 Ruiyun Li1
Ruiyun Li1 Bin Wang
Bin Wang Jing He
Jing He- 1College of Forestry, Gansu Agricultural University, Lanzhou, China
- 2Wolfberry Harmless Cultivation Engineering Research Center of Gansu Province, Lanzhou, China
Fusarium solani is a widely distributed pathogenic fungus that can cause soil borne diseases in various plants and is also one of the main pathogenic bacteria of Lycium barbarum root rot. This study employed tandem mass labeling (TMT) quantitative proteomics technology to investigate the antifungal mechanism of potato glycoside alkaloids (PGA) against Fusarium solani. We elucidated the antifungal mechanism of PGA from the perspective of mitochondrial proteome molecular biology. Furthermore, we identified and annotated the differentially expressed proteins (DEP) of F. solani under PGA stress. A total of 2,412 DEPs were identified, among which 1,083 were significantly up-regulated and 1,329 significantly down-regulated. Subsequent analysis focused on five DEPs related to energy metabolism for verification at both protein and gene levels. Gene Ontology (GO) function analysis revealed that the DEPs were primarily involved in the integral component of the membrane, intrinsic component of the membrane, pyridine-containing compound metabolic processes, carbon-oxygen lyase activity, and the endoplasmic reticulum, with a notable enrichment in membrane components. Furthermore, a total of 195 pathways were identified through KEGG analysis, with significant enrichment in critical pathways including pentose and glucuronate interconversions, propanoate metabolism, various types of N-glycan biosynthesis, the pentose phosphate pathway, and carbon fixation in photosynthetic organisms. The results from both parallel reaction monitoring (PRM) and real-time RT-qPCR were consistent with the overall trends observed in TMT proteomics, thereby confirming the validity of the TMT proteomics analysis. These findings indicate that PGA inhibits the growth of F. solani by impacting mitochondrial energy metabolism. This study reveals the antifungal mechanism of PGA from the perspective of energy metabolism, providing a theoretical basis for the development and application of PGA as a biopesticide.
1 Introduction
Lycium barbarum is a perennial shrub that belongs to the Solanaceae family and the Lycium genus. It is a main economic tree species in Gansu Province. It is widely distributed in the northwest saline alkali desert areas (Zeng et al., 2023). The fruit of L. barbarum is sweet and juicy, commonly consumed raw, juiced, or brewed into tea in local traditions. It possesses high nutritional and medicinal value (Zeng et al., 2023; Li et al., 2018; Liu et al., 2022). However, root rot is one of the primary diseases that hinders the development of the wolfberry exhibition industry, leading to a decline in the quality of wolfberry (Bai et al., 2020). L. barbarum root rot is a prevalent soil-borne disease that primarily affects the roots and stems of the plant, impeding the transport and absorption of water and nutrients. This results in poor plant growth, reduced quality and yield of L. barbarum fruit, and in severe cases, can lead to plant wilting and death. It is classified as a typical vascular disease (He et al., 2023). Fusarium is identified as the main pathogen responsible for root rot (El-Saadony et al., 2021), with Fusarium solani being the most widely distributed species. Currently, the prevention and control of L. barbarum root rot rely primarily on chemical methods. Chemical fungicides not only disrupt the ecological balance of soil, but also lead to excessive pesticide residues in L. barbarum berries and induce resistance of pathogenic fungi. Therefore, developing green, safe, and efficient biological antifungal agents to combat L. barbarum root rot is of significant importance.
Potato glycoside alkaloids (PGA) are a class of steroidal alkaloids characterized by the presence of 1–4 monosaccharides. These compounds are secondary metabolites predominantly found in the green parts of potato tubers, stems, and leaves (Song et al., 2023). The concentration of glycoside alkaloids in potato tubers decreases progressively as the potatoes mature (Koffi et al., 2017). PGA has garnered significant attention due to its abundant availability, cost-effectiveness, environmental safety, and broad-spectrum antifungal properties. Previous research has identified α-solanine and α-theophylline as the primary antifungal constituents of PGA. Notably, α-solanine exhibits a potent antifungal effect against Botrytis cinerea, with its inhibitory intensity being dose-dependent (Sun et al., 2014). Previous studies have utilized the method of inhibiting the growth of flat mycelium to evaluate the antifungal activity of PGA against Cercosporella brassicae, Alternaria porri, A. solani, and F. solani. The results indicated that PGA exhibited inhibitory effects on the growth of all four pathogens. Notably, the inhibitory effect on C. brassicae and A. porri was particularly pronounced, while the effects on A. solani and F. solani were comparatively weaker (Zhao et al., 2013). The antifungal efficacy of PGA against F. solani, Marssonina juglandis, and Capnodium leaophilum was assessed using the growth rate method, revealing that PGA possessed notable antifungal activity against these three fungi, with the strongest inhibition observed against F. solani, which also enhanced cell membrane permeability (Duo et al., 2017). Additionally, it has been reported that PGA can inhibit both mycelial growth and spore development of Curvularia trifolii (Xu et al., 2023), diminish the virulence of Pectobacterium brasiliense (Joshi et al., 2021), and inhibit reactive oxygen species (ROS) metabolism in F. sulphureum, thereby exerting antifungal effects (Li et al., 2022). Our previous study demonstrated that PGA inhibited respiration and ROS metabolism in F. solani, disrupted mitochondrial structure, and ultimately suppressed pathogen growth (Ding et al., 2023). Furthermore, PGA can attenuate tricarboxylic acid cycle flux by modulating the expression of key genes, contributing to its antifungal effects (Zhang et al., 2024).
Previous studies on the antifungal effects of PGA have primarily concentrated on physiological and biochemical aspects, with a notable lack of research involving proteomics. However, recent advancements in proteomics research methods have emerged rapidly. Tandem Mass Tag (TMT)-based quantitative proteomics technology is one of the most powerful analytical techniques available, offering the highest throughput, minimal systematic error, and robust capabilities for the quantitative analysis of differentially expressed proteins (DEPs). This technology is characterized by high sensitivity, excellent separation capacity, and substantial throughput. Furthermore, TMT technology has been employed to systematically analyze the expression changes of mitochondrial proteins from a proteomic perspective, enabling the identification of differential proteins and the verification of DEPs expression levels. This approach aims to elucidate the antifungal mechanism of PGA on F. solani from a molecular biology standpoint.
2 Materials and methods
2.1 Test materials
2.1.1 Tested pathogens
F. solani was isolated from diseased L. barbarum plants, which were identified and preserved in the Forest Protection Science Laboratory at Gansu Agricultural University. Prior to use, the isolates were activated on Potato Dextrose Agar (PDA) medium (Potato 200 g, glucose 20 g, agar 20 g and water 1,000 mL) at 25°C and subsequently stored at 0 to 4°C for later application.
2.1.2 Preparation of PGA
The extraction of PGA follows the method described by Huang et al. (2012). First, weigh 100 g of potato sprout powder and add 400 mL of 5% acetic acid (chemically pure). Stir the mixture using a magnetic stirrer (EMS-13, Tianjin Ounuo Instrument Co., Ltd.) for 1 h. Subsequently, employ a vacuum filtration pump [SHZ-D (III), North South Science Instrument (Beijing) Technology Co., Ltd.] to filter the mixture, and extract the residue twice with 200 mL of 5% acetic acid. Combine the filtrate from these three extractions. Adjust the pH to 10.0 using ammonia water, and then perform an extraction with water-saturated n-butanol (200 mL) in three portions. Collect and combine the extracts, and evaporate them using a rotary evaporator (RV10V, Suzhou Sainz Instrument Co., Ltd.) at 65°C and 40 rpm for 1 h. Finally, dissolve the residue in 20 mL of methanol (chemically pure) to obtain the PGA extraction mother liquor with a concentration of 88.10 mg/mL.
According to the principle of color development, PGA (solanine and chalcone) can form a purple-red complex with formaldehyde in an acidic environment. The intensity of the color is positively correlated with the PGA content. The absorbance of the solution at 530 nm is measured, and the PGA concentration of the sample solution is determined using a standard curve based on previous research results from the group. The regression equation for the standard curve is y = 0.3628x+0.0256, with an R2 value of 0.9583. For PGA measurement, 1 mL of the PGA extract is taken, and the method for constructing the standard curve is followed. Methanol (instead of the sample solution) is adjusted to zero, and the absorbance (A) at 530 nm is measured to calculate the PGA content using the formula: (FW)
where V is the volume of the sampled liquid (mL), C is the concentration of the sample solution as determined by the standard curve (mg/mL), and W is the fresh weight of the sample (g).
The repeatability test was conducted on the PGA extract, revealing an average PGA content of 0.503 mg/g and a relative standard deviation (RSD) of 3.25% (< 10%). This indicates good repeatability of the method, based on five replicates, and confirms its suitability for the quantitative analysis of PGA.
2.1.3 Determination of EC50 value
The EC50 was determined using the mycelium growth rate method. Based on the results of the preliminary concentration screening experiment, plates containing different concentrations of PGA (1.0, 2.0, 4.0, 8.0, and 16.0 mg/mL) were prepared. F. solani was inoculated with fungal cake using a sterile puncher (φ = 6 mm), with sterile water serving as the control (CK). The cultures were maintained at 25°C and the experiment was repeated three times. From this, the toxicity regression equation and the half maximal effect concentration (EC50).
Convert the inhibition rate into a probability value and the mass concentration into the logarithm of the mass concentration. Designate the logarithm of the mass concentration of each agent as x and the probability value of the inhibition rate as y. Subsequently, perform data processing to derive the toxicity regression equation y = 0.9385x–0.0511, and the correlation coefficient was R2 = 0.9407. Finally, determine the effective medium mass concentration EC50 value of the test agent.
2.1.4 Observation of mycelium by scanning electron microscope
Samples for scanning electron microscopy were prepared following the method outlined by Vujanovic et al. (2019). A total of 0.1 g of mycelium from both the control group and the PGA treatment group were fixed in a 2.5% glutaraldehyde phosphate buffer (pH 7.2) for 16 h. The samples were then rinsed three times with phosphate buffer, with each rinse lasting 15 min. Gradient dehydration was conducted sequentially using 30, 50, 70, 90, 95, and 100% ethanol. Subsequently, the ethanol in the samples was replaced with isoamyl acetate. The samples were then dried at the critical point, vacuum coated, and finally examined and imaged using a scanning electron microscope (JSM-5600LV, Japan Electron Optics Co., Ltd.).
2.1.5 Preparation of mitochondria
Mitochondria were prepared using the method described by Zhang et al. (2023) and Zhan et al. (2021), employing differential centrifugation. F. solani was inoculated on PDA and cultured at 25°C for 7 d to allow for the maturation of F. solani spores. A spore suspension of F. solani (1 × 106 spores/mL) was prepared, and 50 μL of this suspension was added to 30 mL of Potato Dextrose Broth (PDB) medium (Potato 200 g, glucose 20 g and water 1,000 mL) for shaking culture at 25°C and 160 rpm. After 2 d of culture, the mycelium was transferred to PDB medium with a final concentration of 3.88 mg/mL. The mycelium was collected after an additional 2 d of continuous culture at 25°C and 160 rpm. The mycelium was then filtered through sterile gauze, washed three times with sterile water, and dried using a vacuum pump. The mycelium was ground to crush the cells; following this, centrifugation was performed at 4°C at 1,500 × g for 5 min, after which the supernatant was collected and the precipitate discarded. The supernatant was subsequently centrifuged at 12,000 × g at 4°C for 20 min. The mitochondrial precipitate was then resuspended in mitochondrial extraction buffer.
2.1.6 TMT analysis of mitochondrial protein of F. solani
The TMT analysis of mitochondrial proteins from F. solani was conducted by Shanghai Personal Biotechnology Co., Ltd. Mitochondrial samples of F. solani were extracted using the SDT [4%(w/v) SDS, 100 mM Tris/HCl pH7.6, 0.1M DTT] lysis method, followed by protein quantification using the BCA method. Each sample underwent trypsinization via the Filter Aided Proteome Preparation (FASP) method (Wiśniewski et al., 2009). The resulting peptides were desalted with a C18 cartridge, then lyophilized and reconstituted in 40 μL of a 0.1% formic acid solution. For peptide quantification, 100 μg of peptides were taken from each sample and labeled according to the instructions provided in the Thermo Scientific TMT labeling kit. The samples were then separated using an Easy nLC HPLC system with a nanoliter flow rate. Chromatographic separation was followed by analysis using a Q-Exactive mass spectrometer. The raw data for mass spectrometry analysis are RAW files, and the software Mascot 2.2 and Proteome Discoverer 1.4 are used for inventory identification and quantitative analysis. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium1 via the iProX partner repository with the dataset identifier PXD058927.
2.1.7 Bioinformatics analysis methods
In this experiment, differentially expressed proteins (DEPs) were screened, and a volcano plot was generated based on the criteria of a fold change (FC) greater than 1.2 for up-regulation or less than 0.83 for down-regulation, with a p-value of less than 0.05 (determined by T-test or other methods). The annotation of the target protein set was conducted using Blast2GO, which can be summarized in four main steps: sequence alignment (Blast), Gene Ontology (GO) item extraction (Mapping), GO annotation (Annotation), and supplementary annotation through InterProScan (Annotation Augmentation). Additionally, KEGG pathway annotation analysis of the target protein set was performed using KAAS (KEGG Automatic Annotation Server) software. Fisher’s Exact Test was employed to compare the distribution of each GO category (or KEGG pathway, or Domain) within the target protein set against the overall protein set. Furthermore, enrichment analysis of GO annotations and KEGG pathway or Domain annotations was conducted on the target protein set.
2.1.8 Verification of DEPs
2.1.8.1 Parallel reaction monitoring (PRM) verification
A sample of approximately 100 μg of protein was prepared by adding DTT to achieve a final concentration of 100 mM. The mixture was boiled in a water bath for 15 min and subsequently cooled to room temperature. Next, 200 μL of UA buffer (8 M Urea, 150 mM Tris-HCl, pH 8.0) was added, and the solution was mixed before being transferred to a 10 kDa ultrafiltration tube. Centrifugation was performed at 4°C and 14,000 × g for 30 min. An additional 200 μL of UA buffer was added, and centrifugation was repeated under the same conditions, after which the filtrate was discarded. Subsequently, 100 μL of iodoacetamide [IAA, (50 mM IAA in UA)] was added, shaken at 600 rpm for 1 min, and protected from light at room temperature for 30 min, followed by centrifugation at 4°C and 14,000 × g for 20 min. This washing step was repeated three times with the addition of 100 μL of UA buffer and centrifugation at 4°C and 14,000 × g for 20 min. Following this, 100 μL of NH4HCO3 buffer (50 mM) was added, and centrifugation was conducted at 4°C and 14,000 × g for 20 min, repeated twice. Next, 40 μL of NH4HCO3 buffer (containing Trypsin at an enzyme-to-substrate ratio of 1:50) was added, shaken at 600 rpm for 1 min, and incubated at 37°C for 16 h. After incubation, a new collector tube was used, and the sample was centrifuged at 4°C and 14,000 × g for 15 min. The filtrate was collected after a subsequent centrifugation at 4°C and 14,000 × g for 30 min, with an additional 40 μL of NH4HCO3 buffer (50 mM) added. The peptides resulting from enzymatic hydrolysis were desalted and freeze-dried, then redissolved in 0.1% FA, with the peptide concentration determined by OD280. The peptide information suitable for PRM analysis was imported into the software Xcalibur for PRM method setup. Approximately 1 μg of peptides from each sample was mixed with 20 fmol of standard peptide (PRTC: ELGQSGVDTYLQTK) for detection. The samples were separated using high-performance liquid chromatography and analyzed by PRM mass spectrometry with a Q-Exactive HF mass spectrometer (Thermo Scientific). PRM-related proteins are shown in Table 1.
Table 1. PRM quantitative protein.
2.1.8.2 Real-time fluorescent quantitative PCR
RT-qPCR technology was employed for further functional verification analysis at the transcriptional level. Total RNA was extracted from the control group and the treatment group using RNAsimple Total RNA Kit [DP419, Tiangen Biochemical Technology (Beijing) Co., Ltd], and all RNA samples were reverse transcribed into cDNA using the Hifair AdvanceFast One-step RT-gDNA Digestion SuperMix for qPCR. Primers for each candidate gene were designed using NCBI online software. The mycelium cDNA served as the template in conjunction with the Hieff UNICON Advanced qPCR SYBY Master Mix reagent, with Actin utilized as the internal reference gene. Quantitative analysis was conducted using a fluorescence quantitative PCR instrument, following a reaction procedure of 95°C for 30 s, 95°C for 10 s, and 60°C for 30 s, repeated for 40 cycles. The specificity of the amplification products was assessed based on the dissolution curve, with three replicates for each sample. The relative expression of the target gene was calculated utilizing the 2–ΔΔCt method. RT-qPCR protein primer sequences are shown in Table 2.

Table 2. RT-qPCR primer sequence.
2.2 Data analysis
All data were analyzed by one-way ANOVA using SPSS 22.0, and the data were plotted by Origin 2021.
3 Results
3.1 Determination of EC50
Different concentrations of PGA extracts (1.0, 2.0, 4.0, 8.0, and 16.0 mg/mL) exhibited varying inhibitory effects on F. solani. As the concentration of PGA treatment increased, the colony diameter progressively decreased (Figure 1), with significant differences observed compared to the control group (P < 0.05). The inhibition rate of PGA against F. solani increased in a dose-dependent manner, reaching a maximum of 100% at a concentration of 16.0 mg/mL. The virulence regression equation was expressed as y = 0.9385x–0.0511, with a correlation coefficient of R2 = 0.9407, indicating a strong linear relationship among the variables in the regression equation and confirming the feasibility of the test. When the value of y is 50%, the half-effect concentration (EC50) was determined to be 3.87 mg/mL (Table 3).
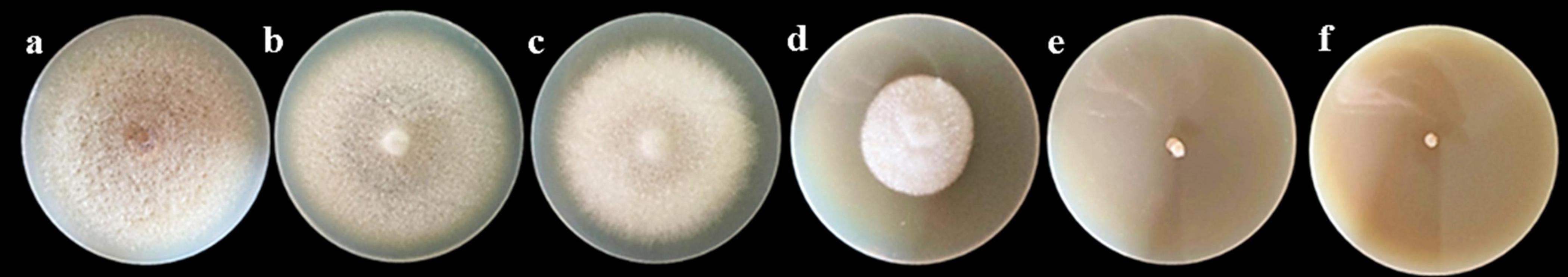
Figure 1. Illustrates the effects of various concentrations of PGA on the growth of F. solani colonies. The concentrations are represented as follows: (A) Control (CK), (B) 1.0 mg/mL, (C) 2.0 mg/mL, (D) 4.0 mg/mL, (E) 8.0 mg/mL, and (F) 16.0 mg/mL.
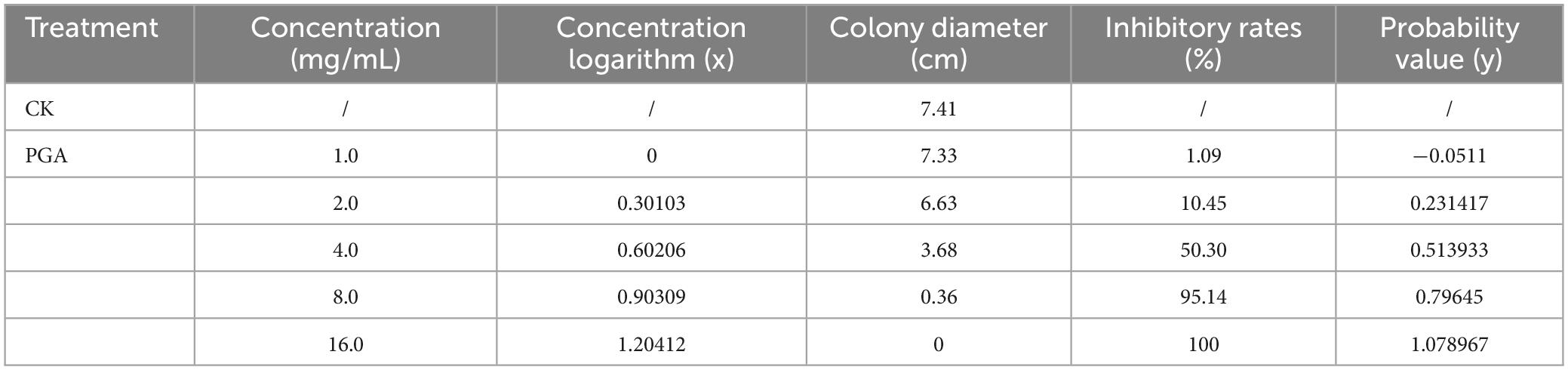
Table 3. Determination of antifungal activity of different concentrations of PGA against F. solani.
3.2 Effects of PGA on the surface morphology of pathogen hyphae
The SEM results indicated that the mycelium of the control group exhibited a uniform thickness and lacked any noticeable defects, while the spore surface morphology appeared normal (Figure 2A). In contrast, following PGA treatment, the mycelium of F. solani displayed signs of swelling and curling, with the spore surface showing pronounced depressions (Figure 2B). These observations suggest that PGA inhibits the normal energy metabolism of F. solani. The energy supply within the hyphae and spore cells is inadequate to fulfill the typical growth and differentiation requirements of these cells. Consequently, their adaptability to environmental changes diminishes, making it challenging for them to adjust their morphology in a timely manner. This results in the development of abnormal hyphae and spore morphology.

Figure 2. Scanning electron micrograph of mycelium morphology. (A) Sterile water treatment control, (B) PG A treatment. (a) hyphae, (b) spore.
3.3 Protein quantification and analysis
By analyzing the expression changes of DEP after PGA treatment, we can deeply understand the functions and interactions of these proteins in F. Solani, and provide new perspectives and ideas for their bacteriostatic mechanism. The protein molecular weight distribution in both the PGA treatment group and the control group was comparable, exhibiting clear and evenly distributed protein bands with no significant diffusion observed (Figure 3). These results indicated that the samples from each group demonstrated good parallelism, maintained protein integrity, and showed no evident degradation. This suggests that the samples are suitable for future proteomic sequencing, protein identification and DEPs analysis. Additionally, the results revealed that, compared to the control group, 1,083 DEPs were up-regulated and 1,329 DEPs were down-regulated following PGA treatment (Figure 4). By analyzing the role of DEPs in metabolic pathway, we can reveal the abnormal changes of metabolic pathway after PGA treatment.
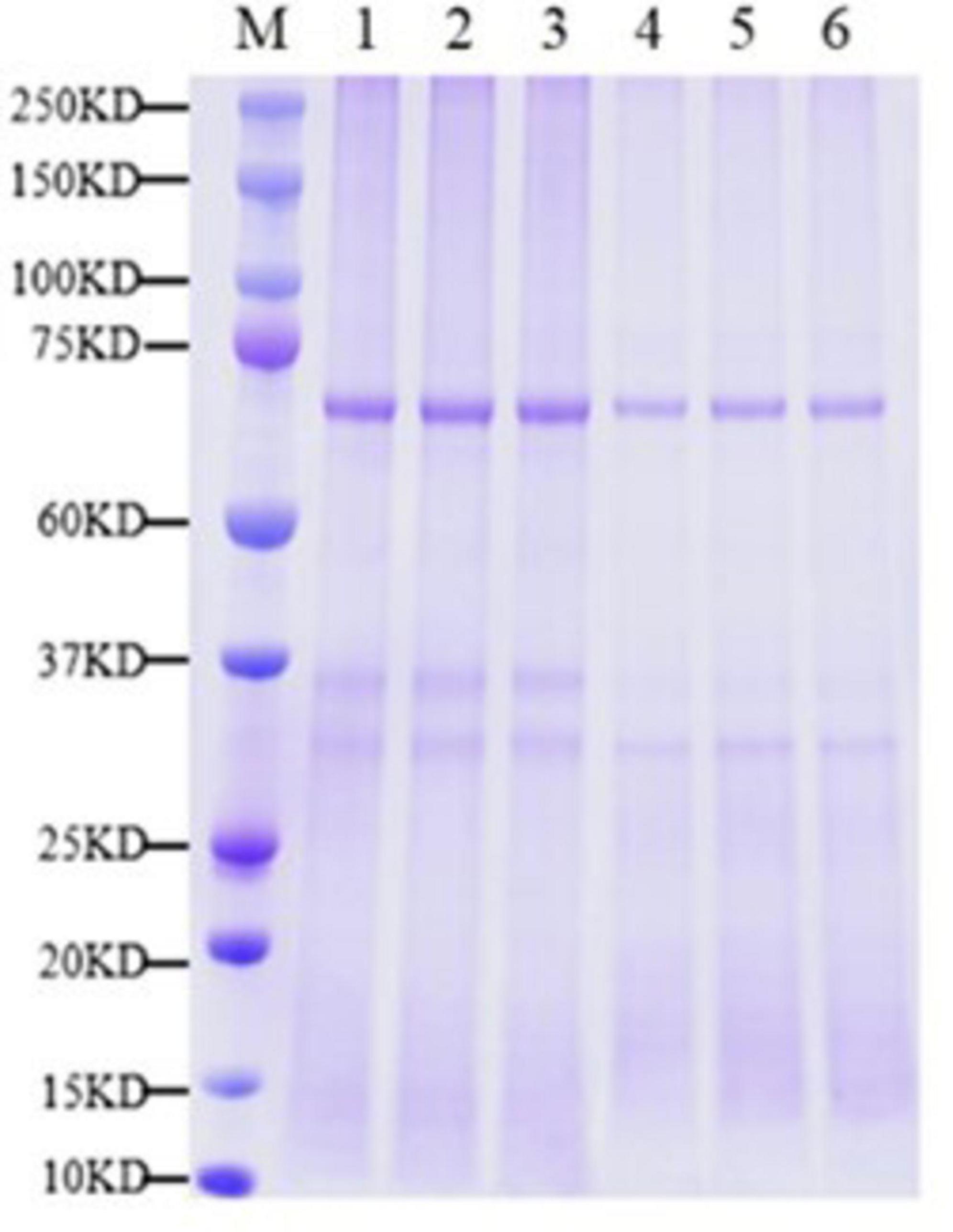
Figure 3. Sample SDS-PAGE. M represents marker; 1–3 were PGA treatment; 4–6 for CK. Each treatment had three biological replicates.
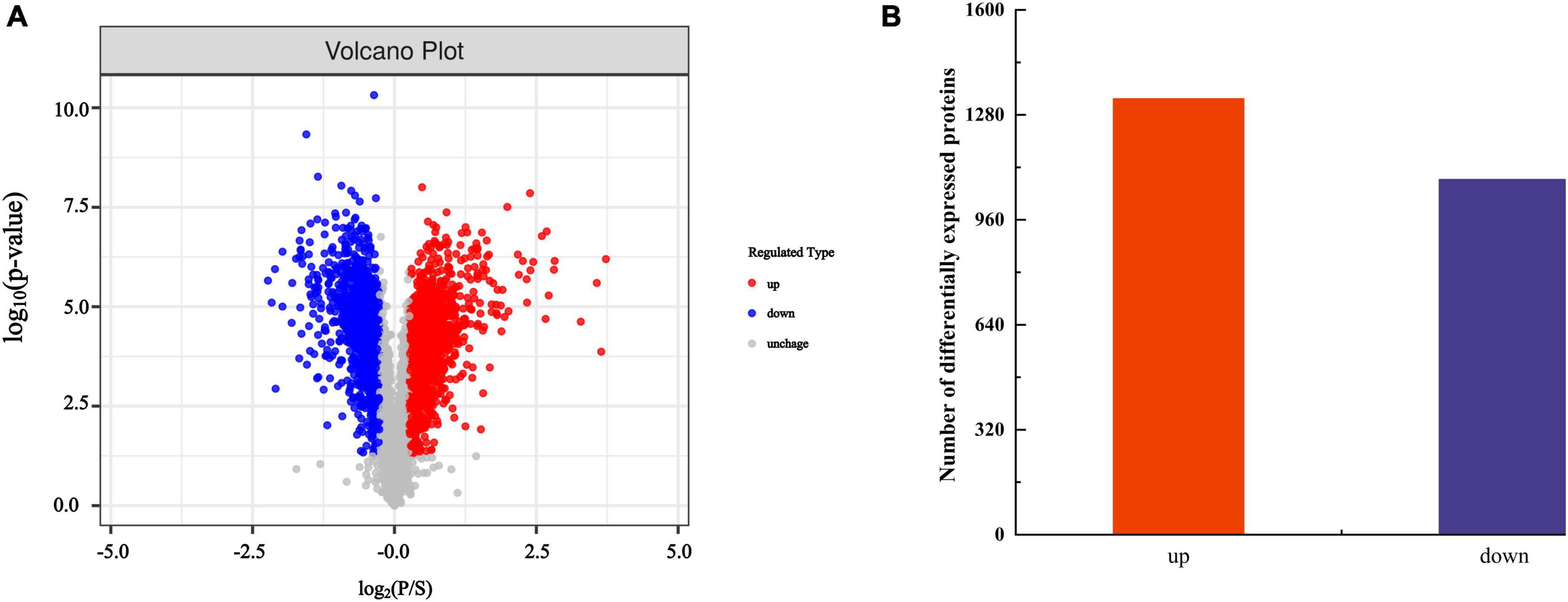
Figure 4. Statistics of DEPs. (A) The volcano map comparing PGA and CK shows red dots representing up-regulated DEPs and blue dots indicating down-regulated DEPs, while gray dots depict proteins with no differential expression changes. (B) Summary statistics of differentially expressed proteins.
3.4 GO functional enrichment analysis of DEPs
GO annotation results indicated that the DEPs were predominantly enriched in biological processes (BP), molecular functions (MF), and cellular components (CC). The biological processes primarily involved pyridine nucleotide metabolism, nicotinamide nucleotide metabolism, and the metabolism of pyridine-containing compounds (Figure 5). These GO results suggest that DEPs are significantly enriched in various nucleotide metabolism biological processes, which may disrupt the energy metabolism rate of F. solani, affect its energy balance and stability, and hinder its ability to maintain normal cellular function.
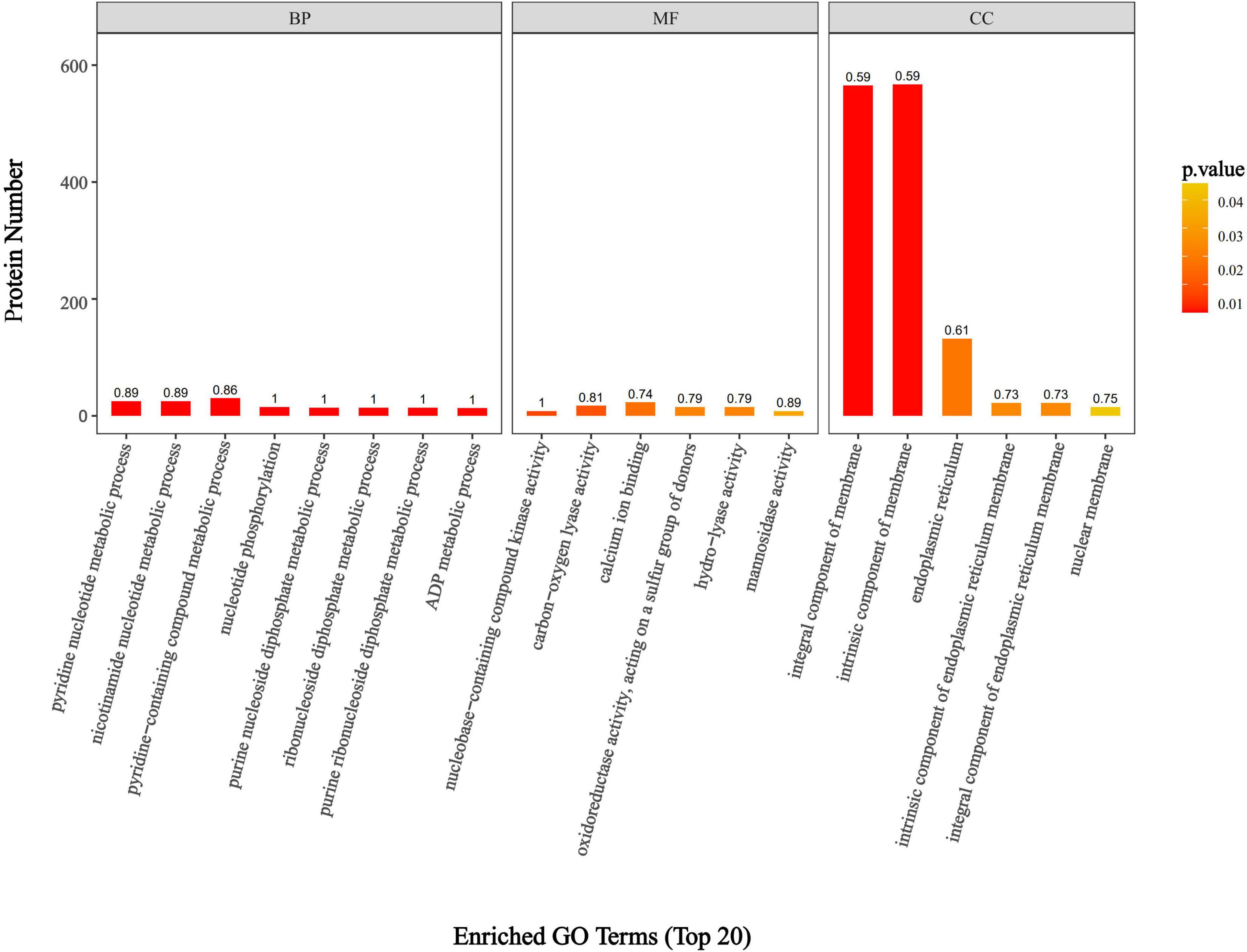
Figure 5. Enrichment statistics of DEPs in PGA vs. CK group.
3.5 KEGG enrichment analysis of DEPs
To elucidate the overall characteristics of metabolic pathway enrichment among all DEPs, we evaluated the significance level of protein enrichment within the KEGG metabolic pathways. The analysis revealed a total of 195 pathways, among which several key pathways exhibited substantial differential protein enrichment, including the mutual transformation of pentose and glucuronic acid, propionic acid metabolism, various types of N-glycan biosynthesis, the pentose phosphate pathway, and carbon fixation in photosynthetic organisms (Figure 6A). Notably, the enrichment of differential proteins was most pronounced in three pathways: the mutual conversion of pentose and glucuronic acid, propionic acid metabolism, and various types of N-glycan biosynthesis, all of which play critical roles in energy metabolism.
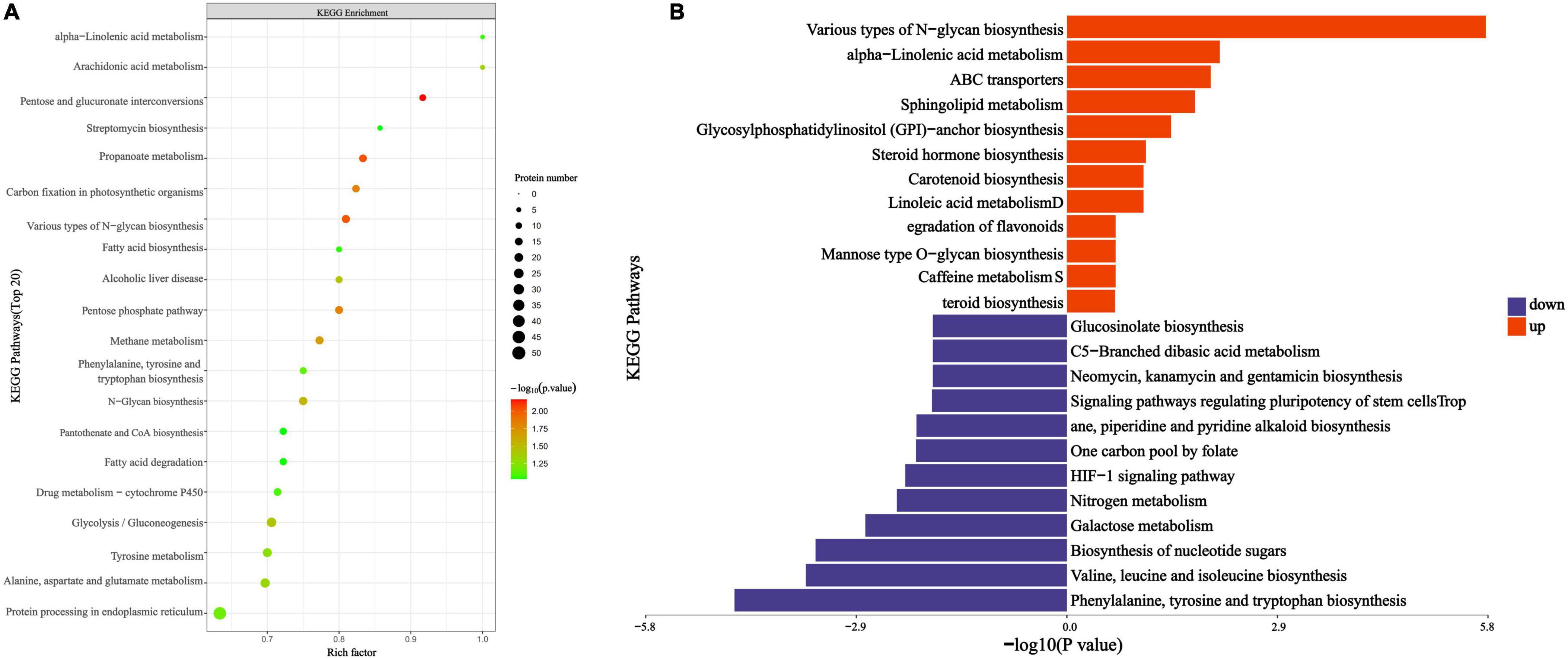
Figure 6. PGA vs. CK KEGG pathway enrichment map. (A) Bubble color indicates the significance of the enriched KEGG pathway. (B) Pathway categories enriched by ordinate KEGG, the statistical results of differential proteins under each KEGG pathway in abscissa; among them, red represents up-regulation and blue represents down-regulation.
Following KEGG metabolic pathway annotation, we analyzed the DEPs that were up-regulated and down-regulated, with the results illustrated in Figure 6B. The up-regulated DEPs were primarily enriched in pathways associated with the biosynthesis of phenylalanine, tyrosine, and tryptophan; the biosynthesis of valine, leucine, and isoleucine; nucleotide sugar biosynthesis; galactose metabolism; and nitrogen metabolism, among others. Conversely, the down-regulated DEPs were mainly enriched in various types of N-glycan biosynthesis and α-linolenic acid metabolism. Our analysis indicates that PGA treatment interferes with the energy metabolism pathways of F. solani, including lactose metabolism, nitrogen metabolism, and various types of N-glycan biosynthesis pathways.
3.6 Pathway analysis related to energy metabolism
Three pathways related to energy metabolism were further examined, including the tricarboxylic acid cycle, glycolysis/gluconeogenesis, oxidative phosphorylation, and pyruvate metabolism, resulting in the identification of a total of 75 differential proteins (Figure 7). The number of down-regulated DEPs in these three pathways exceeded that of the up-regulated DEPs. The specific proteins are shown in Table 4.
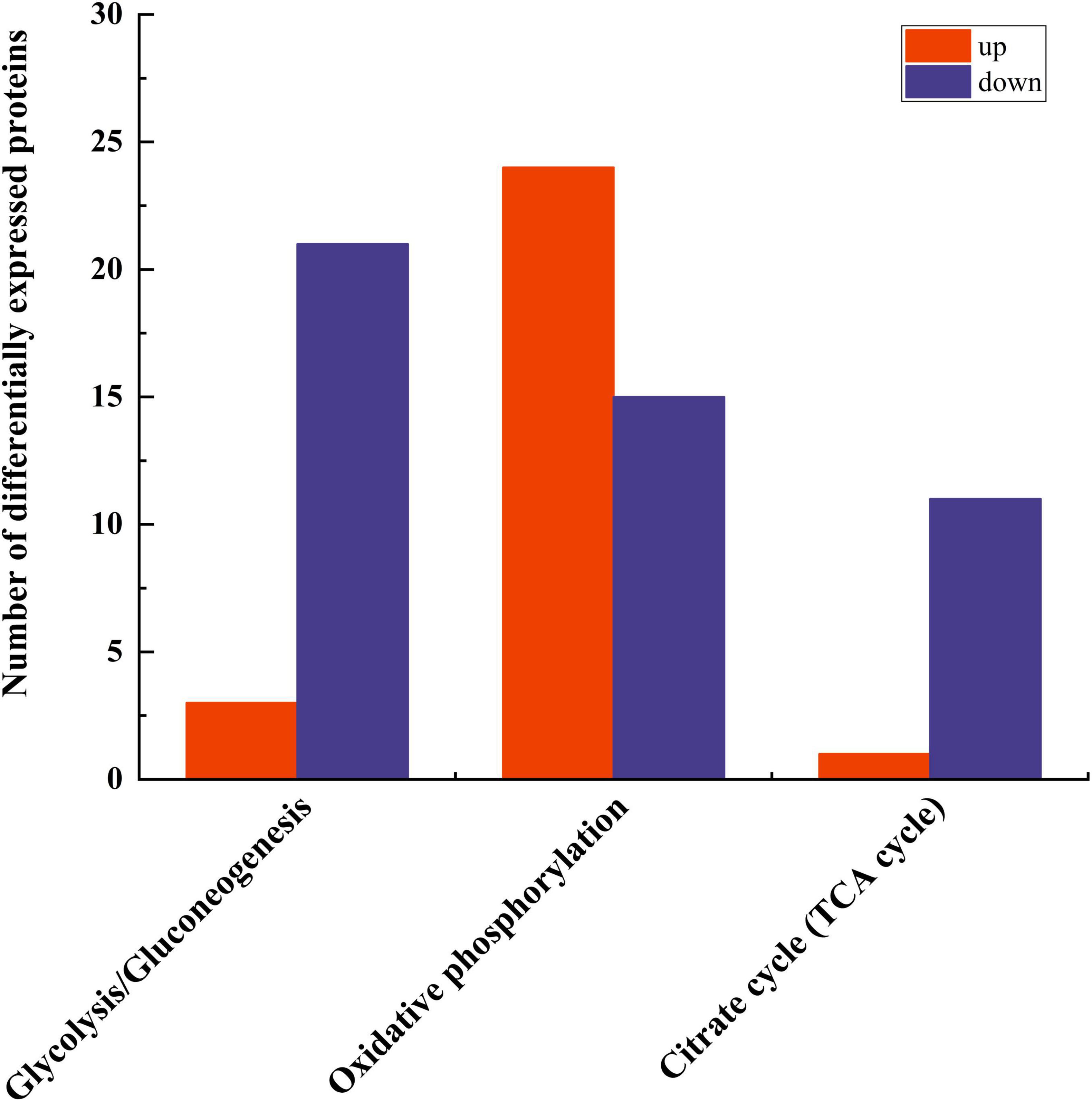
Figure 7. Differential protein statistics of pathways related to energy metabolism.
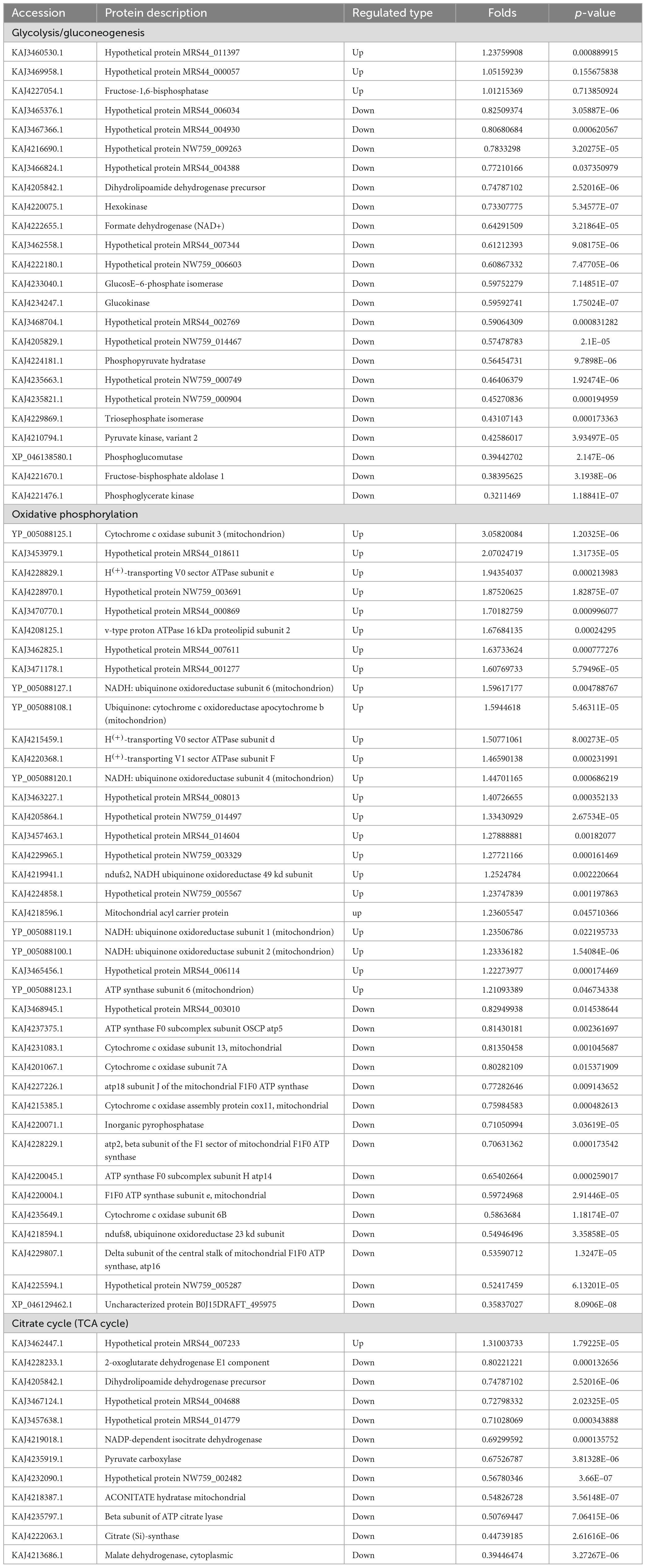
Table 4. Pathway enrichment analysis of differential protein pathway.
3.7 Glycolysis/gluconeogenesis
The glycolysis/gluconeogenesis pathway is essential for the breakdown and synthesis of sugars, with glycolysis a primary pathway for glucose breakdown. This study’s results indicate that following PGA treatment, 24 DEPs were identified within the glycolysis and gluconeogenesis pathways of F. solani, comprising 3 upregulated and 21 downregulated proteins (Figure 8). Specifically, the expression levels of phosphoglucomutase, fructose-bisphosphate aldolase 1, and phosphoglycerate kinase were significantly down-regulated by factors of 0.39, 0.38, and 0.32, respectively, while the expression of fructose-1,6-bisphosphatase was significantly up-regulated by a factor of 1.01. These findings suggest that PGA treatment has a significant impact on the activity of the glycolysis/gluconeogenesis pathway.
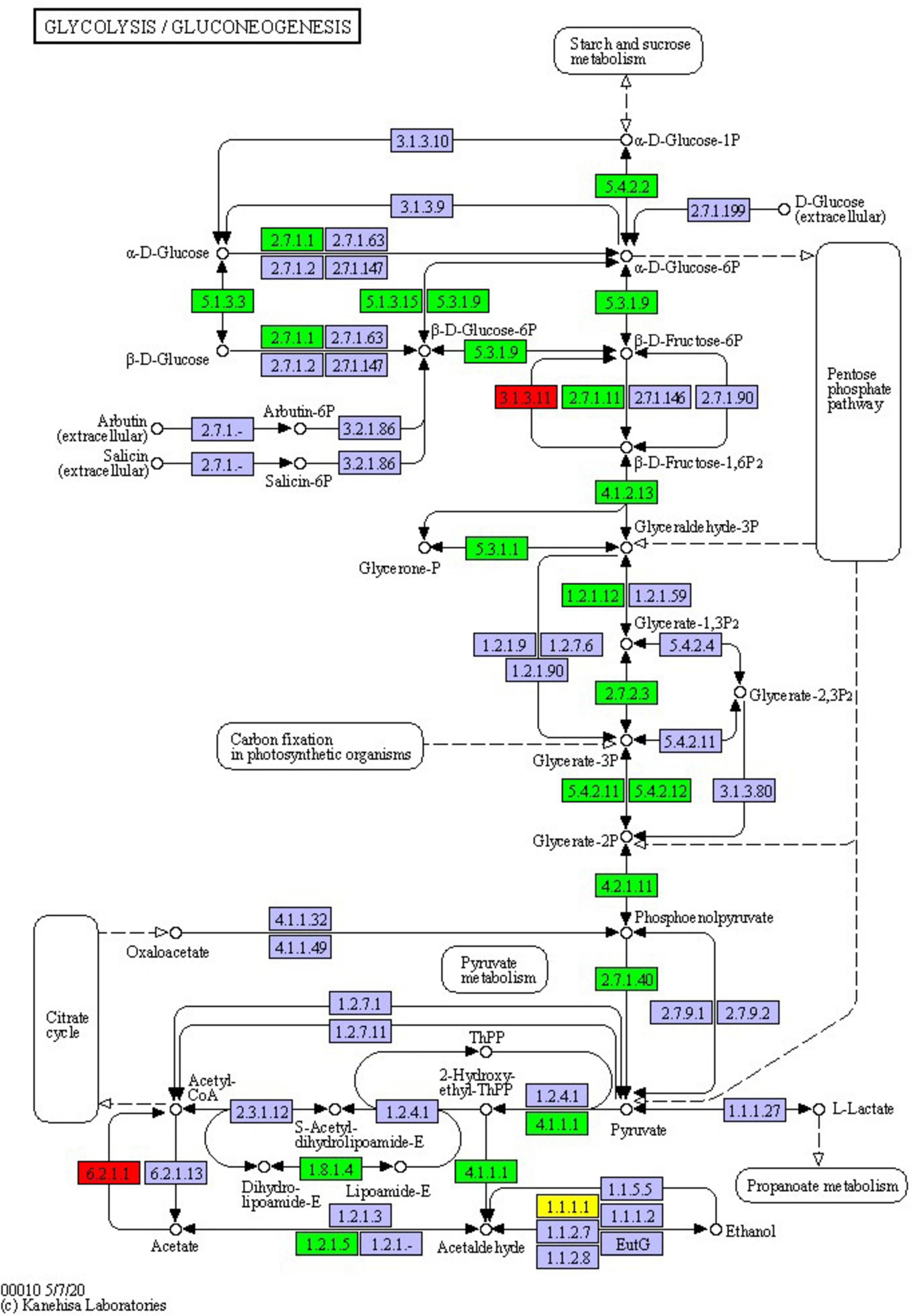
Figure 8. Changes in glycolysis/gluconeogenesis pathway of F. solani treated with PGA. Red and green proteins represent up-regulated and down-regulated protein expression levels.
3.8 Oxidative phosphorylation
Oxidative phosphorylation is the main way for fungi to obtain energy. Through electron transfer in the respiratory chain and the action of ATP synthase, the energy released during the oxidative decomposition of organic matter is converted into ATP which can be directly utilized by cells, providing a lot of energy for the growth, reproduction, metabolism and other life activities of fungi. The results of this study indicated that a total of 39 proteins were differentially expressed in the oxidative phosphorylation pathway of F. solani
following PGA treatment (Figure 9), with 24 proteins up-regulated and 15 proteins down-regulated. Notably, the expression of cytochrome c oxidase subunit 3 was significantly up-regulated by a factor of 3.06, while the ndufs8, ubiquinone oxidoreductase 23 kDa subunit, and delta subunit of the central stalk of mitochondrial F1F0 ATP synthase (atp16) were significantly down-regulated by factors of 0.55 and 0.53, respectively. These findings suggest that PGA treatment has a substantial impact on the oxidative phosphorylation pathway of F. solani.
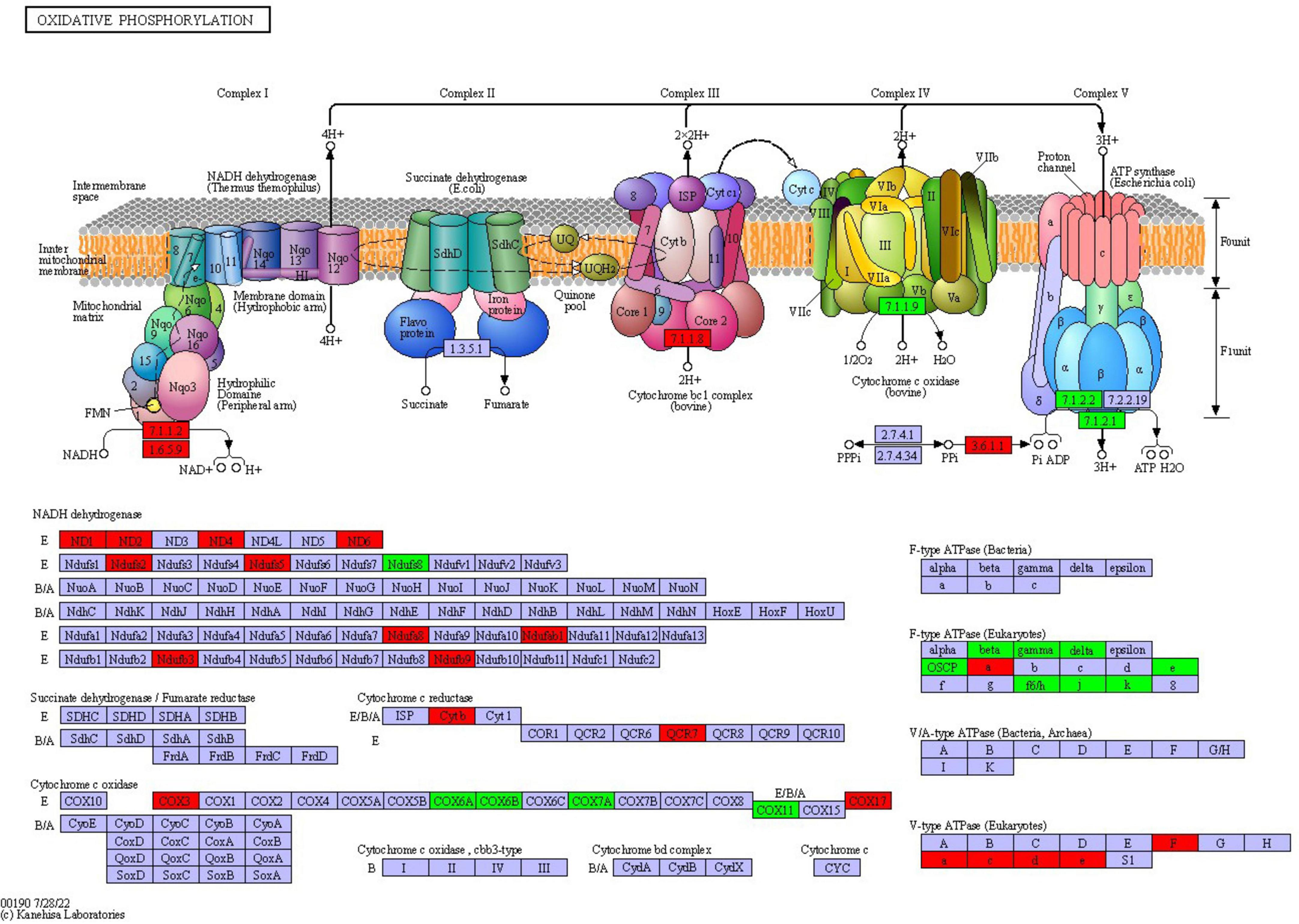
Figure 9. The changes of oxidative phosphorylation pathway of F. solani treated with PGA. Red and green proteins represent up-regulated and down-regulated protein expression levels.
3.9 TCA cycle
Most of the differentially expressed proteins (DEPs) in the tricarboxylic acid (TCA) cycle pathway were found to be up-regulated. This study revealed that a total of 12 proteins exhibited differential expression in the TCA cycle pathway of F. solani following PGA treatment, with 11 proteins being up-regulated and 1 down-regulated (Figure 10). These findings indicate that PGA treatment enhances the expression of the beta subunit of ATP citrate lyase and citrate synthase within the TCA cycle pathway, thereby influencing the overall TCA cycle.
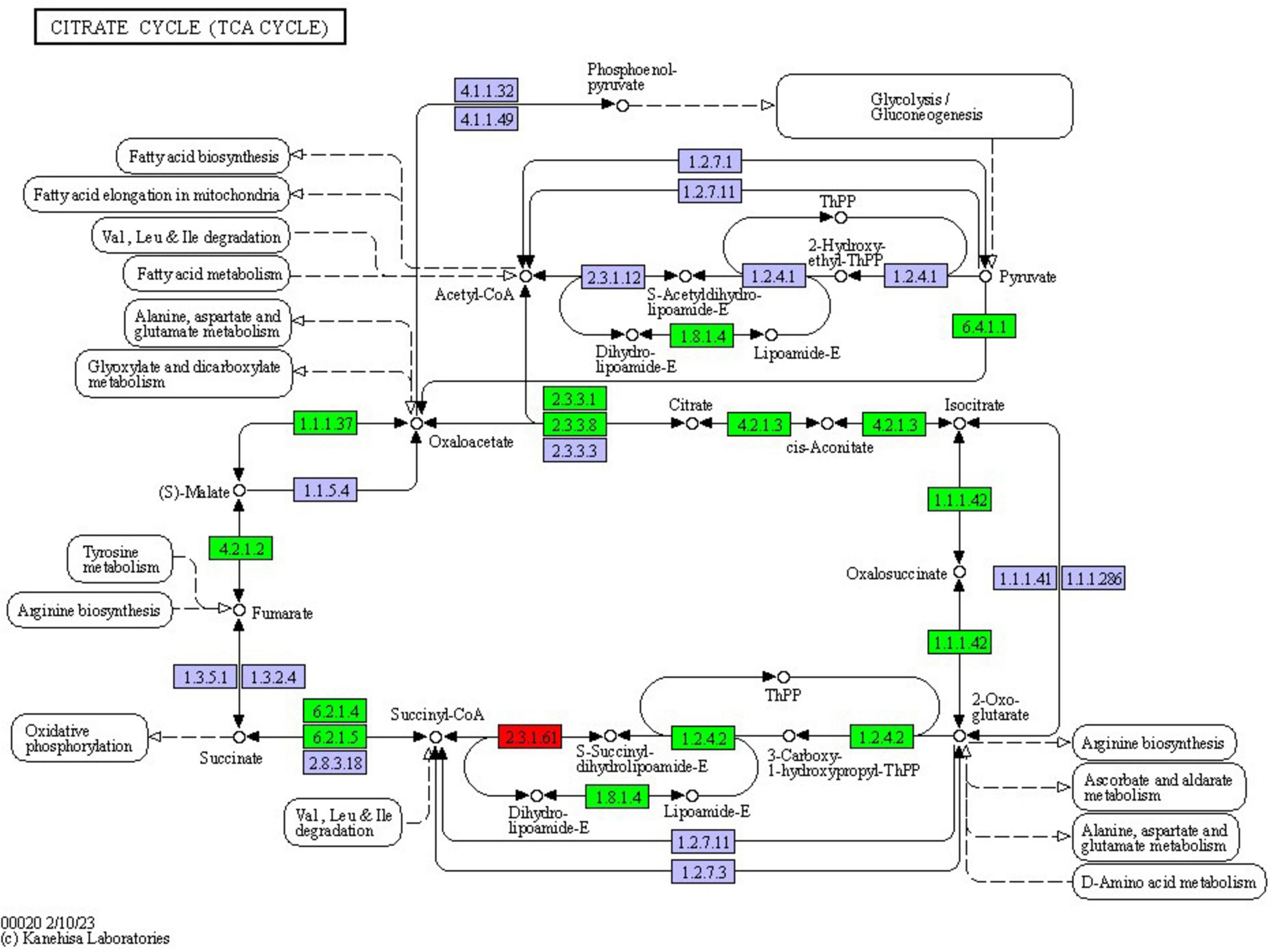
Figure 10. Changes in the tricarboxylic acid cycle pathway of F. solani treated with PGA. Red and green represent up-regulated and down-regulated protein expression levels.
3.10 PRM and PCR validation of differential proteins
According to the results of the pathway analysis related to energy metabolism, this study selected five DEPs for quantitative detection using PRM. The PRM quantitative results were compared with those obtained from TMT. The findings indicate that the quantitative results from PRM align with the overall trends observed in the TMT data; specifically, the down-regulated and up-regulated proteins identified through TMT also exhibited corresponding changes in PRM. Furthermore, the PRM results demonstrated more pronounced expression differences (Figure 11A), underscoring the stability and quantitative accuracy of both the PRM and TMT methodologies, thereby affirming the reliability of the TMT quantitative results.

Figure 11. Validation of DEPs. (A) PRM validation of 5 differential proteins. (B) RT-qPCR verification of 5 genes.
To further validate the accuracy of the experimental outcomes, five proteins associated with energy metabolism were selected for analysis, and reverse transcription quantitative polymerase chain reaction (RT-qPCR) was employed to determine whether the transcription levels of the genes encoding these proteins were consistent with the proteomics findings. The results revealed that the expression trends of the five genes were similar to those observed in the TMT quantitative results, although the expression magnitudes differed (Figure 11B). This suggests that the RT-qPCR analysis corroborated the reliability of the proteomics analysis.
4 Discussions
We employ mitochondrial TMT proteomics technology to elucidate the effects of PGA on F. solani from a molecular biology perspective, specifically focusing on mitochondrial proteomics and its antifungal mechanisms. The results showed that after PGA treatment, the mycelium of F. solani showed swelling and curling, and the spore surface was significantly depressed. PGA has adverse effects on the surface structure of hyphae and spores of F. solani. Furthermore, DEPs were predominantly enriched in pathways related to energy metabolism, including carbohydrate metabolism, membrane components, intrinsic membrane components, and the endoplasmic reticulum, among other cellular components. These findings suggest that PGA inhibits the growth of F. solani by modulating the expression of key proteins involved in processes such as cell membrane synthesis, mitochondrial membrane biosynthesis, and energy metabolism.
Natural metabolite PGA exhibits a wide range of biological activities (Nenaah, 2010), yet its antifungal mechanism remains notably complex. Currently, the antifungal mechanism of PGA against pathogenic fungi has not been fully elucidated. Mitochondria, as critical organelles in eukaryotic cells, play a key role in regulating energy metabolism, biosynthesis, and cell death processes, including apoptosis and programmed necrosis (Fu et al., 2020; Guarente, 2008). They are integral to various physiological processes such as the tricarboxylic acid cycle, oxidative phosphorylation, pyruvate metabolism, fatty acid biosynthesis, and calcium homeostasis (Schrepfer and Scorrano, 2016). Tea tree essential oil disrupts the energy metabolism pathway of Botrytis cinerea by inhibiting glycolysis, impairing the tricarboxylic acid (TCA) cycle, and inducing mitochondrial dysfunction, which subsequently affects cellular life activities (Xu et al., 2017). A proteomics study investigating the inhibitory effects of perilla essential oil on Aspergillus flavus revealed that the DEPs induced by perilla essential oil were primarily enriched in amino acid biosynthesis and metabolism, antioxidant metabolism, fatty acid metabolism, lipid metabolism, and energy metabolism (Hu et al., 2022). Previous research has demonstrated that the antifungal mechanisms of plant-derived extracts are associated with their ability to disrupt the energy metabolism of pathogenic fungi.
Our study found that the majority of DEPs associated with the glycolysis pathway were down-regulated in F. solani treated with PGA, with the exception of fructose-1,6-bisphosphatase, which was up-regulated. The initial step in the glycolytic pathway involves the downregulation of hexokinase expression, followed by the downregulation of ATP-dependent 6-phosphofructokinase (pfka). Subsequently, triose phosphate isomerase (TPIA) is down-regulated, which catalyzes the conversion of dihydroxyacetone phosphate into 3-glyceraldehyde phosphate. Additionally, 3-glyceraldehyde phosphate is down-regulated, affecting the catalysis of 1,3-diphosphate to produce 3-glyceraldehyde 3-phosphate dehydrogenase (gapA) and phosphoglycerate kinase (PGK). All these proteins exhibited a downward trend, indirectly inhibiting the production of 2-phosphoglycerate. The expression of pyruvate kinase is also downregulated at a later stage, thereby limiting pyruvate synthesis. These findings align with previous studies (Lomora et al., 2017; Rashid et al., 2004; Ryu et al., 2006). Furthermore, two key enzymes, phosphoglucomutase (PGM) (Chen et al., 2021) and phosphoglycerate kinase (Chen et al., 2024), were also observed to have down-regulated expression. These results suggest that the antifungal effect of PGA is linked to the down-regulated expression of various key enzymes in the glycolysis pathway. Therefore, we infer that PGA treatment inhibits the normal progression of both glycolysis and gluconeogenesis pathways, thereby disrupting the energy metabolism of F. solani.
Oxidative phosphorylation is a crucial metabolic pathway. Through the phosphorylation of the electron transport chain, the energy released from the dehydrogenation of metabolites is utilized to catalyze the synthesis of ATP, thereby providing essential energy for the organism (Dimroth et al., 2000). In this study, cytochrome c oxidase (Yoshikawa and Shimada, 2015) was identified as a DEP, which is a key enzyme involved in ATP generation within the respiratory electron transport chain. Following PGA treatment, the expression of this enzyme exhibited a significant downward trend. Similar findings were observed in the ATPases detected, which catalyze the decomposition of ATP into ADP and phosphate ions (Kühlbrandt and Davies, 2016). Due to the down-regulation of ATPase and cytochrome c oxidase expression, even the up-regulation of NADH dehydrogenase expression is insufficient to meet ATP demands, thereby disrupting the carbohydrate, protein, and fat metabolism pathways associated with the tricarboxylic acid cycle. Consequently, it can be inferred that the antifungal activity of PGA against F. solani may be linked to the inhibition of ATP synthesis and normal energy metabolism. Additionally, oxidative phosphorylation is closely related to the TCA cycle. Our findings indicate that treatment of F. solani with PGA resulted in the down-regulation of pyruvate carboxylase (ALDH), which slowed the supply of oxaloacetate and disrupted the orderly progression of energy metabolism in F. solani due to the inhibition of the glycolytic pathway. Additionally, we observed that citric acid synthase is also down-regulated within the tricarboxylic acid cycle pathway, limiting the synthesis of citric acid from oxaloacetate and acetyl-CoA, thereby playing a crucial role in energy metabolism. Notably, the down-regulation of isocitrate lyase hampers the conversion of isocitrate into glyoxylic acid and succinic acid, further impeding the orderly progression of the tricarboxylic acid cycle. Glyoxylic acid, under the catalysis of malic acid synthase, reduced the content of malic acid, failing to provide a continuous and effective energy supply for PGA-treated Aspergillus flavus. Zhao et al. (2018) found that the expression of malate dehydrogenase was up-regulated after treatment with shenqinmycin in Phoma sp., and the activity of Phoma was inhibited by the increased activity of this enzyme. In conclusion, the inhibitory effect of PGA on F. solani may be linked to the disruption of its normal energy metabolism.
The trend in enzyme expression observed in RT-qPCR results diverges from that seen in TMT protein quantification. This discrepancy may account for the differences in transcriptional and translational levels of the target gene, with post-translational modification processes also influencing the expression of the final protein.
In summary, morphological and bioinformatics analyses indicate that PGA inhibits the growth of F. solani by modulating the expression of key enzymes involved in various energy metabolism pathways. We have elucidated the antifungal mechanism of PGA on F. solani in terms of energy metabolism from a proteomics perspective, thus providing a theoretical foundation for the application of PGA in plant disease control. Further investigations will be conducted to determine whether additional antifungal mechanisms exist.
5 Conclusion
A total of 2,412 differential proteins were screened by TMT proteomics, of which 1,083 were significantly up-regulated and 1,329 were significantly down-regulated. GO function analysis showed that the DEPs were mainly involved in integral component of membrane, intrinsic component of membrane, pyridine-containing compound metabolic process, carbon-oxygen lyase activity, integral component of membrane, intrinsic component of membrane, and endoplasmic reticulum. The results of KEGG enrichment analysis showed. DEPs were mainly enriched in pentose and glucuronate interconversions, propanoate metabolism, various types of N-glycan biosynthesis, pentose phosphate pathway, carbon fixation in photosynthetic organisms and other important pathways. The quantitative results of PRM were consistent with the overall trend of TMT quantitative results, and the results of PRM verification were more significant in expression, indicating that TMT quantitative results were reliable. The results of RT-qPCR showed that the expression trend of 5 genes was consistent with the results of TMT quantification. Therefore, the results of RT-qPCR analysis supported the reliability of proteomics analysis results. PGA inhibited the growth of F. solani mitochondria by affecting its energy metabolism.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: ProteomeXchange Consortium database (https://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD058927) under the accession number, PXD058927.
Author contributions
RL: Data curation, Investigation, Writing – original draft, Writing – review and editing. BW: Supervision, Validation, Writing – review and editing. WC: Supervision, Writing – review and editing. CZ: Investigation, Writing – review and editing. NL: Data curation, Methodology, Writing – review and editing. YW: Methodology, Writing – review and editing. YY: Data curation, Methodology, Writing – review and editing. YS: Methodology, Writing – original draft. JH: Conceptualization, Funding acquisition, Supervision, Visualization, Writing – review and editing.
Funding
The authors declare that financial support was received for the research, authorship, and/or publication of this article. This research received financial support from the National Natural Science Foundation of China (Grant No. 32060341).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
References
Bai, L., Li, X., Cao, Y., Song, Z., Ma, K., Fan, Y., et al. (2020). Fusarium culmorum and Fusarium equiseti causing root rot disease on Lycium barbarum (goji berry) in China. Plant Dis. 104:3066. doi: 10.1094/PDIS-11-19-2313-PDN
Chen, D., Shi, C., Xu, W., Rong, Q., and Wu, Q. (2024). Regulation of phase separation and antiviral activity of cactin by glycolytic enzyme PGK via phosphorylation in drosophila. mBio 15:e0137823. doi: 10.1128/mbio.01378-23
Chen, L., Zhao, X., He, Y., and Yang, H. (2021). Cloning, purification and characterisation of cytosolic fructose-1,6-bisphosphatase from mung bean (Vigna radiata). Food Chem. 347:128973. doi: 10.1016/j.foodchem.2020.128973
Dimroth, P., Kaim, G., and Matthey, U. (2000). Crucial role of the membrane potential for ATP synthesis by F1F0 ATP synthases. J. Exp. Biol. 203, 51–59. doi: 10.1242/jeb.203.1.51
Ding, D., Zhao, J., Jing, H., Hou, C., Li, N., Zhang, C., et al. (2023). Potato glycoalkaloids affect the respiration and reactive oxygen species metabolism of Fusarium solani. Mic. China 50, 2937–2949. doi: 10.13344/j.microbiol.china.221000
Duo, T., He, J., Chen, W., Zhang, J., Fen, S., Ma, H., et al. (2017). Antifungal activity of potato glycoalkaloid and its effect on fungi cell membrane permeability. J. Gansu Agric. Univer. 52, 50–54. doi: 10.13432/j.cnki.jgsau.2017.05.008
El-Saadony, M. T., Saad, A. M., Najjar, A. A., Alzahrani, S. O., Alkhatib, F. M., Shafi, M. E., et al. (2021). The use of biological selenium nanoparticles to suppress Triticum aestivum L. crown and root rot diseases induced by Fusarium species and improve yield under drought and heat stress. Saudi J. Biol. Sci. 28, 4461–4471. doi: 10.1016/j.sjbs.2021.04.043
Fu, Y., Tigano, M., and Sfeir, A. (2020). Safeguarding mitochondrial genomes in higher eukaryotes. Nat. Struct. Mol. Biol. 27, 687–695. doi: 10.1038/s41594-020-0474-9
Guarente, L. (2008). Mitochondria-a nexus for aging, calorie restriction, and sirtuins? Cell 132, 171–176. doi: 10.1016/j.cell.2008.01.007
He, J., Zhang, X., Wang, Q., Li, N., Ding, D., and Wang, B. (2023). Optimization of the fermentation conditions of Metarhizium robertsii and its biological control of wolfberry root rot disease. Microorganisms 11:2380. doi: 10.3390/microorganisms11102380
Hu, Z., Lu, C., Zhang, Y., Tong, W., Du, L., and Liu, F. (2022). Proteomic analysis of Aspergillus flavus reveals the antifungal action of Perilla frutescens essential oil by interfering with energy metabolism and defense function. Lwt-food Sci. Technol. 154:112660. doi: 10.1016/j.lwt.2021.112660
Huang, X., Zhang, N., Yong, X., Yang, X., and Shen, Q. (2012). Biocontrol of Rhizoctonia solani damping-off disease in cucumber with Bacillus pumilus SQR-N43. Microbiol. Res. 167, 135–143. doi: 10.1016/j.micres.2011.06.002
Joshi, J., Yao, L., Charkowski, A., and Heuberger, A. (2021). Metabolites from wild potato inhibit virulence factors of the soft rot and blackleg pathogen Pectobacterium brasiliense. Mol. Plant Microbe 34, 100–109. doi: 10.1094/MPMI-08-20-0224-R
Koffi, G. Y., Remaud-Simeon, M. A., Due, E., and Combes, D. (2017). Isolation and chemoenzymatic treatment of glycoalkaloids from green, sprouting and rotting Solanum tuberosum potatoes for solanidine recovery. Food Chem. 220, 257–265. doi: 10.1016/j.foodchem.2016.10.014
Kühlbrandt, W., and Davies, K. M. (2016). Rotary ATPases: A new twist to an ancient machine. Trends Biochem. Sci. 41, 106–116. doi: 10.1016/j.tibs.2015.10.006
Li, D., Sun, L., Yang, Y., Wang, Z., Yang, X., and Guo, Y. (2018). Preventive and therapeutic effects of pigment and polysaccharides in Lycium barbarum on alcohol-induced fatty liver disease in mice. CyTA J. Food 16, 938–949. doi: 10.1080/19476337.2018.1512530
Li, H., Li, M., Fang, Y., and Qin, S. (2022). Antifungal activity of potato glycoalkaloids and its potential to control severity of dry rot caused by Fusarium sulphureum. Crop Sci. 63, 801–811. doi: 10.1002/csc2.20874
Liu, P., Zhou, W., Xu, W., Peng, Y., Yan, Y., Lu, L., et al. (2022). The main anthocyanin monomer from Lycium ruthenicum murray fruit mediates obesity via modulating the gut microbiota and improving the intestinal barrier. Foods 11:98. doi: 10.3390/foods11010098
Lomora, M., Gunkel-Grabole, G., Mantri, S., and Palivan, C. G. (2017). Bio-catalytic nanocompartments for in situ production of glucose-6-phosphate. Chem. Commun. 53, 10148–10151. doi: 10.1039/C7CC04739H
Nenaah, G. E. (2010). Toxic and antifeedant activities of potato glycoalkaloids against Trogoderma granarium (Coleoptera: Dermestidae). J. Stored Prod. Res. 47, 185–190. doi: 10.1016/j.jspr.2010.11.003
Rashid, N., Kanai, T., Atomi, H., and Imanaka, T. (2004). Among multiple phosphomannomutase gene orthologues, only one gene encodes a protein with phosphoglucomutase and phosphomannomutase activities in Thermococcus kodakaraensis. J. Bacteriol. 186, 6070–6076. doi: 10.1128/jb.186.18.6070-6076.2004
Ryu, Y., Butler, M. J., Chater, K. F., and Lee, K. J. (2006). Engineering of primary carbohydrate metabolism for increased production of actinorhodin in Streptomyces coelicolor. Appl. Environ. Microb. 72, 7132–7139. doi: 10.1128/AEM.01308-06
Schrepfer, E., and Scorrano, L. (2016). Mitofusins, from mitochondria to metabolism. Mol. Cell 61, 683–694. doi: 10.1016/j.molcel.2016.02.022
Song, F., Li, C., Zhang, N., He, X., Yang, H., Yan, Z., et al. (2023). A novel endophytic bacterial strain improves potato storage characteristics by degrading glycoalkaloids and regulating microbiota. Postharvest Biol. Tec. 196:112176. doi: 10.1016/j.postharvbio.2022.112176
Sun, P., Cao, B., and Han, Y. (2014). Study on antimicrobial activities of α-Solanine against Botrytis cinerea in vitro. Storage Process 14, 12–15. doi: 10.3969/j.issn.1009-6221.2014.02.003
Vujanovic, V., Kim, S. H., Lahlali, R., and Karunakaran, C. (2019). Spectroscopy and SEM imaging reveal endosymbiont-dependent components changes in germinating kernel through direct and indirect coleorhiza-fungus interactions under stress. Sci. Rep. 9:1665. doi: 10.1038/s41598-018-36621-8
Wiśniewski, J. R., Zougman, A., Nagaraj, N., and Mann, M. (2009). Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362. doi: 10.1038/nmeth.1322
Xu, J., Shao, X., Wei, Y., Xu, F., and Wang, H. (2017). iTRAQ proteomic analysis reveals that metabolic pathways involving energy metabolism are affected by tea tree oil in Botrytis cinerea. Front. Microbiol. 8:1989. doi: 10.3389/fmicb.2017.01989
Xu, N., Lu, H., Yi, X., Peng, S., Huang, X., Zhang, Y., et al. (2023). Potential of alpha-(α)-Solanine as a natural inhibitor of fungus causing leaf spot disease in strawberry. Life 13:450. doi: 10.3390/life13020450
Yoshikawa, S., and Shimada, A. (2015). Reaction mechanism of cytochrome c oxidase. Chem. Rev. 115, 1936–1989. doi: 10.1021/cr500266a
Zeng, W., Chen, L., Xiao, Z., Li, Y., Ma, J., Ding, J., et al. (2023). Comparative study on the structural properties and bioactivities of three different molecular weights of Lycium barbarum polysaccharides. Molecules 28:701. doi: 10.3390/molecules28020701
Zhan, J., He, F., Cai, H., Wu, M., Xiao, Y., Xiang, F., et al. (2021). Composition and antifungal mechanism of essential oil from Chrysanthemum morifolium cv. Fubaiju. J. Funct. Foods 87:104746. doi: 10.1016/j.jff.2021.104746
Zhang, C., Chen, W., Wang, B., Wang, Y., Li, N., Li, R., et al. (2024). Potato glycoside alkaloids exhibit antifungal activity by regulating the tricarboxylic acid cycle pathway of Fusarium solani. Front. Microbiol. 15:e1390269. doi: 10.3389/fmicb.2024.1390269
Zhang, C., Ding, D., Wang, B., Wang, Y., Li, N., Li, R., et al. (2023). Effect of potato glycoside alkaloids on energy metabolism of Fusarium solani. J. Fungi 9:777. doi: 10.3390/jof9070777
Zhao, X., Chen, Z., Yu, L., Hu, D., and Song, B. (2018). Investigating the antifungal activity and mechanism of a microbial pesticide Shenqinmycin against Phoma sp. Pestic. Biochem. Phys. 147, 46–50. doi: 10.1016/j.pestbp.2017.08.014
Keywords: Fusarium solani, TMT technology, potato glycoside alkaloids, energy metabolism, mitochondrion
Citation: Li R, Wang B, Chen W, Zhang C, Li N, Wang Y, Yan Y, Sun Y and He J (2025) Effect of potato glycoside alkaloids on mitochondria energy metabolism of Fusarium solani, the root rot pathogen of Lycium barbarum. Front. Microbiol. 15:1512027. doi: 10.3389/fmicb.2024.1512027
Received: 16 October 2024; Accepted: 18 December 2024;
Published: 29 January 2025.
Edited by:
Marie-Joelle Virolle, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Antonio Caporusso, University of Bari Aldo Moro, ItalyPradeep Kumar Singh, Eternal University, India
Copyright © 2025 Li, Wang, Chen, Zhang, Li, Wang, Yan, Sun and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing He, aGVqaW5nMjY4QGFsaXl1bi5jb20=