
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 21 March 2025
Sec. Food Microbiology
Volume 16 - 2025 | https://doi.org/10.3389/fmicb.2025.1501331
 Nereus W. Gunther IV1*
Nereus W. Gunther IV1* Siddhartha Kanrar1Aisha Abdul-Wakeel1
Siddhartha Kanrar1Aisha Abdul-Wakeel1 Michael J. McAnulty2
Michael J. McAnulty2 John Renye2Joseph Uknalis3
John Renye2Joseph Uknalis3 Gaylen A. Uhlich1
Gaylen A. Uhlich1A single nucleotide polymorphism (SNP) in the 126 bp untranslated region (UTR) directly upstream of Campylobacter jejuni’s cysM (cysteine synthase) results in significant effects on gene transcription. UTR sequences, containing the predicted promoter region of cysM, from 264 different strains were compared, and revealed a SNP twenty nucleotides upstream of the cysM translation start site. In 219 strains the UTR sequence contained a guanine at this locus, and the remaining 45 strains had an adenine at the same position. Strains possessing the guanine SNP showed higher amounts of cysM transcripts compared to adenine SNP strains. When both UTR regions were cloned upstream of the major flagellar subunit (flaA) the guanine SNP UTR resulted in significantly greater levels of flaA transcription compared to the adenine SNP containing UTR. Additionally, when the UTR containing the guanine SNP was fused to flaA, motility was restored for a flaAB null mutant. Motility was not rescued initially when flaA was fused to the UTR containing the adenine SNP UTR. However, when the flaAB null mutant, containing a copy of flaA fused to the adenine-containing UTR, was incubated in Brucella broth for a minimum of two consecutive passages each lasting 48 h, transcription of flaA increased and motility was restored. Additional analysis of the flaA mRNA produced by the strain containing the adenine SNP UTR fused to flaA grown in Brucella broth versus agar suggests that the effects on motility occurred through blocking of full-length mRNA production.
Campylobacter jejuni is recognized as the causative agent for the greatest number of bacterial foodborne gastrointestinal disease cases recorded each year in the developed world (Marder et al., 2017; European Food and Safety Authority, 2021). In the United States alone it is estimated that ∼1 million cases of foodborne illness are caused annually by Campylobacter at an economic cost of ∼2 billion dollars (Kaakoush et al., 2015). The bacterium is particularly virulent to humans, requiring an infectious dose of as little as 500 organisms (Black et al., 1988). Additionally, C. jejuni is able to survive and persist in a diverse collection of environments, including both wild and farmed animals and water sources (Levin, 2007; Gillespie et al., 2002). Campylobacter jejuni’s ability to survive, adapt, and infect within diverse environments likely necessitates the organism’s strain to strain genetic variations (Burnham and Hendrixson, 2018).
The advances in the availability and affordability of whole genome sequencing have greatly assisted in the process of identifying the differences between Campylobacter genomes. However, identifying genetic differences is not sufficient to understand their effect on pathogenesis, thus it is necessary to fully characterize gene functions and define the roles that the genetic changes have on Campylobacter physiology. Problematically, there has been a lack of molecular tools available for laboratory research on C. jejuni. In response to this need, a two-plasmid system was developed for the cloning and constitutive expression of exogenous genes inserted within the C. jejuni chromosome (Uhlich et al., 2022). Continuing research has focused on the UTR sequence that is utilized to drive constitutive gene transcription in the two-plasmid system. The UTR originates from a sequence found directly upstream of the cysteine synthase gene (cysM) within the Campylobacter genome, and is responsible for transcription of this gene (Garvis et al., 1997). In C. jejuni, cysM is needed for production of the amino acid cysteine, which occurs from the transfer of sulfur from environmental sources to serine (Hitchcock et al., 2022). C. jejuni require cysteine for growth, therefore cysM is essential under nutrient limiting conditions when cysteine cannot be scavenged from the environment (Alazzam et al., 2011). In analyzing the UTR sequence upstream of cysM, a well-conserved single nucleotide polymorphism (SNP) in the C. jejuni population was identified. The SNP was located near the proposed promoter region of cysM between the putative ribosomal binding site and −10 RNA polymerase site (Garvis et al., 1997). The effects, of this SNP in the UTR sequence, were analyzed to determine the influence upon cysM transcription.
All bacterial strains and plasmids used in this study are listed in Table 1. For long term storage, Campylobacter strain freezer stocks were maintained in Brucella broth (Becton Dickinson, Sparks, USA) plus 15% glycerol frozen at −80°C. Strain cultures were revived from freezer stocks directly onto Brucella agar (1.5%) plates. Kanamycin (15 μg mL1 final concentration) or tetracycline (5 μg mL1 final concentration) was added to Brucella agar plates or Brucella broth when growing strains possessing a cloned aph(3) or tetO gene, respectively. All C. jejuni cultures were incubated in a microaerobic (5% O2, 10% CO2, 85% N2) growth chamber (Concept-M, Baker, Sanford, USA) at 42°C.
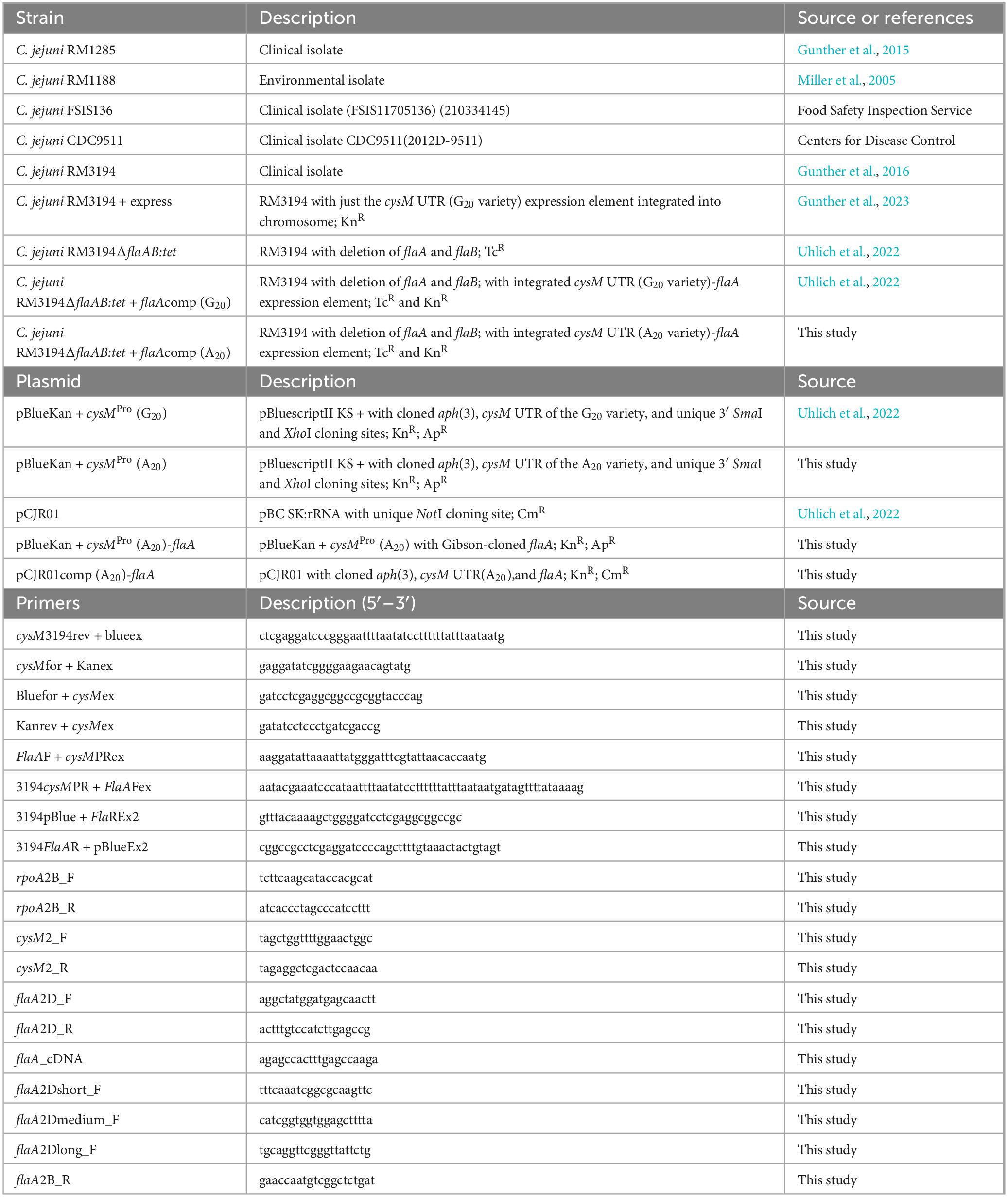
Table 1. Strains, plasmids and primers utilized in research.
Blast analysis of the UTR directly upstream of cysM was performed as follows: the NCBI genome website was searched using “Campylobacter jejuni subsp. jejuni AND (latest OR ‘latest genbank’) AND all NOT anomalous” search words on 05-12-2023 (Altschul et al., 1990). This downloaded 326 genome fasta sequences. A 126-bp UTR sequence was used against the downloaded 326-genome sequences using Blastn + software using default parameters. The subject sequences were selected for 126 bp length and no-gaps in sequences. This produced 264 sequences of 126 bases long. The 264 sequences were aligned with MAAFT software (v7.487) with FFT-NS-i parameter (Katoh and Standley, 2013). The UTR sequence was analyzed for regulatory elements and transcription factor binding sites using bprom software (Solovyev and Salamov, 2011).1
The C. jejuni strain RM3194ΔflaAB:tet + flaAcomp (G20) had been previously constructed (Uhlich et al., 2022). In order to investigate the differences in transcription when an adenine was at the 20th position in the UTR compare to guanine we need to construct a strain the same as RM3194ΔflaAB:tet + flaAcomp (G20), but with the desired single nucleotide change. Plasmid pBlueKan + cysMPro (G20) carrying a guanine at the 20th site in the UTR was PCR amplified using two sets of primers in order to achieve a Gibson style assembly. Primers cysM3194rev + blueex and cysMfor + Kanex were used to amplify the cysM UTR and replace the guanine nucleotide with an adenine. Primers Bluefor + cysMex and Kanrev + cysMex were used to amplify the vector backbone. Using the NEBuilder HiFI DNA Assembly kit (NEB, Ipswitch, MA, USA) and the overlap extensions built into the PCR primers the plasmid pBlueKan + cysMPro (G20) was reconstructed as plasmid pBlueKan + cysMPro (A20) with the adenine in the desired location. Next flaA from strain RM3194 was cloned into the newly constructed plasmid pBlueKan + cysMPro (A20), again using a Gibson style assembly. Primers 3194pBlue + FlaAREx2 and 3194FlaAR + pBlueEx2 were used to clone flaA from RM3194 and primers FlaAF + cysMPRex and 3194cysMPR + FlaAFex were used to amplify the pBlueKan + cysMPro (A20) plasmid. Again using the NEBuilder kit and the overlap extensions built into the primer sets, flaA was cloned into plasmid pBlueKan + cysMPro (A20) producing the new plasmid, pBlueKan + cysMPro (A20)-flaA. The pBlueKan + cysMPro (A20)-flaA plasmid was digested with NotI (NEB) and the fragment containing the kanamycin resistance gene, the A20 cysM UTR and flaA was gel purified away from the plasmid backbone. The purified fragment was ligated with the pCJR01 vector that was also digested with NotI. The resulting plasmid was designated, pCJR01comp (A20)-flaA and subsequently electroporated into strain C. jejuni RM3194ΔflaAB:tet (Davis et al., 2008). Before electroporation the plasmid pCJR01comp (A20)-flaA was sequenced to ensure that no additional changes to the cysM UTR or flaA had occurred during the cloning process. Transformants were selected for on Brucella agar plus kanamycin and cells from the resulting colonies were transferred individually onto Brucella agar plus chloramphenicol and Brucella agar plus kanamycin plates. Successful double crossover integration events of the cysM UTR flaA into the bacteria chromosome were identified by the presence of kanamycin resistance and the absence of chloramphenicol resistance. All successful transformants were directly checked for motility on low agar Brucella plates, no transformants were motile. Several colonies were then chosen at random, stored as freezer stocks and designated, as RM3194ΔflaAB:tet + flaAcomp (A20) strains.
Strains RM3194ΔflaAB:tet + flaAcomp (G20) and RM3194ΔflaAB:tet + flaAcomp (A20) were taken directly from freezer stocks and grown on Brucella agar (1.5%) plus Kanamycin. When either strain was grown (for 16 to 48 h) on solid agar media directly from freezer stocks or passed from one Brucella agar plate to the next, RM3194ΔflaAB:tet + flaAcomp (G20) was referred to as a G20 solid media culture (GSM) and RM3194ΔflaAB:tet + flaAcomp (A20) was referred to as an A20 solid media culture (ASM). Cells from either of these strains taken from agar plates and inoculated into Brucella broth (with kanamycin) and grown for up to 48 h were still classified as GSM or ASM strains since no changes in the motility of the ASM strain was observed at this point. However, when cells were taken from the liquid cultures and placed into fresh Brucella broth plus kanamycin and incubated again for at least 48 h, changes in the motility of the formally non-motile ASM strain began to be observed. Therefore, these cultures were renamed as G20 liquid media cultures (GLM) for RM3194ΔflaAB:tet + flaAcomp (G20) and A20 liquid media cultures (ALM) for RM3194ΔflaAB:tet + flaAcomp (A20). Cells from GLM and ALM cultures after 48 h of growth were routinely transferred into fresh Brucella broth plus kanamycin to maintain the health of the bacterial cultures.
Low concentration agar (0.3%) Brucella plates were used to measure the motility of the C. jejuni strains in this study, with kanamycin or tetracycline being added to the plates where necessary (Barrero-Tobon and Hendrixson, 2012). Bacterial strains to be evaluated were initially grown overnight in 3 mL of Brucella media plus kanamycin or tetracycline if appropriate. The following day 1 μL drops of the individual C. jejuni cultures were injected into the center of the appropriate 0.3% agar Brucella plates. Plates were incubated for 16 h in a microaerobic atmosphere chamber at 42°C. Following incubation, the diameters of the resulting circular bacterial migration zones were measured to the closest millimeter. The average motility for individual strains were then calculated from at least three experimental replicates.
Fifty microliters of the appropriate C. jejuni strain, were placed on acetone cleaned 12 mm Micro-cover glass slides (Thermo Scientific Portsmouth, NH, USA) and allowed to adhere for 30 min. Samples were then covered with 2 ml of 2.5% glutaraldehyde, (Electron Microscopy Sciences, Hatfield, PA, USA) and were allowed to fix for 30 min. The samples were then rinsed twice for 30 min each with 2–3 mL of the 0.1 M imidazole, (Electron Microscopy Sciences, Hatfield, PA, USA), followed by 30 min intervals each of 50, 80, 90% ethanol (The Warner-Graham Company, Cockeysville, MD, USA), 2–3 ml each. The samples were then washed and held three times with 2 mL of 100% ethanol before being critically point dried. The samples were stacked in a wire basket, separated by cloth, and placed in a Critical Point Drying Apparatus, (Denton Vacuum, Inc., Cherry Hill, NJ, USA), using liquid carbon dioxide (Welco Co, Allentown, PA, USA) for approximately 20 min. The samples were mounted on stubs and sputter gold coated for 1 min (EMS 150R ES, EM Sciences, Hatfield, PA, USA). Samples were then viewed with a FEI Quanta 200 F Scanning Electron Microscope, (Hillsboro, OR, USA) with an accelerating voltage of 10KV in high vacuum mode.
RNA was isolated from 2 mL aliquots of each of the designated C. jejuni strains incubated in Brucella broth supplemented with kanamycin or tetracycline where appropriate. Samples were collected for RNA isolation once they had reached optical densities of between 0.1 and 0.3 when measured at 600 nm (OD600) using a Genesys 30 spectrometer (ThermoFisher Scientific). Cells were collected by centrifugation (7,000 × g) and RNA was isolated using the Invitrogen PureLink RNA Kit (ThermoFisher Scientific). The RNA samples were next DNase I treated with Invitrogen’s Turbo Dnase kit (ThermoFisher Scientific). Finally, cDNA synthesis was accomplished using Invitrogen’s SuperScript III First-Strand Synthesis Kit (ThermoFisher Scientific) and a Bio-Rad T100 thermocycler (Bio-Rad, Hercules, CA, USA) following manufacturer’s instructions.
Primer sets used for the quantitative PCR reactions were as follows: rpoA2B_F (forward primer) and rpoA2B_R–(reverse primer) were used to amplify the rpoA housekeeping gene transcription, which served as an internal control to normalize the different starting RNA levels between the samples. cysM2_F and cysM2_R were used to measure the native cysM transcription. flaA2D_F and flaA2D_R were used to measure transcription of the cloned flaA. RT-PCR reactions were performed in 8 tube strips using a Roche LightCycler 96 system (Roche Scientific). Reactions were 20 μL in total volume including: 10 μL of FastStart Essential DNA Green Master Mix (Roche), 1 μL of each 10 μM primer forward and reverse, and 8 μL of cDNA previously diluted 1:40. The amplification protocol was as follows: initial denaturation step at 95°C for 10 min, followed by 45 cycles of 95°C for 10 s, 58°C for 10 s, and 72°C for 10 s and concluded with a melting curve analysis. The LightCycler 96 system software was used to assess the PCR results. Each set of RT-PCR experiments also included “no reverse transcriptase” samples to control for genomic DNA contamination as well as “no template” controls lacking only the cDNA template. For each sample the relative transcription level of the target gene (cysM or flaA) was calculated as a ratio to the housekeeping gene (rpoA) to allow for comparisons between strains and conditions. Equation: ratio = ERCqR/ETCqT (ER, quantification efficiency of reference gene, ET, quantification efficiency of target gene, CqR, quantification cycle of reference, CqT, quantification cycle of target).
RNA was isolated from ASM and ALM samples as described above. The resulting RNA concentrations from each sample were measured using a NanoDrop spectrophotometer (ThermoFisher Scientific) and the RNA was then diluted to produce equal concentrations of starting RNA. cDNA was produced from RNA in the same manner as before except a single primer flaA_cDNA, specific to flaA mRNA was used to produce cDNA only from flaA mRNA in the isolated RNA samples. The flaA specific primer (flaA_cDNA) sequence localizes to the 3′ end of the theoretical flaA mRNA. Next the flaA specific cDNA produced in the previous step were used as template in PCR amplification using the following four forward primers flaA2D_F, flaA2Dshort_F, flaA2Dmedium_F and flaA2Dlong_F each localizing at increasing distance from the 5′ of the flaA cDNA and paired separately with the reverse primer, flaA2B_R. PCR conditions were: 95°C for 10 min followed by 35 cycles of 95°C for 30 s, 57°C for 30 s, 72°C for 90 s with a final single 72°C for 10 min. PCR amplification used the Qiagen Multiplex PCR kit following manufacturer’s recommended method. Subsequent PCR products were visualized on a 1% DNA gel subsequently stained with GelRed (Thomas Scientific).
At least three separate experimental replicates were used to produce all mean and standard error values reported in this paper. The one-way analysis of variance (ANOVA) test was used to determine if mean values compared in this study were significantly different. Subsequent to the ANOVA analysis, the Tukey HSD test was used to separate the mean values. Significant differences were defined as P-values ≤ 0.05.
The 126 bp UTR between the cysteine synthase (cysM) gene and DNA-binding protein HU gene is well conserved between sequenced C. jejuni strains. In this study 264 sequences of this region were aligned and observed for sequence variations. Within the 264 sequences only eighteen nucleotide sites were observed to vary over the 126 bp region. Additionally, most of these nucleotide substitutions were rare with any of the nucleotide variations only occurring in a few strains in total. However, at one site a nucleotide substitution was observed in a sizable percentage of the 264 sequences, with 219 sequences having a guanine (G) nucleotide at this location and the remaining 45 sequences alternately having an adenine (A). Nucleotides within the 126 bp region were numbered starting from the nucleotide directly upstream of the first nucleotide of the cysM coding sequence (#1) and ended at the nucleotide immediately downstream of the last nucleotide of the HU DNA-binding gene (#126). The predominant SNP (G/A) was located at position number 20 with the SNPs designated as G20 or A20. Further analysis of the sequences identified a putative ribosomal binding site ATCCTT running from nucleotides 10 to 15 of the UTR (Figure 1). Additionally, a possible fur binding site AATAATGAT (nucleotides 24 to 32) and an integration host factor (IHF) binding site, which contained the G20/A20 SNP [TTTTA(G)TTT, nucleotides 16 to 23] were identified. While it is believed that C. jejuni lack most transcription factors, including IHF, it does possess the gene for HU which appears to perform many of the same functions of IHF (Stojkova et al., 2019).
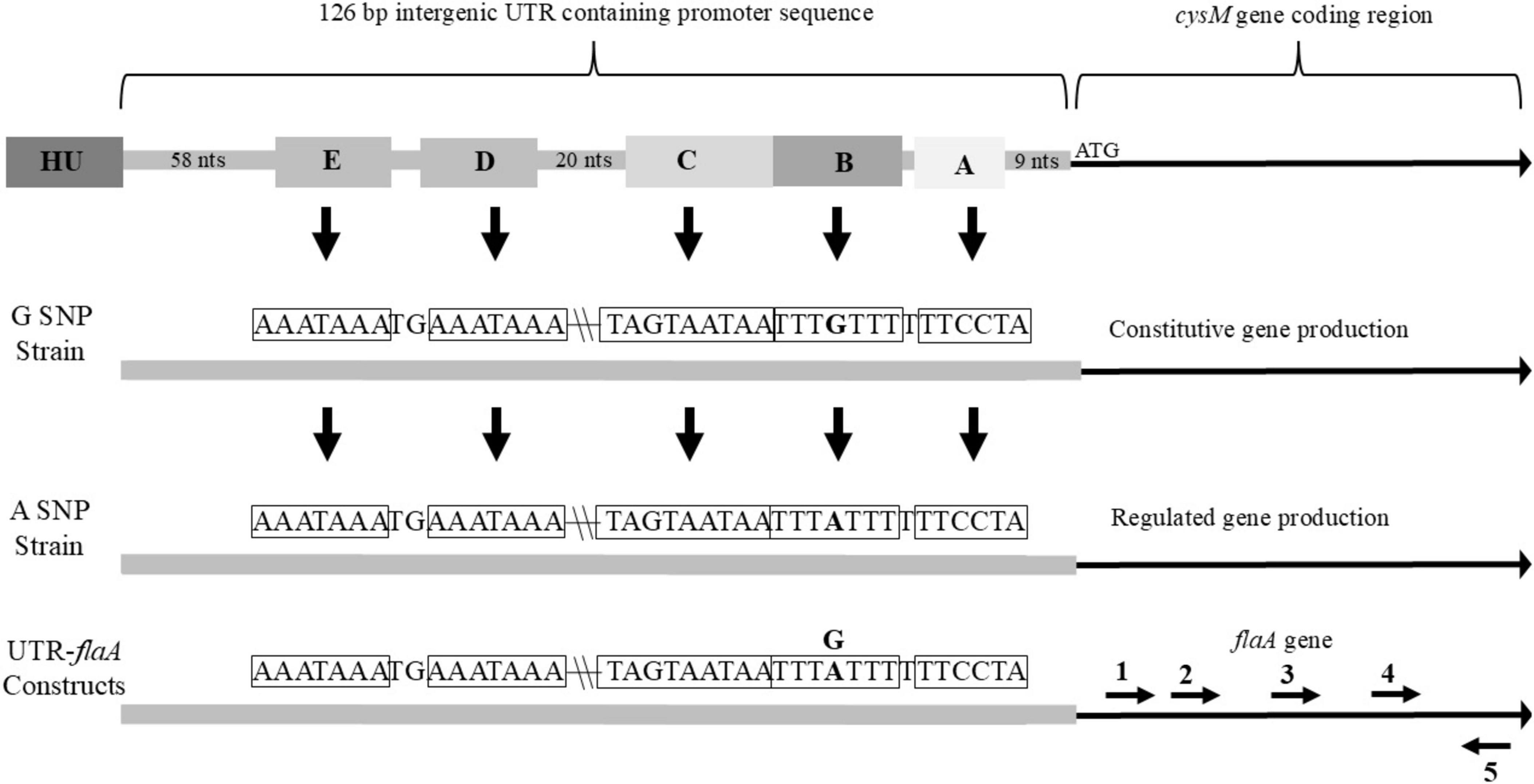
Figure 1. Overview of 126 bp intergenic UTR found upstream of cysM in nature, that was cloned in front of flaA for this study. Important sequence features and a single nucleotide polymorphism (SNP) for strains belonging to the G-SNP or A-SNP groups and their resulting differences in downstream gene production are also noted. (A) predicted ribosome binding site (sequence within corresponding box indicated by arrow); (B) SNP site with surrounding sequence complementary to upstream sequences; (C) predicted fur binding site; (D) first sequence complimentary to sequence B; (E) second sequence complimentary to sequence B. Relative locations of primers (1) flaA2D_F, (2) flaA2Dshort_F, (3) flaA2Dmedium_F, (4) flaA2Dlong_F and (5) flaA2B_R within the flaA sequence of the UTR-flaA constructs. Location of HU gene directly upstream of UTR included in figure.
Sequenced strains with the G20 or A20 SNP in the UTR upstream of cysM were selected from the laboratory strain collection for transcription analysis. The strains were grown in Brucella broth and collected during the exponential phase of growth, at OD600 values of between 0.1 and 0.3. Total RNA was isolated and cDNA produced from the mRNA transcripts. Quantitative RT-PCR was used to measure cysM transcription levels for each strain (Figure 2). The values for the cysM transcription levels were presented as ratios to the transcription levels of the housekeeping gene, rpoA, allowing for normalization of concentration differences between various samples. On average the G20 SNP strains (FSIS136, CDC9511, and RM1285) had higher relative transcription levels of cysM when compared to strains with the A20 SNP (RM3194, RM1188). The average transcription levels of the A20 SNP strains RM3194 and RM1188 did not significantly differ from one another with values of 0.22 and 0.25, respectively. Within the G20 SNP strains, there was variation in the cysM transcription levels. Strain FSIS136 (0.38) differed significantly from CDC9511 (0.51) with regards to cysM transcription, while strain RM1285 (0.47) did not differ significantly from CDC9511 or FSIS136. These results suggested that during exponential growth in a rich medium, the different SNPs contributed to differences in the regulation of cysM transcription.
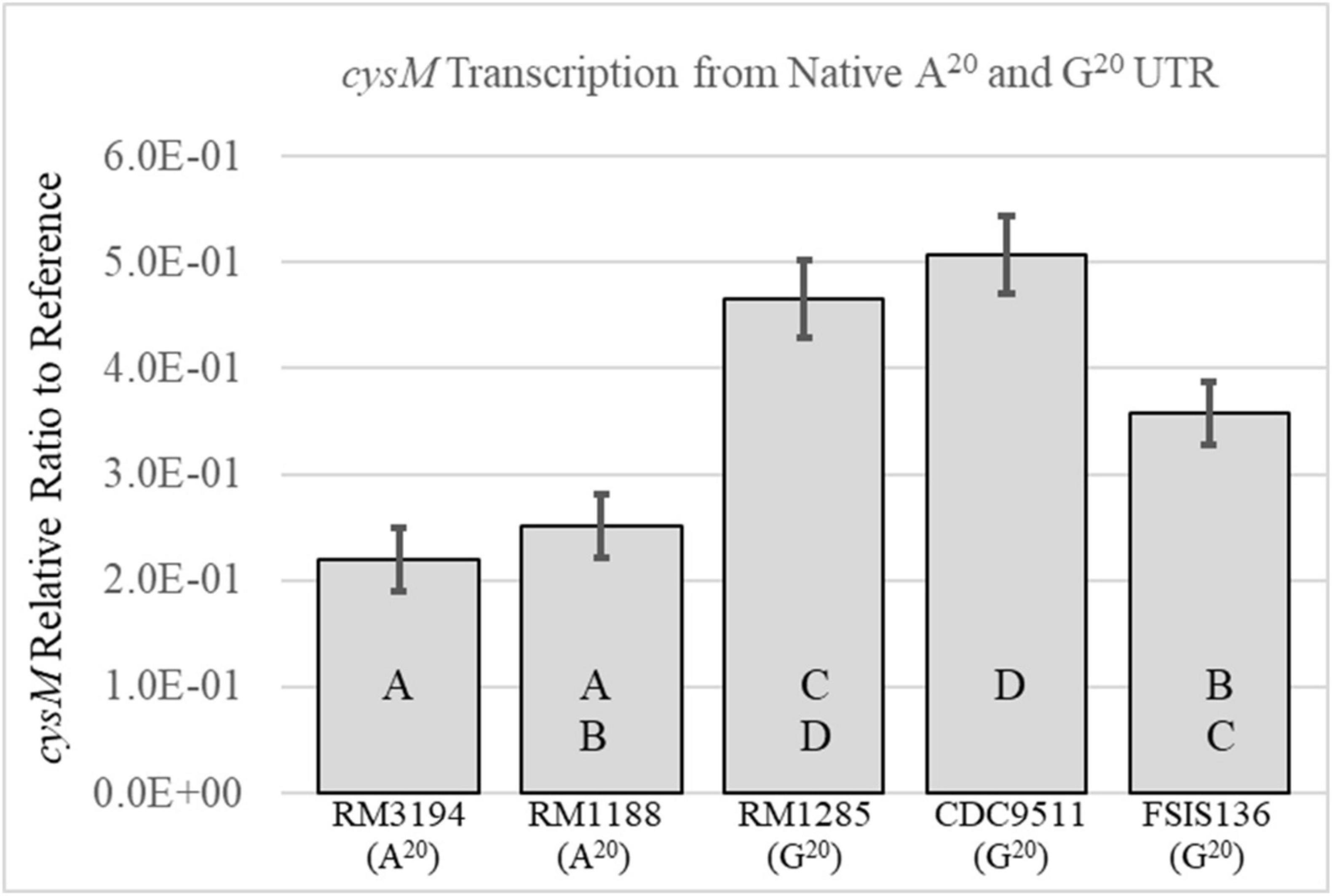
Figure 2. Comparison of the cysM transcription levels between strains with an A20 UTR upstream of cysM—RM3194, RM1188 or a G20 UTR upstream of cysM—RM1285, CDC9511, FSIS136. Significantly different transcription values are indicated by different letter notations.
The UTR upstream of cysM is presumed to contain the promoter region for the gene. The G20 SNP version of the UTR was previously used in research studies as a constitutive promoter to drive expression of exogenous genes cloned into C. jejuni (Uhlich et al., 2022; Gunther et al., 2023). In order to determine how the A20 SNP might affect the transcription of recombinant genes cloned within the C. jejuni chromosome, the cysM UTR contained within of the cloning vector pBlueKan + cysMPro (G20) was changed to adenine (A20). This allowed for the construction of C. jejuni strain RM3194ΔflaAB:tet + flaAcomp (A20). C. jejuni RM3194ΔflaAB:tet is a clinical strain with both flagellar structural genes flaA and flaB removed. The exogenous flaA cloned behind an A20 UTR was inserted within the C. jejuni RM3194ΔflaAB:tet chromosome between the 16S and 23S ribosomal RNA genes. The resulting strain, RM3194ΔflaAB:tet + flaAcomp (A20), was identical to the previously constructed strain, C. jejuni RM3194ΔflaAB:tet + flaAcomp (G20) (Uhlich et al., 2022), with the exception of the A20 SNP modification now upstream of the cloned flaA gene. The C. jejuni RM3194ΔflaAB:tet + flaAcomp (A20) strain was originally believed to be non-motile, however, it was observed to become motile when incubated in Brucella broth for two or more consecutive cultures each lasting 48 h. To illustrate the motility differences produced by the two UTR SNP forms, the strains C. jejuni RM3194ΔflaAB:tet + flaAcomp (A20) and C. jejuni RM3194ΔflaAB:tet + flaAcomp (G20) were grown either on solid media (1.5% agar) to form cultures designated GSM (G20 UTR) and ASM (A20 UTR) or within liquid medium with at least two passages into fresh medium resulting in cultures designated GLM (G20 UTR) and ALM (A20 UTR). The motilities of each of these four cultures, were compared on low percentage agar, along with a positive control strain, C. jejuni RM3194 + express, and a negative control strain, C. jejuni RM3194ΔflaAB:tet (Figure 3). On average the positive control strain with the wild-type copies of both flagella structural genes, driven by their native promoters, produced motility zones of 28.3 mm. The GSM and GLM cultures produced average motility zones of 15.7 and 10.6 mm, respectively, with the different growth conditions having only a small influence on cell motility. This differed significantly from the influence that the growth conditions had on motility when flaA was combined with the A20 UTR. The ALM culture produced an average motility zone of 18.5 mm while the ASM culture showed no motility appearing identical to the negative control strain that lacked any structural flagella genes. Additionally, cells from the ALM culture when placed onto Brucella agar plates and transferred after 24 h of growth onto fresh agar plates twice more, were observed to completely lose their motility. PCR amplification of the UTR locus from both the ASM and ALM cultures confirmed that the adenine SNP was not modified when switching between motile and non-motile states (data not shown). Additionally, since strain RM3194ΔflaAB:tet + flaAcomp (A20) was shown to switch between a motile and non-motile phenotype depending on how it was sub-cultured, suggests that a mutational event in a location other than the UTR is not responsible for the observed changes.

Figure 3. Motility of C. jejuni strains. Strain positive for motility: RM3194 + express. Strain negative for motility: RM3194ΔflaAB:tet. Strains unknown for motility: RM3194ΔflaAB:tet + flaAcomp (A20) grown on solid media (ASM), RM3194ΔflaAB:tet + flaAcomp (A20) grown in liquid media (ALM), RM3194ΔflaAB:tet + flaAcomp (G20) grown on solid media (GSM), RM3194ΔflaAB:tet + flaAcomp (G20) grown in liquid media (GLM). Significantly different motility values are indicated by different letter notations.
The strains and cultures used in the previous motility experiments (Figure 3) were assessed by scanning electron microscopy to identify differences in cell structure that might affect motility. Representative images for each culture condition are presented in Figure 4. Flagella are readily visible on the cells comprising the positive control strain (Figure 4A). Conversely, no flagella were visible on cells of the negative control strain (Figure 4B). The non-motile ASM culture (Figure 4C) appears similar to the negative control without any flagella readily visible on the cells surface. The motile ALM (Figure 4D), GSM (Figure 4E), and GLM (Figure 4F) each have flagella visible on the cells within the respective cultures.
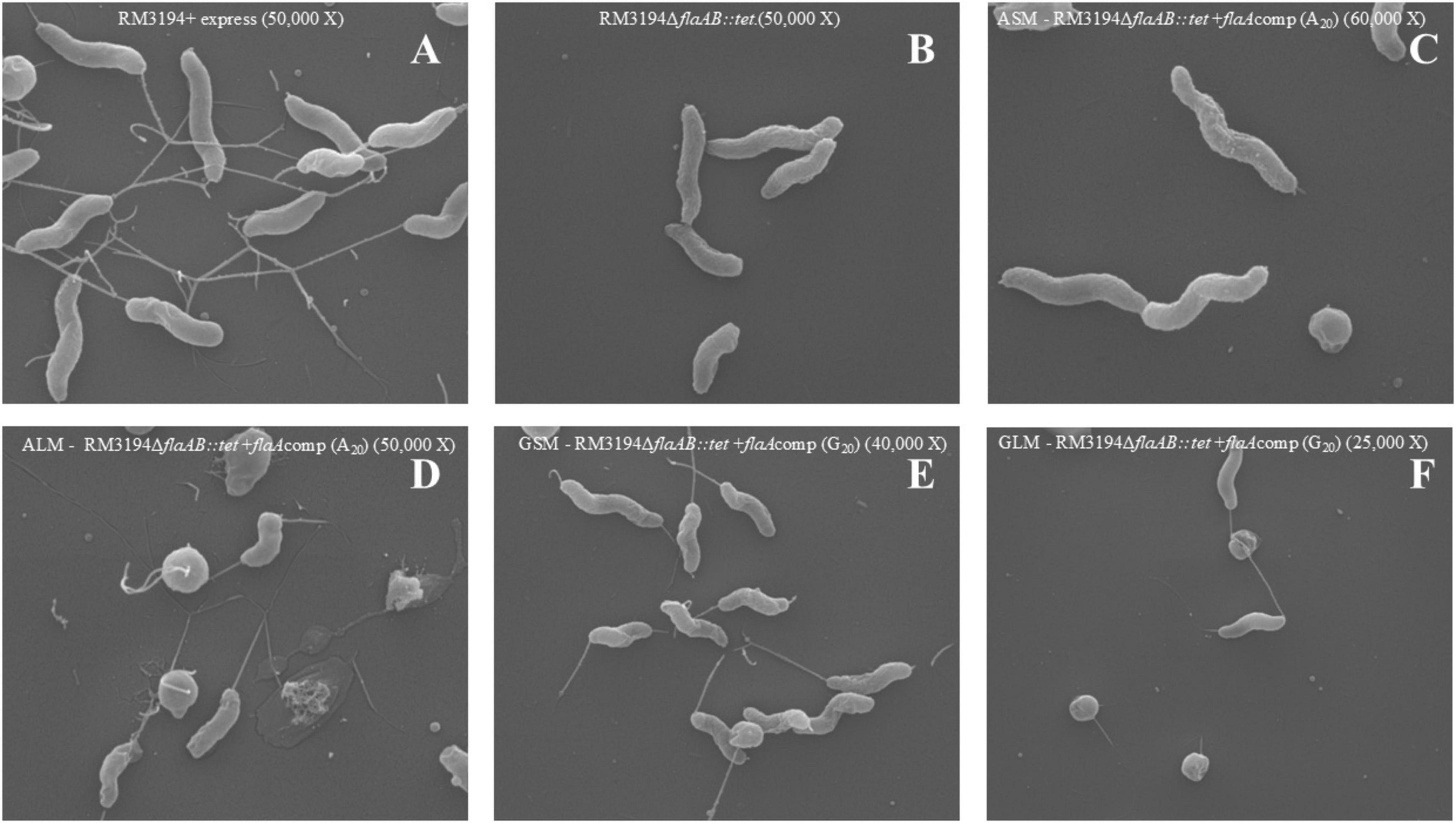
Figure 4. Scanning Electron Microscopy of G20 or A20 UTR plus flaA strains grown to produce different motility. (A) Positive control, RM3194 + express (50,000 X). (B) Negative control, RM3194ΔflaAB:tet (50,000 X). (C) Non-motile strain, ASM–RM3194ΔflaAB:tet + flaAcomp (A20) (60,000 X). (D) Motile strain, ALM–RM3194ΔflaAB:tet + flaAcomp (A20) (50,000 X). (E) Motile strain GSM–RM3194ΔflaAB:tet + flaAcomp (G20) (40,000 X). (F) Motile strain, GLM–RM3194ΔflaAB:tet + flaAcomp (G20) (25,000 X).
Quantitative RT-PCR was used to determine the flaA transcription levels in the positive control, RM3194 + express, and negative control strain, RM3194ΔflaAB:tet; as well as strain, RM3194ΔflaAB:tet + flaAcomp (A20), grown under conditions producing motility and observable flagella (ALM) and the conditions without motility or observable flagella (ASM). The transcription of flaA was also measured for the strain, RM3194ΔflaAB:tet + flaAcomp (G20), grown under GLM and GSM conditions, both of which previously produce motility and observable flagella. Results for these experiments are presented in Figure 5. Not surprisingly strain RM3194 + express had the highest level of flaA transcription since it also previously had demonstrated the greatest motility. The non-motile negative control strain with no flaA or flaB predictably did not show any measurable flaA transcription. The strain and culture conditions that previously demonstrated motility and flagellar expression, ALM, GSM, and GLM all had similar average levels of flaA transcription. Unsurprisingly, these levels of flaA transcription were significantly less than that in the positive control strain given the superior motility demonstrated by the control strain. The non-motile, no observable flagella culture, ASM, had significantly lower flaA transcription levels when compared to ALM, GSM and GLM cultures. In fact the flaA transcription of RM3194ΔflaAB:tet + flaAcomp (A20) grown in the ASM conditions was not significantly different from the negative control strain which corresponded nicely with the observation that both the negative control and ASM samples were non-motile.
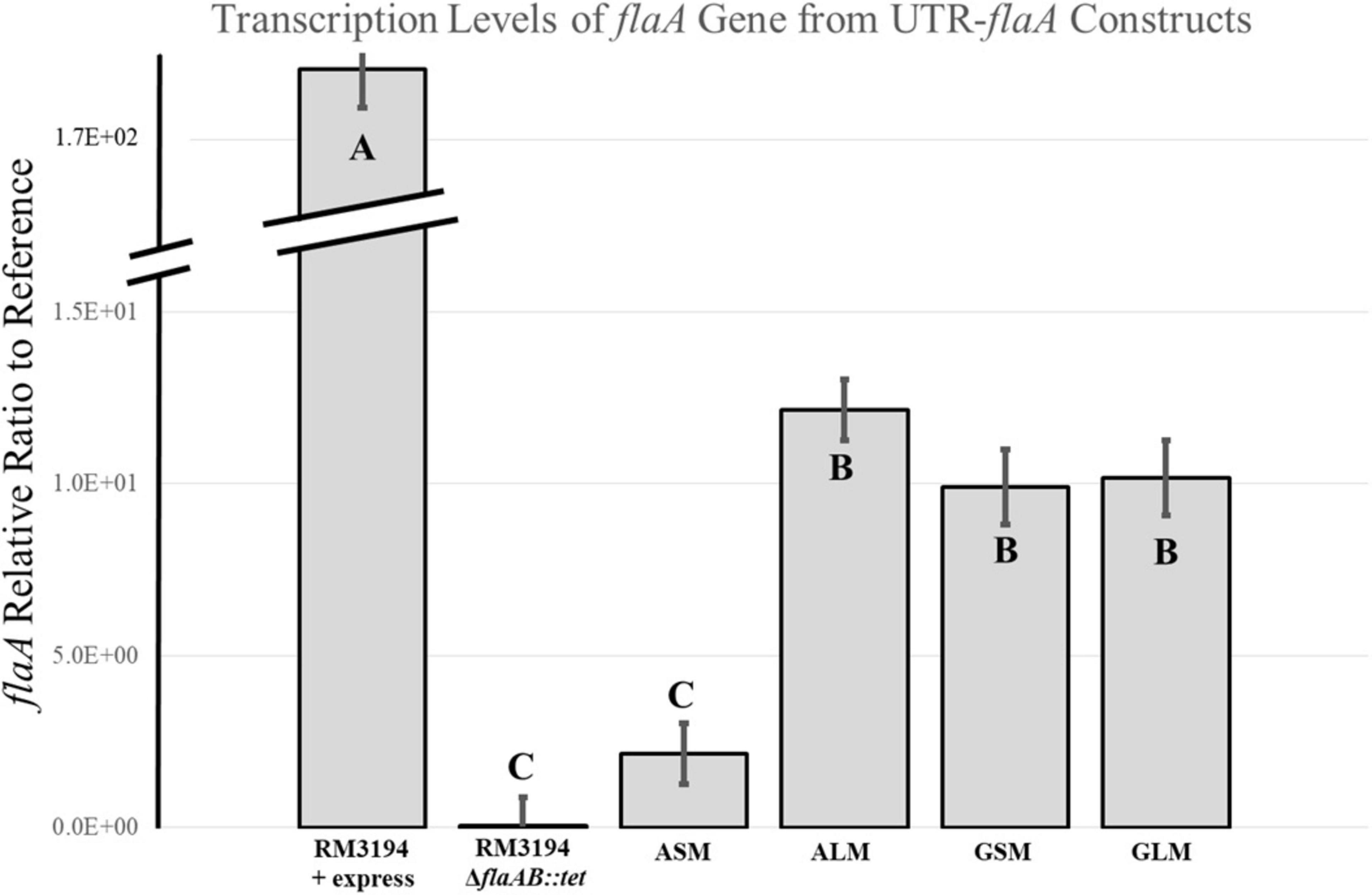
Figure 5. Transcription analysis of flaA downstream of G20 or A20 UTR sequence strains, RM3194ΔflaAB:tet + flaAcomp (G20) or RM3194ΔflaAB:tet + flaAcomp (A20), grown to produce different motility. Results are presented as a relative ratio of flaA transcription to the housekeeping gene rpoA transcription to control for sample-to-sample variations. Significantly different transcription values are indicated by different letter notations.
Strain RM3194ΔflaAB:tet + flaAcomp (A20) grown in ALM or ASM conditions demonstrated differences in motility, observed flagella and flaA transcription levels. Since the parent strain, RM3194, is known to possess an A20 UTR upstream of cysM, quantitative RT-PCR experiments were performed to determine how the ALM and ASM growth conditions affected the transcription of the native cysM. The results of these experiments are presented in Figure 6. The ALM condition, on average, resulted in significantly greater levels of cysM transcription compared to the ASM conditions, which agreed with previous results measuring the expression of recombinant flaA in strain RM3194ΔflaAB:tet + flaAcomp (A20) (Figure 5). This result suggests that transcription of any gene cloned downstream of the A20 UTR will be regulated differently depending on the growth conditions under which the host strain is cultured.
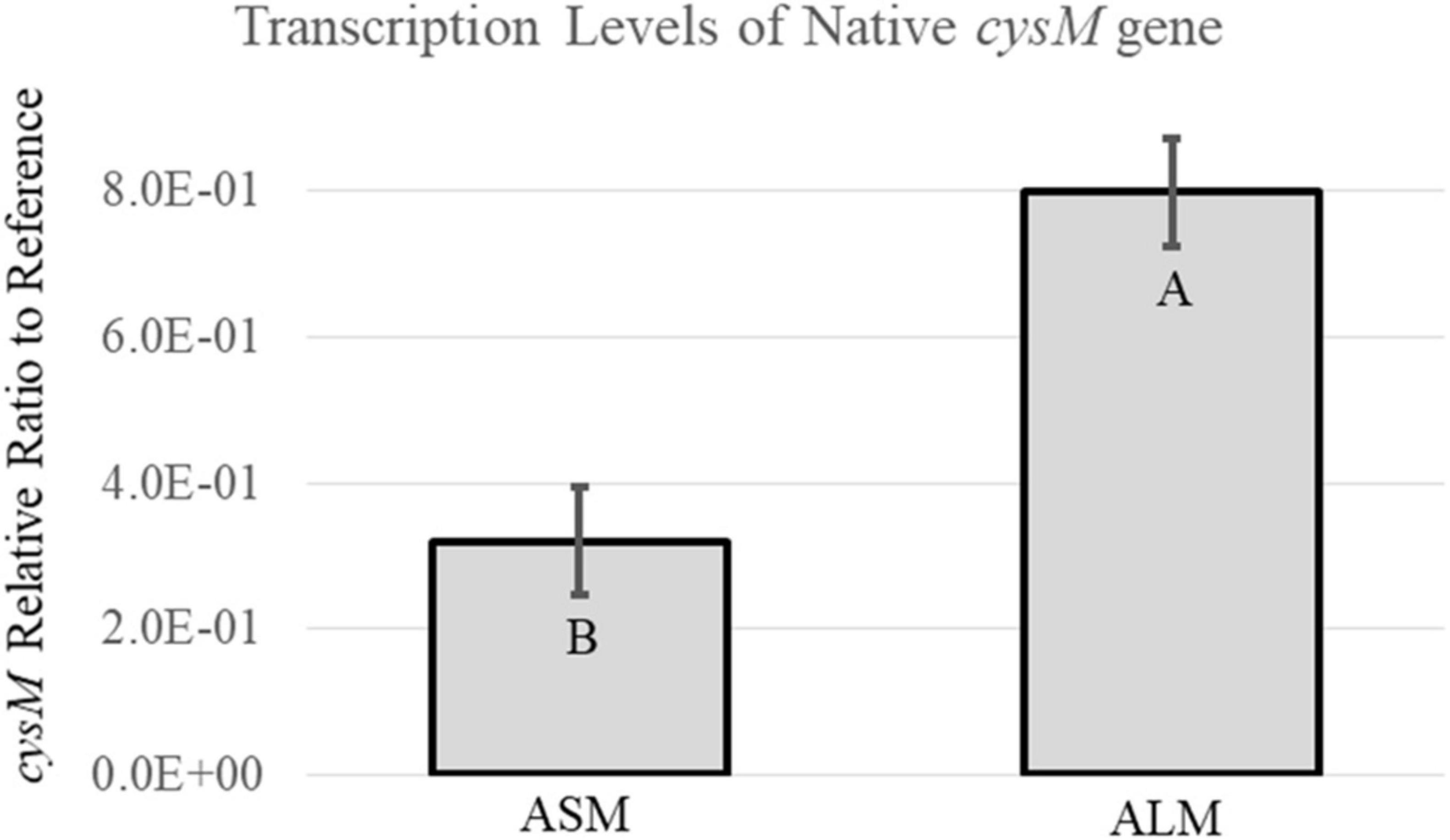
Figure 6. Transcription analysis of native cysM in the A20 UTR sequence strain, RM3194ΔflaAB:tet + flaAcomp (A20), grown to produce different motility. Results are presented as a relative ratio of cysM transcription to the housekeeping gene rpoA transcription to control for sample-to-sample variations. Significantly different transcription values are indicated by different letter notations.
A flaA specific primer was used to only produce flaA cDNA from RNA collected when strain RM3194ΔflaAB:tet + flaAcomp (A20) was grown in ALM or ASM conditions. The total RNA amounts present in the ALM and ASM samples were selectively diluted to equalize the starting RNA concentrations between the samples. The resulting flaA cDNAs isolated from the two different growth conditions were then used as template for PCR amplification. The forward primers used for the PCR amplifications were designed to bind to the flaA cDNA sequences at increasing distances from the 5′ end of the gene. Primer flaA2D_F binds to the sequence starting 239 bps downstream of the ATG start codon of flaA; while flaA2Dshort_F binds 438 bps downstream of the ATG start codon, flaA2Dmedium_F binds 935 bps and flaA2Dlong_F binds 1,223 bps downstream of the gene’s ATG start codon. All forward primers were paired with the same reverse primer flaA2B_R. Therefore, when paired with flaA2B_R for PCR amplification flaA2D_F produces a 1285 bp fragment, flaA2Dshort_F produces a 1,085 bp fragment, flaA2Dmedium_F produces a 588 bp fragment and flaA2Dlong_F produces a 300 bp fragment. The relative location of the primers within flaA are presented in Figure 1. When the resulting PCR amplifications were visualized (Figure 7) only extremely faint bands were visible in lanes 2 and 3, which are the results of amplifying the cDNA from the ASM sample using forward primers flaA2D_F and flaA2Dshort_F, respectively. However, in lanes 4 and 5 the forward primers flaA2Dmedium_F and flaA2Dlong_F produced easily visualized bands amplifying the same ASM sample. With the ALM sample, primers flaA2D_F, flaA2Dshort_F, flaA2Dmedium_F and flaA2Dlong_F (lanes 6–9) all produced easily visualized bands. These results suggest that there were significantly fewer full length flaA cDNAs produced from the ASM growth samples compared to the ALM samples. This would likely result from there being significantly fewer full length flaA mRNA transcripts originally in the ASM samples compared to the ALM samples. The cDNAs, produced from flaA mRNA, appear to exist in similar amounts in the ASM (lanes 4 and 5) and ALM (lanes 8 and 9) samples when the PCR reactions amplify the 3′ region of flaA cDNA present in the samples. Conversely, it appears for the most part that only the 5′ portions of the flaA mRNA sequences from the ALM sample are present and accessible to reverse transcriptase allowing for production of the full length flaA cDNA as demonstrated by the larger PCR products manufactured from the PCR reactions from ALM (lanes 6 and 7) but not ASM (lanes 2 and 3) samples. This suggests that the 5′ region of the flaA mRNA transcripts present in the ASM sample are missing or in some way not accessible to the reverse transcriptase that would normally produce full length flaA cDNA. This same type of experiment was repeated using primers specific for native cysM. Similarly, we observed that the 5′ portions of cysM mRNA sequences from the ALM samples appeared to be more accessible to reverse transcriptase; allowing for the production of more full length cysM cDNA compared to the samples from the ASM conditions. It should be noted that differences observed in full length cysM cDNA between ALM and ASM conditions were not as dramatic as that observed with flaA cDNA (data not shown).
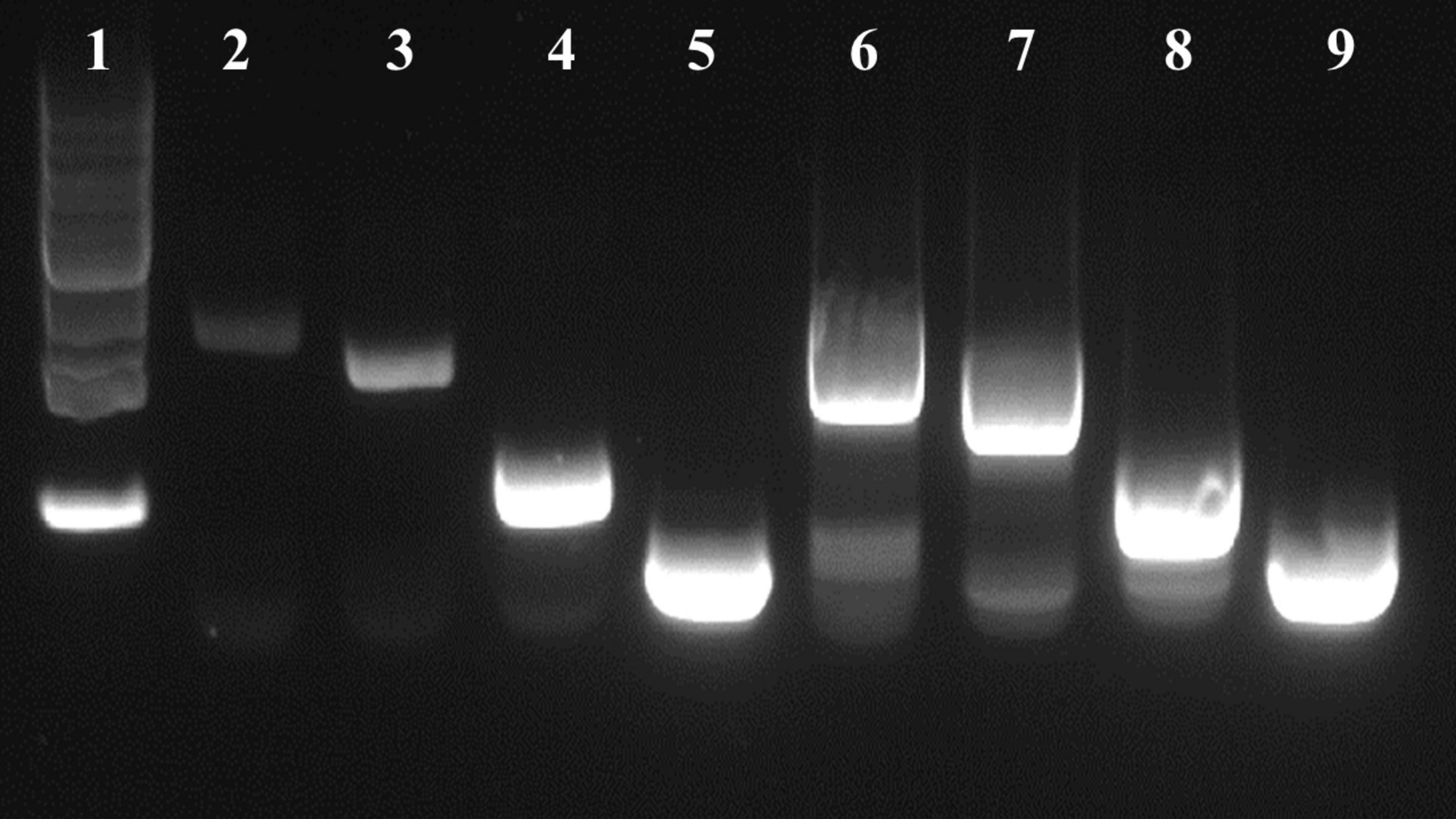
Figure 7. DNA gel visualization of PCR products from amplifications of flaA cDNA produced from strain RM3194ΔflaAB:tet + flaAcomp (A20) grown in ASM or ALM conditions, using the following primer combinations. Lane (1) 1kb DNA Ladder (Invitrogen), lane (2) ASM with flaA2D_F/flaA2B_R, lane (3) ASM with flaA2Dshort_F/flaA2B_R, lane (4) ASM with flaA2Dmedium_F/flaA2B_R, lane (5) ASM with flaA2Dlong_F/flaA2B_R, lane (6) ALM with flaA2D_F/flaA2B_R, lane (7) ALM with flaA2Dshort_F/flaA2B_R, lane (8) ALM with flaA2Dmedium_F/flaA2B_R, lane (9) ALM with flaA2Dlong_F/flaA2B_R.
The observation and subsequent analysis of the SNP that alters gene promoter activity of the UTR upstream of cysM produces two different areas for consideration. The first is the evolutionary utility of the existence of two different approaches to regulating cysM production in C. jejuni. The second is the practical utility of a gene promoting sequence that can be switched between active and inactive states, in terms of its use as a molecular tool for studying gene expression in C. jejuni.
The cysteine synthase gene (cysM) is required for C. jejuni’s production of the amino acid cysteine when it cannot be collected from the bacterium’s surrounding environment (Garvis et al., 1997; Hitchcock et al., 2022). The function of cysM is to facilitate the transfer of sulfur to O-acetyl-L-serine to produce cysteine. The sulfur in this process can be derived from hydrogen sulfide which is produced by sulfate reducing bacteria in the human gut (Dordevic et al., 2021). Those same gut bacteria often utilize cysteine in their production of hydrogen sulfide or other metabolites (Braccia et al., 2021; Louis and Flint, 2017). Additionally, a significant portion of the normal human intestinal flora are cysteine auxotrophs and must utilize available cysteine to survive (Starke et al., 2023). Therefore, a C. jejuni strain infecting the human gut would likely be in a highly competitive, nutrient limited environment and would likely have to scavenge cysteine or a sulfur source in order to survive (Vorwerk et al., 2014; Stahl et al., 2012). It is therefore possible to think of cysM as a virulence factor within the human gut. The gene is necessary for virulence if the organism cannot produce the cysteine it needs to survive from environmental sulfur sources, since it would die and be unable to cause disease (Shayya et al., 2023).
In the presented experiments, C. jejuni with the G20 SNP containing UTR was observed to produce similar levels of downstream gene transcripts under the two different growth conditions tested (ASM and ALM). Therefore, the G20 SNP containing strains should already be producing some level of cysteine synthase when it encounters an environment lacking available cysteine. Conversely, those same G20 SNP containing UTR strains would be theoretically wasting valuable resources to always make cysteine synthase even when the C. jejuni strain finds itself in an environment abundant in cysteine where production of the synthase would be unnecessary (Anand et al., 2020; Geisel, 2011). In the case of an A20 SNP UTR strain, it initially appeared to produce downstream gene transcripts at levels significantly less than the G20 SNP strain. However, after incubation in at least two consecutive cultures each lasting 48 h, the A20 SNP strains showed a significant increase in the transcription of cysM. The potential of the A20 SNP UTR to regulate gene transcription could allow those strains with the A20 SNP to only produce cysteine synthase when needed. This would help solve any potential issue with wasting energy through constitutive transcription of the synthase gene, as appears could be the case with the G20 SNP strain. However, in a direct competition for limited sulfur in the absence of cysteine, the G20 SNP strains, which already produce cysteine synthase, might be able to outcompete A20 SNP strains, due to the time required for these strains to induce their synthase production. Conversely, in an environment rich in available cysteine the A20 SNP stains may be able to conserve energy for growth by not increasing transcription of the cysteine synthase, an option that the G20 strains do not possess, potentially leading to the A20 strains outcompeting the G20 strains.
The exact mechanism utilized by the A20 SNP stains to regulate cysM transcription is currently unknown. Obviously, the mechanism depends to some degree on the UTR SNP (G20 or A20) which is the focus of this study. Based on the results presented in Figure 7, it appears that transcriptional differences observed for strains carrying the A20 SNP, following exposure to different growth conditions (ASM and ALM), may be due to the use of alternate transcription start sites, which lead to differences in the length of transcripts produced. In the ALM condition mRNA transcripts covered the entire gene length while growth in the ASM condition produced truncated mRNA transcripts covering a fraction of the gene’s coding region. The mechanism therefore could involve the blocking of RNA polymerase from starting mRNA transcription at the proper transcription start site within the UTR upstream of the gene, resulting in the polymerase starting mRNA transcription at alternative sites internal to the gene. It is possible that the SNP change from adenine to guanine could alter the binding of a cofactor which could ultimately prevent proper RNA polymerase binding. This proposed mechanism could also depend on interaction with a putative fur binding site (sequence C, Figure 1) almost directly adjacent to the SNP site (Grabowska et al., 2011; Holmes et al., 2005). Previous research has described single nucleotide changes in intergenic regions that altered the binding of regulatory factors and affected transcription (Cagliero et al., 2007; Pérez-Boto et al., 2015; Zeng et al., 2014). These single nucleotide changes most often blocked binding of repressor elements or restored RNA polymerase recognition sites and in both cases led to constitutive gene transcription. Other research described a change in the regulatory region of a gene that altered the gene’s transcription going from inducible to constitutive, but this change required large sequence deletions or additions and not a SNP (Deng et al., 2015). Alternately, there could be a hairpin structure formed through the binding of the UTR sequence B (Figure 1), which contains the SNP, to the complimentary upstream sequences D or E. This could again block binding of the RNA polymerase to the proper transcription start site. The SNP change from adenine to guanine could be sufficient to reduce the potential hairpin formed by these sequences, thus allowing for the observed constitutive transcription.
While the exact mechanism of transcriptional control remains to be determined, there are immediate and practical uses for the A20 SNP UTR sequence. The presented research originated from work developing new cloning/expression tools for C. jejuni which included using the G20 SNP UTR for driving constitutive transcription of specific genes (Uhlich et al., 2022). Additionally, there exists previously developed gene cloning and constitutive transcription systems (Karlyshev and Wren, 2005; Cameron and Gaynor, 2014). However, with the A20 SNP UTR a target gene can now be cloned downstream of the UTR and significantly different amounts of the gene transcribed depending on the growth conditions used, made possible by only a single nucleotide change in the UTR sequence. The ability to toggle gene transcription levels up and down while simultaneously observing the subsequent phenotypic and genotypic changes to the C. jejuni strain should constitute a useful research tool. While currently the switching between gene transcription levels is accomplished by varying growth conditions, it seem likely that there remains a more specific induction method to be discovered. This could potentially lead to a titratable induction model where multiple concentrations of an inducer can produce multiple levels of gene transcription. Further investigations of how the upstream UTR controls cysM transcription will increase the understanding of how C. jejuni can adapt to changes in its immediate environment as well as provide improvements to molecular research tools.
The original contributions presented in this study are included in this article/supplementary material, further inquiries can be directed to the corresponding author.
NG: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review and editing. SK: Data curation, Formal Analysis, Investigation, Methodology, Software, Writing – original draft, Writing – review and editing. A-AW: Data curation, Investigation, Methodology, Validation, Writing – review and editing. MM: Formal Analysis, Investigation, Methodology, Resources, Software, Writing – review and editing. JR: Conceptualization, Formal Analysis, Resources, Writing – review and editing. JU: Investigation, Software, Visualization, Writing – review and editing. GU: Conceptualization, Formal Analysis, Investigation, Methodology, Resources, Writing – original draft, Writing – review and editing.
The authors declare that financial support was received for the research and/or publication of this article. This research was supported by congressional appropriated funds assigned to the USDA–Agricultural Research Service, National Program 108 Food Safety [Project # 8072-42000-094-000-D]. Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the United States Department of Agriculture.
We would like to thank Dr. George Paoli for his helpful review of this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alazzam, B., Bonnassie-Rouxin, S., Dufour, V., and Ermel, G. (2011). MCLMAN, a new minimal medium for Campylobacter jejuni NCTC 11168. Res. Microbiol. 162, 173–179. doi: 10.1016/j.resmic.2010.09.024
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410.
Anand, A., Chen, K., Catoiu, E., Sastry, A. V., Olson, C. A., Sandberg, T. E., et al. (2020). OxyR Is a convergent target for mutations acquired during adaptation to oxidative stress-prone metabolic states. Mol. Biol. Evol. 37, 660–667. doi: 10.1093/molbev/msz251
Barrero-Tobon, A. M., and Hendrixson, D. R. (2012). Identification and analysis of flagellar coexpressed determinants (Feds) of Campylobacter jejuni involved in colonization. Mol. Microbiol. 84, 352–369. doi: 10.1111/j.1365-2958.2012.08027.x
Black, R. E., Levine, M. M., Clements, M. L., Hughes, T. P., and Blaser, M. J. (1988). Experimental Campylobacter jejuni infection in humans. J. Infect. Dis. 157, 472–479.
Braccia, D. J., Jiang, X., Pop, M., and Hall, A. B. (2021). The capacity to produce hydrogen sulfide (H(2)S) via cysteine degradation is ubiquitous in the human gut microbiome. Front. Microbiol. 12:705583. doi: 10.3389/fmicb.2021.705583
Burnham, P. M., and Hendrixson, D. R. (2018). Campylobacter jejuni: collective components promoting a successful enteric lifestyle. Nat. Rev. Microbiol. 16, 551–565. doi: 10.1038/s41579-018-0037-9
Cagliero, C., Maurel, M. C., Cloeckaert, A., and Payot, S. (2007). Regulation of the expression of the CmeABC efflux pump in Campylobacter jejuni: identification of a point mutation abolishing the binding of the CmeR repressor in an in vitro - selected multidrug-resistant mutant. Fems Microbiol. Lett. 267, 89–94. doi: 10.1111/j.1574-6968.2006.00558.x
Cameron, A., and Gaynor, E. C. (2014). Hygromycin B and apramycin antibiotic resistance cassettes for use in Campylobacter jejuni. PLoS One 9:e95084. doi: 10.1371/journal.pone.0095084
Davis, L., Young, K., and Dirita, V. (2008). Genetic manipulation of Campylobacter jejuni. Curr. Protoc. Microbiol. 8, 8A.2.1–8A.2.17.
Deng, F., Shen, J., Zhang, M., Wu, C., Zhang, Q., and Wang, Y. (2015). Constitutive and inducible expression of the rRNA Methylase Gene erm(B) in Campylobacter. Antimicrob. Agents Chemother. 59, 6661–6664. doi: 10.1128/AAC.01103-15
Dordevic, D., Jancikova, S., Vitezova, M., and Kushkevych, I. (2021). Hydrogen sulfide toxicity in the gut environment: Meta-analysis of sulfate-reducing and lactic acid bacteria in inflammatory processes. J. Adv. Res. 27, 55–69. doi: 10.1016/j.jare.2020.03.003
European Food, and Safety Authority. (2021). The European union one health 2019 zoonoses report. EFSA J. 19:e06406.
Garvis, S. G., Tipton, S. L., and Konkel, M. E. (1997). Identification of a functional homolog of the Escherichia coli and Salmonella typhimurium cysM gene encoding O-acetylserine sulfhydrylase B in Campylobacter jejuni. Gene 185, 63–67. doi: 10.1016/s0378-1119(96)00631-2
Geisel, N. (2011). Constitutive versus responsive gene expression strategies for growth in changing environments. PLoS One 6:e27033. doi: 10.1371/journal.pone.0027033
Gillespie, I. A., O’brien, S. J., Frost, J. A., Adak, G. K., Horby, P., Swan, A. V., et al. (2002). A case-case comparison of Campylobacter coli and Campylobacter jejuni infection: A tool for generating hypotheses. Emerg. Infect. Dis. 8, 937–942. doi: 10.3201/eid0809.010817
Grabowska, A. D., Wandel, M. P., Lasica, A. M., Nesteruk, M., Roszczenko, P., Wyszynska, A., et al. (2011). Campylobacter jejuni dsb gene expression is regulated by iron in a Fur-dependent manner and by a translational coupling mechanism. BMC Microbiol. 11:166. doi: 10.1186/1471-2180-11-166
Gunther, N. W. T., Bono, J. L., and Needleman, D. S. (2015). Complete genome sequence of Campylobacter jejuni RM1285, a rod-shaped morphological variant. Genome Announc. 3:e01361–15. doi: 10.1128/genomeA.01361-15
Gunther, N. W., Nunez, A., Bagi, L., Abdul-Wakeel, A., Ream, A., Liu, Y., et al. (2023). Butyrate decreases Campylobacter jejuni motility and biofilm partially through influence on LysR expression. Food Microbiol. 115:104310. doi: 10.1016/j.fm.2023.104310
Gunther, N. W., Reichenberger, E. R., and Bono, J. L. (2016). Complete genome sequence of UV-resistant Campylobacter jejuni RM3194. Including an 81.08-Kilobase Plasmid. Genome Announc. 4, e305–e316. doi: 10.1128/genomeA.00305-16
Hitchcock, N., Kelly, D. J., Hitchcock, A., and Taylor, A. J. (2022). Cysteine biosynthesis in Campylobacter jejuni: Substrate specificity of CysM and the dualism of sulfide. Biomolecules 13:86. doi: 10.3390/biom13010086
Holmes, K., Mulholland, F., Pearson, B. M., Pin, C., Mcnicholl-Kennedy, J., Ketley, J. M., et al. (2005). Campylobacter jejuni gene expression in response to iron limitation and the role of Fur. Microbiology Sgm 151, 243–257.
Kaakoush, N. O., Castano-Rodriguez, N., Mitchell, H. M., and Man, S. M. (2015). Global epidemiology of Campylobacter infection. Clin. Microbiol. Rev. 28, 687–720.
Karlyshev, A. V., and Wren, B. W. (2005). Development and application of an insertional system for gene delivery and expression in Campylobacter jejuni. Appl. Environ. Microbiol. 71, 4004–4013. doi: 10.1128/AEM.71.7.4004-4013.2005
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Levin, R. E. (2007). Campylobacter jejuni: A review of its characteristics, pathogenicity, ecology, distribution, subspecies characterization and molecular methods of detection. Food Biotechnol. 21, 271–347.
Louis, P., and Flint, H. J. (2017). Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 19, 29–41.
Marder, E. P., Cieslak, P. R., Cronquist, A. B., Dunn, J., Lathrop, S., Rabatsky-Ehr, T., et al. (2017). Incidence and trends of infections with pathogens transmitted commonly through food and the effect of increasing use of culture-independent diagnostic tests on surveillance - foodborne diseases active surveillance network, 10 US sites, 2013-2016. Mmwr Morbidity Mortality Weekly Rep. 66, 397–403. doi: 10.15585/mmwr.mm6615a1
Miller, W. G., Pearson, B. M., Wells, J. M., Parker, C. T., Kapitonov, V. V., and Mandrell, R. E. (2005). Diversity within the Campylobacter jejuni type I restriction - modification loci. Microbiology Sgm 151, 337–351.
Pérez-Boto, D., Acebo, P., García-Peña, F. J., Abad, J. C., Echeita, M. A., and Amblar, M. (2015). Isolation of a point mutation associated with altered expression of the CmeABC efflux pump in a multidrug-resistant Campylobacter jejuni population of poultry origin. J. Glob. Antimicrob. Resist. 3, 115–122. doi: 10.1016/j.jgar.2015.03.010
Shayya, N. W., Bandick, R., Busmann, L. V., Mousavi, S., Bereswill, S., and Heimesaat, M. M. (2023). Metabolomic signatures of intestinal colonization resistance against Campylobacter jejuni in mice. Front. Microbiol. 14:1331114. doi: 10.3389/fmicb.2023.1331114
Solovyev, V., and Salamov, A. (2011). “Automatic annotation of microbial genomes and metagenomic sequences,” in Metagenomics and its Applications in Agriculture, Biomedicine, and Environmental Studies, ed. R. W. LI (Hauppauge, NY: Nova Science Publisher’s).
Stahl, M., Butcher, J., and Stintzi, A. (2012). Nutrient acquisition and metabolism by Campylobacter jejuni. Front. Cell. Infect. Microbiol. 2:5. doi: 10.3389/fcimb.2012.00005
Starke, S., Harris, D. M. M., Zimmermann, J., Schuchardt, S., Oumari, M., Frank, D., et al. (2023). Amino acid auxotrophies in human gut bacteria are linked to higher microbiome diversity and long-term stability. ISME J. 17, 2370–2380. doi: 10.1038/s41396-023-01537-3
Stojkova, P., Spidlova, P., and Stulik, J. (2019). Nucleoid-associated protein HU: A lilliputian in gene regulation of bacterial virulence. Front. Cell. Infect. Microbiol. 9:159. doi: 10.3389/fcimb.2019.00159
Uhlich, G. A., Bagi, L., and Gunther, N. W. (2022). Cloning vectors for gene delivery, integration and expression in Campylobacter jejuni. Biotechniques 72, 255–262.
Vorwerk, H., Mohr, J., Huber, C., Wensel, O., Schmidt-Hohagen, K., Gripp, E., et al. (2014). Utilization of host-derived cysteine-containing peptides overcomes the restricted sulphur metabolism of Campylobacter jejuni. Mol. Microbiol. 93, 1224–1245. doi: 10.1111/mmi.12732
Keywords: Campylobacter, transcription, mRNA, regulation, SNP, cysteine
Citation: Gunther IV NW, Kanrar S, Abdul-Wakeel A, McAnulty MJ, Renye J, Uknalis J and Uhlich GA (2025) A single nucleotide polymorphism produces different transcription profiles in Campylobacter jejuni’s cysM. Front. Microbiol. 16:1501331. doi: 10.3389/fmicb.2025.1501331
Received: 24 September 2024; Accepted: 27 February 2025;
Published: 21 March 2025.
Edited by:
Sonia Almeria, United States Food and Drug Administration, United StatesReviewed by:
Young Min Kwon, University of Arkansas, United StatesCopyright © 2025 Gunther IV, Kanrar, Abdul-Wakeel, McAnulty, Renye, Uknalis and Uhlich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nereus W. Gunther, amFjay5ndW50aGVyQHVzZGEuZ292
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.