
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 25 February 2025
Sec. Antimicrobials, Resistance and Chemotherapy
Volume 16 - 2025 | https://doi.org/10.3389/fmicb.2025.1473150
 Junwan Lu1
Junwan Lu1 Lei Zhang2,3
Lei Zhang2,3 Chunxia Yan1
Chunxia Yan1 Naru Lin3
Naru Lin3 Yuan Zhang3
Yuan Zhang3 Yuning Sha3
Yuning Sha3 Jingxuan Zhao3Jun Lu2*Qiyu Bao1,3*
Jingxuan Zhao3Jun Lu2*Qiyu Bao1,3* Guozhi Zhang3,4*
Guozhi Zhang3,4*Background: Multidrug-resistant strains of the genus Aeromonas can produce various β-lactamases that confer resistance to a broad spectrum of β-lactams, which poses a significant public health threat due to their emergence and spread in clinical settings and natural environments. Therefore, a comprehensive investigation into the antibiotic resistance mechanisms of Aeromonas is scientifically significant.
Methods: Between 2018 and 2021, 78 clinical Aeromonas isolates were collected from human clinical specimens. The MicroScan WalkAway system and average nucleotide identity (ANI) analyses were used to classify the bacterial species. Antibiotic susceptibility was determined through the minimum inhibitory concentration (MIC) test via the agar dilution method. To determine the resistance mechanism and the structure of the resistance gene-related sequences, molecular cloning, whole-genome sequencing and bioinformatic analysis were performed.
Results: Among the 78 Aeromonas isolates studied in this work, obtained from various specimens from different clinical departments, 77 were classified into seven known species by ANI analysis. Most of the isolates were A. caviae (34.6%, 27/78), followed by A. hydrophila (25.6%, 20/78). Multilocus sequence typing (MLST) revealed that they belonged to 72 sequence types (STs), including 52 new STs. A total of 334 resistance genes of 30 antibiotic resistance genotypes were identified from the genomes, more than half (55.99%, 187/334) of which were β-lactamase genes. The isolates showed much higher rates of resistance to penicillins (penicillin G, 98.7%) and first-generation cephalosporins (cefazolin, 96.2%), but lower resistance rates to fourth-generation cephalosporins (cefepime, 6.4%), monobactams (aztreonam, 5.1%), and carbapenems (imipenem, 1.3% and meropenem, 5.1%). Structural analyses of some β-lactamase genes (such as blaNDM-1 and blaPER-3) related sequences revealed that they were generally associated with mobile genetic elements.
Conclusion: The investigation of the correlation between the distribution of β-lactamase genes and Aeromonas resistance phenotypes in this study suggested an urgent need for rigorous monitoring and control to counteract the escalating public health threat posed by the increase in Aeromonas strains harboring extended-spectrum β-lactamase and metallo-β-lactamase genes.
The genus Aeromonas constitutes a ubiquitous group of Gram-negative rods found worldwide in natural environments, particularly in aquatic habitats (Parker and Shaw, 2011). The classification of Aeromonas species is complex, and accurate laboratory identification remains a significant challenge. Traditional biochemical tests, 16S rRNA gene homology analysis and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) are unreliable for identifying Aeromonas species (Pérez-Sancho et al., 2018). For example, Aeromonas dhakensis (previously known as Aeromonas aquariorum) is frequently misidentified as A. hydrophila by conventional biochemical methods. Accurate identification can be achieved through core genome phylogenetic analysis (Chen et al., 2014a).
Aeromonas species produce a variety of virulence factors, including adhesins, cytotoxins, hemolysins, lipases, and proteases; exhibit biofilm formation capacity; use specific metabolic pathways; and exhibit virulence factor expression mediated through quorum sensing (Janda and Abbott, 2010). The pathogenicity of Aeromonas appears to differ at the species level. The role of Aeromonas species as human pathogens has been highlighted by their occurrence after natural disasters, such as the 2004 tsunami in Thailand, where they were identified as the most common pathogens causing skin or soft tissue infections (Hiransuthikul et al., 2005). Aeromonas species can cause a variety of infections in humans, ranging from common conditions such as gastroenteritis to more serious illnesses such as septicemia, peritonitis, cholangitis, and catheter-related infections (Janda and Abbott, 2010; Wu et al., 2007). Both immunocompromised and immunocompetent individuals can be infected with Aeromonas, usually via ingestion of or direct mucocutaneous contact with contaminated water or food (Janda and Abbott, 2010). The reported mortality rates of patients with Aeromonas bacteremia range from 24 to 63% (Chen et al., 2014b). Three main Aeromonas species are associated with human disease: A. hydrophila, A. veronii, and A. caviae. While many Aeromonas infections are self-limiting, invasive infections can progress rapidly and become life-threatening for those with underlying conditions or immune system impairments (Zhang et al., 2023; Zhou et al., 2019). In recent years, the widespread presence of Aeromonas species such as A. dhakensis has been identified to be associated with human infections, and these species may be more deadly than other Aeromonas species (Igbinosa et al., 2012).
Furthermore, with the overuse of antibiotics in agriculture, aquaculture, and clinical settings, Aeromonas resistance to antimicrobial agents continues to increase. Antibiotic susceptibility varies depending on the geographical region and the Aeromonas species tested. Appropriate antimicrobial therapy is necessary for controlling the development of infection. The incidence and severity of Aeromonas infections appear to be underestimated, and underreporting remains prevalent in many countries (Kosikowska et al., 2022).
This study aimed to investigate the molecular epidemiology of Aeromonas species from a hospital in Quzhou, which is located in southeastern China and has a subtropical climate, over a four-year period. The clinical features, virulence and antimicrobial resistance mechanisms of the clinical Aeromonas strains were analyzed, and the findings will advance our understanding of Aeromonas infections and help establish appropriate treatment strategies.
A total of 78 clinical Aeromonas isolates were collected from Quzhou People’s Hospital in Zhejiang Province, China, between 2018 and 2021. They were isolated from different sources: excrement (n = 35), fester (n = 14), bile (n = 12), whole blood (n = 6), urine (n = 6), sputum (n = 4), and catheter (n = 1). The age of the patients ranged between 9 months and 93 years with an average age 54.8 years. 70.5% (55/78) were from male patients (with an average age of 56.4 years) and 29.5% (23/78) were from female patients (with an average age of 50.5 years). The isolation of Aeromonas species was performed by directly streaking the fecal specimen onto the starch ampicillin agar (SAA) plate supplemented with 10 mg/L ampicillin (Solarbio, Beijing, China) and incubated at 37°C for 24 h. For the other specimens, enrichment culture was performed over night in tryptic soy broth (TSB, Solarbio, Beijing, China) and the culture was then streaked onto the SAA plates. Typical yellow colonies indicative of Aeromonas species were selected for further identification. Oxidase test was performed using the DrySlide Oxidase kit (BD, Oslo, Norway). The tryptic soy agar (TSA, Solarbio, Beijing, China) plates were used to culture the Aeromonas isolates and the bacteria were then preserved in TSB containing 20% glycerol at −80°C for subsequent analysis. Preliminary species identification of these isolates was performed with a MicroScan WalkAway®, an automated bacterial identification platform (Siemens AG FWB, Germany) (Khan et al., 2015). All 78 isolates were classified as Aeromonas complex isolates. Species/subspecies identification of the Aeromonas complex was further performed by ANI analysis using FastANI v1.31, and a value >95% was used as the threshold for species definition (Jain et al., 2018).
According to the previous publication (Weinstein and Lewis, 2020), the breakpoint criteria of the Clinical and Laboratory Standards Institute (CLSI M100) for Enterobacteriaceae were used for penicillin G, cefazolin, and cefoxitin. Antimicrobial susceptibility was determined using the agar dilution method on Mueller–Hinton (MH) agar plates supplemented with different concentrations of antibiotics ranging from 0.004 to 2,048 μg/mL (Table 1). The test was repeated three times to ensure accuracy. The minimum inhibitory concentrations (MICs) were interpreted following the recommended breakpoint criteria for the Aeromonas hydrophila complex applicable to other non-Enterobacterales, as outlined by the CLSI (2021). For cefotaxime, ceftriaxone, cefepime, aztreonam, imipenem, and meropenem, the MIC breakpoints for other non-Enterobacterales were applied.
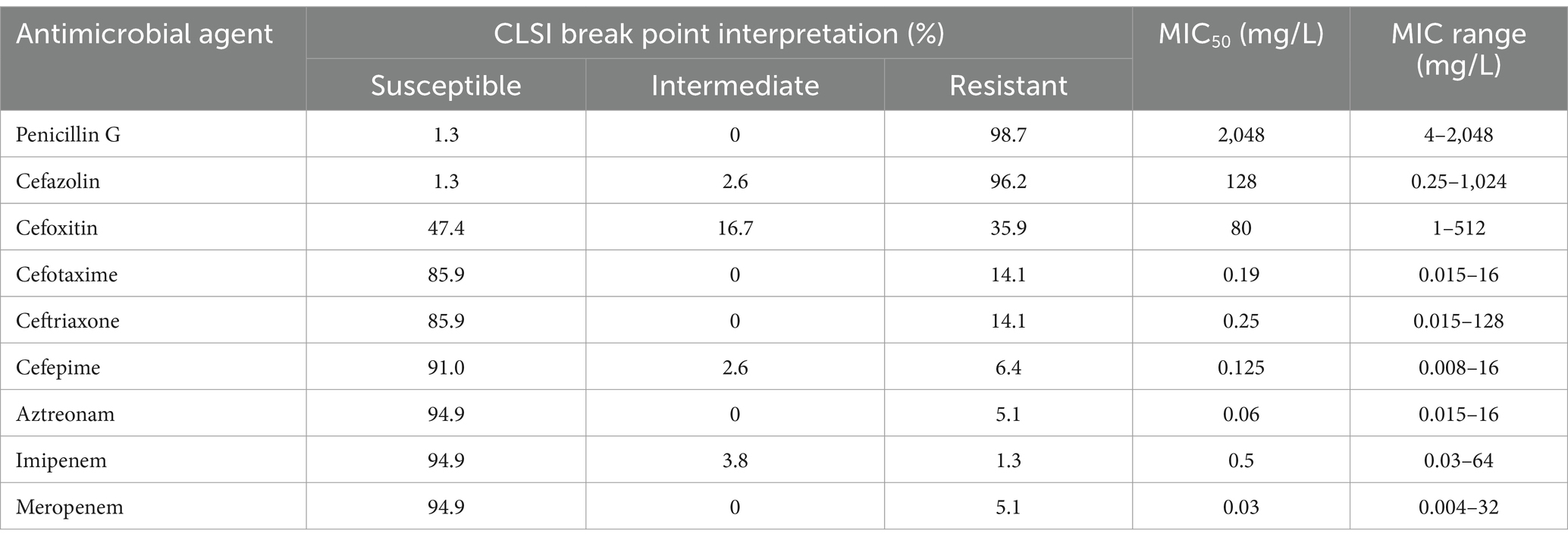
Table 1. Susceptibility profiles and MICs for 78 Aeromonas strains.
The bacterial genomic DNA was extracted by using an AxyPrep Bacterial Genomic DNA Miniprep Kit (Axygen Scientific, Union City, CA, United States). Genome sequencing was performed by the Illumina HiSeq 2500 and PacBio Sequel IIe platforms by Shanghai Personal Biotechnology Co., Ltd. (Shanghai, China). A library with an average insert size of 400 bp was prepared using the NEBNext Ultra II DNA Library Preparation Kit for Illumina HiSeq 2500 sequencing (paired-end run; 2 × 150 bp). Sequence assembly was conducted de novo for Illumina short reads using SPAdes v.3.14.0 (Bankevich et al., 2012). The genomes of isolates carrying metallo-β-lactamase (MBL) genes were further sequenced by PacBio RS II instruments (Pacific Biosciences, CA, United States), and the long reads from PacBio sequencing were assembled by HIFIasm using the data obtained from Illumina sequencing as reference input (Cheng et al., 2021). ORFs present in the genome sequence were predicted by Prokka v1.14.6 (Seemann, 2014), while BLAST analysis against the NCBI protein sequence database helped annotate their function with an e-value threshold of 1e-5. Resistance Gene Identifier v5.2.0 (Jia et al., 2017) and the comprehensive antibiotic resistance database (CARD, McArthur et al., 2013) were used to identify antimicrobial resistance genes. MLST analysis was performed using the database,1 with the MLST scheme developed by Martino et al. (2011). This scheme is based on six loci including gltA, groL, gyrB, metG, ppsA and recA. Gene distribution was visualized using the ComplexHeatmap package in R (Gu et al., 2016). The single-copy core gene phylogenetic tree was generated using Roray v3.11.2 (Page et al., 2015) and then visualized with ggtree v3.2.0 (Yu et al., 2018).
According to the minimal inhibitory concentration (MIC) of meropene, piperacillin + tazobactam and ceftazidime to A. caviae QZ63 or A. hydrophila QZ124, the bactericidal effects of meropene, piperacillin + tazobactam, ceftazidime at various concentrations of 0.5 ×, 1 ×, 2 × MIC were studied by time-kill assay. Following the methods recommended in the previous publicatins (Norden et al., 1979; Paevskiĭ, 1993), two isolates A. caviae QZ63 with blaNDM-1 and A. hydrophila QZ124 with blaPER-3 were selected as test strains, and two isolates A. caviae QZ54 free of blaNDM-1 and A. hydrophila QZ87 without blaPER-3 were used as control strains to study the synergistic bactericidal effects. The experiment procedure is briefly described as follows: cation-adjusted Mueller–Hinton broth (CAMHB) containing 1 × 105 CFU/mL bacteria is mixed with single or combined antimicrobial agents incubated overnight with consecutive shacking at 37°C. Meanwhile, the same broth without antibiotics was served as a control. Broth samples were serially sampled at times of 0, 1, 2, 4, 6, 8, 12 and 24 h and plated onto a Mueller–Hinton plate, respectively. After overnight incubation at 37°C, the colonies were counted.
June, July, and August were the months with the highest incidence rates, 46.2% (36/78) of the isolates were obtained in these 3 months. It was possibly due to favorable environmental conditions for the proliferation and growth of Aeromonas during this period of the year (Figure 1). Consequently, this led to an increase in Aeromonas levels in the environment and, thus, a greater risk of infection (Janda and Abbott, 2010). Aeromonas species are widely distributed aquatic Gram-negative bacteria found in natural environments worldwide. They have become the third most common enteric bacterial pathogens, following the genera Campylobacter and Salmonella (Yuwono et al., 2021). In recent years, more bacteria of the genus Aeromonas than those of the other genera causing diarrhea have been isolated from diarrhea specimens in the outpatient department of Quzhou People’s Hospital, Quzhou, China.
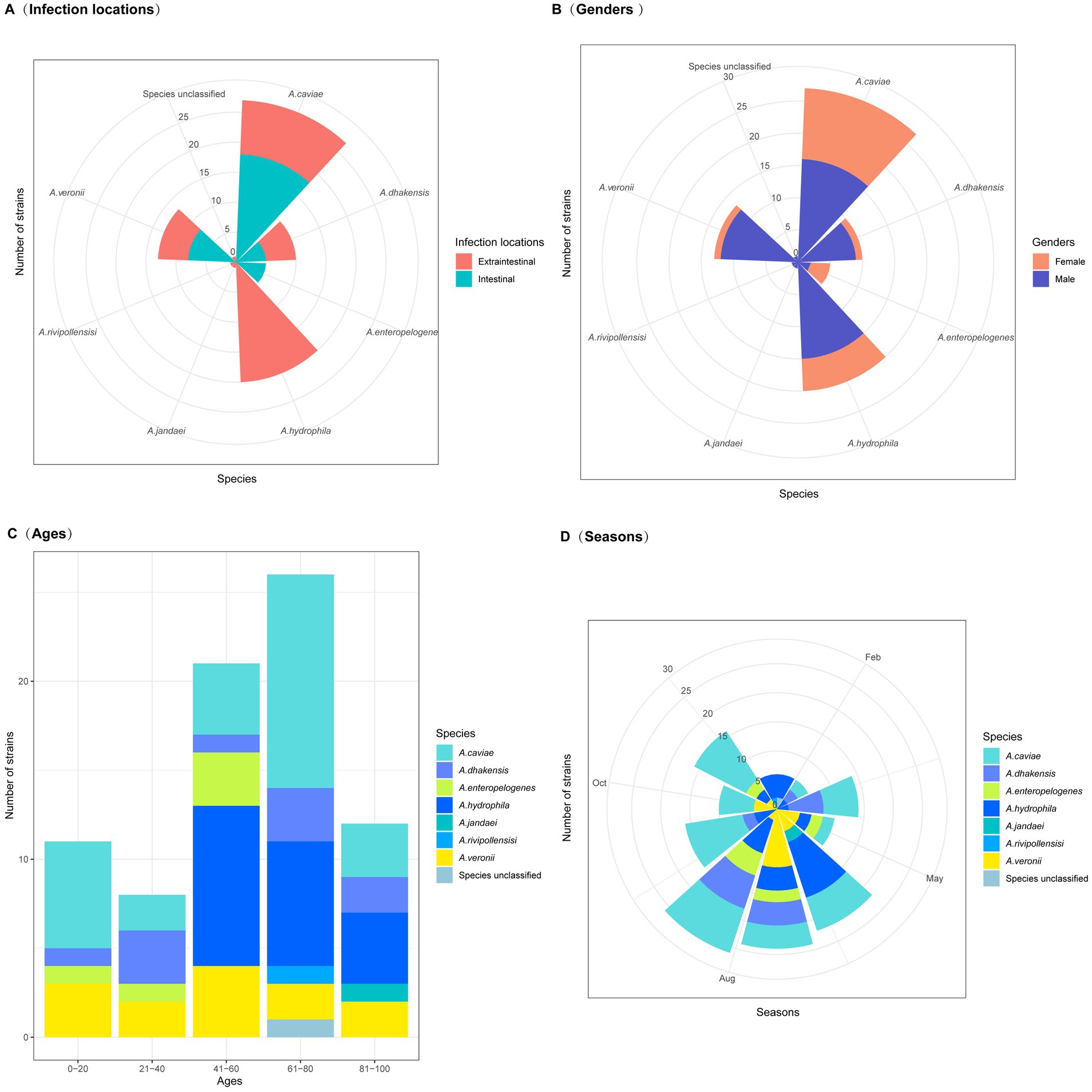
Figure 1. Clinical characteristics of patients with Aeromonas infections. (A) Infection locations (extraintestinal and intestinal infections). (B) Patient gender. (C) Patient ages. (D) Seasons in which the isolates were obtained.
The ANI analysis result of the 78 isolates revealed that the predominant species was A. caviae (n = 27), followed by A. hydrophila (n = 20), A. veronii (n = 13), A. dhakensis (n = 10), A. enteropelogenes (n = 5), A. rivipollensisi (n = 1), A. jandaei (n = 1) and an unclassified isolate temporarily designated Aeromonas sp. 130, which showed the highest ANI of 94.63% with A. veronii CIP 107763 (GenBank Accession No. GCF_000820285.1) (according to the criteria of the ANI analysis, an isolate that showed an ANI below the threshold of 95% for any classified species was considered a novel species) (Figure 2). Notably, there were discrepancies between the identification results from the ANI analysis and the MicroScan WalkAway system (MSWS). Some isolates identified as A. caviae by ANI were A. enteropelogenes or A. hydrophila by MSWS. Similar results occured in other species. Several isolates of A. hydrophila, A. veronii and A. rivpollensi by ANI were identified as A. veronii, A. hydrophila, and A. veronii by MSWS, respectively. Furthermore, since the MSWS database does not contain A. dhakensis data, the strains of the species A. dhakensis identified by the ANI analysis were previously identified as A. hydrophila, A. jandaei or A. caviae by MSWS (Supplementary Table S1). It has been recognized that bacterial species classification by ANI analysis was a gold standard (Jain et al., 2018). Difference did exist between species identification results by ANI and by other methods such as traditional biochemical tests, commercial identification kits, or automatic or semiautomatic systems, which might lead confusion of different species of the genus or misidentification of one species as a member of other genera (Deng et al., 2014; Janda and Abbott, 2010). The whole-genome sequencing-based method exhibited relatively high accuracy in identifying bacterial species (Beaz-Hidalgo et al., 2010; Martínez-Murcia et al., 2005). In this work, the overall concordance rate between the ANI and MSWS analyses for the classification of all Aeromonas species was 71.79%. If the A. dhakensis isolates were excluded because there is no information for this species available in the MSWS database, the concordance rate between the two methods reached 83.58%.
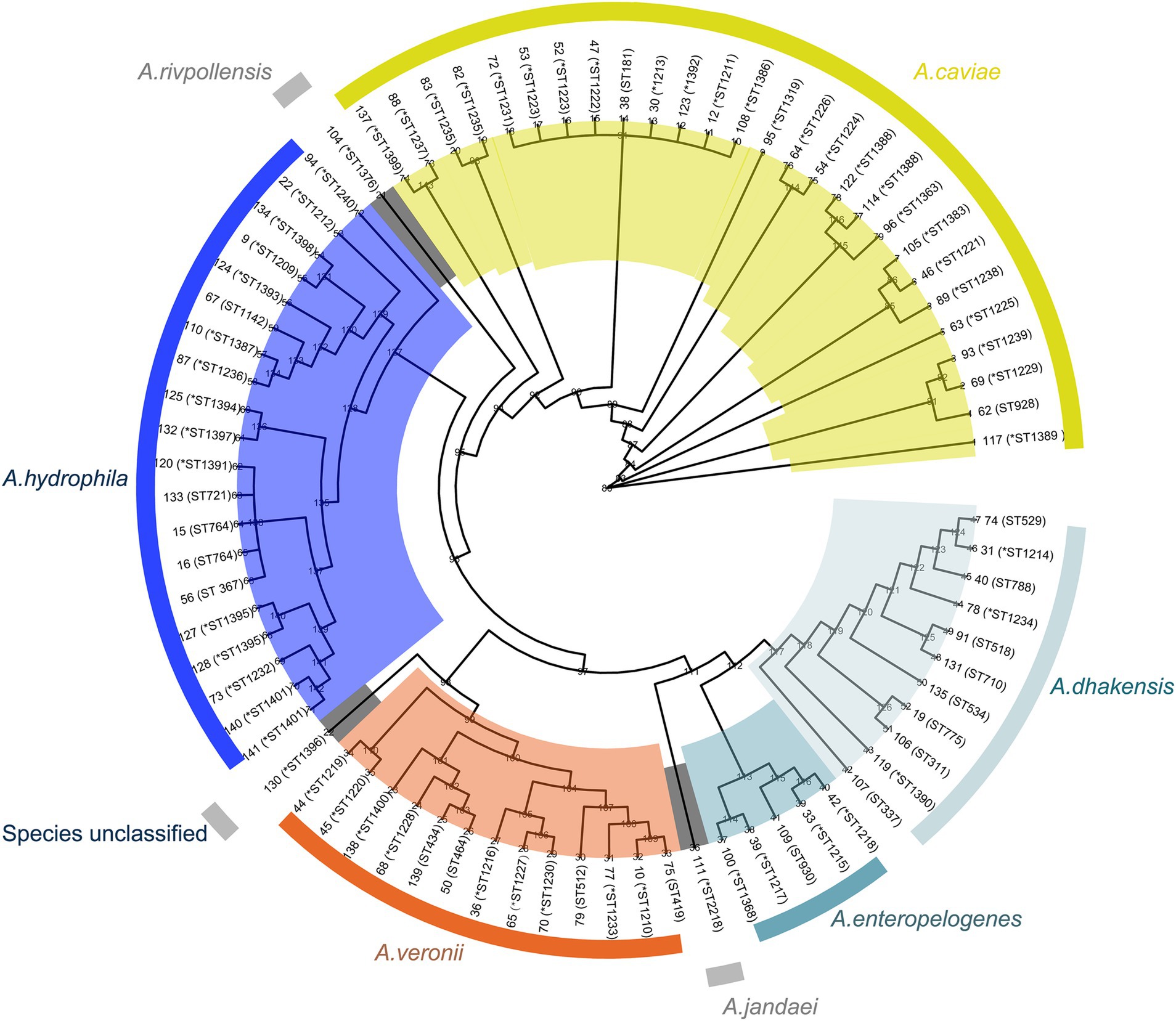
Figure 2. Phylogenetic tree of the genomes of 78 Aeromonas complex isolates. The species and sequence types (STs) are depicted in concentric circles, with “*” representing new STs. The numbers on the branches indicate the bootstrap values.
Among the 35 Aeromonas isolates that cause intraintestinal infections, A. caviae accounted for the greatest proportion (51.43%, 18/35), followed by A. veronii (20.0%, 7/35), A. enteropelogenes (14.29%, 5/35), and A. dhakensis (14.29%, 5/35). In contrast, among the Aeromonas isolates that cause extraintestinal infections, A. hydrophila accounted for the largest proportion (46.51%, 20/43), followed by A. caviae (20.93%, 9/43), A. dhakensis (13.95%, 6/43), A. veronii (11.63%, 5/43), A. jandaei (2.33%, 1/43), A. rivipollensisi (2.33%, 1/43), and the unclassified Aeromonas sp. isolate 130 (2.33%, 1/43) (Figure 2). Although Aeromonas species primarily cause intraintestinal infections (Zhou et al., 2019), there have been an increasing number of reports in recent years about extraintestinal infections caused by Aeromonas (Sinclair et al., 2016). Similar to the results of this study, extraintestinal infections were found to be caused mainly by A. hydrophila (Chen et al., 2021), while intraintestinal infections were caused primarily by A. caviae (Zhou et al., 2019).
These 78 Aeromonas isolates exhibited diverse sequence types (STs) according to multilocus sequence typing (MLST), and 72 STs were identified, including 53 novel STs (ST1209-1240, ST1319, ST1363, ST1368, ST1383 and ST1386-1401) (Supplementary Table S1). Among these STs, new STs were predominant, accounting for 73.61% (53/72). Most STs (91.67%, 66/72) had only one isolate each, and the six STs with more than one isolate were ST1388 (n = 3), ST1223 (n = 2), ST1235 (n = 2), ST1401 (n = 2), ST764 (n = 2) and ST311 (n = 2) (Supplementary Table S1).
Among the nine β-lactams tested, most isolates were resistant to penicillin G (98.7%, 77/78) and the first-generation cephalosporin cefazolin (96.2%, 75/78). A total of 35.9% (28/78) of the isolates were resistant to the second-generation cephalosporin cefoxitin. A total of 14.1% (11/78) of the isolates were resistant to both the third-generation cephalosporins, cefotaxime and ceftriaxone. High susceptibility to fourth-generation antibiotics, namely, a cephalosporin (cefepime), a monobactam (aztreonam), and two carbapenems (imipenem and meropenem), was detected, with resistance rates of 6.4% (5/78), 5.1% (4/78), 1.3% (1/78) and 5.1% (4/78), respectively (Table 1). Only 7.69% (6/78) of the Aeromonas strains showed resistance to one or more of the four antimicrobial agents cefepime, aztreonam, imipenem or meropenem, and uncommon β-lactamase genes were present in these strains. Among the four imipenem-resistant strains, three A. cavese strains (isolates QZ63, QZ114, QZ122) carried blaNDM-1, and one A. cavese strain (isolate QZ62) carried blaIMP-26. The other two strains (A. cavese QZ117 and A. hydrophila QZ124) carrying blaPER-3 showed resistance to cefepime and/or aztreonam. Except for five isolates of the species A. enteropelogenes with one blaTRU gene each, the other (73) isolates generally encoded two or more genotypes of the β-lactamase genes (Table 1; Supplementary Table S3).
A total of 334 resistance genes (>80% aa similarity with the functionally characterized resistance genes) of 30 genotypes (with 65 subgenotypes) associated with eight antimicrobial agent categories were identified in the genomes of the 78 Aeromonas isolates. The category with the most genes (or genotypes) was β-lactams. More than a half (55.99%, 187/334) of the resistance genes were β-lactamase genes from 15 genotypes, including blaPER, blaRSA, blaCTX-M, blaTEM, blacphA, blaIMP, blaAFM, blaimiH, blaNDM, blaVIM, blaMOX, blaCEPS, blaAQU, blaTRU, and blaOXA. Seven genotypes of aminoglycoside genes [aph(3″), aph(3′), aph(6), aac(6′), aac(3), aadA, and ant(2″)] (11.97%, 40/334), and three genotypes of phenicol resistance genes (floR, cmlA, and cat) were found, while for the remaining five antimicrobial categories, only one genotype each was present, which included fluoroquinolone (qnr), tetracycline (tet), sulfonamide (sul), peptide (mcr) and glycylcycline (bleMBL) resistance genes (Figure 3).
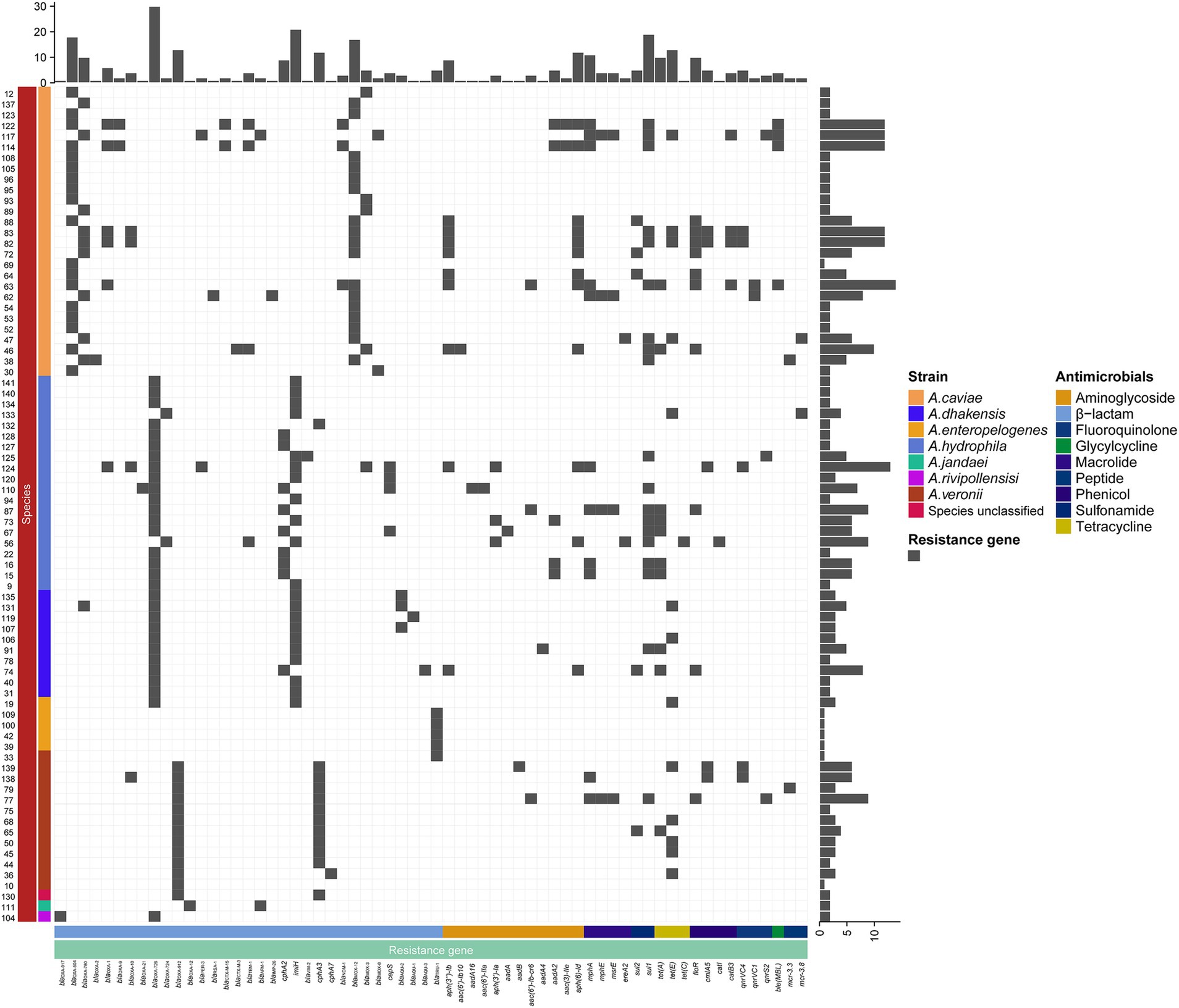
Figure 3. Distributions of the β-lactamase genes in the 78 isolates. The blue and white squares represent the presence and absence of the β-lactamase gene, respectively.
As β-lactams are the most important antimicrobials for treating bacterial infectious diseases, this study focused mainly on β-lactam resistance mechanisms. As mentioned above, of the 334 resistance genes identified in the 78 genomes, more than a half (55.99%, 187/334) were β-lactamase genes of 15 genotypes (34 subgenotypes), which covered all four classes [A (5.3%, 10/187), B (26.7%, 50/187), C (20.4%, 38/187), D (89/187, 47.59%)] (Figure 4). According to the Ambler (1980) classification system, among the last four genotypes, except for blaVIM, which is found in A. hydrophila, the other three are all found in A. caviae. Class C contained four genotypes, namely, blaMOX [n = 26, with 96.2% (25/26) in A. caviae and 3.8% (1/26) in A. hydrophila], blaCEPS (n = 4), blaAQU (n = 5), and blaTRU (n = 5), with the last three being uniquely identified in A. hydrophila, A. dhakensis, and A. enteropelogenes, respectively. However, class D had only one genotype, blaOXA (n = 89) (Supplementary Table S2). Notably, the class D β-lactamase gene blaOXA was the most prevalent (89/187, 47.59%) and was identified in all 78 Aeromonas isolates, except for five isolates of the species A. enteropelogenes. A. cavese and A. hydrophila had nine and eight genotypes, respectively, from all four classes of Ambler β-lactamase genes. In A. dhakensis, four genotypes of three classes (B, C, and D) were present. In the A. veronii isolates, A. jandaei 111 and Aeromonas sp. 130, two genotypes (blacphA3 of class B and blaOXA of class D) were found, while in the A. enteropelogenes isolates and A. rivipollensisi 104, only blaTRU of class C and blaOXA-912 of class D were identified. Among the 15 genotypes, nine were present in only one species. Interestingly, the species A. enteropelogenes (previously known as A. tructior/A. trota) consistently demonstrated susceptibility to ampicillin and is also the only known Aeromonas species that produces the single class C β-lactamase blaTRU-1 (Chen et al., 2012). This finding aligns with the result observed in this study.
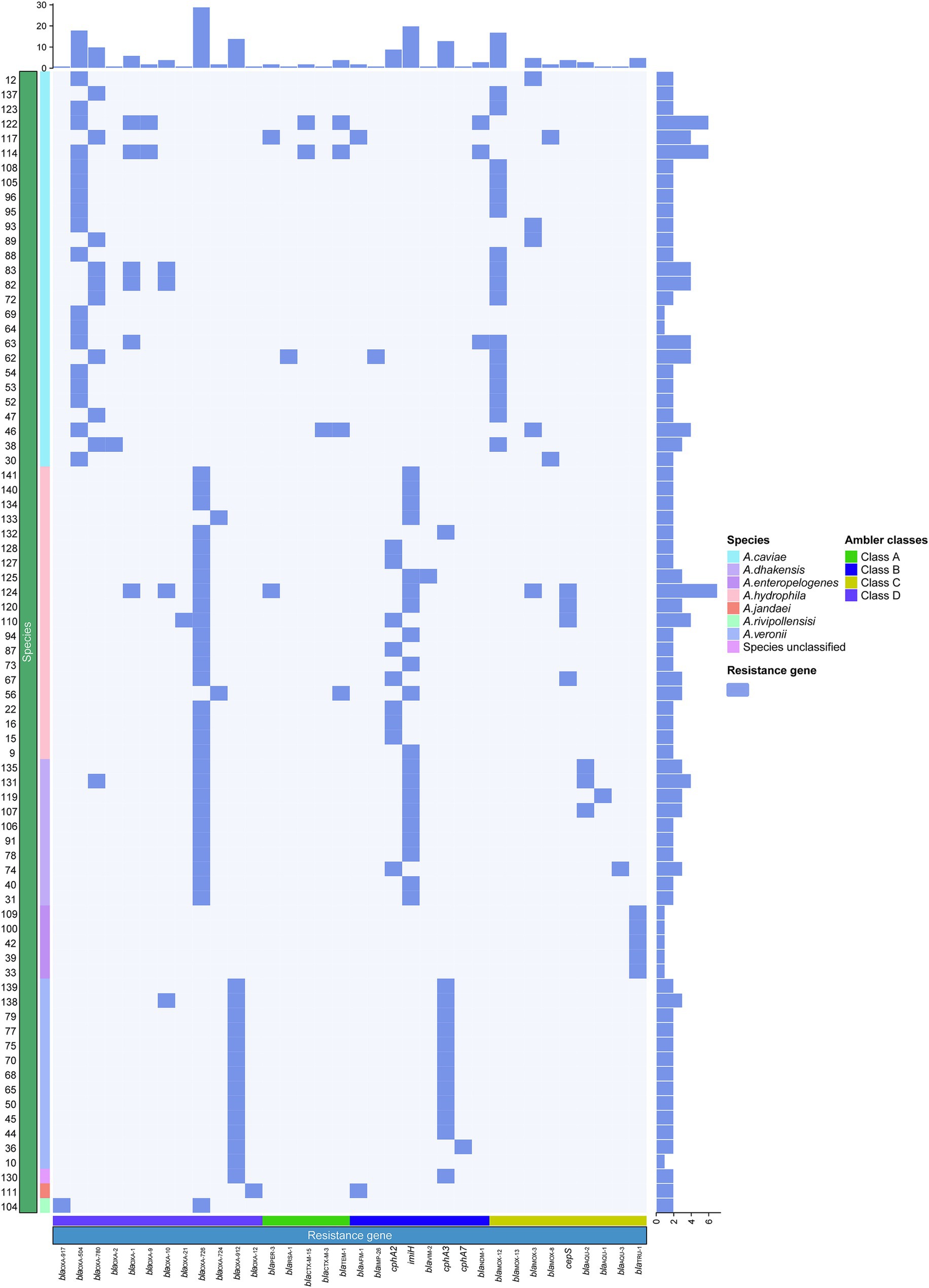
Figure 4. Synteny analysis of the 31.7 kbp MDR region in the chromosome of A. caviae QZ63. hp, hypothetical protein. Accession numbers: Enterobacter hormaechei 22KM1314 chromosome (CP126807.1), Enterobacter hormaechei 22KM0858 chromosome (CP126811.1), plasmid pS44-1 of Aeromonas salmonicida S44 (CP102110.1), Aeromonas caviae GD21SC2284TT chromosome (AP021927.1), Aeromonas hydrophila WP2-W18-ESBL chromosome (CP102110.1), plasmid P2 of Klebsiella pneumoniae 1,083 (OW967807.1), and plasmid pLZ135-NDM from an Escherichia coli isolate (MF353156.1). orfA, orfB: two fusion protein formed by translational frame-shifting which were the transposase for IS1133. orf: DUF3363 domain-containing protein.
To analyze the molecular mechanism underlying aztreonam and carbapenem resistance, the complete genomes of two isolates, A. hydrophila QZ124 (resistant to imipenem), encoding a blaNDM-1 gene, and A. caviae QZ63 (resistant to meropenem and cefepime), encoding a blaPER-3 gene, were sequenced. A. hydrophila QZ124 had a plasmid designated p124-100, however A. caviae QZ63 was free of a plasmid. The A. caviae QZ63 and A. hydrophila QZ124 genomes encoded 18 (13 genotypes) and 23 resistance genes (16 genotypes), respectively (Tables 2, 3), which showed ≥95% similarity to the functionally characterized resistance genes in the CARD. Interestingly, 83.33% (15/18) of the resistance genes of A. caviae QZ63, including three β-lactamase genes (two blaNDM-1 genes and one blaOXA-1 gene), were clustered in an approximately 31.7 kb multidrug resistance (MDR) region.

Table 2. Resistance genes identified in the A. caviae QZ63 genome.
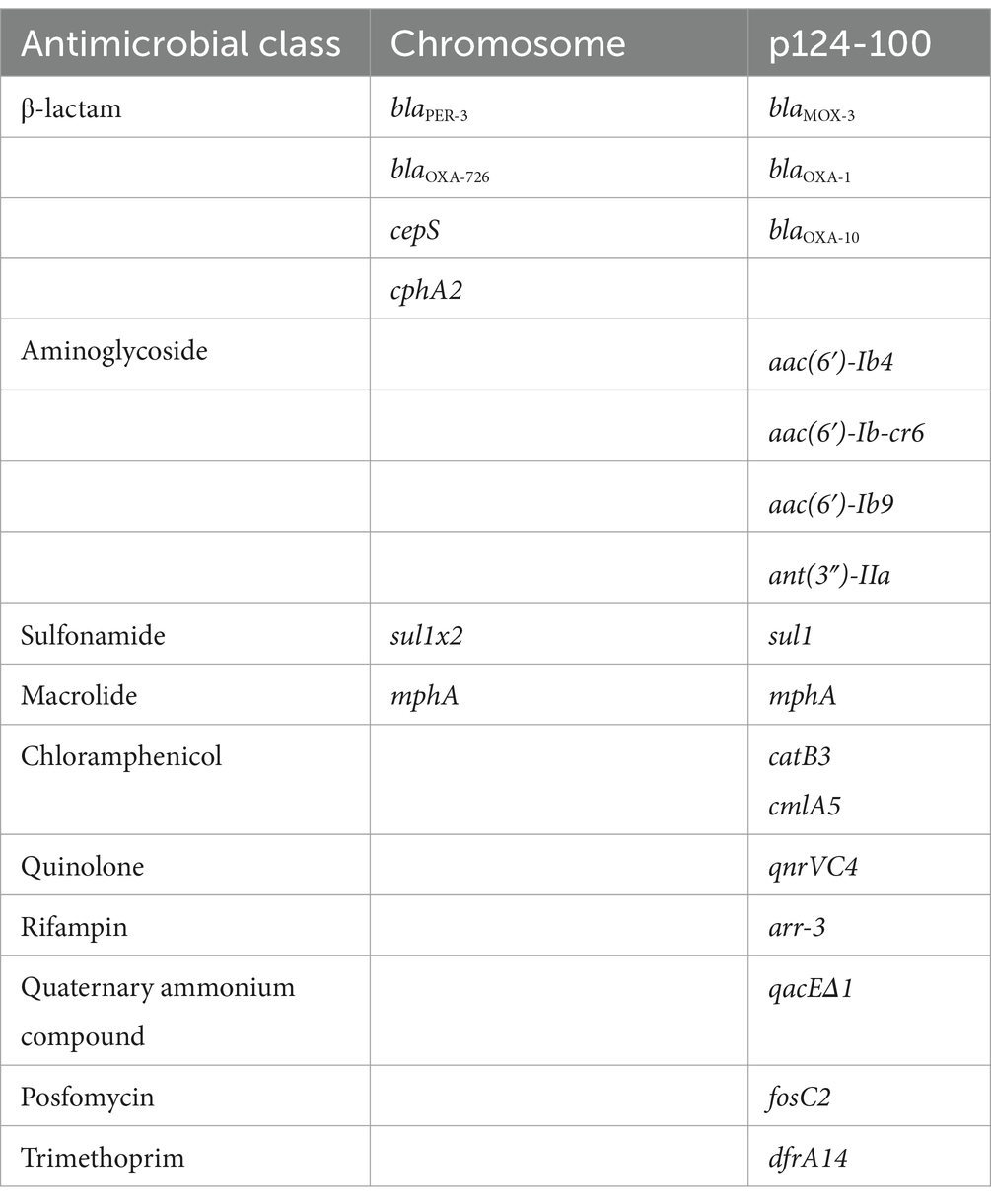
Table 3. Resistance genes identified in the A. hydrophila QZ124 genome.
The analysis of this MDR region of A. caviae QZ63 revealed that it consisted of three main parts encoding resistance genes associated with the mobile genetic elements (MGEs): an unit (named AAFT unit) encoding four resistance genes aph(6)-Id, aph(3″)-Ib, floR and tet(A), a class I integron (hp-gnat-sul1-catB3-blaOXA-1-ant(3″)-IIa-qnrVC1-intl1), and a transposon IS26-blaNDM-1-ble-tfrp-dsbD-hp-blaNDM-1-ble-tfrp-dsbD-hp-IS6100 (TnBB). Further analysis of the structure of the AAFT unit revealed that it was composed mainly of three MGEs or MGE-like fragments, which included an aph(6)-Id and aph(3″)-Ib-encoding Tn5393-like fragment, a floR-encoding fragment (floR-fragment) and a tetA and dhfr1-encoding transposon-like fragment (Tn-tetA-like). When using the 31.7 kb-MDR region as a query to search for similar sequences in the NCBI nonredundant nucleotide database, five sequences carrying two fragments (Tn5393-like and floR-fragment) of the AAFT unit were retrieved (Figure 5). These sequences included one from the same species as in this work (GenBank Accession No. AP021927.1, Aeromonas caviae str. WP2-W18-ESBL-01, isolated from wastewater treatment plant effluent in Japan, with a coverage of 75.42% and an identity of 99.98%), two from different species but the same genus as one of this work (GenBank Accession No. CP022176.1, Aeromonas salmonicida S44, isolated from Atlantic salmon from the RAS Atlantic Salmon facility in China, with a coverage of 75.33% and an identity of 99.98%; GenBank Accession No. CP102110.1, Aeromonas hydrophila strain GD21SC2284TT, isolated from fish in China, with a coverage of 75.31% and an identity of 99.98%). Another two were from the genus Enterobacter of the family Enterobacteriaceae (GenBank Accession No. CP126807.1, Enterobacter hormaechei, isolated from cat urine in Switzerland, with a coverage of 75.31% and an identity of 99.98%; GenBank Accession No. CP126811.1, Enterobacter hormaechei, isolated from a swab of a dog’s hip joint in Switzerland, with a coverage of 75.29% and an identity of 99.98%). In contrast to these five sequences, the sequence of A. caviae QZ63 in this work carried an additional Tn-tetA-like fragment, and no Tn-tetA-like sequence was available in the NCBI nucleotide database.2
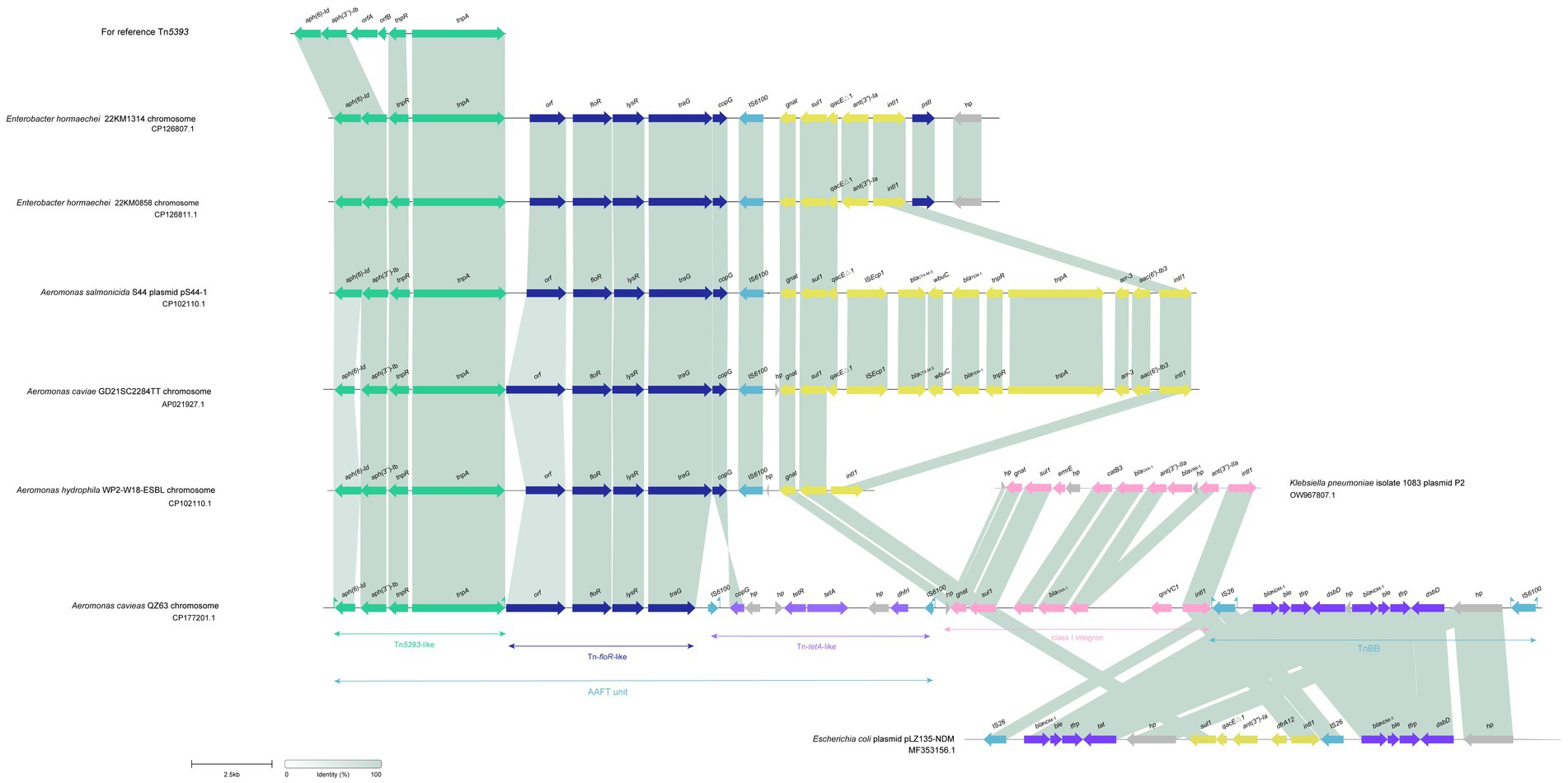
Figure 5. Distributions of resistance genes in the 78 isolates. The blue and white squares represent the presence and absence of the resistance gene, respectively. The categories of resistance genes and the species of the isolates are marked with different colors.
Next to the AAFT unit was an integron that carried five resistance gene, and the sequence sharing the highest similarity with it was from the plasmid P2 of Klebsiella pneumoniae 1,083 isolated from a patient at the Hospital Clinic (Barcelona), Spain (GenBank Accession No. OW967807.1, coverage 74.13% and identity 100%). Notably, even though there were integrons (or a truncated integron) next to the AAFT unit, such as fragments of the five sequences mentioned above, the gene from this work were completely different from any of the other five (Figure 5). The right side transposon (TnBB) of the 31.7 kb MDR region mainly consisted of a repeat sequence (blaNDM-1-ble-tfrp-dsbD-hp-blaNDM-1-ble-tfrp-dsbD-hp) flanked by two insert sequences (ISs), namely, IS26 and IS6100. TnBB showed the highest similarity to a sequence from the Escherichia coli plasmid pLZ135-NDM isolated from a patient in Hong Kong, China (GenBank Accession No. MF353156.1, coverage 74.56% and identity 99.95%).
From the analysis above, it could be concluded that similar sequences of the 31.7 kb MDR region of A. caviae QZ63 could be found to be located on plasmids and/or in the chromosomes of bacteria of different species or genera, isolated from different sources (such as human beings, animals, fishes and the environment) worldwide. This indicated that the resistance genes carried by MGEs spread among bacteria of different species or genera by means of horizontal gene transfer, resulting in the worldwide dissemination of resistance.
blaPER-3 is located in a variable region of the A. hydrophila QZ124 chromosome, and this region is approximately 18 kb in length and harbors several MGE-related sequences, including an intact integron carrying five resistance gene (sul1, qacEΔ1, blaPER-3, Δsul1, qacEΔ1, aadA), ISs (IS6100 and ISCR1) and tnp genes (tnpR and tnpA). blaPER-3 was found immediately downstream an insertion sequence ISCR1 (Figure 6). When searching for similar sequences in the nonredundant nucleotide database of NCBI, four sequences with greater similarity were retrieved. These sequences included one from A. media K521 (GenBank Accession No. CP118993.1, with a coverage of 83.29% and an identity of 99.93%), one from A. caviae 71,485 (GenBank Accession No. CP085468.1, with a coverage of 79.53% and an identity of 99.93%), one from A. caviae SS332 (GenBank Accession No. CP071151.1, with a coverage of 85.31% and an identity of 99.91%), and one from Acinetobacter johnsonii XBB1 (GenBank Accession No. KF017283.1, with a coverage of 72.76% and an identity of 99.92%) (Figure 6).
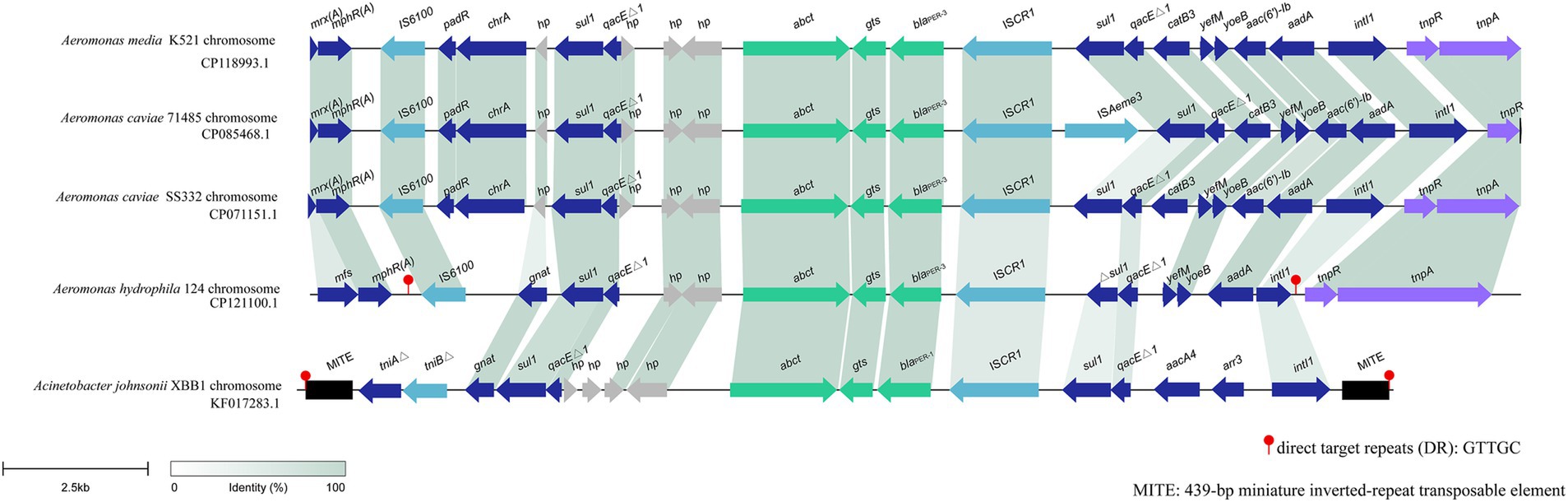
Figure 6. Synteny analysis of the 18 kbp region in the chromosome of A. hydrophila 124. hp, hypothetical protein. Accession numbers: A. media K521 chromosome (CP118993.1), A. caviae 71,485 chromosome (CP085468.1), A. caviae SS332 chromosome (CP071151.1), Aeromonas caviae GD21SC2284TT chromosome (AP021927.1), Acinetobacter johnsonii XBB1 chromosome (KF017283.1).
The bacteria harboring these four sequences were all isolated from human specimens in China. The three isolates with the highest similarities (CP118993.1, CP085468.1, and CP071151.1) to the sequence in this study were all from the same genus, Aeromonas, and were all isolated from the same province (Zhejiang) in China. The one (KF017283.1) with the lowest similarity was from a different bacterial family (Acinetobacter johnsonii) and was isolated from a different province (Sichuan) in China. This finding suggested that this blaPER-containing fragment may be carried mainly by Aeromonas strains and has also spread to bacteria with distant phylogenetic relationships in China. blaPER has been found to be related to MGEs, which facilitate the mobility of this gene. The association of blaPER with ISPa12 and Tn1213 (a composite transposon composed of ISPa12 and its close relative ISPa13) in Pseudomonas aeruginosa RNL-1 and Klebsiella pneumoniae CS1711 has been reported (Bae et al., 2011; Nordmann et al., 1993), and ISCR1 with blaPER was also found in A. johnsonii XBB1 and A. baumannii NF812784 (Wang et al., 2012; Zong, 2014). Several variants of Tn1213 have also been identified, including ISPa12 fragmented by IS6100 or ISPpu17 and ISPrst1 inserted into Tn1213 (Mantengoli and Rossolini, 2005).
The results of time-kill assays demonstrated that A. caviae QZ63 harboring blaNDM-1 resumed growth 2 h after exposure to meropenem at 0.5 ×, 1 × and 2 × MIC. In contrast, the control strain QZ54 ceased growth within 1–2 h under meropenem concentrations of 0.5 ×, 1 ×, and 2 × MIC. The strain QZ124 (carrying the blaPER-3 gene) resumed growth after 1 h of exposure to ceftazidime at 0.5 × MIC. When treated with 1 × MIC, growth was delayed and resumed only after 2 h, whereas treatment with 2 × MIC resulted in a further delay, with growth resuming after 4 h. Conversely, the control strain QZ87 ceased growth within 1–2 h at all tested concentrations (0.5 ×, 1 ×, and 2 × MIC). The antimicrobial combination of piperacillin + tazobactam exhibited no inhibitory effects on QZ63 (harboring blaNDM-1) and QZ124 (harboring blaPER-3). In contrast, the control strains QZ54 and QZ87 ceased growth within 1–2 h across all tested concentrations (0.5 ×, 1 ×, and 2 × MIC) (Figure 7). We observed that piperacillin + tazobactam was unable to inhibit the activity of metallo-β-lactamases (MBL) and extended-spectrum beta-lactamase (ESBL), a phenomenon that has also been reported in other isolates producing NDM-1 (Davido et al., 2017). Therefore, monotherapy with piperacillin + tazobactam or similar agents should be avoided in infections caused by A. caviae harboring blaNDM-1. Moreover, the resistance genes blaNDM-1 and blaPER-3 in QZ63 and QZ124, respectively, were found to be associated with mobile genetic elements (MGEs), suggesting the potential for horizontal transfer. This warrants further attention.
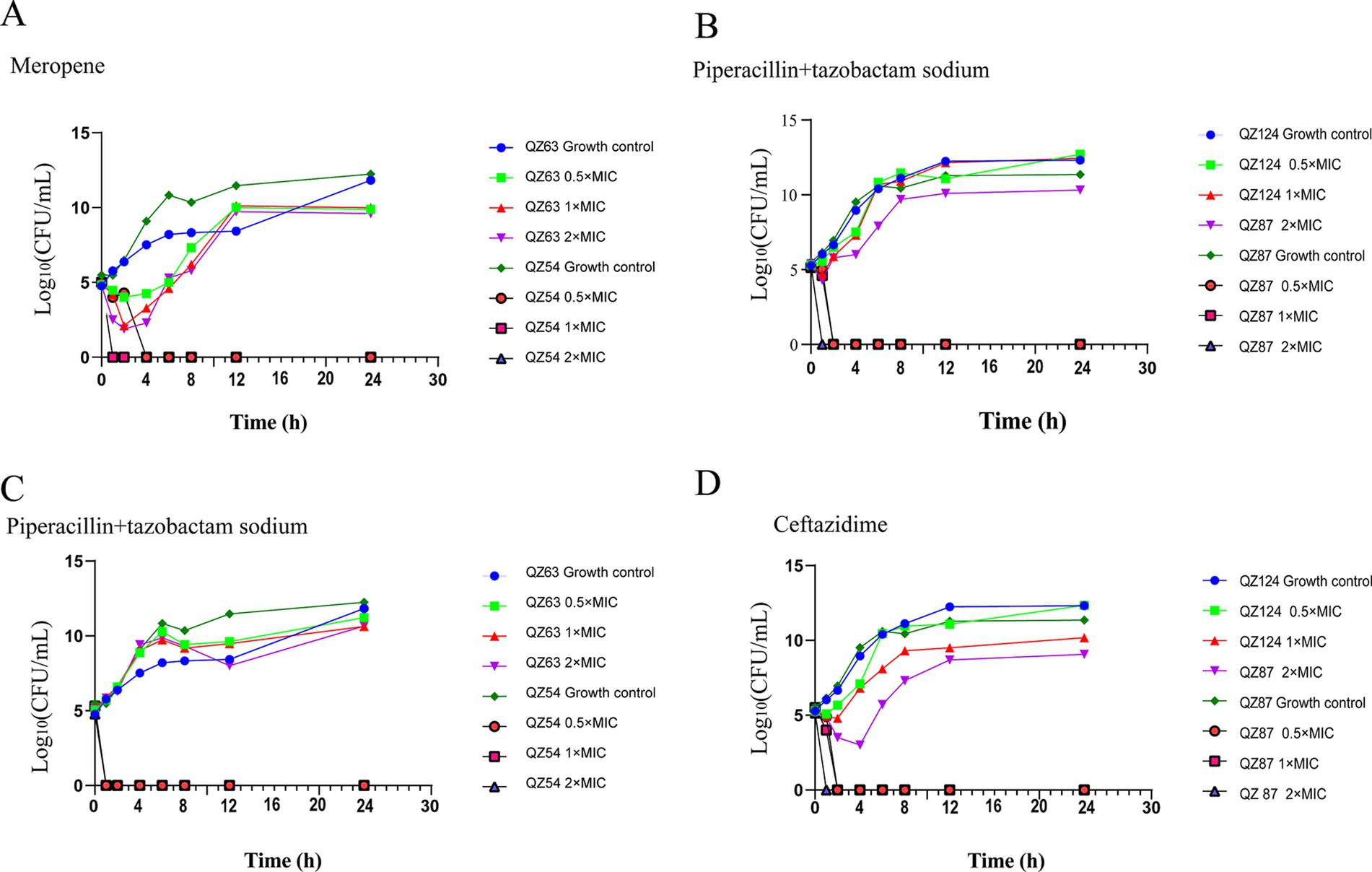
Figure 7. Time-kill curves showing the antibacterial effects. (A) meropenem (MIC: 2 µg/mL), (B,C) piperacillin + tazobactam (MIC: 32 µg/mL), and (D) ceftazidime (MIC: 32 µg/mL) at 0.5 × MIC, 1 × MIC, and 2 × MIC.
This study reported the correlation between the distribution of β-lactamase genes and the resistance phenotypes of clinical Aeromonas isolates, as well as the clinical characteristics of Aeromonas isolates. The increasing emergence of Aeromonas strains harboring extended-spectrum β-lactamase and metallo-β-lactamase genes poses a severe threat to public health. Close attention should be given to monitoring and controlling the spread of bacterial resistance.
The original contributions presented in the study are publicly available. This data can be found here: https://www.ncbi.nlm.nih.gov/genbank/. The GenBank accession numbers for the chromosome and pQZ124-211 of A.hydrophila QZ124, and the chromosome of A. caviae QZ63, are CP121100, CP121101 and CP177201, respectively.
Individual patient data were not involved, and only anonymous clinical residual samples during routine hospital laboratory procedures were used in this study. This study was approved by the Ethics Committee of the Quzhou Affiliated Hospital of Wenzhou Medical University, Quzhou, Zhejiang, China.
JL: Writing – original draft. LZ: Writing – original draft. CY: Writing – original draft. NL: Writing – original draft. YZ: Writing – original draft. YS: Writing – original draft. JZ: Writing – original draft. JL: Writing – review & editing. QB: Writing – review & editing. GZ: Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Science & Technology Project of Jinhua City, China (2022-2-013 and 2021-4-245), and the Science & Technology Project of Wenzhou City, China (N20210001).
The authors would like to acknowledge all study participants and individuals who contributed to this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1473150/full#supplementary-material
Ambler, R. P. (1980). The structure of beta-lactamases. Philos. Trans. R. Soc. B 289, 321–331. doi: 10.1098/rstb.1980.0049
Bae, I. K., Jang, S. J., Kim, J., Jeong, S. H., Cho, B., and Lee, K. (2011). Interspecies dissemination of the bla gene encoding PER-1 extended-spectrum β-lactamase. Antimicrob. Agents Chemother. 55, 1305–1307. doi: 10.1128/AAC.00994-10
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Beaz-Hidalgo, R., Alperi, A., Buján, N., Romalde, J. L., and Figueras, M. J. (2010). Comparison of phenotypical and genetic identification of Aeromonas strains isolated from diseased fish. Syst. Appl. Microbiol. 33, 149–153. doi: 10.1016/j.syapm.2010.02.002
Chen, P.-L., Chi-Jung, W., Tsai, P.-J., Tang, H.-J., Chuang, Y.-C., Lee, N.-Y., et al. (2014b). Virulence diversity among bacteremic Aeromonas isolates: ex vivo, animal, and clinical evidences. PLoS One 9:e111213. doi: 10.1371/journal.pone.0111213
Chen, P.-L., Ko, W.-C., and Chi-Jung, W. (2012). Complexity of β-lactamases among clinical Aeromonas isolates and its clinical implications. J. Microbiol. Immunol. Infect. 45, 398–403. doi: 10.1016/j.jmii.2012.08.008
Chen, Y.-W., Shu-Li, S., Li, C.-W., Tsai, C.-S., Lo, C.-L., Syue, L.-S., et al. (2021). Pancreaticobiliary cancers and Aeromonas isolates carrying type III secretion system genes ascF-ascG are associated with increased mortality: an analysis of 164 Aeromonas infection episodes in southern Taiwan. Front. Cell. Infect. Microbiol. 11:749269. doi: 10.3389/fcimb.2021.749269
Chen, P.-L., Wu, C. J., Chen, C. S., Tsai, P. J., Tang, H. J., and Ko, W. C. (2014a). A comparative study of clinical Aeromonas dhakensis and Aeromonas hydrophila isolates in southern Taiwan: A. dhakensis is more predominant and virulent. Clin. Microbiol. Infect. 20, O428–O434. doi: 10.1111/1469-0691.12456
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H., and Li, H. (2021). Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175. doi: 10.1038/s41592-020-01056-5
Davido, B., Senard, O., de Truchis, P., Salomon, J., and Dinh, A. (2017). Monotherapy of ceftazidime-avibactam and ceftolozane-tazobactam: two effective antimicrobial agents against multidrug-resistant organisms except for NDM-1 isolates. Int. J. Infect. Dis. 62, 124–125. doi: 10.1016/j.ijid.2017.06.021
Deng, J., Liang, F., Wang, R., Nan, Y., Ding, X., Jiang, L., et al. (2014). Comparison of MALDI-TOF MS, gene sequencing and the Vitek 2 for identification of seventy-three clinical isolates of enteropathogens. J. Thorac. Dis. 6, 539–544. doi: 10.3978/j.issn.2072-1439.2014.02.20
Gu, Z., Eils, R., and Schlesner, M. (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. doi: 10.1093/bioinformatics/btw313
Hiransuthikul, N., Tantisiriwat, W., Lertutsahakul, K., Vibhagool, A., and Boonma, P. (2005). Skin and soft-tissue infections among tsunami survivors in southern Thailand. Clin. Infect. Dis. 41, e93–e96. doi: 10.1086/497372
Igbinosa, I. H., Igumbor, E. U., Aghdasi, F., Tom, M., and Okoh, A. I. (2012). Emerging Aeromonas species infections and their significance in public health. Sci. World J. 2012:625023. doi: 10.1100/2012/625023
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114. doi: 10.1038/s41467-018-07641-9
Janda, J. M., and Abbott, S. L. (2010). The genus Aeromonas: taxonomy, pathogenicity, and infection. Clin. Microbiol. Rev. 23, 35–73. doi: 10.1128/CMR.00039-09
Jia, B., Raphenya, A. R., Alcock, B., Waglechner, N., Guo, P., Tsang, K. K., et al. (2017). CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 45, D566–D573. doi: 10.1093/nar/gkw1004
Khan, M. A., Faiz, A., and Ashshi, A. M. (2015). Maternal colonization of group B Streptococcus: prevalence, associated factors and antimicrobial resistance. Ann. Saudi Med. 35, 423–427. doi: 10.5144/0256-4947.2015.423
Kosikowska, U., Stec, J., Andrzejczuk, S., Mendrycka, M., Pietras-Ożga, D., and Stępień-Pyśniak, D. (2022). Plasmid-mediated fluoroquinolone resistance genes in quinolone-susceptible Aeromonas spp. phenotypes isolated from recreational surface freshwater reservoir. Front. Cell. Infect. Microbiol. 12:885360. doi: 10.3389/fcimb.2022.885360
Mantengoli, E., and Rossolini, G. M. (2005). Tn5393d, a complex Tn5393 derivative carrying the PER-1 extended-spectrum beta-lactamase gene and other resistance determinants. Antimicrob. Agents Chemother. 49, 3289–3296. doi: 10.1128/AAC.49.8.3289-3296.2005
Martínez-Murcia, A. J., Soler, L., Saavedra, M. J., Chacón, M. R., Guarro, J., Stackebrandt, E., et al. (2005). Phenotypic, genotypic, and phylogenetic discrepancies to differentiate Aeromonas salmonicida from Aeromonas bestiarum. Int. Microbiol. 8, 259–269
Martino, M. E., Fasolato, L., Montemurro, F., Rosteghin, M., Manfrin, A., Patarnello, T., et al. (2011). Determination of microbial diversity of Aeromonas strains on the basis of multilocus sequence typing, phenotype, and presence of putative virulence genes. Appl. Environ. Microbiol. 77, 4986–5000. doi: 10.1128/AEM.00708-11
McArthur, A. G., Waglechner, N., Nizam, F., Yan, A., Azad, M. A., Baylay, A. J., et al. (2013). The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 57, 3348–3357. doi: 10.1128/AAC.00419-13
Norden, C. W., Wentzel, H., and Keleti, E. (1979). Comparison of techniques for measurement of in vitro antibiotic synergism. J. Infect. Dis. 140, 629–633. doi: 10.1093/infdis/140.4.629
Nordmann, P., Ronco, E., Naas, T., Duport, C., Michel-Briand, Y., and Labia, R. (1993). Characterization of a novel extended-spectrum beta-lactamase from Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 37, 962–969. doi: 10.1128/AAC.37.5.962
Paevskiĭ, S. A. (1993). A means for determining the bactericidal activity of the tissues during the treatment of orthopedic patients by transosseous osteosynthesis methods. Klin. Lab. Diagn. 5, 25–29.
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Parker, J. L., and Shaw, J. G. (2011). Aeromonas spp. clinical microbiology and disease. J. Infect. 62, 109–118. doi: 10.1016/j.jinf.2010.12.003
Pérez-Sancho, M., Cerdá, I., Fernández-Bravo, A., Domínguez, L., Figueras, M. J., Fernández-Garayzábal, J. F., et al. (2018). Limited performance of MALDI-TOF for identification of fish Aeromonas isolates at species level. J. Fish Dis. 41, 1485–1493. doi: 10.1111/jfd.12837
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Sinclair, H. A., Heney, C., Sidjabat, H. E., George, N. M., Bergh, H., Anuj, S. N., et al. (2016). Genotypic and phenotypic identification of Aeromonas species and CphA-mediated carbapenem resistance in Queensland, Australia. Diagn. Microbiol. Infect. Dis. 85, 98–101. doi: 10.1016/j.diagmicrobio.2016.02.005
Wang, F., Kuihai, W., Sun, J., Wang, Q., Chen, Q., Shouyi, Y., et al. (2012). Novel ISCR1-linked resistance genes found in multidrug-resistant Gram-negative bacteria in southern China. Int. J. Antimicrob. Agents 40, 404–408. doi: 10.1016/j.ijantimicag.2012.06.016
Weinstein, M. P., and Lewis, J. S. (2020). The clinical and laboratory standards institute subcommittee on antimicrobial susceptibility testing: background, organization, functions, and processes. J. Clin. Microbiol. 58, e01864–e01819. doi: 10.1128/JCM.01864-19
Wu, C.-J., Jiunn-Jong, W., Yan, J.-J., Lee, H.-C., Lee, N.-Y., Chang, C.-M., et al. (2007). Clinical significance and distribution of putative virulence markers of 116 consecutive clinical Aeromonas isolates in southern Taiwan. J. Infect. 54, 151–158. doi: 10.1016/j.jinf.2006.04.002
Yu, G., Lam, T. T.-Y., Zhu, H., and Guan, Y. (2018). Two methods for mapping and visualizing associated data on phylogeny using Ggtree. Mol. Biol. Evol. 35, 3041–3043. doi: 10.1093/molbev/msy194
Yuwono, C., Wehrhahn, M. C., Liu, F., Riordan, S. M., and Zhang, L. (2021). The isolation of Aeromonas species and other common enteric bacterial pathogens from patients with gastroenteritis in an Australian population. Microorganisms 9:1440. doi: 10.3390/microorganisms9071440
Zhang, D., Li, W., Xin, H., Huang, H., and Zhang, X. (2023). Requiring reconsideration of differences of Aeromonas infections between extra-intestinal and intestinal in hospitalized patients. Infect. Drug Resist. 16, 487–497. doi: 10.2147/IDR.S393347
Zhou, Y., Li, Y., Nan, Z., Zhang, P., Kan, B., Yan, D., et al. (2019). Taxonomy, virulence genes and antimicrobial resistance of Aeromonas isolated from extra-intestinal and intestinal infections. BMC Infect. Dis. 19:158. doi: 10.1186/s12879-019-3766-0
Keywords: Aeromonas complex, antimicrobial resistance mechanism, whole-genome sequencing, species classification, multilocus sequence typing
Citation: Lu J, Zhang L, Yan C, Lin N, Zhang Y, Sha Y, Zhao J, Lu J, Bao Q and Zhang G (2025) Whole-genome sequencing-based species classification, multilocus sequence typing, and antibiotic resistance mechanisms of the clinical Aeromonas complex. Front. Microbiol. 16:1473150. doi: 10.3389/fmicb.2025.1473150
Edited by:
Asad U. Khan, Aligarh Muslim University, IndiaReviewed by:
Miranda Kirchner, Animal and Plant Health Agency, United KingdomCopyright © 2025 Lu, Zhang, Yan, Lin, Zhang, Sha, Zhao, Lu, Bao and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Lu, Y2F0aHk5MzI5NkAxNjMuY29t; Qiyu Bao, YmFvcXlAZ2Vub21pY3MuY24=; Guozhi Zhang, MTMwNTQ2MDI0MUBxcS5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.