 Magdalena Szuplewska1*
Magdalena Szuplewska1* Dorota Sentkowska1Robert Lasek1
Dorota Sentkowska1Robert Lasek1 Przemysław Decewicz2Mateusz Hałucha1
Przemysław Decewicz2Mateusz Hałucha1 Łukasz Funk1Cora Chmielowska1
Łukasz Funk1Cora Chmielowska1 Dariusz Bartosik1
Dariusz Bartosik1- 1Department of Bacterial Genetics, Institute of Microbiology, Faculty of Biology, University of Warsaw, Warsaw, Poland
- 2Department of Environmental Microbiology and Biotechnology, Institute of Microbiology, Faculty of Biology, University of Warsaw, Warsaw, Poland
Introduction: Paracoccus yeei is the first species in the genus Paracoccus to be implicated in opportunistic infections in humans. As a result, P. yeei strains provide a valuable model for exploring how bacteria shift from a saprophytic to a pathogenic lifestyle, as well as for investigating the role of horizontally transferred DNA in this transition. In order to gain deeper insights into the unique characteristics of this bacterium and the molecular mechanisms underlying its opportunistic behavior, a comparative physiological and genomic analysis of P. yeei strains was performed.
Results: Complete genomic sequences of 7 P. yeei isolates (both clinical and environmental) were obtained and analyzed. All genomes have a multipartite structure comprising numerous extrachromosomal replicons (59 different ECRs in total), including large chromids of the DnaA-like and RepB families. Within the mobile part of the P. yeei genomes (ECRs and transposable elements, TEs), a novel non-autonomous MITE-type element was identified. Detailed genus-wide comparative genomic analysis permitted the identification of P. yeei-specific genes, including several putative virulence determinants. One of these, the URE gene cluster, determines the ureolytic activity of P. yeei strains—a unique feature among Paracoccus spp. This activity is induced by the inclusion of urea in the growth medium and is dependent on the presence of an intact nikR regulatory gene, which presumably regulates expression of nickel (urease cofactor) transporter genes.
Discussion: This in-depth comparative analysis provides a detailed insight into the structure, composition and properties of P. yeei genomes. Several predicted virulence determinants (including URE gene clusters) were identified within ECRs, indicating an important role for the flexible genome in determining the opportunistic properties of this bacterium.
1 Introduction
Bacteria of the genus Paracoccus (family Paracoccaceae, order Rhodobacterales, class Alphaproteobacteria) are known for their great metabolic diversity and flexibility. Many of them can switch between growth modes, using different carbon and energy sources, and employing various final electron acceptors (Czarnecki and Bartosik, 2019). There are currently 105 known species of the genus Paracoccus (NCBI Taxonomy, 17 July 2024), including Paracoccus denitrificans—the type strain of the genus (isolated in 1910) —commonly used as a model denitrifying organism (Beijerinck and Minkman, 1910; Bordel et al., 2024). A large number of these species were identified only recently and many have yet to be the subject of detailed studies.
Paracoccus spp. inhabit diverse marine and terrestrial environments, such as soil, sediments, sludge, brines or groundwater (e.g., Urakami et al., 1990; Siller et al., 1996; Tsubokura et al., 1999; Berry et al., 2003; Lee et al., 2004; Liu et al., 2006, 2008). In addition, some strains have been isolated from the rhizosphere or from the surface of other organisms, e.g., ticks, marine bryozoans and corals (Pukall et al., 2003; Ghosh et al., 2006; Carlos et al., 2017). The ubiquity of these bacteria is further demonstrated by their presence in house dust (Thompson et al., 2021).
Among these environmental bacteria, Paracoccus yeei (formerly classified as a eugonic oxidizer in group 2; strain EO-2) is of particular interest because it is the first species of the genus Paracoccus to be implicated in opportunistic infections in humans (Daneshvar et al., 2003). This bacterium (naturally occurring in soil) is not associated with a specific disease (Fosso et al., 2021). P. yeei isolates have been recovered from several clinical conditions, e.g., from the dialysate of a patient with peritonitis, myocarditis in a transplanted heart, corneal transplantation, bacteremia, keratitis, otitis and dermatological lesions (Lasek et al., 2018). The number of cases of infection by P. yeei appears to be increasing (Funke et al., 2004; Kanis et al., 2010; Schweiger et al., 2011; Courjaret et al., 2014; Arias and Clark, 2019; Aliste-Fernández et al., 2020; Fosso et al., 2021; Shifera et al., 2021; Bhikoo et al., 2022). However, this reported incidence is likely to be an underestimate since current common diagnostic tests do not detect this bacterium (Sack et al., 2017). Moreover, P. yeei cells can appear under-decolorized after Gram staining, so may be inadvertently reported or dismissed as gram-positive cocci (Dyer and Harris, 2020).
Analysis of a larger number of clinical cases suggests that immunocompromised patients are at increased risk of infection with this bacterium (Dyer and Harris, 2020). However, the natural reservoir of this opportunistic pathogen as well as the molecular bases of its pathogenicity remain unclear.
Numerous P. yeei strains have been isolated from everyday objects in hospital environments, which may play an important role in its transmission. This bacterium was found on the surface of medical equipment, such as operating tables, endoscopes or laryngoscopes (in direct contact with the mucosa, saliva and blood of patients) (Choi et al., 2017; Sartoretti et al., 2017; Pasquale et al., 2021), as well as on the surface of mobile phones of hospital employees and patients (Murgier et al., 2016; Cantais et al., 2020). Moreover, in non-hospital settings P. yeei strains were detected on the surface of benches, doors and walls of schools and kindergartens (Kruszewska et al., 2021), and on hand-drying devices in public toilets (Huesca-Espitia et al., 2018). Interestingly, this bacterium was also identified as a dominant member of microbial consortia contributing to the biodegradation of pre-Columbian canvasses in a museum collection (Pietrzak et al., 2017).
P. yeei represents a convenient model organism to study the switch from a saprophytic to a pathogenic lifestyle and to determine the role of horizontally acquired DNA in this process. Comparative genomic analyses of P. yeei and other members of the genus Paracoccus should yield a great deal of information on the mechanisms involved in this transition. Our previous analysis of the genomes of 3 P. yeei isolates (CCUG 32053, FDAARGOS_252 and TT13) gave the first insight into the genome composition, mobilome and metabolic potential of these bacteria (Lasek et al., 2018). However, the small number of strains analyzed was a major limitation of this study.
The main objective of the present study was to define the specific properties and genetic information of P. yeei species that are potentially involved in the process of pathogenesis, as well as to characterize mobile genetic elements—natural gene carriers that may be responsible for the horizontal transmission of virulence determinants. For 7 P. yeei strains available in our laboratory, we performed various physiological tests and detailed analyses of their mobile genetic elements. To identify P. yeei-specific genes, we conducted a comparative analysis of the 11 genomes of P. yeei strains available in the NCBI database (both clinical and environmental isolates) and four genomes of other members of the genus Paracoccus.
2 Materials and methods
2.1 Strains and culture conditions
P. yeei strains CCUG 13493, CCUG 17731, CCUG 32052, CCUG 32054, CCUG 46822, and CCUG 54214 were purchased from the Culture Collection of the University of Gothenburg (CCUG) (Sweden). Strain LM20 was isolated from black shale collected within the Lubin underground copper mine in Poland (Dziewit et al., 2015). P. yeei CCUG 13493R, CCUG 17731R, CCUG 32052R, CCUG 32054R, CCUG 46822R, CCUG 54214R, and LM20R, rifampicin-resistant derivatives of the respective wild-type strains, and Escherichia coli TG1 (Sambrook and Russell, 2001), were used as plasmid recipients. E. coli DH5α (Hanahan, 1983) was the host strain of helper plasmid pRK2013, used in triparental matings (Ditta et al., 1980) (Supplementary Table S1).
All strains were cultured in lysogeny broth (LB) medium (Sambrook and Russell, 2001) at 37°C for E. coli or 30°C for the other strains. Liquid cultures were grown under shaking conditions. When required, the medium was supplemented with 10% sucrose and the following concentrations of selective antibiotics: kanamycin, 50 μg/ml for all strains except Achromobacter sp. LM16R (300 μg/ml) (Dziewit et al., 2015); rifampicin, 50 μg/ml; tetracycline, 2 μg/ml for P. yeei and 20 μg/ml for E. coli strains.
2.2 Physiological analyses
The response to various growth conditions (temperature, pH and salinity) of P. yeei strains was determined as described previously by Dziewit et al. (2013). Motility was tested according to the method previously outlined by Dziewit et al. (2015). The hemolytic ability of different strains was tested by growth on blood agar plates that were incubated at 30 and 37°C under aerobic conditions—the results were read after 24 and 48 h of incubation. Siderophore production was assessed using the chrome azurol S (CAS) agar plate method, as described previously (Dziewit et al., 2015). The plates were incubated in the dark at 30°C for 72 h, after which halo formation around the colonies was assessed. Each isolate was phenotypically characterized using (i) the API 50 CH test system (to differentiate strains based on their ability to metabolize different carbohydrates) and (ii) the API ZYM test system (to detect and identify enzymatic activities); according to the recommendations of the supplier (bioMerieux, Marcy l'Etoile, France). Ureolytic activity was detected by cultivation on Christensen's urea medium, containing urea and a pH indicator (phenol red) (Christensen, 1946) or using rapid urease tests. Proteus vulgaris and/or Klebsiella pneumoniae strains (University of Warsaw collection) were used as positive controls, and P. aminophilus JCM 7686 and/or P. aminovorans JCM 7685 as negative controls (Urakami et al., 1990). The minimal inhibitory concentrations (MIC) of antimicrobial agents were determined for P. yeei strains by using Mueller Hinton II agar diffusion with E-tests as recommended by the supplier (bioMérieux). E. coli ATCC 25922 was used as a control. To confirm reproducibility, antimicrobial susceptibility testing was repeated at least twice. Since no interpretative guidelines for P. yeei susceptibility have been published, we used the criteria proposed by Arias and Clark (2019). MIC values for selected heavy metal ions were established on titration plates using a procedure described previously (Dziewit et al., 2013). The following heavy metal salts were used to prepare appropriate stock solutions in water: NaAsO2; 3CdSO4 × 8H2O; CoSO4 × 7H2O; K2Cr2O7; CuSO4; HgCl2; NiCl2 × 6H2O; NaO3V; ZnSO4 × 7H2O. Each microplate was monitored for growth using an automated microplate reader at 24-h intervals for 3 days. Isolates that demonstrated growth at the following minimum metal ion concentrations were classified as resistant using previously described criteria (Dziewit et al., 2015): (i) 20 mM V5+, (ii) 1 mM As3+, Cd2+, Co2+, Cu2+, Ni2+, Zn2+ or Cr6+, and (iii) 0.1 mM Hg2+.
2.3 DNA isolation, standard molecular biology procedures and PCR conditions
Plasmid DNA was isolated using the alkaline lysis procedure (Birnboim and Doly, 1979) or purified by CsCl-ethidium bromide gradient centrifugation (Sambrook and Russell, 2001). The visualization of mega-sized replicons was achieved by in-gel lysis and DNA electrophoresis (Eckhardt, 1978; Hynes and McGregor, 1990). Plasmid DNA was also isolated using a Plasmid Mini kit (A&A Biotechnology), GeneMATRIX Miniprep DNA Purification Kit (EURx) and GeneJET Plasmid Miniprep Kit (Thermo Fisher Scientific). Routine DNA manipulation was performed using standard methods (Sambrook and Russell, 2001). DNA amplification by PCR was performed in a Mastercycler (Eppendorf) using synthetic oligonucleotides (Supplementary Table S1), High Fidelity Taq DNA polymerase (Qiagen) or Phusion Hi-Fidelity DNA polymerase (Thermo Fisher Scientific), dNTPs and appropriate template DNAs, as described previously (Bartosik et al., 2003).
2.4 Introduction of plasmid DNA into bacterial cells
Chemical transformation of E. coli cells was performed by a standard method (Kushner, 1978). Plasmid DNA was introduced into Paracoccus spp. strains by triparental mating using helper E. coli strain DH5α carrying plasmid pRK2013 (containing the transfer system of plasmid RP4) (Ditta et al., 1980), as described previously (Lasek et al., 2018).
2.5 Plasmid host range testing
REP systems of extrachromosomal replicons of the analyzed strains were cloned in vector pABW1 (Bartosik et al., 1997) which cannot replicate in Paracoccus strains or other tested recipients (Supplementary Table S1). These shuttle plasmid constructs were introduced into the following strains: (i) class Alphaproteobacteria—Agrobacterium tumefaciens LBA 288R (Hooykaas et al., 1980), Paracoccus aminophilus JCM 7686R (without plasmids pAMI2, pAMI3) (Dziewit et al., 2014), Paracoccus aminovorans JCM 7686R (Czarnecki et al., 2017), Paracoccus pantotrophus KL100 (Jordan et al., 1997), (ii) class Betaproteobacteria—Achromobacter sp. LM16R (Dziewit et al., 2015) and (iii) class Gammaproteobacteria—Cronobacter sakazakii ATCC 29544 (Iversen et al., 2008) and Pseudomonas sp. LM6R (Dziewit et al., 2015).
2.6 Identification of functional transposable elements
Trap plasmids pMAT1 and pMEC1 (Bartosik et al., 2003; Szuplewska and Bartosik, 2009; Gay et al., 1985; Schneider et al., 2000) were used for the identification of functional TEs of P. yeei (Supplementary Table S1). These plasmids were transferred from E. coli TG1 to P. yeei strains by triparental mating. The “capture” of TEs was verified by PCR using primers specific to the selection cassettes, with DNA isolated from Tcr and Sucr mutants as the template (Supplementary Table S1). A total of 1,304 clones were tested this way. The amplified DNA fragments were subjected to electrophoretic analysis, which permitted the detection of DNA elements (TEs), ranging in size from ~0.8 to 1.5 kb, embedded in the cassettes. DNA sequencing of the termini of individual TEs was used to identify the complete elements within the genome sequences of their parental strains. All trapped TEs were sequenced and compared with the ISfinder database (Siguier et al., 2006).
2.7 Mutational analysis of URE modules
To confirm that the identified URE modules are responsible for ureolytic activity, mutational analysis was performed using the gene replacement method. Mutations (insertion of a Kmr cassette) were introduced into URE type 1 modules, either in the 5′ region of the Ureα subunit gene or within the nikR gene. Both mutations were created in the strain CCUG 32053 (which contains only URE type 1) and the strain CCUG 13493 (which also contains a URE type 2 module). The following procedure was used to obtain ureα::Kmr and nikR::Kmr mutants: (i) DNA fragments containing ureα-ureβ-ureγ or part of the region encoding NikR and nickel transporters were amplified by PCR using CCUG 32053 DNA as the template, (ii) these DNA fragments were cloned into the SacI and SphI sites of the mobilizable suicide vector pDS132 (Philippe et al., 2004), (iii) the Kmr cassette from plasmid pDIY-Km (Dziewit et al., 2011) was inserted into the AleI site within these fragments to obtain constructs pDS132-ureα and pDS132-nikR (Supplementary Table S1); (iv) these plasmids were then introduced into strains CCUG 32053R and CCUG 13493R by conjugation. Mutant clones ureα::Kmr and nikR::Kmr were selected on medium supplemented with kanamycin (for recombinant selection) and sucrose (for counter-selection). The introduced mutations were confirmed by DNA sequencing. Ureolytic activity of the obtained mutants was tested following growth on minimal medium supplemented with 1% urea, using rapid urease tests. Complementation of the mutants was performed using plasmids pBBR-ureα or pBBR-nikR, which were prepared by cloning PCR fragments containing ureα-ureβ-ureγ or part of the region encoding NikR and nickel transporters into vector pBBR-MCS5 (Gmr) (Kovach et al., 1995) (Supplementary Table S1).
2.8 Genome sequencing
The P. yeei genomes were sequenced using a combination of Oxford Nanopore and Illumina technologies, as described previously (Lasek et al., 2018).
2.9 Bioinformatic analyses
Sequence annotation and bioinformatic analyses (identification of tRNA genes and rRNA operons, relaxases—MOB, toxin-antitoxin modules, transposable elements—TE) were performed as described previously (Lasek et al., 2018). Potential ICE and IME elements were predicted using ICEfinder (Liu et al., 2019).
Categories for clusters of orthologous groups (COGs) were assigned to each protein by performing a local RPS-BLAST search against the COG database (last updated on January 22, 2015). A threshold e-value of 1e-5 was used, and only the top BLAST hits were taken into account (Tatusov et al., 2003).
The core genome of Paracoccus spp. and P. yeei species was defined based on the complete genomic sequences of P. aminophilus JCM 7686 (Dziewit et al., 2014), P. aminovorans JCM 7685 (Czarnecki et al., 2017), P. contaminans RKI16-01929T (Aurass et al., 2017) and P. denitrificans PD1222 (NC_008686-8), and eleven P. yeei strains: CCUG 32053 (Lasek et al., 2018), FDAARGOS_252 (NZ_CP020440–47), FDAARGOS_643 (NZ_CP044078-82), TT13 (Lim et al., 2018) and CCUG 13493, CCUG 17731, CCUG 32052, CCUG 32054, CCUG 46822, CCUG 54214, LM20 (Dziewit et al., 2015). Proteins encoded within the genomes were used in all-against-all BLASTp searches utilized by a stand-alone version of OrthoVenn2 (Xu et al., 2019), using an e-value of 1e-15 as the threshold and inflation level of 1.5. Based on this analysis, all proteins were clustered into groups reflecting similarity and designated as core proteins when encoded by all eleven genomes or as singletons when the respective gene was identified in only a single genome. A similar approach with a different manner of grouping was applied to identify strain-specific gene clusters.
Protein datasets from the Virulence Factor Database VFDB (Chen et al., 2016), the Comprehensive Antibiotic Resistance Database CARD (Alcock et al., 2020) and Virulence Factors Database VICTORS (Sayers et al., 2019), all downloaded on April 1 2021, were used for BLASTp searches (e-value threshold of 1e-30 and 80% of query coverage per HSP) with the proteomes of P. yeei and four other Paracoccus spp. strains listed above to identify putative P. yeei species virulence determinants.
EasyFig (Sullivan et al., 2011) was used to perform comparative genomic analyses (including genomes and plasmidomes comparisons) and visualize the results. RNA secondary structures were predicted by in silico folding using Mfold software (Zuker, 2003).
Phylogenetic analysis of the genus Paracoccus was based on alignment of concatenated nucleotide sequences of selected core genes: atpD, dnaA, dnaK, gyrB, recA, rpoB, and thrC (homologous genes of Roseobacter denitrificans OCh 114 were used as an outgroup), as described previously (Lasek et al., 2018). Nucleotide alignments were obtained with Mafft (Katoh et al., 2019). Then, concatenated genes were analyzed with ModelTest-NG (Darriba et al., 2020), checking all models to select the best-fit nucleotide substitution model. The selected substitution model was applied in RaxML-NG (Kozlov et al., 2019) with 2,000 regular bootstrap replicates performed on the best Maximum Likelihood (ML) tree selected from 100 independently generated ML starting trees.
2.10 Nucleotide sequence accession numbers
The nucleotide sequences of the P. yeei chromosomes and extrachromosomal replicons were deposited in GenBank (NCBI), with the following accession numbers: (i) CCUG 13493—CP038080 (chromosome), CP038073 (plasmid pYEE13493P1), CP038074 (pYEE13493P2), CP038075 (pYEE13493P3), CP038076 (pYEE13493P4), CP038077 (pYEE13493P5), CP038078 (pYEE13493P7), CP038079 (pYEE13493P8), CP038081 (pYEE13493P6); (ii) CCUG 17731—CP038042 (chromosome), CP038035 (pYEE17731P1), CP038036 (pYEE17731P2), CP038037 (pYEE17731P3), CP038038 (pYEE17731P4), CP038039 (pYEE17731P5), CP038040 (pYEE17731P6), CP038041 (pYEE17731P7); (iii) CCUG 32052—CP038090 (chromosome), CP038082 (pYEE32052P1), CP038083 (pYEE32052P2), CP038084 (pYEE32052P3), CP038085 (pYEE32052P4), CP038086 (pYEE32052P5), CP038087 (pYEE32052P6), CP038088 (pYEE32052P7), CP038089 (pYEE32052P8); (iv) CCUG 32054—CP038095 (chromosome), CP038091 (pYEE32054P6), CP038092 (pYEE32054P7), CP038093 (pYEE32054P8), CP038094 (pYEE32054P9), CP038096 (pYEE32054P1), CP038097 (pYEE32054P2), CP038098 (pYEE32054P3), CP038099 (pYEE32054P4), CP038100 (pYEE32054P5); (v) CCUG 46822—CP038056 (chromosome), CP038043 (pYEE46822P1), CP038044 (pYEE46822P2), CP038045 (pYEE46822P3), CP038046 (pYEE46822P4), CP038047 (pYEE46822P5), CP038048 (pYEE46822P6), CP038049 (pYEE46822P7), CP038050 (pYEE46822P8), CP038051 (pYEE46822P9), CP038052 (pYEE46822P10), CP038053 (pYEE46822P11), CP038054 (pYEE46822P12), CP038055 (pYEE46822P13); (vi) CCUG 54214—CP038061 (chromosome), CP038057 (pYEE51214P1), CP038058 (pYEE51214P2), CP038059 (pYEE51214P3), CP038060 (pYEE51214P4); (vii) LM20—CP038072 (chromosome), CP038062 (pLM20P1), CP038063 (pLM20P2), CP038064 (pLM20P3), CP038065 (pLM20P4), CP038066 (pLM20P5), CP038067 (pLM20P6), CP038068 (pLM20P7), CP038069 (pLM20P8), CP038070 (pLM20P9), CP038071 (pLM20P10). The nucleotide sequences of newly identified ISs (ISPye73—ISPye80) and MITEPye1 were deposited in the ISfinder database (Siguier et al., 2006).
3 Results
3.1 P. yeei strains selected for characterization
Seven (five clinical and two environmental) isolates of P. yeei were selected for detailed characterization (Table 1). Clinical strains were sourced from two continents—they were isolated in different geographical locations, at different times and from different clinical cases (Table 1). The other strains were isolated either from an anthropogenic environment (CCUG 54214; a metal surface), or from the natural environment (LM20; organic-rich black shale from the Lubin copper mine in Poland; Dziewit et al., 2015). The clinical isolate CCUG 32053, characterized in a previous study (Lasek et al., 2018), was used as a reference strain.

Table 1. Paracoccus yeei strains analyzed in this study.
3.1.1 Physiological and phenotypic characterization
P. yeei is a non-motile, oxidase- and catalase-positive gram-negative bacterium. The analyzed strains were facultative aerobes capable of nitrate reduction and denitrification (except for CCUG 32052). The strains were mesophilic, capable of growth at temperatures ranging from 21 to 37°C (except LM20, which grew from 15 to 37°C) (Table 2). All strains grew in LB medium at pH values close to 7 (ranging from 5 to 8 or 9), typical for neutrophilic bacteria. Salinity tolerance testing showed that the isolates could tolerate NaCl concentrations of 4–6%. These bacteria were non-hemolytic, did not produce bacteriocins, and exhibited weak adhesion to polystyrene. However, all produced siderophores, as determined by the chrome azurol S (CAS) agar plate assay. They demonstrated the ability to degrade 27 carbohydrates (out of 49 tested), with identical profiles for all strains except LM20, which is unable to ferment mannitol, sorbitol or arabitol (Table 2). The enzymatic capabilities of these strains were assessed by testing for 19 hydrolytic enzymes from various groups. All strains produced alkaline phosphatase (except CCUG 13493), esterase, lipase esterase, leucine arylamidase, acid phosphatase, naphthol-AS-BI phosphohydrolase, and α-glucosidase, with only CCUG 17731 producing β-galactosidase (Table 2). The strains also displayed ureolytic activity, which is a unique feature among Paracoccus spp.
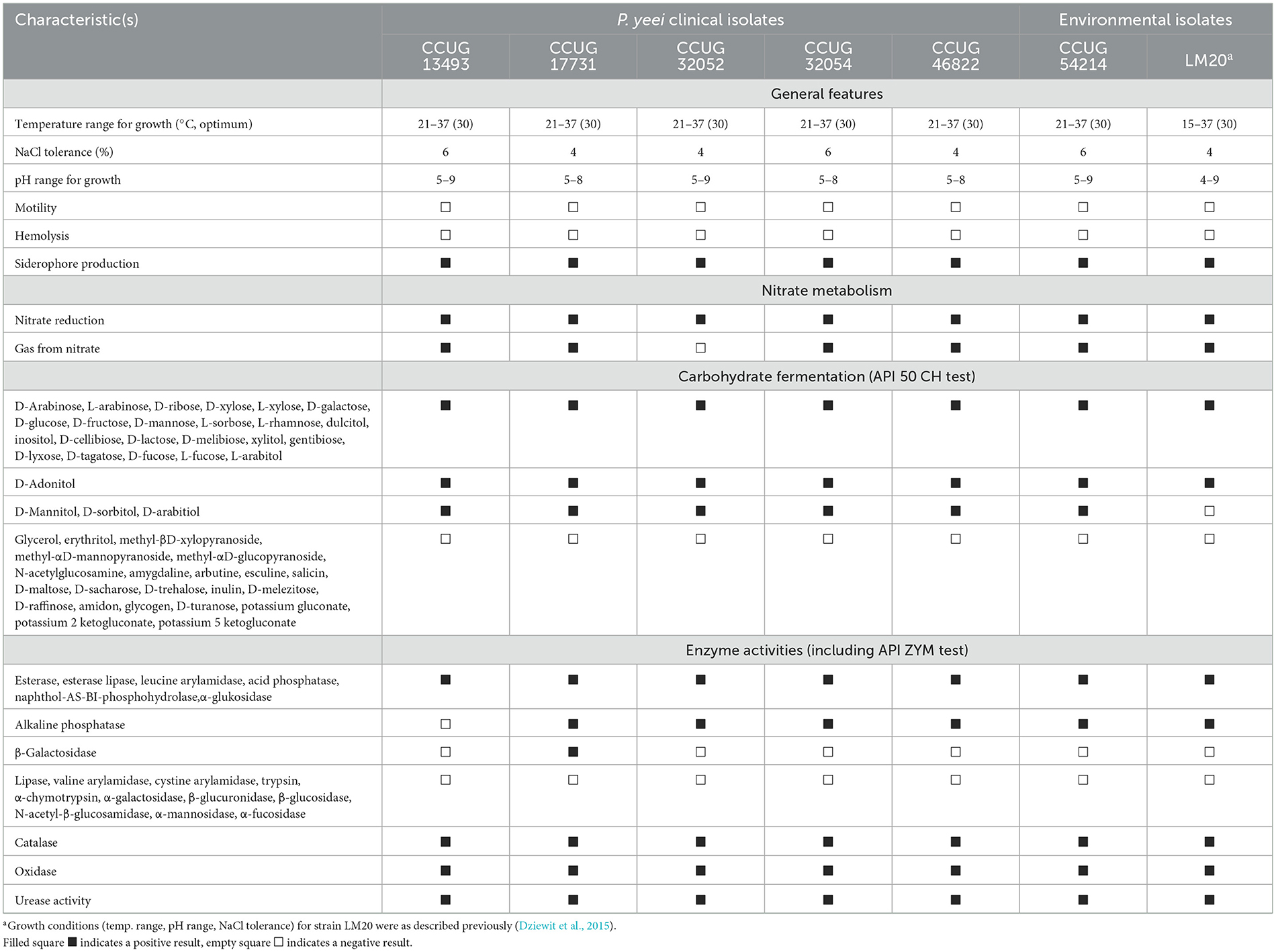
Table 2. Phenotypic and physiological characteristics of P. yeei strains.
MIC values for selected antibiotics (ampicillin, ciprofloxacin, erythromycin, gentamicin, tetracycline, and vancomycin) were determined for each strain (Supplementary Table S2). The data were analogous to those from previous studies of clinical isolates of P. yeei, showing sensitivity to beta-lactams, especially aminopenicillins, as well as macrolides and aminoglycosides (Funke et al., 2004; Wallet et al., 2010; Schweiger et al., 2011; Sastre et al., 2016; Arias and Clark, 2019; Aliste-Fernández et al., 2020). Nevertheless, we observed variations in the susceptibility of the isolates, e.g., CCUG 13493 was most sensitive to the tested antibiotics, CCUG 32054 was the most tolerant, and some strains (CCUG 32052, CCUG 32054, CCUG 46822, LM20) were less sensitive to ciprofloxacin (Supplementary Table S2).
MIC values for selected heavy metal ions were similar for all strains, with no major differences between clinical and environmental isolates. CCUG 54214 showed higher tolerance to As3+, Ni2+, V5+, and Zn2+, while LM20 showed higher tolerance to As3+, Cd2+, and Hg2+, reflecting the adaptation of these strains to the environments from which they were isolated (Supplementary Table S2).
3.2 Genomic features
The genomes of the seven strains of P. yeei were fully sequenced and characterized. Their genome sizes range from 4,423,927 to 4,829,807 bp (average 4,610,003 bp). All are multipartite, containing from 4 (CCUG 54214) to 13 (CCUG 46822) extrachromosomal replicons (ECRs), that range in size from 3.5 kb (pYEE46822P1 of CCUG 46822) to 485 kb (pYEE17731P7 of CCUG 17731) (Table 3; Figure 1B; Supplementary Figure S1). In total, the strains contain 59 ECRs (11,665,529 bp), which on average constitute ~25% of the size of each genome (Table 3; Figure 1B). The physicochemical parameters of the genome sequences were determined, including the %GC content (average 67%), the number of protein-coding sequences (CDSs) (average 4,200) and pseudogenes (average 190). The GC content of the ECRs ranges from 53% (pYEE46822P4, CCUG 46822) to 69% (pYEE13493P5, CCUG 13493). The number of CDSs present in ECRs ranges from 849 (CCUG 54214), accounting for 20% of the entire genome, to 1,270 (CCUG 17731) (28%) (Supplementary Table S3). The rRNA operons and most of the tRNA genes are localized within the chromosomes, although some additional tRNA genes (2–3) were identified within ECRs (Table 3).
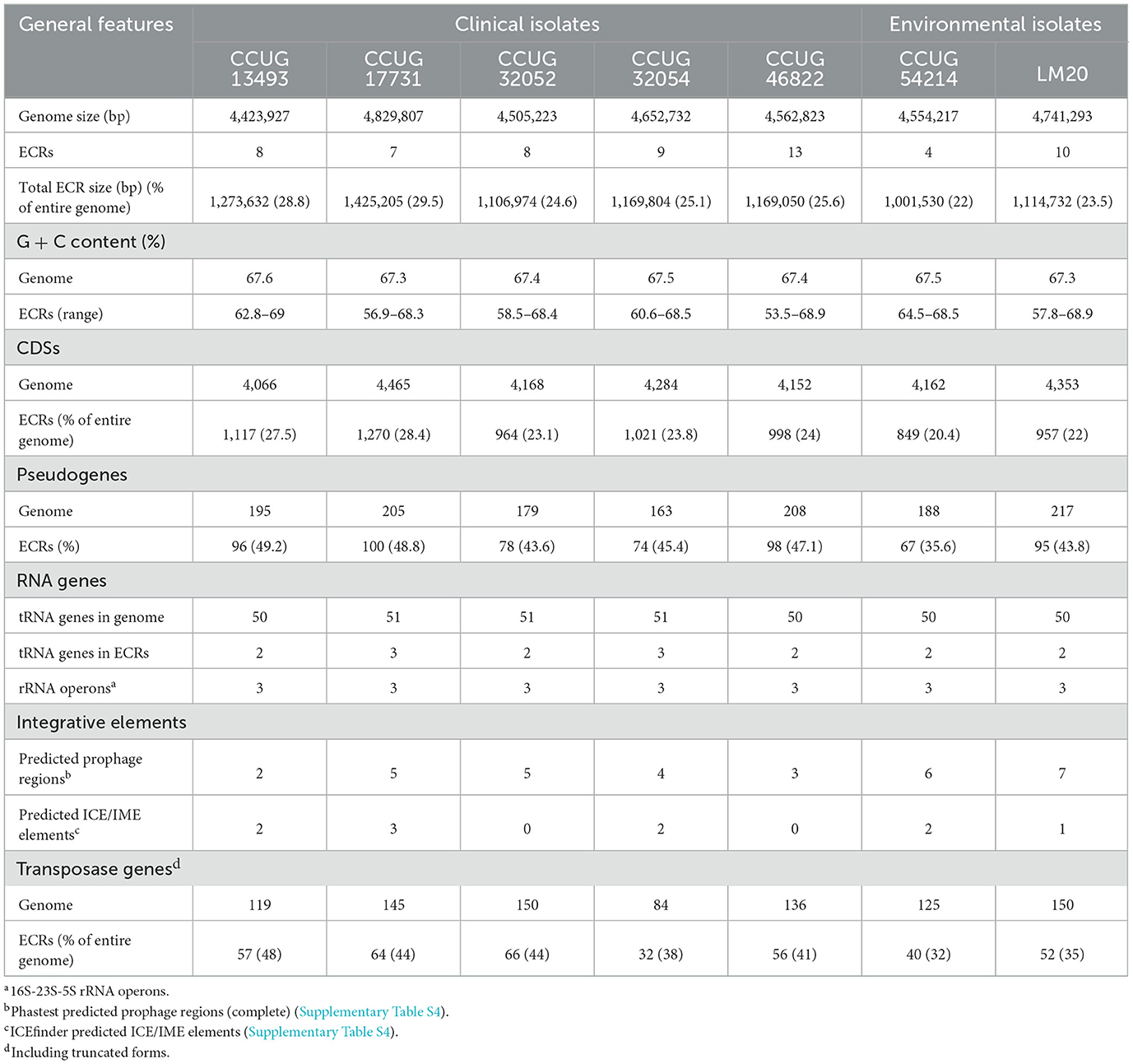
Table 3. General features of P. yeei genomes.
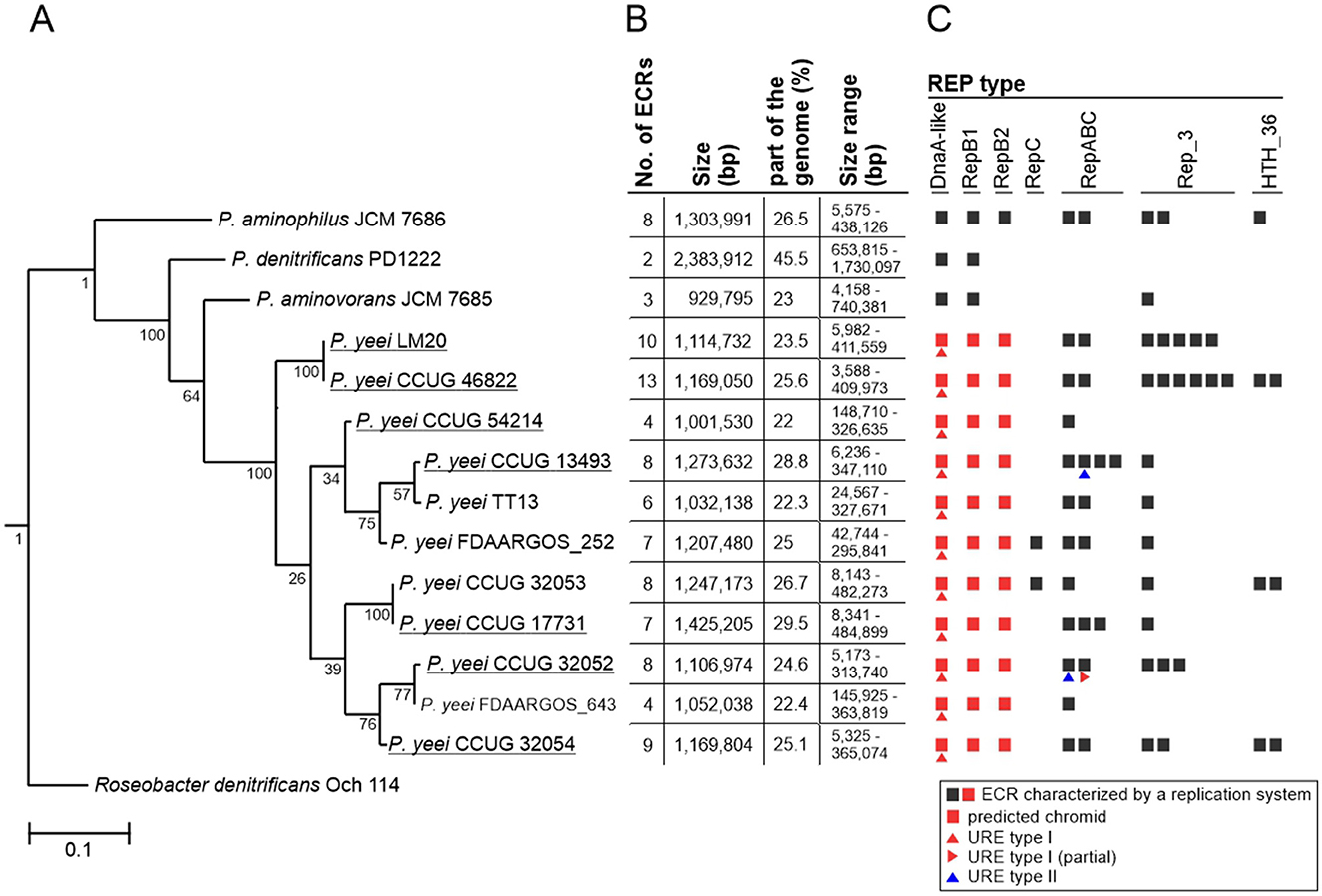
Figure 1. Distribution of ECRs in P. yeei genomes. (A) Phylogenetic tree of P. yeei constructed using core gene nucleotide sequences. Three other members of the genus Paracoccus—P. aminophilus JCM 7686, P. aminovorans JCM 7685, and P. denitrificans PD1222—as well as Roseobacter denitrificans Och 114 (as an outgroup) were included in the analysis. Bar represents 0.1 nucleotide substitutions per position. (B) Summary of data for ECRs of the analyzed Paracoccus spp. strains. (C) Distribution of different REP module types among ECRs of Paracoccus spp.
3.3 Extrachromosomal replicons
The analyzed P. yeei strains contain numerous ECRs (59 in total)—from 4 (CCUG 54214) to 13 (CCUG 46822) (Figure 1B; Supplementary Figure S1). These are mainly large DNA molecules, constituting a significant proportion of their host strain genomes—from 22% (CCUG 54214) to >28% (CCUG 13493) (Figure 1B). The replication system (REP) and the gene encoding the replication initiation protein (Rep) within each ECR were identified. The REP modules (and entire plasmids) were classified based on the presence of specific amino acid (aa) sequence signatures in the Rep proteins (Figure 2A). Replicons of the DnaA-like, RepB (RepB1 and RepB2), RepABC, RepC, Rep_3 and HTH_36 families were identified, all of which are common among Paracoccus spp. and other Alphaproteobacteria (Figures 1C, 2).
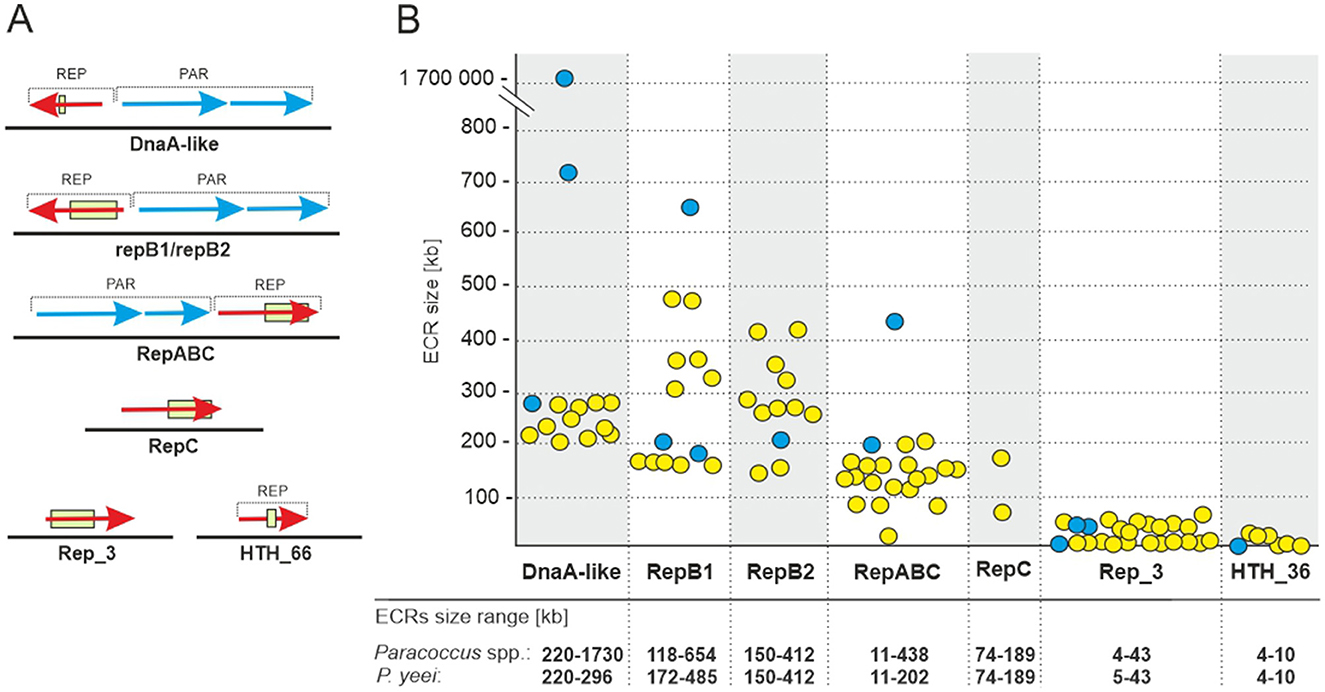
Figure 2. ECRs identified in the genomes of P. yeei strains. (A) Genetic organization of the replication (REP) and partitioning modules of the ECRs. The box indicates the location of sequence motifs specific to each type of replication initiation protein. (B) Size range of P. yeei replicons carrying a given REP module type (yellow). The blue color indicates replicons present in the genomes of P. aminophilus JCM 7686, P. aminovorans JCM 7685, and P. denitrificans PD1222.
The ECRs carrying REPs of a given type are generally similar in size, with the exception of RepB1 and RepB2 replicons, in which greater size variation is observed (Figure 2B). The REPs of the DnaA-like, RepB and RepABC types (occurring in each P. yeei strain) are characteristic for large replicons, which is consistent with previous observations (e.g., Lasek et al., 2018). Detailed characterization of individual ECRs, including their size, nucleotide sequence GC content, coding capacity and tRNA gene distribution, is presented in Supplementary Table S3.
To test the host range of individual ECRs representing the different replicon families, their REP regions were cloned in mobilizable shuttle plasmids and introduced by triparental mating into recipient strains from different classes of Proteobacteria (listed in Section 2). This analysis revealed that all of the analyzed REPs function solely in Alphaproteobacteria, so have a relatively narrow host range.
The P. yeei ECRs contain numerous loci enabling their stable maintenance in bacterial cells and populations. The DnaA-like, RepB, and RepABC replicons all carry type I partitioning systems, encoding Walker-type ATPases (Thomas, 1981) (Figure 2A). The vast majority of ECRs also have class II toxin-antitoxin (TA) systems, mainly encoding toxins of the VapC family (18 toxins), as well as toxins of the RelE/ParE (14), PhD/YefM (14) or parDE (8) families. None of the ECRs encode a complete type IV secretion system (T4SS), suggesting that these replicons are not self-transmissible. However, 16 of them contain genes encoding predicted relaxases of the MobC (14), MobQ (1), and MobHEN (1) families (Garcillán-Barcia et al., 2009), which are typical components of genetic modules, enabling plasmid mobilization for conjugative transfer (MOB).
Further analysis was conducted to verify whether the P. yeei genomes contain ECRs that meet the criteria for chromids (secondary chromosomes)—essential replicons of plasmid origin. This revealed that all the DnaA-like, RepB1 and RepB2 replicons can be considered chromids (Figure 1C), since they (i) carry plasmid-type REP modules, (ii) have a nucleotide sequence composition that is close to that of the chromosome (< 1% difference in GC content), and (iii) possess a set of core genes (in one copy in the genome) characteristic for the entire genus (Harrison et al., 2010). The genes carried by these chromids participate in several important metabolic pathways, e.g., encoding acetyl-CoA C-acyltransferase, and proteins likely to be involved in biosynthesis of the lipoyl cofactor, in oxidative metabolism, leucine metabolism, and electron transport via the respiratory chain (complex I).
The Rep proteins of the DnaA-like, RepB1 and RepB2 chromids display a high level of aa sequence identity within a given replicon group. These replicons are also more highly conserved in terms of their structure and genetic load (Supplementary Table S5). In contrast, the replication initiators of the RepABC, Rep_3 and HTH_36 families (and their entire replicons) show much greater sequence variability. Many of these replicons appear to be unique among P. yeei strains, suggesting their recent horizontal acquisition (Supplementary Table S5).
3.4 Comparative analysis of P. yeei genomes
Phylogenetic analysis showed that the two environmental isolates (CCUG 54214 and LM20) are situated on different branches of the P. yeei phylogenetic tree and are closely related to the clinical isolates. Strains LM20 and CCUG 46822 form a separate cluster, while the other five strains can be divided into two phylogenetic groups: (i) CCUG 54214 and CCUG 13493, and (ii) CCUG 17731, CCUG 32052 and CCUG 32054 (Figure 1A).
Comparative genomic analysis of the P. yeei strains (including clinical isolates CCUG 32053, FDAARGOS_643, FDAARGOS_252, and TT13) revealed a conserved genome structure, preserved ECR composition and synteny of gene arrangement in this species (Supplementary Table S5). There is a particularly high degree of synteny between the genomes of the strains LM20 and CCUG 46822 (Figure 3). The ECRs of these strains are nearly identical, although they display minor rearrangements (Supplementary Table S5).
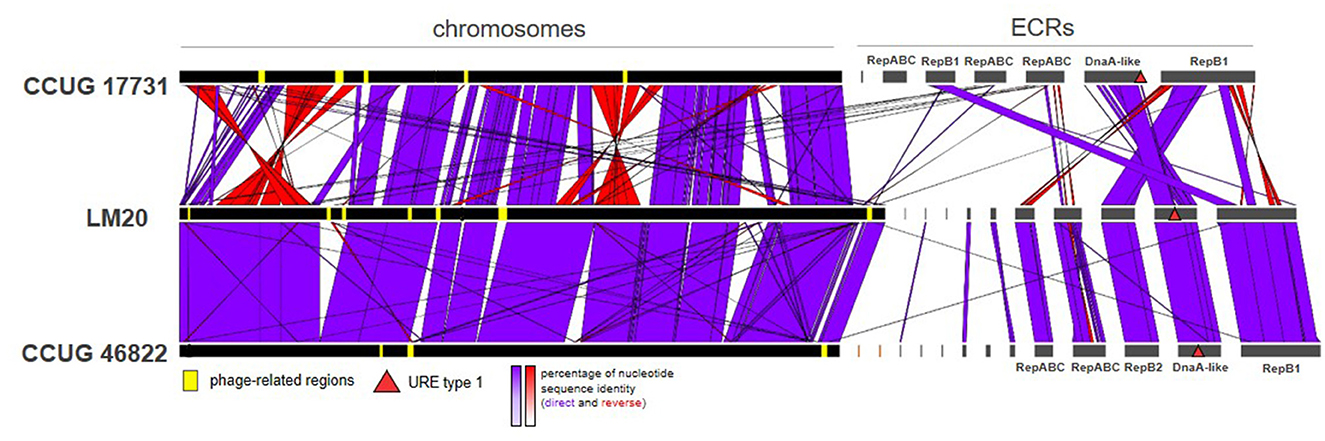
Figure 3. Comparative genomic analysis of three P. yeei strains, CCUG 17731, LM20, and CCUG 46822, illustrating the two chromosome structural variants identified in this species.
Our analysis identified two chromosome structure variants (one found in strains CCUG 17731, CCUG 32052, CCUG 32053, CCUG 32054, CCUG 54214, FDAARGOS_643, TT13, and the other in CCUG 13493, CCUG 46822, LM20, FDAARGOS_252), which correlates with the phylogeny of the strains (Figure 3; Supplementary Table S5). The differences result mainly from inversions or translocations of fairly large DNA segments (Figure 3).
The DnaA-like replicons show a high degree of conservation, although with some rearrangements, e.g., translocation of DNA segments in strains CCUG 32053 and CCUG 13493, sometimes with inversion of the transferred DNA segment (Supplementary Table S5).
3.5 Genetic load of P. yeei genomes
Analysis of the proteome of P. yeei determined in silico permitted the assignment of individual proteins to functional COG groups. For all genomes a similar proportion of the encoded proteins belong to each category, indicating a conserved genome structure and limited variability within the species. Interestingly, significant similarity in the distribution of COG groups was found between the chromosomes and ECRs, suggesting that ECRs may contain a large amount of genetic information of chromosomal origin (Figure 4).
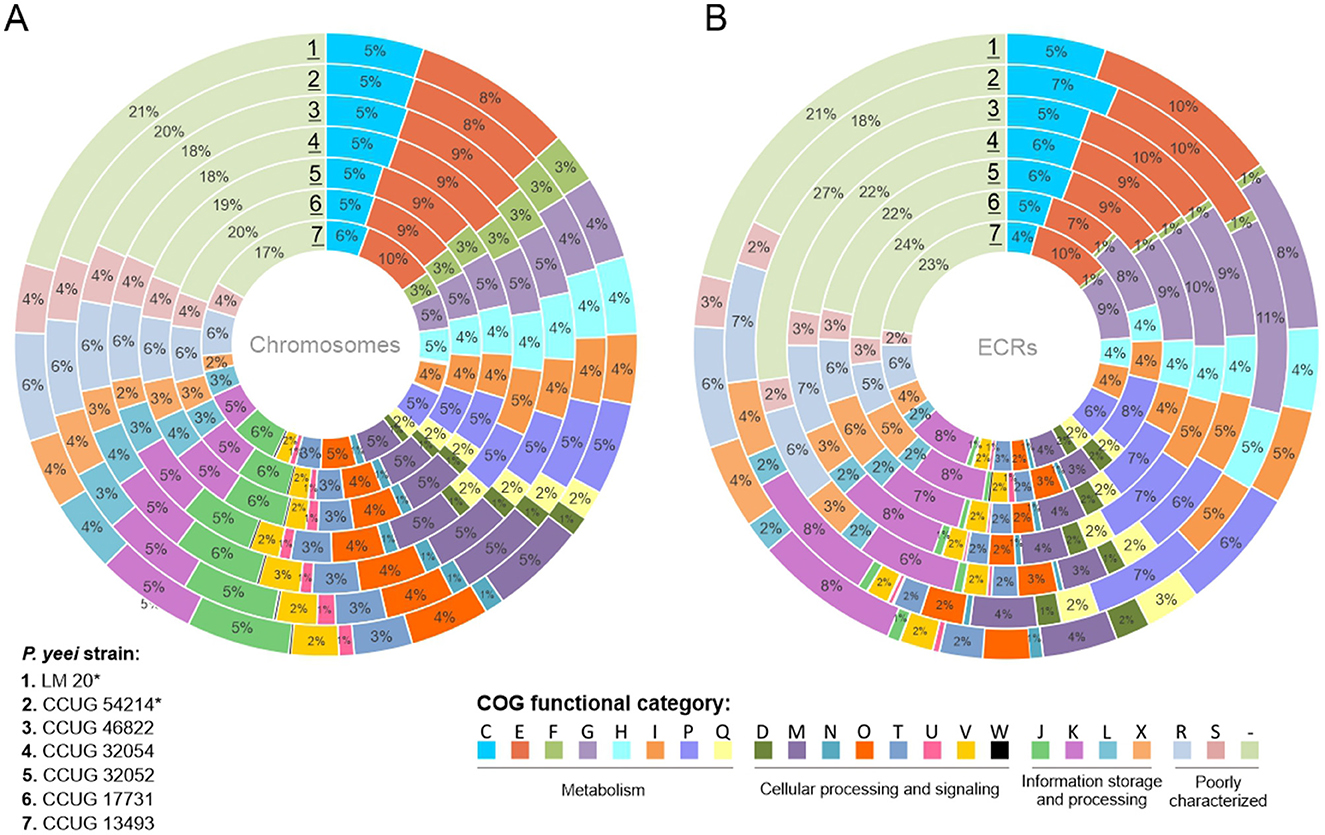
Figure 4. Distribution of genes encoding proteins belonging to different COG functional categories within (A) the chromosomes and (B) ECRs of the analyzed P. yeei strains (*environmental isolates). Each colored segment indicates the relative contribution of a functional category as a percentage of total COGs. Each ring represents a different strain of P. yeei. COG functional categories: C, energy production and conversion; E, amino acid transport and metabolism; F, nucleotide transport and metabolism; G, carbohydrate transport and metabolism; H, coenzyme transport and metabolism; I, lipid transport and metabolism; P, inorganic ion transport and metabolism; Q, secondary metabolite biosynthesis, transport and catabolism; D, cell cycle control, cell division, chromosome partitioning; M, cell wall/membrane/envelope biogenesis; N, cell motility; O, posttranslational modification, protein turnover, chaperones; T, signal transduction mechanisms; U, intracellular trafficking, secretion and vesicular transport; V, defense mechanisms; W, extracellular structures; Z, cytoskeleton; J, translation, ribosomal structure and biogenesis; K, transcription; L, replication, recombination and repair; X, mobilome, i.e., prophages, transposons; R, general function, prediction only; S, function unknown, COG not assigned.
In all cases, a considerable number of genes are of unknown function (R and S categories) or could not be classified into any COG group: 27–31% for chromosomes and 27–32% for ECRs. The largest group of classified proteins comprises those related to amino acid transport and metabolism (category E; 9–10%), followed by carbohydrate metabolism (G; 5–6%), transcription (K; 6%), transport and metabolism of inorganic ions (P; 5–6%), energy acquisition processes (C; 5%), and the synthesis of cell wall, membrane, and envelope components, including capsules (M; 5%) (Supplementary Figure S2). In the case of ECRs, the largest fraction of classified proteins was assigned to categories E (7–10%), G (8–11%), P (6–8%), and K (6–8%) (Figure 4B). There are no significant differences between clinical and environmental isolates in the distribution of COG categories.
Transport-related genes constitute a dominant group of functional genes in the P. yeei genomes—from 530 (CCUG 32052) to over 600 CDSs (CCUG 17731). These genes encode various types of transporters: ABC, MSF (Major Facilitator Superfamily), P-type, RND (Resistance-Nodulation-Division), TRAP (Tripartite ATP-independent Periplasmic), and TTT (Tripartite Tricarboxylate Transporters) (Table 4; Supplementary Table S6). ECRs (mainly DnaA-like and RepB chromids) contain over 35% of the ABC transporter genes. Less common in ECRs are MFS, RND and TTT transporter genes. Only a few genes encoding proteins with similarity to P-type ATPase integral membrane transporters were found in the analyzed genomes.
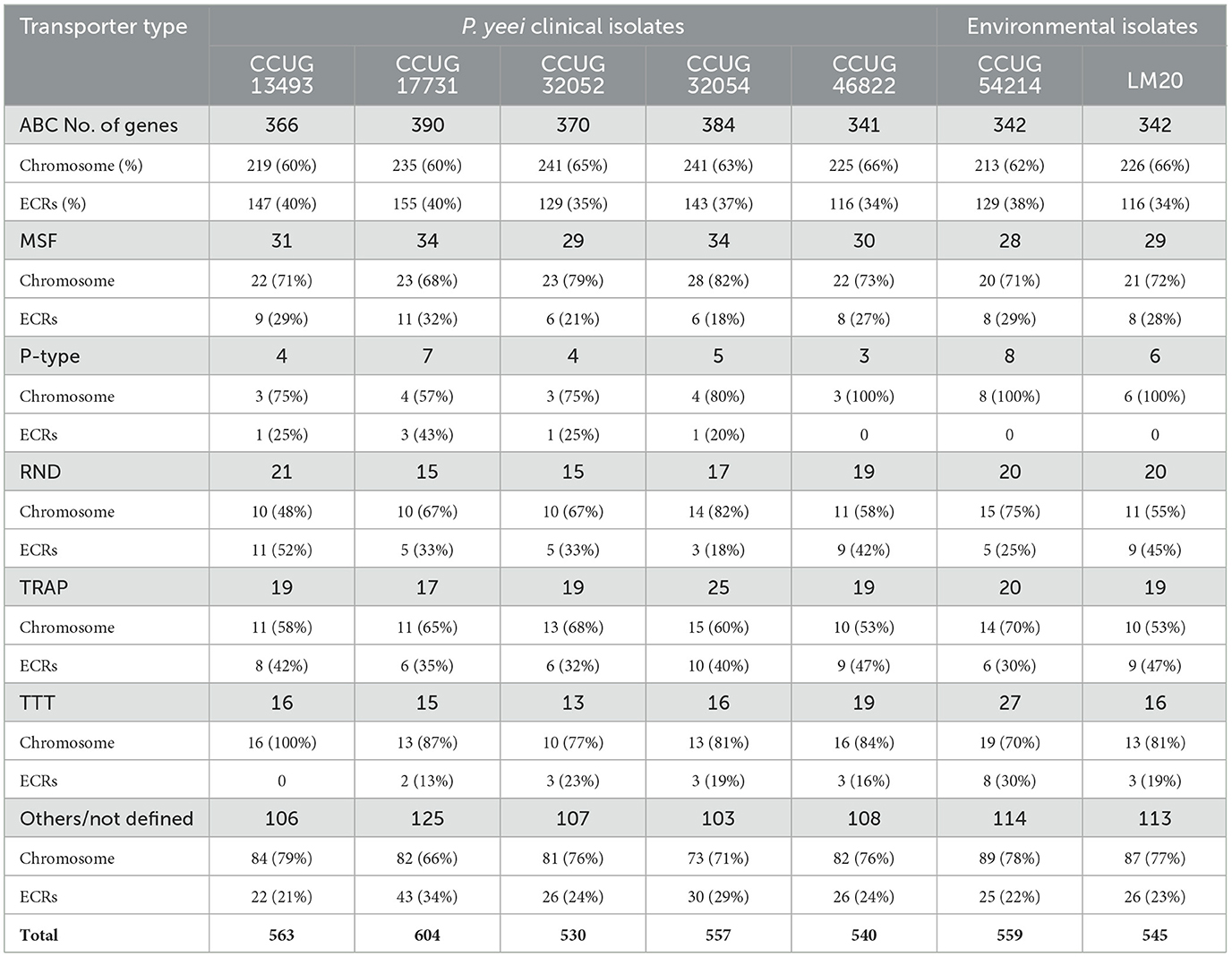
Table 4. Distribution and characterization of transport-related genes in P. yeei genomes.
The ECRs also carry numerous genes that may increase the adaptability of their host strains in the environment. All types of chromids and RepABC plasmids encode potential iron or carbohydrate transporters. Siderophore receptor genes were identified in all RepB2 plasmids, including the FhuADCB ferric hydroxamate transporter, which is essential for the uptake of Fe3+-aerobactin. All RepB1 replicons carry a mauMGJCBDEAF gene cluster involved in the utilization of methylamine (Baker et al., 1998). Organisms that utilize C1 compounds as their sole carbon and energy sources are described as methylotrophs and this trait is common in bacteria of the genus Paracoccus (Czarnecki and Bartosik, 2019). Methylotrophs play an important role in global carbon, nitrogen and sulfur cycling, and they have been successfully employed in the bioremediation of contaminated soils (Chistoserdova, 2011).
The RepB1 replicons of three phylogenetically closely related P. yeei strains (CCUG 17731, CCUG 32052, CCUG 32054) (Figure 1A) contain a putative operon (phnCDEFGHIJKLMNOP) encoding carbon-phosphorus lyase, which permits the utilization of phosphorus from a wide range of stable phosphonate compounds containing a C-P bond (Makino et al., 1991; Horsman and Zechel, 2017; Amstrup et al., 2023). In numerous environments, inorganic phosphate, a vital nutrient, is available in very limited amounts, compelling microorganisms to rely on alternative phosphorus sources for survival (Podzelinska et al., 2009). Therefore, the ability to uptake and breakdown phosphonates would be advantageous.
Enzymes catalyzing rhamnose biosynthesis (RfbABCD) are encoded by all RepB2 replicons (CCUG 32052 has an additional copy of the rfbABCD operon in Rep_3 plasmid pYEE32052P3). Rhamnose is present in the O-antigens of many gram-negative bacteria, forming part of the lipopolysaccharide (LPS). This deoxy-hexose sugar is also present in capsular polysaccharides, covalently bound to the cell wall, and in exopolysaccharides that are loosely associated with the cell wall (Marolda and Valvano, 1995). The P. yeei strains analyzed in this study display characteristic mucoid growth, probably related to the presence of a polysaccharide capsule (data not shown).
The ECRs also carry genes for proteins involved in (i) the cobalamin (vitamin B12) biosynthetic pathway (cobAWNGHIJKLMBF; DnaA-like chromids), (ii) ectoine biosynthesis (ehuABCD; RepB1 chromids), (iii) propionate catabolism (mmgE/prpD; several RepB1, RepB2, and RepABC replicons), (iv) disulfide bond formation (dsbA, dsbB, and dsbE), and (v) resistance to arsenic compounds (permease Arc3, arsenate reductase ArsC, flavoprotein ArsH, repressor ArsR, and transporter ArsJ). Notably, all RepB1 chromids carry a complete conserved set of crt genes (crtXYIBZE-idi) for carotenoid synthesis (Maj et al., 2013). However, colonies of the strains with these chromids lack the characteristic color associated with carotenoid pigment production and these compounds could not be detected by HPLC analysis (data not shown).
In summary, the DnaA-like, RepB1 and RepB2 chromids and the RepABC plasmids carry the largest number of genes of adaptive potential. The genetic load of smaller replicons (HTH_36 or Rep_3) consists mainly of genes of unknown function. The distribution of selected genes in individual ECRs is presented in Supplementary Table S3.
3.6 Transposable elements
A search for transposable elements (TEs) in the P. yeei genomes revealed the presence of over 900 tnp genes (complete or partial) encoding transposases from 18 families of insertion sequences (ISs): IS3, IS5, IS6, IS21, IS30, IS66, IS91, IS110, IS256, IS481, IS630, IS701, IS1182, IS1202, IS1380, IS1595, ISL3, and ISNCY (Figure 5; Supplementary Table S7). The number of tnp genes in individual genomes ranges from 84 (CCUG 32054) to 150 (CCUG 32052 and LM20). The dominant elements are representatives of the IS3, IS5, IS110, and IS256 families. Several IS families were identified in only one or a few isolates, e.g., IS6—CCUG 17731 and CCUG 32052, IS91—CCUG 46822, IS630—CCUG 32054 and LM20, IS1202—CCUG 46822, IS1380—CCUG 46822, IS1595—CCUG 13493, CCUG 46822 and LM20, and ISNCY—CCUG 13493 and CCUG 32054 (Supplementary Table S7).
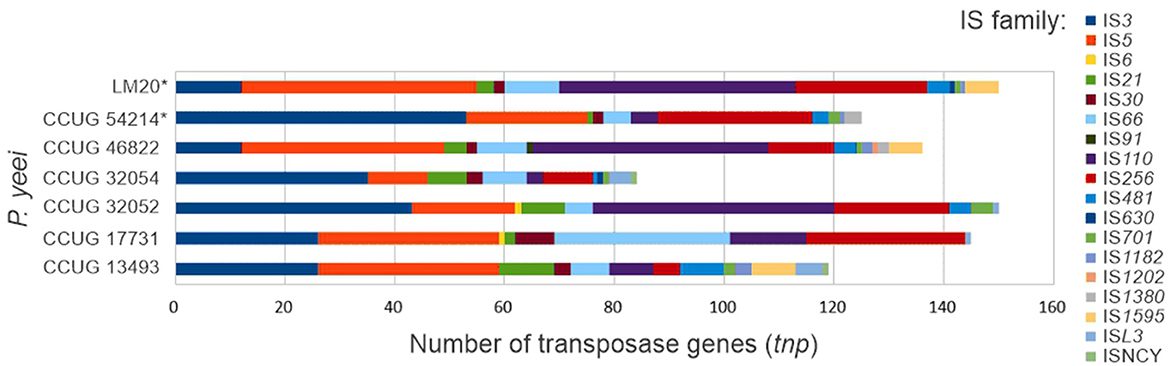
Figure 5. Distribution of insertion sequences in P. yeei genomes (*environmental isolates).
Numerous ISs are localized within ECRs. Surprisingly, most of the largest ECRs (DnaA-like and RepB chromids), except for those from CCUG 46822 and LM20, contain relatively few ISs, indicating a more conserved replicon structure and the presence of essential genes. The replicons richest in these elements are RepABC plasmids, which in most cases carry almost 50% of the tnp genes of a given strain (Supplementary Table S7).
In the chromosomes and ECRs of strains CCUG 46822 and LM20 several repetitive IS arrays were identified with a structure resembling composite transposon (Supplementary Table S7). These DNA regions are flanked by divergently oriented complete isoforms of ISPye46 (1361 bp; IS110 family) and contained within them are tnp genes of the IS256 family (related to ISPye43) and/or the IS5 family (related to ISPye12). Each copy of ISPye46 is flanked by identical DRs (5′-TG-3′), so it is not clear whether these IS110-IS5/IS256-IS110 arrays constitute functional TEs or are the result of preferential transposition of ISPye46 into ISs representing the IS256 or IS5 families. Interestingly, ISPye46 is dominant in the analyzed P. yeei genomes (47 copies in total); however, most of the copies are present in strains CCUG 46822 (19 copies) and LM20 (24 copies) (Figure 5; Supplementary Table S7). No related IS110-IS5/IS256-IS110 arrays were identified in the genomes of other bacteria by BLASTn analysis.
Interestingly, a 159-bp-long sequence element was identified in most chromosomes of the analyzed P. yeei strains (Supplementary Table S7). These elements show high sequence similarity (96–100% identity), lack any open reading frames, occur in intergenic regions at different locations, and are flanked by short (4 bp) direct repeats (DRs), suggesting their acquisition through transposition (Supplementary Table S7). Additionally, they contain inverted repeats at both termini (TIRs; IRL and IRR) (18 bp) with significant similarity to the TIRs of insertion sequences of the IS1182 family, including ISPye18, previously identified in the CCUG 32053 genome (Lasek et al., 2018) (Figure 6A). Analysis of the distribution of ISPye18 in P. yeei genomes revealed the presence of intact isoforms of this element in strains FDAARGOS_252 (1 copy; plasmid 4) and CCUG 32053 (1 copy; plasmid pYEE3). However, related ISs from the IS1182 family were also identified in the genomes of CCUG 13493, CCUG 46822, CCUG 54214, and LM20 (Supplementary Figure S3).
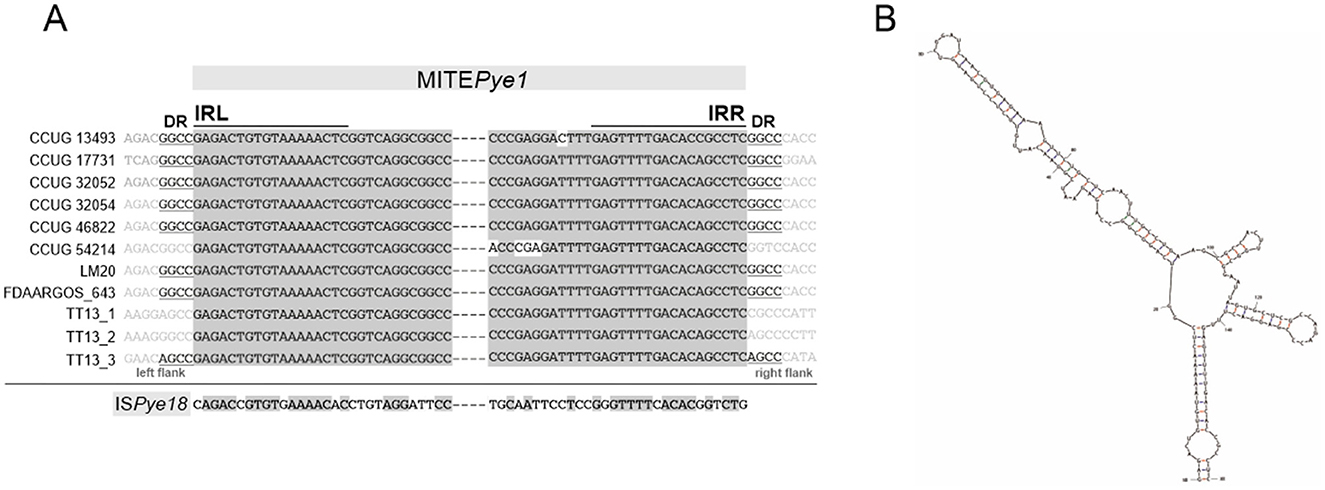
Figure 6. Non-autonomous transposable element MITEPye1 identified in P. yeei strains. (A) Alignment of terminal inverted repeat nucleotide sequences (IRL, left IR; IRR, right IR) of complete MITEPye1 elements identified in P. yeei genomes and ISPye18. Identical residues are indicated by gray shading. Nucleotide sequences of direct repeats (DRs) generated by MITEPye1 elements during transposition are indicated by underlining. (B) RNA secondary structures predicted by in silico folding using Mfold software for MITEPye1. The minimum folding energy (ΔG −47.93) of the predicted secondary structures was calculated by Mfold.
Considering (i) the potential mobility of these DNA regions, (ii) their small size, (iii) the presence of TIRs, (iv) DRs, and (v) the absence of a transposase gene, it seems likely that they constitute a novel group of MITE-type non-autonomous TEs (Delihas, 2011), that are most probably mobilized for transposition by ISs of the IS1182 family. This element was named MITEPye1 (ISfinder).
The activity of TEs of P. yeei was investigated in vivo using trap plasmids that allow positive selection of transposition events. Two trap plasmids were used, pMEC1 and pMAT1, containing the cI-tetA and sacB selection cassettes, respectively. Transposition of TEs into these cassettes resulted in the appearance of new phenotypes—the cells carrying a mutated plasmid become resistant to tetracycline (pMEC1) or to sucrose (pMAT1) (see Section 2 for details).
Despite the presence of a very large number of transposase genes in the P. yeei genomes, use of the entrapment vectors led to the identification of only six distinct elements: (i) three ISs previously identified in silico in the CCUG 32053 genome—ISPye2 (IS5 family, IS5 group), ISPye38 (IS5 family) and ISPye41 (IS5 family, IS903 group), (ii) an isoform of IS1247 of Xanthobacter autotrophicus (IS1380 family), and (iii) two novel elements, designated ISPye79 and ISPye80, representing the IS5 and IS427 groups within the IS5 family, respectively.
The distribution of every defined IS of P. yeei (ISfinder and this study) across all P. yeei genomes is shown in Supplementary Figure S3. The most abundant elements are (i) ISPye46−47 copies in 5 strains (most copies present in CCUG 46822 and LM20), (ii) ISPye53−45 copies in seven strains, and (iii) ISPye41−30 copies in six strains (most copies present in CCUG 17731 and CCUG 32053). Many of the ISs are present in only 1 copy, e.g., (i) ISPye13 (chromosome FDAARGOS_252), (ii) ISPye63 (chromosome of CCUG 32052), and (iii) IS1247a (chromosome of CCUG 46822).
3.7 P. yeei strain- and species-specific genes
The core genomes of Paracoccus spp. and of P. yeei were defined based on the complete genomic sequences of P. aminophilus JCM 7686 (Dziewit et al., 2014), P. aminovorans JCM 7685 (Czarnecki et al., 2017), P. contaminans RKI16-01929T (Aurass et al., 2017), P. denitrificans PD1222, plus eleven P. yeei strains. Comparative genomic analysis was used to define (i) the core genome of bacteria of the genus Paracoccus spp., (ii) genes specific to P. yeei (not found in other Paracoccus spp. with complete genomes), and (iii) genes that are unique to individual P. yeei strains (singletons). The collected data, also showing the distribution of these genes in the individual replicons of each P. yeei strain, are presented in Supplementary Table S8.
This analysis revealed that the core genome of Paracoccus spp. consists of 1,647 protein-encoding genes, of which 116 are located within ECRs of P. yeei. The total number of P. yeei-specific genes is 250 and the number of singletons ranges from 138 (CCUG 46822) to 332 (FDAARGOS 252) (Supplementary Table S8). In some strains, the majority of singletons occur in the chromosome, while in others (CCUG 13493, CCUG 17731) they are predominantly in ECRs. The vast majority of singletons encode proteins of unknown function, although a predicted role in adaptation could be assigned to some genes from the P. yeei environmental isolates. The chromosome of LM20 encodes (i) the membrane protein YeeE, which mediates the uptake of thiosulfate as an inorganic sulfur source for cysteine synthesis (Tanaka et al., 2020), (ii) mercury transporter Mer, and (iii) the sensor kinase KdpD, which senses potassium levels, and the kdpABC operon, which encodes a high-affinity potassium uptake system crucial for bacterial survival in low potassium environments (Laermann et al., 2013; Ali et al., 2017). In the CCUG 54214 genome, many transport-related genes (ABC, RND, TTT types) were distinguished among the singletons, as well as genes for toxin-antitoxin systems (e.g., HipA-, PrlF-, Yha- family), similarly to clinical strains.
The largest group of P. yeei-specific genes (found in chromosomes and DnaA-like, RepB1, RepB2 and RepABC replicons) encode transposases from different IS families. The second largest group of genes encodes transporters, mostly of the ABC type, which are located in chromosomes and in DnaA-like and RepB1 chromids. Other abundant genes, found mainly in RepB1 chromids, encode transcription regulators of the LuxR, CopG, TetR/AcrR, MarR, LacI, AraC, TetR, DeoR/GlpR, and NikR families (Supplementary Table S8).
Examples of proteins encoded by other P. yeei-specific genes include a urease enzyme, components of the trehalose, rhamnose, and cobalamin biosynthetic pathways, chaperone proteins, and factors possibly involved in the cell's response to stress conditions (Supplementary Table S8).
3.8 Putative virulence determinants
The proteomes of a number of Paracoccus strains were analyzed in order to identify potential virulence factors: (i) the seven P. yeei strains described in this study, (ii) other P. yeei strains whose complete genomic sequences are available (CCUG 32053, TT13, FDAARGOS_262, FDAARGOS_643), as well as (iii) several environmental isolates of the genus Paracoccus, including the type strain P. denitrificans PD1222. These protein sets were compared against databases containing bacterial virulence factors and antibiotic resistance genes (see Section 2 for details). Relatively few of the identified determinants were unique to P. yeei (all clinical isolates). The pool of identified genes includes those encoding six types of protein, that were previously mentioned by Lasek et al. (2018), i.e., (i) methionine sulfoxide reductase (msrA1, msrA2, msrB), (ii) diguanylate cyclase, (iii) superoxide dismutase, (iv) sugar transferase, (v) type IV secretion system components, and (vi) urease (Supplementary Tables S3, S9).
The chromosomally-located msr genes of CCUG 32052 and CCUG 46822 encode predicted proteins sharing 61–100% aa sequence similarity, while the homologous proteins encoded by other P. yeei strains are less well-conserved (38–55% aa identity). Msr proteins participate in the detoxification of reactive oxygen intermediates (both prokaryotic and eukaryotic cells lacking Msr are sensitive to oxidative stress) (Denkel et al., 2011). Moreover, MsrA has been shown to be critical for the survival of Erwinia chrysanthemi (El Hassouni et al., 1999), Mycoplasma genitalium (Das et al., 2012), and Helicobacter pylori (Alamuri and Maier, 2006) in their infected hosts. Genes encoding components of a putative type IV secretion system (T4SS) were identified only in CCUG 17731 (30–50% aa identity) and in CCUG 13493 (25–50% aa identity)—within RepABC plasmids in both cases. However, these were not complete modules, unlike the one previously identified in CCUG 32053 (Lasek et al., 2018).
The remaining virulence factors, present in all analyzed genomes, include the gene encoding diguanylate cyclase, an enzyme synthesizing cyclic diguanylate (c-di-GMP)—an important bacterial second messenger. This signaling molecule is involved in the regulation of a number of complex physiological processes, including biofilm formation and motility, which affect the pathogenesis of many bacteria (Tamayo et al., 2007).
Genes encoding several other putative virulence factors were identified within ECRs, although they were not unique to P. yeei. These include proteins participating in the following: (i) numerous ABC and RND transport systems (DnaA-like, RepB, RepABC replicons), (ii) siderophore synthesis (RepB2 replicons, chromosomes), (iii) polysaccharide capsule production (RepB2 replicons), and (iv) trehalose synthesis (DnaA-like type replicons).
All P. yeei strains contain a set of genes (URE) involved in the synthesis of the metalloenzyme urease, which hydrolyzes urea to ammonia and CO2. These genes are located within essential replicons—DnaA-like chromids. They display synteny and a high degree of nucleotide sequence identity (98%), indicating their biological importance. The role of urease in bacterial pathogenesis is well-documented (e.g., Mobley, 1996; Dupuy et al., 1997; Burne and Chen, 2000; Berutti et al., 2014; Rutherford, 2014; Graham and Miftahussurur, 2018; Zhou et al., 2019; Minami et al., 2021).
3.9 URE gene clusters
Besides genes for the main urease subunits (UreA—γ subunit, UreB—β subunit, and UreC—α subunit) and accessory proteins (ureD, ureE, ureF, and ureG), the URE gene cluster also contains the nikR gene (encoding a predicted nickel-dependent transcription factor) and numerous ABC transporter genes, likely to be involved in the uptake of Ni2+ (an essential cofactor for urease; Rutherford, 2014). It is probable that NikR regulates the expression of the transporter genes (Figure 7). Interestingly, in P. yeei strain CCUG 32052, part of the URE gene cluster, containing the ureABC and ureD genes, has been duplicated and transferred to another ECR (RepABC; pYEE32052P4) (99% nucleotide sequence identity to the corresponding URE genes) (Figure 7).
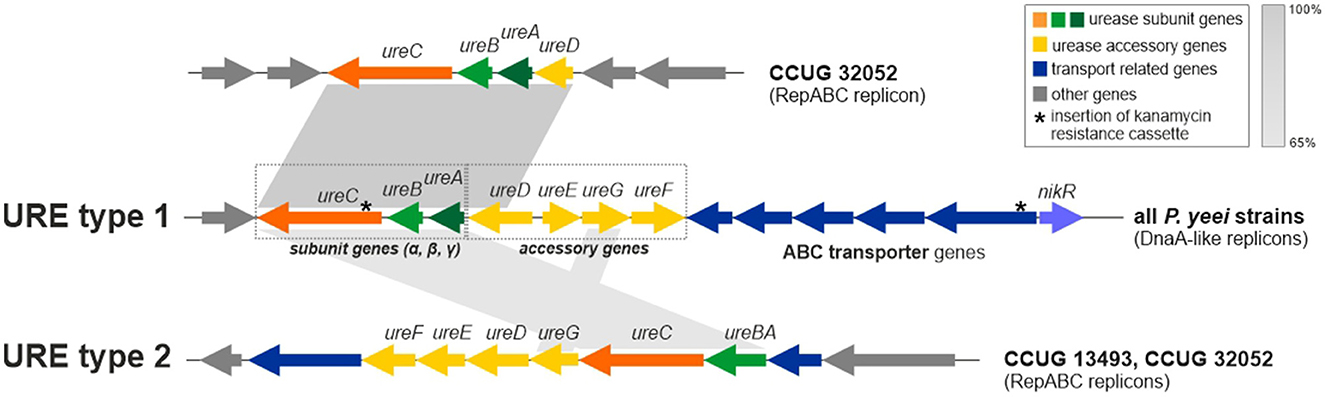
Figure 7. Genetic organization of urease gene clusters (URE) located in P. yeei ECRs—URE type 1 (present in all DnaA-like chromids) and URE type 2 (present in two RepABC plasmids of P. yeei CCUG 13493 and CCUG 32052). Shaded areas connect homologous DNA regions.
The URE gene cluster of DnaA-like replicons (designated URE type 1) (Figure 7) is unique to and characteristic of P. yeei strains. However, in two strains (CCUG 13493 and CCUG 32052) another set of ure genes (URE type 2; Figure 7), possibly associated with urease synthesis, was identified. These genes are present in RepABC plasmids in both strains (pYEE13493P4 and pYEE32052P5, respectively) and their genetic organization is different from that of URE type 1. In the URE type 2 cluster, the γ and β urease subunits are encoded by a single fusion gene ureBC, the order and transcriptional orientation of the accessory genes ureFEDG is different, there are fewer transporter genes and the nikR gene is absent (Figure 7).
URE type 2 represents a novel type of ureolytic gene cluster which has a different evolutionary origin from URE type 1. The two clusters share some sequence similarity between the genes encoding the main urease subunits, but no similarity was found in case of the accessory genes and their arrangement is different.
The ureolytic activity of three P. yeei strains (CCUG 32053, CCUG 32052—containing the duplicated part of URE type 1, and CCUG 32052—also carrying URE type 2; Figure 7) was examined using Christensen's differential medium without (control) and with the addition of urea (1%) (see Section 2 for details). All the strains displayed ureolytic activity, which was induced by the presence of urea in the medium.
To unequivocally confirm the role of the type 1 URE gene cluster in the observed ureolytic activity, mutations were introduced by insertion of a kanamycin resistance cassette into the ureC (encoding the α subunit of urease) or nikR genes of CCUG 32053 and CCUG 13493. These mutations completely abolished ureolytic activity in these strains, indicating the importance of both ureC and nikR in determining this phenotype. The lack of ureolytic activity in mutated strain CCUG 13493, which also contains URE type 2, indicates that this second cluster does not produce an active urease under the tested conditions and its genes are unable to complement mutations introduced in URE type 1.
4 Discussion
These comprehensive comparative genomic analyses of a pool of strains of the opportunistic bacterium P. yeei, have provided valuable information on (i) the structural diversity of genomes within this species, (ii) the components of the mobilome, their impact on genome structure and properties, and (iii) the genetic information determining the opportunistic properties of this species. Importantly, the analyzed strains originated from different environments (clinical and natural) and were isolated at different times. Therefore, the collected data better reflects the extent of strain variability within this species, and also permits the identification of genes conserved during evolution, including those involved in the process of pathogenesis (Table 1).
The physiological and phenotypic properties of the analyzed strains were found to be very similar. High levels of similarity were also evident when the structure and genetic content of the genomes of clinical and environmental isolates were compared. The relatedness of these strains is reflected in the topology of the P. yeei phylogenetic tree (Figure 1A).
All strains contain numerous ECRs of varying sizes, ranging from 4 to 485 kbp. It should be noted that strain CCUG 46822 carries the largest number of ECRs (13) among Paracoccus spp. strains analyzed so far. The main ECRs are large replicons representing the DnaA-like, RepB (RepB1 and RepB2 subgroups) and RepABC families, which is consistent with our previous observations (Lasek et al., 2018) (Figures 1B, C). All of the DnaA-like, RepB1 and RepB2 replicons (which meet the criteria of chromids) contain numerous adaptive genes (facilitating survival in challenging environmental conditions) plus a set of conserved genes of the core Paracoccus spp. genome. However, the genetic load of these replicons is not equal. DnaA-like replicons can be considered major chromids due to (i) the presence of the largest number of core genes, (ii) the relatively low variability in their structure and size (especially in relation to DnaA-like replicons of other Paracoccus species) (Figure 3; Supplementary Table S5), and (iii) the presence of the URE gene cluster, which is chromosomally located in other pathogenic bacteria.
DnaA-like chromids are characteristic for Paracoccus spp. and play an important role in the biology of these bacteria. Related essential replicons were previously identified and analyzed e.g., in P. denitrificans PD1222, P. aminophilus JCM 7686 and P. aminovorans JCM 7685 (Dziewit et al., 2014; Czarnecki et al., 2017). The removal of DnaA-like and RepB chromids from P. aminophilus cells was attempted (Dziewit et al., 2014), but this was only successful in the latter case. The strain lacking RepB replicons grew much more slowly on complete medium and was unable to grow at all on minimal media (regardless of the type of carbon source). Based on these observations, two classes of chromids were proposed: obligatorily essential primary chromids (e.g., DnaA-like) and facultatively essential secondary chromids (e.g., RepB).
It should be noted that DnaA-like chromids have yet to be identified in other taxonomic groups of bacteria, which is consistent with the assertion of Harrison et al. (2010) that particular chromid types are characteristic for a specific taxonomic group of bacteria (mainly genus). This claim seems reasonable given that these types of replicons are likely to have arisen from plasmids (potentially with a narrow host range) specific to a particular group of bacteria. DnaA-like replicons therefore have some aptitude to generate chromids. However, it has yet to be explained why, for example, RepABC replicons, which are the main chromids of e.g.. Rhizobium spp. and Agrobacterium spp. (Landeta et al., 2011; diCenzo et al., 2013; Döhlemann et al., 2017), despite also being common in Paracoccus spp., do not play the same role in these bacteria.
The flexible genome of P. yeei also comprises transposable elements. A previous study examining strain CCUG 32053 highlighted an unusual diversity of insertion sequences, not seen in other Paracoccus spp. strains (Lasek et al., 2018). This analysis resulted in the examination of the prevalence of 80 ISs (ISPye1–ISPye80) sequences in the genomes of P. yeei, that had not been previously characterized in the transposable mobilome of Paracoccus spp. (Dziewit et al., 2012). In this study, we searched for putative TEs in all available complete genomic sequences of P. yeei. The results of these analyses showed (i) the widespread presence of ISs in this species (although there is great variability in their number in individual strains), (ii) the predominance of ISs belonging to the IS5, IS3, IS110, and IS256 families (Figure 5; Supplementary Figure S3), and the absence of (iii) P. yeei-specific elements conserved in all genomes or (iv) transposons that could be linked with potential determinants of pathogenesis. A more global comparative analysis using data collected in the ISfinder database showed that the overall distribution pattern of IS families in the P. yeei genomes corresponds to their distribution across the genus Paracoccus. This also highlighted the ubiquity and diversity of these elements in Paracoccus spp. in comparison to other genera of Alphaproteobacteria (Supplementary Figure S4).
The presence of multiple copies of particular ISs in P. yeei genomes may promote homologous recombination, resulting in genomic structural rearrangements. It is likely that such events led to the generation of the two chromosome structural variants identified in this study (Figure 3). Notably, the large number of TEs predicted in silico did not correlate with the number of active elements identified by the application of trap plasmids. Only a few elements that were the most dynamic in the process of transposition were captured. The low activity of TEs may be due to the presence of multiple regulatory systems keeping this process in check to reduce the likelihood of lethality caused by insertional inactivation of host housekeeping genes (Lipszyc et al., 2022).
Analysis of the P. yeei genomes also identified a novel group of non-autonomous TEs (MITE-type; MITEPye1). These elements contain TIRs and are bordered by DRs, so resemble functional TEs in this respect (Figure 6A). Their TIRs share significant sequence similarity with terminal sequences of IS1182-family elements, which suggests that they originate from defective ISs and their transposition may be trans-activated by compatible transposases. In the chromosomes, these MITE-type elements occur in low numbers (1–3) and are preferentially located within intergenic regions (Supplementary Table S7). Mfold analysis revealed that these elements are able to fold into long stem-loop structures at the RNA level (Figure 6B). Therefore, their co-transcription with upstream genes, may influence the conformation and stability of the resulting transcripts. The altered expression may result in various phenotypes depending on the specific gene function (Szuplewska and Bartosik, 2009; Dziewit et al., 2012; Szuplewska et al., 2014).
Particular attention was given to the identification of genes responsible for the opportunistic phenotype of P. yeei. Collections of P. yeei species-specific genes were searched for potential virulence factors. A noteworthy finding of this analysis was the very large number of transporter genes, which is a unique feature among Paracoccus spp. Although it is difficult to directly link their presence to the ability of these bacteria to cause opportunistic infections, they could potentially play an important role in pathogenesis.
The URE gene clusters of P. yeei identified in the screen for potential pathogenicity determinants were examined in more detail. These clusters are involved in the synthesis of urease, an enzyme that acts as a virulence factor in many pathogenic bacteria, including Staphylococcus spp. (90% of methicillin-resistant S. aureus display ureolytic activity), Helicobacter pylori, Mycobacterium tuberculosis, Mycobacterium bovis, anaerobic Clostridium perfringens and Vibrio parahaemolyticus (Dupuy et al., 1997; Berutti et al., 2014; Graham and Miftahussurur, 2018; Zhou et al., 2019; Minami et al., 2021). In the case of H. pylori, urease is one of the major virulence factors, since strains unable to produce this enzyme cannot colonize the gastric mucosa. The ammonia released by urease action affects the tissue, causing damage to epithelial cells. Urease activity also enables pathogens to survive and proliferate in macrophages (Fu et al., 2018). This enzyme elicits a response from the human immune system by stimulating the production of antibodies (Konieczna et al., 2012).
Two diverse URE gene clusters were identified in the P. yeei genomes: type 1—species specific, and type 2—present only in two RepABC plasmids. We showed that URE type 1 determined the urease activity, which was induced by the presence of urea in the growth medium. Activity of the URE type 2 cluster could not be demonstrated. It may be speculated that these URE module variants require different substrates/activators to initiate the synthesis of urease. Nevertheless, it is highly probable that the presence of additional URE gene clusters may enhance the ureolytic capacity of the bacteria, which highlights the biological importance of this phenotype. It is important to note that urea is present in large quantities in the human body, mainly in the kidneys, but is also found in the stomach, blood serum, sweat and milk (Fu et al., 2018; Graham and Miftahussurur, 2018; Schimmel et al., 2021).
Mutations introduced into the ureC (encoding a subunit of urease) or nikR (encoding a putative regulator of nickel transporter gene expression) genes of URE type 1 completely abolished urease activity. This effect was previously observed in analogous mutants of H. pylori. One of the main phenotypes of an H. pylori nikR mutant was the absence of nickel-responsive induction of urease expression (Van Vliet et al., 2002). Further studies showed that NikR binds to the ureA promoter (in a nickel-dependent manner), which results in nickel-induced transcription and expression of a ureolytic phenotype (Ernst et al., 2005). We hypothesize that the P. yeei ure genes may be regulated in a similar way.
As the conserved URE type 1 gene cluster of P. yeei is unique among Paracoccus spp., it is highly probable that urease plays an important role in the pathogenesis of this species. The verification of this hypothesis is an immediate goal of our future studies.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
MS: Conceptualization, Formal analysis, Funding acquisition, Investigation, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. DS: Formal analysis, Investigation, Visualization, Writing – review & editing. RL: Data curation, Formal analysis, Visualization, Writing – review & editing. PD: Data curation, Formal analysis, Methodology, Software, Writing – review & editing. MH: Formal analysis, Investigation, Writing – review & editing. ŁF: Investigation, Writing – review & editing. CC: Formal analysis, Investigation, Writing – review & editing. DB: Conceptualization, Formal analysis, Methodology, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by the National Science Center (NCN), Poland (grant no. 2016/21/D/NZ2/03824).
Acknowledgments
We would like to thank Aleksandra Pawłot for her experimental assistance with physiological characterization of bacteria, Albert Roethel for his help with the identification of transposable elements, and Anna Rokowska and Kamil Krakowski for their contribution to genomic data analysis.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1483110/full#supplementary-material
References
Alamuri, P., and Maier, R. J. (2006). Methionine sulfoxide reductase in Helicobacter pylori: interaction with methionine-rich proteins and stress-induced expression. J. Bacteriol. 188:5839. doi: 10.1128/JB.00430-06
Alcock, B. P., Raphenya, A. R., Lau, T. T. Y., Tsang, K. K., Bouchard, M., Edalatmand, A., et al. (2020). CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucl. Acids Res. 48, D517–D525. doi: 10.1093/nar/gkz935
Ali, M. K., Li, X., Tang, Q., Liu, X., Chen, F., Xiao, J., et al. (2017). Regulation of inducible potassium transporter KdpFABC by the KdpD/KdpE two-component system in Mycobacterium smegmatis. Front. Microbiol. 8:570. doi: 10.3389/fmicb.2017.00570
Aliste-Fernández, M., Sanfeliu-Sala, I., and Sánchez-Delgado, J. (2020). Bacteremia caused by Paracoccus yeei in patient with compensated cirrhosis of the liver. Enferm. Infecc. Microbiol. Clin. 38, 451–452. doi: 10.1016/j.eimc.2020.01.013
Amstrup, S. K., Ong, S. C., Sofos, N., Karlsen, J. L., Skjerning, R. B., Boesen, T., et al. (2023). Structural remodelling of the carbon–phosphorus lyase machinery by a dual ABC ATPase. Nat. Commun. 14, 1–12. doi: 10.1038/s41467-023-36604-y
Arias, M. A., and Clark, J. (2019). Paracoccus yeei as a cause of peritoneal dialysis peritonitis in the United Kingdom. IDCases 15:e00486. doi: 10.1016/j.idcr.2019.e00486
Aurass, P., Karste, S., Trost, E., Glaeser, S. P., Kämpfer, P., and Flieger, A. (2017). Genome sequence of Paracoccus contaminans LMG 29738T, isolated from a water microcosm. Genome Announc. 5, 487–504. doi: 10.1128/genomeA.00487-17
Baker, S. C., Ferguson, S. J., Ludwig, B., Page, M. D., Richter, O.-M. H., and van Spanning, R. J. M. (1998). Molecular genetics of the genus Paracoccus: metabolically versatile bacteria with bioenergetic flexibility. Microbiol. Mol. Biol. Rev. 62, 1046–1078. doi: 10.1128/MMBR.62.4.1046-1078.1998
Bartosik, D., Sochacka, M., and Baj, J. (2003). Identification and characterization of transposable elements of Paracoccus pantotrophus. J. Bacteriol. 185:3753. doi: 10.1128/JB.185.13.3753-3763.2003
Bartosik, D., Wlodarczyk, M., and Thomas, C. M. (1997). Complete nucleotide sequence of the replicator region of Paracoccus (Thiobacillus) versutus pTAV1 plasmid and its correlation to several plasmids of Agrobacterium and Rhizobium species. Plasmid 38, 53–59. doi: 10.1006/plas.1997.1295
Beijerinck, M. W., and Minkman, D. C. J. (1910). Bildung und verbrauch von stickoxydul durch bakterien. Zentbl. Bakteriol. Parasitenkd. Infektionskr. Hyg. Abt. II 25, 30–63.
Berry, A., Janssens, D., Hümbelin, M., Jore, J. P. M., Hoste, B., Cleenwerck, I., et al. (2003). Paracoccus zeaxanthinifaciens sp. nov., a zeaxanthin-producing bacterium. Int. J. Syst. Evol. Microbiol. 53, 231–238. doi: 10.1099/ijs.0.02368-0
Berutti, T. R., Williams, R. E., Shen, S., Taylor, M. M., and Grimes, D. J. (2014). Prevalence of urease in Vibrio parahaemolyticus from the Mississippi Sound. Lett. Appl. Microbiol. 58, 624–628. doi: 10.1111/lam.12237
Bhikoo, R., Chong, S., McLeod, K., Heyworth, P., and McAllister, I. L. (2022). Paracoccus yeei, a novel bacterial cause of endophthalmitis following intravitreal injection. Ocul. Immunol. Inflamm. 30, 995–997. doi: 10.1080/09273948.2020.1836233
Birnboim, H. C., and Doly, J. (1979). A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 7, 1513–1523. doi: 10.1093/nar/7.6.1513
Bordel, S., Martín-González, D., Börner, T., Muñoz, R., and Santos-Beneit, F. (2024). Genome-scale metabolic model of the versatile bacterium Paracoccus denitrificans Pd1222. mSystems 9:e0107723. doi: 10.1128/msystems.01077-23
Burne, R. A., and Chen, Y. Y. M. (2000). Bacterial ureases in infectious diseases. Microbes Infect. 2, 533–542. doi: 10.1016/S1286-4579(00)00312-9
Cantais, A., Grattard, F., Gagnaire, J., Mory, O., Plat, A., Lleres-Vadeboin, M., et al. (2020). Longitudinal study of viral and bacterial contamination of hospital pediatricians' mobile phones. Microorganisms 8, 1–12. doi: 10.3390/microorganisms8122011
Carlos, C., Pereira, L. B., and Ottoboni, L. M. M. (2017). Comparative genomics of Paracoccus sp. SM22M-07 isolated from coral mucus: insights into bacteria-host interactions. Curr. Genet. 63, 509–518. doi: 10.1007/s00294-016-0658-3
Chen, L., Zheng, D., Liu, B., Yang, J., and Jin, Q. (2016). VFDB 2016: hierarchical and refined dataset for big data analysis−10 years on. Nucleic Acids Res. 44, D694–D697. doi: 10.1093/nar/gkv1239
Chistoserdova, L. (2011). Modularity of methylotrophy, revisited. Environ. Microbiol. 13, 2603–2622. doi: 10.1111/j.1462-2920.2011.02464.x
Choi, J. H., Cho, Y. S., Lee, J. W., Shin, H. B., and Lee, I. K. (2017). Bacterial contamination and disinfection status of laryngoscopes stored in emergency crash carts. J. Prev. Med. Public Health 50:158. doi: 10.3961/jpmph.17.013
Christensen, W. B. (1946). Urea decomposition as a means of differentiating proteus and paracolon cultures from each other and from Salmonella and Shigella Types. J. Bacteriol. 52, 461–466. doi: 10.1128/jb.52.4.461-466.1946
Courjaret, J. C., Drancourt, M., and Hoffart, L. (2014). Paracoccus yeei keratitis in a contact lens wearer. Eye Contact Lens 40, e21–e22. doi: 10.1097/ICL.0b013e31829e8fc7
Czarnecki, J., and Bartosik, D. (2019). Diversity of methylotrophy pathways in the genus Paracoccus (Alphaproteobacteria). Curr. Issues Mol. Biol. 33, 117–131. doi: 10.21775/cimb.033.117
Czarnecki, J., Dziewit, L., Puzyna, M., Prochwicz, E., Tudek, A., Wibberg, D., et al. (2017). Lifestyle-determining extrachromosomal replicon pAMV1 and its contribution to the carbon metabolism of the methylotrophic bacterium Paracoccus aminovorans JCM 7685. Environ. Microbiol. 19, 4536–4550. doi: 10.1111/1462-2920.13901
Daneshvar, M. I., Hollis, D. G., Weyant, R. S., Steigerwalt, A. G., Whitney, A. M., Douglas, M. P., et al. (2003). Paracoccus yeeii sp. nov. (formerly CDC Group EO-2), a novel bacterial species associated with human infection. J. Clin. Microbiol. 41:1289. doi: 10.1128/JCM.41.3.1289-1294.2003
Darriba, D.i., Posada, D., Kozlov, A. M., Stamatakis, A., Morel, B., and Flouri, T. (2020). ModelTest-NG: a new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 37, 291–294. doi: 10.1093/molbev/msz189
Das, K., de la Garza, G., Maffi, S., Saikolappan, S., and Dhandayuthapani, S. (2012). Methionine sulfoxide reductase A (MsrA) deficient Mycoplasma genitalium shows decreased interactions with host cells. PLoS ONE 7:e0036247. doi: 10.1371/journal.pone.0036247
Delihas, N. (2011). Impact of small repeat sequences on bacterial genome evolution. Genome Biol. Evol. 3:959. doi: 10.1093/gbe/evr077
Denkel, L. A., Horst, S. A., Rouf, S. F., Kitowski, V., Böhm, O. M., Rhen, M., et al. (2011). Methionine sulfoxide reductases are essential for virulence of Salmonella typhimurium. PLoS ONE 6:e0026974. doi: 10.1371/journal.pone.0026974
diCenzo, G., Milunovic, B., Cheng, J., and Finan, T. M. (2013). The tRNAarg gene and engA are essential genes on the 1.7-Mb pSymB megaplasmid of Sinorhizobium meliloti and were translocated together from the chromosome in an ancestral strain. J. Bacteriol. 195, 202–212. doi: 10.1128/JB.01758-12
Ditta, G., Stanfield, S., Corbin, D., and Helinski, D. R. (1980). Broad host range DNA cloning system for gram-negative bacteria: construction of a gene bank of Rhizobium meliloti. Proc. Natl. Acad. Sci. U. S. A. 77, 7347–7351. doi: 10.1073/pnas.77.12.7347
Döhlemann, J., Wagner, M., Happel, C., Carrillo, M., Sobetzko, P., Erb, T. J., et al. (2017). A family of single copy repABC-type shuttle vectors stably maintained in the alpha-proteobacterium Sinorhizobium meliloti. ACS Synth. Biol. 6, 968–984. doi: 10.1021/acssynbio.6b00320
Dupuy, B., Daube, G., Popoff, M. R., and Cole, S. T. (1997). Clostridium perfringens urease genes are plasmid borne. Infect. Immun. 65:2313. doi: 10.1128/iai.65.6.2313-2320.1997
Dyer, J., and Harris, P. (2020). Paracoccus yeei – An emerging pathogen or incidental finding? Pathology 52:S123. doi: 10.1016/j.pathol.2020.01.419
Dziewit, L., Adamczuk, M., Szuplewska, M., and Bartosik, D. (2011). DIY series of genetic cassettes useful in construction of versatile vectors specific for Alphaproteobacteria. J. Microbiol. Methods 86, 166–174. doi: 10.1016/j.mimet.2011.04.016
Dziewit, L., Baj, J., Szuplewska, M., Maj, A., Tabin, M., Czyzkowska, A., et al. (2012). Insights into the transposable mobilome of Paracoccus spp. (Alphaproteobacteria). PLoS ONE 7:e0032277. doi: 10.1371/journal.pone.0032277
Dziewit, L., Czarnecki, J., Wibberg, D., Radlinska, M., Mrozek, P., Szymczak, M., et al. (2014). Architecture and functions of a multipartite genome of the methylotrophic bacterium Paracoccus aminophilus JCM 7686, containing primary and secondary chromids. BMC Genom. 15, 1–16. doi: 10.1186/1471-2164-15-124
Dziewit, L., Pyzik, A., Matlakowska, R., Baj, J., Szuplewska, M., and Bartosik, D. (2013). Characterization of Halomonas sp. ZM3 isolated from the Zelazny Most post-flotation waste reservoir, with a special focus on its mobile DNA. BMC Microbiol. 13:59. doi: 10.1186/1471-2180-13-59
Dziewit, L., Pyzik, A., Szuplewska, M., Matlakowska, R., Mielnicki, S., Wibberg, D., et al. (2015). Diversity and role of plasmids in adaptation of bacteria inhabiting the Lubin copper mine in Poland, an environment rich in heavy metals. Front. Microbiol. 6:152. doi: 10.3389/fmicb.2015.00152
Eckhardt, T. (1978). A rapid method for the identification of plasmid desoxyribonucleic acid in bacteria. Plasmid 1, 584–588. doi: 10.1016/0147-619X(78)90016-1
El Hassouni, M., Chambost, J. P., Expert, D., Van Gijsegem, F., and Barras, F. (1999). The minimal gene set member msrA, encoding peptide methionine sulfoxide reductase, is a virulence determinant of the plant pathogen Erwinia chrysanthemi. Proc. Natl. Acad. Sci. U. S. A. 96, 887–892. doi: 10.1073/pnas.96.3.887
Ernst, F. D., Kuipers, E. J., Heijens, A., Sarwari, R., Stoof, J., Penn, C. W., et al. (2005). The nickel-responsive regulator NikR controls activation and repression of gene transcription in Helicobacter pylori. Infect. Immun. 73, 7252–7258. doi: 10.1128/IAI.73.11.7252-7258.2005
Fosso, C., Maillart, E., Beun, B., Touzani, F., Mahadeb, B., and Clevenbergh, P. (2021). Opportunistic peritonitis in peritoneal dialysis: the example of Paracoccus yeei. Clin. Case Rep. 9:4176. doi: 10.1002/ccr3.4176
Fu, M. S., Coelho, C., De Leon-Rodriguez, C. M., Rossi, D. C. P., Camacho, E., Jung, E. H., et al. (2018). Cryptococcus neoformans urease affects the outcome of intracellular pathogenesis by modulating phagolysosomal pH. PLoS Pathog. 14:7144. doi: 10.1371/journal.ppat.1007144
Funke, G., Frod, R., and Sommer, H. (2004). First comprehensively documented case of Paracoccus yeei infection in a human. J. Clin. Microbiol. 42, 3366–3368. doi: 10.1128/JCM.42.7.3366-3368.2004
Garcillán-Barcia, M. P., Francia, M. V., and De La Cruz, F. (2009). The diversity of conjugative relaxases and its application in plasmid classification. FEMS Microbiol. Rev. 33, 657–687. doi: 10.1111/j.1574-6976.2009.00168.x
Gay, P., Le Coq, D., Steinmetz, M., Berkelman, T., and Kado, C. I. (1985). Positive selection procedure for entrapment of insertion sequence elements in gram-negative bacteria. J. Bacteriol. 164:918. doi: 10.1128/JB.164.2.918-921.1985
Ghosh, W., Mandal, S., and Roy, P. (2006). Paracoccus bengalensis sp. nov., a novel sulfur-oxidizing chemolithoautotroph from the rhizospheric soil of an Indian tropical leguminous plant. Syst. Appl. Microbiol. 29, 396–403. doi: 10.1016/j.syapm.2005.10.004
Graham, D. Y., and Miftahussurur, M. (2018). Helicobacter pylori urease for diagnosis of Helicobacter pylori infection: a mini review. J. Adv. Res. 13, 51–57. doi: 10.1016/j.jare.2018.01.006
Hanahan, D. (1983). Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166, 557–580. doi: 10.1016/S0022-2836(83)80284-8
Harrison, P. W., Lower, R. P. J., Kim, N. K. D., and Young, J. P. W. (2010). Introducing the bacterial ‘chromid': not a chromosome, not a plasmid. Trends Microbiol. 18, 141–148. doi: 10.1016/j.tim.2009.12.010
Hooykaas, P. J. J., den Dulk-Ras, H., and Schilperoort, R. A. (1980). Molecular mechanism of Ti plasmid mobilization by R plasmids: isolation of Ti plasmids with transposon-insertions in Agrobacterium tumefaciens. Plasmid 4, 64–75. doi: 10.1016/0147-619X(80)90083-9
Horsman, G. P., and Zechel, D. L. (2017). Phosphonate biochemistry. Chem. Rev. 117, 5704–5783. doi: 10.1021/acs.chemrev.6b00536
Huesca-Espitia, L., del, C., Aslanzadeh, J., Feinn, R., Joseph, G., Murray, T. S., et al. (2018). Deposition of bacteria and bacterial spores by bathroom hot-air hand dryers. Appl. Environ. Microbiol. 84, e00044-18. doi: 10.1128/AEM.00044-18
Hynes, M. F., and McGregor, N. F. (1990). Two plasmids other than the nodulation plasmid are necessary for formation of nitrogen-fixing nodules by Rhizobium leguminosarum. Mol. Microbiol. 4, 567–574. doi: 10.1111/j.1365-2958.1990.tb00625.x
Iversen, C., Mullane, N., McCardell, B., Tall, B. D., Lehner, A., Fanning, S., et al. (2008). Cronobacter gen. nov., a new genus to accommodate the biogroups of Enterobacter sakazakii, and proposal of Cronobacter sakazakii gen. nov., comb. nov., Cronobacter malonaticus sp. nov., Cronobacter turicensis sp. nov., Cronobacter muytjensii sp. nov., Cronobacter dublinensis sp. nov., Cronobacter genomospecies 1, and of three subspecies, Cronobacter dublinensis subsp. dublinensis subsp. nov., Cronobacter dublinensis subsp. lausannensis subsp. nov. and Cronobacter dublinensis subsp. lactaridi sub. Int. J. Syst. Evol. Microbiol. 58, 1442–1447. doi: 10.1099/ijs.0.65577-0
Jordan, S. L., McDonald, I. R., Kraczkiewicz-Dowjat, A. J., Kelly, D. P., Rainey, F. A., Murrell, J. C., et al. (1997). Autotrophic growth on carbon disulfide is a property of novel strains of Paracoccus denitrificans. Arch. Microbiol. 168, 225–236. doi: 10.1007/s002030050492
Kanis, M. J., Oosterheert, J. J., Lin, S., Boel, C. H. E., and Ekkelenkamp, M. B. (2010). Corneal graft rejection complicated by Paracoccus yeei infection in a patient who had undergone a penetrating keratoplasty. J. Clin. Microbiol. 48:323. doi: 10.1128/JCM.01798-09
Katoh, K., Rozewicki, J., and Yamada, K. D. (2019). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 20, 1160–1166. doi: 10.1093/bib/bbx108
Konieczna, I., Żarnowiec, P., Kwinkowski, M., Kolesińska, B., Fraczyk, J., Kamiński, Z., et al. (2012). Bacterial urease and its role in long-lasting human diseases. Curr. Protein Pept. Sci. 13:789. doi: 10.2174/138920312804871094
Kovach, M. E., Elzer, P. H., Steven Hill, D., Robertson, G. T., Farris, M. A., Roop, R. M., et al. (1995). Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166, 175–176. doi: 10.1016/0378-1119(95)00584-1
Kozlov, A. M., Darriba, D., Flouri, T., Morel, B., and Stamatakis, A. (2019). RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35, 4453–4455. doi: 10.1093/bioinformatics/btz305
Kruszewska, E., Grze,ś, H., Czupryna, P., Pancewicz, S., Groth, M., Wondim, M., et al. (2021). Fogging with peracetic acid in schools and kindergartens. Front. Public Health 9:697917. doi: 10.3389/fpubh.2021.697917
Kushner, S. (1978). “Genetic engineering,” in Proc. Symp. on Genetic Engineering, eds. H. Boyer, and S. Nicosia (Amsterdam: Elsevier).
Laermann, V., Cudić, E., Kipschull, K., Zimmann, P., and Altendorf, K. (2013). The sensor kinase KdpD of Escherichia coli senses external K+. Mol. Microbiol. 88, 1194–1204. doi: 10.1111/mmi.12251
Landeta, C., Dávalos, A., Cevallos, M. Á., Geiger, O., Brom, S., and Romero, D. (2011). Plasmids with a chromosome-like role in rhizobia. J. Bacteriol. 193, 1317–1326. doi: 10.1128/JB.01184-10
Lasek, R., Szuplewska, M., Mitura, M., Decewicz, P., Chmielowska, C., Pawlot, A., et al. (2018). Genome structure of the opportunistic pathogen Paracoccus yeei (Alphaproteobacteria) and identification of putative virulence factors. Front. Microbiol. 9:2553. doi: 10.3389/fmicb.2018.02553
Lee, J. H., Kim, Y. S., Choi, T. J., Lee, W. J., and Kim, Y. T. (2004). Paracoccus haeundaensis sp. nov., a Gram-negative, halophilic, astaxanthin-producing bacterium. Int. J. Syst. Evol. Microbiol. 54, 1699–1702. doi: 10.1099/ijs.0.63146-0
Lim, J. Y., Hwang, I., Ganzorig, M., Pokhriyal, S., Singh, R., and Lee, K. (2018). Complete genome sequence of Paracoccus yeei TT13, isolated from human skin. Genome Announc. 6, e01514-17. doi: 10.1128/genomeA.01514-17
Lipszyc, A., Szuplewska, M., and Bartosik, D. (2022). How do transposable elements activate expression of transcriptionally silent antibiotic resistance genes? Int. J. Mol. Sci. 23:63. doi: 10.3390/ijms23158063
Liu, M., Li, X., Xie, Y., Bi, D., Sun, J., Li, J., et al. (2019). ICEberg 2.0: an updated database of bacterial integrative and conjugative elements. Nucl. Acids Res. 47, D660–D665. doi: 10.1093/nar/gky1123
Liu, X. Y., Wang, B. J., Jiang, C. Y., Liu, S. J., and Liu, S. J. (2006). Paracoccus sulfuroxidans sp. nov., a sulfur oxidizer from activated sludge. Int. J. Syst. Evol. Microbiol. 56, 2693–2695. doi: 10.1099/ijs.0.64548-0
Liu, Z. P., Wang, B. J., Liu, X. Y., Dai, X., Liu, Y. H., and Liu, S. J. (2008). Paracoccus halophilus sp. nov., isolated from marine sediment of the South China Sea, China, and emended description of genus Paracoccus Davis 1969. Int. J. Syst. Evol. Microbiol. 58, 257–261. doi: 10.1099/ijs.0.65237-0
Maj, A., Dziewit, L., Czarnecki, J., Wlodarczyk, M., Baj, J., Skrzypczyk, G., et al. (2013). Plasmids of carotenoid-producing Paracoccus spp. (Alphaproteobacteria) - structure, diversity and evolution. PLoS ONE 8:e0080258. doi: 10.1371/journal.pone.0080258
Makino, K., Kim, S. K., Shinagawa, H., Amemura, M., and Nakata, A. (1991). Molecular analysis of the cryptic and functional phn operons for phosphonate use in Escherichia coli K-12. J. Bacteriol. 173, 2665–2672. doi: 10.1128/jb.173.8.2665-2672.1991
Marolda, C. L., and Valvano, M. A. (1995). Genetic analysis of the dTDP-rhamnose biosynthesis region of the Escherichia coli VW187 (O7:K1) rfb gene cluster: identification of functional homologs of rfbB and rfbA in the rff cluster and correct location of the rffE gene. J. Bacteriol. 177, 5539–5546. doi: 10.1128/jb.177.19.5539-5546.1995
Minami, M., Hashikawa, S., Ando, T., Kobayashi, H., Goto, H., and Ohta, M. (2021). Involvement of CO2 generated by urease in multiplication of Helicobacter pylori. GSC Adv. Res. Rev. 7, 045–053. doi: 10.30574/gscarr.2021.7.3.0123
Mobley, H. L. T. (1996). The role of Helicobacter pylori urease in the pathogenesis of gastritis and peptic ulceration. Aliment. Pharmacol. Ther. 10(Suppl. 1), 57–64. doi: 10.1046/j.1365-2036.1996.22164006.x
Murgier, J., Coste, J. F., Cavaignac, E., Bayle-Iniguez, X., Chiron, P., Bonnevialle, P., et al. (2016). Microbial flora on cell-phones in an orthopedic surgery room before and after decontamination. Orthop. Traumatol. Surg. Res. 102, 1093–1096. doi: 10.1016/j.otsr.2016.09.014
Pasquale, L., Maurano, A., Cengia, G., Da Massa Carrara, P., German,à, B., Graziani, M. G., et al. (2021). Infection prevention in endoscopy practice: comparative evaluation of re-usable vs single-use endoscopic valves. Infect. Prev. Pract. 3:100123. doi: 10.1016/j.infpip.2021.100123
Philippe, N., Alcaraz, J. P., Coursange, E., Geiselmann, J., and Schneider, D. (2004). Improvement of pCVD442, a suicide plasmid for gene allele exchange in bacteria. Plasmid 51, 246–255. doi: 10.1016/j.plasmid.2004.02.003
Pietrzak, K., Puchalski, M., Otlewska, A., Wrzosek, H., Guiamet, P., Piotrowska, M., et al. (2017). Microbial diversity of pre-Columbian archaeological textiles and the effect of silver nanoparticles misting disinfection. J. Cult. Herit. 23, 138–147. doi: 10.1016/j.culher.2016.07.007
Podzelinska, K., He, S. M., Wathier, M., Yakunin, A., Proudfoot, M., Hove-Jensen, B., et al. (2009). Structure of PhnP, a phosphodiesterase of the carbon-phosphorus lyase pathway for phosphonate degradation. J. Biol. Chem. 284:17216. doi: 10.1074/jbc.M808392200
Pukall, R., Laroche, M., Kroppenstedt, R. M., Schumann, P., Stackebrandt, E., and Ulber, R. (2003). Paracoccus seriniphilus sp. nov., an L-serine-dehydratase-producing coccus isolated from the marine bryozoan Bugula plumosa. Int. J. Syst. Evol. Microbiol. 53, 443–447. doi: 10.1099/ijs.0.02352-0
Rutherford, J. C. (2014). The emerging role of urease as a general microbial virulence factor. PLoS Pathog. 10:e1004062. doi: 10.1371/journal.ppat.1004062
Sack, J., Peaper, D. R., Mistry, P., and Malinis, M. (2017). Clinical implications of Paracoccus yeeii bacteremia in a patient with decompensated cirrhosis. IDCases 7:9. doi: 10.1016/j.idcr.2016.11.008
Sambrook, J., and Russell, D. (2001). Molecular Cloning: A Laboratory Manual, 3rd Edn. New York, NY: Cold Spring Harbor Laboratory Press.
Sartoretti, T., Sartoretti, E., Bucher, C., Doert, A., Binkert, C., Hergan, K., et al. (2017). Bacterial contamination of ultrasound probes in different radiological institutions before and after specific hygiene training: do we have a general hygienical problem? Eur. Radiol. 27, 4181–4187. doi: 10.1007/s00330-017-4812-1
Sastre, A., González-Arregoces, J., Romainoik, I., Mariño, S., Lucas, C., Monfá, E., et al. (2016). Paracoccus yeei peritonitis in peritoneal dialysis. Nefrología 36, 445–446. doi: 10.1016/j.nefroe.2016.09.011
Sayers, S., Li, L., Ong, E., Deng, S., Fu, G., Lin, Y., et al. (2019). Victors: a web-based knowledge base of virulence factors in human and animal pathogens. Nucleic Acids Res. 47:D693. doi: 10.1093/nar/gky999
Schimmel, P., Kleinjans, L., Bongers, R. S., Knol, J., and Belzer, C. (2021). Breast milk urea as a nitrogen source for urease positive Bifidobacterium infantis. FEMS Microbiol. Ecol. 97:fiab019. doi: 10.1093/femsec/fiab019
Schneider, D., Faure, D., Noirclerc-Savoye, M., Barrière, A. C., Coursange, E., and Blot, M. (2000). A broad-host-range plasmid for isolating mobile genetic elements in gram-negative bacteria. Plasmid. 44, 201–207. doi: 10.1006/PLAS.2000.1483
Schweiger, M., Stiegler, P., Scarpatetti, M., Wasler, A., Sereinigg, M., Prenner, G., et al. (2011). Case of Paracoccus yeei infection documented in a transplanted heart. Transplant Infectious Disease 13, 200–203. doi: 10.1111/j.1399-3062.2010.00571.x
Shifera, A. S., Pockrandt, C., Rincon, N., Ge, Y., Lu, J., Varabyou, A., et al. (2021). Identification of microbial agents in tissue specimens of ocular and periocular sarcoidosis using a metagenomics approach. F1000Res 10:820. doi: 10.12688/f1000research.55090.1
Siguier, P., Perochon, J., Lestrade, L., Mahillon, J., and Chandler, M. (2006). ISfinder: the reference centre for bacterial insertion sequences. Nucl. Acids Res. 34:gkj014. doi: 10.1093/nar/gkj014
Siller, H., Rainey, F. A., Stackebrandt, E., and Winter, J. (1996). Isolation and characterization of a new gram-negative, acetone-degrading, nitrate-reducing bacterium from soil, Paracoccus solventivorans sp. nov. Int. J. Syst. Bacteriol. 46, 1125–1130. doi: 10.1099/00207713-46-4-1125
Sullivan, M. J., Petty, N. K., and Beatson, S. A. (2011). Easyfig: a genome comparison visualizer. Bioinformatics 27:1009. doi: 10.1093/bioinformatics/btr039
Szuplewska, M., and Bartosik, D. (2009). Identification of a mosaic transposable element of Paracoccus marcusii composed of insertion sequence ISPmar4 (ISAs1 family) and an IS1247a-driven transposable module (TMo). FEMS Microbiol. Lett. 292, 216–221. doi: 10.1111/j.1574-6968.2009.01495.x
Szuplewska, M., Ludwiczak, M., Lyzwa, K., Czarnecki, J., and Bartosik, D. (2014). Mobility and generation of mosaic non-autonomous transposons by Tn3-derived inverted-repeat miniature elements (TIMEs). PLoS ONE 9:0105010. doi: 10.1371/journal.pone.0105010
Tamayo, R., Pratt, J. T., and Camilli, A. (2007). Roles of cyclic diguanylate in the regulation of bacterial pathogenesis. Annu. Rev. Microbiol. 61, 131–148. doi: 10.1146/annurev.micro.61.080706.093426
Tanaka, Y., Yoshikaie, K., Takeuchi, A., Ichikawa, M., Mori, T., Uchino, S., et al. (2020). Crystal structure of a YeeE/YedE family protein engaged in thiosulfate uptake. Sci. Adv. 6:aba7637. doi: 10.1126/sciadv.aba7637
Tatusov, R. L., Fedorova, N. D., Jackson, J. D., Jacobs, A. R., Kiryutin, B., Koonin, E. V., et al. (2003). The COG database: an updated vesion includes eukaryotes. BMC Bioinf. 4, 1–14. doi: 10.1186/1471-2105-4-41
Thomas, C. M. (1981). Complementation analysis of replication and maintenance functions of broad host range plasmids RK2 and RP1. Plasmid 5, 277–291. doi: 10.1016/0147-619X(81)90005-6
Thompson, J. R., Argyraki, A., Bashton, M., Bramwell, L., Crown, M., Hursthouse, A. S., et al. (2021). Bacterial diversity in house dust: characterization of a core indoor microbiome. Front. Environ. Sci. 9:754657. doi: 10.3389/fenvs.2021.754657
Tsubokura, A., Yoneda, H., and Mizuta, H. (1999). Paracoccus carotinifaciens sp. nov., a new aerobic Gram-negative astaxanthin-producing bacterium. Int. J. Syst. Bacteriol. 49, 277–282. doi: 10.1099/00207713-49-1-277
Urakami, T., Araki, H., Oyanagi, H., Suzuki, K. I., and Komagata, K. (1990). Paracoccus aminophilus sp. nov. and Paracoccus aminovorans sp. nov., which utilize N,N-dimethylformamide. Int. J. Syst. Bacteriol. 40, 287–291. doi: 10.1099/00207713-40-3-287
Van Vliet, A. H. M., Poppelaars, S. W., Davies, B. J., Stoof, J., Bereswill, S., Kist, M., et al. (2002). NikR mediates nickel-responsive transcriptional induction of urease expression in Helicobacter pylori. Infect. Immun. 70:2846. doi: 10.1128/IAI.70.6.2846-2852.2002
Wallet, F., Blondiaux, N., de Sainte Foy, C. L., Loïez, C., Armand, S., Pagniez, D., et al. (2010). Paracoccus yeei: a new unusual opportunistic bacterium in ambulatory peritoneal dialysis. Int. J. Infect. Dis. 14:30. doi: 10.1016/j.ijid.2009.03.030
Xu, L., Dong, Z., Fang, L., Luo, Y., Wei, Z., Guo, H., et al. (2019). OrthoVenn2: a web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucl. Acids Res. 47, W52–W58. doi: 10.1093/nar/gkz333
Zhou, C., Bhinderwala, F., Lehman, M. K., Thomas, V. C., Chaudhari, S. S., Yamada, K. J., et al. (2019). Urease is an essential component of the acid response network of Staphylococcus aureus and is required for a persistent murine kidney infection. PLoS Pathog. 15:1007538. doi: 10.1371/journal.ppat.1007538
Keywords: Paracoccus yeei, opportunistic pathogen, multipartite genome, chromid, evolution of pathogenic bacteria, transposable element, non-autonomous transposable element, urease
Citation: Szuplewska M, Sentkowska D, Lasek R, Decewicz P, Hałucha M, Funk Ł, Chmielowska C and Bartosik D (2024) Genome-wide comparative analysis of clinical and environmental strains of the opportunistic pathogen Paracoccus yeei (Alphaproteobacteria). Front. Microbiol. 15:1483110. doi: 10.3389/fmicb.2024.1483110
Received: 19 August 2024; Accepted: 14 October 2024;
Published: 06 November 2024.
Edited by:
Xuanyu Tao, University of Oklahoma, United StatesCopyright © 2024 Szuplewska, Sentkowska, Lasek, Decewicz, Hałucha, Funk, Chmielowska and Bartosik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Magdalena Szuplewska, bS5zenVwbGV3c2thQHV3LmVkdS5wbA==