 Qiuxu Li1†
Qiuxu Li1† Yingjie Lian1†
Yingjie Lian1† Ketong Zhang2†Jinchao Chen1Long Chen1Jiandong Wu1Yangyang Zhang1Minyi Chen1Weize Zhang1Mengke Lu1
Ketong Zhang2†Jinchao Chen1Long Chen1Jiandong Wu1Yangyang Zhang1Minyi Chen1Weize Zhang1Mengke Lu1 Jun Ma1*Aiquan Bai1*
Jun Ma1*Aiquan Bai1*- 1School of Animal Science and Technology, Foshan University, Foshan, China
- 2College of Horticulture and Landscape Architecture, Zhongkai University of Agriculture and Engineering, Guangzhou, China
The red imported fire ant (RIFA) has made China its habitat for approximately 25 years, but few reports have described the species and amount of virus circulating in it. Researchers are currently exploring viruses associated with RIFAs as potential biological control agents against invasive ants. The present meta-transcriptome analysis revealed the virome of red imported fire ants in Guangdong, southern China, which included 17 viruses, including Solenopsis invicta virus 4-GD (SINV-4) and Guangdong Polycipiviridae ant virus 1 (GPAV1) in the Polycipiviridae family; Solenopsis invicta virus 1-GD (SINV-1), and Guangdong Dicistroviridae ant virus 2-3 (GDAV2-3) in the Dicistroviridae family; Guangdong Iflaviridae ant virus 4-9 (GIAV4-9) in the Iflaviridae family; Guangdong Parvoviridae ant virus 10 (GPAV10) in the Parvoviridae family; and Guangdong ant virus 11-15 (GAV11-15). A total of 15 novel viruses and 2 known viruses were identified in this study. These findings reveal the virome of red imported fire ants in Guangdong Province and present a different result from that of a similar study reported in the United States, providing more choices for potential classical biological control agents against red imported fire ants in China.
1 Introduction
The red imported fire ant (RIFA) is an invasive species originating from South America that exhibits wide adaptability, strong stress resistance, and a rapid reproductive rate it poses significant risks to both humans and animals because of its painful stings and bites, which can cause burns, severe shock, or even death (Kundrotas, 1993). This species can damage biodiversity and ecological stability by hunting other insects, small mammals, and birds (Vinson, 2013). Moreover, the production of crops, orchards, and horticultural plants is disrupted when red imported fire ant (RIFA) activity is located near farms (Wan and Yang, 2016; Xu et al., 2022).
Solenopsis invicta, colloquially known as the red imported fire ant (RIFA), is a species of ant that is notorious for its aggressive behavior and painful stings. It first invaded China in 1999 in the Taiwan Province and then quickly expanded to four cities, with a total area of more than 6,000 hectares (Lin et al., 2021). At the end of 2004, the RIFA was transmitted into mainland China and was discovered in Wuchuan city, Guangdong Province (Zeng et al., 2005). Presently, their distribution spans the southern and eastern provinces of China, such as Hainan, Guangdong, Guangxi, Fujian, Zhejiang, Guizhou, Yunnan, and Jiangxi (Bamisile et al., 2023; Wang et al., 2023). According to a Chinese article, between 1998 and 2020, a total of 862 human cases of ant stings were reported from hospitals, mostly from Quanzhou city of Fujian Province, Yangjiang city of Guangdong Province, and Guangzhou city of Guangdong Province. The clinical symptoms mainly include itch pain (100%), ruddy (99.2%), blain (98.8%), and pimple (59.0%), and severe symptoms such as systemic hypersusceptibility (0.8%) and shock (0.8%) rarely occur (Que Maoqi et al., 2021).
The exploration and utilization of biological control agents and populations have proven instrumental in the management and suppression of fire ants (Manfredini et al., 2016). Viruses are recognized as significant biological control agents against insect populations and have been studied extensively. High-throughput sequencing technology has emerged as a valuable tool for investigating viromes to diagnose unknown infectious diseases (Shi et al., 2016). Guangdong Province in China is acknowledged as a global hotspot for emerging zoonotic diseases and vector-borne diseases because of its unique climate, environment, and biodiversity (Wang et al., 2008; Allen et al., 2017). However, the viromes of RIFAs within Guangdong, southern China, remain inadequately understood.
2 Materials and methods
2.1 Sample collection and preprocessing
Nest soil from ants was collected in September 2022 from villages in Jieyang and Yunfu cities, Guangdong Province, China. After collection, the samples were transported to the laboratory within 24 h and stored at −80°C immediately. After being extracted from the nest soil, the ants were washed with 95% ethanol. Ant species were morphologically identified by an experienced technician and confirmed by sequencing the mitochondrial 16S ribosomal RNA (16S rRNA) gene. To better identify the viruses present in the samples, the ants from the two locations were synthesized into one group. After being washed with phosphate-buffered saline (PBS), the ants were homogenized in PBS, followed by centrifugation at 2,500 × g for 5 min at 4°C. The supernatants were collected for RNA extraction with TRIzol LS reagent (Invitrogen, Carlsbad, CA, United States).
2.2 Meta-transcriptomic and bioinformatics analyses
Meta-transcriptome sequencing was conducted as previously described (Shi et al., 2016). In brief, after the ribosomal RNA (rRNA) was removed, the RNA was fragmented, reverse-transcribed, and adapted, followed by paired-end (150 bp) sequencing on the Illumina HiSeq 2500 platform. All library preparation and sequencing were performed by Tianjin Novogene Bioinformatics Technology Co., Ltd., Tianjin, China.
The sequencing reads were adaptor-and quality-trimmed via the FASTP program, followed by de novo assembly via the Megahit program, and the resulting contigs were compared against the nr database via the diamond BLASTX program. The confirmed viral contigs with assembly overlaps, or from the same scaffold, were merged via the SeqMan program (version 7.1, DNAstar, Madison, WI, United States). Nested RT–PCR was conducted to fill gaps and verify the obtained sequence. The genomic terminus of the target viruses was determined via 5′/3′ RACE kits (TaKaRa, Dalian, China). The primers used are shown in Supplementary Table S2. The reads were mapped to the target contigs via Bowtie 2, and an integrated genomics viewer (IGV) was used to check for assembly faults to validate the assembly results.
2.3 Virus classification
The discovered viruses were classified on the basis of their nucleotide (nt) and amino acid (aa) identities. If the species demarcation criteria remain unclear within a genus, a novel viral species is defined if it holds less than 80% nt identity across the complete genome, or less than 90% aa identity of the RNA-dependent RNA polymerase (RdRp) domain with known viruses. All the novel viruses were named “Guangdong ant,” followed by common viral names according to their taxonomy. Viruses classified into established taxonomies were marked with “Guangdong (GD)” to distinguish them from other viral strains.
2.4 Phylogenetic analysis
Reference virus sequences were acquired from the GenBank database and examined via MAFFT v7.450 to determine the phylogenetic relationships of the identified viruses (Katoh et al., 2019). After testing with ProtTest 3.4, phylogenetic trees were constructed via the maximum-likelihood method in PhyML v3.0 with the Le and Gascuel substitution model for amino acid sequence analysis, which is based on a bootstrap value of 1,000 replicates (Darriba et al., 2011; Guindon et al., 2005).
3 Results
3.1 Viral diversity in ants in Guangdong
A total of 432 ants were collected from two distinct locations within Guangdong Province, China Jieyang (n = 324) and Yunfu (n = 108). Species identification was conducted through a comprehensive amalgamation of morphological scrutiny and 16S PCR techniques. Among the collected samples, 411 were unequivocally identified as Solenopsis invicta, and the remaining 21 were characterized as Polyrhachis dives and were excluded from this study (Supplementary Table S1; Supplementary Figure S1).
Ants were pooled into one group for RNA library construction and sequencing. After quality control and adapter trimming, a total of 85,900,504 paired-end clean reads were generated in these libraries, resulting in 79,054 viral reads, which accounted for 0.1% of the total RNA reads, and were assembled into 95 viral contigs. After alignment via BLAST, the viral contigs were ultimately annotated to 17 viruses from the viral families Polycipiviridae, Dicistroviridae, Iflaviridae, Parvoviridae, and unclassified families. The raw data and obtained virus sequences have been uploaded to NCBI with Bioproject Accessions: PRJNA1061181; Biosample Accessions: SAMN39259552; and SRA accessions: SRR27458542.
3.2 Viral genome organization and phylogenetic characterization
The genomes of 17 viruses, including 15 previously unknown viruses, were obtained through contig-based PCR and rapid amplification of cDNA ends (RACE). The complete genomes of 14 viruses were obtained and named Guangdong Dicistroviridae ant virus 2–3 (GDAV2-3), Guangdong Iflaviridae ant virus 4–9 (GIAV4-9), Guangdong Parvoviridae ant virus 10 (GPAV10), Guangdong ant virus 12,13,15 (GAV12,13,15), Solenopsis invicta virus 1-GD (SINV1 GD) and Solenopsis invicta virus 4-GD (SINV4 GD). Additionally, three viruses with partial sequences were identified and named Guangdong Polycipiviridae ant virus 1 (GPAV1), Guangdong ant virus 11 (GAV11) and Guangdong ant virus 14 (GAV14) (Table 1).
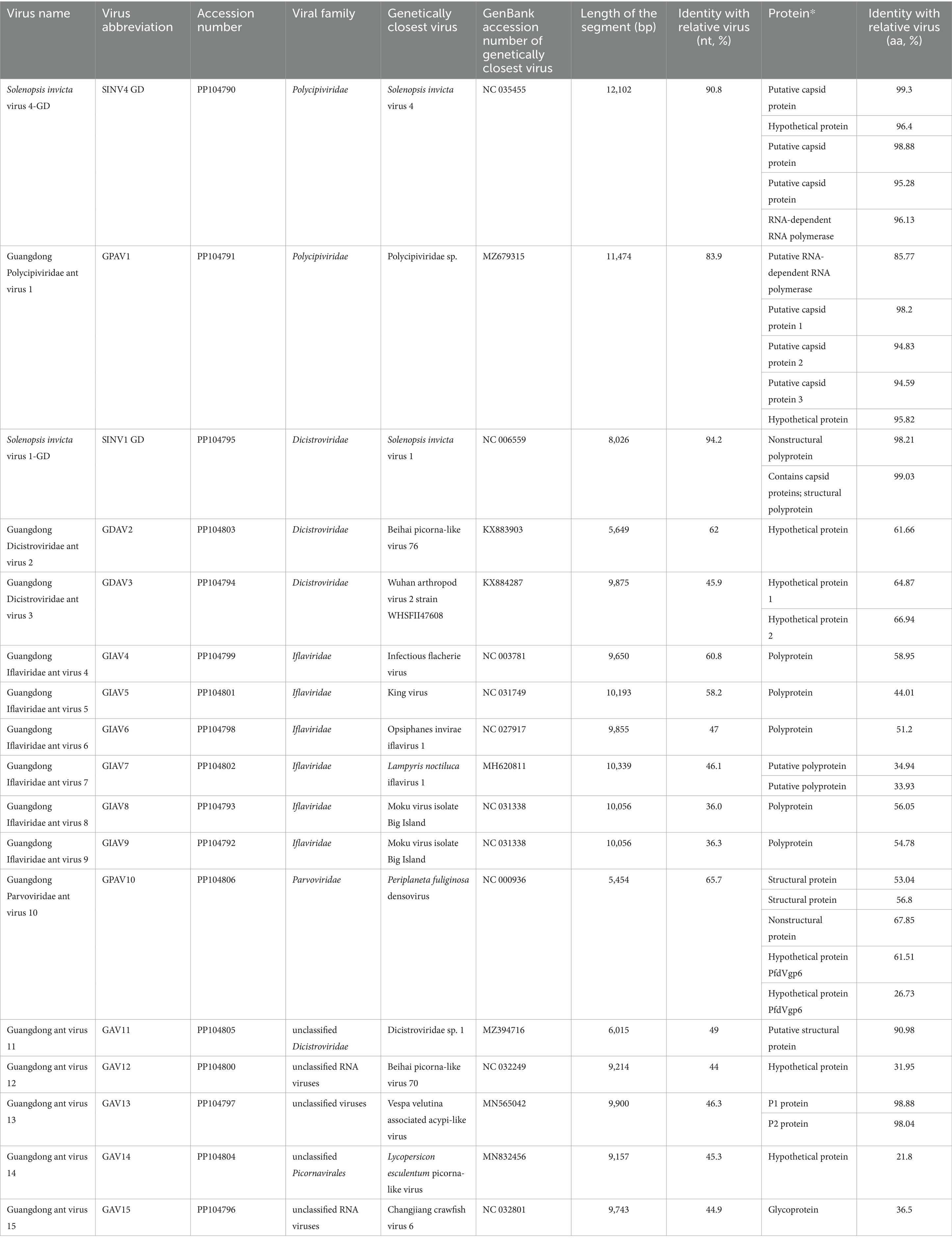
Table 1. Information on the viral genome obtained in this study.
3.3 Polycipiviridae: SINV4 GD, GPAV1
Metagenomic analysis revealed that approximately 2.2% (1,722/79,054) and 0.2% (195/79,054) of the total reads mapped to the genomes of SINV4 GD and GAV7, respectively (Supplementary Table S3). We obtained two complete viral genomes belonging to Polycipiviridae and designated them SINV4 GD and GAV7.
The genome of SINV4 GD includes five ORFs (Figure 1) encoding the RdRp and RNA helicase. The length of the SINV4 GD is 11,965 bp, and it shares high identity (96.1% of the RdRp aa sequence, 90.8% of the complete nt sequence) with Solenopsis invicta virus 4, which was identified in the United States (Olendraite et al., 2017). The GPAV1 genome is 10,504 bp in length, and its RdRp gene shares 83.9 and 85.8% nt and deduced aa sequence identity with the closest viral strain, Polycipiviridae sp., which was identified in China (Chen et al., 2022). Phylogenetically, both SINV4 GD and GPAV1 identified in this study were grouped into the Polycipiviridae family.
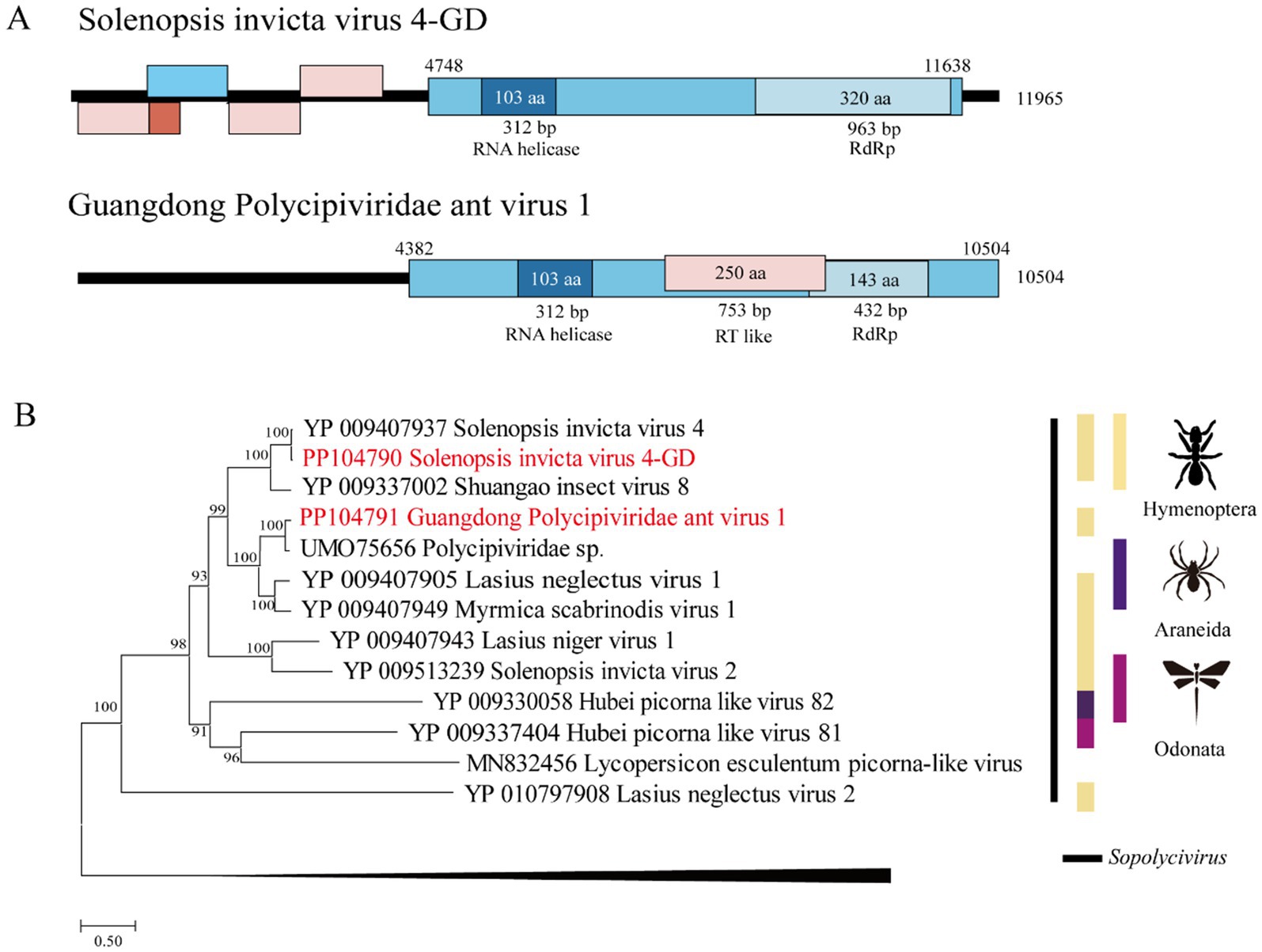
Figure 1. Genome characterization and phylogenetic analysis of Polycipiviridae. (A) Genome organization and putative coding regions of Solenopsis invicta virus 4-GD (SINV4) and Guangdong Polycipiviridae ant virus 1 (GPAV1). Both viral genomes include RNA helicases and RNA-dependent RNA polymerase (RdRp). (B) Phylogenetic analyses of Polycipiviridae. Phylogenetic trees were constructed on the basis of the RdRp amino acid sequences of representative viruses in the family Polycipiviridae. The viruses obtained in this study are highlighted in red. The scale bar at the bottom indicates the amino acid substitutions per site. The vertical color bars at the right of the tree indicate the order of the virus host. The vertical black line at the right of the tree indicates the virus genus.
3.4 Dicistroviridae: SINV1 GD, GDAV2, and GDAV3
In total, 28,419 reads (35.9%; 28,419/79,054) were mapped to viruses in the Dicistroviridae family. A total of 0.3% of the reads (209/79,054) in the pools were matched with SINV1 GD, 0.2% of the reads (261/79,054) were mapped with GDAV2, and 35.4% of the reads (27,994/79,054) were matched with GDAV3 (Supplementary Table S4).
The length of the SINV1 GD was 9,944 bp, with high identity (98.2% of the RdRp aa sequence and 94.2% of the complete nt sequence) to Solenopsis invicta virus 1, which was identified in the United States (Valles et al., 2013). The length of GDAV3 is 10,144 bp, and its RdRp gene shares 45.9 and 66.9% nt and deduced aa sequence identity, respectively, with the Wuhan arthropod virus 2 strain WHSFII47608 (Shi et al., 2016). The length of GDAV2 was 7,418 bp, and its RdRp gene shares 62.0 and 61.7% nt and deduced aa sequence identity with the strain Beihai picorna-like virus 76 (Supplementary Table S4).
Phylogenetically, the SINV1 GD, GDAV2, and GDAV3 genes identified in this study were grouped within the Dicistroviridae clade (Figure 2).
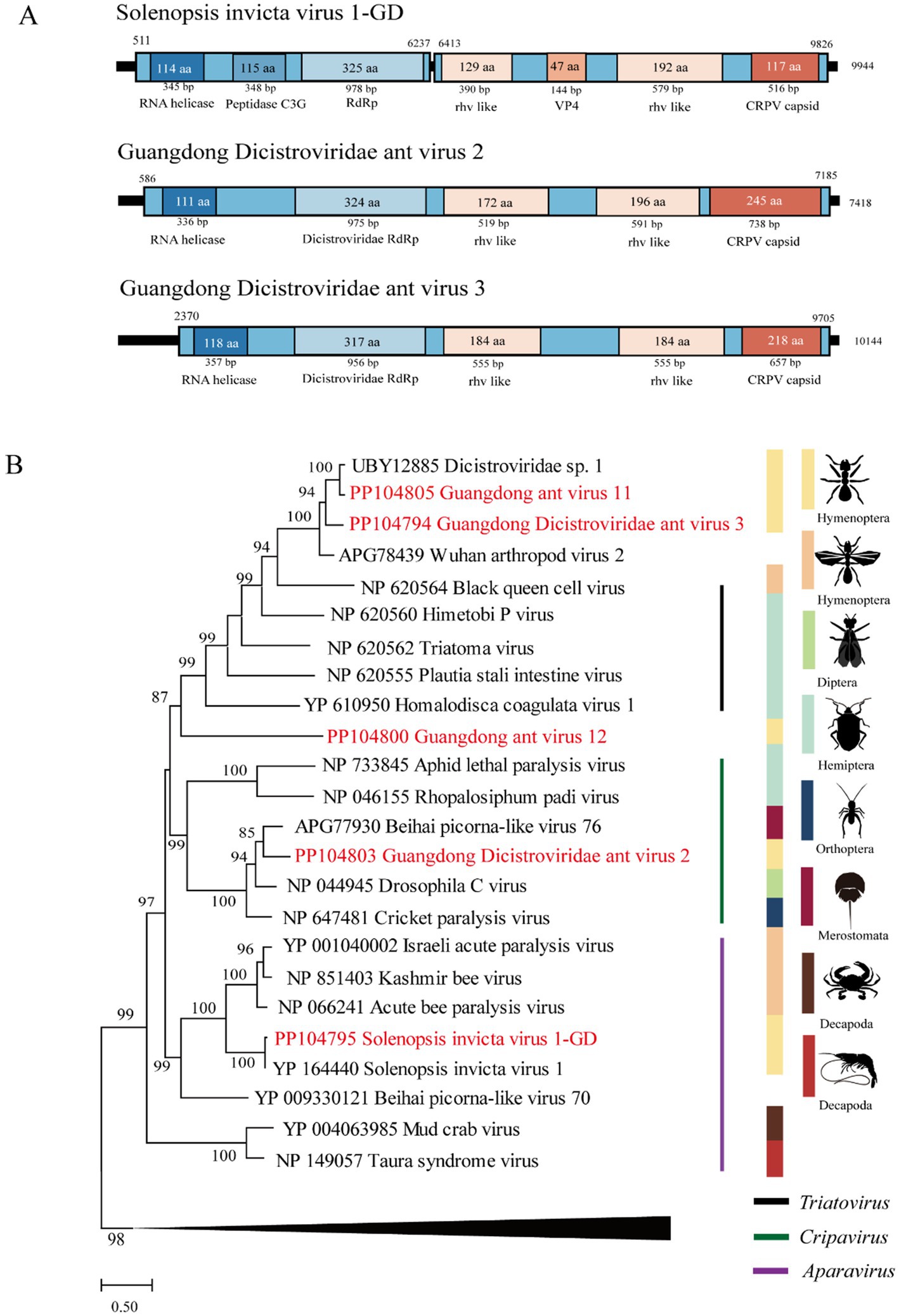
Figure 2. The genome structure and phylogenetic analysis of Dicistroviridae. (A) Genome organization and putative coding regions of Solenopsis invicta virus 1-GD (SINV1), Guangdong Dicistroviridae ant virus 2 (GDAV2), and Guangdong Dicistroviridae ant virus 3 (GDAV3). The viral genome contains segments encoding the RdRp and RNA helicases. (B) Phylogenetic analyses of Dicistroviridae. Phylogenetic trees were constructed on the basis of the RdRp protein sequences of representative viruses in the family Dicistroviridae. The viruses obtained in this study are highlighted in red. The scale bar at the bottom indicates the amino acid substitutions per site. The vertical color bars at the right of the tree indicate the order of the virus host. The vertical color lines at the right of the tree indicate the virus genera.
3.5 Iflaviridae: GIAV4-9
Metagenomic analysis revealed that a total of 6.8% (5,408/79,054) of the viral reads mapped to the genomes of the iflavirus, which included 1.4% (1,114/79,054) GIAV4, 0.4% (345/79,054) GIAV5, 0.2% (172/79,054) GIAV6, 0.6% (506/79,054) GIAV7, 2.2% (1,743/79,054) GIAV8, and 1.9% GIAV9 (1,528/79,054) reads.
The length of GIAV4 is 9,727 bp, and its RdRp gene shares 61 and 59% nt and deduced aa sequence identity with the infectious flounder virus strain (Isawa et al., 1998). The length of GIAV5 was 8,459 bp, and its RdRp gene shares 58.2 and 44% nt and deduced aa sequence identity with the strain King virus. The length of GIAV6 is 9,827 bp, and its RdRp gene shares 47 and 51.2% nt and deduced aa sequence identity with the strain Opsiphanes invirae iflavirus 1 (Silva et al., 2015). The length of GIAV7 is 7,799 bp, and the RdRp gene of GIAV7 is 46.1% nucleotide and has amino acid sequence identities with the Lampyris noctiluca iflavirus 1 strain, with amino acid identities ranging from 33.9 to 34.9%. Compared with those of the Moku virus isolate Big Island, the lengths of GIAV8 and GIAV9 are 10,320 bp and 10,336 bp, indicating nucleotide identities of 36 and 36.3%, respectively, and amino acid identities of 56.1 and 54.8%, respectively, in the RdRp region (Supplementary Table S5) (Mordecai et al., 2016).
Phylogenetically, the six viruses identified in this study were grouped into the Iflaviridae clade (Figure 3).
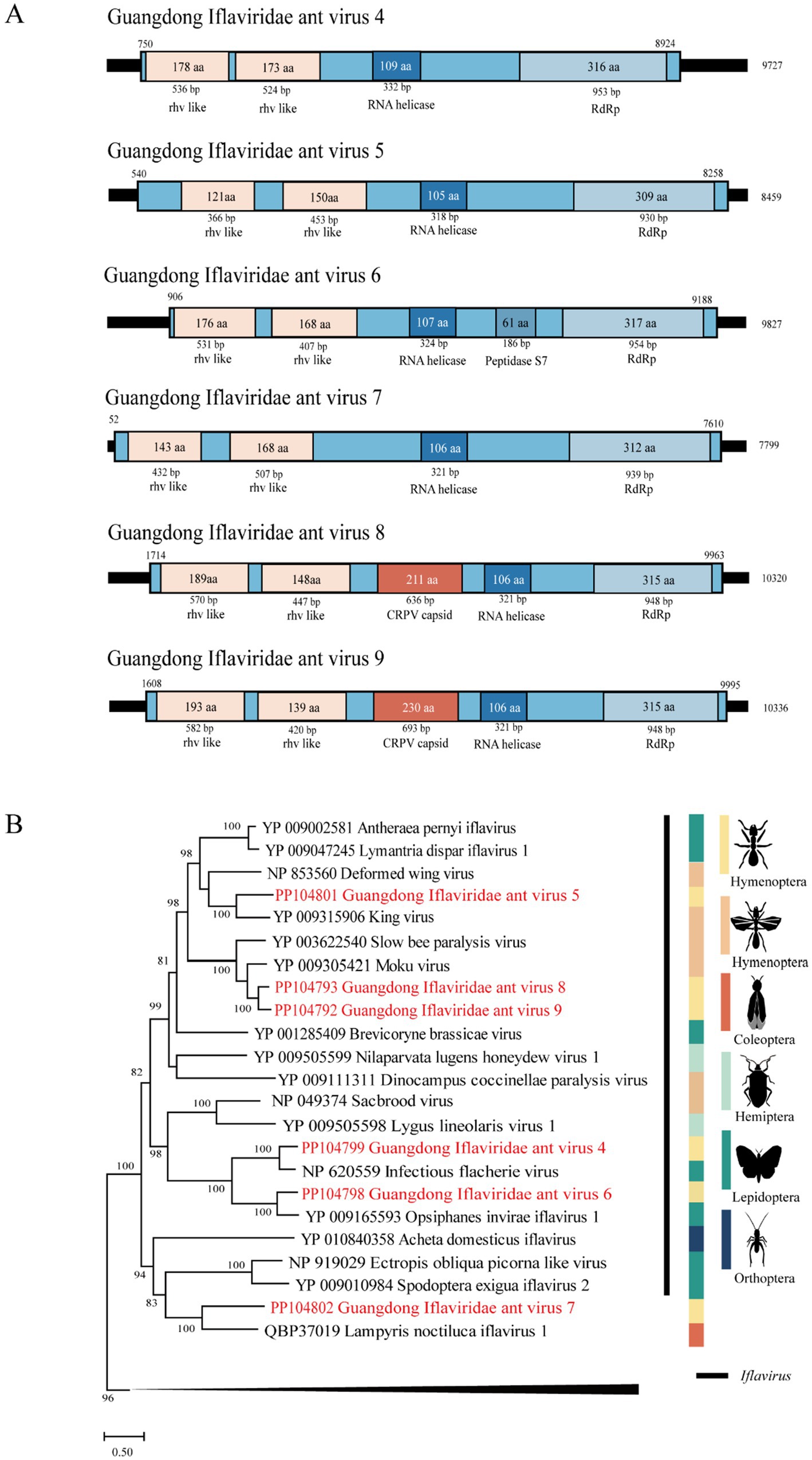
Figure 3. The genome structure and phylogenetic analysis of Iflaviridae. (A) Genome organization and putative coding regions of Guangdong Iflaviridae ant virus 4–9 (GIAV4-9). The viral genome contains segments that encode RdRp, rhv-like, and RNA helicases. (B) Phylogenetic analyses of members of the family Iflaviridae. Phylogenetic trees were constructed on the basis of the RdRp protein sequences of representative viruses in the family Iflaviridae. The viruses obtained in this study are highlighted in red. The scale bar at the bottom indicates the amino acid substitutions per site. The vertical color bars at the right of the tree indicate the order of the virus host. The vertical black line at the right of the tree indicates the virus genus.
3.6 Parvoviridae: GPAV10
Approximately 44.4% of the reads (35,116/79,054) were mapped to the genome of Guangdong Parvoviridae ant virus 8 (GPAV10), which is located in the genus Pefuambidensovirus in the phylogenetic tree. We designated this virus GPAV10, whose genome shares 65.7% and 26.7 ~ 67.9% nt and deduced aa sequence identity with Periplaneta fuliginosa densovirus (Guo et al., 2000) (Figure 4).
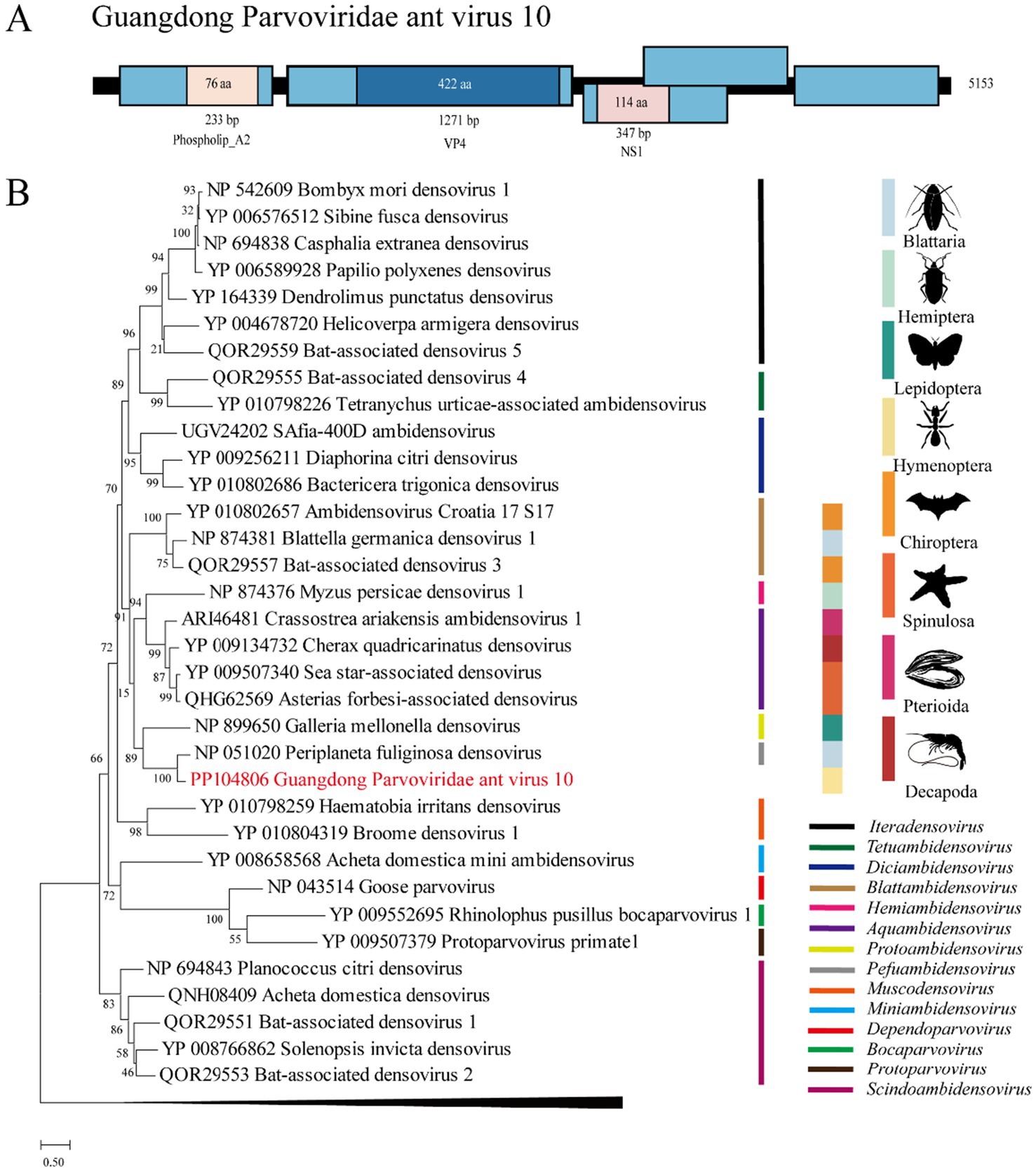
Figure 4. The genome structure and phylogenetic analysis of Parvoviridae. (A) Genome organization and putative coding regions of Guangdong ant Parvoviridae virus 8 (GPAV10). The viral genome contains segments that encode NS1, VP4, and phospholip A2. (B) Phylogenetic analyses of Parvoviridae. Phylogenetic trees were constructed on the basis of the NS1 protein sequences of representative viruses in the Parvoviridae family. The viruses obtained in this study are highlighted in red. The scale bar at the bottom indicates the amino acid substitutions per site. The vertical color bars at the right of the tree indicate the order of the virus host. The vertical color lines at the right of the tree indicate the virus genera.
3.7 Unclassified viruses: GAV11-15
The length of GAV11 is 5,249 bp, and its RdRp gene shares 49 and 91% nt and deduced aa sequence identity with the strain Dicistroviridae sp. 1. The length of GAV14 is 7,345 bp, and its genome shares 45.3% nt identity with that of the Lycopersicon esculentum picorna-like virus. The length of GAV12 is 9,640 bp, and its RdRp gene shares 44% nt and 32% nt and deduced aa sequence identity with the strain Beihai picorna-like virus 70 (Shi et al., 2016). For GAV11, GAV14 and GAV12 may only have partial RdRp information or high diversity with their closest viruses; thus, these viruses were deemed unclassified viruses in this study.
Vespa velutina-associated acypi-like virus, first discovered in the Asian yellow-legged hornet Vespa velutina nigrithorax, is an unclassified virus (Dalmon et al., 2019). In this study, a total of 344 (0.4%, 344/79,054) reads were mapped to the Vespa velutina-associated acypi-like virus. We named this virus Guangdong ant virus 13 (GAV13), which has 9,860 nt and shares 46.3% and 98 ~ 98.9% nt and deduced aa sequence similarity with the Vespa velutina-associated acypi-like virus.
Changjiang crawfish virus 6 is an unclassified RNA virus (Shi et al., 2016). In this study, a total of 3,214 (4.1%, 3,214/79,054) reads were mapped with the Changjiang crawfish virus 6. We named this virus Guangdong ant virus 15 (GAV15), whose genome is 9,901 nt long and shares 44.9 and 36.5% nt and whose aa sequence similarity is with that of the Changjiang crawfish virus 6.
4 Discussion
RIFA originates from South America, but studies indicate that the United States may be the main source of its invasion in China (Ascunce et al., 2011). This species can be transported to new areas through human transport, such as ships, trains and trucks (Wan and Yang, 2016). This study reported the results of a metagenomic analysis of RNA viruses associated with RIFA in Guangdong, southern China, revealing high viral diversity within this region, which is different from the results of a similar study reported in the United States (Valles and Rivers, 2019) (Supplementary Table S6). Seventeen different viruses belonging to four families, namely, Polycipiviridae, Dicistroviridae, Iflaviridae, Parvoviridae, and unclassified families, were identified.
Polycipiviridae is a family of picorna-like viruses with nonsegmented, linear, positive-sense RNA genomes of approximately 10 ~ 12 kb. All the members of the species within the family have been derived from arthropods, mostly from ants (Olendraite et al., 2017). The Solenopsis invicta virus 4 found in Guangdong Province is closely related to the virus found in the United States, and this result is consistent with the previous conclusion that RIFAs in China are from the United States (Ascunce et al., 2011). We also found that a novel virus, GPAV1, clustered in the Polycipiviridae family, and formed a distinct group from other polycipiviruses discovered in ants in the phylogenetic tree.
Dicistroviridae is a family of small nonenveloped viruses with RNA genomes of approximately 8 ~ 10 kilobases (Valles et al., 2017a). Two novel viruses, GDAV2 and GDAV3, were identified and phylogenetically clustered in the Dicistroviridae family. The genome of Solenopsis invicta virus 1 identified in this study is highly similar to that of strains reported in the United States, and is considered a potential tool for preventing the expansion of RIFAs (Hashimoto and Valles, 2007; Valles and Rivers, 2019).
Iflaviridae is a family of small nonenveloped viruses with RNA genomes of approximately 9 ~ 11 kilobases in length encoding a single polyprotein. Currently, iflaviruses are identified mainly from arthropods and primarily from insects (Valles et al., 2017b). In this study, six novel viruses belonging to this family were identified in Guangdong, China, suggesting that diverse iflaviruses circulate in RIFAs in this area.
Parvoviridae is a family of nonenveloped, round, icosahedral symmetric viruses with an approximately 4-to 6-kb-long single-stranded DNA genome (Cotmore et al., 2019). In the ICTV classification, Parvoviridae is divided into two subfamilies: Parvovirinae, which infect mammals and birds, and Densovirinae, which infect arthropods. In this study, we found that a novel virus, GPAV10, which is phylogenetically located in Densovirinae, occupied approximately half of the viral reads in the pool of RIFAs. This result may indicate that GPAV10 is highly infectious to RIFAs.
5 Conclusion
This study investigated the virome of red imported fire ants in Guangdong, southern China, and detected 17 viruses, including 14 viruses with complete genomes and 3 viruses with partial genomes. A total of 15 novel viruses and 2 known viruses were identified in this study. These findings provide information on viruses circulating in red imported fire ants in Guangdong Province and offer more options for potential classical biological control agents against red imported fire ants.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Ethics statement
Ethical approval was not required for the study involving animals in accordance with the local legislation and institutional requirements because given that our research solely involved data analysis and did not conduct any animal experimentation, we were exempt from requiring ethical review, according to the guidelines stipulating ethical approval is primarily necessary for studies involving human or animal subjects.
Author contributions
QL: Data curation, Formal analysis, Investigation, Validation, Writing – original draft, Writing – review & editing. YL: Data curation, Formal analysis, Software, Writing – review & editing. KZ: Resources, Writing – review & editing. JC: Formal analysis, Software, Writing – review & editing. LC: Writing – review & editing. JW: Writing – review & editing. YZ: Writing – review & editing. MC: Writing – review & editing. WZ: Writing – review & editing. ML: Writing – review & editing. JM: Conceptualization, Methodology, Resources, Writing – review & editing. AB: Resources, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was funded by the Guangdong Modern Agricultural Industry Technology System Innovation Team Construction Project 2023, grant number BKS209152, and the Guangdong Basic and Applied Basic Research Foundation, grant number 2022A1515110357.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflicts of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1479934/full#supplementary-material
References
Allen, T., Murray, K. A., Zambrana-Torrelio, C., Morse, S. S., Rondinini, C., Di Marco, M., et al. (2017). Global hotspots and correlates of emerging zoonotic diseases. Nat. Commun. 8:1124. doi: 10.1038/s41467-017-00923-8
Ascunce, M. S., Yang, C. C., Oakey, J., Calcaterra, L., Wu, W. J., Shih, C. J., et al. (2011). Global invasion history of the fire ant Solenopsis invicta. Science 331, 1066–1068. doi: 10.1126/science.1198734
Bamisile, B. S., Siddiqui, J. A., Nie, L., Idrees, A., Aguila, L. C. R., Jia, C., et al. (2023). Baseline analysis of Endophytic fungal associates of Solenopsis invicta Buren from mounds across five counties of Guangdong Province, China. J. Fungi (Basel) 9:377. doi: 10.3390/jof9030377
Chen, Y. M., Sadiq, S., Tian, J. H., Chen, X., Lin, X. D., Shen, J. J., et al. (2022). RNA viromes from terrestrial sites across China expand environmental viral diversity. Nat. Microbiol. 7, 1312–1323. doi: 10.1038/s41564-022-01180-2
Cotmore, S. F., Agbandje-McKenna, M., Canuti, M., Chiorini, J. A., Eis-Hubinger, A. M., Hughes, J., et al. (2019). ICTV virus taxonomy profile: Parvoviridae. J. Gen. Virol. 100, 367–368. doi: 10.1099/jgv.0.001212
Dalmon, A., Gayral, P., Decante, D., Klopp, C., Bigot, D., Thomasson, M., et al. (2019). Viruses in the invasive hornet Vespa velutina. Viruses 11:1041. doi: 10.3390/v11111041
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2011). Prot test 3: fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165. doi: 10.1093/bioinformatics/btr088
Guindon, S., Lethiec, F., Duroux, P., and Gascuel, O. (2005). PHYML online--a web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res. 33 (Web Server issue): W557-559, W557–W559. doi: 10.1093/nar/gki352
Guo, H., Zhang, J., and Hu, Y. (2000). Complete sequence and organization of Periplaneta fuliginosa densovirus genome. Acta Virol. 44, 315–322
Hashimoto, Y., and Valles, S. M. (2007). Solenopsis invicta virus-1 tissue tropism and intracolony infection rate in the red imported fire ant: a quantitative PCR-based study. J. Invertebr. Pathol. 96, 156–161. doi: 10.1016/j.jip.2007.04.006
Isawa, H., Asano, S., Sahara, K., Iizuka, T., and Bando, H. (1998). Analysis of genetic information of an insect picorna-like virus, infectious flacherie virus of silkworm: evidence for evolutionary relationships among insect, mammalian and plant picorna(−like) viruses. Arch. Virol. 143, 127–143. doi: 10.1007/s007050050273
Katoh, K., Rozewicki, J., and Yamada, K. D. (2019). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 20, 1160–1166. doi: 10.1093/bib/bbx108
Kundrotas, L. (1993). Images in clinical medicine. Sting of the fire ant (Solenopsis). N. Engl. J. Med. 329:1317.
Lin, C. H., Wen, T. H., Liu, Y. H., Huang, R. N., and Liu, H. K. (2021). Elucidating how the red imported fire ant (Solenopsis invicta) diffused spatiotemporally among different landscapes in North Taiwan, 2008-2015. Ecol. Evol. 11, 18604–18614. doi: 10.1002/ece3.8465
Manfredini, F., Shoemaker, D., and Grozinger, C. M. (2016). Dynamic changes in host–virus interactions associated with colony founding and social environment in fire ant queens (Solenopsis invicta). Ecol. Evol. 6, 233–244. doi: 10.1002/ece3.1843
Mordecai, G. J., Brettell, L. E., Pachori, P., Villalobos, E. M., Martin, S. J., Jones, I. M., et al. (2016). Moku virus; a new Iflavirus found in wasps, honey bees and Varroa. Sci. Rep. 6:34983. doi: 10.1038/srep34983
Olendraite, I., Lukhovitskaya, N. I., Porter, S. D., Valles, S. M., and Firth, A. E. (2017). Polycipiviridae: a proposed new family of polycistronic picorna-like RNA viruses. J. Gen. Virol. 98, 2368–2378. doi: 10.1099/jgv.0.000902
Que Maoqi, Y. K., Yongwu, L., Chuanlin, W., Xinjun, L., Qingjun, C., Hongzhi, Z., et al. (2021). Research status and bibliometric analysis of ant stings in China from 1998 to 2020. Chin. J. Emerg. Resuscitat. Disast. Med. 16, 1311–1314. doi: 10.3969/j.issn.1673-6966.2021.11.027
Shi, M., Lin, X. D., Tian, J. H., Chen, L. J., Chen, X., Li, C. X., et al. (2016). Redefining the invertebrate RNA virosphere. Nature 540, 539–543. doi: 10.1038/nature20167
Silva, L. A., Ardisson-Araujo, D. M., Tinoco, R. S., Fernandes, O. A., Melo, F. L., and Ribeiro, B. M. (2015). Complete genome sequence and structural characterization of a novel iflavirus isolated from Opsiphanes invirae (Lepidoptera: Nymphalidae). J. Invertebr. Pathol. 130, 136–140. doi: 10.1016/j.jip.2015.08.001
Valles, S. M., Chen, Y., Firth, A. E., Guerin, D. M. A., Hashimoto, Y., Herrero, S., et al. (2017a). ICTV virus taxonomy profile: Dicistroviridae. J. Gen. Virol. 98, 355–356. doi: 10.1099/jgv.0.000756
Valles, S. M., Chen, Y., Firth, A. E., Guerin, D. M. A., Hashimoto, Y., Herrero, S., et al. (2017b). ICTV virus taxonomy profile: Iflaviridae. J. Gen. Virol. 98, 527–528. doi: 10.1099/jgv.0.000757
Valles, S. M., Oi, D. H., Plowes, R. M., Sanchez-Arroyo, H., Varone, L., Conant, P., et al. (2013). Geographic distribution suggests that Solenopsis invicta is the host of predilection for Solenopsis invicta virus 1. J. Invertebr. Pathol. 113, 232–236. doi: 10.1016/j.jip.2013.04.006
Valles, S. M., and Rivers, A. R. (2019). Nine new RNA viruses associated with the fire ant Solenopsis invicta from its native range. Virus Genes 55, 368–380. doi: 10.1007/s11262-019-01652-4
Vinson, S. B. (2013). Impact of the invasion of the imported fire ant. Insect. Sci. 20, 439–455. doi: 10.1111/j.1744-7917.2012.01572.x
Wan, F. H., and Yang, N. W. (2016). Invasion and Management of Agricultural Alien Insects in China. Annu. Rev. Entomol. 61, 77–98. doi: 10.1146/annurev-ento-010715-023916
Wang, X., Qin, Y., Xu, Y., Feng, X., Zhao, S., Lu, Y., et al. (2023). Surveillance and invasive risk of the red imported fire ant, Solenopsis invicta Buren in China. Pest Manag. Sci. 79, 1342–1351. doi: 10.1002/ps.7297
Wang, L., Wang, Y., Jin, S., Wu, Z., Chin, D. P., Koplan, J. P., et al. (2008). Emergence and control of infectious diseases in China. Lancet 372, 1598–1605. doi: 10.1016/S0140-6736(08)61365-3
Xu, Y., Vargo, E. L., Tsuji, K., and Wylie, R. (2022). Exotic ants of the Asia-Pacific: invasion, National Response, and ongoing needs. Annu. Rev. Entomol. 67, 27–42. doi: 10.1146/annurev-ento-060721-085603
Keywords: virome, metagenomic analysis, RNAseq, red imported fire ant, virus
Citation: Li Q, Lian Y, Zhang K, Chen J, Chen L, Wu J, Zhang Y, Chen M, Zhang W, Lu M, Ma J and Bai A (2024) Virome of red imported fire ants by metagenomic analysis in Guangdong, southern China. Front. Microbiol. 15:1479934. doi: 10.3389/fmicb.2024.1479934
Edited by:
Subir Sarker, James Cook University, AustraliaReviewed by:
Pedro Luis Ramos-González, Biological Institute of São Paulo, BrazilQianzhuo Mao, Ningbo University, China
Copyright © 2024 Li, Lian, Zhang, Chen, Chen, Wu, Zhang, Chen, Zhang, Lu, Ma and Bai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Ma, bWFqdW5AZm9zdS5lZHUuY24=; Aiquan Bai, YmFpYWlxdWFuMjAwOEAxNjMuY29t
†These authors have contributed equally to this work and share first authorship