 Nagham Shiekh Suliman
Nagham Shiekh Suliman Reza Talaei-Hassanloui
Reza Talaei-Hassanloui Hamid Abachi
Hamid Abachi Sadegh Zarei
Sadegh Zarei Ebrahim Osdaghi
Ebrahim Osdaghi- 1Department of Plant Protection, College of Agriculture, University of Tehran, Karaj, Iran
- 2Department of Plant Protection, College of Agriculture, Isfahan University of Technology, Isfahan, Iran
Bacillus thuringiensis is the most important biological control agent against various agricultural pests. The bacterium taxonomically belongs to the Bacillus cereus group, which also contains human pathogenic species, e.g., Bacillus anthracis. Thus, precise identification and taxonomic delineation of candidate strains for agricultural usage is of high importance in terms of both public health and biosecurity. By October 2023, whole genome sequences (WGS) of 885 bacterial strains were labeled as B. thuringiensis in the NCBI GenBank database. This study investigates the taxonomic authenticity of those strains using DNA similarity indexes, i.e., average nucleotide identity (ANI) and digital DNA–DNA hybridization (dDDH). All strains were compared with the type strain of B. thuringiensis ATCC 10972T. WGS-based phylotaxonomic investigations showed that out of 885 strains 803 strains authentically belonged to B. thuringiensis while 82 strains were mislabeled as B. thuringiensis having dDDH and ANI values less than the acceptable threshold of 70 and 95% respectively, for prokaryotic species definition in comparison with the B. thuringiensis type strain. Among these 82 mislabeled strains, 39 strains need to be reclassified within the B. cereus group in the species B. anthracis (33 strains), Bacillus toyonensis (five strains), and Bacillus mycoides (one strain). Furthermore, four strains were identified as Bacillus tropicus while one strain belonged to each of the species Bacillus licheniformis, Bacillus paranthracis, and Bacillus weidmannii. The remaining 36 strains did not match with any known Bacillus species nor the species of other bacterial genera, thus they could be assigned to hypothetical new species. Results of the present study, on the one hand, pave the way of comprehensive taxonomic refinements within B. thuringiensis species. On the other hand, highlight the role of taxonomic investigations in targeting authentic B. thuringiensis strains for biological control purposes.
Introduction
Bacillus thuringiensis is a Gram-positive, rod-shaped, spore-forming bacterium, found usually in soil, grain dust, water, and dead insect bodies (Lambert and Peferoen, 1992). This entomopathogenic species (causeing disease on insects) has been used as a successful biological insecticide for more than 80 years being a specific, safe, and effective tool for controlling a wide variety of insect pests (Nazarian et al., 2009). Bacillus thuringiensis belongs to Bacillus cereus group, which also contains seven other species, i.e., Bacillus cereus sensu stricto, Bacillus anthracis, Bacillus weihenstephanensis, Bacillus cytotoxicus, Bacillus mycoides, Bacillus pseudomycoides, and Bacillus toyonensis (Guinebretière et al., 2013; Jiménez et al., 2013). To distinguish various species of this group from one another, phylogenetic analysis based on the sequences of housekeeping genes and investigation of virulence plasmids can play an important role. However, as virulence plasmids may be transferred or lost in evolution, they are not suitable for the classification (Liu et al., 2015). Furthermore, sole reliance on insecticidal crystal protein genes (cry) for identification of B. thuringiensis has some limitations. Comparative genomics data indicates that B. thuringiensis is not the only species that possesses these genes (Meric et al., 2018; Baek et al., 2019; Castillo-Esparza et al., 2019). Until recently, B. thuringiensis strains that were commercialized as pesticides did not have an available complete genome sequence in the public database (Rang et al., 2015; Zhu et al., 2015). Bacillus thuringiensis is classified into more than 85 subspecies, which are distinguished by the antigenic characteristics of the flagellar H-antigen (Lecadet et al., 1999; Jakhar et al., 2017).
Classification, nomenclature, and phylogenetic characterization are the three interrelated parts of prokaryotic taxonomy that have historically been recognized (Garrity, 2005). Classification of the genus Bacillus has changed significantly over time because of advancements in molecular biology techniques (Maughan and Van der Auwera, 2011). Based on phenotypic similarity metrics, 16S rRNA sequences, rep-PCR (repetitive extragenic palindromic), and DNA–DNA hybridization (DDH) in addition to the presence or absence of virulent plasmids, the bacteria in this genus have historically been divided into several species (Rasko et al., 2005). DNA–DNA hybridization (DDH) has been used since 1960s to elucidate taxonomic relationships between bacteria. This method was widely used as it provides comprehensive comparisons between organisms. Wayne et al. (1987) suggested a 70% DDH value as the gold standard for defining prokaryotic species. For species circumscription, the new gold standards are digital whole genome comparisons using average nucleotide identity (ANI) or genome-to-genome-distance calculations (GGDCs) (Konstantinidis and Tiedje, 2005; Richter and Rosselló-Móra, 2009; Meier-Kolthoff et al., 2013). Next-generation sequencing technologies have led to a change in the approaches used in microbial taxonomy. According to Patil and McHardy (2013), digital DNA–DNA hybridization (dDDH) has replaced classical DDH. For instance, using the dDDH approach Liu et al. (2015) found that some strains previously classified as B. cereus or B. thuringiensis were B. anthracis. Furthermore, considering the evolutionary relationships of bacteria, multilocus sequence analysis (MLSA) is a valuable tool for classifying related strains (Gevers et al., 2005). Because it is based on the allelic differences between several conserved housekeeping genes, MLSA has become a strong tool for classifying bacterial strains (Clarke et al., 2002).
Altogether, correct and precise identification of B. thuringiensis within a well-defined taxonomic framework is a prerequisite for initiating biological application of the bacterium. Further, differentiation of B. thuringiensis strains from taxonomically closely related species, i.e., B. cereus and B. anthracis is crucial for biotechnology advancement, food health, and bio-pesticides industry. Hence, this study aims to investigate if all genome sequences deposited as B. thuringiensis in the NCBI GenBank database authentically belong to this species. To meet this goal, we have retrieved all publicly available whole genome resources labeled as B. thuringiensis in the NCBI GenBank database. The genome sequences were subjected to phylotaxonomic analyses using standard criteria in the taxonomy of prokaryotic species. Results showed that among 885 genome sequences named as B. thuringiensis, 82 strains (9.26%) did not belong to this species and were identified as members of either previously defied Bacillus species or hypothetical new species within the genus.
Materials and methods
Data collection
In October 2023, all publicly available genome sequences labeled as B. thuringiensis in the NCBI GenBank database were retrieved and subjected to phylotaxonomic analyses. Furthermore, the gyrB gene sequence in B. thuringiensis ATCC 10972T was used to find putative mislabeled B. thuringiensis strains in the NCBI GenBank database with the BLAST engine. In total, 885 whole-genome sequences that were classified as B. thuringiensis were obtained from the NCBI GenBank database and subjected to the following analyses (Supplementary Table S1). The genome quality evaluation was conducted using the BUSCO software, a widely recognized tool for assessing genome assembly completeness by examining the presence of conserved single-copy orthologs (Seppey et al., 2019). The version employed in this study was v5.2.2, which offers robust and standardized metrics for genome quality assessment Manni et al. (2021).
Phylogenetic analyses
All genome sequences were uploaded to the Galaxy Europe platform 1for phylogenetic analysis and comparative genomics. Prokka v1.14.6 was used to reannotate all genome sequences on the Galaxy Europe platform. The resulting gff3 files were then utilized to build a pan-genome using Roary v3.13.0 (Goecks et al., 2010; Page et al., 2015; Seemann, 2014; Stein, 2013). MEGA software v7.0 was used to visualize the resulting phylogenetic trees (Kumar et al., 2016). For each clade of the core genome-based phylogenetic tree, dDDH and ANI were calculated in comparison with the type strain of B. thuringiensis ATCC 10972T. Members of the clades that had a strain with ANI and dDDH less than 95 and 70% respectively, with B. thuringiensis ATCC 10972T were selected for further analyses.
In order to elucidate if the results of core genome-based phylotaxonomic analyses were in congruence with the routine MLSA, all 885 whole-genome sequences were subjected to the five-gene-based phylogenetic analyses as recommended for Bacillus spp. (Guinebretière et al., 2013). Hence, sequences of the five housekeeping genes atpD, dnaK, gyrB, rpoB, and rpoD were extracted from the whole genome sequences of all strains using the BLAST engine of NCBI. The sequences were aligned using MAFFT online service v7.490 2 (Katoh et al., 2019) and subjected to phylogenetic analyses either alone or in concatenation of five housekeeping genes. The phylogenetic trees were constructed and visualized via MEGA software v7.0 using the recommended procedure.
Taxonomic refinements
Preliminary phylogenetic analyses showed that out of 885 whole-genome sequences retrieved from the NCBI GenBank with B. thuringiensis label, 82 strains clustered separately from the core members of the species. To determine the taxonomic relationships of these 82 strains that were possibly incorrectly classified as B. thuringiensis, and define a taxonomic status for a particular taxon, the ANI and dDDH indices were calculated for all these strains. The latter analyses showed that the 82 strains had dDDH and ANI less than the accepted threshold for the definition of prokaryotic species with the type strain of B. thuringiensis ATCC 10972T (Kim et al., 2014). Thus, for these 82 strains, ANI was calculated by comparing them with each other and with type strains of Bacillus spp. using ANIm (Pritchard et al., 2016). dDDH was also calculated using the Genome-to-Genome Distance Calculator v.3.1 (Meier-Kolthoff et al., 2022).
According to the list of prokaryotic names with standing in nomenclature (LPSN)3, by November 2023, 110 Bacillus spp. species were validly published in the literature. Out of these 110 species, whole genome sequences of 97 species were publicly available as shown in Supplementary Table S2. Thus, all the latter 97 strains were included in the analyses. For whole-genome sequence-based classification of the bacterial strains, the genome assemblies were used as input for the pyANI v0.2.11 Python pipeline (Pritchard et al., 2016), with the ANIm parameter to calculate pairwise distances using MUMmer (nucmer). The pipeline generates a distance matrix and a double hierarchical clustered heatmap, in which a red color indicates ANI percent identities above 95%. In the result of pyANI, if one strain in comparison with a type strain had ANI and dDDH values more than 95 and 70% respectively, it was considered to belong to the species of the corresponding type strain.
Type Strain Genome Server (TYGS)4 by comparing the user genomes with its collection of type strains consisting of currently 21,334 microbial type strain genomes helps to understand taxonomic status of the query strain. The results are presented in the form of a 16S rRNA-based phylogenetic tree and whole genome-based phylogeny (Meier-Kolthoff and Göker, 2019). To determine the taxonomic status of the strains with low similarity to the B. thuringiensis type strain, and to verify the validity of the results reached by the WGS-based phylogenetic tree, MLSA-based phylogenetic tree, and pyANI, the genomes of 82 suspected strains were uploaded to the TYGS website and the resulting data was presented in the form of phylogenetic trees.
Results
By October 2023, the NCBI GenBank database contained 885 whole-genome sequences labeled as B. thuringiensis (Supplementary Table S1). Using ANI, dDDH, core genome-based phylogeny, and MLSA taxonomic authenticity of those 885 strains was investigated. Supplementary Table S1 highlights the strains with lower genome quality. These strains are also indicated within the MLSA-based phylogenetic tree presented in Supplementary Figure S1. Notably, four strains, i.e., SRR5713945-bin, XL6, F14-1, and s1783 are absent from this tree. Their exclusion is due to their failure to meet the minimum genomic criteria for constructing the phylogenetic tree, as they lack one or more of the five housekeeping genes atpD, dnaK, gyrB, rpoB, and rpoD used in MLSA-based phylogenetic analysis. This limitation underscores the importance of genome quality in ensuring the reliability and comprehensiveness of phylogenetic and comparative analyses. The strains missing from the MLSA phylogenetic tree further exemplify how incomplete genomic assemblies can hinder accurate evolutionary interpretations.
Out of 885 strains, 803 strains were confirmed to belong to B. thuringiensis, while 82 strains were incorrectly labeled as B. thuringiensis considering the above-mentioned criteria. The latter 82 strains had low DNA similarity indices with the B. thuringiensis type strain (Table 1). Out of 82 strains incorrectly classified as B. thuringiensis, 39 strains should be transferred to other species belonging to the B. cereus group including B. anthracis, B. toyonensis, and B. mycoides. Taxonomic status of 46 strains was clarified as follows: 33 strains belonged to B. anthracis (Supplementary Table S3). Five strains belonged to B. toyonensis (Table 2), and four strains were identified as B. tropicus (Table 3). Four strains were related to different species as follows: Et10/1 belonged to Bacillus licheniformis with 99.81 and 97.60% ANI and dDDH, respectively, BGSC 4BM1 belonged to B. mycoides with 97.37 and 74.80% ANI and dDDH, respectively, 4I3 belonged to Bacillus paranthracis with 97.43 and 77.30% ANI and dDDH respectively, and BGSC 4BV1 belonged to Bacillus weidmannii with 96.95 and 72.20% ANI and dDDH, respectively.
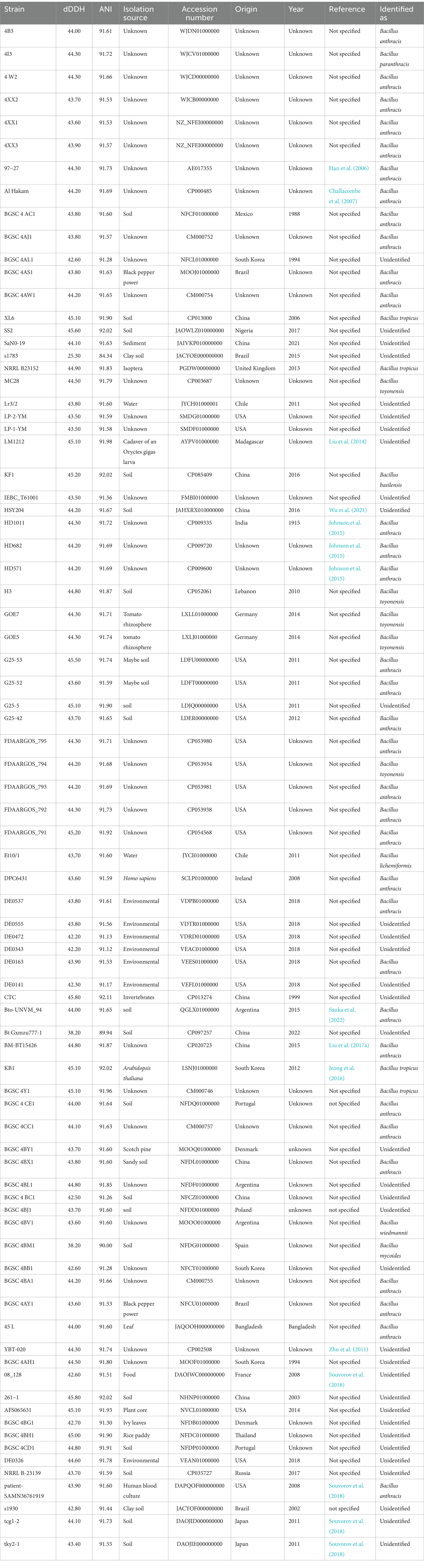
Table 1. The list of 82 strains mislabeled as Bacillus thuringiensis with ANI and DDH less than 95 and 70%, respectively, compared to type strain of the species.
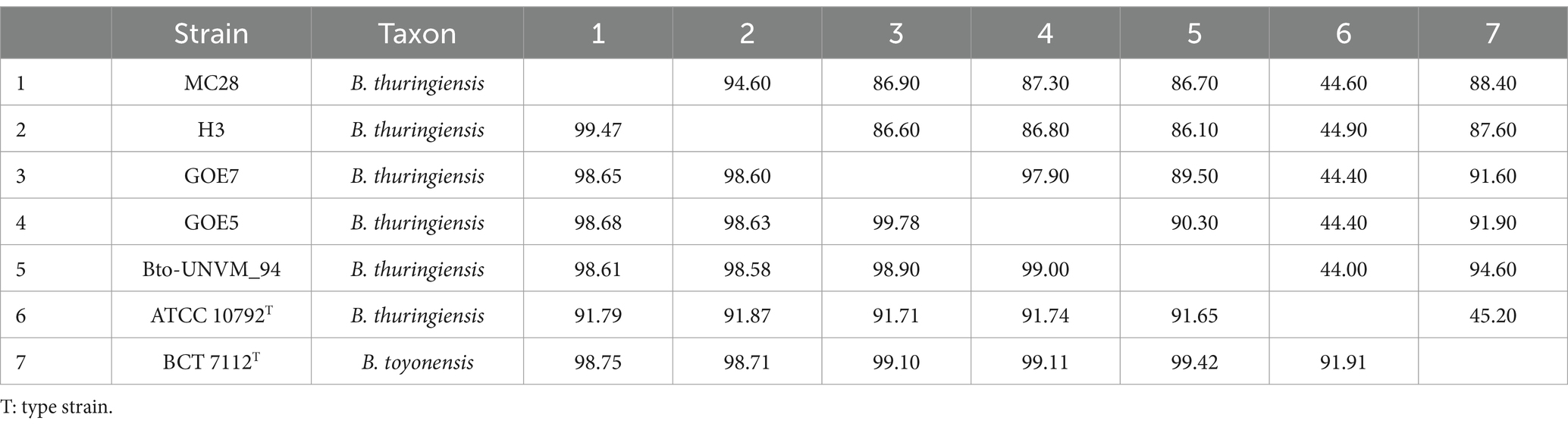
Table 2. Digital DNA–DNA hybridization (dDDH; upper diagonal) and average nucleotide identity (ANIm; lower diagonal) values generated from the DNA sequence similarity comparisons among different mislabeled Bacillus thuringiensis strains and the Bacillus toyonensis type strain.
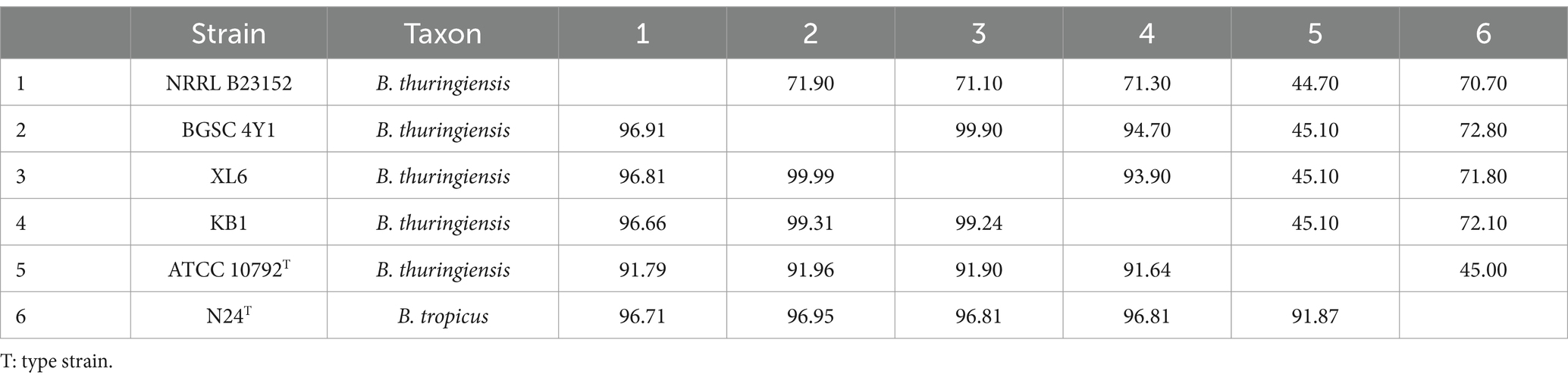
Table 3. Digital DNA–DNA hybridization (dDDH; upper diagonal) and average nucleotide identity (ANIm; lower diagonal) values generated from the DNA sequence similarity comparisons among different mislabeled Bacillus thuringiensis strains and type strain of Bacillus tropicus.
The remaining 36 strains did not match with any known Bacillus species nor the species of other bacterial genera, thus they could be assigned to hypothetical new species (Table 1). In the resulting core genome- and MLSA-based phylogenetic trees, all 82 strains that did not belong to B. thuringiensis were clustered together, demonstrating their distinct phylogenetic status (Supplementary Figure S1; Figure 1). On the other hand, BLAST-based explorations showed that three strains that were incorrectly labeled as B. toyonensis in the NCBI GenBank should belong to B. thuringiensis (Table 4).
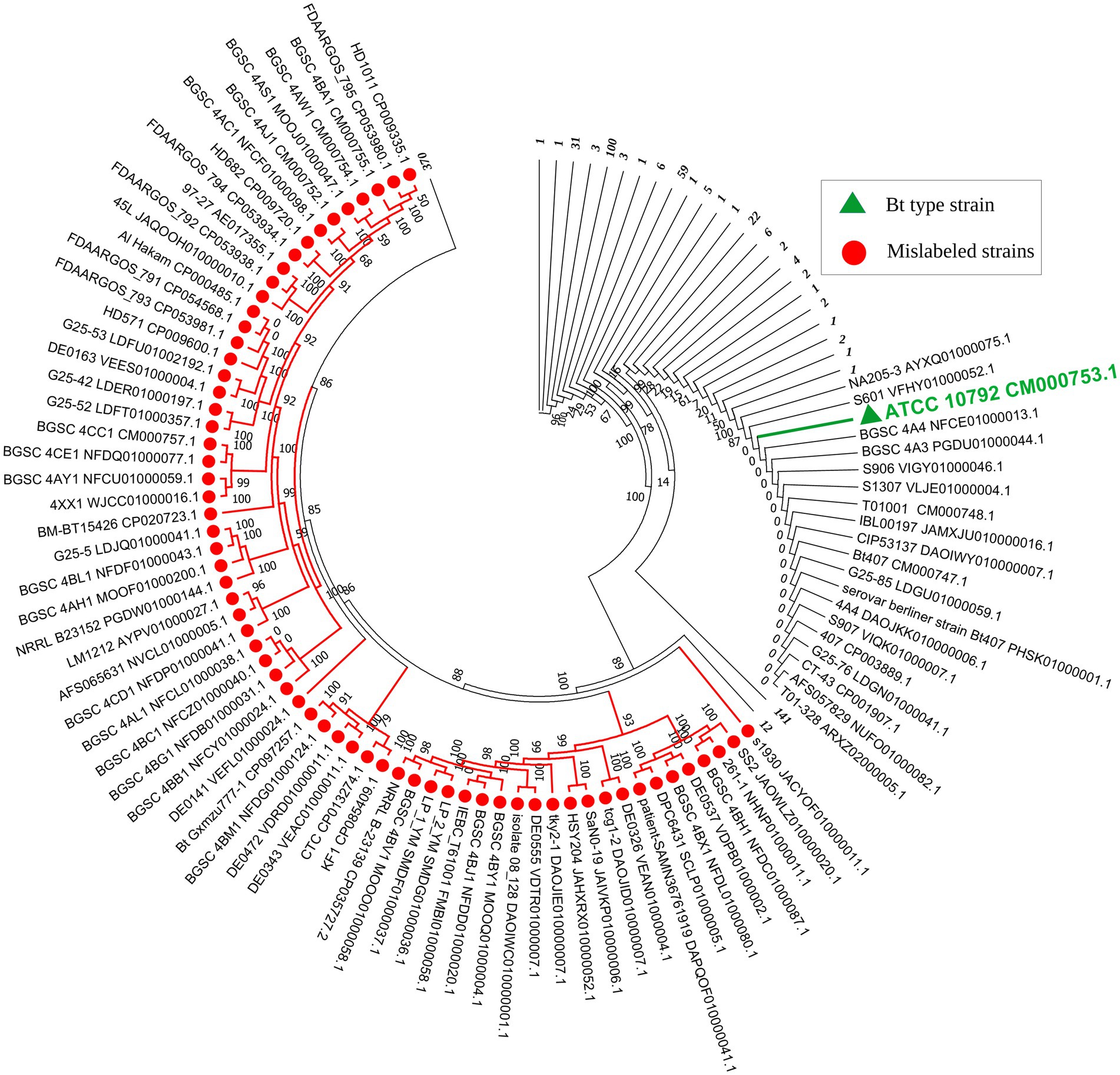
Figure 1. Circular representation of the MLSA-based phylogenetic tree of Bacillus thuringiensis strains retrieved from the NCBI database. The number of strains in each compressed branch is shown in italics and bold in front of the corresponding branch. A comprehensive view of the collapsed strains can be found in Supplementary Figure S1. The 82 strains that did not belong to Bacillus thuringiensis are shown in red, while the type strain of the species is shown in green.
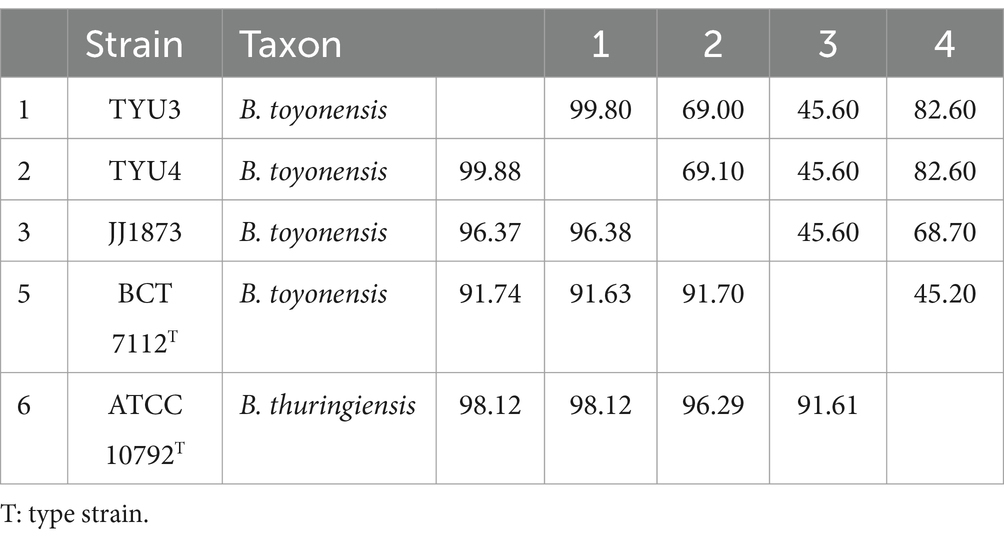
Table 4. Digital DNA–DNA hybridization (dDDH; upper diagonal) and average nucleotide identity (ANIm; lower diagonal) values generated from the DNA sequence similarity comparisons among different mislabeled Bacillus toyonensis strains and type strain of that species.
All 82 mislabeled B. thuringiensis strains, i.e., 46 strains that were assigned to a valid species and 36 strains with undetermined taxonomic status were subjected to a taxonomic investigation in TYGS. Regarding the former 46 strains, the results of TYGS were in congruence with the pyANI data (Figure 2; Supplementary Figure S2). As for the 36 strains that do not belong to any species of the genus Bacillus, the results of TYGS were similar to what was described above (Figure 2; Supplementary Figure S3), except for the strain KF1 where TYGS results showed that this strain belongs to B. basilensis. However, WGS of the type strain of B. basilensis is not publicly available, thus, we could not confirm the TYGS results using another WGS-based approach. Since the TYGS platform contains a very large number of type strains, including type strain of B. basilensis, it was possible to determine the taxonomic status of the strain based on the 16S rDNA data.

Figure 2. Average nucleotide identity-based pyANI matrix demonstrating nucleotide-level genomic similarity between the 82 mislabeled Bacillus thuringiensis strains and type strains of all validly described Bacillus species (Pritchard et al., 2016). B.p, Bacillus pretiosus; B.w, Bacillus wiedmannii; B.pa, Bacillus paranthracis; B.tr, Bacillus tropicus; B.l, Bacillus licheniformis; B.an, Bacillus anthracis; B.to, Bacillus toyonensis; B.m, Bacillus mycoides.
Out of 36 mislabeled strains that did not belong to any species, the strains LP_2_YM and LP_1_YM are members of the same species, while the strains SaN0-19 and HSY204 belong to the same species. The strains BGSC 4AL1, BGSC 4BB1, BGSC 4 BC1, and BGSC 4BG1 were members of the same species, the strains DE0472 and DE0343 are classified within the same species, the strains LM1212, BGSC 4CD1, AFS065631, BGSC 4BH1, and 261–1 fall into the same species, while the strains G25-5, BGSC 4BL1, and BGSC 4AH1 belong to the same species. The strains tcg1-2 and DE0326 were members of the same species, and the strains YBT-020 and Lr3/2 belonged to the same species. Strains BGSC 4BY1, IEBC_T61001, BGSC 4BJ1, and NRRL B-23139 were confirmed to belong to Bacillus pretiosus, as they showed dDDH values >70% and ANI values >95% with the latter species. These results support the classification of these strains within the species B. pretiosus. Additionally, the type strain of B. pretiosus SAICEU11T showed no significant genomic similarity with the other type strains in the genus Bacillus and this species continues to be listed as a species with an invalid name. Similarly, the strain s1783 is assigned to Lysinibacillus pinottii; however, this species is considered to have an invalid name. In conclusion, although genomic similarities clearly indicated the assignment of the mentioned examined strains to the species B. pretiosus and L. pinottii, these two species were considered invalid at the time of this study. This underscores the necessity for more comprehensive taxonomic evaluations and validation of their nomenclature. It is concluded that the number of possible new species to which these 36 strains belong is 18 novel species. The analysis conducted in TYGS confirmed the accuracy of the WGS-based results as shown in Supplementary Figures S2, S3.
Discussion
Classification of the B. cereus group has always been a controversial issue considering the high possibility of error. This is due in part to the fact that taxonomy of species belonging to this group has been conducted based on phenotypic characteristics (e.g., virulence repertories) while corresponding genes of those phenotypic characteristics are located in most cases on the plasmid, which can be transmitted between close species via conjugation process. In this study, we used phylogenomics and comparative genomics on all available whole genome resources of B. thuringiensis to refine taxonomy of the species. Our results showed that out of 885 strains deposited in the NCBI GenBank database, 803 are taxonomically genuine belonging to the core clade of the species regardless of their sources, biological characteristics, and pathogenicity status on the host of isolation. However, 82 strains do not belong to the species B. thuringiensis because of the low ANI and dDDH similarities between the type strain of the species and those 82 WGS. Among the mislabeled 82 strains, 39 strains belonged to the B. cereus group including B. anthracis, B. toyonensis, and B. mycoides. In addition to these three taxa, mislabeled strains of B. thuringiensis were scattered within the genus belonging to several hypothetical novel species, which need formal descriptions.
Although the species belonging to the B. cereus group are phylogenetically closely related, they exhibit notable differences in biological characteristics and phenotypic features. This group includes two species B. anthracis and B. cereus which are recognized as human pathogens, along with the well-known B. thuringiensis, widely used as a biological control agent. These distinctions highlight the importance of accurate classification within this group, particularly given its relevance to public health, medicine, and agriculture. Liu et al. (2015) demonstrated the utility of whole-genome sequence-based Genome BLAST Distance Phylogeny (GBDP) approach to determine taxonomic affiliations of 224 strains within the B. cereus group, identifying 11 known and 19–20 putative novel species. Their findings highlighted the pressing need to re-evaluate the classification and biosafety of strains within this group. Moreover, their study revealed the potential for misclassifications, where B. anthracis was incorrectly identified as B. cereus or B. thuringiensis. Extending this line of inquiry, our study uncovers further misclassifications, including strains labeled as B. thuringiensis that are, in fact, B. anthracis, the causative agent of anthrax. This misidentification raises serious biosafety concerns, particularly when such strains are employed in biological control programs under the assumption that they are harmless B. thuringiensis strains. These findings underscore the urgent need for rigorous taxonomic and biosafety reassessment to mitigate potential risks to public health and environmental safety.
By October 2023, 885 bacterial strains were labeled as B. thuringiensis in the NCBI database. After downloading the genomes and phylotaxonomic analyses, it was concluded that out of 885 strains 803 strains were labeled with the correct name. However, 82 strains did not belong to the species B. thuringiensis because of low ANI and dDDH similarities between the type strain of the species and those strains. To deal with this taxonomic problem, for the strains mentioned in Supplementary Table S3, we suggest changing the classification from the species B. thuringiensis to the species B. anthracis. The ANI and dDDH calculations showed that the latter strains had ANI less than 92% and dDDH less than 45% with the type strain of B. thuringiensis, while those strains had more than 97 and 77% similarities in ANI and dDDH values with B. anthracis type strain, respectively (Supplementary Table S3).
Among Gram-positive bacteria, Bacillus thuringiensis is well known for its beneficial features in agriculture. However, other members of Gram-positive bacteria are economically important plant pathogens which are subjected to strict quarantine rules and inspections (Osdaghi et al., 2020a, 2022; Haghverdi et al., 2024). Hence, precise identification and taxonomic delineation of the latter bacteria is also of high importance in terms of plant health and international food trade. For instance, it has recently been noted that plant pathogenic members of Clavibacter sp. and Curtobacterium sp. needed to be taxonomically refined to meet the current criteria in bacterial taxonomy (Osdaghi et al., 2020b, 2024). This would ease decision making in plant inspections and quarantine ports. Fundamentals of taxonomic studies are similar in both Gram-negative and Gram-positive bacteria. However, sight modifications and adjustments might be proposed for certain groups of bacteria to increase the preciseness and accuracy of the analyses. The 95% ANI threshold was adopted in this study; since, the investigated strains were not only compared to type strain of B. thuringiensis but also with all type strains of species within the Bacillus genus. The threshold of 96.2% ANI proposed by Liu et al. (2017b) for the B. cereus group was not employed because the strains in our study were compared not only with the type strain of B. thuringiensis, which belongs to the B. cereus group but also with type strains from other species within the Bacillus genus that do not belong to the B. cereus group. Furthermore, ANI was not used as a sole criteria in this study. Instead, it was combined with dDDH, MLSA-based phylogeny, and core genome-based phylogeny criteria for a more comprehensive species definition. Thus, a strain was classified as belonging to a specific species when both ANI and dDDH exceeded 95 and 70%, respectively, compared to the type strain of that species. This integrated method ensures a more robust classification. Additionally, our observations indicated that for strains within species related to the Bacillus genus, when the dDDH value exceeds 70%, the ANI value also tends to exceed 96.6% (data not shown). Therefore, in practice,an ANI threshold of 96.6% was effectively used in these cases, which is close to the threshold suggested by Liu et al. (2017b) for the B. cereus group.
However, the 95% ANI threshold is a widely accepted and reliable metric for bacterial species classification, supported by extensive studies validating its efficacy in accurately delineating species boundaries. For instance, Jain et al. (2018) conducted a comprehensive analysis of 90,000 prokaryotic genomes, demonstrating that 99.8% of genome pairs exhibited either >95% ANI within the same species or < 83% ANI across different species, highlighting a clear genetic discontinuity. This robust threshold was consistent across diverse datasets and unaffected by sequencing biases or the predominance of commonly studied species, confirming its universality and applicability. Similarly, Kim et al. (2014) observed a distinct ANI distribution between intra- and interspecies relationships around the 95–96% mark, further reinforcing the validity of the 95% threshold. Moreover, Chan et al. (2012) propose that a combination of core genome phylogenetic analysis and 95% pairwise ANI is an appropriate method for defining bacterial species, providing additional evidence for ANI as a reliable metric. These findings collectively underscore the robustness and utility of the 95% ANI threshold as a foundational criterion for bacterial taxonomy in the genomic era. Even in certain studies, recommendations have been made to lower the ANI threshold below 95% for delineating bacterial species, reflecting the variability in genetic relatedness across different bacterial groups. For instance, Konstantinidis and Tiedje (2005) demonstrated that an ANI of 94% corresponds to the established 70% DNA–DNA reassociation benchmark traditionally used for defining bacterial species, suggesting that a slightly lower threshold can still accurately capture genetic relationships. Similarly, Carroll et al. (2020) investigated the B. cereus group and found that the conventional 95% ANI threshold led to overlapping genomospecies clusters, with many genomes belonging to multiple clusters. They proposed a new threshold of approximately 92.5% ANI, which better aligns with natural genome similarity gaps within this group, resulting in more distinct and minimally overlapping genomospecies clusters. Moreover, the study introduced a taxonomic nomenclature framework that integrates genomic species definitions with clinically and industrially significant phenotypes, emphasizing the need for adaptable ANI thresholds to reflect specific genomic and phenotypic contexts.
Richter and Rosselló-Móra (2009) proposed ANI as a superior alternative to traditional DNA–DNA hybridization (DDH), recommending a threshold of 95–96% for species definition. Our study aligns with this approach, employing ANI and dDDH thresholds to evaluate the taxonomic status of the studied B. thuringiensis strains. Similarly, Konstantinidis and Tiedje (2005) validated the use of ANI coupled with DDH values, demonstrating that an ANI threshold of 94% corresponds to the traditional 70% DNA reassociation benchmark. While their findings emphasize the complementary use of ANI and DDH, our study extends this framework by incorporating phylogenetic methods, including MLSA and core genome phylogenetic trees. These methods not only confirmed the divergence of the misclassified strains but also added resolution to taxonomic relationships within the B. cereus group. Chan et al. (2012) recommended combining ANI with core genome phylogenetic analysis for precise species delineation, which strongly aligns with our methodology. In our study, the integration of core genome analyses with ANI and dDDH enhanced the accuracy of reclassification, demonstrating the necessity of multiple complementary approaches in resolving complex taxonomic challenges. Conversely, Palmer et al. (2020) emphasized the variability in genetic diversity across taxa, advocating for the use of adaptive thresholds and supplementary methods. Our findings align with this perspective, as phylogenetic analyses of misclassified strains consistently revealed distinct clustering patterns in both MLSA- and core genome-based trees. This highlights the importance of employing diverse tools to achieve a comprehensive and accurate species classification. Furthermore, Goris et al. (2007) emphasized the strong correlation between ANI (95%) and DDH (70%) thresholds, advocating their combined application for bacterial species classification. Our results are consistent with this integrative approach, as misclassified strains, including those reclassified as B. anthracis, B. toyonensis, and other species, exhibited ANI and dDDH values below these established thresholds. In summary, while previous studies primarily focused on ANI and DDH, This study highlights the value of combining ANI, DDH, and phylogenetic analyses to improve bacterial taxonomy, particularly within genetically diverse groups like the B. cereus complex.
For the strains MC28, H3, GOE7, GOE5, Bto-UNVM_94, we suggest changing their classification from B. thuringiensis to B. toyonensis because of their low ANI/dDDH similarity with type strain of the former species (ANI less than 92% and dDDH less than 45%). Whereas they show high similarity (ANI more than 98% and dDDH more than 87%) with the B. toyonensis type strain, as shown in Table 2. We propose reclassification of many other strains mislabeled as B. thuringiensis as follows: the strains NRRL B23152, BGSC 4Y1, KB1 and XL6 to B. tropicus (Table 3), the strain Et10/1 to B. licheniformis, BGSC 4BM1 to B. mycoides, 4I3 to B. paranthracis, BGSC 4BV1 to B. weidmannii, and KF1 to B. basilensis. In all cases, ANI and dDDH values for those strains in comparison with the B. thuringiensis type strain were below the accepted threshold for prokaryotic species description (ANI < 95 and dDDH<70).
On the other hand, there were 35 strains, when compared to all type strains of the genus Bacillus, showed a similarity below the acceptable threshold for prokaryotic species definition (ANI < 95, dDDH<70). According to Kumar et al. (2016), when strains did not show similarity higher than the acceptable limit for species definition with all type strains of a particular genus, those strains are likely to be new species. Therefore, we suggest reviewing the classification of the strains mentioned in Table 1. The four strains BGSC 4BY1, IEBC_T61001, BGSC 4BJ1, and NRRL B-23139 were confirmed to relate to Bacillus pretiosus, Furthermore, the type strain of B. pretiosus SAICEU11T was found to have no significant genomic similarity with the other type strains in the genus Bacillus. The species is still listed as having an invalid name, while our findings indicate that its validity as a species should be acknowledged based on genomic evidence, and we propose to consider it an independent species with a reliable name (Figure 2; Supplementary Figure S3).
Through our study and detailed comparative analyses, we concluded that there were no instances where a strain exhibited a dDDH value greater than 70% while simultaneously having an ANI value lower than 95% when compared to the type strain of B. thuringiensis or other type strains within the genus Bacillus. For all species within the genus Bacillus, a dDDH value exceeding 70% typically requires an ANI of 96.6% or higher. This specific finding is a unique outcome of our study and reflects the relationship between these metrics based on our analyzed cases. Conversely, we identified instances where strains had ANI values higher than 95% but dDDH values below 70%. However, these cases were disregarded as they did not simultaneously meet both criteria. This observation was not limited to comparisons involving the type strain of B. thuringiensis alone but was also consistent when comparing misclassified strains with the type strains of the other species within the genus Bacillus. For example, strain Et10/1 exhibited an ANI value of 95.02% and a dDDH value of 59.10% when compared to the type strain of B. anthracis. Thus, it was not classified as belonging to B. anthracis. Further comparisons to determine the species to which it belongs revealed that it is a member of B. licheniformis, with an ANI value of 99.81% and a dDDH value of 97.60%. This example highlights the utility of comprehensive genomic analyses combined with standard criteria in achieving precise taxonomic classifications across species within this genus.
The question that arises here is why these 82 strains were classified as B. thuringiensis, despite their chromosomal similarity to the type strain of the species being minimal and insufficient to consider them part of the species. The main reason for this misclassification is the reliance on the ability of these strains to produce crystal protein, which is commonly used as a Bio-insecticide against various insect pests, as a defining feature of B. thuringiensis strains (Bravo et al., 2013). It has been shown that the gene responsible for encoding crystal protein is, in most cases, located on a plasmid rather than the chromosome. Plasmids are susceptible to transfer between closely related bacteria through a process known as conjugation (González et al., 1982). This has been observed in several strains such as HSY204, Bt Gxmzu777-1, and YBT020. For instance, in the strain HSY204 which shows low similarity (ANI = 91.6, dDDH = 44.2) to B. thuringiensis type strain, the bacterium contains four plasmids, with the PB plasmid harboring the gene encoding the insecticidal delta-endotoxin Cry8Ea1. Whereas, on the pC plasmid, there is a gene encoding the insecticidal crystal protein Cry1Ac. When this strain was first isolated, it was described as a bio-insecticide effective against Aedes aegypti larvae (Wu et al., 2021).
The strain Bt Gxmzu777-1 has been identified as a bacterium belonging to B. thuringiensis despite the very low similarity (ANI = 89.9, dDDH = 38.2) of this strain to type strain of B. thuringiensis (data not shown). This might be due in part to the fact that this strain possesses four plasmids, where two of these plasmids, i.e., pa (262,245 bp) and pd. (259,656 bp) containing the gene encoding the insecticidal delta-endotoxin Cry8Ea1 family protein. The strain YBT-020 possesses two plasmids, and on plasmid pBMB26 (187,880 pb), there are genes encoding the pesticidal crystal proteins cry4Ba and cry4Aa, while on plasmid PBMB28 there is gene cry4Aa. In addition, it is likely that the misclassification of the three strains (TYU3, TYU4, JJ1873) as members of the B. toyonensis species despite their phylogenetic relation to B. thuringiensis, stems from the absence of crystalline protein production in these strains (data not shown).
All this evidence emphasizes that the fundamental principle when working on the classification of B. thuringiensis strains is the production of crystal proteins known for their insecticidal activity. This may lead to inaccurate classification and errors in identification. Therefore, it is essential to strive for the use of more reliable and precise standards, such as those based on chromosomal comparison (calculating dDDH and ANI) and relying on traits encoded by genes present on the chromosome to differentiate between strains, which are less prone to loss or transfer between bacterial species. The important result that was reached through this study is the necessity of searching for modern and approved methods to classify species belonging to the genus Bacillus, and in particular, species belonging to the B. cereus group, which are very similar genetically while very different in terms of phenotypic traits. One of the reliable methods is to use a biomarker specific to the species to be classified. For example, in the case of B. thuringiensis, the gene coding for the transcriptional regulator of insecticidal toxin (xre), xenobiotic response element is commonly observed in strains that are classified as B. thuringiensis and was used as a biomarker by Wei et al. (2019) to distinguish the mentioned species from its close species in the B. cereus group. Out of the 33 mislabeled B. thuringiensis strains, which should be classified as B. anthracis, some strains do not contain xre gene like FDAARGOS_792, 97–27, BM-BT15426, HD682, FDAARGOS_794, HD1011, FDAARGOS_795, 4XX3, 4XX2, 4XX1, 4 W2, BGSC 4BX1, Patient-SAMN36761919, FDAARGOS_793, Al Hakam, HD571, FDAARGOS_791, and 4B3. Since the strains belonging to B. thuringiensis contain the xre gene in their genomes and the mentioned B. anthracis strains do not have that gene, this supports our hypothesis that these strains do not belong to B. thuringiensis species (data not shown). Another example is PLCR-regulated proteins. In B. thuringiensis PLCR is a pleiotropic regulator of extracellular virulence factor gene expression (Lereclus and Agaisse, 2000). None of the 33 strains in this study that must be reclassified as B. anthracis has this protein. This further confirms the validity of the hypothesis that these strains not only do not belong to B. thuringiensis but also belong to the B. anthracis species, because the characteristic of the B. anthracis strains is that they do not contain PLCR-regulated proteins PRP2 and PLCR gene is truncated (Lereclus et al., 2000). Instead, the essential virulence factors such as capsule formation and anthrax toxin are regulated by regulatory genes, acpA and atxA located on plasmids pXO2 and pXO2, respectively, (Bourgogne et al., 2003).
In conclusion, the outcomes of this study are anticipated to enhance the safety and precision of biological control applications of B. thuringiensis strains by ensuring accurate identification and taxonomic delineation. Given the coexistence of B. thuringiensis with human pathogenic species like B. cereus and B. anthracis, the latter possessing a significant biosecurity threat, the refined classification approach outlined here will facilitate reliable selection of strains suitable for biological control. Finally, MLSA-based taxonomic framework drawn in this study would facilitate identification of suspected B. thuringiensis strains in the coming works. These findings underscore the importance of integrating genomic tools for the precise classification of strains to minimize risks to the environment and human health while optimizing the use of microbial agents in agriculture.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
NS: Conceptualization, Investigation, Writing – original draft. RT-H: Software, Supervision, Writing – review & editing. HA: Methodology, Validation, Writing – review & editing. SZ: Investigation, Software, Writing – review & editing. EO: Conceptualization, Data curation, Investigation, Methodology, Software, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work is based upon research funded by Iran National Science Foundation (INSF) under project No. 4038278, and University of Tehran, Iran.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1518307/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | MLSA-based phylogenetic tree using the sequences of five housekeeping genes gyrB, atpD, dnaK, rpoB, and rpoD for all Bacillus thuringiensis strains retrieved from the NCBI database in this study.
SUPPLEMENTARY FIGURE S2 | Phylogenetic tree inferred with FastME 2.1.6.1 (Lefort et al., 2015) of 46 mislabeled Bacillus thuringiensis strains belonging to different species of the genus Bacillus from GBDP distances calculated from genome sequences. The branch lengths are scaled in terms of GBDP distance formula d5. The numbers above branches are GBDP pseudo-bootstrap support values >60% from 100 replications, with an average branch support of 38.4%. The tree was rooted at the midpoint (Farris, 1972).
SUPPLEMENTARY FIGURE S3 | Phylogenetic tree inferred with FastME 2.1.6.1 (Lefort et al., 2015) of the 35 mislabeled Bacillus thuringiensis strains not belonging to any species generated from GBDP distances calculated of genome sequences. The branch lengths are scaled in terms of GBDP distance formula d5. The numbers above branches are GBDP pseudo-bootstrap support values >60% from 100 replications, with an average branch support of 49.7%. The tree was rooted at the midpoint (Farris, 1972).
SUPPLEMENTARY TABLE S1 | List of 885 strains labeled as Bacillus thuringiensis retrieved from the NCBI GenBank database.
SUPPLEMENTARY TABLE S2 | List of type strains of all validly described Bacillus species used in the study according to the LPSN database https://lpsn.dsmz.de/.
SUPPLEMENTARY TABLE S3 | Digital DNA–DNA hybridization (dDDH; upper diagonal) and average nucleotide identity (ANIm; lower diagonal) values generated from the DNA sequence similarity comparisons among different mislabeled Bacillus thuringiensis strains and the Bacillus anthracis type strain.
Footnotes
References
Baek, I., Lee, K., Goodfellow, M., and Chun, J. (2019). Comparative genomic and phylogenomic analyses clarify relationships within and between Bacillus cereus and Bacillus thuringiensis: Proposal for the recognition of two Bacillus thuringiensis genomovars. Front. Microbiol. 10:466325. doi: 10.3389/fmicb.2019.01978
Bourgogne, A., Drysdale, M., Hilsenbeck, S. G., Peterson, S. N., and Koehler, T. M. (2003). Global effects of virulence gene regulators in a Bacillus anthracis strain with both virulence plasmids. Infect. Immun. 71, 2736–2743. doi: 10.1128/IAI.71.5.2736-2743.2003
Bravo, A., Gómez, I., Porta, H., García-Gómez, B. I., Rodriguez-Almazan, C., Pardo, L., et al. (2013). Evolution of Bacillus thuringiensis Cry toxins insecticidal activity. Microb. Biotechnol. 6, 17–26. doi: 10.1111/j.1751-7915.2012.00342.x
Carroll, L. M., Wiedmann, M., and Kovac, J. (2020). Proposal of a taxonomic nomenclature for the Bacillus cereus group which reconciles genomic definitions of bacterial species with clinical and industrial phenotypes. MBio 11, 10–1128. doi: 10.1128/mBio.00034-20
Castillo-Esparza, J. F., Hernández-González, I., and Ibarra, J. E. (2019). Search for Cry proteins expressed by Bacillus spp. genomes, using hidden Markov model profiles. 3 Biotech 9:13. doi: 10.1007/s13205-018-1533-3
Challacombe, J. F., Altherr, M. R., Xie, G., Bhotika, S. S., Brown, N., Bruce, D., et al. (2007). The complete genome sequence of Bacillus thuringiensis Al Hakam. J. Bacteriol. 189, 3680–3681. doi: 10.1128/JB.00241-07
Chan, J. Z., Halachev, M. R., Loman, N. J., Constantinidou, C., and Pallen, M. J. (2012). Defining bacterial species in the genomic era: insights from the genus Acinetobacter. BMC Microbiol. 12, 1–11. doi: 10.1186/1471-2180-12-302
Clarke, S. C., Diggle, M. A., and Edwards, G. F. S. (2002). Multilocus sequence typing and porA gene sequencing differentiates strains of Neisseria meningitidis during case clusters. Br. J. Biomed. Sci. 59, 160–162. doi: 10.1080/09674845.2002.11978034
Farris, J. S. (1972). Estimating phylogenetic trees from distance matrices. Am. Nat. 106, 645–668. doi: 10.1086/282802
Garrity, G. M. (2005) in Bergey's Manual® of Systematic Bacteriology: Volume Two: The Proteobacteria. eds. D. J. Brenner, N. R. Krieg, and J. T. Staley (Springer US).
Gevers, D., Cohan, F. M., Lawrence, J. G., Spratt, B. G., Coenye, T., Feil, E. J., et al. (2005). Re-evaluating prokaryotic species. Nat. Rev. Microbiol. 3, 733–739. doi: 10.1038/nrmicro1236
Goecks, J., Nekrutenko, A., and Taylor, J.Galaxy Team team@ galaxyproject. org (2010). Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 11, 1–13. doi: 10.1186/gb-2010-11-8-r86
González, J. M. Jr., Brown, B. J., and Carlton, B. C. (1982). Transfer of Bacillus thuringiensis plasmids coding for delta-endotoxin among strains of B. thuringiensis and B. cereus. Proc. Natl. Acad. Sci. 79, 6951–6955. doi: 10.1073/pnas.79.22.6951
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Guinebretière, M. H., Auger, S., Galleron, N., Contzen, M., De Sarrau, B., De Buyser, M. L., et al. (2013). Bacillus cytotoxicus sp. nov. is a novel thermotolerant species of the Bacillus cereus group occasionally associated with food poisoning. Int. J. Syst. Evol. Microbiol. 63, 31–40. doi: 10.1099/ijs.0.030627-0
Haghverdi, M., Taghavi, S. M., Zarei, S., Mafakheri, H., Abachi, H., Briand, M., et al. (2024). Pink-pigmented variant of Clavibacter michiganensis expands phenotypic range of tomato bacterial canker pathogen. Phytopathology. [Advance online publication]. doi: 10.1094/PHYTO-07-24-0236-R
Han, C. S., Xie, G., Challacombe, J. F., Altherr, M. R., Bhotika, S. S., Brown, N., et al. (2006). Pathogenomic sequence analysis of Bacillus cereus and Bacillus thuringiensis isolates closely related to Bacillus anthracis. J. Bacteriol. 188, 3382–3390. doi: 10.1128/JB.188.9.3382-3390.2006
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114. doi: 10.1038/s41467-018-07641-9
Jakhar, A., Kashyap, L., Goswami, T., Patel, V., and Sharma, R. (2017). “Bacillus thuringiensis and insect pest management” in Biopesticides and bioagents: novel tools for pest management. ed. Anwer (New York: Apple Academic Press), 418.
Jeong, H., Jo, S. H., Hong, C. E., and Park, J. M. (2016). Genome Sequence of the Endophytic Bacterium Bacillus thuringiensis Strain KB1, a Potential Biocontrol Agent against Phytopathogens. Genome Announc. 4, e00279–e00216. doi: 10.1128/genomeA.00279-16
Jiménez, G., Urdiain, M., Cifuentes, A., López-López, A., Blanch, A. R., Tamames, J., et al. (2013). Description of Bacillus toyonensis sp. nov., a novel species of the Bacillus cereus group, and pairwise genome comparisons of the species of the group by means of ANI calculations. Syst. Appl. Microbiol. 36, 383–391. doi: 10.1016/j.syapm.2013.04.008
Johnson, S. L., Daligault, H. E., Davenport, K. W., Jaissle, J., Frey, K. G., Ladner, J. T., et al. (2015). Complete genome sequences for 35 biothreat assay-relevant bacillus species. Genome Announc. 3, e00151–e00115. doi: 10.1128/genomeA.00151-15
Katoh, K., Rozewicki, J., and Yamada, K. D. (2019). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 20, 1160–1166. doi: 10.1093/bib/bbx108
Kim, M., Oh, H. S., Park, S. C., and Chun, J. (2014). Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J.Syst.Evol. Microbiol. 64, 346–351. doi: 10.1099/ijs.0.059774-0
Konstantinidis, K. T., and Tiedje, J. M. (2005). Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. 102, 2567–2572. doi: 10.1073/pnas.0409727102
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lambert, B., and Peferoen, M. (1992). Insecticidal promise of Bacillus thuringiensis. Bioscience 42, 112–122. doi: 10.2307/1311652
Lecadet, M. M., Frachon, E., Dumanoir, V. C., Ripouteau, H., Hamon, S., Laurent, P., et al. (1999). Updating the H-antigen classification of Bacillus thuringiensis. J. Appl. Microbiol. 86, 660–672. doi: 10.1046/j.1365-2672.1999.00710.x
Lefort, V., Desper, R., and Gascuel, O. (2015). FastME 2.0: a comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 32, 2798–2800. doi: 10.1093/molbev/msv150
Lereclus, D., and Agaisse, H. (2000). Toxin and virulence gene expression in Bacillus thuringiensis. Entomopathogenic Bacteria: From Laboratory to Field Application, 127–142.
Lereclus, D., Agaisse, H., Grandvalet, C., Salamitou, S., and Gominet, M. (2000). Regulation of toxin and virulence gene transcription in Bacillus thuringiensis. Int. J. Med. Microbiol.: IJMM. 290, 295–299. doi: 10.1016/S1438-4221(00)80024-7
Liu, G., Deng, C., Song, L., Peng, Q., Zhang, J., Lereclus, D., et al. (2014). Draft Genome Sequence of Bacillus thuringiensis Strain LM1212, Isolated from the Cadaver of an Oryctes gigas Larva in Madagascar. Genome Announc. 2, e00516–e00514. doi: 10.1128/genomeA.00516-14
Liu, Y., Lai, Q., Du, J., and Shao, Z. (2017b). Genetic diversity and population structure of the Bacillus cereus group bacteria from diverse marine environments. Sci. Rep. 7:689. doi: 10.1038/s41598-017-00817-1
Liu, Y., Lai, Q., Göker, M., Meier-Kolthoff, J. P., Wang, M., Sun, Y., et al. (2015). Genomic insights into the taxonomic status of the Bacillus cereus group. Sci. Rep. 5:14082. doi: 10.1038/srep14082
Liu, J., Li, L., Peters, B. M., Li, B., Chen, D., Xu, Z., et al. (2017a). Complete genome sequence and bioinformatics analyses of Bacillus thuringiensis strain BM-BT15426. Microb. Pathog. 108, 55–60. doi: 10.1016/j.micpath.2017.05.006
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A., and Zdobnov, E. M. (2021). BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654. doi: 10.1093/molbev/msab199
Maughan, H., and Van der Auwera, G. (2011). Bacillus taxonomy in the genomic era finds phenotypes to be essential though often misleading. Infect. Genet. Evol. 11, 789–797. doi: 10.1016/j.meegid.2011.02.001
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H. P., and Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14, 1–14. doi: 10.1186/1471-2105-14-60
Meier-Kolthoff, J. P., and Göker, M. (2019). TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 10:2182. doi: 10.1038/s41467-019-10210-3
Meier-Kolthoff, J. P., Sardà Carbasse, J., Peinado-Olarte, R. L., and Göker, M. (2022). TYGS and LPSN: a database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acid Res. 50, D801–D807. doi: 10.1093/nar/gkab902
Meric, G., Mageiros, L., Pascoe, B., Woodcock, D. J., Mourkas, E., Lamble, S., et al. (2018). Lineage-specific plasmid acquisition and the evolution of specialized pathogens in Bacillus thuringiensis and the Bacillus cereus group. Mol. Ecol. 27, 1524–1540. doi: 10.1111/mec.14546
Nazarian, A., Jahangiri, R., Jouzani, G. S., Seifinejad, A., Soheilivand, S., Bagheri, O., et al. (2009). Coleopteran-specific and putative novel cry genes in Iranian native Bacillus thuringiensis collection. J. Invertebr. Pathol. 102, 101–109. doi: 10.1016/j.jip.2009.07.009
Osdaghi, E., Rahimi, T., Taghavi, S. M., Ansari, M., Zarei, S., Portier, P., et al. (2020b). Comparative genomics and phylogenetic analyses suggest several novel species within the genus Clavibacter, including nonpathogenic tomato-associated strains. Appl. Environ. Microbiol. 86, e02873–e02819. doi: 10.1128/AEM.02873-19
Osdaghi, E., Taghavi, S. M., Hamidizade, M., Kariminejhad, M., Fazliarab, A., Maleki, H. H., et al. (2024). Multiphasic investigations imply transfer of orange−/red-pigmented strains of the bean pathogen Curtobacterium flaccumfaciens pv. flaccumfaciens to a new species as C. aurantiacum sp. nov., elevation of the poinsettia pathogen C. flaccumfaciens pv. poinsettiae to the species level as C. poinsettiae sp. nov., and synonymy of C. albidum with C. citreum. Syst. Appl. Microbiol. 47:126489. doi: 10.1016/j.syapm.2024.126489
Osdaghi, E., van der Wolf, J. M., Abachi, H., Li, X., De Boer, S. H., and Ishimaru, C. A. (2022). Bacterial ring rot of potato caused by Clavibacter sepedonicus: a successful example of defeating the enemy under international regulations. Mol. Plant Pathol. 23, 911–932. doi: 10.1111/mpp.13191
Osdaghi, E., Young, A. J., and Harveson, R. M. (2020a). Bacterial wilt of dry beans caused by Curtobacterium flaccumfaciens pv. flaccumfaciens: A new threat from an old enemy. Mol. Plant Pathol. 21, 605–621. doi: 10.1111/mpp.12926
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Palmer, M., Steenkamp, E. T., Blom, J., Hedlund, B. P., and Venter, S. N. (2020). All ANIs are not created equal: implications for prokaryotic species boundaries and integration of ANIs into polyphasic taxonomy. Int. J. Syst. Evol. Microbiol. 70, 2937–2948. doi: 10.1099/ijsem.0.004124
Patil, K. R., and McHardy, A. C. (2013). Alignment-free genome tree inference by learning group-specific distance metrics. Genome Biol. Evol. 5, 1470–1484. doi: 10.1093/gbe/evt105
Pritchard, L., Glover, R. H., Humphris, S., Elphinstone, J. G., and Toth, I. K. (2016). Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal. Methods 8, 12–24. doi: 10.1039/C5AY02550H
Rang, J., He, H., Wang, T., Ding, X., Zuo, M., Quan, M., et al. (2015). Comparative analysis of genomics and proteomics in Bacillus thuringiensis 4.0718. PLoS One 10:e0119065. doi: 10.1371/journal.pone.0119065
Rasko, D. A., Altherr, M. R., Han, C. S., and Ravel, J. (2005). Genomics of the Bacillus cereus group of organisms. FEMS Microbiol. Rev. 29, 303–329. doi: 10.1016/j.femsre.2004.12.005
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Sauka, D. H., Peralta, C., Pérez, M. P., Onco, M. I., Fiodor, A., Caballero, J., et al. (2022). Bacillus toyonensis biovar Thuringiensis: A novel entomopathogen with insecticidal activity against lepidopteran and coleopteran pests. Biol. Control 167:104838. doi: 10.1016/j.biocontrol.2022.104838
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Seppey, M., Manni, M., and Zdobnov, E. M. (2019). BUSCO: assessing genome assembly and annotation completeness. Methods Mol. Biol., (Clifton, N.J.), 1962, 227–245. doi: 10.1007/978-1-4939-9173-0_14
Souvorov, A., Agarwala, R., and Lipman, D. J. (2018). SKESA: strategic k-mer extension for scrupulous assemblies. Genome Biol. 19:153. doi: 10.1186/s13059-018-1540-z
Wayne, L. G., Brenner, D. J., Colwell, R. R., Grimont, P. A. D., Kandler, O., Krichevsky, M. I., et al. (1987). Report of the ad hoc committee on reconciliation of approaches to bacterial systematics. Int. J. Syst. Evol. Microbiol. 37, 463–464. doi: 10.1099/00207713-37-4-463
Wei, S., Chelliah, R., Park, B. J., Kim, S. H., Forghani, F., Cho, M. S., et al. (2019). Differentiation of Bacillus thuringiensis from Bacillus cereus group using a unique marker based on real-time PCR. Front. Microbiol. 10:883. doi: 10.3389/fmicb.2019.00883
Wu, J., Wei, L., He, J., Fu, K., Li, X., Jia, L., et al. (2021). Characterization of a novel Bacillus thuringiensis toxin active against Aedes aegypti larvae. Acta Trop. 223:106088. doi: 10.1016/j.actatropica.2021.106088
Zhu, L., Peng, D., Wang, Y., Ye, W., Zheng, J., Zhao, C., et al. (2015). Genomic and transcriptomic insights into the efficient entomopathogenicity of Bacillus thuringiensis. Sci. Rep. 5:14129. doi: 10.1038/srep14129
Keywords: Bacillus, biological control, phylotaxonomy, bacterial taxonomy, Bacillus cereus
Citation: Shiekh Suliman N, Talaei-Hassanloui R, Abachi H, Zarei S and Osdaghi E (2025) Taxonomic refinement of Bacillus thuringiensis. Front. Microbiol. 16:1518307. doi: 10.3389/fmicb.2025.1518307
Edited by:
Mariusz Cycoń, Medical University of Silesia, PolandReviewed by:
Toni A. Chapman, Elizabeth Macarthur Agricultural Institute, AustraliaAfef Najjari, Tunis El Manar University, Tunisia
Copyright © 2025 Shiekh Suliman, Talaei-Hassanloui, Abachi, Zarei and Osdaghi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ebrahim Osdaghi, ZW9zZGFnaGlAdXQuYWMuaXI=