
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 01 April 2025
Sec. Food Microbiology
Volume 16 - 2025 | https://doi.org/10.3389/fmicb.2025.1543511
This article is part of the Research Topic Antimicrobial Resistance: Tracking and Tackling in the Food Chain View all articles
 Xinyang Zhu1Yucui Huang1Yuxia Shi1Xiaojie Gao1Duanduan Chen1Cheng Liu1
Xinyang Zhu1Yucui Huang1Yuxia Shi1Xiaojie Gao1Duanduan Chen1Cheng Liu1 Shengliang Cao1Xijuan Xue2
Shengliang Cao1Xijuan Xue2 Yubao Li3*
Yubao Li3*Clostridium perfringens (C. perfringens) is a foodborne pathogen that poses a significant threat to both animal husbandry and public health. In this study, 27 C. perfringens strains were isolated from animal samples and animal-derived food products. Antibiotics resistances among the isolates were phenotypically and genotypically analyzed and Whole genome sequencing (WGS). In combination with the genomes of 141 human-derived C. perfringens strains from public databases, this study conducted comprehensive analyses of antibiotic resistance genes, virulence genes, multilocus sequence typing (MLST), prophage detection, and pan-genome analysis for a total of 168 strains of C. perfringens. Antibiotics resistances among the isolates were phenotypically and genotypically analyzed and found 24 of them (88.9%, 24/27) were identified as multidrug-resistant (MDR). WGS analysis revealed that 13 strains belonged to known sequence types (ST), and the remaining strains represented 10 new STs. By analysis in combination with data of 141 C. perfringens isolates from the database, it was implied that ST221, ST72 and ST370 were present in both animal-derived and human-derived C. perfringens. It is worth noting that 108 out of 168 strains of C. perfringens (64.3%, 108/168) were found to carry prophages, which were found more prevalent in human-derived C. perfringens isolates. Pan-genome and phylogenetic analysis of 168 C. perfringens strains indicated that C. perfringens possesses an open pan-genome with genetic diversity. This study provides genomic insights into C. perfringens from food animals and humans, shedding light on the importance for monitoring the C. perfringens in livestock in China for better public health.
Clostridium perfringens (C. perfringens) is an anaerobic pathogen that belongs to Gram-positive bacteria. It is generally found in environments and animals and is regarded as one of the main determinants of gastrointestinal disorders in both humans and livestock (Scallan et al., 2011). This bacterium is primarily responsible for necrotic enteritis and enterotoxemia in animals especially in poultry, while in humans, it is often associated with food poisoning and gas gangrene (Guillaume et al., 2017). C. perfringens is known to produce over 20 different types of exotoxins which are considered to be their principal virulence factors (Rood et al., 2018), wherein the key toxins including α, β, ε, iota, cpe, and NetB toxin are of primary concerns (Navarro et al., 2018). C. perfringens can be classified into seven types (A - G) based on the carriage of these key toxins.
According to the Centers for Disease Control and Prevention (CDC), C. perfringens is ranked among the five most common food-borne pathogens in the United States (Ha et al., 2019). Outbreaks of C. perfringens have also been reported in meat retail establishments (Wittry et al., 2022). For instance, there were several reports of human food poisoning caused by C. perfringens in the United States in 1983 (Shandera et al., 1983). Additionally, reports indicated the presence of C. perfringens in from retail beef (Jiang et al., 2021), chicken (Xu et al., 2021) and duck (Liu et al., 2020) meat in China. In recent years, there have also been patients infected by C. perfringens that reported in Guangzhou (Huang et al., 2023), Hangzhou (Yan et al., 2024), and central China (Wang et al., 2021). Studies have demonstrated that C. perfringens are able to colonize in the intestines of newborns within 2 days post birth (Huang et al., 2023), and can cause severe neonatal necrotic enteritis (Li et al., 2022). Notably, the prevalence of drug resistance in C. perfringens has been increasing, particularly the animal-derived C. perfringens have been frequently found to be multidrug-resistant (Derongs et al., 2020; Liu et al., 2020). Considering their capability to transmit to anthropogenic community via food chain, these multidrug-resistant strains are thought to pose a substantial threat to human wellbeing and public health (Khandia et al., 2021).
The widespread application of high-throughput sequencing technology has greatly promoted the in-depth understanding toward C. perfringens. For example, a research in 2002 revealed that the relatively low GC contents in the genome of C. perfringens and confirmed that the presence of typical anaerobic fermentation enzymes responsible for gas production on its genome (Shimizu et al., 2002). In addition, further studies elucidating the virulence genes of C. perfringens has improved our understanding to their mechanisms associated with pathogenicity (Fourie et al., 2020; Mehdizadeh Gohari et al., 2021). And based on whole-genome analysis, researchers have established a new multi-locus sequence typing (MLST) method which provides a new perspective for accurately profiling the isolates (Abdel-Glil et al., 2021b). MLST is a well-established and widely used method for bacterial typing and phylogenetic analysis. It is based on the polymorphism of conserved housekeeping genes, which are essential for bacterial survival and are less likely to be affected by horizontal gene transfer (Jolley et al., 2018). This characteristic ensures MLST’s reliability and portability for cross-laboratory strain comparisons. However, MLST exhibits limitations in discriminatory power, particularly when differentiating closely related strains (Hepner et al., 2025). In contrast, genome-wide analyses such as core-genome methods provide greater resolution and accuracy in inferring evolutionary relationships by utilizing single nucleotide variations (SNPs). This approach captures finer genetic distinctions between strains. Compared to MLST, core-genome techniques are more effective at distinguishing strains that share the same sequence type (ST), offering enhanced sensitivity in tracing pathogen transmission routes and conducting epidemiological investigations (Yan et al., 2023). Some studies have pointed out that only 12.6% of the C. perfringens genome is assigned as the core genome, which indicated that C. perfringens possesses a pan-genome diversity (Kiu et al., 2017). Recent genomic and comparative studies on C. perfringens highlight its dynamic evolution and virulence mechanisms (Abdel-Glil et al., 2021a). Advances in pan-genome analyses reveal extensive toxin gene diversity linked to strain pathogenicity, driven by plasmid-mediated horizontal gene transfer (Gulliver et al., 2023). Comparative genomics underscores host-specific adaptations, such as poultry-associated necrotic enteritis strains harboring unique virulence loci (Meniaï et al., 2021). These findings unveil the valuable insights into the diversity and heterogeneity of C. perfringens.
In this study, we investigated the genome sequences of 27 C. perfringens strains isolated from Shandong, China, and comprehensively analyzed the genetic determinants of antibiotic resistances or virulences. Subsequently, the genome data of 141 human-derived C. perfringens were obtained from NCBI which have been subjected to a comparative genomic analysis.
Between 2021 and 2022, C. perfringens isolates were recovered from retail meat products in three cities of Shandong Province, China (Liaocheng, Weifang, and Linyi). Among 88 analyzed samples comprising 36 pork and 52 poultry, C. perfringens was detected in 19.4% (7/36) of pork and 38.5% (20/52) of poultry samples, totaling 27 isolates. The samples were carefully homogenized, then inoculated onto solid tryptone-sulfite-cycloserine agar (TSC, Qingdao Hope Bio-Technology Co., Ltd). The plates were incubated under anaerobic conditions at 37°C for 24 h, after which single colonies exhibiting the morphological characteristics of C. perfringens were selected and re-inoculated onto additional TSC plates. The selected single colonies were then transferred to brain heart infusion (BHI, Qingdao Hope Bio-Technology Co., Ltd) and cultured under anaerobic conditions at 37°C for 12 h. To profile the toxins, DNA were extracted the samples and applied for the polymerase chain reaction (PCR) using primers listed in Supplementary Table 1 (Miyamoto et al., 2012). Finally, the identified strains were kept and stored at −80°C. Additionally, we obtained C. perfringens genomes of Chinese origin deposited in the NCBI database (n = 141) (Supplementary Table 2).
Genomic DNA from C. perfringens was extracted using the FastPure Bacteria DNA Isolation Mini Kit-Box 2 (Nanjing Vazyme Biotechnology Co., Ltd.). Whole-genome sequencing was carried out by Novogene Technology Co., Ltd. (Beijing, China) using Illumina Novaseq-PE150 platform. All data were processed using fastp v0.23.4 to trim adapter and low-quality reads (Chen et al., 2018), and FastQC v0.12.1 (Wingett and Andrews, 2018). The clean reads were then assembled using Shovill v1.1.01 to construct genome assembly. The quality of genome assemblies was determined using Quast v5.2.0 (quast.py contigs.fasta) (Gurevich et al., 2013), and genome completeness/contamination was evaluated using CheckM v1.2.0 (Parks et al., 2015). A high-quality genome is defined as one exceeding 90% completeness with contamination levels below 5% (Parks et al., 2020). Genome annotation was performed using the NCBI Prokaryotic Genome Annotation Pipeline (PGAP v6.4) (Tatusova et al., 2016).
All antibacterial tablets used in this study were obtained from Hangzhou Microbial Reagent Co., Ltd. (Hangzhou, China). The susceptibility of C. perfringens to nine antibiotics from six different classes was assessed: lincomycin (2 μg), cefotaxime (30 μg), neomycin (30 μg), florfenicol (30 μg), amoxicillin (20 μg), tetracycline (30 μg), norfloxacin (10 μg), bacitracin (0.04 IU), and spectinomycin (100 μg). C. perfringens ATCC 13124 was used as a quality control strain. The susceptibility of these strains was categorized as resistant, moderately susceptible, or susceptible according to the Clinical and Laboratory Standards Institute (CLSI) criteria2 (Humphries et al., 2021). Multidrug-resistant bacteria (MDR) was defined as acquired non-susceptibility to at least one agent in three or more antimicrobial categories (Magiorakos et al., 2012).
The comparison of the C. perfringens genome was conducted using the Pathogen Virulence Factors Database (VFDB) database3 to identify the virulence factors (Bo et al., 2019). To predict the antibiotic resistance genes (ARGs), the Resistance Gene Identifier (RGI v6.0.2) tool was employed in combination with the Comprehensive Antibiotic Resistance Database (CARD v4.0.0)4 (Alcock et al., 2023). To minimize the inclusion of mis-assembled or mis-annotated antibiotic resistance genes (ARGs), we excluded protein-coding genes that met either of the following criteria: less than 70% amino acid sequence identity to the corresponding CARD reference ARG, or sequence length falling outside the 90%–130% range relative to the reference sequence (Wang and Dagan, 2024; Supplementary Table 3). Additionally, we utilized the online toolkit PHASTEST5 (David et al., 2016), to predict the loci of prophages within the C. perfringens and genome and to analyze the genomic characteristics of these prophages. PHASTEST scores are determined based on the number of coding sequences (CDS) present in the DNA sequence as well as the presence or absence of phage-related genes. Predicted prophage sequences were categorized into three categories: complete, putative, and incomplete. Heatmap visualization and similarity-based clustering analysis of the data were performed using TBtools-II (Chen et al., 2023).
The genomes of 27 C. perfringens strains were subjected to the PubMLST C. perfringens database (Jolley et al., 2018)6 and compared with the alleles and sequence types (ST) deposited in the database for multi-locus sequence typing (MLST) analysis. The online website CSI Phylogeny 1.47 (Kaas et al., 2014) was used to determine the SNPs and constructs core genome phylogeny through concatenated high-quality SNP alignments. The visualization was achieved using iTOL v68 (Letunic and Bork, 2021).
To study the phylogenetic relationships among C. perfringens, the GFF3 files of 168 isolates we generated using the script bp_genbank2gff3.pl were used as Roary (v3.13.0) (Page et al., 2015). To identify potential false-positive clusters, examine the Min group, Max group, and Avg group columns in the gene_presence_absence.csv file generated by Roary for discrepancies in gene length. If significant discrepancies are detected, extract the target cluster sequences from pan_genome.fasta, perform multiple sequence alignment using MAFFT (v.7.526) (Katoh et al., 2019) to assess consistency, and finally remove confirmed false-positive clusters from the gene_presence_absence.csv file. The input for extracting and aligning core genes (core_gene_alignment.aln) was processed with an 95% threshold to differentiate core genomes from non-essential and strain-specific genomes, for pan-genome and core gene analysis of all strains. A phylogenetic tree of C. perfringens was then constructed based on the core genome sequence using MEGA 11 software (Tamura et al., 2021), subsequently visualized by iTOL v6 (Letunic and Bork, 2021).
All statistical analyzes were performed using the GraphPad Prism 9.0 (GraphPad Software Inc., San Diego, CA, USA) and SPSS version 19.0 (IBM Corporation, USA). The concordance between ARGs and antimicrobial resistance phenotypes was evaluated using the kappa coefficient as documented previously (Landis and Koch, 1977).
Among the 24 of the total C. perfringens strains were isolated from poultry meat and intestines, while the other 3 were identified in the pork samples. Of these, 26 isolates were classified as type A, with only 1 strain was classified as type G (Supplementary Table 4). The genome sizes of the C. perfringens strain from 3.1 to 3.5 Mbp, with GC contents between 28 and 28.5%. The completeness of the genomes ranged from 92.6 to 98.9%, while the contamination levels were observed to be between 2.4 and 4.8%. Additionally, the N50 values varied significantly, ranging from 78,831 to 2,071,785 bp. The sequencing data have been deposited in NCBI, which was accessible from the accession numbers listed in Supplementary Table 5.
A total of nine resistance genes were identified in sequencing data of 27 C. perfringens isolates (Figure 1A). All strains were found to harbor one to five resistance genes, wherein the tetracycline resistance genes tetA (P) and tetB (P) were most prevalent (74.7 and 96.2%). Approximately 48.1% of the strains carried the lnu (P) resistance gene, which is responsible for the inactivation of lincosamide antibiotics. Additionally, there are two types of genes that mediated the resistances to macrolides. Specifically, 70.4% of the isolates were found with erm (Q) gene, while 7.4% of them contained the erm (B) gene. All 27 strains of C. perfringens isolates exhibited high phenotypic resistances to tetracycline, neomycin, and bacitracin, yet demonstrating sensitivities to cefotaxime, amoxicillin, and florfenicol. We also found that 24 isolates were classified as multidrug-resistant (MDR) by mediating resistance to several antibiotics of different classes (Figure 1B). Of note, the tight association between resistance phenotypes and genotypes was only observed in tetracycline class, with a kappa coefficient of approximately 1. However, the remainings scarcely showed the significant correlation (Figure 1C).
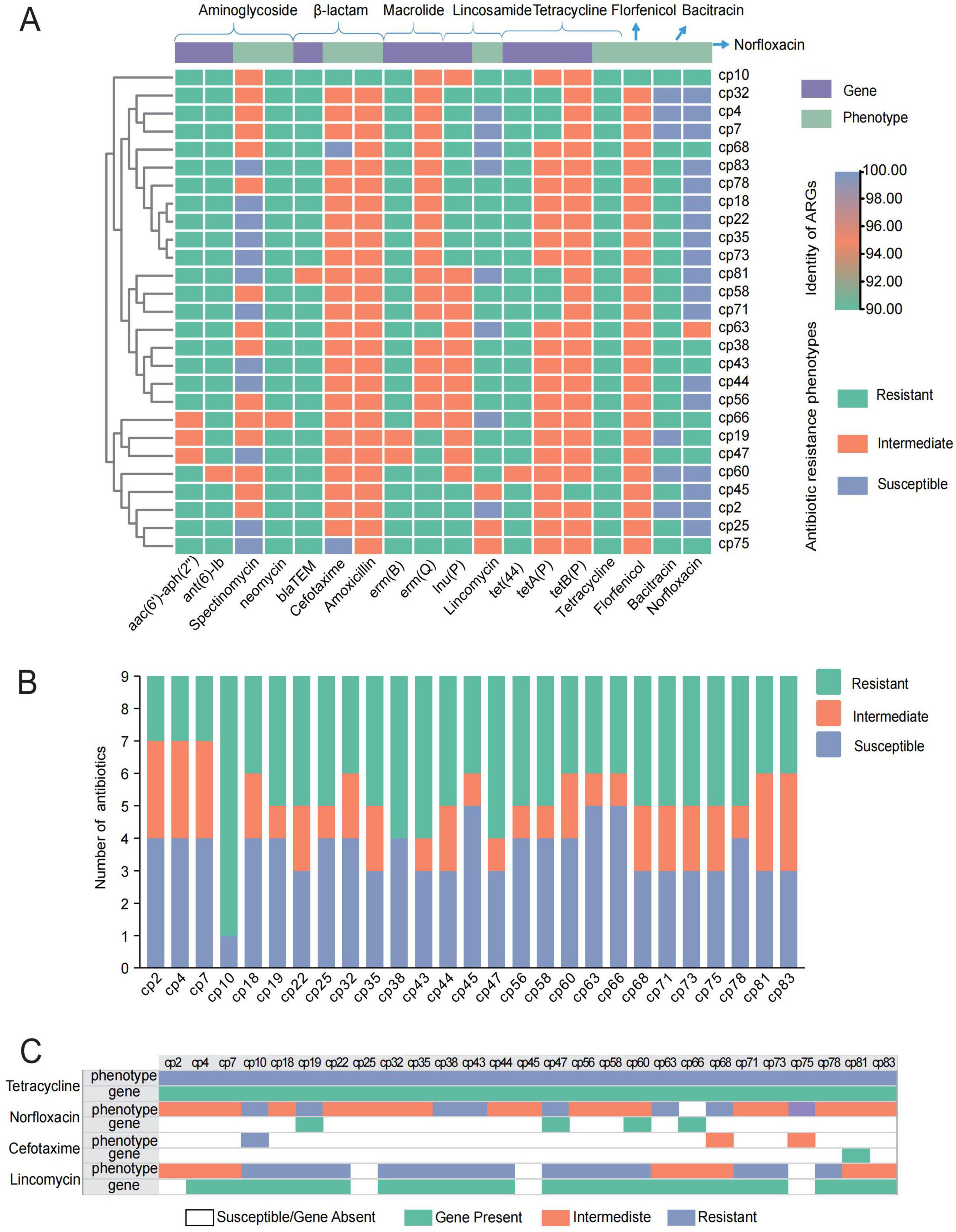
Figure 1. Results of C. perfringens resistance analysis. (A) Heatmap: antimicrobial resistance phenotypes and predicted ARGs; Dendrogram: clustering rows with similar data. (B) Antibiotic resistance of each C. perfringens strain; (C) comparison of drug-resistant phenotypes and ARGs correlations in 27 strains of C. perfringens.
To provide a comprehensive overview of the virulence factors of C. perfringens, we first profiled the virulence genes of isolates in the current study as well as the data from the database (Figure 2A). Among the 1all analyzed strains, type A C. perfringens presented as the majority of the C. perfringens (94.6%, 159/168). A total of 16 genes with significant nucleotide similarity to the known virulence factors among all analyzed strains. Specifically, all strains were identified the genes encoding α-toxin gene (plc), and over 70% of them carried the genes responsible for productions of toxins like α-clostripain (cloSI), k-toxin (colA), θ-toxin (pfo), hyaluronidase (nagH, nagI, nagJ, nagK, nagL), and sialidase (nanH, nanI, and nanJ). It was noteworthy that the 5 strains from human origin were found with abundant virulence factors. However, there was no significant difference (P > 0.05) in the numbers of virulence factors among the C. perfringens from either animal or human origin (Figure 2B). Virulence genes related to typing genes other than plc were detected in 9 strains, including one C. perfringens type D strain, and one C. perfringens type G strain along with seven strains of C. perfringens type F (Figure 2C).
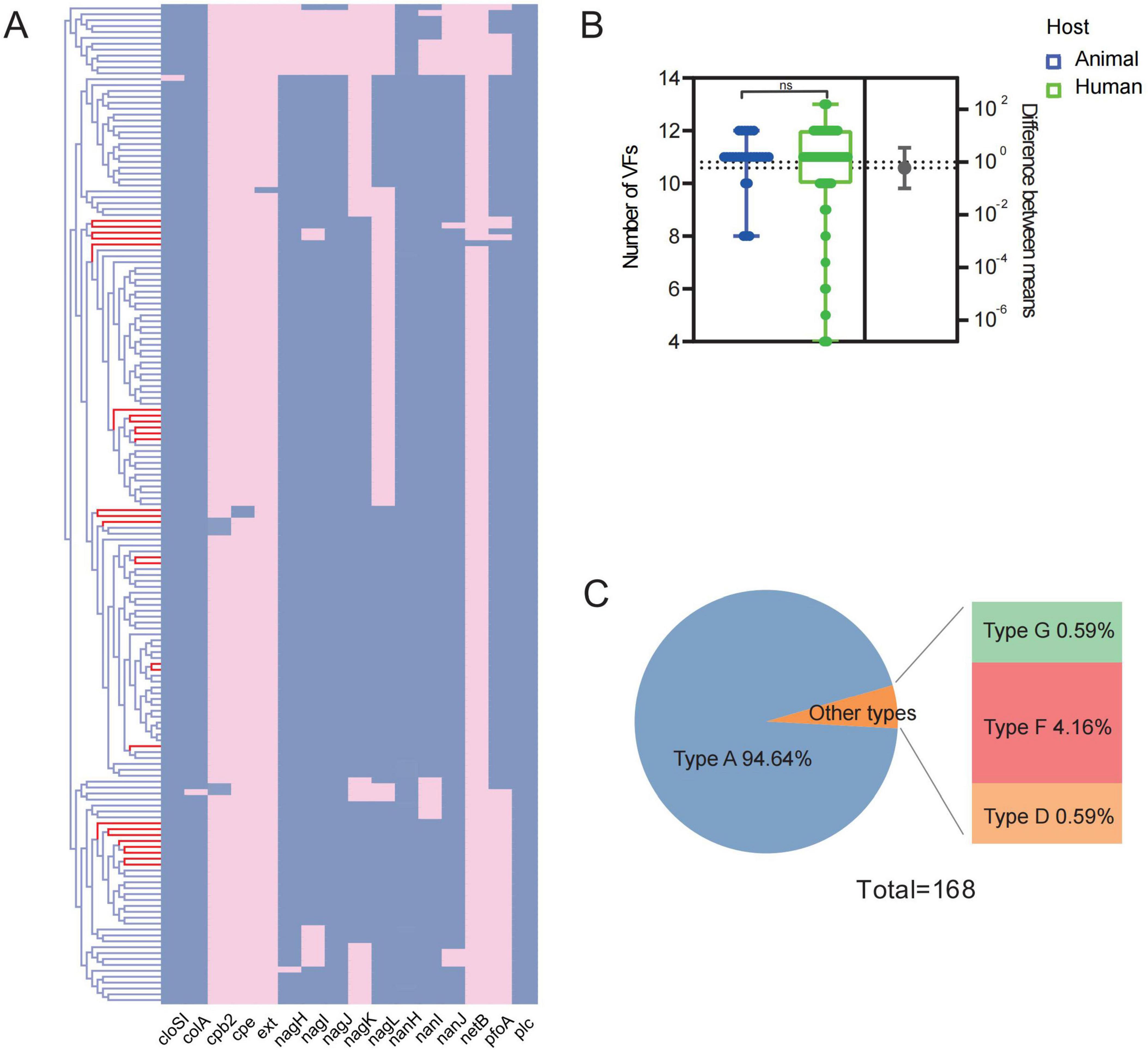
Figure 2. Results of virulence gene analysis. (A) Heatmap: prediction results of virulence genes; Dendrogram: clustering rows with similar data (red branches indicating strains of animal origin); (B) comparison of the number in virulence genes of C. perfringens from human and animal sources; (C) typing ratio of 168 C. perfringens strains.
A further sequence mining on the genome data of 168 C. perfringens was performed and indicated the presence of prophages in 108 of them. This collection includes 142 complete prophages, 23 putative prophages and 15 incomplete prophages. The 180 prophages were categorized into 37 distinct phage types, with the clostridial phage vB CpeS-CP51 as the most prevalent type (29/180, 26.9%). This was the followed by the clostridial phage phiCT19406C (22/108, 20.4%), clostridial phage PhiS63 (21/108, 19.4%) and clostridial phage phi3626 (20/108, 18.5%) (Figure 3A). Further analysis of the prophage sequences (Figures 3B, C) showed that the C. perfringens prophage genome lengths were ranging from 6.7 to 66.4 kb and the GC contents were 21.7%–38.5%, respectively. The lengths of the complete prophage were around 13.4–57.6 Kb, and the GC contents were 21.7%–38.5%. The lengths of the putative prophage were 14–66.4 Kb and the GC contents were 26.9%–43.3%. Incomplete prophages were found with the smallest size of 6.7–30.8 Kb, whereas the GC contents were 22.5%–34.9%. Our statistical analysis, conducted using one-way analysis of variance (ANOVA) and Independent samples t-test in SPSS software, revealed that the numbers of prophages in human-derived strains were significantly higher than those in animal-derived strains (P < 0.01), but there were no significant differences in the lengths and GC contents (P > 0.05). The sequences of complete prophages were significantly longer than that of the suspected prophages and incomplete prophages (P < 0.05), while no significance was observed in terms of the GC contents (P > 0.05).
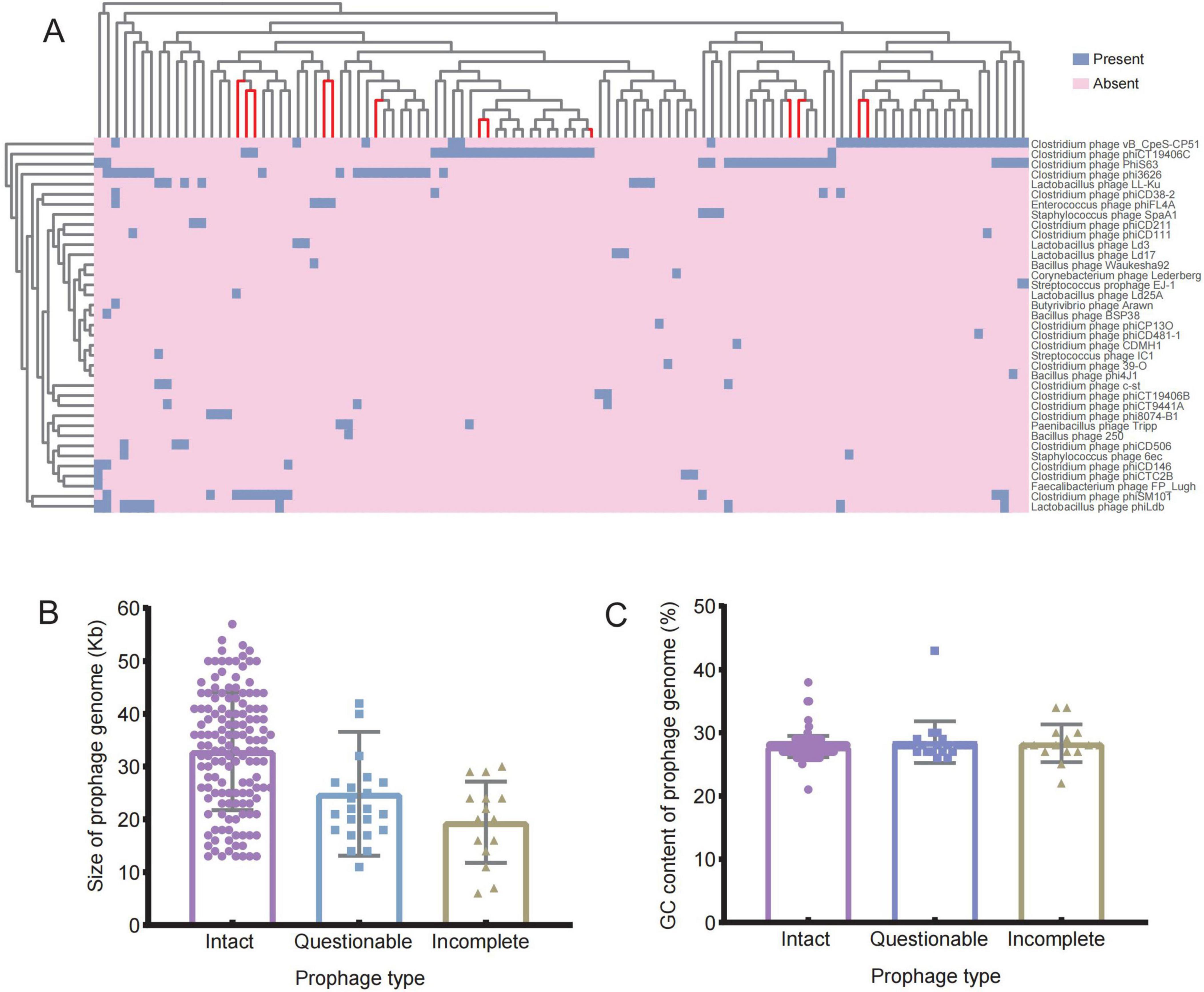
Figure 3. Prophages in the C. perfringens genome by the prophages. (A) Heatmap: the prophage profile of 168 C. perfringens genomes; Dendrogram: clustering rows with similar data (red branches indicate strains of animal origin). (B) Genome sizes of complete, putative and incomplete prophages; (C) GC content of intact, questionable and incomplete prophages.
The genome sequences of C. perfringens isolates were subjected to the comparative genomics using PubMLST C. perfringens database, and 13 of 27 C. perfringens isolates were assigned to be the known ST. Nonetheless, 14 unknown STs have been identified in the C. perfringens isolates from the current study (Supplementary Table 6). In a more comprehensive view, there were a total of 103 STs among the C. perfringens strains including both our isolates and the isolates from the database. There were differences amongst STs between human- or animal-derived C. perfringens isolates. Among human-derived C. perfringens isolates, ST221 (n = 5) was the most dominant ST, followed by ST62 (n = 4) and ST408 (n = 4). the most dominant ST among food-animal-derived strains was ST72 (n = 3). However, strains of the same ST from different sources showed no significant clustering differences (Figure 4). The 10 major STs of animal-derived strains were classified into 6 clusters. ST221, ST72, ST370, ST210, and ST353 were also detected in human-originated strains.
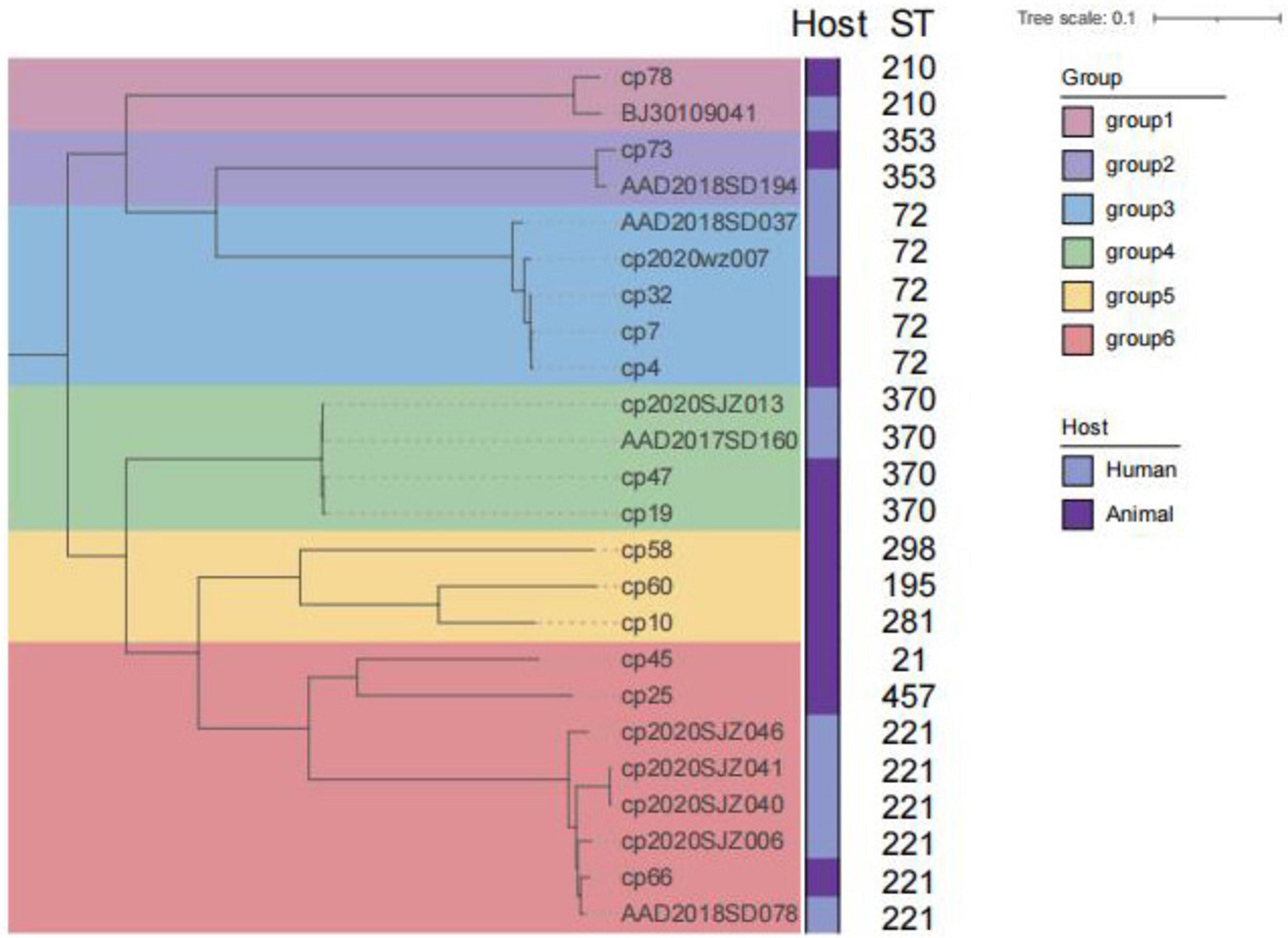
Figure 4. Phylogenetic tree based on the core genome of 24 strains of C. perfringens. The core genome of C. perfringens ATCC 13124 was used as outgroup. The six colors on the tree represent the six groups of analytical identification. The bootstrap values for each branch are all 1 (100%). The scale bar corresponds to 0.1 substitutions per site.
The genomic data of C. perfringens strains from this study and the database were analyzed and an open pan-genomes was determined with the pan-gene counts increasing proportionally (Figure 5A). The pan-genome of C. perfringens strains comprised 19,289 genes, encompassing 1,558 (9.8%) genes in the core genome, 333 (2.1%) genes in the soft core genome, 1,445 (9.1%) genes in the shell genome, and 15,953 (79.1%) genes in the cloud genome (Figure 5B). Genome-based phylogenetic analysis of the core genomes from the tested C. perfringens isolates revealed four major clusters, with the third and fourth clusters further classified into two subclusters (Figure 5C). Food-animal-derived source isolates were distributed within 6 clusters, indicating high diversity. However, there were also some interesting findings. For example, animal-derived isolates cp2, cp63 and cp75 together with human-derived ST421 strains (B021 and cp2020SJZ003) constituted a main lineage. Type A isolates (cp78 and BJ30109041, ST210) were closely related to the type F strain AAD2017SD113, which belongs to ST448 and has been associated with antibiotic-associated diarrhea in humans. It was worth noting that animal-derived C. perfringens cp47 and human-derived AAD2017SD160 showed high identity in Single Nucleotide Polymorphism (SNP). The SNP counts between animal-derived strains cp4, cp7 and cp66 and human-derived strains AAD2018SD078 as well as the cp2020wz007 were less than 395. This finding suggested that food animal served as a potential source of C. perfringens infection in humans. In addition, it was interesting that all animal-derived C. perfringens isolates in this study demonstrated a sharply different genomic traits with differences of more than 11,000 SNPs. This result indicated the potential diversity among C. perfringens genomes within the same region.
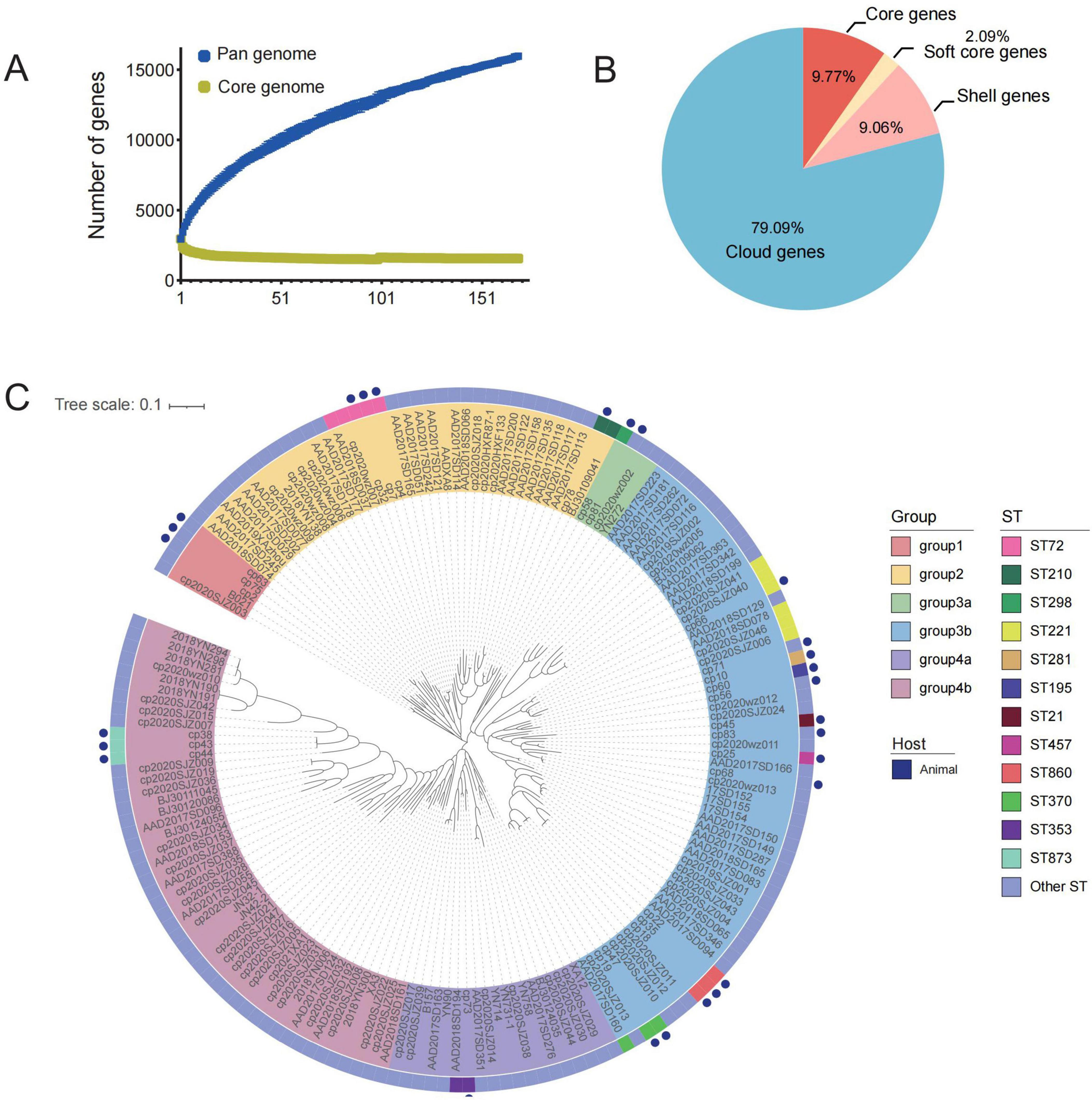
Figure 5. Results of pan-genomic analysis of C. perfringens. (A) Trend map of pan-genome; (B) the pan-genomes of these isolates were determined by comparing the pan-genomes, core genes, shell genes and cloud genes of the isolated C. perfringens; (C) phylogenetic tree analysis of the core genome of 168 strains of C. perfringens.
Food poisoning caused by C. perfringens is frequently associated with contaminated meat products (Hu et al., 2018). In recent years, this pathogen has received increasing attention globally due to its potential threat to both human and animal (Talukdar et al., 2024). With the rapid development of whole-genome sequencing technology, the use of bioinformatics to study the pathogenicity, drug resistance, and genetic diversity of C. perfringens provides insights into the prevention and treatment of C. perfringens (Abdel-Glil et al., 2021a). In this study, we successfully isolated 27 strains of C. perfringens from the markets in Shandong, of which 26 strains were classified as type A and 1 strain as type G. The genome sizes ranged from 3.1 to 3.5 M bp, with GC contents ranging from 28 to 28.5%. This data, combined with the sequencing data of human-derived C. perfringens in China from database, was further employed for phylogenetic and functional analysis.
Numerous studies have revealed that the emergence of MDR C. perfringens poses a serious threat to public health (Khandia et al., 2021; Priya et al., 2023). Our study demonstrated the high levels of antibiotic resistance among C. perfringens isolates to tetracycline (100%, 27/27), lincomycin (59.3%, 16/27) and bacitracin (77.8%, 21/27). These high rates were likely owing to the tremendous usage of such antibiotics in poultry farming (Żbikowska et al., 2020). Among the 27 isolates, 24 (88.9%) exhibited resistance to three or more antibiotics. Previous studies have outlined that many C. perfringens isolates demonstrated resistance to a broad range of antibiotic classes including tetracyclines (2019), lincosamides (Udhayavel et al., 2017), aminoglycosides, and macrolides (Camargo et al., 2022). The global emergence and spread of MDR pathogens pose an unneglected challenge to therapeutic regimen based on antibiotics (Krishnamurthy et al., 2016). Previous investigations demonstrated that tetracycline resistance has been a prevalent drug-resistant phenotype among C. perfringens isolates, and most resistance genes are capable to transfer via plasmid conjugation (Park et al., 2010). Additionally, the resistance of C. perfringens to macrolide antibiotics has been well-documented (Baquero and Reig, 1992). The primary mechanism underlying this resistance is often mediated by the erm and mef genes. In our study, 19 (70.4%) of the 27 were found to carry the erm gene. Notably, the erm gene not only confers the resistance to macrolide antibiotics but also mediates the tolerance to lincosamide and streptogramin B. Furthermore, the dissemination of erm genes between species by mobile genetic elements has also been reported (Soge et al., 2009). Collectively, these findings suggested that tetracycline and macrolide resistance genes are prevalent in C. perfringens from both animal and human origins.
In this study, the discrepancy between phenotypes and genotypes of antibiotic resistances were observed except for tetracyclines and lincosamide drugs. This may be attributed to the presence of multiple resistance mechanisms. The relationship between ARGs and phenotypic resistance is complex and frequently oversimplified in public discourse. Although ARGs are routinely identified genomic sequencing technologies, their presence does not necessarily equate to phenotypic resistance. In fact, many ARGs require specific regulatory conditions to manifest resistance, such as when cloned into expression vectors or mutated to enhance expression (Perry et al., 2022). Moreover, environmental factors and genetic context also play crucial roles in determining whether ARGs confer phenotypic resistance. For example, the mobilization of ARGs by mobile genetic elements can lead to overexpression and resistance, while their presence in chromosomal locations may not result in significant phenotypic changes (Dai et al., 2022). We have learned that the combination of multiple software tools can provide a more comprehensive detection of genes resistance. For example, integrating the results from CARD and AMRFinderPlus (Feldgarden et al., 2021) can enhance the detection of resistance genetic determinants. We plan to incorporate AMRFinderPlus into our future analyses to ensure a more comprehensive detection of resistance genetic elements. This will provide additional support for our further analysis of the relationship between resistance genes and resistance phenotypes.
C.perfringens induces disease through the production of various toxins and enzymes. Notably, they are known to release over 20 distinct toxins, which serve as the primary mechanism for eliciting histotoxic pathogenesis in both humans and animals (Mehdizadeh Gohari et al., 2021). Most genes for encoding toxins in C. perfringens, particularly those relevant to strain typing, are conserved, thereby facilitating identification via PCR and sequencing (Feng et al., 2020). This study corresponded to this point. Type A and Type C of C. perfringens represent a substantial neglected threat to food hygiene and safety, as evidenced by the elucidation from this study. It is widely acknowledged that cpa is the primary toxin responsible for gas gangrene (Stevens et al., 1997), the pfoA gene plays a synergistic role alongside cpa in myonecrosis by promoting tissue destruction and stable persistence (Katayama et al., 2015). The toxin production is the significant traits for C. perfringens pathogenicity in human cases. Studies have shown that pfoA-positive strains are a common cause for the necrotic enteritis in infants (Kiu et al., 2023). Studies have shown that C. perfringens strains carrying cpb2 are often found in the intestinal tracts of livestock and human (van Asten et al., 2010). Among human-derived strains, three strains of them were identified belonging to types D and F, respectively. According to the previous reports, the type D isolates mainly infect goats and sheep (Soge et al., 2009). These findings indicate that humans may be infected through contact with contaminated animals or food.
Prophages are recognized as significant contributors to the evolution of bacterial hosts (Fortier and Sekulovic, 2013). Through horizontal gene transfer, prophages are able to induce functional alterations in host bacteria by modifying the global transcriptomics. In our study, 108 of tested C. perfringens strains (63.9%) were found to carry complete prophages. Previous reports indicated that prophages constitute a relatively substantial portion of bacterial genomes, comprising up to 10%–20% of the genetic material of host bacteria (Casjens, 2003), In contrast, the prophages identified in this study only constitute approximately 1.04% of the entire bacterial genome, which is notably smaller than those observed in E. coli and Salmonella (Bobay et al., 2013). Our findings reveal that the most prevalent prophages within the C. perfringens genome are Clostridium phages that have been previously isolated and documented, aligning with the prior studies (Feng et al., 2020). Notably, the sequences of Clostridium phage vB CpeS-CP51, a prophage induced by mitomycin C (Gervasi et al., 2013), is the most abundant in the C. perfringens genome. This also suggested that tackling the spread of C. perfringens by inducing or lytic actions of prophages through mitomycin C is possible.
In this study, we found that the numbers of prophages in the human-derived strains were significantly higher than those in the animal-derived strains. This may be related to the unique selective pressures of the host environment, as the human body provides a complex and diverse habitat that favors the survival of bacterial strains with enhanced adaptability. Prophages, as mobile genetic elements, can provide bacteria with new genes that aid in survival, such as ARGs and VFGs. For instance, studies have shown that prophages in human-impacted environments, including those from human isolates, often carry a higher load of ARGs and VFGs, which can enhance bacterial fitness and virulence (Liao et al., 2024). Moreover, the high density and diversity of bacterial populations in human-associated habitats promote horizontal gene transfer (HGT). Prophages are known to facilitate HGT through transduction, allowing genes to be exchanged between different bacterial species. This process is particularly prevalent in human-derived isolates due to the close proximity and frequent interactions among diverse bacterial taxa within the human microbiome (Ku et al., 2025).
The identification of 10 novel STs in C. perfringens isolates holds significant epidemiological importance, especially in understanding cross-species transmission dynamics and zoonotic spillover risks. These STs may represent genetic adaptations that enhance pathogen fitness in diverse hosts or environments, potentially altering transmission efficiency between animals and humans (Fu et al., 2020). The emergence of novel STs may also reflect ecological niche expansion, similar to what has been observed in New Campylobacter linked to environmental persistence and low infectious doses (Cookson et al., 2024). From a public health perspective, the identification of these new STs underscores the need for revised diagnostic frameworks capable of detecting these emerging variants and for tailored interventions targeting high-risk interfaces, such as livestock markets or healthcare settings. Future studies should prioritize the functional validation of ST-specific virulence traits and evaluate their impact on outbreak trajectories using agent-based transmission models.
Our pan-genome analysis indicates that C. perfringens possesses an open pan-genome characterized by a relatively small core genome, suggesting high genomic plasticity. This finding is consistent with a recent study analyzing 372 C. perfringens genomes from diverse sources, which identified only 959 core genome genes within a pan-genome of 35,876 genes, accounting for just 2.7% (AlJindan et al., 2023). Similarly, another study demonstrated that C. perfringens strains carrying the becAB genes were distributed across distinct lineages and horizontally transmitted via a conserved Pcp plasmid (Fang et al., 2025), further confirming the species’ genomic plasticity. To address potential annotation errors encountered during our use of Roary, we implemented manual correction of high-frequency false-positive gene clusters. These quality control steps helped reduce inaccuracies in core genome representation. Furthermore, we observed that Panaroo offers significant advantages in resolving annotation errors during pan-genome analysis. Studies indicate that compared to Roary, Panaroo achieves more accurate identification of pairwise SNPs and effectively corrects diverse types of annotation errors through its graph-based algorithm (Tonkin-Hill et al., 2020). We have incorporated Panaroo into our future research plans, particularly for scenarios involving complex genome rearrangements or large-scale pan-genome studies.
This implied that C. perfringens genomes are highly plastic, plausibly acquiring favorable genetic materials from the surrounding environment for enhancing its adaptability (Feng et al., 2020). Notably, the utilization of pan-genome analysis has proven effective in elucidating the evolutionary trajectory of pathogenic bacteria (Stott and Bobay, 2020). Based on the core genome analysis of analyzed C. perfringens strains, we found that shared STs exist between animal-derived and human-derived strains, specifically ST221, ST72, and ST370. Of note, ST72 was isolated from a farm in Tai’an, Shandong province, as reported in previous studies (Xiu et al., 2020). Studies have shown that human-derived isolates are closely related to chicken-derived strains isolated from retail markets (Xu et al., 2021). It is important to highlight that the SNP sites of strains from different origins within the shared ST types only exhibit minor differences. It sheds the light on the possibility for outbreaks caused by C. perfringens (Abdel-Glil et al., 2021b).
This study provides valuable insights into the genetic and epidemiological characteristics of C. perfringens in China, particularly in Shandong Province, the country’s largest poultry-producing region. However, its geographically restricted scope limits the ability to comprehensively assess the pathogen’s global transmission and evolutionary dynamics. Specifically, the global distribution and genetic diversity of C. perfringens are known to vary across regions due to differences in environmental conditions, host interactions, and agricultural practices (Ba et al., 2024). Consequently, reliance on Chinese strains alone may not fully capture the evolutionary trajectories or transmission patterns observed in other parts of the world. For example, strains adapted to China’s intensive farming systems may exhibit distinct genetic or phenotypic traits compared to those from regions with alternative livestock management strategies (Yanxia et al., 2025). Therefore, while this work advances our understanding of C. perfringens within a Chinese context, its relevance to broader global patterns of zoonotic spread and pathogen evolution remains constrained. To fully understand the pathogen’s global adaptability and transmission mechanisms, future studies will include strains from more diverse regions. This approach will improve the generalizability of our findings.
In this study, we conducted a comprehensive whole-genome analysis of 27 C. perfringens isolates from the markets in Shandong, China. They were subjected to a comparative genomic analysis with data of 141 C. perfringens database. By profiling the molecular characteristics, the understanding toward the genetic diversity of antibiotic resistances and virulences among the C. perfringens. Eventually, the in-depth analysis also shed the light on the evolutionary trajectory and transmission dynamics of C. perfringens, as well as the emerging risks of these pathogens to livestock industry and public health.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in this article/Supplementary material.
XZ: Writing – original draft, Conceptualization, Data curation, Writing – review and editing. YH: Conceptualization, Data curation, Writing – original draft. YS: Conceptualization, Data curation, Writing – original draft. XG: Conceptualization, Data curation, Writing – original draft. DC: Writing – review and editing. CL: Writing – review and editing. SC: Writing – review and editing. XX: Writing – review and editing. YL: Funding acquisition, Project administration, Supervision, Writing – review and editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This work was financially supported by the National Natural Science Foundation of China (No. 32372957) and the Natural Science Foundation of Shandong Province (ZR2023QC082).
We thank all the contributors of this Research Topic and we wish you all a good reading.
XX was employed by Shandong Sinder Technology Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1543511/full#supplementary-material
Abdel-Glil, M. Y., Thomas, P., Linde, J., Busch, A., Wieler, L. H., Neubauer, H., et al. (2021a). Comparative in silico genome analysis of Clostridium perfringens unravels stable phylogroups with different genome characteristics and pathogenic potential. Sci. Rep. 11:6756. doi: 10.1038/s41598-021-86148-8
Abdel-Glil, M. Y., Thomas, P., Linde, J., Jolley, K. A., Harmsen, D., Wieler, L. H., et al. (2021b). Establishment of a publicly available core genome multilocus sequence typing scheme for clostridium perfringens. Microbiol. Spectr. 9:e0053321. doi: 10.1128/Spectrum.00533-21
Alcock, B. P., Huynh, W., Chalil, R., Smith, K. W., Raphenya, A. R., Wlodarski, M. A., et al. (2023). CARD 2023: Expanded curation, support for machine learning, and resistome prediction at the comprehensive antibiotic resistance database. Nucleic Acids Res. 51, D690–D699. doi: 10.1093/nar/gkac920
AlJindan, R., AlEraky, D. M., Farhat, M., Almandil, N. B., AbdulAzeez, S., and Borgio, J. F. (2023). Genomic insights into virulence factors and multi-drug resistance in clostridium perfringens IRMC2505A. Toxins (Basel) 15:359. doi: 10.3390/toxins15060359
Ba, X., Jin, Y., Ning, X., Gao, Y., Li, W., Li, Y., et al. (2024). Clostridium perfringens in the intestine: Innocent bystander or serious threat? Microorganisms 12:1610. doi: 10.3390/microorganisms12081610
Baquero, F., and Reig, M. (1992). Resistance of anaerobic bacteria to antimicrobial agents in Spain. Eur. J. Clin. Microbiol. Infect. Dis. 11, 1016–1020. doi: 10.1007/bf01967792
Bo, L., Dandan, Z., Qi, J., Lihong, C., and Jian, Y. (2019). VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 47, D687–D692.
Bobay, L. M., Rocha, E. P., and Touchon, M. (2013). The adaptation of temperate bacteriophages to their host genomes. Mol. Biol. Evol. 30, 737–751. doi: 10.1093/molbev/mss279
Camargo, A., Guerrero-Araya, E., Castañeda, S., Vega, L., Cardenas-Alvarez, M. X., Rodríguez, C., et al. (2022). Intra-species diversity of Clostridium perfringens: A diverse genetic repertoire reveals its pathogenic potential. Front. Microbiol. 13:952081. doi: 10.3389/fmicb.2022.952081
Casjens, S. (2003). Prophages and bacterial genomics: What have we learned so far? Mol. Microbiol. 49, 277–300. doi: 10.1046/j.1365-2958.2003.03580.x
Chen, C., Wu, Y., Li, J., Wang, X., Zheng, Z., Xu, J., et al. (2023). TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 16, 1733–1742. doi: 10.1016/j.molp.2023.09.010
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Cookson, A. L., Burgess, S., Midwinter, A. C., Marshall, J. C., Moinet, M., Rogers, L., et al. (2024). New campylobacter lineages in New Zealand freshwater: Pathogenesis and public health implications. Environ. Microbiol. 26:e70016. doi: 10.1111/1462-2920.70016
Dai, D., Brown, C., Bürgmann, H., Larsson, D. G. J., Nambi, I., Zhang, T., et al. (2022). Long-read metagenomic sequencing reveals shifts in associations of antibiotic resistance genes with mobile genetic elements from sewage to activated sludge. Microbiome 10:20. doi: 10.1186/s40168-021-01216-5
David, A. R., Grant, J., Ana, M., Tanvir, S., Allison, P., Yongjie, L., et al. (2016). PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21.
Derongs, L., Druilhe, C., Ziebal, C., Le Maréchal, C., and Pourcher, A. M. (2020). Characterization of Clostridium Perfringens isolates collected from three agricultural biogas plants over a one-year period. Int. J. Environ. Res. Public Health 17:5450. doi: 10.3390/ijerph17155450
Fang, M., Yuan, Y., Fox, E. M., Wu, K., Tian, X., Zhang, L., et al. (2025). Prevalence and genomic characteristics of becAB-carrying Clostridium perfringens strains. Food Microbiol. 125:104640. doi: 10.1016/j.fm.2024.104640
Feldgarden, M., Brover, V., Gonzalez-Escalona, N., Frye, J. G., Haendiges, J., Haft, D. H., et al. (2021). AMRFinderPlus and the reference gene catalog facilitate examination of the genomic links among antimicrobial resistance, stress response, and virulence. Sci. Rep. 11:12728. doi: 10.1038/s41598-021-91456-0
Feng, Y., Fan, X., Zhu, L., Yang, X., Liu, Y., Gao, S., et al. (2020). Phylogenetic and genomic analysis reveals high genomic openness and genetic diversity of Clostridium perfringens. Microb. Genom. 6:mgen000441. doi: 10.1099/mgen.0.000441
Fortier, L. C., and Sekulovic, O. (2013). Importance of prophages to evolution and virulence of bacterial pathogens. Virulence 4, 354–365. doi: 10.4161/viru.24498
Fourie, J. C. J., Bezuidenhout, C. C., Sanko, T. J., Mienie, C., and Adeleke, R. (2020). Inside environmental Clostridium perfringens genomes: Antibiotic resistance genes, virulence factors and genomic features. J. Water Health 18, 477–493. doi: 10.2166/wh.2020.029
Fu, S., Wei, D., Yang, Q., Xie, G., Pang, B., Wang, Y., et al. (2020). Horizontal plasmid transfer promotes the dissemination of asian acute hepatopancreatic necrosis disease and provides a novel mechanism for genetic exchange and environmental adaptation. mSystems 5:e0799-19. doi: 10.1128/mSystems.00799-19
Gervasi, T., Curto, R. L., Narbad, A., and Mayer, M. J. (2013). Complete genome sequence of ΦCP51, a temperate bacteriophage of Clostridium perfringens. Arch. Virol. 158, 2015–2017. doi: 10.1007/s00705-013-1647-1
Guillaume, D., Jacques, T., Naike, B., and Eric, M. (2017). Clostridium perfringens related spleen gangrene. Intensive Care Med. 43, 1730–1731.
Gulliver, E. L., Adams, V., Marcelino, V. R., Gould, J., Rutten, E. L., Powell, D. R., et al. (2023). Extensive genome analysis identifies novel plasmid families in Clostridium perfringens. Microb. Genom. 9:mgen000995. doi: 10.1099/mgen.0.000995
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: Quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Ha, E., Chun, J., Kim, M., and Ryu, S. (2019). Capsular polysaccharide is a receptor of a Clostridium perfringens bacteriophage CPS1. Viruses 11:1002.
Hepner, S., Jolley, K. A., Castillo-Ramirez, S., Mourkas, E., Dangel, A., Wieser, A., et al. (2025). A core genome MLST scheme for Borrelia burgdorferi sensu lato improves insights into the evolutionary history of the species complex. Cell Rep. Methods 5:100935. doi: 10.1016/j.crmeth.2024.100935
Hu, W. S., Kim, H., and Koo, O. K. (2018). Molecular genotyping, biofilm formation and antibiotic resistance of enterotoxigenic Clostridium perfringens isolated from meat supplied to school cafeterias in South Korea. Anaerobe 52, 115–121. doi: 10.1016/j.anaerobe.2018.06.011
Huang, K. Y., Liang, B. S., Zhang, X. Y., Chen, H., Ma, N., Lan, J. L., et al. (2023). Molecular characterization of Clostridium perfringens isolates from a tertiary children’s hospital in Guangzhou, China, establishing an association between bacterial colonization and food allergies in infants. Gut Pathog. 15:47. doi: 10.1186/s13099-023-00572-x
Humphries, R., Bobenchik, A. M., Hindler, J. A., and Schuetz, A. N. (2021). Overview of changes to the clinical and laboratory standards institute performance standards for antimicrobial susceptibility testing, M100, 31st edition. J. Clin. Microbiol. 59:e0021321. doi: 10.1128/jcm.00213-21
Jiang, H., Qin, Y. M., Yang, X. T., Li, Q. L., Shen, Q. C., Ding, J. B., et al. (2021). Bacteriological and molecular typing of Clostridium perfringens strains isolated in retail beef in Beijing, China. J. Vet. Med. Sci. 83, 1593–1596. doi: 10.1292/jvms.21-0129
Jolley, K. A., Bray, J. E., and Maiden, M. C. J. (2018). Open-access bacterial population genomics: BIGSdb software, the website and their applications. Wellcome Open Res. 3:124. doi: 10.12688/wellcomeopenres.14826.1
Kaas, R. S., Leekitcharoenphon, P., Aarestrup, F. M., and Lund, O. (2014). Solving the problem of comparing whole bacterial genomes across different sequencing platforms. PLoS One 9:e104984. doi: 10.1371/journal.pone.0104984
Katayama, S., Tagomori, M., Morita, N., Yamasaki, T., Nariya, H., Okada, M., et al. (2015). Determination of the Clostridium perfringens-binding site on fibronectin. Anaerobe 34, 174–181. doi: 10.1016/j.anaerobe.2014.11.007
Katoh, K., Rozewicki, J., and Yamada, K. D. (2019). MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 20, 1160–1166. doi: 10.1093/bib/bbx108
Khandia, R., Puranik, N., Bhargava, D., Lodhi, N., Gautam, B., and Dhama, K. (2021). Wound infection with multi-drug resistant Clostridium perfringens: A case study. Arch. Razi Inst. 76, 1565–1573. doi: 10.22092/ari.2021.355985.1757
Kiu, R., Caim, S., Alexander, S., Pachori, P., and Hall, L. J. (2017). Probing genomic aspects of the multi-host pathogen Clostridium perfringens reveals significant pangenome diversity, and a diverse array of virulence factors. Front. Microbiol. 8:2485. doi: 10.3389/fmicb.2017.02485
Kiu, R., Shaw, A. G., Sim, K., Acuna-Gonzalez, A., Price, C. A., Bedwell, H., et al. (2023). Particular genomic and virulence traits associated with preterm infant-derived toxigenic Clostridium perfringens strains. Nat. Microbiol. 8, 1160–1175. doi: 10.1038/s41564-023-01385-z
Krishnamurthy, M., Moore, R. T., Rajamani, S., and Panchal, R. G. (2016). Bacterial genome engineering and synthetic biology: Combating pathogens. BMC Microbiol. 16:258. doi: 10.1186/s12866-016-0876-3
Ku, H., Kelk, D., Bauer, D. C., and Sidhu, J. P. S. (2025). Phage-plasmid hybrids as vectors for antibiotic resistance in environmental Escherichia coli. Sci. Total Environ. 959:178157. doi: 10.1016/j.scitotenv.2024.178157
Landis, J. R., and Koch, G. G. (1977). The measurement of observer agreement for categorical data. Biometrics 33, 159–174.
Letunic, I., and Bork, P. (2021). Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Li, Z., Yan, C., Gong, X., and Wang, J. (2022). Severe intravascular hemolysis from Clostridium perfringens septicemia in a neonate with necrotizing enterocolitis in China: A case report. Infect. Drug Resist. 15, 1461–1465. doi: 10.2147/idr.S355621
Liao, H., Liu, C., Zhou, S., Liu, C., Eldridge, D. J., Ai, C., et al. (2024). Prophage-encoded antibiotic resistance genes are enriched in human-impacted environments. Nat. Commun. 15:8315. doi: 10.1038/s41467-024-52450-y
Liu, Y., Xiu, L., Miao, Z., and Wang, H. (2020). Occurrence and multilocus sequence typing of Clostridium perfringens isolated from retail duck products in Tai’an region, China. Anaerobe 62:102102. doi: 10.1016/j.anaerobe.2019.102102
Magiorakos, A. P., Srinivasan, A., Carey, R. B., Carmeli, Y., Falagas, M. E., Giske, C. G., et al. (2012). Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 18, 268–281. doi: 10.1111/j.1469-0691.2011.03570.x
Mehdizadeh Gohari, I., Navarro, M., Li, J., Shrestha, A., Uzal, F., and McClane, B. (2021). Pathogenicity and virulence of Clostridium perfringens. Virulence 12, 723–753. doi: 10.1080/21505594.2021.1886777
Meniaï, I., Thibodeau, A., Quessy, S., Parreira, V. R., Fravalo, P., Beauchamp, G., et al. (2021). Putative antigenic proteins identified by comparative and subtractive reverse vaccinology in necrotic enteritis-causing Clostridium perfringens isolated from broiler chickens. BMC Genomics 22:890. doi: 10.1186/s12864-021-08216-7
Miyamoto, K., Li, J., and McClane, B. A. (2012). Enterotoxigenic Clostridium perfringens: Detection and identification. Microbes Environ. 27, 343–349. doi: 10.1264/jsme2.me12002
Navarro, M. A., McClane, B. A., and Uzal, F. A. (2018). Mechanisms of action and cell death associated with Clostridium perfringens toxins. Toxins 10:212.
Page, A., Cummins, C., Hunt, M., Wong, V., Reuter, S., Holden, M., et al. (2015). Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693.
Park, M., Rooney, A. P., Hecht, D. W., Li, J., McClane, B. A., Nayak, R., et al. (2010). Phenotypic and genotypic characterization of tetracycline and minocycline resistance in Clostridium perfringens. Arch. Microbiol. 192, 803–810. doi: 10.1007/s00203-010-0605-5
Parks, D. H., Chuvochina, M., Chaumeil, P. A., Rinke, C., Mussig, A. J., and Hugenholtz, P. (2020). A complete domain-to-species taxonomy for Bacteria and Archaea. Nat. Biotechnol. 38, 1079–1086. doi: 10.1038/s41587-020-0501-8
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Perry, E. K., Meirelles, L. A., and Newman, D. K. (2022). From the soil to the clinic: The impact of microbial secondary metabolites on antibiotic tolerance and resistance. Nat. Rev. Microbiol. 20, 129–142. doi: 10.1038/s41579-021-00620-w
Priya, G. B., Srinivas, K., Shilla, H., and Milton, A. A. P. (2023). High prevalence of multidrug-resistant, biofilm-forming virulent Clostridium perfringens in broiler chicken retail points in Northeast India. Foods 12:4185. doi: 10.3390/foods12224185
Rood, J. I., Adams, V., Lacey, J., Lyras, D., McClane, B. A., Melville, S. B., et al. (2018). Expansion of the Clostridium perfringens toxin-based typing scheme. Anaerobe 53, 5–10.
Scallan, E., Hoekstra, R., Angulo, F., Tauxe, R., Widdowson, M., et al. (2011). Foodborne illness acquired in the United States–Major pathogens. Emerg. Infect. Dis. 17, 7–15. doi: 10.3201/eid1701.p11101
Shandera, W. X., Tacket, C. O., and Blake, P. A. (1983). Food poisoning due to Clostridium perfringens in the United States. J. Infect. Dis. 147, 167–170. doi: 10.1093/infdis/147.1.167
Shimizu, T., Ohtani, K., Hirakawa, H., Ohshima, K., Yamashita, A., Shiba, T., et al. (2002). Complete genome sequence of Clostridium perfringens, an anaerobic flesh-eater. Proc. Natl. Acad. Sci. U S A. 99, 996–1001. doi: 10.1073/pnas.022493799
Soge, O. O., Tivoli, L. D., Meschke, J. S., and Roberts, M. C. (2009). A conjugative macrolide resistance gene, mef(A), in environmental Clostridium perfringens carrying multiple macrolide and/or tetracycline resistance genes. J. Appl. Microbiol. 106, 34–40. doi: 10.1111/j.1365-2672.2008.03960.x
Stevens, D. L., Tweten, R. K., Awad, M. M., Rood, J. I., and Bryant, A. E. (1997). Clostridial gas gangrene: Evidence that alpha and theta toxins differentially modulate the immune response and induce acute tissue necrosis. J. Infect. Dis. 176, 189–195. doi: 10.1086/514022
Stott, C. M., and Bobay, L. M. (2020). Impact of homologous recombination on core genome phylogenies. BMC Genomics 21:829. doi: 10.1186/s12864-020-07262-x
Talukdar, P. K., Banawas, S., and Sarker, M. R. (2024). Clostridium perfringens. Trends Microbiol. 7, 104–110. doi: 10.1016/j.tim.2024.09.009
Tamura, K., Stecher, G., and Kumar, S. (2021). MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. doi: 10.1093/molbev/msab120
Tatusova, T., DiCuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Tonkin-Hill, G., MacAlasdair, N., Ruis, C., Weimann, A., Horesh, G., Lees, J. A., et al. (2020). Producing polished prokaryotic pangenomes with the Panaroo pipeline. Genome Biol. 21:180. doi: 10.1186/s13059-020-02090-4
Udhayavel, S., Thippichettypalayam Ramasamy, G., Gowthaman, V., Malmarugan, S., and Senthilvel, K. (2017). Occurrence of Clostridium perfringens contamination in poultry feed ingredients: Isolation, identification and its antibiotic sensitivity pattern. Anim. Nutr. 3, 309–312. doi: 10.1016/j.aninu.2017.05.006
van Asten, A. J., Nikolaou, G. N., and Gröne, A. (2010). The occurrence of cpb2-toxigenic Clostridium perfringens and the possible role of the beta2-toxin in enteric disease of domestic animals, wild animals and humans. Vet. J. 183, 135–140. doi: 10.1016/j.tvjl.2008.11.005
Wang, B., Dong, W., Ma, L., Dong, Y., Wang, S., Yuan, Y., et al. (2021). Prevalence and genetic diversity of Clostridium perfringens Isolates in Hospitalized diarrheal patients from Central China. Infect. Drug Resist. 14, 4783–4793. doi: 10.2147/idr.S338593
Wang, Y., and Dagan, T. (2024). The evolution of antibiotic resistance islands occurs within the framework of plasmid lineages. Nat. Commun. 15:4555. doi: 10.1038/s41467-024-48352-8
Wingett, S. W., and Andrews, S. (2018). FastQ Screen: A tool for multi-genome mapping and quality control. F1000Res 7:1338. doi: 10.12688/f1000research.15931.2
Wittry, B. C., Holst, M. M., Anderberg, J., and Hedeen, N. (2022). Operational antecedents associated with Clostridium perfringens outbreaks in retail food establishments, United States, 2015-2018. Foodborne Pathog. Dis. 19, 209–216. doi: 10.1089/fpd.2021.0068
Xiu, L., Liu, Y., Wu, W., Chen, S., Zhong, Z., and Wang, H. (2020). Prevalence and multilocus sequence typing of Clostridium perfringens isolated from 4 duck farms in Shandong province. China. Poult. Sci. 99, 5105–5117. doi: 10.1016/j.psj.2020.06.046
Xu, W., Zhang, H., Hu, Z., Miao, Z., Zhang, Y., and Wang, H. (2021). Prevalence and multilocus sequence typing of Clostridium perfringens isolated from retail chicken products and diseased chickens in Tai’an region. China. Vet. Med. Sci. 7, 2339–2347. doi: 10.1002/vms3.616
Yan, S., Jiang, Z., Zhang, W., Liu, Z., Dong, X., Li, D., et al. (2023). Genomes-based MLST, cgMLST, wgMLST and SNP analysis of Salmonella Typhimurium from animals and humans. Comp. Immunol. Microbiol. Infect. Dis. 96:101973. doi: 10.1016/j.cimid.2023.101973
Yan, Z., Fu, B., Zhu, Y., Zhang, Y., Wu, Y., Xiong, P., et al. (2024). High intestinal carriage of Clostridium perfringens in healthy individuals and ICU patients in Hangzhou. China. Microbiol. Spectr. 12:e0338523. doi: 10.1128/spectrum.03385-23
Yanxia, S., Xuewei, W., Gang, L., and Wei, J. (2025). Analysis on characteristics and multilocus sequence typing of Clostridium perfringens in western China. J. Antimicrob. Chemother. 80, 216–226. doi: 10.1093/jac/dkae399
Keywords: Clostridium perfringens, whole genome sequencing, multidrug-resistant bacteria, toxinotypes, pangenome analysis
Citation: Zhu X, Huang Y, Shi Y, Gao X, Chen D, Liu C, Cao S, Xue X and Li Y (2025) Comparative genomic analysis of food-animal-derived and human-derived Clostridium perfringens isolates from markets in Shandong, China. Front. Microbiol. 16:1543511. doi: 10.3389/fmicb.2025.1543511
Received: 11 December 2024; Accepted: 05 March 2025;
Published: 01 April 2025.
Edited by:
Dario De Medici, Italian National Institute of Health (ISS), ItalyReviewed by:
Mostafa Y. Abdel-Glil, Friedrich-Loeffler-Institut, GermanyCopyright © 2025 Zhu, Huang, Shi, Gao, Chen, Liu, Cao, Xue and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yubao Li, bGl5dWJhb0BsY3UuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.