
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Microbiol., 12 February 2025
Sec. Infectious Agents and Disease
Volume 16 - 2025 | https://doi.org/10.3389/fmicb.2025.1483895
 Monique Cristina dos Santos1,2
Monique Cristina dos Santos1,2 Natalia Fintelman-Rodrigues1,2
Natalia Fintelman-Rodrigues1,2 Aline de Paula Dias da Silva1,2
Aline de Paula Dias da Silva1,2 Rodolfo Leandro Nascimento Silva3
Rodolfo Leandro Nascimento Silva3 Victor Corrêa Seixas3
Victor Corrêa Seixas3 Amanda A. Batista-da-Silva4,5
Amanda A. Batista-da-Silva4,5 Marcelo Alves Ferreira2
Marcelo Alves Ferreira2 Patrícia T. Bozza1
Patrícia T. Bozza1 Fernando A. Bozza3,6,7*
Fernando A. Bozza3,6,7* Thiago Moreno L. Souza1,2*
Thiago Moreno L. Souza1,2*Because of growing inequalities, more than one-third of the worldwide population is expected to live in slums by 2050. Although slum dwellers are at increased risk of infectious diseases, this population may have been overlooked with respect to the sustainability of virus evolution. In this study, we aimed to analyze the genetic diversity and evolution of SARS-CoV-2 in the Complexo de Favelas da Maré slum, Rio de Janeiro, Brazil, and assess its impact on the global spread of the virus. We found that this slum harbored multiple strains of SARS-CoV-2, and its amplification and genetic diversity connected with the global circulation from 2020 to 2022. Thus, enhancing surveillance in slums could be important for future epidemic/pandemic preparedness by connecting virus genetic diversity in this region with its circulation at divergent locations.
From the early 2019 coronavirus disease 2019 (COVID-19) pandemic outbreak to the subsequent dissemination of severe acute respiratory coronavirus 2 (SARS-CoV-2), certain scenarios may have favored global virus spread (Brainard et al., 2023). These scenarios are complex and not fully understood. Both developed and developing countries suffer from high mortality and the emergence of variants of concern (VoC) (Earnest et al., 2022; Gräf et al., 2021; Tallarita et al., 2022; Trombetta et al., 2022; Worobey, 2021), possibly because of the lack of understanding of the disease’s natural history, virus routes of transmission among asymptomatic/presymptomatic individuals, susceptibility and transmissibility of specific age groups, difficulty in implementing preventive measures (Lee et al., 2020), and the inability to reduce the chains of transmission (Brainard et al., 2023). Nevertheless, in low- and middle-income countries (LMICs), the poorest live in slums and are at greater risk of respiratory pathogens, such as Mycobacterium tuberculosis (Noykhovich et al., 2019), measles virus (Raoot et al., 2016), and SARS-CoV-2 (Friesen and Pelz, 2020). An in-depth analysis of SARS-CoV-2 circulation in these areas is important because accurate data collection in slums can be challenging. Informal settlements, along with a lack of access to diagnose COVID-19 and underreporting of cases (Bakibinga et al., 2019) make SARS-CoV-2 circulation in slums encrypted. Overcrowded homes make social distancing impossible (Von Seidlein et al., 2020). Poor sanitation increases the overall risk of disease transmission (Health Equity, n.d.). Limited healthcare access leads to delayed case management, resulting in increased mortality. Economic insecurity may also have caused the poorest people to avoid lockdowns to continue working (King et al., 2023).
These socioeconomic and demographic limitations, constant health vulnerabilities, and the rise of anti-science movements in a less educated population may have translated into an increased case–fatality ratio (CFR) in slums. Because of growing inequalities worldwide, one to two-thirds of the global population is predicted to live in slums by 2050 (Perlman, 2010). Thus, it is pivotal to better comprehend the dynamics of pathogen circulation in slums as part of pandemic preparedness and epidemic responses.
In the city of Rio de Janeiro, state of Rio de Janeiro, Brazil, the largest slum is Complexo de Favelas da Maré (in Portuguese: Tide Slum Complex; henceforth referred to as Maré). At Maré, the COVID-19 CFR reached 16%, 2- to 3-fold higher than that of the rest of the city (Batista-Da-Silva et al., 2023; Canal e Boletim, n.d.; Painéis Epidemiológicos Painel Rio COVID-19, n.d.). Because of this high mortality ratio, massive virus circulation may have occurred at Maré. Thus, we aimed to catalog SARS-CoV-2 genetic diversity and evolution at this slum and investigate whether the viral strains we identified remained there or whether they were linked to the global spread of the virus. In fact, Maré harbored multiple strains of SARS-CoV-2, and its amplification and genetic diversity connected with the global circulation from 2020 to 2022. Thus, enhancing surveillance in slums could be important for future epidemic/pandemic preparedness by connecting virus genetic diversity in this region with its circulation at divergent locations.
Nasopharyngeal swabs (NSs) were collected from residents of Maré under ethical approval 30650420.4.1001.0008 (by the National Research Ethics Commission, CONEP). Informed consent was obtained from all participants or patient representatives. Data anonymization was used in this study. The samples were collected during 2020 (August to October), 2021 (March to October), and 2022 (January to November), when a multidisciplinary task force was mobilized to medical assistance at Maré, and when the pandemic waves were peaking in Brazil (Brazil-COVID-19 Overview-Johns Hopkins, n.d.). Samples were collected from patients with fever (>37°C) and cough who presented to the task force on the basis of spontaneous demand. The task force was composed of a multidisciplinary team of health professionals called the Dados do Bem (in English data for good) project (Batista-Da-Silva et al., 2023; Dantas et al., 2021), which involved the Instituto D’or de Pesquisa e Ensino (IDOR) and the Instituto Nacional de Infectologia Evandro Chagas Filho (INI), Fiocruz.
NS total RNA was extracted with a QIAamp Viral RNA Kit (Qiagen, Hilden, Germany). RT-PCR was performed according to our previous publication (Fintelman-Rodrigues et al., 2021).
SARS-CoV-2 complete genomes were obtained via ATOPlex SARS-CoV-2 Panel v2.0 (MGI Tech Co., Shenzhen, China), as previously described (Fintelman-Rodrigues et al., 2021). RNA from samples with Ct ≤ 25 was reverse-transcribed into cDNA and amplified for targeted enrichment and dual indexing. Using samples with Ct ≤ 25 indicates that we selected samples with high viral loads to facilitate sequencing. DNA nanoballs were amplified by rolling circles and paired-end sequenced (150 nt) on the MGISEQ-g400 platform (MGI Tech Co., Shenzhen, China).
The raw genomic data were processed via the UseGalaxy platform1 with a SARS-CoV-2 reference sequence (Wuhan-hu-1 isolate, GenBank MN908947.3) and a BED file containing primer coordinates from the MGI ATOPlex panel.2 FASTQ files were preprocessed by FASTP v.0.20.1 to remove adapters and reads shorter than 50 bp (−l 50). Reference mapping and genome assembly were carried out with BWA-MEM v. 0.7.17 in default mode. BAM files were filtered by quality (−q 20) to exclude (−F) unmapped reads (and their mate pairs) and non-primary alignments (SAMtools view v.1.13). The primer binding sites were trimmed (iVar trim v.1.3.1; −m 1 −q 0 −s 4 −e), and the file was aligned to the reference genome with LoFreq v.2.1.5 (with indel filtering by the Dindel algorithm). Variants were called with iVar variants v.1.3.1 (−q 30 −t 0.51 –pass_only), and output VCF files were used to call consensus with BCFtools v.1.10. SARS-CoV-2 consensus genomes were aligned and assigned to global outbreak lineage next-strain clades with NextClade v.1.5.1.3 The latter also provided quality check reports for each consensus sequence and generated an output JSON file for subsequent phylogenetic assessment in Auspice v.0.8.0.4
Neutrality tests and haplotype data were generated with DnaSP v6.12.03. The haplotype network was inferred with TCS networks (Clement et al., 2000) and drawn with PopART v1.7. The haplotypic (h) and nucleotide (π) diversities were calculated via the DNAsp application v.6. The calculations of F’s for Fu and D for Tajima were performed by Arlequin v.3.5.2.2 within the R program v.4.0.3. Finally, the modeling of the haplotype network was performed in PopArt v.1.7 through the TCS network with statistical parsimony and a confidence level of 95%.
In total, we diagnosed and sequenced NS samples from 501 patients (GenBank Accession codes #OR538895–OR539182 and PQ567385–PQ567517, PQ567519, and PQ577902–PQ577980) from 2020, 2021, and 2022 from Maré residents [53% female and aged 37 ± 24 years (median ± SD)]. Although patients are recruited on the basis of spontaneous demand, their ages and ethnic backgrounds are similar to those of dwellers from Maré, who were censored in 2022 (Batista-Da-Silva et al., 2023; Canal e Boletim, n.d.; IBGE | Biblioteca, n.d.) (Supplementary Figure S1).
During the 2020–2021 COVID-19 pandemic, Maré had a CFR of 10–16% (Batista-Da-Silva et al., 2023; Canal e Boletim, n.d.), which is twice as high as the CFR in the city of Rio de Janeiro (Painéis Epidemiológicos Painel Rio COVID-19, n.d.). Because of the high mortality ratio at Maré, we were interested in better comprehending the viral dynamics in this slum region. With respect to this period of early SARS-CoV-2 introduction, prior to vaccination campaigns, we looked for chains of transmission within the community and built a haplotype network for SARS-CoV-2 genomes. Haplotypes that clustered independently of the residence of the dweller within 5 km2 were called Maré (Figure 1). In fact, the SARS-CoV-2 haplotypes were distributed according to the variant of concern to which they belong (VoC) (Figure 1A). We identified 265 haplotypes, with 6 shared pairs and 4 detected in the same location (Figure 1A), suggesting some level of intraslum transmission. Among these strains at Maré, we also found high haplotype and nucleotide diversity indices, along with significant Fu and Li parameters (Table 1), indicating that new SARS-CoV-2 genomes entered the community and evolved there, which is consistent with massive virus introduction and circulation in this setting.
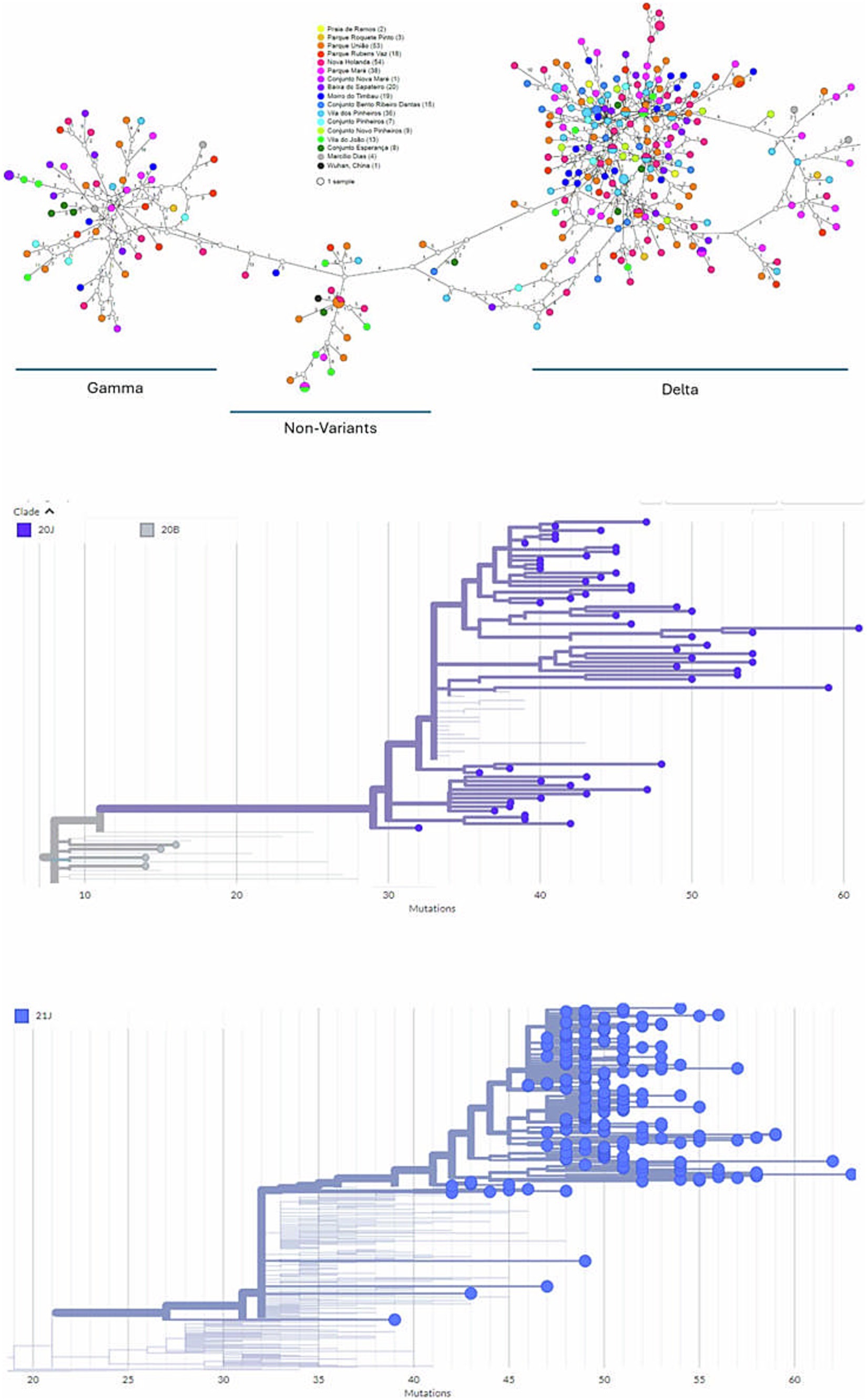
Figure 1. Positive nasopharyngeal swabs from dwellers of Complexo de Favelas da Maré (Maré), Rio de Janeiro, Brazil, from 2020 to 2022 were sequenced (Atoplex version 2.0, using the pair-end of 150 nucleotides in the DNBSEQ-G50 sequencer; MGI Tech, https://en.mgi-tech.com/). Full-length consensus genomes were generated with quality scores >30 and depths >10. (A) The color represents the place of residence among Maré sublocations and was analyzed in a haplotype network inferred via the TCS method (statistical parsimony). For phylogenetic analysis, the maximum-likelihood method was used with consensus sequences and publicly available datasets from NextClade v.1.5.1. Diagrams of mutations across the genomes of the emphasizing strains 20J (B) and 21J (C) are presented. The original sequences are marked as spheres (GenBank Accession codes #OR538895–OR539182).
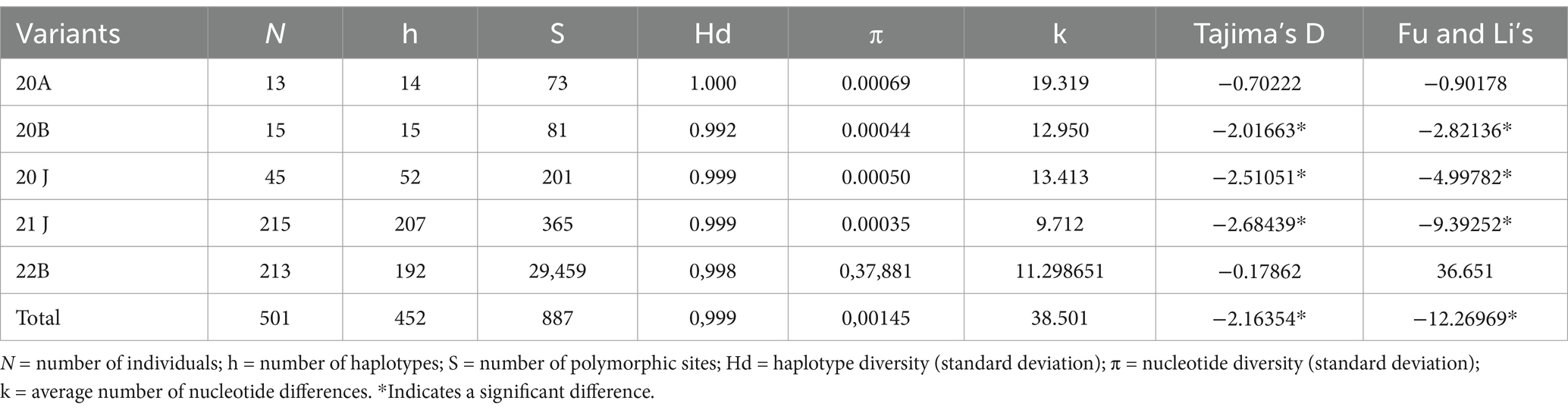
Table 1. SARS-CoV-2 diversity at Complexo de Favela da Maré.
We also performed phylogenetic analysis to compare the SARS-CoV-2 genomes from Maré with those found elsewhere. Both branches of the Gamma (clade 20 J) and Delta (clade 21 J) VoCs indicate that SARS-CoV-2 genomes from Maré not only root additional sequences of this virus in the slum region but are also ancestral to pandemic coronavirus genomes from other countries (Figures 1B,C; Supplementary Tables S1, S2). SARS-CoV-2 gamma VoC from Favela da Maré is associated with cases in Brazil, the USA, and Switzerland (Supplementary Table S1). Delta VoC from the Brazilian slum region is associated with other cases from this country, the USA, India, South Africa, China, and Switzerland (Supplementary Table S2). Delta samples from Maré early presented the spike mutations D614G, P681R, and L452R, which are associated with increased infectivity and immune escape. The discovery of SARS-CoV-2 genomes with more competitive features from Maré is also supported by a tendency toward directional selection depicted by Tajima’s D parameter (Table 1).
During 2022, omicron VoC took over previous strains at Maré (Figure 2). Indeed, the SARS-CoV-2 omicron strains 21 L, 22A, 22B, and 22E and a new out-group were observed at Maré (Figure 2A). SARS-CoV-2 is constantly mutating, and the emergence of a new out-group means that the virus has accumulated enough mutations to make it genetically different from the dominant circulating variants. The accumulation of genetic evidence is necessary for an out-group to be defined as a new omicron subvariant. In this sense, the study of SARS-CoV-2 evolution in the poor slum of Maré adds new sequences to a SARS-CoV-2 out-group derived from the BA.2 subvariant of Omicron, which was previously underrepresented (Figure 2B). This new BA.2 out-group is similar to sequences observed in all continents; sequences from Maré are initially rooted in a virus strain from Wales, and next, other strains from Maré are ancestral to cases in Brazil and Latin America (Figure 2C).
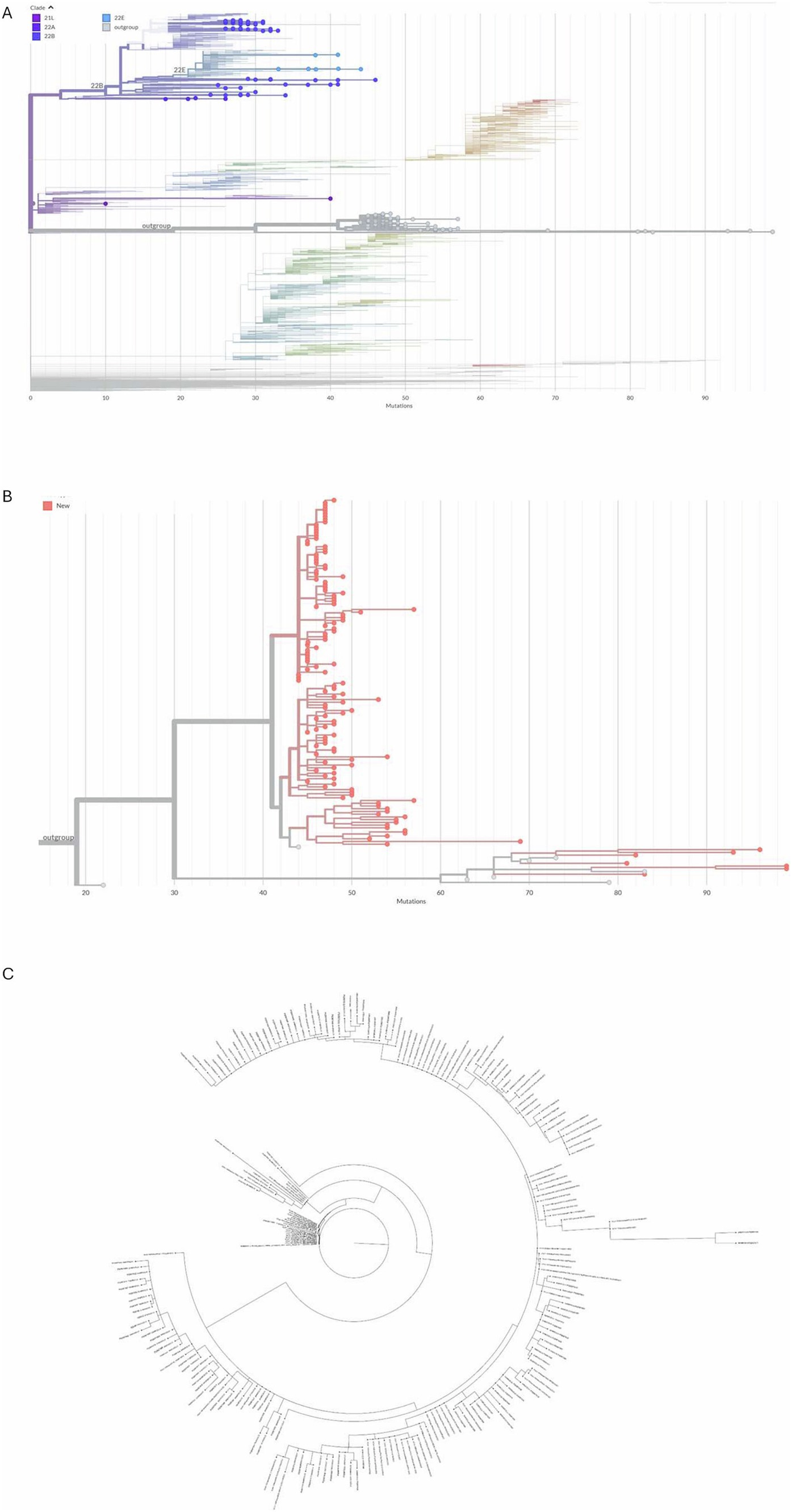
Figure 2. Positive nasopharyngeal swabs from dwellers of Complexo de Favelas da Maré (Maré), Rio de Janeiro, Brazil, from 2022 were sequenced (Atoplex version 2.0, using the pair-end of 150 nucleotides in the DNBSEQ-G50 sequencer; MGI Tech, https://en.mgi-tech.com/). Full-length consensus genomes were generated with quality scores >30 and depths >10. For phylogenetic analysis, the maximum-likelihood method was used with consensus sequences and publicly available datasets from NextClade v.1.5.1 highlighting the strains 21L, 22A, 22B, 22E, and an out-group (A). A phylogeny of mutations across the genomes of the emphasizing strains in the new omicron out-group (in red) compared with predecessor strains from this same out-group (in gray) is displayed (B). Further phylogenetic analysis was performed with samples from the out-group found in Brazil and other territories (C). The original sequences are marked as spheres (GenBank Accession codes #Q567385–PQ567517, PQ567519, and PQ577902–PQ577980).
With increasing global inequalities, more than one-third of the world’s population is projected to reside in slums by 2050 (Perlman, 2010). These communities, characterized by high population density, poverty, and inadequate sanitation, create environments where infectious diseases can easily spread (Neiderud, 2015). Studying disease transmission in these settings is vital because these areas often have a higher prevalence of infectious diseases than other urban areas do.
Understanding the dynamics of infectious agents in slums is crucial for public health interventions. However, these poor communities may have been overlooked in studies concerning the sustainability of virus evolution. Research focusing on slums such as the Complexo de Favelas da Maré in Rio de Janeiro, Brazil, reveals crucial insights into the genetic diversity and evolution of viruses such as SARS-CoV-2. These findings indicate that slums harbor multiple introductions of the virus, contributing to its amplification and genetic diversity and connecting with global circulation patterns.
In Brazil, approximately 8% of the population lives in slums (IBGE | Biblioteca, n.d.). Like other territories around the world, many Brazilian slums are in the central urban areas of cosmopolitan cities and megacities, leading to the hypothesis that a pathogen spread in these areas could reach wider dissemination. Maré is located just by Guanabara Bay (the 2nd largest bay in Brazil and an area of intense naval activity), is 7 km away from Rio’s International Airport, and is crossed by three of the most used highways in the city. More than 140,000 residents of this slum are distributed in more than 43,000 homes in 16 sublocations encompassing a total area of 5 km2, an area ranked as one of the last neighborhoods among Rio de Janeiro’s districts according to the Human Development Index (Rio em Síntese, n.d.).
In the early SARS-CoV-2 circulation at Maré, a diverse array of SARS-CoV-2 strains, including multiple variants of concern (VOCs), were found. This diversity suggests that the slum may have acted as an environmental reservoir for the virus, facilitating the evolution of new strains. This is represented by massive virus circulation/replication at Maré from 2020 to 2021, which is related to epidemics in distinct areas of the world: the USA, Switzerland, India, South Africa, and China.
By the end of 2021 and the beginning of 2022, the Brazilian population had broader immunity as the result of natural exposure and vaccination campaigns, which led to the decline of the Brazilian CFR in 2022 (Boletins Epidemiológicos COVID-19, n.d.). Nevertheless, several important features of the molecular epidemiology of SARS-CoV-2 were observed at Maré. During 2022, Omicron VoC circulated, and at Maré, we found a new out-group of genotype BA.2, which was similar to sequences observed worldwide. The SARSC-CoV-2 sequences from Maré subsequently became ancestral to the virus strains found in Brazil, Latin American countries, and Wales. These findings from all the years analyzed suggest that Maré may have played a role in the global dissemination of the virus.
The dynamic environment of slums, with overcrowding and limited resources, makes it difficult to capture a comprehensive picture of viral diversity and inform effective public health interventions. By analyzing viral genetic diversity in Maré via high viral load samples, we can potentially over represent dominant strains and miss less prevalent variants. Moreover, genomic tools and diversity metrics can introduce biases due to assumptions of neutral evolution. Thus, it is plausible that more longitudinal sampling strategies and universal sequencing result in greater genetic diversity.
The data from 2022 from Maré on the circulation of a new Omicron out-group reinforce that the pandemic’s impact on slums underscores the urgent need for several actions. Improved infrastructure and basic services, such as clean water, sanitation, and healthcare in slums, are crucial for improving public health and resilience to future crises. Social protection programs are needed because stronger social safety nets are needed to protect vulnerable populations from economic shocks and ensure access to essential needs. Community empowerment is important for supporting community-led initiatives, and organizations can strengthen local responses to crises and promote long-term development. Therefore, enhanced surveillance in slums is critical for future epidemic and pandemic preparedness as it links viral genetic diversity in these regions with its circulation in diverse locations.
Studies conducted in India, specifically in Mumbai, have revealed that crowding plays a significant role in the transmission of COVID-19. Research has shown that the seroprevalence in slum areas is considerably greater than that in non-slum areas, highlighting the impact of close proximity on virus spread (Malani et al., 2021). In response, experts have suggested implementing the “4 Ts” approach—tracing, tracking, testing, and treating—which is effective in interrupting the chain of transmission in Dharavi (Kaushal and Mahajan, 2021). Similarly, a multicomponent intervention in Maré, Brazil, incorporating community engagement, mobile surveillance, mass testing, and telemedicine, resulted in a substantial decrease in the COVID-19 fatality rate (Batista-Da-Silva et al., 2023). However, genomic analysis in Maré indicated that most infections originated outside the community, emphasizing the influence of resident mobility and external exposure on virus circulation. This finding underscores the need for urban planning strategies that account for human movement to effectively mitigate transmission in densely populated urban slums.
Bangladesh, with its high population density and a significant portion of its urban population residing in slums with inadequate living conditions, faces challenges in controlling the spread of COVID-19. Strict lockdown measures were difficult to maintain, resulting in the introduction of various virus clades from different parts of the world, including Europe, the United States, the United Kingdom, and Australia (Islam et al., 2021). While genomic studies have tracked the evolution of the virus in Bangladesh, further research is needed to understand how individual-level genomic changes influence the overall dynamics of transmission at the population level. The study in Maré provides valuable genomic data from slums over time, which can help assess viral dynamics and inform strategies for addressing future pandemics.
Comparing Maré to slums in India and Bangladesh reveals important differences in healthcare systems, population densities, and mobility patterns (Osuh et al., 2024). While Brazil has a universal healthcare system, access to and quality of care remain challenges. India and Bangladesh have more fragmented systems with greater reliance on private providers and non-governmental organizations. Slums in India and Bangladesh often have significantly higher population densities than those in Brazil do, posing greater challenges for public health interventions. Mobility restrictions, while present in all settings, may be more difficult to enforce in Brazilian slums because of safety concerns and geographical barriers. Indian and Bangladeshi slums experience high levels of internal migration, which can create challenges in accessing healthcare and social services.
To improve the resilience of slums to future health crises, it is crucial to invest in improved infrastructure and basic services such as clean water, sanitation, and healthcare. Community health worker programs have also been shown to be effective in increasing access to healthcare and health education in slums (Javanparast et al., 2018). By addressing these multifaceted challenges and investing in comprehensive interventions, we can better protect vulnerable slum communities from the devastating impacts of pandemics.
We endorse robust surveillance systems in underserved communities to track the emergence and spread of pathogens. This includes genomic sequencing, epidemiological monitoring, community-based participatory surveillance, and a priori epidemiological design. We also recognize that countries may face distinct challenges and often explore tailored interventions; that is, public health interventions developed in light of slum specificities may be necessary. This may involve culturally appropriate health education campaigns, community health worker programs, and improved access to healthcare services. Global public health agencies could reinforce their initiatives toward underserved communities and establish a tone for politicians around the world.
The population at Complexo de Favelas da Maré in Rio de Janeiro, Brazil, is not only at higher risk for SARS-CoV-2, as documented by multiple virus introductions within this slum, but also the virus that circulated there evolved and correlated with epidemics in other countries/continents. Considering that a substantial part of the world population will live in slums in the next few decades, we reinforce the necessity of prioritizing these areas for health initiatives and sanitation and further advocate that slums should be prioritized to receive funds and public health efforts in epidemic/pandemic periods.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, OR538895–OR539182; https://www.ncbi.nlm.nih.gov/genbank/, PQ567385–PQ567517; https://www.ncbi.nlm.nih.gov/genbank/, PQ567519; https://www.ncbi.nlm.nih.gov/genbank/, PQ577902–PQ577980.
The studies involving humans were approved by Ethical approval 30650420.4.1001.0008 by the National Research Ethics Commission, CONEP. Informed consent was obtained from all participants or patient representatives. Data analysis was deidentified. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
MS: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. NF-R: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. AS: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. RS: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. VS: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. AB-S: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. MF: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. PB: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. FB: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. TS: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The work was funded in part by the National Institutes of Science and Technology Program (INCT) on Diseases of Neglected Populations (INCT-IDPN), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), Coordination for the Improvement of Higher Education Personnel (CAPES) and Coordenação Geral de Planejamento Estratégico (COGEPLAN), Fiocruz.
The authors thank Drs. Ricardo Godoi and Marco Alberto Medeiros, from Instituto de Tecnologia em Imunobiológicos/Biomanguinhos, Dr. Erika de Carvalho, Unidade de Diagnóstico (Unadig), Fiocruz, and Drs. João Gesto and Antônio Fidalgo-Neto from Instituto Senai de Inovação em Química Verde, Firjan, for assessment on sequencing platforms. We deeply appreciate the MGI, a partner in the implementation of next-generation sequencing through collaborations with our center.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1483895/full#supplementary-material
Bakibinga, P., Kabaria, C., Kyobutungi, C., Manyara, A., Mbaya, N., Mohammed, S., et al. (2019). A protocol for a multi-site, spatially-referenced household survey in slum settings: methods for access, sampling frame construction, sampling, and field data collection. BMC Med. Res. Methodol. 19:109. doi: 10.1186/s12874-019-0732-x
Batista-Da-Silva, A. D. A., Moraes, C. B., Bozza, H. R., Bastos, L. D. S. L., Ranzani, O. T., Hamacher, S., et al. (2023). Effectiveness of a multicomponent intervention to face the COVID-19 pandemic in Rio de Janeiro’s favelas: difference-in-differences analysis. BMJ Glob. Health 8:e009997. doi: 10.1136/bmjgh-2022-009997
Boletins Epidemiológicos COVID-19. Minist. Saúde. Available at: https://www.gov.br/saude/pt-br/centrais-de-conteudo/publicacoes/boletins/epidemiologicos/covid-19/boletins-epidemiologicos-covid-19 (Accessed 11.6.24).
Brainard, J., Jones, N. R., Harrison, F. C. D., Hammer, C. C., and Lake, I. R. (2023). Superspreaders of novel coronaviruses that cause SARS, MERS and COVID-19: a systematic review. Ann. Epidemiol. 82, 66–76.e6. doi: 10.1016/j.annepidem.2023.03.009
Brazil-COVID-19 Overview-Johns Hopkins. Johns Hopkins coronavirus Resour. Cent. Available at: https://coronavirus.jhu.edu/region/brazil (accessed 12.20.24).
Canal e Boletim. Available at: https://www.redesdamare.org.br/br/artigo/90/canal-e-boletim-de-olho-no-corona (accessed 11.6.24).
Clement, M., Posada, D., and Crandall, K. A. (2000). TCS: a computer program to estimate gene genealogies. Mol. Ecol. 9, 1657–1659. doi: 10.1046/j.1365-294x.2000.01020.x
Dantas, L. F., Peres, I. T., Bastos, L. S. L., Marchesi, J. F., de Souza, G. F. G., Gelli, J. G. M., et al. (2021). App-based symptom tracking to optimize SARS-CoV-2 testing strategy using machine learning. PLoS One 16:e0248920. doi: 10.1371/journal.pone.0248920
Earnest, R., Uddin, R., Matluk, N., Renzette, N., Turbett, S. E., and Siddle, K. J. (2022). Comparative transmissibility of SARS-CoV-2 variants Delta and alpha in New England, USA. Cell Rep. Med. 3:100583. doi: 10.1016/j.xcrm.2022.100583
Fintelman-Rodrigues, N., da Silva, A. P. D., Dos Santos, M. C., Saraiva, F. B., Ferreira, M. A., Gesto, J., et al. (2021). Genetic evidence and host immune response in persons Reinfected with SARS-CoV-2, Brazil. Emerg. Infect. Dis. 27, 1446–1453. doi: 10.3201/eid2705.204912
Friesen, J., and Pelz, P. F. (2020). COVID-19 and slums: a pandemic highlights gaps in knowledge about urban poverty. JMIR Public Health Surveill. 6:e19578. doi: 10.2196/19578
Gräf, T., Bello, G., Venas, T. M. M., Pereira, E. C., Paixão, A. C. D., Appolinario, L. R., et al. (2021). Identification of a novel SARS-CoV-2 P.1 sublineage in Brazil provides new insights about the mechanisms of emergence of variants of concern. Virus Evol 7:veab091. doi: 10.1093/ve/veab091
Health Equity. Available at: https://www.who.int/teams/environment-climate-change-and-health/healthy-urban-environments/housing/health-equity (accessed 11.6.24).
IBGE | Biblioteca. IBGE Bibl. Available at: https://biblioteca.ibge.gov.br/index.php/biblioteca-catalogo?view=detalhes&id=2102062 (accessed 11.6.24).
Islam, A., Sayeed, M. A., Rahman, M. K., Zamil, S., Abedin, J., Saha, O., et al. (2021). Assessment of basic reproduction number (R0), spatial and temporal epidemiological determinants, and genetic characterization of SARS-CoV-2 in Bangladesh. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 92:104884. doi: 10.1016/j.meegid.2021.104884
Javanparast, S., Windle, A., Freeman, T., and Baum, F. (2018). Community health worker programs to improve healthcare access and equity: are they only relevant to low- and middle-income countries? Int. J. Health Policy Manag. 7, 943–954. doi: 10.15171/ijhpm.2018.53
Kaushal, J., and Mahajan, P. (2021). Asia’s largest urban slum-Dharavi: a global model for management of COVID-19. Cities 111:103097. doi: 10.1016/j.cities.2020.103097
King, B., Adepoju, O. E., Woodard, L., Oluyomi, A. O., Zhang, X., Amos, C. I., et al. (2023). The effects of COVID-19 lockdown on social connectedness and psychological distress in U.S. adults with chronic diseases. Int. J. Environ. Res. Public Health 20:6218. doi: 10.3390/ijerph20136218
Lee, E. C., Wada, N. I., Grabowski, M. K., Gurley, E. S., and Lessler, J. (2020). The engines of SARS-CoV-2 spread. Science 370, 406–407. doi: 10.1126/science.abd8755
Malani, A., Shah, D., Kang, G., Lobo, G. N., Shastri, J., Mohanan, M., et al. (2021). Seroprevalence of SARS-CoV-2 in slums versus nonslums in Mumbai, India. Lancet Glob. Health 9, e110–e111. doi: 10.1016/S2214-109X(20)30467-8
Neiderud, C.-J. (2015). How urbanization affects the epidemiology of emerging infectious diseases. Infect. Ecol. Epidemiol. 5:27060. doi: 10.3402/iee.v5.27060
Noykhovich, E., Mookherji, S., and Roess, A. (2019). The risk of tuberculosis among populations living in slum settings: a systematic review and Meta-analysis. J. Urban health bull. N. Y. Acad. Med. 96, 262–275. doi: 10.1007/s11524-018-0319-6
Osuh, M. E., Oke, G. A., Lilford, R. J., Osuh, J. I., Harris, B., Owoaje, E., et al. (2024). Systematic review of oral health in slums and nonslum urban settings of low and middle-income countries (LMICs): disease prevalence, determinants, perception, and practices. PLoS One 19:e0309319. doi: 10.1371/journal.pone.0309319
Painéis Epidemiológicos Painel Rio COVID-19, (n.d.). EpiRio. Available at: https://epirio.svs.rio.br/painel/painel-rio-covid-19/(accessed 11.6.24).
Perlman, J. (2010). Favela: Four decades of living on the edge in Rio de Janeiro. New York, USA: Oxford University Press.
Raoot, A., Dewan, D. K., Dubey, A. P., Batra, R. K., and Seth, S. (2016). Measles outbreak in high risk areas of Delhi: epidemiological investigation and laboratory confirmation. Indian J. Pediatr. 83, 200–208. doi: 10.1007/s12098-015-1845-9
Rio em Síntese. (n.d.). Available at: https://www.data.rio/pages/rio-em-sntese (accessed 11.6.24).
Tallarita, M., Giardina, F., Novazzi, F., Gaiarsa, S., Batisti Biffignandi, G., Paolucci, S., et al. (2022). Spread of multiple SARS-CoV-2 lineages April-august 2020 anticipated the second pandemic wave in Lombardy (Italy). Pediatr. Allergy Immunol. Off. Publ. Eur. Soc. Pediatr. Allergy Immunol. 33, 89–92. doi: 10.1111/pai.13641
Trombetta, C. M., Marchi, S., Viviani, S., Manenti, A., Casa, E., Dapporto, F., et al. (2022). A serological investigation in southern Italy: was SARS-CoV-2 circulating in late 2019? Hum. Vaccin. Immunother. 18:2047582. doi: 10.1080/21645515.2022.2047582
Seidlein, L.Von, Alabaster, G., Deen, J., and Knudsen, J. (2020). Crowding has consequences: prevention and management of COVID-19 in informal urban settlements. Build. Environ. 188:107472. doi: 10.1016/j.buildenv.2020.107472
Keywords: COVID-19, SARS-CoV-2, slum, Rio de Janeiro, Brazil
Citation: dos Santos MC, Fintelman-Rodrigues N, Silva AdPDd, Silva RLN, Seixas VC, Batista-da-Silva AA, Ferreira MA, Bozza PT, Bozza FA and Souza TML (2025) Does viral circulation in slums have a global impact? The lesson learned from SARS-CoV-2 circulation in Complexo de favelas da Maré, Rio de Janeiro, Brazil. Front. Microbiol. 16:1483895. doi: 10.3389/fmicb.2025.1483895
Edited by:
Silvia Spoto, Fondazione Policlinico Universitario Campus Bio-Medico, ItalyReviewed by:
Johid Malik, University of Nebraska Medical Center, United StatesCopyright © 2025 dos Santos, Fintelman-Rodrigues, Silva, Silva, Seixas, Batista-da-Silva, Ferreira, Bozza, Bozza and Souza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thiago Moreno L. Souza, dGhpYWdvLm1vcmVub0BmaW9jcnV6LmJy; Fernando A. Bozza, ZmVybmFuZG8uYm96emFAZmlvY3J1ei5icg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.