 Gabriele Arcari
Gabriele Arcari Alice De Francesco
Alice De Francesco Riccardo Polani3
Riccardo Polani3 Valerio Capitani
Valerio Capitani
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 31 October 2024
Sec. Infectious Agents and Disease
Volume 15 - 2024 | https://doi.org/10.3389/fmicb.2024.1493621
This article is part of the Research Topic Insights in Infectious Agents and Disease: 2023/2024 View all 34 articles
Introduction: The Providencia genus is assuming greater clinical relevance among infections caused by Enterobacterales also because of its intrinsic and acquired resistance to last-resort antibiotics. However, despite having been known and studied for over 50 years, genomics and taxonomy of the Providencia genus are currently undergoing a deep rearrangement. In this study we aim to outline and characterized the P. stuartii species.
Methods: We retrieved from the GenBank database all genomes labelled as Providencia and performed a comprehensive genome-based species definition founded on average nucleotide identity (ANI) and on alignment-free approaches.
Results: After defining the genomes assuredly identifiable as P. stuartii, we devised a MultiLocus Sequence Typing (MLST) and a core-genome MLST (cgMLST) schemes, based on 7 and 2,296 loci respectively.
Discussion: This work hence provides a framework for understanding the role of P. stuartii and of other members of this genus, which should be considered as emerging multidrug-resistant pathogens.
The genus Providencia has traditionally been classified as part of the Proteeae tribe (Ewing, 1962). Nonetheless, based on a recent proposal for the classification of genera previously assigned to the Enterobacteriaceae family, this genus is now recognized as a member of the Morganellaceae family within the Enterobacterales order (Adeolu et al., 2016).
Strains belonging to the family Morganellaceae have a significant impact not only as environmental bacteria (Ferheen et al., 2023) or opportunistic pathogens (Liu et al., 2016) but also as infectious agents (Salas et al., 2023) in both human (Minnullina et al., 2019) and animal settings (Palmieri et al., 2020). In fact, members of this family are characterized by virulence traits, such as biofilm formation (Hola et al., 2012), motility, and cell invasion (Kurmasheva et al., 2018).
These virulence traits are invariably coupled with intrinsic resistance to several last-resort antibiotics such as colistin [due to the carriage of the arnBCADTEF and eptB genes (Gogry et al., 2021)], tigecycline (Yaghoubi et al., 2022), and imipenem (Clinical and Laboratory Standards Institute, 2024) and, occasionally, with acquired resistance genes [mostly blaNDM and blaVIM (Mondol et al., 2024; Abdallah and Balshi, 2018)]. Hence, the use of these antimicrobial agents to treat multidrug-resistant (Magiorakos et al., 2012) strains is significantly promoting the spread of members of the family Morganellaceae (Hayakawa et al., 2012) through bystander selection (Tedijanto et al., 2018).
As of June 2023, the genus Providencia accounted for 11 different recognized species such as Providencia alcalifaciens, Providencia burhodogranariea, Providencia heimbachae, Providencia manganoxydans, Providencia rettgeri, Providencia rustigianii, Providencia sneebia, Providencia stuartii, Providencia thailandensis, Providencia vermicola, and Providencia huaxiensis, and a twelfth species, the candidatus Providencia siddallii, was under evaluation. However, the taxonomy of Providencia is an ever-changing field, and, as of June 2024, the P. thailandensis species has been redefined as P. stuartii (Dong et al., 2024) and two novel species, namely, Providencia hangzhouensis (Dong et al., 2023) and Providencia zhijiangensis (Dong et al., 2024), were enlisted among the members of this genus.1 Of these species, P. alcalifaciens, P. rettgeri, and P. stuartii are the most notorious since they are more frequently ascribed to clinically relevant events (Mnif et al., 2013; Shah et al., 2019; Liu et al., 2023). However, owing to the contemporary genomic-based revision of the genus Providencia, its taxonomy is undergoing several extensive refinements, which highlighted the presence of species yet to be clearly defined (Wu et al., 2023; Yuan et al., 2020).
In this context, a major change is the above-mentioned proposal to consider “Providencia thailandensis” as a heterotypic synonym of “Providencia stuartii.” Indeed, the two type strains of these species display a low average nucleotide identity (ANI) difference (Dong et al., 2024). Under an epidemiology point of view, this change increased the relevance of P. stuartii and emphasized the necessity of a typing scheme for this species.
In this manuscript, we started by outlining the genomes that should be attributed to P. stuartii (i.e., displaying an ANI > 95% with the reference genome) and developed a core-genome multilocus sequence typing and a 7-gene multilocus sequence typing for this species.
As of 1 June 2022, the NCBI genome downloading script (ncbi-genome-download bacteria --genera Providencia --assembly-levels all --formats fasta)2 was used to download all (386) genomes from isolates identified as Providencia species. To these, three genomes sequenced in a previous study were added (Capitani et al., 2023) for a total of 389. The only genome assemblies of Providencia manganoxydans and of the candidatus Providencia siddallii were discarded, being the first contaminated and the latter partial.
The quality of the remaining 387 genomes was assessed by the BUSCO tool version 5.3.2 (Simão et al., 2015), using the Enterobacterales_odb10 set created on 23 February 2021. A total of 7 genomes displaying a BUSCO score lower than 95% were excluded from further analyses, resulting in a 380 genome dataset.
Genome relatedness between the P. stuartii type strain genome (NCBI RefSeq assembly no. GCF_029075745.1, isolate ATCC 29914) and the Providencia genome was assessed by ANI using the CompareM tool v0.0.23.3
The genome labeled as P. stuartii according to ANI was annotated using Prokka (Seemann, 2014), and the resulting. gff files were used as input for Roary (Page et al., 2015). A core-genome alignment was generated using standard Roary parameters (95% identity for BLASTp and presence in 99% of isolates to be defined as “core”) and used to infer a phylogenetic tree using the IQ-TREE (Minh et al., 2020) [GTR + F + R4 model (Kalyaanamoorthy et al., 2017)] and 10,000 ultrafast bootstraps (Hoang et al., 2018).
This procedure was repeated twice with the same parameters using the two P. stuartii datasets (1 June 2022 and 23 June 2023).
The POPulation Partitioning Using Nucleotide Kmers (PopPUNK) tool version 2.5.0 (Lees et al., 2019) was used to analyze the relative pairwise genetic distances of the core (π) and accessory (a) genomes and infer the population structure of Providencia. The PopPUNK quality control, using a π distance of 0.17, a distance of 0.9, and a length differing of 3 or more standard deviations from the mean as thresholds, discarded 43 isolates. A refined dbscan fit model was deployed and visualized using Cytoscape (version 3.10.2).
The final versions of the PopPUNK dbscan graph and of the phylogenetic trees were adjusted using the open-source InkScape software (version 1.3.2).
A core-genome multilocus sequence typing (cgMLST) and MLST schemes were devised for the P. stuartii species complex. Specifically, the cgMLST scheme was developed using the chewBBACA tool (Silva et al., 2018) on a subset of 51 P. stuartii genomes [48 submitted to the GenBank database as of 1 June 2022, and 3 genomes sequenced in a previous study (Capitani et al., 2023)].
Single-nucleotide polymorphisms (SNPs, determined using the SNP distance tool matrix)4 on the core genes among genomes of the subset were used to define the different sequence types (STs).
We checked the selected seven MLST loci for homoplasy with SplitsTree (Huson and Bryant, 2006), assessed the presence of potential selection pressure with the FUBAR, aBSREL, SLAC, and BUSTED tools from the HyPhy suite (Kosakovsky Pond et al., 2020), calculated Tajima’s D and dN/dS ratio with MEGA (Oxford academic, 2021) and evaluated several additional genomic metrics (i.e., haplotype diversity, nucleotide diversity, and segregating sites) with DnaSP6 (Rozas et al., 2017). Finally, single-linkage hierarchical clustering of the cgMLST alleles was performed across various thresholds (5, 10, 50, 100, 250, 500, 750, and 1,000) using Reportree (Mixão et al., 2023), and all were compared with the seven-loci MLST scheme using the Comparing Partitions online tool (Carriço et al., 2006) to define Simpson’s diversity index and estimating a 95% confidence interval using jackknife (Severiano et al., 2011).
As of 23 June 2023, the NCBI genome downloading script was used again with the same parameters as of 1 June 2022 (ncbi-genome-download bacteria --genera Providencia --assembly-levels all --formats fasta), and 202 more genomes were downloaded. The quality of these remaining 202 genomes was assessed by the BUSCO tool version 5.3.2 (Simão et al., 2015), and no genome was excluded from further analyses according to its BUSCO score.
Out of the 202 novel Providencia genomes submitted in GenBank between 1 June 2022 and 23 June 2023, we identified 26 genomes displaying an ANI > 95% with the P. stuartii reference genome and hence attributable to this species. These genomes were used to assess in silico the newly devised MLST scheme.
We also developed a PCR-based MLST scheme based on seven housekeeping genes to be applied when genomic facilities are not available. Amplification reactions contained 3 U Taq DNA polymerase (Meridian Bioscience, Cincinnati, OH, United States of America), 10 μL 5× PCR buffer, 2 μL amplification primers (10 μM each, Supplementary Table S1), 34 μL deionized H2O, and 1.5 μL bacterial template. We obtained bacterial template by the boiled DNA from a fully sequenced P. stuartii isolate [isolate 41, genome acc. No. CP142095 (Capitani et al., 2023)]. Amplification was performed starting with 2 min at 95°C to activate the DNA polymerase, followed by 29 cycles consisting of denaturation (94°C, 25 s), annealing (55°C, 25 s), and extension (72°C, 1 min and 15 s, Supplementary Table S1).
Amplicons were analyzed using Sanger sequencing. While most genes were sequenced using amplification primers, the znuA amplicon (owing to its size and to the distance of allele-defining SNPs from the primers) needed two additional primers (znuA_seq_1 and znuA_seq_2) specifically for sequencing the variable region (Supplementary Table S1). The resulting sequences were then aligned to the corresponding P. stuartii allele using BLASTn.
Recent genomic analyses offered a detailed classification of species and lineages belonging to the genus Providencia, highlighting a wide species diversity (Dong et al., 2024; Wang et al., 2023).
A total of 335 Providencia genomes (386 from the GenBank database as of 23 June 2023 and 3 from our collection, deprived of 7 genomes displaying a low BUSCO score, the P. manganoxydans and the P. siddallii reference genomes, and 43 genomes that did not pass the PopPUNK quality module) were included in the PopPUNK population analysis. These genomes defined 23 clusters (5 of which constituted only by one singleton) (Lees et al., 2019) (Figure 1). Two of the singletons were the P. burhodogranariea and P. vermicola reference strain genomes. Of note, all other eight genomes submitted in GenBank as P. vermicola clustered with five genomes labeled as P. stuartii and with genomes of unclassified Providencia spp.
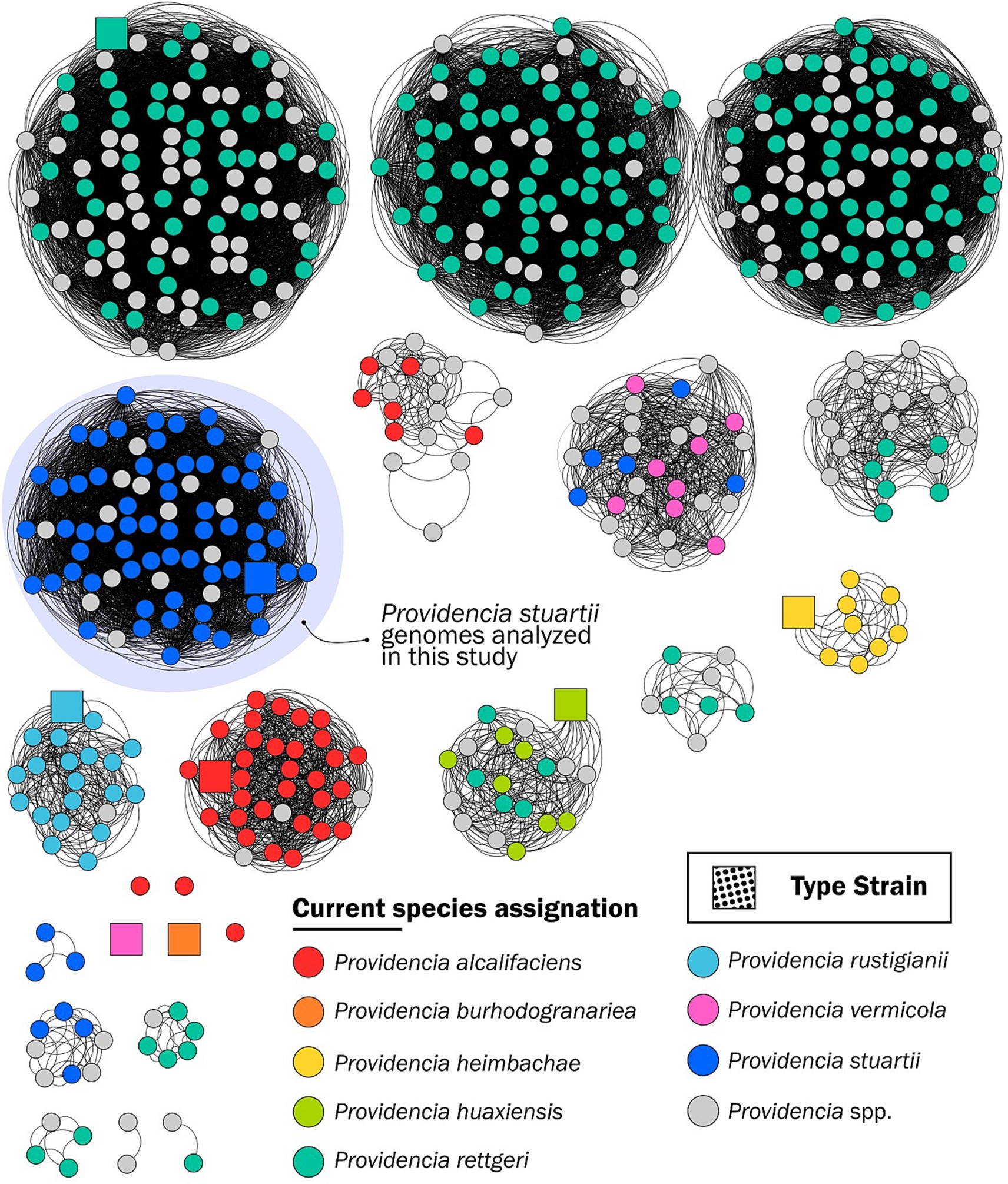
Figure 1. Grouping of 581 Providencia spp. genomes downloaded from the GenBank database as of 23 June 2023, generated by PopPUNK (Lees et al., 2019). The nodes are color-coded according to the legend and indicate the current NCBI assignation of the genome; square nodes represent the reference strain genomes.
The P. heimbachae and P. rustigianii clusters contain genomes that are coherently assigned to the respective Providencia species. Genomes assigned to P. rettgeri exhibit the highest rate of potential misclassification, with a dispersed distribution in nine different clusters, where only one comprising the P. rettgeri reference genome strain.
Since the P. zhijiangensis and the P. hangzhouensis genomes were released in November and September 2023, respectively, they were not included in this analysis.
We devised a specific cgMLST for the P. stuartii species complex. This was based on 51 genomes downloaded from the GenBank database as of 1 June 2022, displaying an ANI > 95% with the P. stuartii genome of the reference strain (NCBI RefSeq assembly no. GCF_029075745.1, isolate ATCC 29914). Out of these 51 genomes, 3 were from a previous study (Capitani et al., 2023) and 48 were retrieved from the NCBI RefSeq database, not taking into consideration if they were submitted in GenBank as P. stuartii or Providencia spp.
The chewBBACA tool version 3.2.0 (Silva et al., 2018) was deployed, using the NCBI RefSeq assembly no. GCF_029075745.1 (isolate ATCC 29914) as a training file. After the removal of 1,594 loci (32 containing ambiguous or invalid characters and 1,562 shorter than 201 bp), a total of 7,561 loci were identified, showing a total of 53,725 alleles. Of these loci, 2,296 corresponded to the genes of the cgMLST (identified as present in every isolate).
The SNP distance matrix generated starting from the Roary core-genome alignment displayed either <1,100 SNPs or > 7,500 SNPs; hence, we defined each P. stuartii ST as a group of genomes showing less than 1,100 SNPs in the apart one from the other (Supplementary Dataset 1).
The 7-gene MLST was determined starting from the cgMLST alleles as determined by chewBBACA (Silva et al., 2018). The P. stuartii SNP-based phylogenetic tree on the core genome from the 51 genomes displayed a comb-like disposition with 22 branches (Supplementary Figure S1). Each branch corresponded to one of the 22 STs above defined.
After removal of the genes whose alleles did not fit with the 22 assigned ST, 471 loci remained. To ensure the stability and the discriminatory power of the MLST scheme, only 184 loci displaying more than 4 but less than 10 different alleles were considered. Of these, a representative of each protein was annotated using the UniprotFinder module of chewBBACA (Silva et al., 2018). After removal of genes coding for hypothetical or not fully characterizable proteins, non-structural proteins (e.g., fimbriae), and subjectable to selective pressure (e.g., porins or antimicrobial efflux systems), 15 suitable candidate genes were selected.
To assess species specificity, we performed a BLASTn screening of the 15 candidate genes against the Providencia genus collection. These genes showed a nucleotide identity >99.9% only with genomes displaying an ANI > 95% with the P. stuartii reference genome, whereas their identity dropped to <90% with other Providencia species.
Finally, using Bandage 0.8.1 (Wick et al., 2015), we mapped these genes on the circular ST3 P. stuartii genome 41 (genome acc. No. CP142095) to assess their distribution alongside the genome (Supplementary Figure S2). Using the gene position criterion to minimize the risk that a single recombination event would affect multiple genes, we limited the number of genes to seven.
The proteins encoded by the genes chosen as representatives of the MLST scheme are as follows:
i. GreA, encoding a transcription elongation factor that is highly conserved among prokaryotes and that regulates gene expression (Yuzenkova et al., 2014; Feng et al., 2020).
ii. FtsH, a protease involved in the cell division process (Ito and Akiyama, 2005).
iii. TolR, the stator protein of the Tol-Pal system, is involved in the outer membrane invagination during cell division (Wojdyla et al., 2015).
iv. ArnE, part of the ArnE/ArnF heterodimer, by transporting the 4-amino-4-deoxy-L-arabinose (L-Ara4N) is involved in lipid A modification (Yan et al., 2007).
v. ZnuA, responsible (together with ZnuB and ZnuC) for the intracellular Zn++ homeostasis (Ammendola et al., 2007).
vi. YciA, a hexameric broad specificity acyl-CoA thioesterase plausibly involved in membrane biogenesis (Willis et al., 2008).
vii. RseA, an anti-sigma-E factor protein that averts the transcription of the sigma-E factor induced by heat-shock promoters (de Las Peñas et al., 1997).
We performed multiple tests to assess the quality of the chosen loci. Positive selection examinations did not report any significant positive selection on all branches, and no loci displayed a dN/dS ratio greater than 1 (Supplementary Dataset 1). Bayesian analyses confirmed that most codon positions in all seven genes are neither positively nor negatively selected, with six of the seven MLST genes displaying a variable number of codons conserved under purifying selection with low Bayes factor values (ranging from 0.008 to 0.093), except for codon 16 in the znuA gene, which showed potential positive selection with a posterior probability of 0.944 and a Bayes factor of 19.7. Site-specific analysis with SLAC did not detect any significant pervasive positive or negative selection, and no homoplasy event was identified in five of the seven genes (greA and tolR could not be evaluated due to the low number of informative characters).
Moreover, we compared the seven-loci MLST genes with the 2,289 resting cgMLST scheme genes across multiple analyses. MLST genes were significantly shorter than the cgMLST ones, a feature needed for Sanger sequencing, and showed significantly lower variability in terms of segregation sites, nucleotide diversity, and haplotype number (p = 0.044, p = 0.048, and p = 0.019, respectively). Simpson’s index analysis confirmed the robustness of the seven-loci MLST scheme compared to the cgMLST scheme, highlighting how the resolution level of the seven-loci MLST scheme is higher than the resolution level of single-linkage hierarchical clustering at 250 mismatches but lower when compared to the one of single-linkage hierarchical clustering at 100 mismatches (Supplementary Dataset 2). Tajima’s D test was carried out for all cgMLST genes, and the results indicated no strong selective pressure on the seven MLST genes, apart from yciA, which had a Tajima’s D value of −1.36, suggesting some degree of purifying selection (Supplementary Dataset 1).
To validate the discriminative power of our newly devised MLST scheme, we blind-tested it on an additional dataset composed of 24 P. stuartii genomes, downloaded between 1 June 2022 and 23 June 2023, which were not used to develop the scheme (marked by a * in Supplementary Table S2).
First, we performed a BLASTn analysis of the 7 designated genes to assign the respective alleles, which identified 9 different profiles in the 25 genomes from the additional dataset. While 7 of these profiles coincided with previously defined STs, 2 were unmatched and were associated with novel STs (named ST23 and ST24).
A second P. stuartii SNP-based phylogenetic tree was built starting from the 76 genomes (51 from the first dataset and 25 from the validation dataset), and the 7-loci MLST correctly predicted the core-genome distribution of the isolates assigned to each ST. Compared to the 51-genome tree (Supplementary Figure S1), the updated 76-genome tree displayed two novel branches, which corresponded to the genomes typed as ST23 and ST24 (Figure 2; Supplementary Table S2).
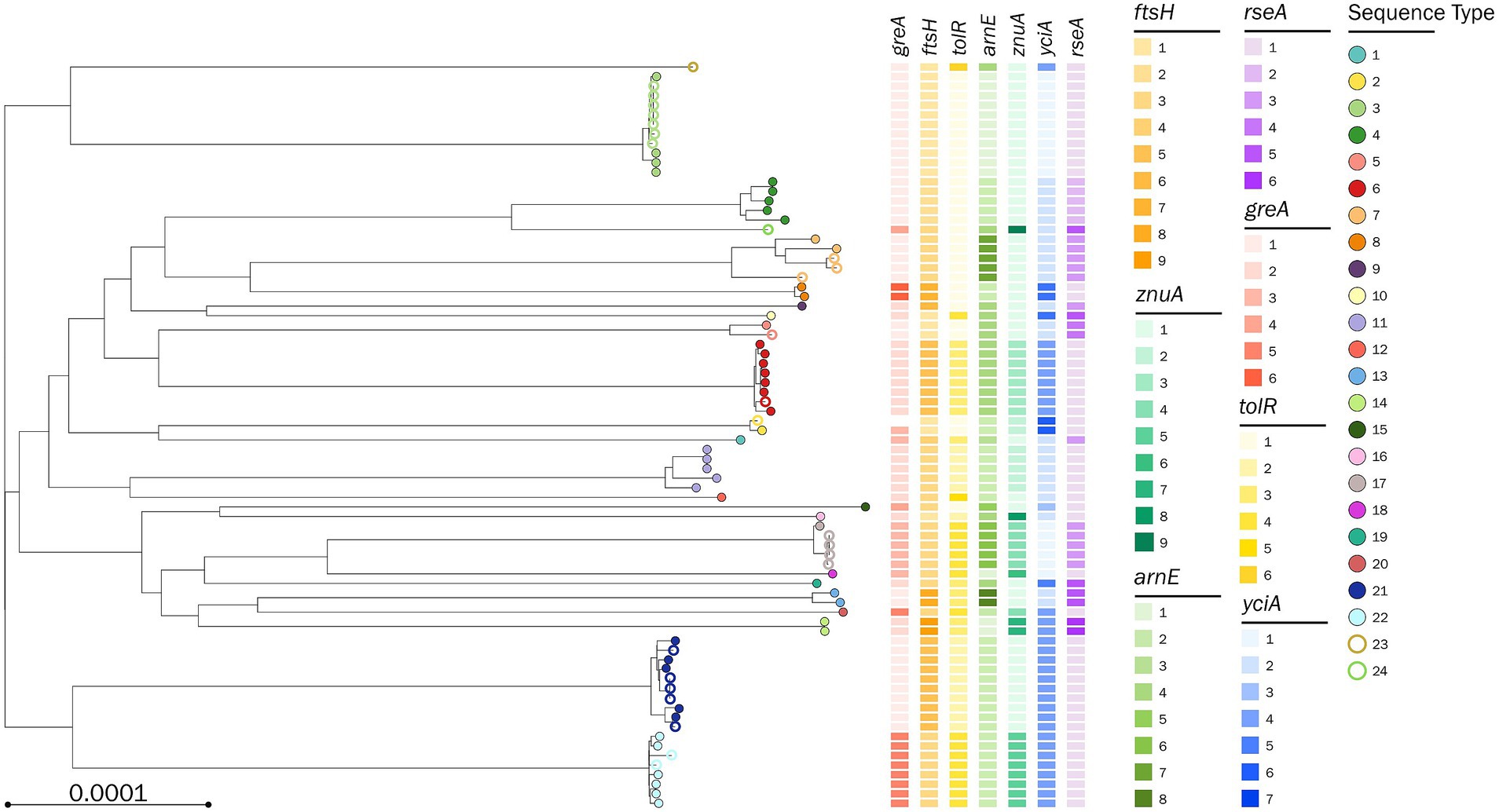
Figure 2. Phylogenetic tree based on a core-genome alignment of 71 Providencia stuartii genomes. The tips are color-coded according to the assigned sequence type (ST); full circles represent the genomes used to define the 7-loci multilocus sequence type (MLST); empty circles represent the genomes used to validate the 7-loci MLST; the allelic variants defining the MLST combination are color-coded according to the legend.
To make the P. stuartii MLST independent from WGS, we also devised a set of PCR primers to amplify and sequence the seven housekeeping genes (Supplementary Table S1) and tested both amplification and Sanger sequencing procedures using strain 41 as a template (Figure 3).
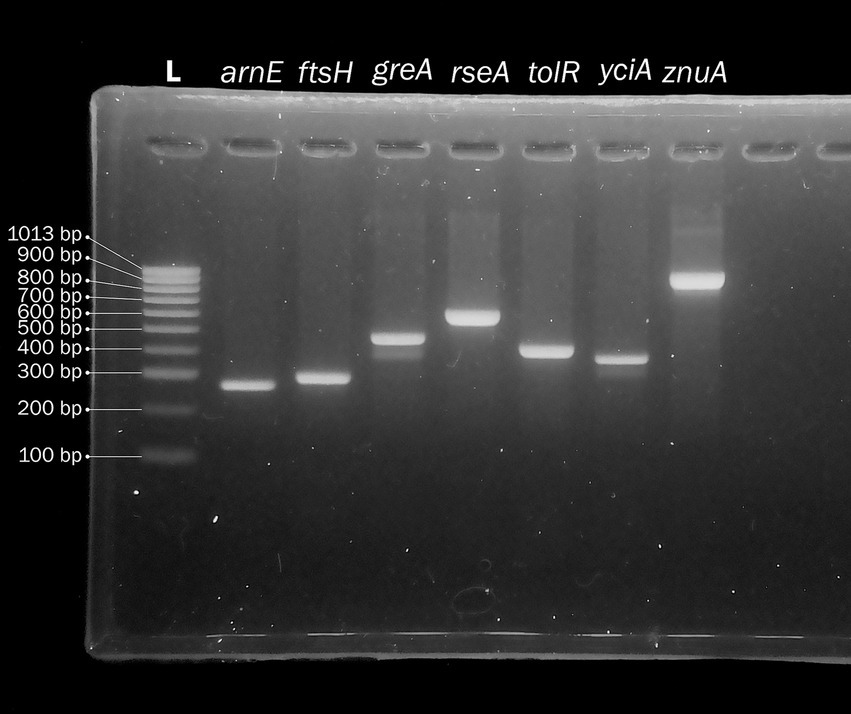
Figure 3. Agarose gel electrophoresis of a DNA ladder (lane 1) and the PCR amplification product of the seven genes used to define the Providencia stuartii multilocus sequence type (MLST, lanes 2–8).
The current taxonomic classification of the Providencia genus is marked by substantial disarray. While a subset of our findings corroborates previous reports of incorrect reference assignments, within this manuscript, we did not attempt to resolve these issues. The focus of our study, instead, was to offer a robust framework for the identification and typing of the emerging species P. stuartii. Future research should aim to address and rectify the broader taxonomic ambiguities within the Providencia genus to enhance our understanding and management of these pathogens.
This study underscores the importance of P. stuartii as an emerging pathogen and provides a method for its accurate identification and typing through the development of MLST and cgMLST schemes.
Of note, to develop the 7-gene MLST scheme, we made use of a reverse approach: We first performed a core-genome-based phylogenetic analysis of P. stuartii genomes, which served as a cornerstone to define the STs, and then, we found the genes whose alleles fitted our a priori assignment. This approach could be applied to swiftly develop 7-gene MLST schemes for emerging species that cannot be typed with portable and scalable methods.
A BioProject has been released at DDBJ/ENA/GenBank, No: PRJNA948429. Circular complete genome and plasmid of isolate 41 have been released under Acc. Nos. CP142095–CP142096. Reference Providencia stuartii NCBI RefSeq assembly no. GCF_029075745.1, isolate ATCC 29914. Single-nucleotide polymorphism (SNP) core-genome distance matrix of the 51 genomes used for the construction of the Providencia stuartii multilocus sequence typing scheme is supplied as Supplementary Dataset 1.
GA: Conceptualization, Data curation, Formal analysis, Methodology, Project administration, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. AF: Conceptualization, Investigation, Methodology, Writing – review & editing. RP: Methodology, Validation, Writing – original draft. AC: Funding acquisition, Investigation, Resources, Writing – original draft, Writing – review & editing. VC: Data curation, Formal analysis, Investigation, Methodology, Validation, Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was supported by the European Union funding A. C. from the NextGeneration EU-MUR PNRR Extended Partnership Initiative on Emerging Infectious Diseases (project no. PE00000007, PE13 INF-ACT, Spoke 3). R. P. was supported by the PNRR PHD scholarship (EX M.D. 351/22) financed by the Rome Technopole project.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1493621/full#supplementary-material.
SUPPLEMENTARY FIGURE S1 | Phylogenetic tree based on a core-genome alignment of the 51 Providencia stuartii genomes used to define the first 22 sequence types (STs).
SUPPLEMENTARY FIGURE S2 | Bandage (Wick et al., 2015) representation of the closed genome of the Providencia stuartii isolate 41 (Capitani et al., 2023). The position of the 7 genes chosen for the MultiLocus Sequence Type (MLST) definition is represented by colored, thick rectangles; the position of the 8 genes discarded is represented by grey, thin rectangles.
SUPPLEMENTARY DATASET | Single Nucleotide Polymorphisms (SNPs) core-genome distance matrix of the 51 Providencia stuartii genomes used to define the relative MultiLocus Sequence Type (MLST) scheme. Genomes displaying less than 1,100 SNPs on the core genome are defined as belonging to the same Sequence Type (ST) and are highlighted in green.
1. ^https://www.ncbi.nlm.nih.gov/data-hub/taxonomy/586/
2. ^https://github.com/kblin/ncbi-genome-download
Abdallah, M., and Balshi, A. (2018). First literature review of carbapenem-resistant Providencia. New Microb. New Infect 25, 16–23. doi: 10.1016/j.nmni.2018.05.009
Adeolu, M., Alnajar, S., Naushad, S., and S Gupta, R. (2016). Genome-based phylogeny and taxonomy of the 'Enterobacteriales': proposal for Enterobacterales Ord. Nov. divided into the families Enterobacteriaceae, Erwiniaceae fam. Nov., Pectobacteriaceae fam. Nov., Yersiniaceae fam. Nov., Hafniaceae fam. Nov., Morganellaceae fam. Nov., and Budviciaceae fam. Nov. Int. J. Syst. Evol. Microbiol. 66, 5575–5599. doi: 10.1099/ijsem.0.001485
Ammendola, S., Pasquali, P., Pistoia, C., Petrucci, P., Petrarca, P., Rotilio, G., et al. (2007). High-affinity Zn2+ uptake system ZnuABC is required for bacterial zinc homeostasis in intracellular environments and contributes to the virulence of Salmonella enterica. Infect. Immun. 75, 5867–5876. doi: 10.1128/IAI.00559-07
Capitani, V., Arcari, G., Oliva, A., Sacco, F., Menichincheri, G., Fenske, L., et al. (2023). Genome-based retrospective analysis of a Providencia stuartii outbreak in Rome, Italy: broad Spectrum IncC plasmids spread the NDM Carbapenemase within the hospital. Antibiotics 12:943. doi: 10.3390/antibiotics12050943
Carriço, J. A., Silva-Costa, C., Melo-Cristino, J., Pinto, F. R., de Lencastre, H., Almeida, J. S., et al. (2006). Illustration of a common framework for relating multiple typing methods by application to macrolide-resistant Streptococcus pyogenes. J. Clin. Microbiol. 44, 2524–2532. doi: 10.1128/JCM.02536-05
Clinical and Laboratory Standards Institute. (2024). M100Ed33 | performance standards for antimicrobial susceptibility testing, 33rd edition. Available at:https://clsi.org/standards/products/microbiology/documents/m100/ Accessed 9 January 2024.
de Las Peñas, A., Connolly, L., and Gross, C. A. (1997). The sigmaE-mediated response to extracytoplasmic stress in Escherichia coli is transduced by RseA and RseB, two negative regulators of sigmaE. Mol. Microbiol. 24, 373–385. doi: 10.1046/j.1365-2958.1997.3611718.x
Dong, X., Jia, H., Yu, Y., Xiang, Y., and Zhang, Y. (2024). Genomic revisitation and reclassification of the genus Providencia. mSphere 9, e00731–e00723. doi: 10.1128/msphere.00731-23
Dong, X., Yu, Y., Liu, J., Cao, D., Xiang, Y., Bi, K., et al. (2023). Whole-genome sequencing provides insights into a novel species: Providencia hangzhouensis associated with urinary tract infections. Microbiol Spectr 11:e0122723. doi: 10.1128/spectrum.01227-23
Ewing, W. H. (1962). The tribe Proteeae: its nomenclature and taxonomy. Int. Bull. Bacteriol. Nomen. Taxon. 12, 93–102. doi: 10.1099/0096266X-12-3-93
Feng, S., Liu, Y., Liang, W., el-Sayed Ahmed, M. A. E. G., Zhao, Z., Shen, C., et al. (2020). Involvement of transcription elongation factor GreA in Mycobacterium viability, antibiotic susceptibility, and intracellular fitness. Front. Microbiol. 11:413. doi: 10.3389/fmicb.2020.00413
Ferheen, I., Spurio, R., Mancini, L., and Marcheggiani, S. (2023). Detection of Morganella morganii bound to a plastic substrate in surface water. J Glob Antimicrob Resist 32, 104–107. doi: 10.1016/j.jgar.2023.01.008
Gogry, F. A., Siddiqui, M. T., Sultan, I., and QMR, H. (2021). Current update on intrinsic and acquired Colistin resistance mechanisms in Bacteria. Front. Med. 8:677720. doi: 10.3389/fmed.2021.677720
Hayakawa, K., Marchaim, D., Divine, G. W., Pogue, J. M., Kumar, S., Lephart, P., et al. (2012). Growing prevalence of Providencia stuartii associated with the increased usage of colistin at a tertiary health care center. Int. J. Infect. Dis. 16, e646–e648. doi: 10.1016/j.ijid.2012.05.1029
Hoang, D. T., Chernomor, O., von Haeseler, A., Minh, B. Q., and Vinh, L. S. (2018). UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522. doi: 10.1093/molbev/msx281
Hola, V., Peroutkova, T., and Ruzicka, F. (2012). Virulence factors in Proteus bacteria from biofilm communities of catheter-associated urinary tract infections. FEMS Immunol Med Microbiol 65, 343–349. doi: 10.1111/j.1574-695X.2012.00976.x
Huson, D. H., and Bryant, D. (2006). Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267. doi: 10.1093/molbev/msj030
Ito, K., and Akiyama, Y. (2005). Cellular functions, mechanism of action, and regulation of FtsH protease. Ann. Rev. Microbiol. 59, 211–231. doi: 10.1146/annurev.micro.59.030804.121316
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Kosakovsky Pond, S. L., Poon, A. F. Y., Velazquez, R., Weaver, S., Hepler, N. L., Murrell, B., et al. (2020). HyPhy 2.5—a customizable platform for evolutionary hypothesis testing using phylogenies. Mol. Biol. Evol. 37, 295–299. doi: 10.1093/molbev/msz197
Kurmasheva, N., Vorobiev, V., Sharipova, M., Efremova, T., and Mardanova, A. (2018). The potential virulence factors of Providencia stuartii: motility, adherence, and invasion. Biomed. Res. Int. 2018, 1–8. doi: 10.1155/2018/3589135
Lees, J. A., Harris, S. R., Tonkin-Hill, G., Gladstone, R. A., Lo, S. W., Weiser, J. N., et al. (2019). Fast and flexible bacterial genomic epidemiology with PopPUNK. Genome Res. 29, 304–316. doi: 10.1101/gr.241455.118
Liu, M., Yi, N., Wang, X., and Wang, R. (2023). Analysis of resistance genes of carbapenem-resistant Providencia rettgeri using whole genome sequencing. BMC Microbiol. 23:283. doi: 10.1186/s12866-023-03032-3
Liu, H., Zhu, J., Hu, Q., and Rao, X. (2016). Morganella morganii, a non-negligent opportunistic pathogen. Int. J. Infect. Dis. 50, 10–17. doi: 10.1016/j.ijid.2016.07.006
Magiorakos, A.-P., Srinivasan, A., Carey, R. B., Carmeli, Y., Falagas, M. E., Giske, C. G., et al. (2012). Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 18, 268–281. doi: 10.1111/j.1469-0691.2011.03570.x
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., von Haeseler, A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. doi: 10.1093/molbev/msaa015
Minnullina, L., Pudova, D., Shagimardanova, E., Shigapova, L., Sharipova, M., and Mardanova, A. (2019). Comparative genome analysis of Uropathogenic Morganella morganii strains. Front. Cell. Infect. Microbiol. 9:167. doi: 10.3389/fcimb.2019.00167
Mixão, V., Pinto, M., Sobral, D., di Pasquale, A., Gomes, J. P., and Borges, V. (2023). ReporTree: a surveillance-oriented tool to strengthen the linkage between pathogen genetic clusters and epidemiological data. Genome Med. 15:43. doi: 10.1186/s13073-023-01196-1
Mnif, B., Ktari, S., Chaari, A., Medhioub, F., Rhimi, F., Bouaziz, M., et al. (2013). Nosocomial dissemination of Providencia stuartii isolates carrying blaOXA-48, blaPER-1, blaCMY-4 and qnrA6 in a Tunisian hospital. J. Antimicrob. Chemother. 68, 329–332. doi: 10.1093/jac/dks386
Mondol, S. M., Islam, I., Islam, M. R., Shakil, S. K., Rakhi, N. N., Mustary, J. F., et al. (2024). Genomic landscape of NDM-1 producing multidrug-resistant Providencia stuartii causing burn wound infections in Bangladesh. Sci. Rep. 14:2246. doi: 10.1038/s41598-024-51819-9
Oxford academic. (2021). MEGA11: molecular evolutionary genetics analysis version 11 | molecular biology and evolution. Available at:https://academic.oup.com/mbe/article/38/7/3022/6248099 Accessed 9 October 2024.
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Palmieri, N., Hess, C., Hess, M., and Alispahic, M. (2020). Sequencing of five poultry strains elucidates phylogenetic relationships and divergence in virulence genes in Morganella morganii. BMC Genomics 21:579. doi: 10.1186/s12864-020-07001-2
Rozas, J., Ferrer-Mata, A., Sánchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Salas, B., Conway, H. E., Vacek, D. C., Vitek, C., and Schuenzel, E. L. (2023). Pathogenicity of multiple Providencia species (Enterobacteriales: Morganellaceae) to the mass-reared Mexican fruit fly (Diptera: Tephritidae). J. Insect Sci. 23:4. doi: 10.1093/jisesa/iead024
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Severiano, A., Carriço, J. A., Robinson, D. A., Ramirez, M., and Pinto, F. R. (2011). Evaluation of jackknife and bootstrap for defining confidence intervals for pairwise agreement measures. PLoS One 6:e19539. doi: 10.1371/journal.pone.0019539
Shah, M. M., Odoyo, E., and Ichinose, Y. (2019). Epidemiology and pathogenesis of Providencia alcalifaciens infections. Am. J. Trop. Med. Hyg. 101, 290–293. doi: 10.4269/ajtmh.18-0376
Silva, M., Machado, M. P., Silva, D. N., Rossi, M., Moran-Gilad, J., Santos, S., et al. (2018). chewBBACA: a complete suite for gene-by-gene schema creation and strain identification. Microb Genom 4:e000166. doi: 10.1099/mgen.0.000166
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Tedijanto, C., Olesen, S. W., Grad, Y. H., and Lipsitch, M. (2018). Estimating the proportion of bystander selection for antibiotic resistance among potentially pathogenic bacterial flora. Proc. Natl. Acad. Sci. USA 115, E11988–E11995. doi: 10.1073/pnas.1810840115
Wang, P., Li, C., Yin, Z., Jiang, X., Li, X., Mu, X., et al. (2023). Genomic epidemiology and heterogeneity ofProvidenciaand theirblaNDM-1-carrying plasmids. Emerg. Microb. Infect 12:2275596. doi: 10.1080/22221751.2023.2275596
Wick, R. R., Schultz, M. B., Zobel, J., and Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352. doi: 10.1093/bioinformatics/btv383
Willis, M. A., Zhuang, Z., Song, F., Howard, A., Dunaway-Mariano, D., and Herzberg, O. (2008). Structure of YciA from Haemophilus influenzae (HI0827), a hexameric broad specificity acyl-coenzyme a thioesterase. Biochemistry 47, 2797–2805. doi: 10.1021/bi702336d
Wojdyla, J. A., Cutts, E., Kaminska, R., Papadakos, G., Hopper, J. T. S., Stansfeld, P. J., et al. (2015). Structure and function of the Escherichia coli Tol-pal stator protein TolR. J. Biol. Chem. 290, 26675–26687. doi: 10.1074/jbc.M115.671586
Wu, R., Zhu, Z., and Wang, G.-H. (2023). Genome sequence of Providencia stuartii prov-sta1, isolated from the wasp Nasonia vitripennis. Microbiol. Res. Announc. 12, e00430–e00423. doi: 10.1128/MRA.00430-23
Yaghoubi, S., Zekiy, A. O., Krutova, M., Gholami, M., Kouhsari, E., Sholeh, M., et al. (2022). Tigecycline antibacterial activity, clinical effectiveness, and mechanisms and epidemiology of resistance: narrative review. Eur. J. Clin. Microbiol. Infect. Dis. 41, 1003–1022. doi: 10.1007/s10096-020-04121-1
Yan, A., Guan, Z., and Raetz, C. R. H. (2007). An undecaprenyl phosphate-aminoarabinose flippase required for polymyxin resistance in Escherichia coli. J. Biol. Chem. 282, 36077–36089. doi: 10.1074/jbc.M706172200
Yuan, C., Wei, Y., Zhang, S., Cheng, J., Cheng, X., Qian, C., et al. (2020). Comparative genomic analysis reveals genetic mechanisms of the variety of pathogenicity, antibiotic resistance, and environmental adaptation of Providencia genus. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.572642
Keywords: Providencia stuartii, MLST, phylogenomics, gene, allele
Citation: Arcari G, De Francesco A, Polani R, Carattoli A and Capitani V (2024) Development of a Providencia stuartii multilocus sequence typing scheme. Front. Microbiol. 15:1493621. doi: 10.3389/fmicb.2024.1493621
Edited by:
Axel Cloeckaert, Institut National de recherche pour l’agriculture, l’alimentation et l’environnement (INRAE), FranceReviewed by:
German Matias Traglia, Universidad de La Republica, UruguayCopyright © 2024 Arcari, De Francesco, Polani, Carattoli and Capitani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gabriele Arcari, Z2FicmllbGUuYXJjYXJpQHVuaW5zdWJyaWEuaXQ=
†ORCID: Gabriele Arcari, https://orcid.org/0000-0001-9629-4637
Alessandra Carattoli, https://orcid.org/0000-0002-6120-6526
Valerio Capitani, https://orcid.org/0009-0008-4569-1972
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.