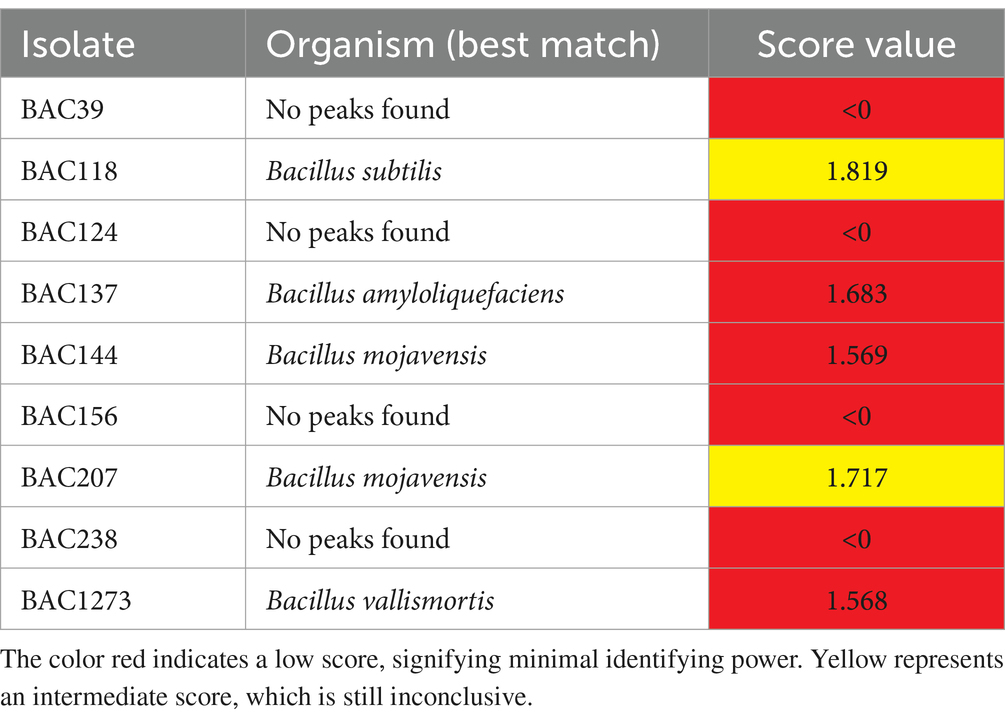
Table 2. Identification of isolates by MALDI-TOF MS.
The quality of the assembly, according to the QUAST report, is shown in Table 3. Isolates BAC1273 and BAC207 had the largest and smallest genomes, with 4,176,055 and 4,029,220 base pairs, respectively. All had 100% completeness, between 0.07 and 0.22 contamination by CheckM2, and did not show chimerism as predicted by GUNC (Supplementary Table 3). In addition, the genomes had between ~3,952 and 4,125 coding sequences (CDS), 3 to 5 rRNA genes, and 76 to 84 tRNA genes which were predicted by Prokka.
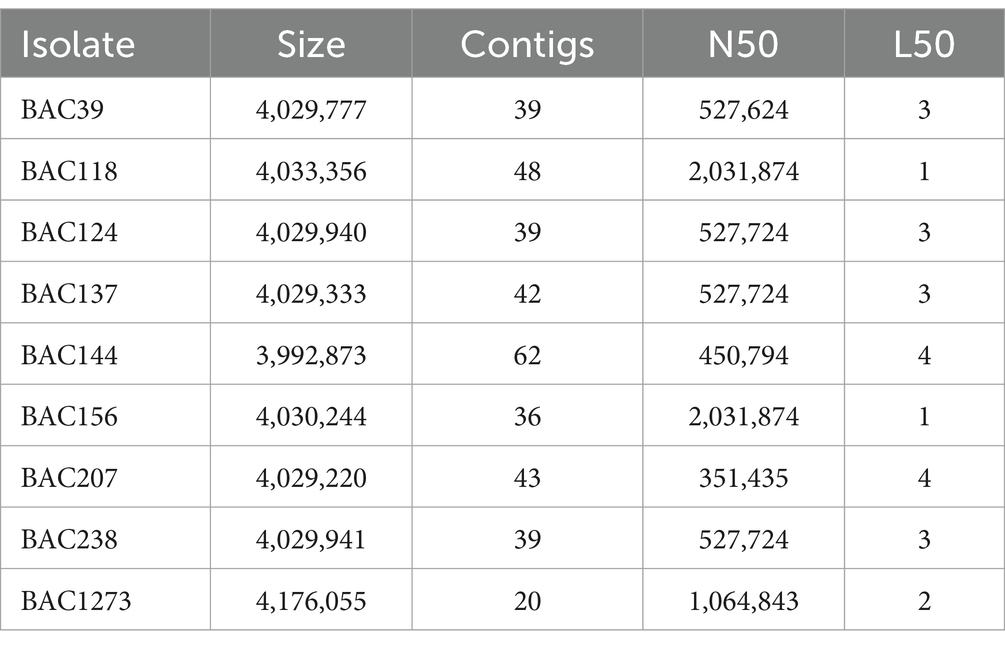
Table 3. Quality metrics for Bacillus isolation.
All the strains were classified as B. velezensis by GTDBtk, with an ANI value above 95% (Table 4). Two isolates, BAC1273 and BAC144, were classified as B. amyloliquefaciens subsp. plantarum with strain FZB4 (GCF_001461825.1) as reference by TYGS with a dDDH of 91.8%. Nevertheless, the latter strain was reclassified as B. velezensis by Dunlap et al. (2016), and by the List of Prokaryotic names with Standing in Nomenclature (LPSN) (Dunlap et al., 2016).12 All the other strains were classified as B. velezensis with dDDH ranging from 89.3 to 91.8% with high similarity to strain NRRL B-41580 (GCF_001461825), which was selected as the reference for the subsequent analyses in this study (Table 4).
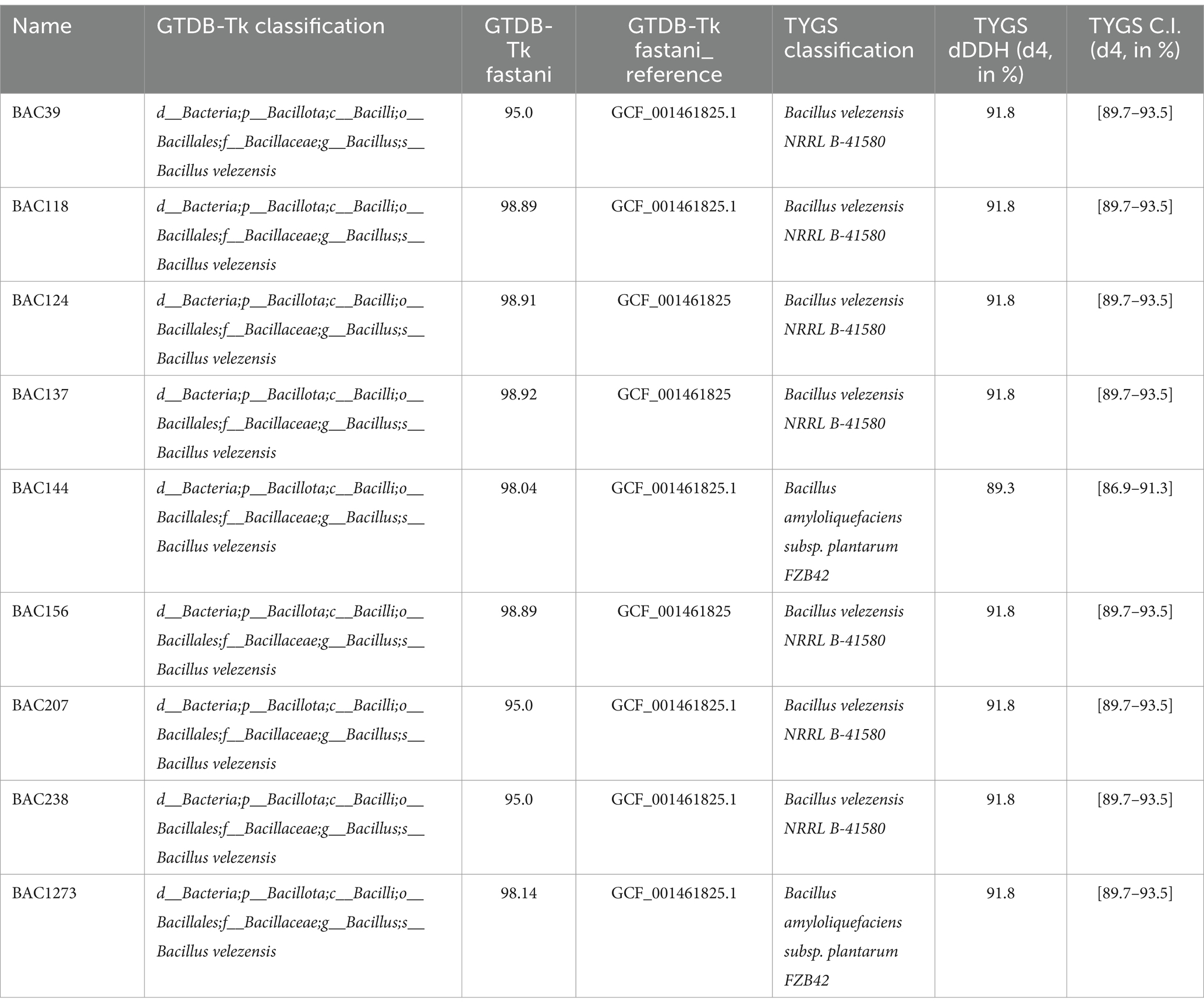
Table 4. Taxonomic classification of genomes using GTDB-Tk and TYGS.
The genomes identified by TYGS analysis (Supplementary Table 2) were downloaded by RefSeq NCBI in “.fna” format for ANI analyses by pyANI (Figure 2; Supplementary Figure 1). The genomes were compared, including the genome sequence of the 9 strains isolated in this study and 16 identified by TYGS (Supplementary Data 1). The clustering had a high similarity, values greater than 95%, with B. velezensis NRRL B-41580, as predicted in the subsequent analyses. In particular, isolates BAC144 and BAC1273 also showed high similarity with Bacillus oryzicola KACC 18228 T (GCF_001461835.1), B. amyloliquefaciens subsp. plantarum FZB42 and Bacillus methylotrophicus, which were subsequently reclassified as B. velezensis (Dunlap et al., 2016; Adeniji et al., 2019).13 This reclassification is also supported by studies that address issues related to genome identification in databases, particularly those identifications based solely on 16S rRNA sequences (Diabankana et al., 2022).
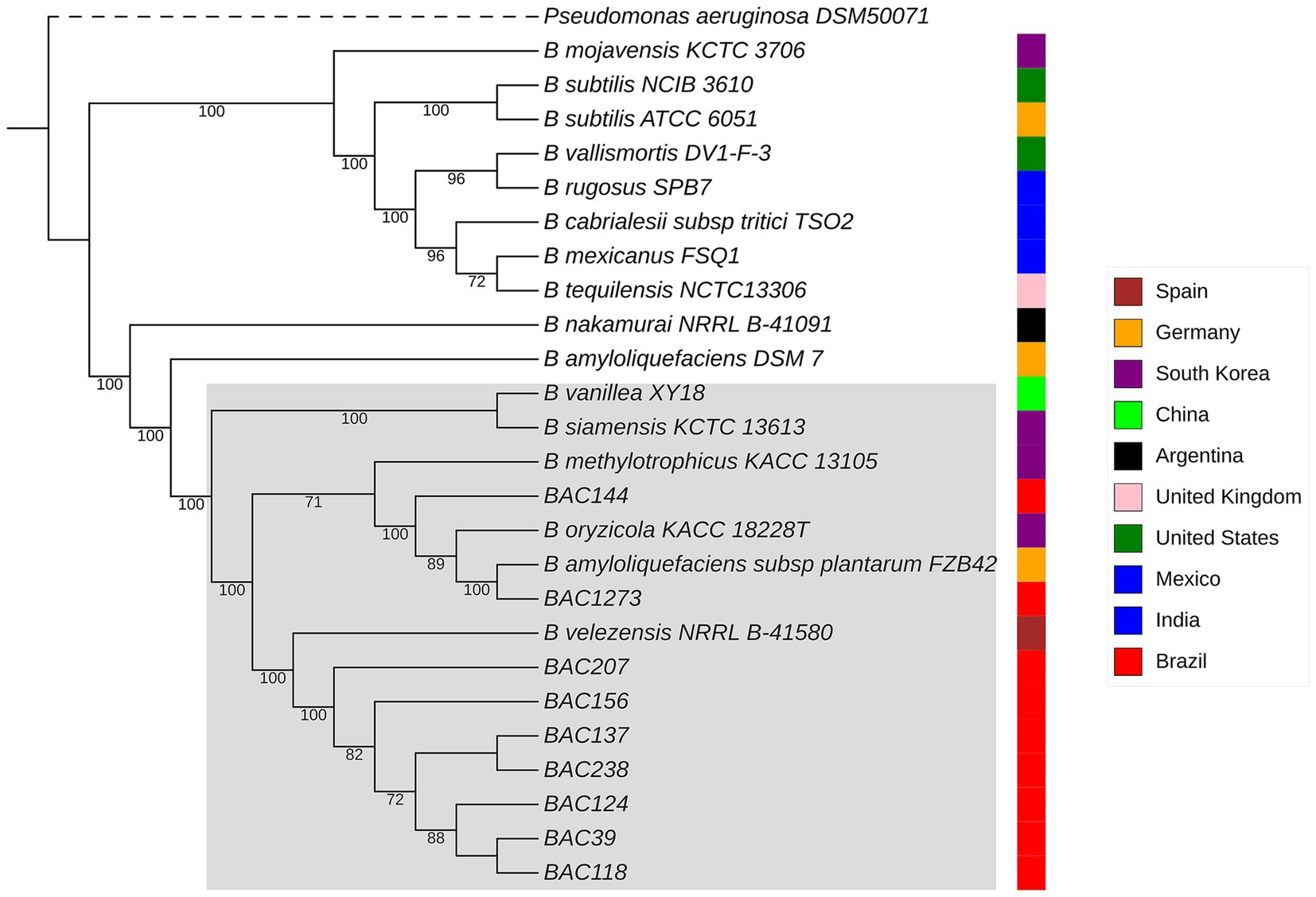
Figure 2. The phylogenetic tree shows the relationships between all related genomes predicted by TYGS for B. velezensis strains, along with their geographical locations of isolation. Bootstrap values, based on 1,000 replicates, range from 71 (lowest) to 100 (highest). The same clusters observed in the ANI heatmap can also be visualized here. B. velezensis strains that group together are highlighted within a gray square. Additionally, Bacillus oryzicola KACC 18228 T, B. amyloliquefaciens subsp. plantarum FZB42, and B. methylotrophicus were subsequently reclassified as B. velezensis.
The isolated strains’ sequence type (ST) was determined using the MLST scheme defined in PubMLST. The strains exhibited a pattern consistent with that of B. subtilis for the ST-91 complex, yet they displayed a novel combination of alleles, warranting their classification as a distinct “new_ST” variant. The identified housekeeping genes and their respective alleles were glpF, ilvD, pta, purH, pycA, rpoD, and tpiA (Supplementary Table 4). Different alleles were found only in BAC144 and BAC1273.
The phylogenetic tree was constructed by maximum likelihood with PhyloPhlAn (Asnicar et al., 2020) to determine their genetic relationships using the MUSCLE algorithm (Edgar, 2004) for multiple sequence alignment (MSA) and phylogenetic reconstruction with FastTree.
The first dataset used for the analysis was with strains of the genus Bacillus present in NCBI and GTDB, with the two similar predicted by TYGS (25 genomes), as mentioned in Methodology Section 2.1, and with Pseudomonas aeruginosa DSM50071T as outgroup (Asnicar et al., 2020). The same pattern was observed in the ANI heatmap, with strains BAC144 and BAC1273 clustering with B. amyloliquefaciens subsp. plantarum and the rest with B. velezensis NRRLB-41580. In addition, a clade with B. velezensis, B. amyloliquefaciens, Bacillus siamensis and Bacillus nakamurai was observed (Supplementary Figure 2). However, the phylogenetic analysis based on the 16S rRNA gene using the TYGS dataset did not provide reliable resolution of the branching positions, as indicated by the low bootstrap values, indicating weak statistical support for the branching positions (Supplementary Figure 3).
The second dataset was based on similar data used in the plane analyses, with the genomes predicted by TYGS, as explained in Section 2.1, and using the same outgroup. The same clustering division between strains was observed, with an additional clade formation with the B. velezensis from the database and identified in this study, consisting of B. velezensis, B. amyloliquefaciens, B. siamensis, B. nakamurai, Bacillus vanillea, B. methylotrophicus and B. oryzicola (reclassified as B. velezensis), which can be seen in Figure 1. The data were analyzed, and it was revealed that the bacterial samples collected from different regions did not group together according to their geographical location. However, the samples isolated in this study grouped together phylogenetically closely, except for BAC144 and BAC 1273, which were close to samples from South Korea (KACC 18228 T) and Mexico (FZB42), respectively.
The 25 genomes used in the subsequent analyses were analyzed to obtain a distance matrix related to the similarity of the species to each other, where greenish colors show high similarity, and reddish colors have low similarity. The genomes that formed a cluster with the isolates from this study in the previous analyses showed the same pattern of similarity with a range of 93 to 100%, and the formation of the same group with high similarity was observed as seen in the phylogenetic trees, with a range of 81 to 96% (Figure 3). The same high similarity between the strains isolated in this study and B. velezensis NRRL B-41580 was observed in a range of 93 to 94%, presented by BAC144 and BAC1273, up to 96 to 100% with the others (Supplementary Figure 4).
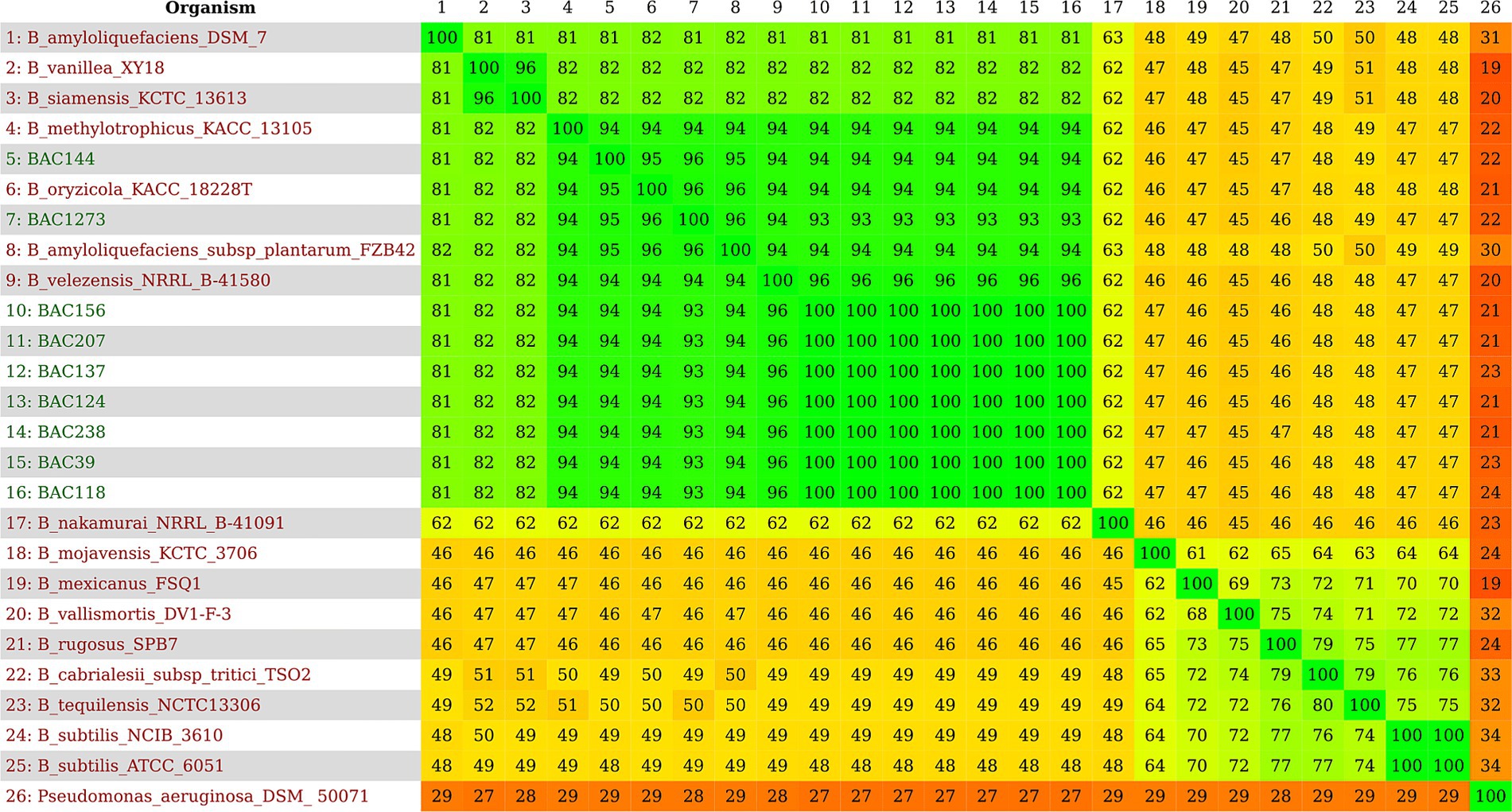
Figure 3. Heatmap with nine genomes identified as B. velezensis in this study and 16 representative genomes according to TYGS. In green, a high similarity can be observed, ranging from 81 to 100%; in orange, a median similarity ranging from 46 to 65%; and finally, a low similarity in red, ranging from 19 to 34%.
The pangenome analysis revealed the existence of an open pangenome, wherein the number of shared genes among sequenced genomes increases with an α value of 0.784. The number of gene families identified in the persistent, shell, and cloud was 3,196, 2,384, and 5,404 genes, respectively. Figure 4 illustrates the rarefaction curve, which demonstrates the open pangenome. It depicts the evolution of the number of gene families as more genomes are incorporated into the pangenome. For each partition, multiple representations of the observed data are provided, including the observed means, medians, 1st and 3rd quartiles of the number of gene families per number of genomes, as well as the best fitting of the data by Heaps’ law, which is commonly used to represent this evolution of diversity in terms of gene families.
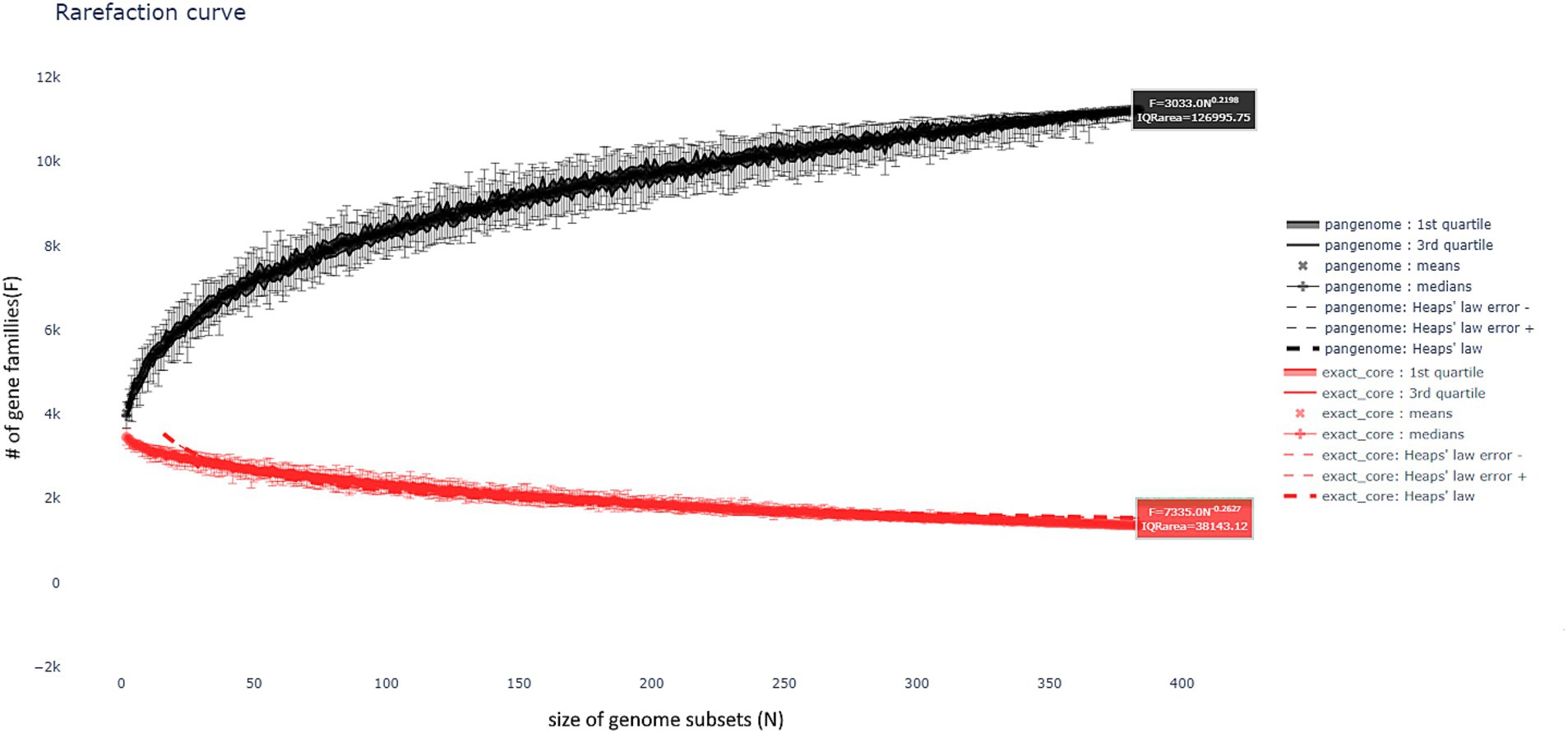
Figure 4. Rarefaction curve of the pangenome of isolated genomes compared to the NCBI database genome of B. velezensis, illustrating the open pangenome. The curve shows the increasing number of gene families as more genomes are included, indicating the continuous expansion of gene diversity.
A multiple alignment was carried out between the strains isolated in this study and B. velezensis NRRL B-41580 from the database, with the high similarity shown in the previous results (Figure 5) and one without this strain (Supplementary Figure 5). We analyzed the synteny between them and whether the one in the database interfered with this collinearity, of which there was no difference. It was possible to observe a high level of conservation between the blocks, with some inversion and translocation processes and some deletion blocks, especially at the end of these genomes. In addition, there was more significant conservation between the strains in this study than in the database. In both analyses, collinearity was also observed between the groups formed: the first formed with only BAC118; another group with BAC124, 137, 238, 1,273, and 39; and one with BAC144, 156, and 207.

Figure 5. Comparison between the genomes identified in this study and B. velezensis NRRL B-41580 according to synteny between the blocks, in which each color corresponds to a specific region that has undergone evolutionary events such as inversions, translocations, or deletions.
Based on the clustering observed in the phylogenetic tree (Figure 2), a functional analysis of exclusive genes was performed using pangenome data and annotation with eggNOG-mapper. Three groups were analyzed: a subset of seven genomes (BAC156, BAC207, BAC238, BAC39, BAC124, BAC137, and BAC118), BAC144, and BAC1273, each compared to the remaining genomes. Exclusive genes were identified through the gene presence/absence matrix from PPanGGOLiN and annotated using COG, KEGG, and PFAM domains.
In the subset of seven genomes, 19 exclusive genes were primarily associated with transcriptional regulation, phage presence, genetic recombination, phosphate metabolism, stress response, and energy metabolism. The most prevalent COG categories were L (Replication, recombination, and repair), K (Transcription), C (Energy production and conversion), S (Function unknown), and G (Carbohydrate transport and metabolism). In the BAC144 genome, four exclusive genes were predominantly linked to transcriptional regulation, nucleotide metabolism, and DNA synthesis, represented mainly by COG categories F, K, and L. In BAC1273, nine genes were identified, which are associated with a variety of functions, including transcriptional regulation, sulfur group transfer reactions, genetic recombination, detoxification, resistance, nucleotide metabolism, DNA synthesis, and carbohydrate metabolism. The most prevalent COG categories were K, L, C, G, and S. A comprehensive summary of these findings can be found in Figure 6 and Supplementary Table 5.
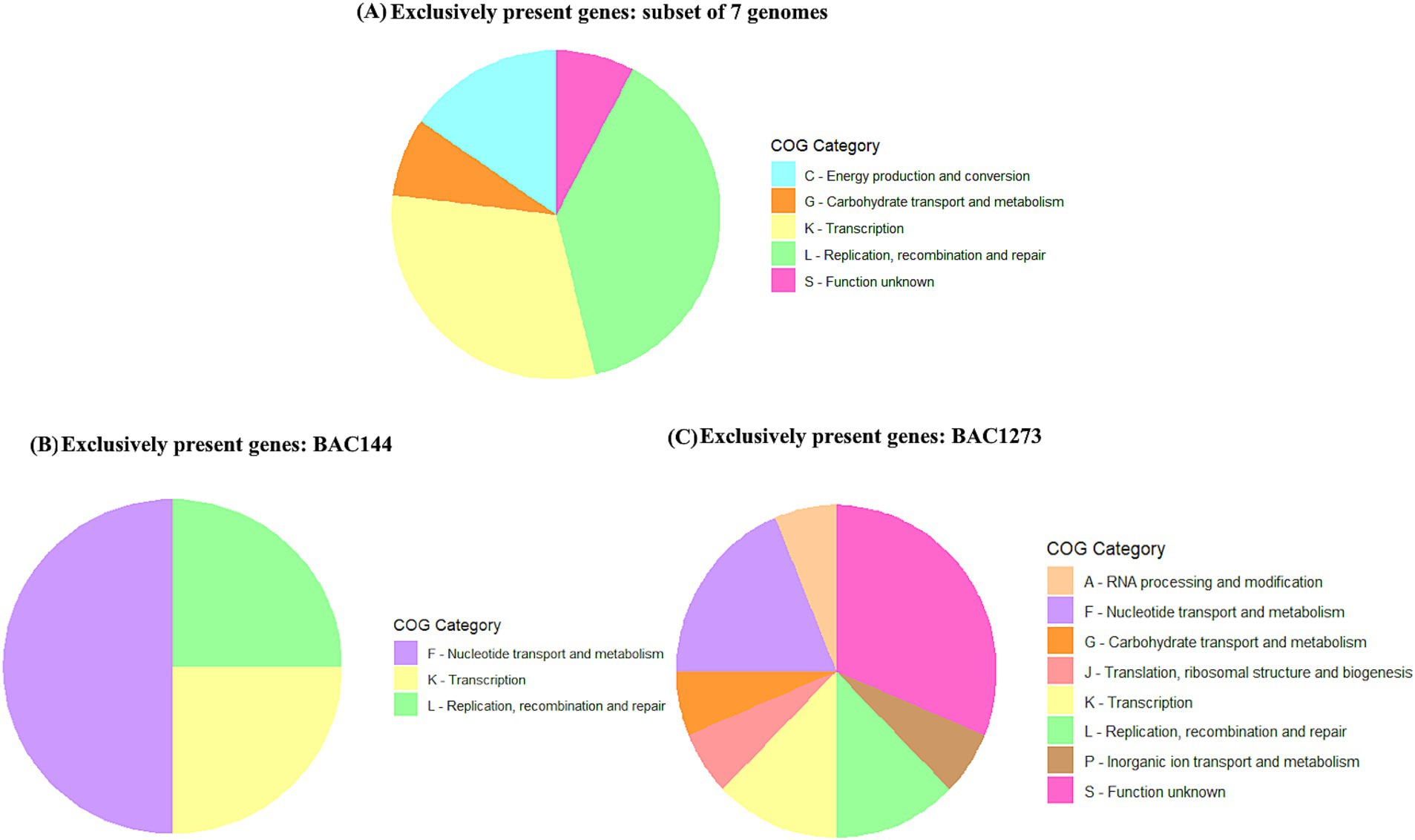
Figure 6. Distribution of COG categories among exclusive genes identified in the subsets analyzed. The three groups include (A) the subset of seven genomes (BAC156, BAC207, BAC238, BAC39, BAC124, BAC137, and BAC118), (B) BAC144, and (C) BAC1273, compared against the remaining genomes. Exclusive genes were classified into their respective COG functional categories.
The taxonomic identification of strains by whole genome sequencing is crucial for determining the diversity between bacterial genera and species and differentiating strains. In bacterial research, 16S rRNA sequencing has been extensively studied for its utility in phylogenetic inference. However, this approach has inherent limitations in accurately differentiating the taxonomic group of bacterial lineages. This is because some areas of microorganisms are often conserved, and the forces that shape the evolution of bacterial genomes act with varying degrees of influence on different parts of the genome. Furthermore, it does not provide sufficient evolutionary information (Janda and Abbott, 2007; Gupta et al., 2023). In contrast, the average nucleotide identity (ANI) and dDDH demonstrated superior resolution in differentiating genomes, including those of reference sequences from other species.
In this same context, MALDI-TOF MS technology is widely used for bacterial identification (Han S. S. et al., 2021). However, here, we show that the identification of B. velezensis isolates was inconclusive, with very low scores, and a lack of resolution at the strain level. A similar result was obtained with a MLST approach based on genetic diversity of housekeeping genes. The results demonstrated the existence of a novel sequence type (ST), hitherto unrepresented in the PubMLST database. This highlights the necessity for the utilization of whole genome sequencing in conjunction with ANI and dDDH for a robust and more precise identification of bacterial strains. Furthermore, the incorporation of spectral profiles of novel bacterial species and strains into the database could facilitate the accurate identification of bacterial isolates through MALDI-TOF MS technology.
Methods based on whole-genome sequencing data such as ANI and dDDH approaches enabled all nine isolates to be assigned to the B. velezensis species (Table 4). Note that two strains (BAC144 and BAC1273) exhibited a high similarity to a B. amyloliquefaciens subsp. plantarum strain that was reclassified as B. velezensis (Dunlap et al., 2016). The ANI comparison heatmap (Figure 2) demonstrates the clustering of various strains of the Bacillus genus, including those isolated and sequenced in this study and belonging to the B. velezensis species, as well as strains that have been reclassified within the same species, formerly namely B. oryzicola, B. amyloliquefaciens subsp. plantarum and B. methylotrophicus. Furthermore, other strains exhibited high similarity to B. velezensis, with an ANI of less than 95%, namely B. siamensis and B. methylotrophicus, which were also reclassified as B. velezensis (Dunlap et al., 2016).
The identification and genomic comparison analyses indicate that all strains in this study exhibit high similarity, suggesting that they were derived from a single clone. However, the percentage of similarity was lower for strains BAC144 and BAC1273. This behavior can be explained by analyzing the housekeeping genes, which revealed that only these two strains had different alleles. The sequence type (ST) of the isolated strains was determined using the MLST scheme defined in PubMLST, showing a pattern consistent with the B. subtilis ST-91 complex, yet they displayed a novel combination of alleles, warranting their classification as a distinct “new_ST” variant. The identified housekeeping genes and their respective alleles were glpF, ilvD, pta, purH, pycA, rpoD, and tpiA (Supplementary Table 4), with different alleles found only in BAC144 and BAC1273.
In the gene synteny analyses, which demonstrate a high degree of collinearity between the strains in this study (Supplementary Figure 5) and a slightly lower degree of collinearity between the strains and B. velezensis NRRL B-41580 (Figure 5). The latter was selected for comparison due to its high ANI value compared to the strains in this study. This discrepancy is likely attributable to the fact that the strains were isolated from disparate geographical locations: the strain from the Spanish database and the strains from Brazil.
In the phylogenomic analyses, a maximum likelihood approach was used to investigate the formation of clades between the strains in the study and those in the database. This revealed that the isolated genomes had similar clustering behavior with the same Bacillus strains. A monophyletic clade was formed between seven of the strains and another between two of them. These were the same strains that clustered with B. amyloliquefaciens subsp. plantarum in all the analyses (Figure 1; Supplementary Figure 2). These were BAC 144 and 1,273. A similar pattern is evident in the similarity heatmap generated using the distance matrix (Figure 3; Supplementary Figure 4). Interestingly, the data revealed that the bacterial samples collected from different geographical regions did not group together based solely on their location. However, the samples isolated in this study clustered phylogenetically closely, except for BAC144 and BAC1273. BAC144 clustered closely with B. velezensis strains from South Korea (KACC 18228 T), while BAC1273 showed close phylogenetic ties with a strain from Mexico (FZB42).
The phylogenetic tree constructed using 16S rRNA gene sequences did not provide a reliable resolution, suggesting that the 16S rRNA gene alone may not be sufficient for precise taxonomic placement (Supplementary Figure 3). The lack of strong statistical support in the tree further reinforces the need for whole-genome-based approaches to achieve a more robust classification. Moreover, previous studies have discussed the limitations of using 16S rRNA for analyzing evolutionary relationships within the Bacillus genus, as this approach may not always yield consistent or conclusive results (Alcaraz et al., 2010; Velsko et al., 2019; Hassler et al., 2022).
The pangenome of the B. velezensis strains isolated in this study was analyzed and compared with the reference genomes in the NCBI database. The results revealed the presence of an open pangenome (Figure 4). This phenomenon is distinguished by the sustained growth in the number of gene families as the analysis incorporates an increasing number of genomes. An open pangenome indicates that, as the number of sequenced genomes increases, the genetic diversity observed continues to grow, as evidenced by the corresponding increase in the number of shared genes. With regard to the strains isolated in this study, it is evident that the number of genes shared between the isolates and the genomes in the NCBI database continues to increase. This suggests a considerable degree of genetic diversity within the B. velezensis populations, including previously reclassified variants such as B. amyloliquefaciens subsp. plantarum and B. methylotrophicus. These findings are consistent with the hypothesis that the B. velezensis pangenome is not yet fully closed, but is still dynamic, allowing for the acquisition of new genes as more strains are sequenced.
A comparative analysis of the number of genes shared between the isolated strains and the reference genomes in the database indicates that, while substantial similarities exist, some of the isolated strains exhibited slightly greater genetic diversity. This is exemplified by strains BAC144 and BAC1273. These strains may represent significant genetic variations within the B. velezensis species. This pattern was also observed in the phylogenomic analyses, which demonstrated a distinct grouping of these strains, indicating that they may derive from a distinct clone.
As illustrated in Figure 4, the pangenome rarefaction curve demonstrates that the diversity of gene families continues to expand with the addition of new genomes, thereby supporting the concept of an open pangenome. This phenomenon also underscores the necessity of using whole genome sequencing in conjunction with alternative identification techniques, such as ANI and dDDH, for a more precise characterization of strains and to enhance comprehension of the phylogenetic relationships between disparate isolates. This type of analysis is fundamental to understanding the differences and similarities between isolates and reference strains, which has direct implications for the taxonomic classification and biotechnological potential of the strains under study.
Building upon these taxonomic and phylogenomic insights, a deeper functional analysis was performed to investigate the diversity of exclusive gene repertoires within the three identified groups: Subset 7 (BAC156, BAC207, BAC238, BAC39, BAC124, BAC137, and BAC118), BAC144, and BAC1273 (Figure 6; Supplementary Table 5). These analyses revealed distinct adaptations and ecological roles reflected in the predominant COG categories and exclusive gene functions. For Subset 7, the exclusive genes highlight adaptations related to genetic recombination and mobility, such as the integration of mobile genetic elements (e.g., phages and plasmids) through genes like yqaS (DNA packaging) with PFAM domain of phage terminase (Médigue et al., 1995; Kimura et al., 2010) and RecU (resolution of Holliday junctions) (Cañas et al., 2008). Also, yobL (nuclease activity), that is a toxin-immunity that module enhances competitive fitness during biofilm formation by mediating strain segregation and preventing conflict, it ensures survival under competitive conditions (Holberger et al., 2012). The COG category L (Replication, recombination, and repair) predominates, underscoring the genetic flexibility of this group. Additionally, genes like phoD (phosphate metabolism) (Mingchao et al., 2010; Huang et al., 2024) and ydhN3 (carbohydrate transport) (Mingchao et al., 2010) reflect metabolic adaptations, with significant representation in COG categories C (Energy production and conversion) and G (Carbohydrate transport and metabolism). The presence of stress response genes, such as csbD, and defense mechanisms, including yhdJ (DNA methylation) (Kunst et al., 1997; Adhikari and Curtis, 2016), suggests that this group is well-adapted to environmental challenges, with functions aligned to COG categories S (Function unknown) and K (Transcription).
In contrast, BAC144 exhibited exclusive genes focused on nucleotide metabolism and DNA synthesis, as exemplified by yncF (dUTPase domain) (Sznyter et al., 1987; Dervyn et al., 2023) and nrdE (ribonucleotide reductase), which maintain genomic stability. The COG category F (Nucleotide transport and metabolism) was particularly enriched, reflecting the group’s emphasis on metabolic precision and DNA replication. Additionally, the gene licT (transcriptional antiterminator) highlights the importance of dynamic transcriptional regulation, supported by genes in COG category K (Transcription). The presence of ddeI (DNA methyltransferase) further suggests active epigenetic defense mechanisms, aligning with COG category L (Sznyter et al., 1987). For BAC1273, the exclusive gene repertoire indicates a strong focus on genome defense and environmental adaptation. Genes such as yhdJ (methyltransferase) and mcrA (restriction endonuclease) point to robust systems for protection against phages and other mobile genetic elements (Mulligan and Dunn, 2008; Bourgeois et al., 2022). This is complemented by the presence of phage-related genes (Phage_capsid and Transposase IS66), indicating interactions with mobile genetic elements. The COG categories K (Transcription) and L (Replication, recombination, and repair) predominate, reflecting the group’s focus on genomic stability and transcriptional control. Additionally, metabolic flexibility is suggested by genes like dut (dUTPase) which plays an essential role in nucleotide metabolism, and yrkH (Rhodanese) associated with catalytic functions related to sulfur group transfer, classified under COG categories F and P, respectively and indicate the ability to maintain a stable and efficient genome, even under challenging environmental conditions, and provide an advantage in agricultural soils or other environments rich in sulfur and related compounds, enhancing bacterial survival and competitiveness (Huang et al., 1999; Adhikari and Curtis, 2016; Tang et al., 2023; Sisodia et al., 2024). As well as the presence of exuT (carbohydrate transport) under COG category G. The gene xpaC, associated with detoxification of halogenated compounds (Huang et al., 1999; Hassan, 2021), highlights a unique adaptation to toxic environments, reinforcing the group’s ecological versatility.
These findings align with the phylogenomic analyses, which revealed distinct clustering patterns for the isolates. While most strains formed a cohesive monophyletic clade, BAC144 and BAC1273 consistently grouped separately in all analyses. This divergence is consistent with the functional differences observed in their exclusive gene repertoires, indicating distinct ecological roles and adaptive strategies. The results underscore the importance of integrating functional and phylogenomic analyses to fully understand the genetic diversity and adaptive potential of bacterial populations. Taxonomic studies are of great importance for the identification of strains based on their genomes, given that the taxonomy of prokaryotes has undergone significant changes and reclassifications as a result of the advancement of more sophisticated technologies (Ferraz Helene et al., 2022). Several reclassifications have been proposed for the genus Bacillus following comparative phylogenomic analysis, including the reclassification of B. cereus and B. subtilis (Xu and Kovács, 2024). This study presents the classification of several B. velezensis strains and their high similarity to strains reclassified to the same species.
A further study by Fan et al. (2017) and Elshaghabee et al. (2017) demonstrates the formation of an operational group of B. amyloliquefaciens, as were the taxonomic status of related strains of B. amyloliquefaciens, as evidenced by comparative and phylogenomic analyses. The operational group B. amyloliquefaciens comprises the following strains: B. amyloliquefaciens subsp. plantarum FZB42, B. velezensis, B. methylotrophicus KACC 13105, B. siamensis KCTC 13613, and B. amyloliquefaciens DSM7, the same strains used in this study. It should be noted that this classification does not designate these strains as a species but rather as a group. The same strains used in this study were also used to demonstrate the formation of the same monophyletic group, with a high degree of similarity between them. This suggests a reclassification of these strains as the same species due to the high degree of similarity in this study and similar applications in the agricultural industry. For example, B. velezensis is a heterotypic synonym of B. methylotrophicus, B. amyloliquefaciens subsp. plantarum, and B. oryzicola, which are used for the control of plant fungal diseases (Ngalimat et al., 2021).
The identification of these strains is crucial for a more comprehensive understanding of their potential applications and contributions in fields such as microbiology, genetics, bioinformatics, and biotechnology. The results of the comparative analysis revealed a close grouping and similarity between the isolates and the strains of the previously mentioned operational group, as well as between the isolates and B. vanillea XY18 and B. oryzicola KACC 18228 T. A comparative study by Xu et al. (2020) demonstrates and compares the applicability of strain FZB42 as a biofertiliser, biocontrol and probiotic (Xu et al., 2020). The study also considers the production of secondary metabolites and the antifungal and antibacterial activities of the strain. Additionally, other studies have demonstrated its capacity for fengycin production (Medeot et al., 2023) and probiotic activity (Reva et al., 2019). Moreover, our studies revealed that the FZB42 strain exhibited a high degree of similarity to the isolates.
Bacillus velezensis is a bacterium of significant importance in several industrial sectors, including food, farming, and biomedical. It has a variety of applications, including as a potential probiotic. Classifying these strains is essential to subsequently evaluate their gene repertoire for possible applications (Khalid et al., 2021; Huang et al., 2023; Sam-on et al., 2023).
Bacillus velezensis is an important specie but its identification is not yet correctly identified by MALDI-TOF MS. Identifying and classifying bacterial strains of this species is essential to facilitate differentiation from other strains within the Bacillus genus. This is due to the advent of more robust technologies for taxonomic analysis. The present study identified nine strains belonging to B. velezensis. It demonstrated the necessity for reclassifying other species of the same genus through next-generation sequencing and comparative genomic and phylogenomic analyses. Additionally, the functional annotation of exclusive genes provided critical insights into the ecological roles and adaptive capacities of the strains. The observed functional diversity, reflected in the predominant COG categories, revealed specific adaptations across the analyzed groups. Future analyses will be necessary to characterize each of these strains to better understand the applications of these isolates, given the high applicability of the species.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
ES: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. GC: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. MV: Conceptualization, Formal analysis, Investigation, Methodology, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. GG: Methodology, Writing – review & editing. DR: Data curation, Methodology, Writing – review & editing. FA: Data curation, Methodology, Writing – review & editing. BF: Writing – review & editing. MA: Writing – review & editing. MC: Writing – review & editing. EG: Conceptualization, Investigation, Writing – review & editing. BB: Investigation, Methodology, Writing – review & editing. SS: Conceptualization, Data curation, Formal analysis, Methodology, Resources, Supervision, Visualization, Writing – review & editing. VA: Conceptualization, Data curation, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – review & editing.
The author(s) declare that no financial support was received for the research and/or publication of this article.
The authors would like to acknowledge the Pró-Reitoria de Pesquisa—Universidade Federal de Minas Gerais, Rede de Ciências Ômicas (RECOM), the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG) for their financial support and fellowships.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
The authors declare that no Gen AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1544934/full#supplementary-material
2. ^https://github.com/OpenGene/fastp
3. ^https://quast.sourceforge.net/
4. ^https://github.com/chklovski/CheckM2
5. ^https://github.com/grp-bork/gunc
6. ^https://github.com/tseemann/prokka
8. ^https://github.com/Ecogenomics/GTDBTk
9. ^https://github.com/EnzoAndree/FastMLST
10. ^https://github.com/biobakery/phylophlan
12. ^https://lpsn.dsmz.de/subspecies/bacillus-amyloliquefaciens-plantarum
Adeniji, A. A., Loots, D. T., and Babalola, O. O. (2019). Bacillus velezensis: phylogeny, useful applications, and avenues for exploitation. Appl. Microbiol. Biotechnol. 103, 3669–3682. doi: 10.1007/s00253-019-09710-5
Adhikari, S., and Curtis, P. D. (2016). DNA methyltransferases and epigenetic regulation in bacteria. FEMS Microbiol. Rev. 40, 575–591. doi: 10.1093/FEMSRE/FUW023
Ågren, J., Sundström, A., Håfström, T., and Segerman, B. (2012). Gegenees: fragmented alignment of multiple genomes for determining Phylogenomic distances and genetic signatures unique for specified target groups. PLoS One 7:e39107. doi: 10.1371/JOURNAL.PONE.0039107
Alcaraz, L. D., Moreno-Hagelsieb, G., Eguiarte, L. E., Souza, V., Herrera-Estrella, L., and Olmedo, G. (2010). Understanding the evolutionary relationships and major traits of Bacillus through comparative genomics. BMC Genomics 11, 1–17. doi: 10.1186/1471-2164-11-332/FIGURES/4
Asnicar, F., Thomas, A. M., Beghini, F., Mengoni, C., Manara, S., Manghi, P., et al. (2020). Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0. Nat. Commun. 11, 1–10. doi: 10.1038/s41467-020-16366-7
Babraham Bioinformatics – FastQC A Quality Control tool for High Throughput Sequence Data (n.d.). Available online at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed January 30, 2024).
Barale, S. S., Ghane, S. G., and Sonawane, K. D. (2022). Purification and characterization of antibacterial surfactin isoforms produced by Bacillus velezensis SK. AMB Express 12, 1–20. doi: 10.1186/S13568-022-01348-3/TABLES/3
Bourgeois, J. S., Anderson, C. E., Wang, L., Modliszewski, J. L., Chen, W., Schott, B. H., et al. (2022). Integration of the Salmonella Typhimurium methylome and transcriptome reveals DNA methylation and transcriptional regulation are largely decoupled under virulence-related conditions. MBio 13:e03464-21. doi: 10.1101/2021.11.11.468322
Brutscher, L. M., Gebrechristos, S., Garvey, S. M., and Spears, J. L. (2024). Genetic and phenotypic characterization of Bacillus velezensis strain BV379 for human probiotic applications. Microorganisms 12:436. doi: 10.3390/microorganisms12030436
Cañas, C., Carrasco, B., Ayora, S., and Alonso, J. C. (2008). The RecU Holliday junction resolvase acts at early stages of homologous recombination. Nucleic Acids Res. 36, 5242–5249. doi: 10.1093/NAR/GKN500
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P., and Huerta-Cepas, J. (2021). eggNOG-mapper v2: functional annotation, Orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829. doi: 10.1093/MOLBEV/MSAB293
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P., and Parks, D. H. (2020). GTDB-Tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics 36, 1925–1927. doi: 10.1093/BIOINFORMATICS/BTZ848
Chen, S. (2023). Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using fastp. iMeta 2:e107. doi: 10.1002/IMT2.107
Chklovski, A., Parks, D. H., Woodcroft, B. J., and Tyson, G. W. (2023). CheckM2: a rapid, scalable and accurate tool for assessing microbial genome quality using machine learning. Nat. Methods 208, 1203–1212. doi: 10.1038/s41592-023-01940-w
Christensen, H., and Olsen, J. E. (2018). Sequence-based classification and identification of prokaryotes, 121–134. doi: 10.1007/978-3-319-99280-8_7
Ciufo, S., Kannan, S., Sharma, S., Badretdin, A., Clark, K., Turner, S., et al. (2018). Using average nucleotide identity to improve taxonomic assignments in prokaryotic genomes at the NCBI. Int. J. Syst. Evol. Microbiol. 68, 2386–2392. doi: 10.1099/ijsem.0.002809
Darling, A. C. E., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/GR.2289704
Dervyn, E., Planson, A. G., Tanaka, K., Chubukov, V., Guérin, C., Derozier, S., et al. (2023). Greedy reduction of Bacillus subtilis genome yields emergent phenotypes of high resistance to a DNA damaging agent and low evolvability. Nucleic Acids Res. 51, 2974–2992. doi: 10.1093/NAR/GKAD145
Diabankana, R. G. C., Shulga, E. U., Validov, S. Z., and Afordoanyi, D. M. (2022). Genetic characteristics and enzymatic activities of Bacillus velezensis KS04AU as a stable biocontrol agent against Phytopathogens. Int. J. Plant Biol. 13, 201–222. doi: 10.3390/ijpb13030018
Dong, X., Tu, C., Xie, Z., Luo, Y., Zhang, L., and Li, Z. (2022). The genome of Bacillus velezensis SC60 provides evidence for its plant probiotic effects. Microorganisms 10:767. doi: 10.3390/microorganisms10040767
Dos Santos, R. G., Seyffert, N., Dorneles, E. M. S., Aguiar, E. R. G. R., Ramos, C. P., Haas, D. J., et al. (2022). Exploring the MALDI Biotyper for the identification of Corynebacterium pseudotuberculosis biovar Ovis and Equi. J. Am. Soc. Mass Spectrom. 33, 2055–2062. doi: 10.1021/jasms.2c00174
Dunlap, C. A., Kim, S. J., Kwon, S. W., and Rooney, A. P. (2016). Bacillus velezensis is not a later heterotypic synonym of Bacillus amyloliquefaciens; Bacillus methylotrophicus, Bacillus amyloliquefaciens subsp. Plantarum and “Bacillus oryzicola” are later heterotypic synonyms of Bacillus velezensis based on phylogenomics. Int. J. Syst. Evol. Microbiol. 66, 1212–1217. doi: 10.1099/IJSEM.0.000858/CITE/REFWORKS
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/NAR/GKH340
Elshaghabee, F. M. F., Rokana, N., Gulhane, R. D., Sharma, C., and Panwar, H. (2017). Bacillus as potential probiotics: status, concerns, and future perspectives. Front. Microbiol. 8:271541. doi: 10.3389/FMICB.2017.01490/BIBTEX
Fan, B., Blom, J., Klenk, H. P., and Borriss, R. (2017). Bacillus Amyloliquefaciens, Bacillus Velezensis, and Bacillus Siamensis Form an “Operational Group B. Amyloliquefaciens” within the B. Subtilis Species Complex. Front. Microbiol. 8. doi: 10.3389/FMICB.2017.00022
Ferraz Helene, L. C., Klepa, M. S., and Hungria, M. (2022). New insights into the taxonomy of Bacteria in the genomic era and a case study with rhizobia. Int. J. Microbiol. 2022, 4623713–4623719. doi: 10.1155/2022/4623713
Gautreau, G., Bazin, A., Gachet, M., Planel, R., Burlot, L., Dubois, M., et al. (2020). PPanGGOLiN: depicting microbial diversity via a partitioned pangenome graph. PLoS Comput. Biol. 16:e1007732. doi: 10.1371/journal.pcbi.1007732
Guerrero-Araya, E., Muñoz, M., Rodríguez, C., and Paredes-Sabja, D. (2021). FastMLST: a multi-core tool for multilocus sequence typing of draft genome assemblies. Bioinform. Biol. Insights 15:11779322211059238. doi: 10.1177/11779322211059238
Gupta, R. K., Fuke, P., Khardenavis, A. A., and Purohit, H. J. (2023). In silico genomic characterization of Bacillus velezensis strain AAK_S6 for secondary metabolite and biocontrol potential. Curr. Microbiol. 80, 1–12. doi: 10.1007/S00284-022-03173-0/TABLES/4
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/BIOINFORMATICS/BTT086
Han, S. S., Jeong, Y. S., and Choi, S. K. (2021). Current scenario and challenges in the direct identification of microorganisms using MALDI TOF MS. Microorganisms 9:1917. doi: 10.3390/MICROORGANISMS9091917
Han, X., Shen, D., Xiong, Q., Bao, B., Zhang, W., Dai, T., et al. (2021). The plant-beneficial rhizobacterium Bacillus velezensis FZB42 controls the soybean pathogen phytophthora sojae due to bacilysin production. Appl. Environ. Microbiol. 87:e0160121. doi: 10.1128/AEM.01601-21
Hassan, M. K. (2021). In vitro pectate lyase activity and carbon uptake assays and whole genome sequencing of Bacillus amyloliquefaciens subsp. plantarum strains for a pectin defective pathway. bioRxiv. doi: 10.1101/2021.01.03.425148
Hassler, H. B., Probert, B., Moore, C., Lawson, E., Jackson, R. W., Russell, B. T., et al. (2022). Phylogenies of the 16S rRNA gene and its hypervariable regions lack concordance with core genome phylogenies. Microbiome 10, 1–18. doi: 10.1186/S40168-022-01295-Y
Holberger, L. E., Garza-Sánchez, F., Lamoureux, J., Low, D. A., and Hayes, C. S. (2012). A novel family of toxin/antitoxin proteins in Bacillus species. FEBS Lett. 586, 132–136. doi: 10.1016/J.FEBSLET.2011.12.020
Huang, X., Gaballa, A., Cao, M., and Helmann, J. D. (1999). Identification of target promoters for the Bacillus subtilis extracytoplasmic function σ factor, σW. Mol. Microbiol. 31, 361–371. doi: 10.1046/J.1365-2958.1999.01180.X
Huang, Y., Zhai, L., Chai, X., Liu, Y., Lv, J., Pi, Y., et al. (2024). Bacillus B2 promotes root growth and enhances phosphorus absorption in apple rootstocks by affecting MhMYB15. Plant J. 119, 1880–1899. doi: 10.1111/TPJ.16893
Huang, T., Zhang, Y., Yu, Z., Zhuang, W., and Zeng, Z. (2023). Bacillus velezensis BV01 has broad-Spectrum biocontrol potential and the ability to promote plant growth. Microorganisms 11:2627. doi: 10.3390/MICROORGANISMS11112627
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9, 1–8. doi: 10.1038/s41467-018-07641-9
Janda, J. M., and Abbott, S. L. (2007). 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. J. Clin. Microbiol. 45, 2761–2764. doi: 10.1128/JCM.01228-07
Jolley, K. A., Bliss, C. M., Bennett, J. S., Bratcher, H. B., Brehony, C., Colles, F. M., et al. (2012). Ribosomal multilocus sequence typing: universal characterization of bacteria from domain to strain. Microbiology 158, 1005–1015. doi: 10.1099/MIC.0.055459-0
Keshmirshekan, A., de Souza Mesquita, L. M., and Ventura, S. P. M. (2024). Biocontrol manufacturing and agricultural applications of Bacillus velezensis. Trends Biotechnol. 42, 986–1001. doi: 10.1016/J.TIBTECH.2024.02.003
Khalid, F., Khalid, A., Fu, Y., Hu, Q., Zheng, Y., Khan, S., et al. (2021). Potential of Bacillus velezensis as a probiotic in animal feed: a review. J. Microbiol. 59, 627–633. doi: 10.1007/s12275-021-1161-1
Kimura, T., Amaya, Y., Kobayashi, K., Ogasawara, N., and Sato, T. (2010). Repression of sigK intervening (skin) element gene expression by the CI-like protein SknR and effect of SknR depletion on growth of Bacillus subtilis cells. J. Bacteriol. 192, 6209–6216. doi: 10.1128/JB.00625-10
Kumar, S., Stecher, G., Suleski, M., Sanderford, M., Sharma, S., Tamura, K., et al. (2024). MEGA12: molecular evolutionary genetic analysis version 12 for adaptive and green computing. Mol. Biol. Evol. 41, 1–9. doi: 10.1093/MOLBEV/MSAE263
Kunst, F., Ogasawara, N., Moszer, I., Albertini, A. M., Alloni, G., Azevedo, V., et al. (1997). The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature 390, 249–256. doi: 10.1038/36786
Letunic, I., and Bork, P. (2024). Interactive tree of life (iTOL) v6: recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. 52, W78–W82. doi: 10.1093/NAR/GKAE268
Logan, N. A., and Vos, P. D. (2015). “Bacillus” in Bergey's manual of systematics of archaea and bacteria. Bergey’s Man. Syst. Archaea Bact. 1–163. doi: 10.1002/9781118960608
Medeot, D., Sannazzaro, A., Estrella, M. J., Torres Tejerizo, G., Contreras-Moreira, B., Pistorio, M., et al. (2023). Unraveling the genome of Bacillus velezensis MEP218, a strain producing fengycin homologs with broad antibacterial activity: comprehensive comparative genome analysis. Sci. Rep. 13, 1–14. doi: 10.1038/s41598-023-49194-y
Médigue, C., Moszer, I., Viari, A., and Danchin, A. (1995). Analysis of a Bacillus subtilis genome fragment using a co-operative computer system prototype. Gene 165, GC37–GC51. doi: 10.1016/0378-1119(95)00636-K
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H. P., and Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14, 1–14. doi: 10.1186/1471-2105-14-60
Meier-Kolthoff, J. P., and Göker, M. (2019). TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 10, 1–10. doi: 10.1038/s41467-019-10210-3
Meier-Kolthoff, J. P., Klenk, H. P., and Göker, M. (2014). Taxonomic use of DNA G+C content and DNA-DNA hybridization in the genomic age. Int. J. Syst. Evol. Microbiol. 64, 352–356. doi: 10.1099/ijs.0.056994-0
Mingchao, M., Wang, C., Ding, Y., Li, L., Shen, D., Jiang, X., et al. (2010). Complete genome sequence of Paenibacillus polymyxa SC2, a strain of plant growth-promoting Rhizobacterium with broad-Spectrum antimicrobial activity. J. Bacteriol. 193, 311–312. doi: 10.1128/JB.01234-10
Mulligan, E. A., and Dunn, J. J. (2008). Cloning, purification and initial characterization of E. coli McrA, a putative 5-methylcytosine-specific nuclease. Protein Expr. Purif. 62, 98–103. doi: 10.1016/J.PEP.2008.06.016
Ngalimat, M. S., Yahaya, R. S. R., Baharudin, M. M. A. A., Yaminudin, S. M., Karim, M., Ahmad, S. A., et al. (2021). A review on the biotechnological applications of the operational group Bacillus amyloliquefaciens. Microorganisms 9:614. doi: 10.3390/MICROORGANISMS9030614
Orakov, A., Fullam, A., Coelho, L. P., Khedkar, S., Szklarczyk, D., Mende, D. R., et al. (2021). GUNC: detection of chimerism and contamination in prokaryotic genomes. Genome Biol. 22:178. doi: 10.1186/s13059-021-02393-0
Pritchard, L., Cock, P., and Esen, Ö. (2019). Pyani v0.2.8: average nucleotide identity (ANI) and related measures for whole genome comparisons.
Ramasamy, D., Mishra, A. K., Lagier, J. C., Padhmanabhan, R., Rossi, M., Sentausa, E., et al. (2014). A polyphasic strategy incorporating genomic data for the taxonomic description of novel bacterial species. Int. J. Syst. Evol. Microbiol. 64, 384–391. doi: 10.1099/ijs.0.057091-0
Reva, O. N., Swanevelder, D. Z. H., Mwita, L. A., Mwakilili, A. D., Muzondiwa, D., Joubert, M., et al. (2019). Genetic, epigenetic and phenotypic diversity of four Bacillus velezensis strains used for plant protection or as probiotics. Front. Microbiol. 10:489002. doi: 10.3389/FMICB.2019.02610/BIBTEX
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 106, 19126–19131. doi: 10.1073/pnas.0906412106
Sam-on, M. F. S., Mustafa, S., Mohd Hashim, A., Yusof, M. T., Zulkifly, S., Malek, A. Z. A., et al. (2023). Mining the genome of Bacillus velezensis FS26 for probiotic markers and secondary metabolites with antimicrobial properties against aquaculture pathogens. Microb. Pathog. 181:106161. doi: 10.1016/J.MICPATH.2023.106161
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/BIOINFORMATICS/BTU153
Sisodia, R., Sarmadhikari, D., Mazumdar, P. A., Asthana, S., and Madhurantakam, C. (2024). Molecular analysis of dUTPase of Helicobacter pylori for identification of novel inhibitors using in silico studies. J. Biomol. Struct. Dyn. 42, 8598–8623. doi: 10.1080/07391102.2023.2247080
Sznyter, L. A., Slatko, B., Moran, L., O’Donnell, K. H., and Brooks, J. E. (1987). Nuclotide sequence of the Dde I restriction-modification system and characterization of the methylase protein. Nucleic Acids Res. 15, 8249–8266. doi: 10.1093/NAR/15.20.8249
Tang, C., Li, J., Shen, Y., Liu, M., Liu, H., Liu, H., et al. (2023). A sulfide-sensor and a sulfane sulfur-sensor collectively regulate sulfur-oxidation for feather degradation by Bacillus licheniformis. Commun. Biol. 6, 1–16. doi: 10.1038/s42003-023-04538-2
Velsko, I. M., Perez, M. S., and Richards, V. P. (2019). Resolving phylogenetic relationships for Streptococcus mitis and Streptococcus oralis through Core-and Pan-genome analyses. Genome Biol. Evol. 11, 1077–1087. doi: 10.1093/GBE/EVZ049
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:e1005595. doi: 10.1371/journal.pcbi.1005595
Xu, X., and Kovács, Á. T. (2024). How to identify and quantify the members of the Bacillus genus? Environ. Microbiol. 26:e16593. doi: 10.1111/1462-2920.16593
Keywords: identification, characterization, sequencing, Bacillus velezensis, genomic
Citation: Sousa EG, Campos GM, Viana MVC, Gomes GC, Rodrigues DLN, Aburjaile FF, Fonseca BB, de Araújo MRB, da Costa MM, Guedon E, Brenig B, Soares S and Azevedo V (2025) The research on the identification, taxonomy, and comparative genomics analysis of nine Bacillus velezensis strains significantly contributes to microbiology, genetics, bioinformatics, and biotechnology. Front. Microbiol. 16:1544934. doi: 10.3389/fmicb.2025.1544934
Edited by:
Shrabanti Chowdhury, Icahn School of Medicine at Mount Sinai, United StatesReviewed by:
Waheed Akram, University of the Punjab, PakistanCopyright © 2025 Sousa, Campos, Viana, Gomes, Rodrigues, Aburjaile, Fonseca, de Araújo, da Costa, Guedon, Brenig, Soares and Azevedo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eduarda Guimarães Sousa, ZWR1YXJkYWd1aW1hcmFlc3NvdXNhQGdtYWlsLmNvbQ==; Vasco Azevedo, dmFzY29hcmlzdG9uQGdtYWlsLmNvbQ==
†ORCID: Eduarda Guimarães Sousa, orcid.org/0000-0001-8326-3612
Gabriela Munis Campos, orcid.org/0000-0002-6903-8184
Marcus Vinícius Canário Viana, orcid.org/0000-0002-7017-6437
Gabriel Camargos Gomes, orcid.org/0009-0004-5048-7264
Diego Lucas Neres Rodrigues, orcid.org/0000-0003-2812-3072
Flavia Figueira Aburjaile, orcid.org/0000-0002-1067-1882
Belchiolina Beatriz Fonseca, orcid.org/0000-0001-8485-078X
Max Roberto Batista de Araújo, orcid.org/0000-0002-3293-8496
Mateus Matiuzzi da Costa, orcid.org/0000-0002-9884-2112
Eric Guedon, orcid.org/0000-0002-0901-4447
Bertram Brenig, orcid.org/0000-0002-7635-9656
Siomar Soares, orcid.org/0000-0001-7299-3724
Vasco Azevedo, orcid.org/0000-0002-4775-2280
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.