 Juan Li1,2
Juan Li1,2 Qingqing Fang
Qingqing Fang Zhiyong Zong
Zhiyong Zong- 1Center of Infectious Diseases, West China Hospital, Sichuan University, Chengdu, China
- 2Division of Infectious Diseases, State Key Laboratory of Biotherapy, Chengdu, China
- 3Laboratory of Pathogen Research, West China Hospital, Sichuan University, Chengdu, China
- 4Department of General Practice, General Practice Medical Center, West China Hospital, Sichuan University, Chengdu, China
- 5State Key Laboratory of Respiratory Health and Multimorbidity, Chengdu, China
Lytic bacteriophages (phages) are promising clinically viable therapeutic options against carbapenem-resistant Klebsiella pneumoniae (CRKP). In China, the predominant strains are those assigned to sequence type 11 and capsular type 64 (ST11-KL64). The emergence of phage resistance is a major bottleneck hindering effective phage therapy, requiring more new phages to provide the flexibility for creating different phage cocktails. However, the majority of phages against ST11-KL64 CRKP belong to the genus Przondovirus of the family Autographiviridae, which limits the options for constructing cocktails. We recovered a novel lytic phage of the genus Taipeivirus within the family Ackermannviridae against ST11-KL64 CRKP from a river in China. We phenotypically characterized this phage and obtained its genome sequence for analysis. This phage can inhibit the growth of ST11-KL64 CRKP for 6.5 h at a 0.1 multiplicity of infection and exhibits a narrow host range, being unable to attack CRKP strains of the other 30 capsular types. This phage carries no genes encoding antimicrobial resistance, virulence, or lysogeny. It is stable across a wide range of temperatures and pH values, making it suitable for phage therapy. Unlike other Taipeivirus phages, P01 has two tail spike proteins and a unique tail fiber protein. The distinct tail composition of this phage contributes to its activity against ST11-KL64 CRKP and its narrow host range. Taken together, we recovered a phage of a novel viral species with the potential for therapy, which expands the phage biobank against CRKP.
Introduction
Antimicrobial resistance in clinically significant bacteria such as carbapenem-resistant Klebsiella pneumoniae (CRKP), has emerged as a severe global problem (World Health Organization, 2024). The limited pipeline in the development of new antimicrobial agents indicates the urgent need for alternative therapies. Bacteriophages (phages) are viruses that attack bacteria, and some phages can lyse bacteria and can be used for treating bacterial infections (Oechslin, 2018; Nikolich and Filippov, 2020). However, bacteria commonly develop resistance to phages after exposure. One strategy for countering phage resistance is to obtain more phages against the target bacteria to constructing different phage cocktails (Labrie et al., 2010; Tkhilaishvili et al., 2019; Wang et al., 2021). K. pneumoniae of sequence type 11 and capsular type 64 (ST11-KL64) is the predominant type of CRKP in China (Zhou et al., 2020). Lytic phages against ST11-KL64 CRKP typically belong to the genus Przondovirus of the family Autographiviridae within the order Caudovirales (Fang et al., 2022; Yin and Zong, 2022). We have previously demonstrated that the combination of multiple phages belonging to different species of the genus Przondovirus does not prolong the inhibition of the emerging phage resistance, which is likely due to the competition for the same receptor (Yin and Zong, 2023). Therefore, new phages belonging to different genera or families are needed to expand the phage biobank against CRKP. Here, we report a new phage belonging to the genus Taipeivirus of the family Ackermannviridae that effectively lyses ST11-KL64 CRKP.
Methods
Bacterial strain, genome sequencing, and in vitro susceptibility testing
We used a clinical CRKP strain, 135080, for the study and subjected it to whole-genome sequencing using the HiSeq X10 system (Illumina, San Diego, CA). We assembled reads into contigs using SPAdes v3.15.3 (Bankevich et al., 2012) in isolate mode. We performed precise species identification based on average nucleotide identity (ANI) using JSpeciesWS based on BLAST (Peplies et al., 2016) with a ≥ 95% cutoff to define a bacterial species (Richter and Rossello-Mora, 2009). We assigned the strain to a sequence type (ST) using MLST 2.0 (Larsen et al., 2012) by querying the PubMLST database1 and determined the capsular type using Kleborate v2.2.0 (Lam et al., 2021). We screened for β-lactamase encoding genes using AMRFinderPlus v3.10.23 (Feldgarden et al., 2021). We determined minimum inhibitory concentrations (MICs) of meropenem and imipenem using the Clinical and Laboratory Standards Institute (CLSI) microdilution method (CLSI, 2022).
Phage isolation
After we determined that strain 135080 was an ST11-KL64 CRKP (see Results section), we used it for phage isolation. We obtained a 200-mL water sample from a river in Shanghai in May 2022. We filtered the water sample through a 0.22-μm membrane and mixed 17 mL of filtered sewage, 2 mL 10× LB broth, and 1 mL overnight culture of strain 135080 for incubation at 37°C for 4 h. We centrifuged the co-culture at 12,000×g at 4°C for 2 min, filtered the supernatant through a 0.22-μm membrane, and used Tris–HCl-MgSO4 (TM buffer) to dilute the supernatant. We obtained individual plaques by tenfold dilution of the supernatant against strain 135080 using the double-layer agar method as described previously (Green and Sambrook, 2014; Chen et al., 2019). We further purified the isolated phage plaque five times to ensure single phage purification. We therefore obtained a phage capable of lysing 135080 and named it P01 (also called PH1).
Phage genome sequencing and taxonomic assignment
We prepared the genomic DNA of phage P01 using a Phage DNA isolation Kit (Norgen Biotek; Thorold, Canada) following the manufacturer’s protocol. The preparation included treatment with DNase I and RNase to remove nucleic acids from host bacterial cells when preparing phage particles, and another round of DNase I treatment before lysing the intact phages using proteinase K. We sequenced the genome of P01 using the HiSeq X10 platform. Given the large quantity of sequencing data obtained (see Results section), we minimized host genome contamination by randomly subsampling 1,000,000 reads. This provided approximately 940× coverage of the target phage, which was then used for genome assembly using Unicycler v0.5.0 (Wick et al., 2017). We also checked for contamination and discarded contigs that did not belong to the phage using CheckV (Nayfach et al., 2021). We annotated the phage genome using pharokka v1.7.2 (Bouras et al., 2023) and phold v0.1.4 (https://github.com/gbouras13/phold). We predicted the lifestyle of P01 using PhaTYP (Shang et al., 2023). We searched for the phage sharing the highest overall DNA similarity (identity × coverage) with P01 using BLAST.2 We determined the taxonomy of P01 according to the rule defined by the International Committee on Taxonomy of Viruses (ICTV). For the genus Taipeivirus, species demarcation is ≥95% overall DNA similarity.3 In addition to overall DNA similarity, we also generated a heatmap of mutual intergenomic similarities among Taipeivirus phages using VIRIDIC (Moraru et al., 2020). We also performed a maximum-likelihood phylogenetic analysis based on the amino acid sequences of two critical viral components, i.e., the DNA polymerase and the major capsid protein, for all Taipeivirus phages (n = 23) following the recommendation of the ICTV.
Comparison with other Taipeivirus phages
For further exploration of the genetic features of P01, we retrieved all complete assemblies of Taipeivirus (n = 23) from the NCBI GenBank collection (accessed 13 June 2024). We annotated these genomes using pharokka v1.7.2 (Bouras et al., 2023) and phold v0.1.4.4 We clustered orthologous genes using PIRATE v1.0.5 (Bayliss et al., 2019), aligned genes present in all genomes using MAFFT v7.526 (Katoh et al., 2002), and concatenated the sequences for phylogenetic inference. We inferred a maximum likelihood phylogenetic tree using IQ-TREE v2.3.4 (Minh et al., 2020) under the GTR + G + I model with 1,000 ultra-fast bootstraps. We annotated the trees using iTOL v6.9 (Letunic and Bork, 2024) and generated circular genome alignments using the Proksee server (Grant et al., 2023) with P01 as the reference sequence.
Phenotypic characterization of phage P01
We characterized P01 for its morphological and phenotypic profiles, including host range, phage titer, optimal multiplicity of infection (MOI), stability at various pH values and temperatures, and in vitro phage bacteriolytic assay using methods as described previously (Fang et al., 2022; Yin and Zong, 2022). We observed the morphology of P01 using a JEM-1400PLUS transmission electron microscope (JEOL; Tokyo, Japan) at an accelerating voltage of 80 kV. We determined the host range of P01 against a collection of 32 CRKP strains belonging to 30 non-KL64 capsular types and 9 additional CRKP strains of KL64 (Table 1) using spot testing on double-layer agar plates. We tested the thermal stability of P01 by 1 h incubation of 108 PFU per ml in TM Buffer at 4, 25, 37, 50, 60, or 70°C, followed by the double-layer agar method to check phage viability. Similarly, we also examined the stability of P01 at pH 1 to 13 in increments of 1.0 by 1 h incubation of 108 PFU/ml in TM buffer at 37°C. We incubated P01 at 104, 105, 106, 107, or 108 plague-forming units (PFU)/mL at 37°C for 3 h at a MOI of 0.01, 0.1, 1, or 10 to determine the optimal MOI.
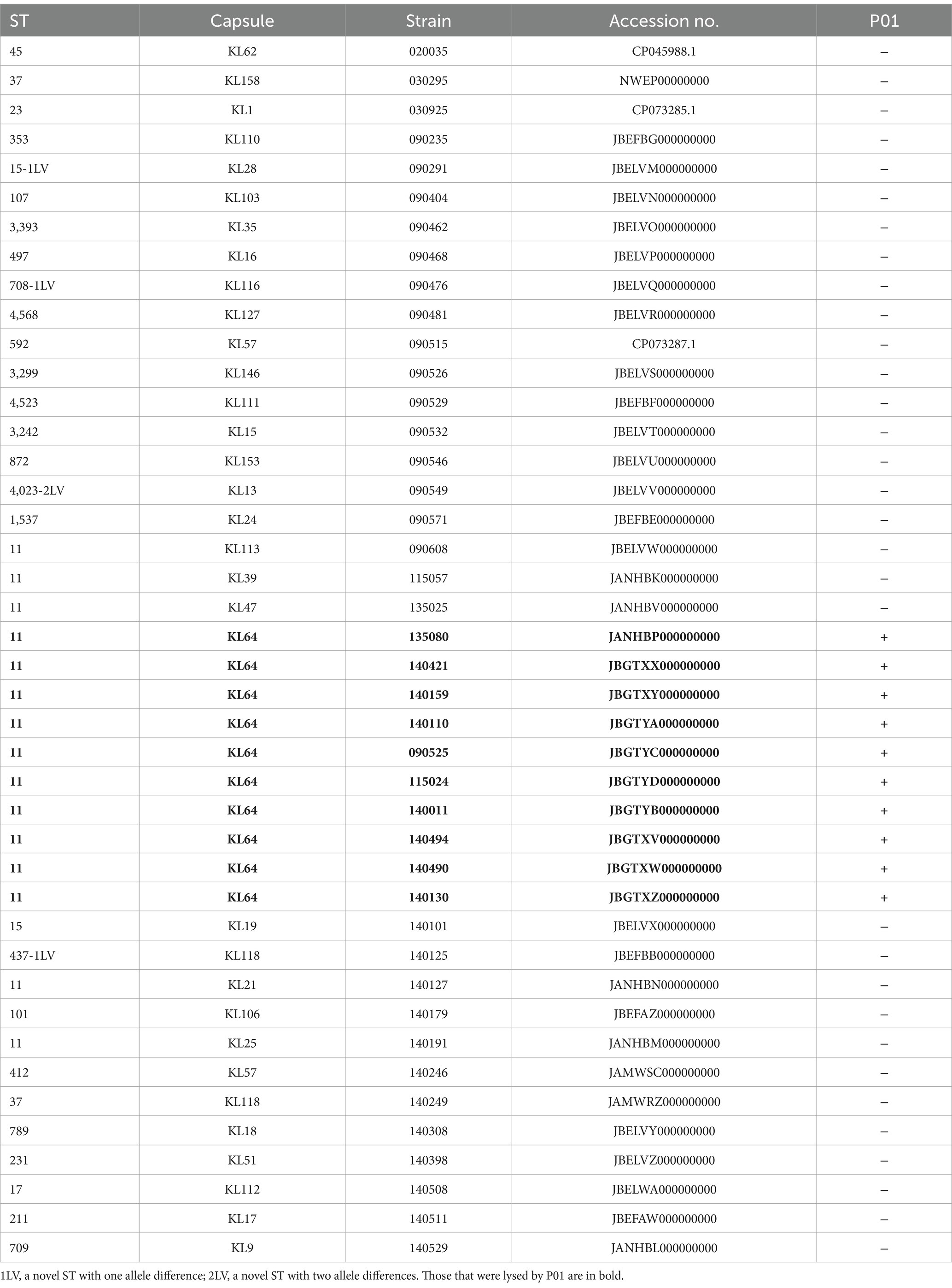
Table 1. Host range of phage P01.
We investigated the in vitro bacteriolytic activity of P01 by adding phage at the optimal MOI (0.1) to the culture of strain 135080 in the pre-logarithmic phase, with an OD600 of 0.20, equivalent to approximately 108 colony-forming units (CFU) per ml. We used the culture of strain 135080 without phages and LB broth without the bacterial strain as controls. We also determined the latent period and the burst size of P01 using the one-step growth experiments as described previously (Abedon and Katsaounis, 2018).
Results
Strain 135080 is an ST11-KL64 CRKP
The genome sequencing of strain 135080 generated 1.39 clean gigabases (Gb, 243× coverage), which were de novo assembled into 147 contigs (N50, 188,881 bp). This strain was identified as K. pneumoniae (sensu stricto) as it shares 98.86% ANI with the K. pneumoniae type strain ATCC 13883T (Accession No. CP064368). This strain has blaKPC-2, a carbapenemase gene, and was indeed resistant to meropenem (MIC, 256 μg/mL) and imipenem (MIC, 128 μg/mL).
Phage P01 lyses ST11-KL64 CRKP and represents a novel species of the genus Taipeivirus within the family Ackermannviridae
We obtained phage P01 from the river sample, which can lyse strain 135080. The genome sequencing of P01 produced 4,056,080 pairs of 150 bp reads, totaling 1.22 Gb. The complete genome of P01 was found to be 158,784 bp in length, with a 46.26% G + C content. It contained 237 genes (Supplementary material S1), along with four tRNA genes and one ncRNA. Notably, no known antimicrobial resistance or virulence genes were present in the genome of P01. In addition, no lysogeny genes were detected in P01, indicating its lytic nature. This was further supported by PhaTYP predictions, which classified P01 as a virulent phage with a full score. The abovementioned findings indicate that this phage is suitable for phage therapy (Suh et al., 2022). Using BLAST, the genome of P01 shows the highest overall DNA similarity (89.19%, identity × coverage) to Klebsiella phage vB_KqM-Westerburg (accession no. LR881137). This phage belongs to the genus Taipeivirus of the family Ackermannviridae within the order Caudovirales of the class Caudoviricetes. Notably, the 89.19% overall DNA similarity is below the 95% cutoff for defining species within the genus Taipeivirus. Consistent with the overall DNA similarity, the intergenomic similarity between P01 and all other Taipeivirus phages ranged from 73.7 to 89.0% (Figure 1). P01 is clustered within the Taipeivirus phages in maximum-likelihood phylogenetic trees based on the amino acid sequences of the DNA polymerase (Supplementary Figure S1) and the major capsid protein (Supplementary Figure S2). We also inferred a maximum-likelihood phylogenetic tree based on all orthologous genes, which also demonstrates the clustering of P01 with Taipeivirus phages (Figure 2). All the above analyses confirm its Taipeivirus membership, and according to the rule defined by the ICTV, P01 represents a novel species of the genus Taipeivirus.
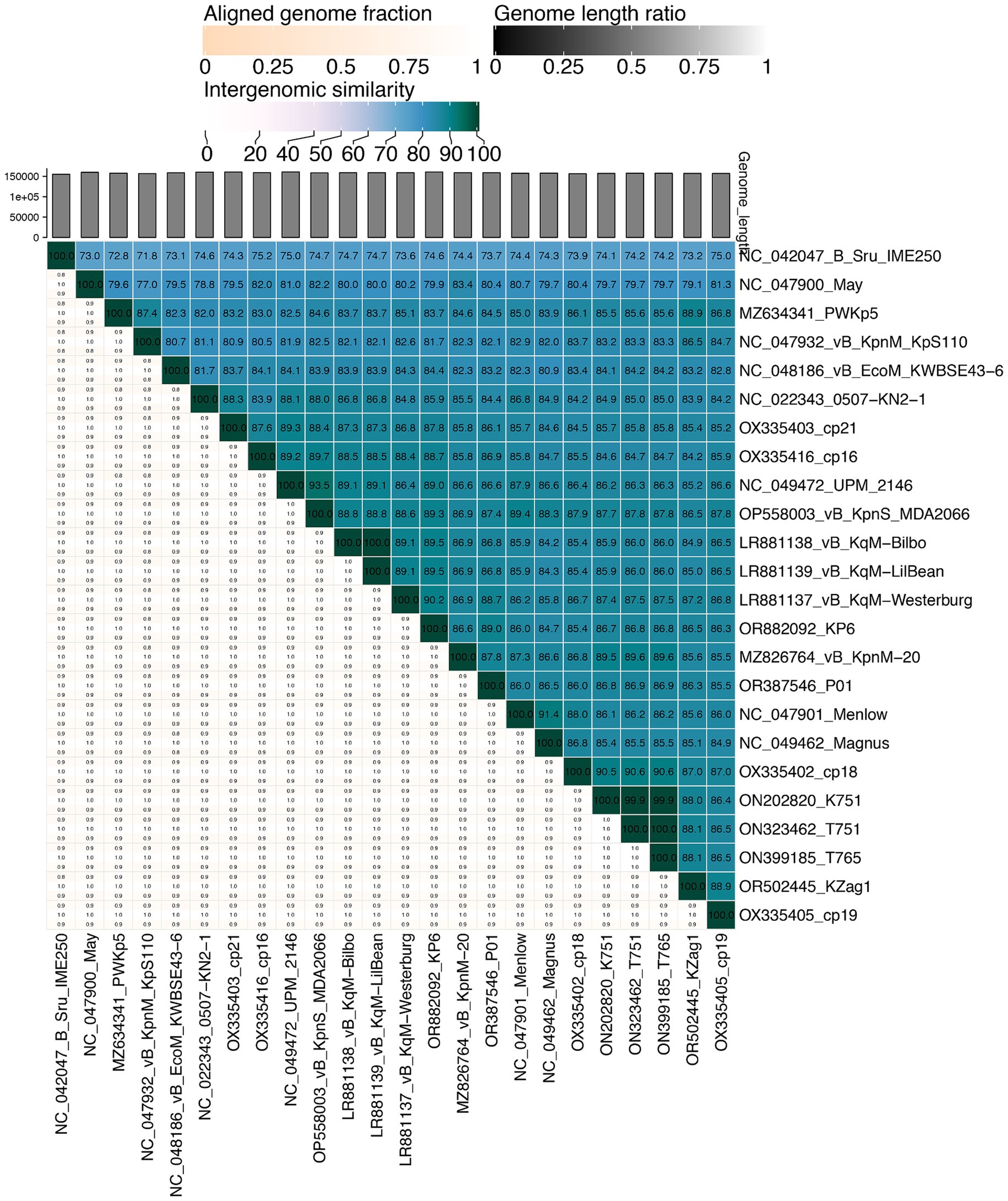
Figure 1. Mutual intergenomic similarities among Taipeivirus phages. This heatmap was generated using VIRIDIC (Moraru et al., 2020). Intergenomic similarities (%) are depicted in blue boxes and alignment genome fractions (0.8, 0.9, and 1.0) are present underneath. The phage names are shown with the accession numbers of their genome sequences.
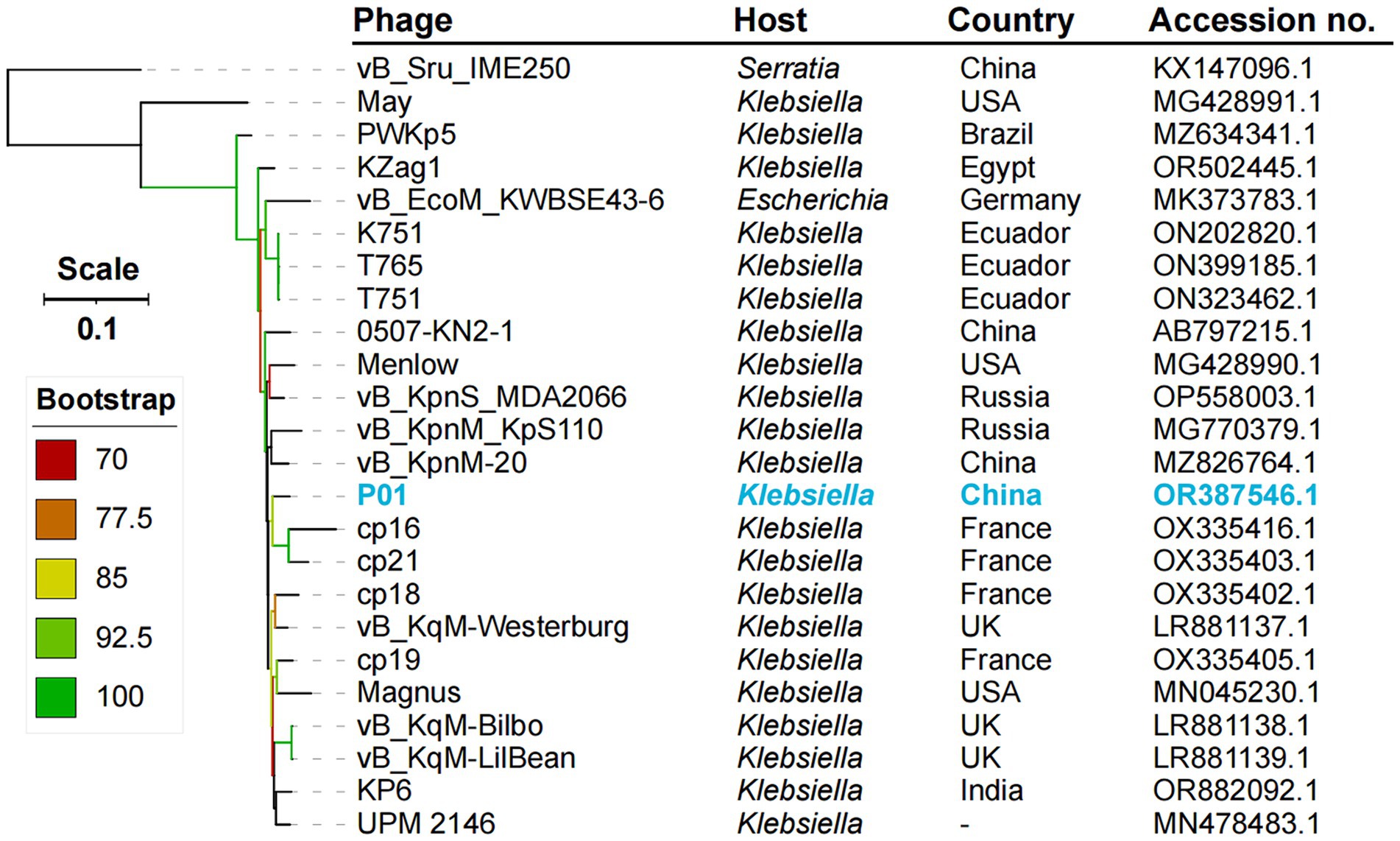
Figure 2. Phylogenetic tree of Taipeivirus phages based on the genome sequences. The maximum likelihood tree was constructed using IQ-TREE v2.3.4 (Minh et al., 2020) under the GTR + G + I model with 1,000 ultra-fast bootstraps and was annotated using iTOL v6.9 (Letunic and Bork, 2024). –, not available.
P01 has a distinct tail composition compared to other Taipeivirus phages
Pairwise comparison of P01 with the 23 Taipeivirus genomes in GenBank revealed the overall similarity (identity × coverage), ranging from 73.61% (with phage vB_Sru_IME250, accession no. KX147096) to 89.19% (with vB_KqM-Westerburg, accession no. LR881137) (Supplementary material S2). Although the genome alignments showed a generally high average coverage of 88% against P01 (Supplementary material S1), we detected several genomic regions across the genome with varying degrees of similarity (Figure 3). These regions contained genes encoding proteins with diverse roles and functions. Specifically, we uncovered two adjacent genes, which are involved in ribose phosphate diphosphokinase activity when coupled with Mg2+, in 14 Taipeivirus phages, including P01 (Supplementary material S1). The two genes encode a pyrophosphokinase and a nicotinamide phosphoribosyl transferase, respectively, with an average nucleotide identity of 97.76 and 96.11% within the 14 Taipeivirus phages (Supplementary material S1). We also found an integrase gene in only six Taipeivirus phages including P01, sharing an average nucleotide identity of 98.57% (Supplementary material S1). Notably, we found that regions with genes classified as tail components were highly variable. Strikingly, P01 encodes two tail spike proteins. In contrast, other Taipeivirus phages typically lack genes encoding tail spikes, with only one such gene identified in phage May (accession no. MG428991) (Nguyen et al., 2019). Phage May shares an 85.26% nucleotide identity with one tail spike gene of P01 (Supplementary material S1). The other tail spike gene of P01 has no similar counterparts within Taipeivirus, and its encoded protein is most similar to a tail spike of an Alcyoneusvirus phage (named K64-1, accession no. NC_027399), sharing a 63.0% coverage and 56.25% amino acid identity. In addition, P01 also has two tail fiber protein genes, one of which is also present in all other 23 Taipeivirus phages with the highest nucleotide identity of 98.08% (Supplementary material S1). The other tail fiber protein gene has no similar counterpart within Taipeivirus but encodes a protein that is most similar to a tail fiber protein of Alcyoneusvirus phage K64-1 sharing a 100% coverage and 57.16% amino acid identity.
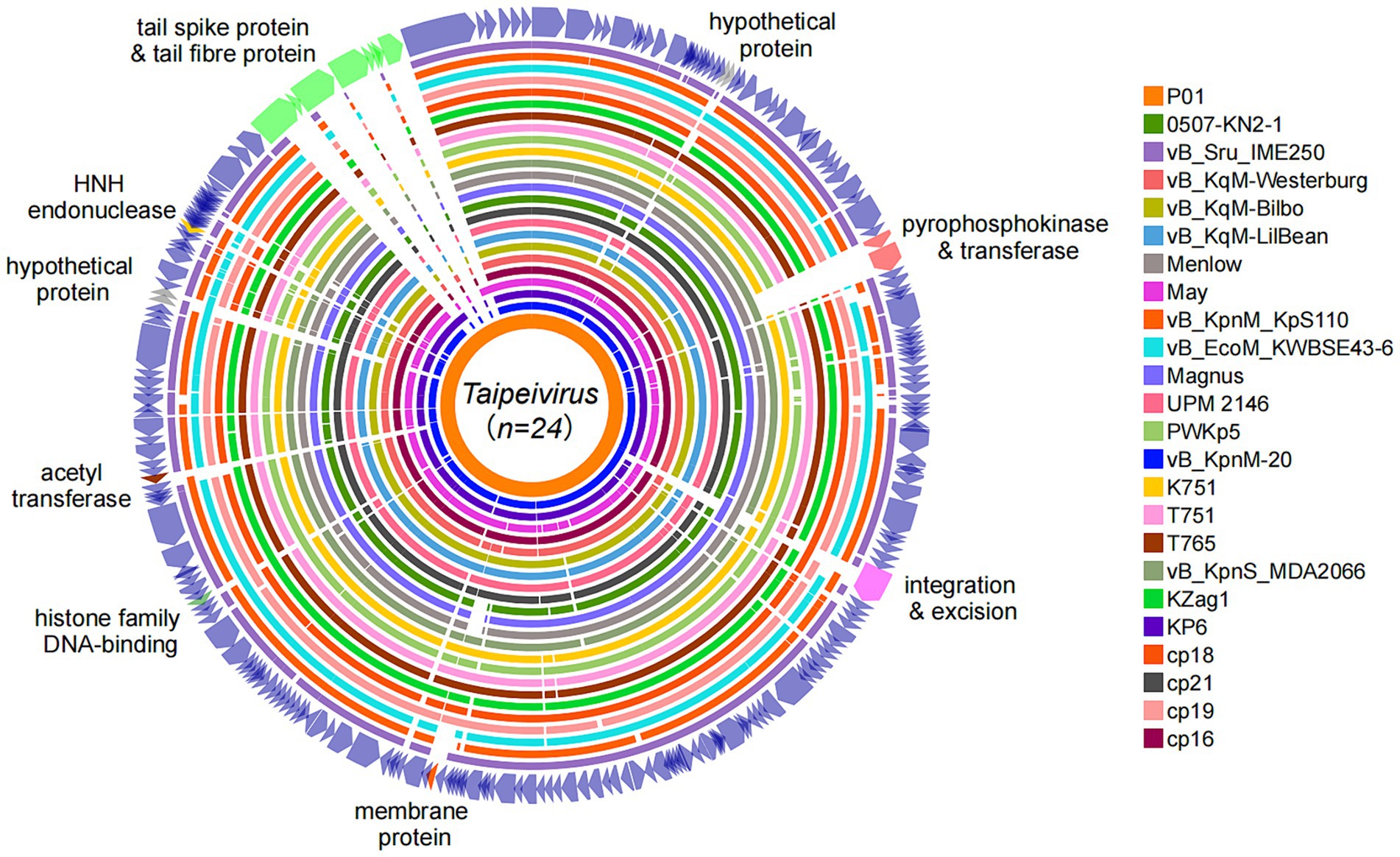
Figure 3. Genome alignments of Taipeivirus phages. Circular genome alignments were generated using the Proksee server (Grant et al., 2023) with P01 as the reference sequence and were annotated using iTOL v6.9 (Letunic and Bork, 2024).
P01 exhibits a Myovirus-like morphology, has a narrow host range, and is stable over a wide range of temperatures and pH values
Under a transmission electron microscope, P01 has a head of an isometric polyhedral structure, with approximately 93.3 nm in diameter and a long, rod-shaped, contractile tail of approximately 72.3 nm in length (Figure 4). Like other Taipeivirus phages, the appearance of P01 is consistent with Myovirus-like morphology (Townsend et al., 2021). In addition to strain 135080, P01 lysed the 9 additional CRKP strains of KL64, while it was unable to lyse non-ST11 strains or ST11 strains of a non-KL64 capsular type, exhibiting a narrow host range. P01 had a burst size of 143 progeny phages per infected bacterial cell and a latent period of 30 min. We found this phage to be stable in the temperature range of 4–50°C and at pH 3–12. We observed that the phage titer was all of 107 PFU/mL at a MOI of 0.01, 0.1, 1, or 10, but was slightly lower (106 PFU/mL) at a MOI of 100. P01 was able to inhibit the growth of strain 135080 for 6.5 h at a 0.1 MOI (Figure 5).
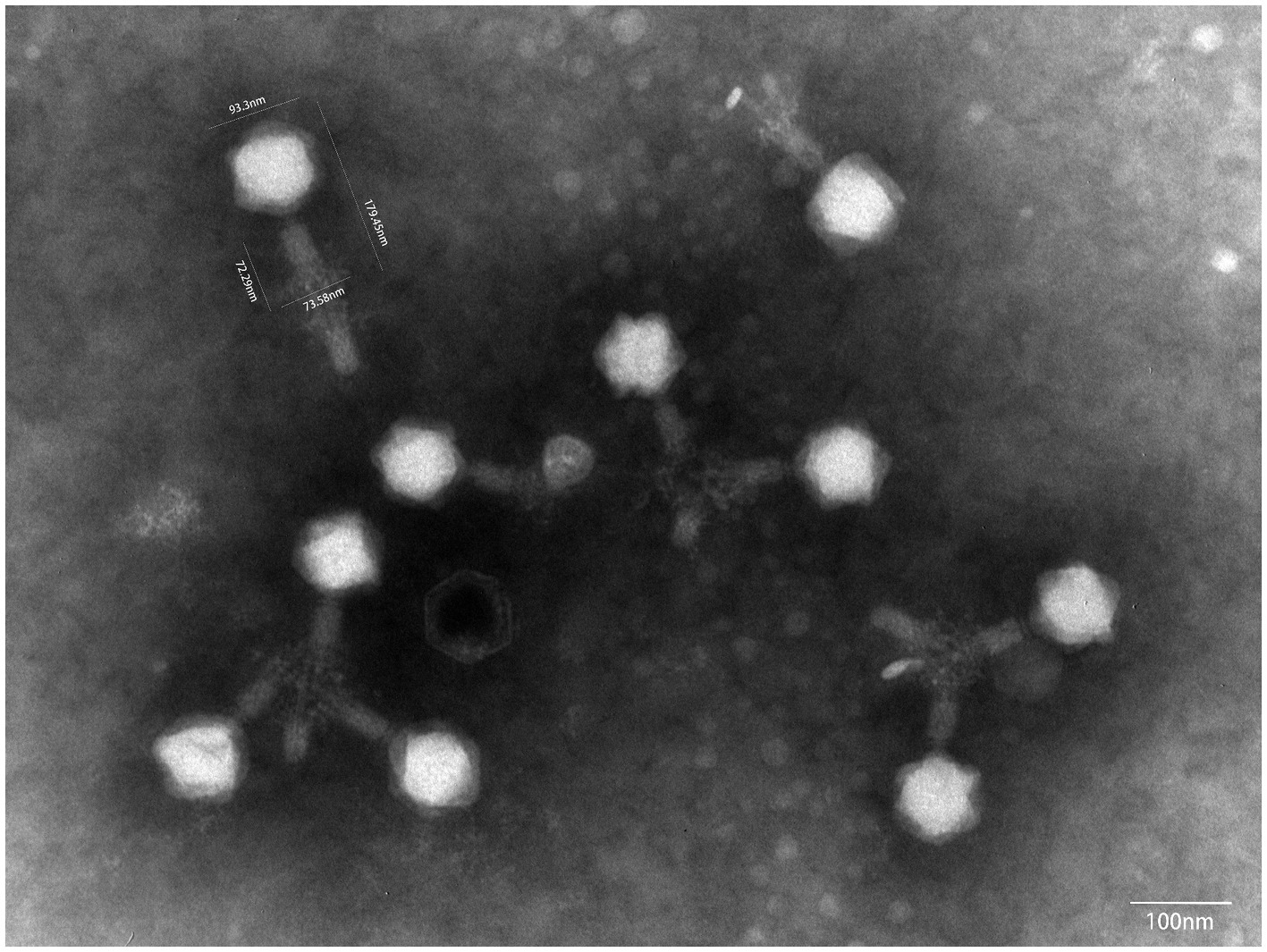
Figure 4. The transmission electron microscope image of P01. The image was obtained using a JEM-1400PLUS transmission electron microscope at an accelerating voltage of 80 kV.
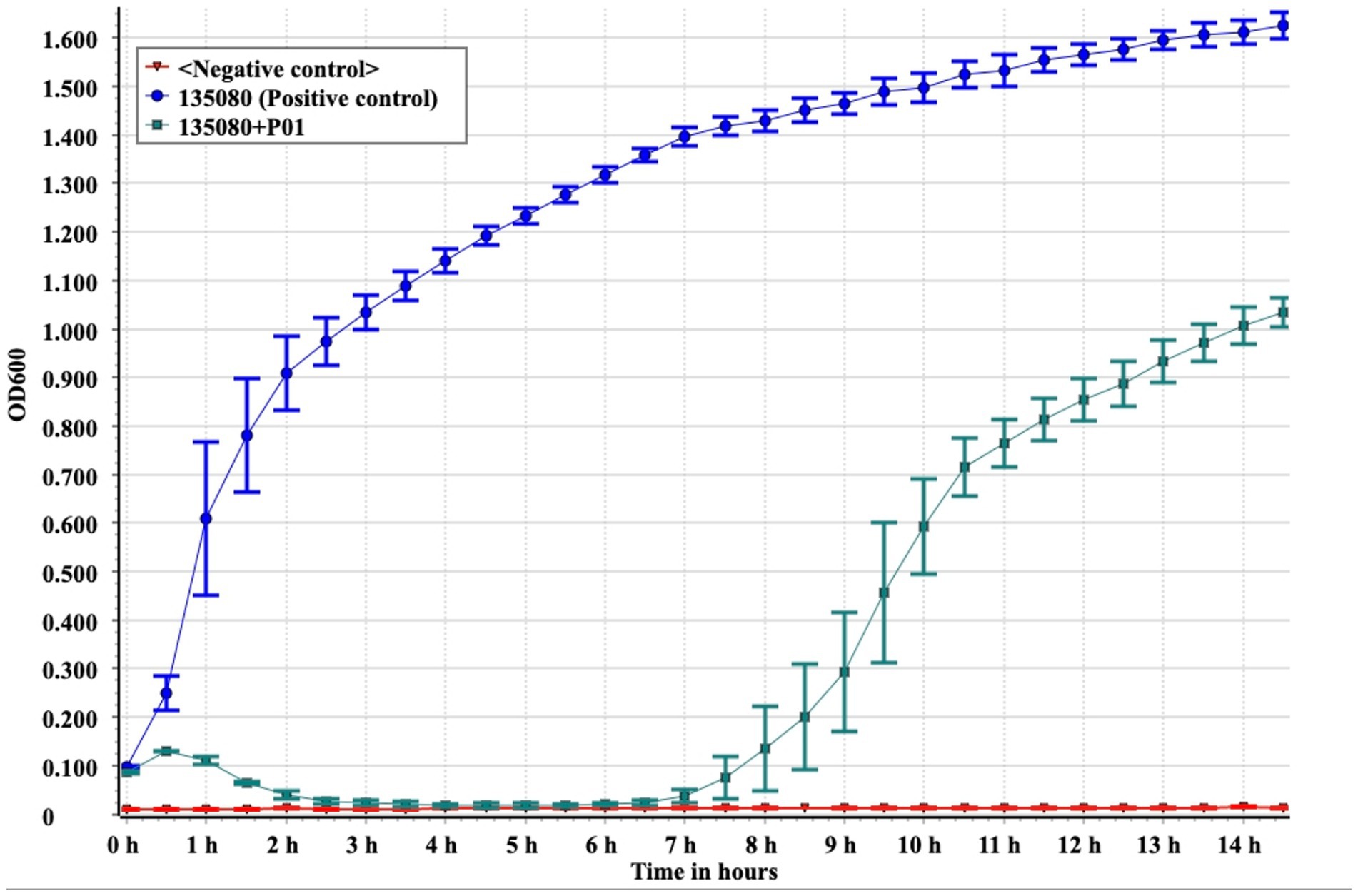
Figure 5. In vitro bactericidal assays of P01 against strain 135080. Assays were performed in triplicate. The median and standard deviation are shown. Negative control, LB broth.
Discussion
In this study, we recovered and characterized a novel phage with the ability to efficiently lyse the predominant CRKP type, ST11-KL64, in China. This phage carries no antimicrobial resistance nor virulence genes and is deemed suitable for phage therapy, thus expanding the biobank of phages viable against CRKP, a difficult-to-treat pathogen. In contrast to the majority of phages recovered so far against ST11-KL64 CRKP, which are members of the genus Przondovirus within the family Autographiviridae, P01 belongs to a novel species of the genus Taipeivirus within the family Ackermannviridae. This suggests that phages from distinct taxonomic positions can target a common bacterial host species.
The genus Taipeivirus was first proposed in 2019 (Korf et al., 2019), while the isolation of the first Taipeivirus phage can be traced back to 2013 (Hsu et al., 2013). As mentioned above, in addition to P01, there are 23 identified Taipeivirus phages: 21 isolated using Klebsiella host strains, one with E. coli, and one with S. rubidaea. This indicates that Taipeivirus phages are viruses that infect Enterobacterales. Taipeivirus phages typically exhibit a narrow host range with the ability to attack a certain species or Klebsiella strains of one or more specific capsular types (Acevedo Ugarriza et al., 2019; Korf et al., 2019; Martins et al., 2021, 2022; Lourenco et al., 2023; Tisalema-Guanopatin et al., 2023; Wu et al., 2023). For instance, phage vB_EcoM_KWBSE43-6, which was recovered using an E. coli strain, cannot infect K. pneumoniae and Klebsiella oxytoca (Korf et al., 2019). Furthermore, phages vB_KqM-LilBean, vB_KqM-Bilbo, and vB_KqM-Westerburg were isolated using Klebsiella strains of the KL35 capsular type (Townsend et al., 2021), while P01 cannot lyse KL35 Klebsiella (Table 1). In addition, vB_KpnM-20 cannot lyse KL64 Klebsiella (Wu et al., 2023) in contrast to P01. Capsular-specific activity indicates that capsules provide the receptor for Taipeivirus phages. The phage host range is determined by the receptor-binding proteins that form the baseplate-attached tail spike or tail fiber proteins (Dunne et al., 2018; Yehl et al., 2019). Taipeivirus phages contain one to four (median, three) tail fiber proteins but typically lack tail spike proteins (Supplementary material S1). Compared to other Taipeivirus phages, P01 has a distinct composition comprising two tail spike proteins plus two tail fiber proteins. Notably, one tail spike protein and one tail fiber protein of P01 are not present in other Taipeivirus phages, but both are most similar to the counterparts of an Alcyoneusvirus phage. Alcyoneusvirus is an independent genus within the class Caudoviricetes, but without being assigned to a specific family or an order.5 It is likely that the two P01-unique tail protein-encoding genes were acquired from other, as yet identified, non-Ackermannviridae phages within the class Caudoviricetes. Nevertheless, the highly variable tail composition provides the flexibility for Taipeivirus phages to adapt to different Enterobacterales strains. The differences between the tail spike and fiber proteins highlight that phage tails are the key evolutionary hotspots in the arms race of phages with their bacterial hosts.
Data availability statement
The draft genome sequence of strain 135080 and the complete sequence of phage P01 have been deposited in GenBank under accession numbers JANHBP000000000 and OR387546, respectively.
Author contributions
JL: Investigation, Project administration, Writing – original draft. YF: Investigation, Writing – original draft, Data curation, Visualization. HL: Writing – original draft, Formal analysis, Validation. QF: Validation, Writing – original draft, Investigation. YY: Investigation, Writing – original draft, Funding acquisition. ZZ: Funding acquisition, Conceptualization, Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The study was supported by grants from the National Natural Science Foundation of China (Project nos. 81861138055 and 82372290), the National Key Research and Development Program of China (Grant nos. 2022YFC2303900 and 2023YFC2308800), the Natural Science Foundation of Sichuan Province (2023NSFSC1467), and grants from the West China Hospital of Sichuan University (1.3.5 Project for Disciplines Of Excellence, Grant nos. ZYYC08006 and ZYGD22001).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1462459/full#supplementary-material
Footnotes
1. ^https://pubmlst.org/organisms/klebsiella-aerogenes
2. ^https://blast.ncbi.nlm.nih.gov/Blast.cgi
3. ^https://ictv.global/taxonomy/taxondetails?taxnode_id=202308036&taxon_name=Taipeivirus
4. ^https://github.com/gbouras13/phold
5. ^https://ictv.global/taxonomy/taxondetails?taxnode_id=202106653
References
Abedon, S. T., and Katsaounis, T. I. (2018). Basic phage mathematics. Methods Mol. Biol. 1681, 3–30. doi: 10.1007/978-1-4939-7343-9_1
Acevedo Ugarriza, L. E., Michalik-Provasek, J., Newkirk, H., Liu, M., Gill, J. J., and Ramsey, J. (2019). Complete genome sequence of Klebsiella pneumoniae Myophage Magnus. Microbiol. Resour. Announc. 8, e01049–e01019. doi: 10.1128/MRA.01049-19
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bayliss, S. C., Thorpe, H. A., Coyle, N. M., Sheppard, S. K., and Feil, E. J. (2019). PIRATE: a fast and scalable pangenomics toolbox for clustering diverged orthologues in bacteria. Gigascience 8:giz119. doi: 10.1093/gigascience/giz119
Bouras, G., Nepal, R., Houtak, G., Psaltis, A. J., Wormald, P. J., and Vreugde, S. (2023). Pharokka: a fast scalable bacteriophage annotation tool. Bioinformatics 39:btac776. doi: 10.1093/bioinformatics/btac776
Chen, L., Fan, J., Yan, T., Liu, Q., Yuan, S., Zhang, H., et al. (2019). Isolation and characterization of specific phages to prepare a cocktail preventing Vibrio sp. Va-F3 infections in shrimp (Litopenaeus vannamei). Front. Microbiol. 10:02337. doi: 10.3389/fmicb.2019.02337
CLSI (2022). Methods for dilution antimicrobial susceptibility tests for Bacteria that grow aerobically. 11th Edn. Wayne, PA, USA: Clinical and Laboratory Standards Institute.
Dunne, M., Denyes, J. M., Arndt, H., Loessner, M. J., Leiman, P. G., and Klumpp, J. (2018). Salmonella phage S16 tail Fiber Adhesin features a rare Polyglycine rich domain for host recognition. Structure 26, 1573–1582.e4. doi: 10.1016/j.str.2018.07.017
Fang, Q., Feng, Y., Mcnally, A., and Zong, Z. (2022). Characterization of phage resistance and phages capable of intestinal decolonization of carbapenem-resistant Klebsiella pneumoniae in mice. Commun. Biol. 5:48. doi: 10.1038/s42003-022-03001-y
Feldgarden, M., Brover, V., Gonzalez-Escalona, N., Frye, J. G., Haendiges, J., Haft, D. H., et al. (2021). AMRFinderPlus and the reference gene catalog facilitate examination of the genomic links among antimicrobial resistance, stress response, and virulence. Sci. Rep. 11:12728. doi: 10.1038/s41598-021-91456-0
Grant, J. R., Enns, E., Marinier, E., Mandal, A., Herman, E. K., Chen, C. Y., et al. (2023). Proksee: in-depth characterization and visualization of bacterial genomes. Nucleic Acids Res. 51, W484–w492. doi: 10.1093/nar/gkad326
Green, M., and Sambrook, J. (2014). Molecular cloning: a laboratory manual. Fourth Edn. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Hsu, C. R., Lin, T. L., Pan, Y. J., Hsieh, P. F., and Wang, J. T. (2013). Isolation of a bacteriophage specific for a new capsular type of Klebsiella pneumoniae and characterization of its polysaccharide depolymerase. PLoS One 8:e70092. doi: 10.1371/journal.pone.0070092
Katoh, K., Misawa, K., Kuma, K., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Korf, I. H. E., Meier-Kolthoff, J. P., Adriaenssens, E. M., Kropinski, A. M., Nimtz, M., Rohde, M., et al. (2019). Still something to discover: novel insights into Escherichia coli phage diversity and taxonomy. Viruses 11:454. doi: 10.3390/v11050454
Labrie, S. J., Samson, J. E., and Moineau, S. (2010). Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 8, 317–327. doi: 10.1038/nrmicro2315
Lam, M. M. C., Wick, R. R., Watts, S. C., Cerdeira, L. T., Wyres, K. L., and Holt, K. E. (2021). A genomic surveillance framework and genotyping tool for Klebsiella pneumoniae and its related species complex. Nat. Commun. 12:4188. doi: 10.1038/s41467-021-24448-3
Larsen, M. V., Cosentino, S., Rasmussen, S., Friis, C., Hasman, H., Marvig, R. L., et al. (2012). Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 50, 1355–1361. doi: 10.1128/jcm.06094-11
Letunic, I., and Bork, P. (2024). Interactive tree of life (iTOL) v6: recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. 52, W78–W82. doi: 10.1093/nar/gkae268
Lourenco, M., Osbelt, L., Passet, V., Gravey, F., Megrian, D., Strowig, T., et al. (2023). Phages against noncapsulated Klebsiella pneumoniae: broader host range, slower resistance. Microbiol. Spectr. 11:e0481222. doi: 10.1128/spectrum.04812-22
Martins, W., Cino, J., Li, M., Lenzi, M., Sands, K., Portal, E., et al. (2021). Lytic bacteriophages against Mutidrug-resistant Klebsiella pneumoniae: development of an effective phage-based approach to combat multidrug resistance. Res. Sq. [Preprint] doi: 10.21203/rs.21203.rs-850585/v850581
Martins, W., Li, M., Sands, K., Lenzi, M. H., Portal, E., Mathias, J., et al. (2022). Effective phage cocktail to combat the rising incidence of extensively drug-resistant Klebsiella pneumoniae sequence type 16. Emerg. Microbes. Infect. 11, 1015–1023. doi: 10.1080/22221751.2022.2051752
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., Von Haeseler, A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. doi: 10.1093/molbev/msaa015
Moraru, C., Varsani, A., and Kropinski, A. M. (2020). VIRIDIC-A novel tool to calculate the Intergenomic similarities of prokaryote-infecting viruses. Viruses 12:1268. doi: 10.3390/v12111268
Nayfach, S., Camargo, A. P., Schulz, F., Eloe-Fadrosh, E., Roux, S., and Kyrpides, N. C. (2021). CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 39, 578–585. doi: 10.1038/s41587-020-00774-7
Nguyen, K. T., Bonasera, R., Benson, G., Hernandez-Morales, A. C., Gill, J. J., and Liu, M. (2019). Complete genome sequence of Klebsiella pneumoniae Myophage May. Microbiol. Resour. Announc. 8, e00252–e00219. doi: 10.1128/MRA.00252-19
Nikolich, M. P., and Filippov, A. A. (2020). Bacteriophage therapy: developments and directions. Antibiotics (Basel) 9:135. doi: 10.3390/antibiotics9030135
Oechslin, F. (2018). Resistance development to bacteriophages occurring during bacteriophage therapy. Viruses 10:351. doi: 10.3390/v10070351
Peplies, J., Oliver Glöckner, F., Rosselló-Móra, R., and Richter, M. (2016). JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 32, 929–931. doi: 10.1093/bioinformatics/btv681
Richter, M., and Rossello-Mora, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 106, 19126–19131. doi: 10.1073/pnas.0906412106
Shang, J., Tang, X., and Sun, Y. (2023). PhaTYP: predicting the lifestyle for bacteriophages using BERT. Brief. Bioinform. 24:bbac487. doi: 10.1093/bib/bbac487
Suh, G. A., Lodise, T. P., Tamma, P. D., Knisely, J. M., Alexander, J., Aslam, S., et al. (2022). Considerations for the use of phage therapy in clinical practice. Antimicrob. Agents Chemother. 66:e0207121. doi: 10.1128/aac.02071-21
Tisalema-Guanopatin, E., Cabezas-Mera, F., Nolivos-Rodriguez, K., Fierro, I., Pazmino, L., Garzon-Chavez, D., et al. (2023). New bacteriophages members of the Ackermannviridae family specific for Klebsiella pneumoniae ST258. Phage (New Rochelle) 4, 99–107. doi: 10.1089/phage.2022.0039
Tkhilaishvili, T., Winkler, T., Muller, M., Perka, C., and Trampuz, A. (2019). Bacteriophages as adjuvant to antibiotics for the treatment of Periprosthetic joint infection caused by multidrug-resistant Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 64, 00924–00919. doi: 10.1128/AAC.00924-19
Townsend, E. M., Kelly, L., Gannon, L., Muscatt, G., Dunstan, R., Michniewski, S., et al. (2021). Isolation and characterization of Klebsiella phages for phage therapy. Phage (New Rochelle) 2, 26–42. doi: 10.1089/phage.2020.0046
Wang, X., Loh, B., Gordillo Altamirano, F., Yu, Y., Hua, X., and Leptihn, S. (2021). Colistin-phage combinations decrease antibiotic resistance in Acinetobacter baumannii via changes in envelope architecture. Emerg. Microbes Infect. 10, 2205–2219. doi: 10.1080/22221751.2021.2002671
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:e1005595. doi: 10.1371/journal.pcbi.1005595
World Health Organization (2024). Bacterial priority pathogens list. Geneva: World Health Organization.
Wu, J. W., Wang, J. T., Lin, T. L., Liu, Y. Z., Wu, L. T., and Pan, Y. J. (2023). Identification of three capsule depolymerases in a bacteriophage infecting Klebsiella pneumoniae capsular types K7, K20, and K27 and therapeutic application. J. Biomed. Sci. 30:31. doi: 10.1186/s12929-023-00928-0
Yehl, K., Lemire, S., Yang, A. C., Ando, H., Mimee, M., Torres, M. T., et al. (2019). Engineering phage host-range and suppressing bacterial resistance through phage tail Fiber mutagenesis. Cell 179, 459–469.e9. doi: 10.1016/j.cell.2019.09.015
Yin, X., and Zong, Z. (2022). Interruption of capsular polysaccharide biosynthesis gene wbaZ by insertion sequence IS903B mediates resistance to a lytic phage against ST11 K64 carbapenem-resistant Klebsiella pneumoniae. mSphere 7, 00518–00522. doi: 10.1128/msphere.00518-22
Yin, X., and Zong, Z. (2023). Safety and efficacy of phage therapy in difficult-to-treat infections: the taxonomic concern. Romanian Arch. Microbiol. Immunol. 82, 16–24. doi: 10.54044/RAMI.2023.01.03
Keywords: antimicrobial resistance, phage therapy, Klebsiella pneumoniae, microbiology, bacteriophages, phage biology
Citation: Li J, Feng Y, Luo H, Fang Q, Yang Y and Zong Z (2024) Ackermannviridae bacteriophage against carbapenem-resistant Klebsiella pneumoniae of capsular type 64. Front. Microbiol. 15:1462459. doi: 10.3389/fmicb.2024.1462459
Edited by:
Steve Petrovski, La Trobe University, AustraliaReviewed by:
Aylin Uskudar Guclu, Başkent University, TürkiyePhitchayapak Wintachai, Walailak University, Thailand
Copyright © 2024 Li, Feng, Luo, Fang, Yang and Zong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhiyong Zong, em9uZ3poaXlAc2N1LmVkdS5jbg==