 Sujatha Kandasamy
Sujatha Kandasamy Kil-Ho Lee†
Kil-Ho Lee† Jun-Sang Ham
Jun-Sang Ham
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 26 September 2024
Sec. Food Microbiology
Volume 15 - 2024 | https://doi.org/10.3389/fmicb.2024.1458221
This article is part of the Research Topic Probiotics for Global Health: Advances, Applications and Challenges View all 23 articles
Lacticaseibacillus casei KACC92338 was originally isolated from Korean raw milk. The antioxidant activities and protective effect in vitro of this strain were evaluated extensively. The results showed that KACC92338 can tolerate hydrogen peroxide up to 2 mM and cell-free supernatant (CFS) had higher scavenging rates for DPPH, hydroxyl radical, reducing power, and iron chelating activities with 95.61 ± 1.59%, 34.10 ± 1.93%, 2.220 ± 0.82 and 81.06 ± 1.06%, respectively. Meanwhile, the CFS showed a protective effect on yeast cells against 10 mM hydrogen peroxide with a survival rate of 76.05 ± 5.65%. To explore the probiotic potential of KACC92338, whole genome assembly and gene clusters with probiotic properties were further analyzed. The genome size was 3,050,901 bp with a 47.96% GC ratio, and 63 contigs. The genome contains 3,048 genes composed of 2,981 coding sequences and 67 RNAs (including 57 tRNAs +9 rRNAs +1 tmRNA). Average Nucleotide Identity and genome-based taxonomy showed that the KACC92338 genome had close similarity with L. casei strains with 96% ANI. Functional annotation by EggNOG and KEGG revealed the presence of numerous genes putatively involved in carbohydrate- and amino acid-transport and metabolism, genetic information processing, and signaling and cellular processes. Additionally, several genes conferring probiotic characteristics such as tolerance to stress, heat, cold, acid, bile salts, oxidative stress, immunomodulation, and adhesion to intestinal epithelium were identified. Notably absent were acquired antibiotic resistance genes, virulence, and pathogenic factors, that prove KACC92338 is a safe strain. Besides, the defense mechanisms of KACC92338 include six prophage regions and three clustered regularly interspaced short palindromic repeat (CRISPR) arrays as acquired immune systems against mobile elements. Further, the BAGEL4 database determined antimicrobial bacteriocin clusters of class IIb: sakacin-P, Enterolysin_A, sactipeptides, and Enterocin X, which suggests the strain could exhibit a wide range of antimicrobial functions. Together, these findings show that the L. casei KACC92338 strain can be a potential probiotic candidate in producing functional fermented foods-, health care- and skin care products- with antioxidant properties. However, a few more mechanistic studies are necessary on the safety assurance and potential application of the strain as a probiotic agent.
Oxidative stress refers to an imbalance between the over production of reactive oxygen species (ROS) and reduction of antioxidant defense mechanisms (Feng and Wang, 2020). Consequently, it is pivotal to protect the human body from attack of ROS, which is related to the onset and or progression of several diseases (i.e., atherosclerosis, chronic diseases, cardiovascular diseases, metabolic disorders, and neurogenerative diseases) (Valko et al., 2007; Zhou et al., 2008; Sharifpanah and Sauer, 2016). Supplementation of antioxidants have become an important means of controlling the oxidative stress. Particularly, natural antioxidants are now more in line with demand. In recent years, lactic acid bacteria (LAB) with antioxidant function have been gaining attention as an effective natural antioxidant because of their potential to alleviate the ROS accumulation in the host (Feng and Wang, 2020; Tian et al., 2022). To develop and commercialize such antioxidant LAB as supplements, they would also have to exhibit probiotic properties. A great quantity of researches reported that probiotic LAB such as L. plantarum KCTC 3099, L. fermentum ME-3, and L. brevis DSMZ 23034 (Lee et al., 2005; Mikelsaar and Zilmer, 2009; Amaretti et al., 2013; Mishra et al., 2015) have positive antioxidant effects in body or cells.
LAB strains have been extensively studied as probiotics because of their desired properties like consumer safety, stability, persistence in the gastrointestinal tract and antioxidation. Traditionally, the selection of probiotic strains relies on studies of their physiological (e.g., acid tolerance) and biochemical properties (e.g., enzyme production) to ensure their safety and potential health benefits. With current advances in next-generation sequencing technologies and platform, the approach has shifted to study the molecular mechanisms and genetic characteristics which can provide more precise confirmation of the taxonomy, lifestyle, and industrial potential of novel LAB strains from their genomes (Guinane et al., 2016). Combining in silico analysis of whole genome sequencing and genome mining tools can provide insights into the complete genetic information and presence of genes related to probiotic properties, metabolic capacities, virulence factors, antibiotic resistance genes, and hazardous metabolites (Guinane et al., 2016; Kandasamy et al., 2022), as well assuring the safety of each probiotic strain at the genome level (Toh et al., 2013; Kang et al., 2017; Chokesajjawatee et al., 2020). However, it is important to note that the genomic information cannot guarantee but only identify the key molecular players involved in the functional role/ mechanisms of the strains (Guinane et al., 2016). In accordance, (Chokesajjawatee et al., 2020) stated that the presence of macrolide (msrA) and beta lactamase (penP) resistance genes in L. plantarum BCC 9546 genome did not confer resistance to erythromycin and ampicillin, respectively. Similarly, identification of hemolysin III protein, the product of hly gene, is another example. Therefore, the combination of characteristic experiments and genetic information of candidate strains can turn out to be very effective to promote the evaluation system of probiotics.
Lacticaseibacillus casei (previously Lactobacillus casei) is a facultative heterofermentative LAB with significant research interest due to its industrial value and potential health benefits. L. casei strains has been extensively used worldwide for manufacturing milk products and other commercial purposes (Hill et al., 2018; Zheng et al., 2020). L. casei-01 strain has traditionally been employed as a probiotic in dairy products. L. casei-01 has recently been incorporated into whey-protein isolate or polysaccharide-based edible films used in meat and bakery products or fruits (Odila Pereira et al., 2018; Dianin et al., 2019; Pruksarojanakul et al., 2020). Although several functionalities of L. casei have been described, to date only few reports have been published describing antioxidative effects and only one have integrated functional genomic data with properties of L. casei (Kim et al., 2022).
To date, there is not a single L. casei strain from raw milk reported for functional genomic data with antioxidant properties. Therefore, it is critically important to build a comprehensive profile of the characteristics and genomic data of the newly isolated L. casei strain. Here, we report the antioxidant properties, whole-genome sequence, and in silico analysis that unravel the probiotic and technological traits of Lacticaseibacillus casei KACC92338 strain, isolated from raw milk samples collected from Republic of Korea (ROK) dairy farms.
Lactic acid bacteria (LAB) strain used in this study were originally isolated from raw milk collected from dairy farm in the ROK. Serial decimal dilutions were prepared from the raw milk and plated on MRS agar (Difco, USA) at 37°C for 2 days in an anaerobic atmosphere. The single colony was subjected to colony purification twice and identification was primarily based on cell morphology, gram staining, and catalase reactions. The cryostocks of the strain was prepared in MRS broth with 20% (v/v) glycerol and stored at −80°C.
The resistance of LAB strain to H2O2 was determined as described by Li et al. (2012), with some modifications. The overnight grown strain was inoculated at 2% (v/v) into MRS broth (control) and MRS broth supplemented with 2 mM H2O2 (sample). Every 1 h, the plate was shaken briefly and the OD600 was measured by a microplate reader (TECAN Infinite M200 Pro, San Jose, USA).
where Asample and Acontrol are optical absorbance of sample and control at 600 nm, respectively.
The strain KACC 92338 was cultured in MRS broth for 24 h at 37°C and the supernatant was collected by centrifugation at 6000 g, 20 min, 4°C. The supernatant was filter sterilized using a 0.22 μm filter membrane (Millipore, MA, USA) and the resulting cell-free supernatant (CFS) was used in for antioxidant assays.
The 1,1-diphenyl-2-picrylhydrazyl (DPPH) free radical-scavenging capacity of KACC 92338 was determined according to the method described by Kao and Chen (2006), with some modifications. Briefly, 1.0 mL CFS was added to same volume of 0.2 mmol/L methanol solution of DPPH. The mixture was mixed vigorously and kept in the dark for 30 min. Then absorbance was measured at 517 nm and scavenging ability is calculated as:
where As is the absorbance of sample (CFS + DPPH), Ab is absorbance of blank (methanol + DPPH), and Ac is absorbance of control (deionized water + DPPH).
The hydroxyl radical scavenging assay was conducted as described by Li et al. (2013) with slight modifications. In brief, 0.5 mL CFS was mixed into a reaction mixture containing 1.0 mL of PBS (20 mM, pH 7.4), 0.5 mL of 1,10-phenanthroline (2.5 mM, Sigma-Aldrich) and 0.5 mL of FeSO4 (2.5 mM). Then 0.5 mL H2O2 (2.5 mM) was added to activate the reaction and kept in waterbath at 37°C for 90 min. The absorbance was measured at 536 nm and free radical scavenging activity is expressed as:
where As is the absorbance in presence of sample (CFS), Ac is the absorbance without sample (deionized water replaced CFS), and Ab is the absorbance of blank (deionized water replaced CFS and H2O2).
The reducing power was determined according to the method of Oyaizu (1986) with few modifications. Briefly, CFS (500 μL) was mixed with an equal volume of each 0.2 M sodium phosphate buffer (pH 6.6) and 1% potassium ferricyanide, and the mixture was incubated at 50°C for 20 min. After cooling rapidly, 10% trichloroacetic acid (0.5 mL) was added and the mixture was centrifuged at 3000 g for 10 min. The obtained supernatant was mixed with an equal volume of deionized water and 0.1% ferric chloride solution (200 μL). The absorbance was read at 700 nm after 10 min.
The Fe2 + −chelating ability was estimated using the method of Lin and Yen (1999). In brief, 0.1 mL sample was added with 0.05 mL of 2 mM FeCl2, 1.85 mL of deionized water, and 0.1 mL ferrozine (5 mM), mixed vigorously and then incubated for 10 min. The absorbance was read at 562 nm and the Fe2 + −chelating ability is calculated as:
where As and Ac are absorbance of sample and control (deionized water replaced sample), respectively.
To measure the protective effect of CFS against oxidative toxicity in eukaryotic cells, Saccharomyces cerevisiae belonged to our lab was incubated with Glucose Peptone Yeast Extract Broth (GPY) at 28°C for 48 h with shaking at 180 rpm. The S. cerevisiae (0.05 mL) was inoculated into 0.95 mL of CFS and incubated for 1 h at 30°C with shaking at 180 rpm. Then H2O2 was added to the suspension at a final concentration 10 mM. After 1 h of incubation at 28°C with shaking, serial dilutions were immediately made and the surviving yeast cells were counted with Potato dextrose agar at 30°C for 48 h. Survival rate was calculated as the ratio of CFU of surviving cells to the CFU without hydrogen peroxide.
All experimental data in this study were obtained from at least three replicates and are expressed as the mean ± standard deviation (SD).
The whole genome sequencing of KACC92338 was performed at TK Biotech and science (Jeonbuk, Republic of Korea) as mentioned in our previous study (Kandasamy et al., 2022), In brief, the genomic DNA from KACC92338 strain (grown in MRS broth aerobically at 37°C, 16 h) was extracted using Exgene™ Cell SV Kit (Cambio, Reading, UK) and its DNA concentration and purity was measured using the Quant-iT™ BR assay Kit (Invitrogen, Waltham, MA, USA). A standard genomic Illumina 350 bp paired-end library was constructed and sequenced using Illumina Novaseq 6,000 platform. The resulting sequence reads were uploaded in Galaxy1, and removal of adapter and low-quality reads was done using the Trimmomatic tool v0.38.12. Then de novo assembly was carried out with Shovill v1.1.0 under default parameters by excluding contigs <100 bp. Finally, the quality of assembled sequence was assessed through the Quast v5.2.0 tool.
The presence /absence of plasmid was determined through Plasmid Finder 2.0 (Carattoli et al., 2014). A graphical genome map of the strain was visualized in the Proksee server (Grant et al., 2023). 2.2. Genome-based identification and taxonomy analysis.
Species identification of KACC92338 was determined via calculation of the Average Nucleotide Identity (ANI) and tetra indices using the JSpecies web server tool with default parameters (Richter et al., 2016). In addition, genome-based taxonomical affiliation at the species level was illustrated through a bootstrapped phylogenetic tree constructed in the Type (Strain) Genome Server (TYGS) server (Meier-Kolthoff and Göker, 2019).
The gene prediction and annotation were performed using the Prokaryotic Genome Annotation System (Prokka) pipeline tool, v1.14.6 in Galaxy (Seemann, 2014) and the Rapid Annotations using Subsystems Technology (RAST) webserver (Aziz et al., 2008) with default parameters. The cluster of orthologous groups (COG) for the protein-coding genes was obtained using Egg-NOG mapper version (2.1.12) tool (Cantalapiedra et al., 2021) from the online Egg-NOG database (version 5.0). Further, a complementary functional analysis was performed using the Kyoto Encyclopedia of Genes and Genomes (KEGG) mapper/BLASTKOALA tool (Kanehisa et al., 2016). Prediction of Carbohydrate-active enzymes (CAZymes) families within the genome was annotated using the dbCAN3 meta server (Huang et al., 2018).
Screening of the annotated genes putative for important probiotic properties was manually predicted through Prokka, RAST, and KEGG–derived annotations, based on previous studies. The bacteriocins in the KACC92338 genome were identified using the BAGEL 4 online server3 (Van Heel et al., 2018).
The presence of antibiotic resistance genes in the genome was analyzed using three databases, i.e., ResFinder tool v.4.1. of the Center for Genomic Epidemiology (Bortolaia et al., 2020), Resistance Gene Identifier (RGI) tool in Comprehensive Antibiotic Resistance Database (CARD) (Alcock et al., 2020) and BlastKOALA tool in KEGG database (Kanehisa et al., 2016).
Putative virulence factors and toxin genes were searched using the Virulence finder v.2.0.3 (Joensen et al., 2014) and Virulence Factor of Bacterial Pathogen database (VFDB) (Liu et al., 2022). In addition, the BlastKOALA tool in KEGG database (Kanehisa et al., 2016) was used for inspection of virulence factors and undesirable genes. The MobileElementFinder (version 1.03) was used to detect mobile genetic elements and their relation to antimicrobial resistance genes and virulence factors (Johansson et al., 2021).
Bacterial insertion sequences were identified using ISfinder with an E-value threshold of 1e−5 (Siguier et al., 2006). The genomic plasticity analysis was performed in Island Viewer 4 platform to determine the presence of genes related to virulence factors and antibiotic resistance genes (Bertelli et al., 2017). The presence of Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated (Cas) genes were identified using the CRISPRCasFinder (Couvin et al., 2018). Prophage regions within the genome were detected using the PHAge Search Tool Enhanced Release (PHASTER) webserver (Arndt et al., 2016).
Hydrogen peroxide is a weak oxidant than hydroxyl, but can easily penetrate the cell membrane and form more active ROS and hydroxyl radicals, causing oxidative damage to DNA, proteins, and lipids (Mishra et al., 2015). The survival rate of KACC92338 strain at various concentrations of H2O2 is presented in Table 1. This result indicated that strains had a considerable tolerance toward H2O2. The addition of H2O2 hindered the growth of the strain and duration of the lag phase was prolonged with increase in H2O2 concentration (data not shown), implying that the presence of H2O2 caused oxidative damage in bacterial cells. The observations were similar to growth pattern reported earlier with L. plantarum MA2 (Tang et al., 2017). It could be observed that KACC92338 strain could survive the challenges until 2 mM H2O2, with a survival rate of 95.99% within 48 h, higher than that reported elsewhere (Vijayakumar et al., 2015; Mu et al., 2018). The higher survival rate might be caused by the extracellular polysaccharides produced by the bacteria (Li et al., 2023). Therefore, the cell free supernatant (CFS) of the strain was further used to explore its in-vitro antioxidant potential.
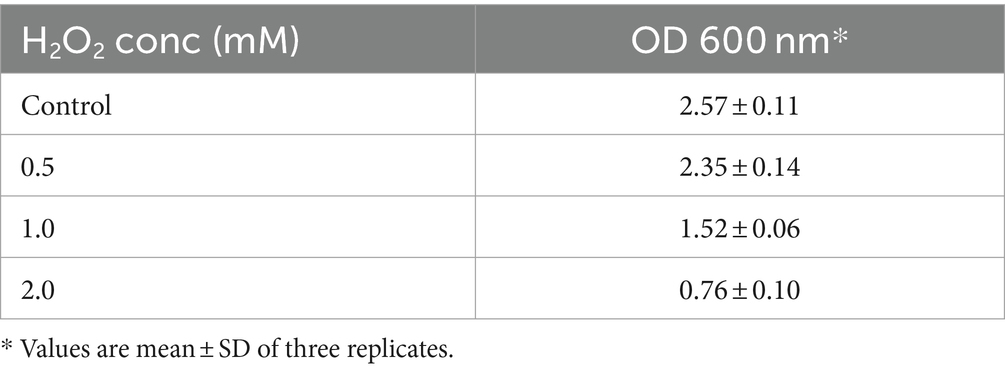
Table 1. Survival of L. casei KACC92338 under the stress of different concentrations of H2O2.
Several methods have been developed to evaluate the antioxidant potential of LAB strains (Gulcin, 2020). In the present study, four indices (DPPH radical, hydroxyl radical, reducing power, and Fe-chelating ability) were chosen to evaluate the antioxidant activity of KACC92338 in vitro in order to gather more complete information for a better understanding of the topic (Table 2). The CFS exhibited stronger scavenging activity for DPPH (95.61 ± 1.59%), reducing power (2.220 ± 0.82), hydroxyl radical (81.06 ± 1.06%) and Fe-chelating ability (34.10 ± 1.93%). The DPPH and hydroxyl radical scavenging activity of this study was much higher than to previous reports by L. plantarum MA2 (40.42 ± 2.19%) (Tang et al., 2017) and L. plantarum KM1 (Tian et al., 2022). Similarly, the iron chelating activity (81.06 ± 1.06%) of KACC92338 was much higher than reported elsewhere. The reducing power which serve as a significant indicator for potential antioxidant capacity was also seems to be remarkable for our strain. These results conclude that the CFS of KACC92338 possess good antioxidant capacity.
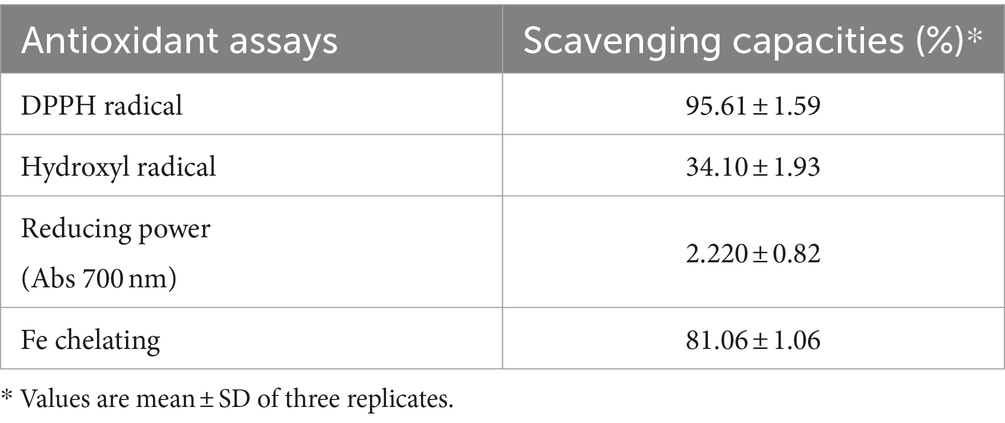
Table 2. Antioxidant activity of cell-free supernatant from L. casei KACC92338 determined using different assays.
Since L. casei KACC92338 has good antioxidant capacity in-vitro, the protective effect of CFS against the H2O2 toxicity was determined using S. cerevisiae. Yeast cells was chosen as it was the simplest eukaryote and closest to the human body cells. Supplementation of antioxidants can effectively reduce the H2O2 damage to DNA, by directly reacting with H2O2 and the intermediate products of H2O2 or inhibit the peroxidase in conjunction with H2O2 can reduce the damage to cells (Tang et al., 2017). The results (Figure 1) showed that compared to control (42 ± 3.26%), cells treated with CFS showed higher viability (76.05 ± 5.65%), indicating the strongest protective effect against H2O2. These results suggested that KACC92338 is an effective scavenger for free radicals and protective agents that could help to reduce damages in human body caused by oxidative stress. Previous reports have shown that CFS supernatants from LAB are rich in antioxidant elements such as bioactive peptides, antioxidant enzymes, lipoteichoic acid or extracellular polysaccharides, carotenoids, histamine and magnesium ions (Kim et al., 2021). Therefore, whether KACC92338 can produce antioxidant molecules to improve its antioxidant performance needs to be further verified.
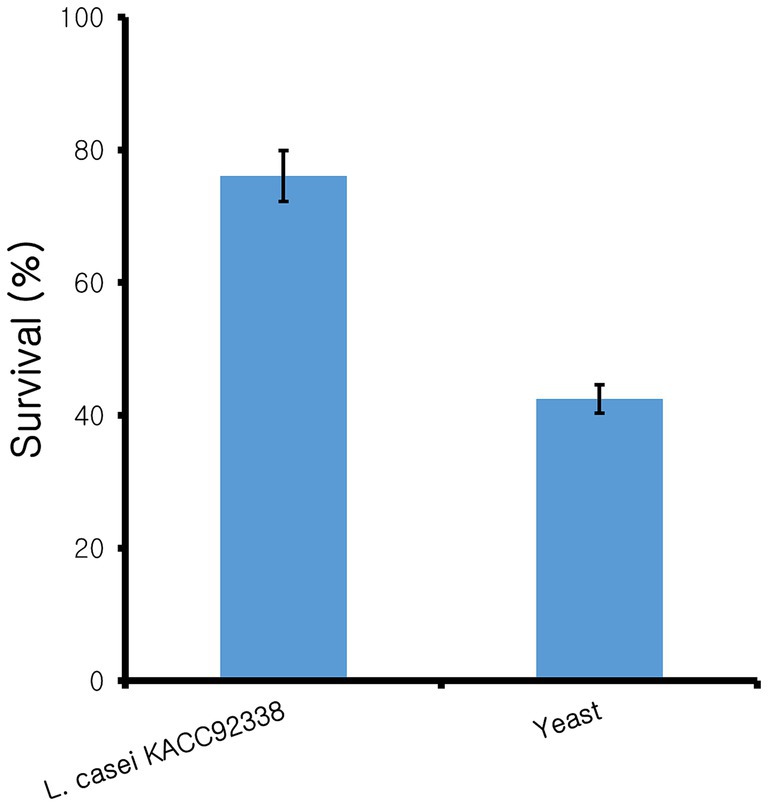
Figure 1. Protective effect of cell-free supernatant from L. casei KACC92338 on yeast cells from H2O2 oxidative damage. Values are mean ± SD of three replicates.
In conclusion, KACC92338 exhibits great in-vitro antioxidant activity, and signifies promising antioxidant potentials for therapeutic benefits to human health. Hence, to further understand its genetic characteristics and evaluate its probiotic potential of KACC92338 at genetic levels, whole-genome sequencing was performed.
The sequenced whole genome of KACC92338 consists of a circular chromosome of length 3,050,901 bp (Genbank accession number SRR14601783), with an overall guanine-cytosine (GC) content of 47.96% (Figure 2), and devoid of plasmid DNA. The general features of the KACC92338 genome are presented in Table 3. The L. casei genomes are the second largest Lactobacillus genomes sequenced to date. Notably, the genome of KACC92338 was higher than L. casei ATCC 334 (Cai et al., 2009) and L. casei ATCC 393 (Toh et al., 2013), while GC content ratio was similar to the foresaid L. casei strains. In previous studies, a positive correlation was reported between the genome size and ubiquity, indicating strains with larger genomes adapt to a wider ecological niches (Cai et al., 2009; Guinane et al., 2016).
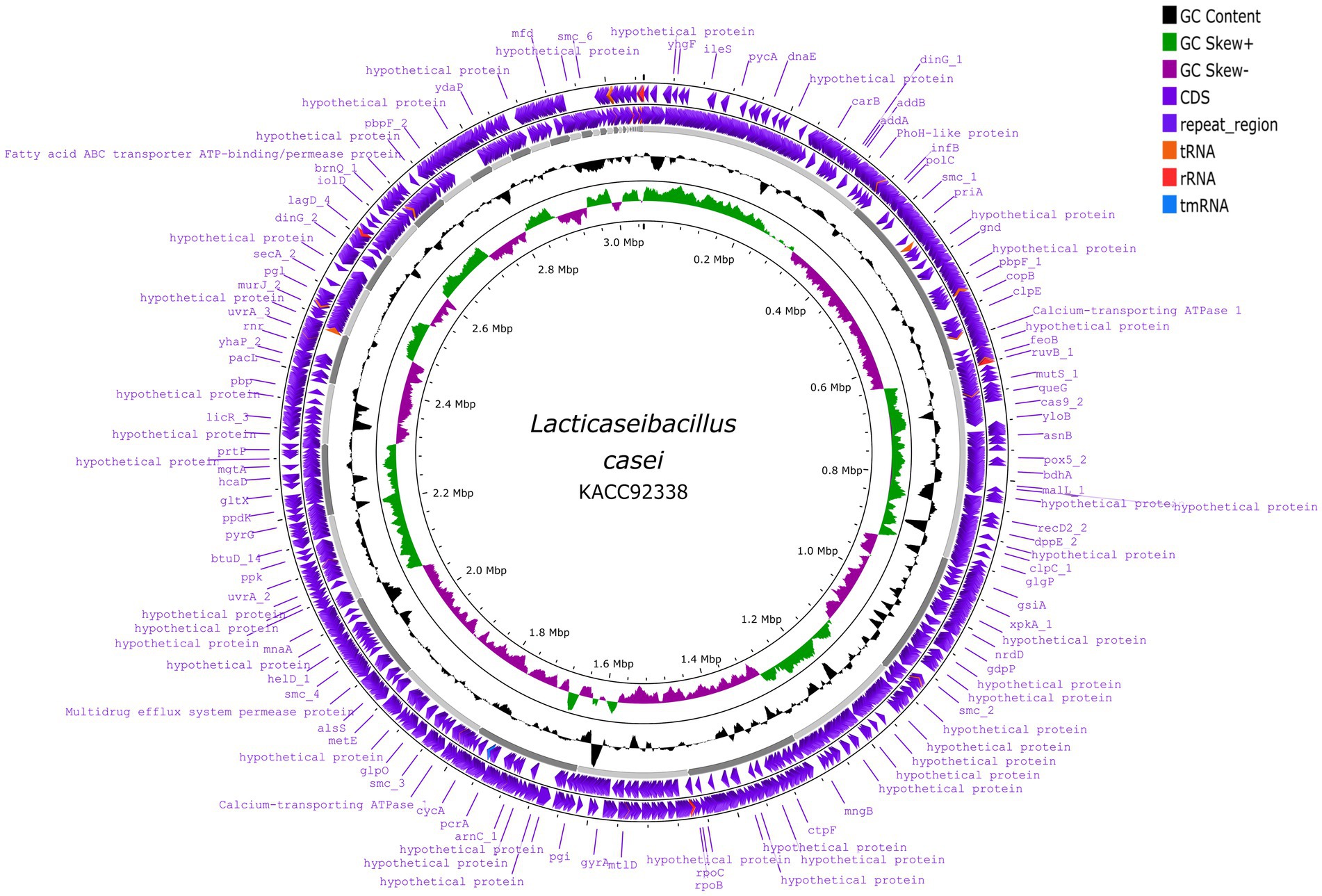
Figure 2. Circular genome map of L. casei KACC92338 (visualized using Proksee genome visualization tool). From the outer to inside: the first and second circles are forward and reverse CDS (coding sequences) annotated using Prokka, respectively, with tRNA, rRNA, tmRNA and Cas elements; the third circle is the GC content; the fourth circle depicts the GC skew (G-C)/(G + C), and the fifth circle represented the genome size (3,050,901 bp).
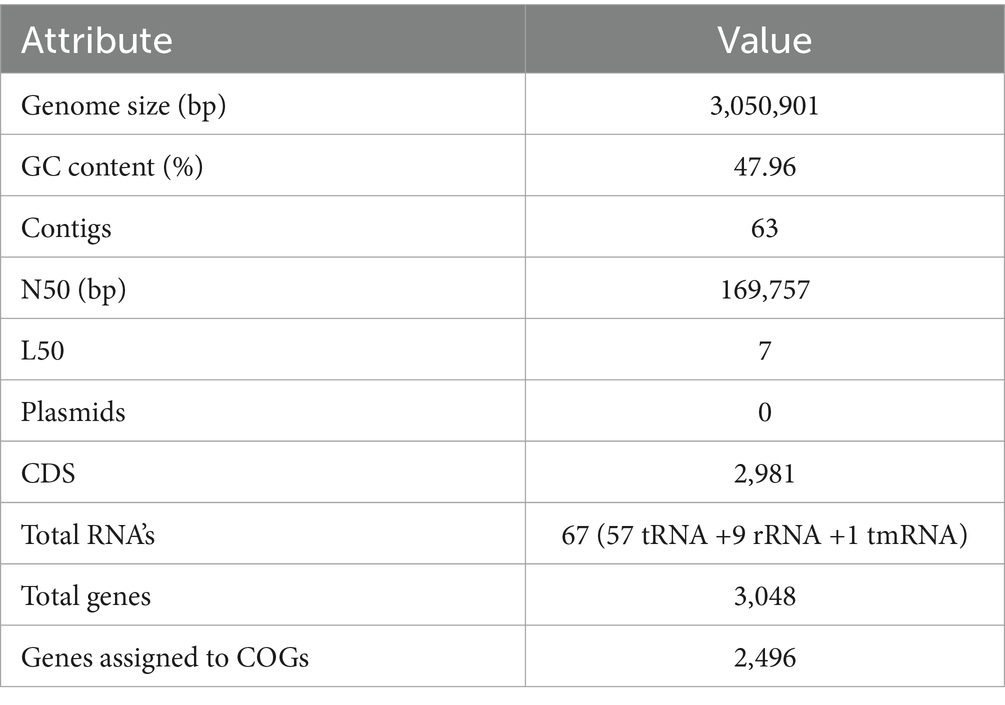
Table 3. Genomic information of L. casei KACC92338.
The ANI-based taxonomy revealed that the genome of KACC92338 strain was most closely related with Lacticaseibacillus casei N87 (ANIb 96.55%; ANIm 96.92) and L. casei ATCC 393 (Tetra 0.99779).
In addition, the genome-based taxonomic analysis conducted in TYGS web server indicated that KACC92338 strain showed similarity with other strains such as Lacticaseibacillus casei DSM 20011 and L. casei ATCC 393 (Figure 3). Therefore, the results of TYGS confirmed that KACC92338 indeed belongs to L. casei species.
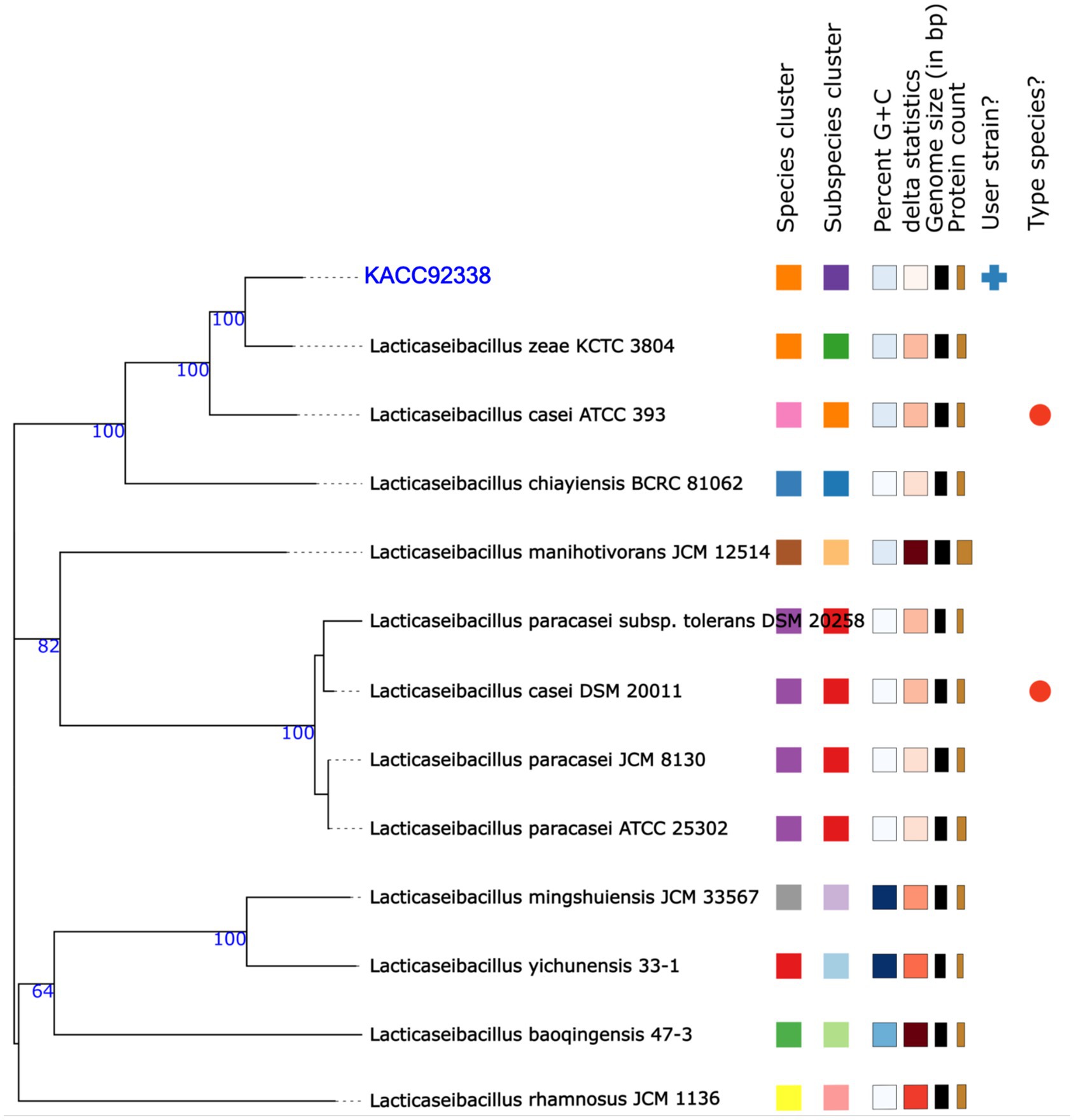
Figure 3. Phylogenetic tree based on TYGS result for the whole genome data set of L. casei KACC92338 with representative complete genomes of other Lacticaseibacillus strains. The tree was inferred with FASTME 2.1.6.1. from GBDP (genome BLAST distance phylogeny method) distances calculated from 16S rDNA gene sequences. The bootstrap support value before each node represents the confidence degree of each branch.
The genome annotation performed by Prokka revealed a total of 3,048 genes, with 2,981 protein-coding sequences (CDS), 57 tRNAs, 9 rRNAs, and 1 tmRNA (Table 3). Among the predicted CDS, 2135 genes (71.62%) were annotated with known functions, and the rest 846 genes (28.38%) were annotated as hypothetical proteins without any known functions. The gene coding for 57 tRNAs correspond to 20 natural amino acids: Arg (6 sequences); Gly, and Leu (5); Ala, Met, and Ser (4); Asn, Glu, Lys, and Thr (3); Asp., Gln, Ile, Phe, Pro, and Tyr (2); and Cys, His, Trp, and Val (1).
The analysis obtained from the RAST/SEED provided an overview of the coded biological features with a subsystem coverage of 42%, distributed in 232 subsystems (Table 4). The most represented subsystem features were involved in the metabolism of carbohydrates (25.43%), proteins (10.73%), amino acids and derivatives (7.38%), DNA (6.52%), and RNA (5.88%). Besides, 92 genes (5.31%) that participate in the production of a plethora of cofactors, vitamins, prosthetic groups and pigments were unveiled. The presence of genes encoding the biosynthesis of complex B vitamins (biotin, folate, pyridoxine, riboflavin, and thiamin) is a desirable trait of a probiotic, as humans are unable to synthesize B vitamins and is required as an external supplement. Further characterization of the biomolecules can prove advantageous in the food industry and can add to the nutritional value of food products.
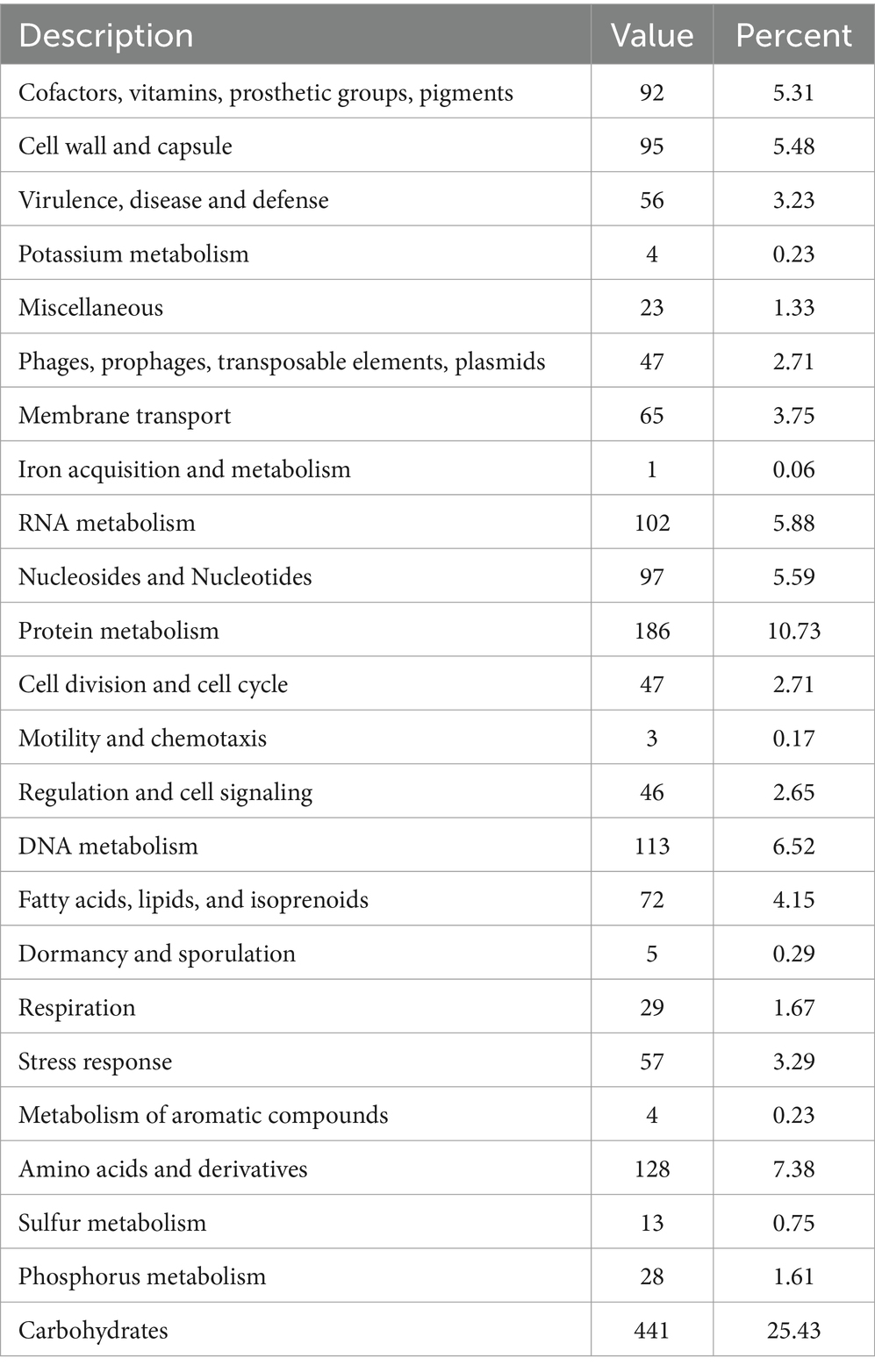
Table 4. General overview of the RAST annotation and subsystem categories for the L. casei KACC92338 genome.
To further understand the functional properties of KACC92338, the protein-encoding genes in the genome were annotated using COG and KEGG databases. Of the 2,981 CDS, 2496 (83.73%) genes were assigned to COG families comprising 19 functional categories by the EggNOG mapper, with the rest being functionally unknown. As shown in Figure 4, the most abundant COGs are carbohydrate transport and metabolism (G: 283); transcription (K: 227); amino acid transport and metabolism (E: 197); translation, ribosomal structure and biogenesis (J: 166); inorganic ion transport and metabolism (P: 150); replication, recombination and repair (L: 140); cell wall/membrane/envelope biogenesis (M: 129); nucleotide transport and metabolism (F: 109) and energy production and conversion (C: 103). Other than the above, defense mechanisms (V: 87); coenzyme transport and metabolism (H: 70); transduction mechanisms (T: 58); signal lipid transport and metabolism (I: 56); posttranslational modification, protein turnover, chaperones (O: 54); cell cycle control, cell division, chromosome partitioning (D: 46); intracellular trafficking, secretion, and vesicular transport (U: 46); secondary metabolites biosynthesis 23), and Cell motility (N: 10) were found to be enriched in KACC92338. The highest proportion of genes were under the function unknown (S: 542) category which contains several proteins related to phages, CRISPR, transport, and stress. The functional assignment was consistent with the previous studies (Wuyts et al., 2017; Fontana et al., 2018), highlighting the necessity for further enhancements in functional gene prediction. The higher number of genes involved in carbohydrate and amino acids metabolisms indicate our strains ability to utilize and degrade a large number of carbohydrate and proteins (Kim et al., 2022). The former attributes are mainly correlated to the large genome size similar to L. plantarum (Fontana et al., 2018).
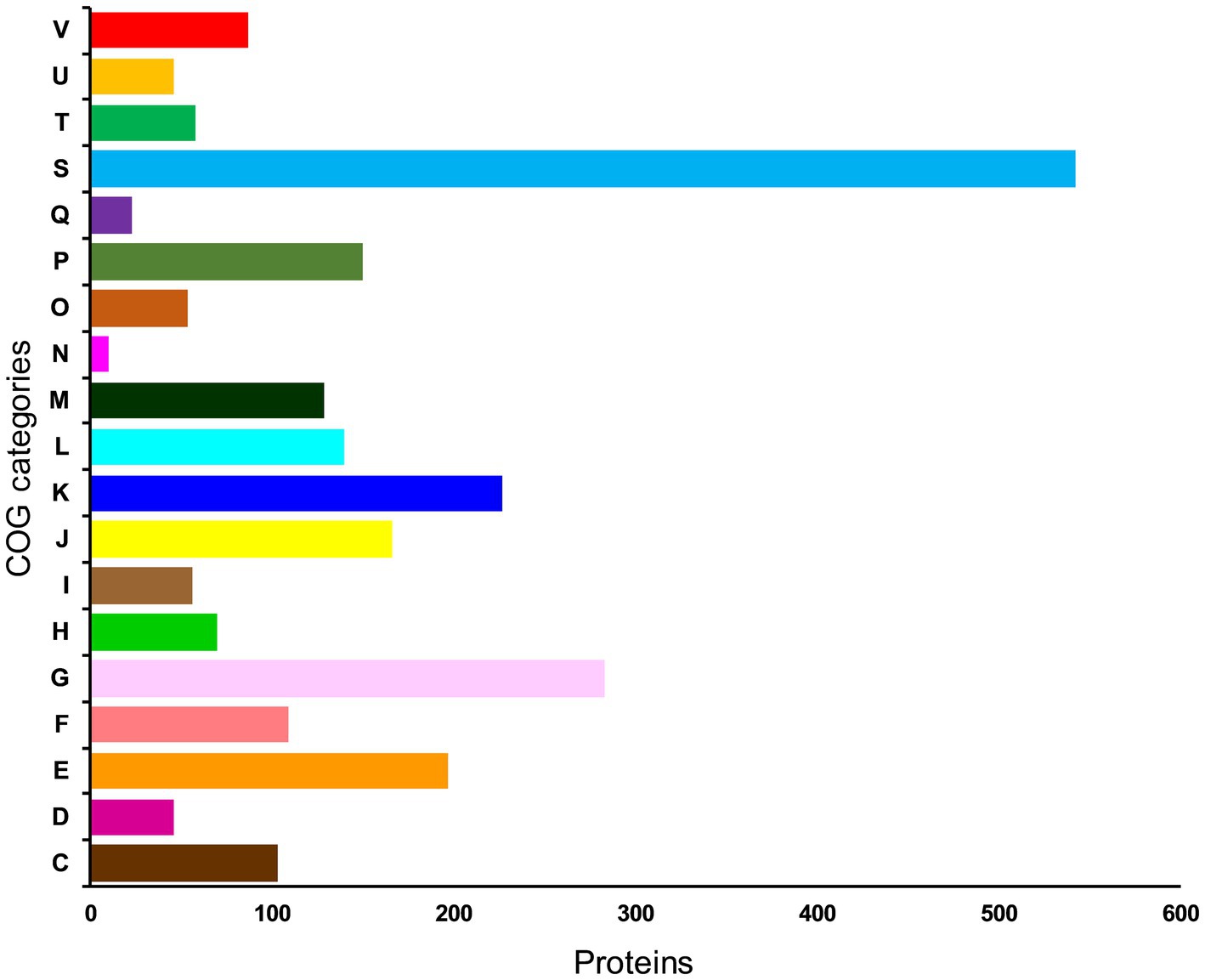
Figure 4. Distribution of genes across COG functional categories in the genome of L. casei KACC92338. In y-axis, C denotes energy production and conversion, D—cell cycle control, cell division, and chromosome partitioning; E—amino acid transport and metabolism; F—nucleotide transport and metabolism; G—carbohydrate transport and metabolism; H—coenzyme transport and metabolism; I—lipid transport and metabolism; J-translation, ribosomal structure and biogenesis; K—transcription; L—replication, recombination and repair; cell wall/membrane/envelope biogenesis; N—Cell motility; O—posttranslational modification, protein turnover, chaperones; P—inorganic ion transport and metabolism; Q—secondary metabolites biosynthesis, transport and catabolism; S—function unknown; T—signal transduction mechanisms; U—intracellular trafficking, secretion, and vesicular transport; and V—defense mechanisms.
Concerning the KEGG pathway database, approximately half of the CDSs (49.39%, 1,410 genes) were classified into 22 different functional categories (Table 5). Among them, the most abundant belong to carbohydrate metabolism (8.55%), protein families: genetic information processing (7.04%), protein families: signaling and cellular processes (5.95%), genetic information processing (5.71%), environmental information processing (4.76%), unclassified: metabolism (2.77%), amino acid metabolism (2.35%), and nucleotide metabolism (2.17%).
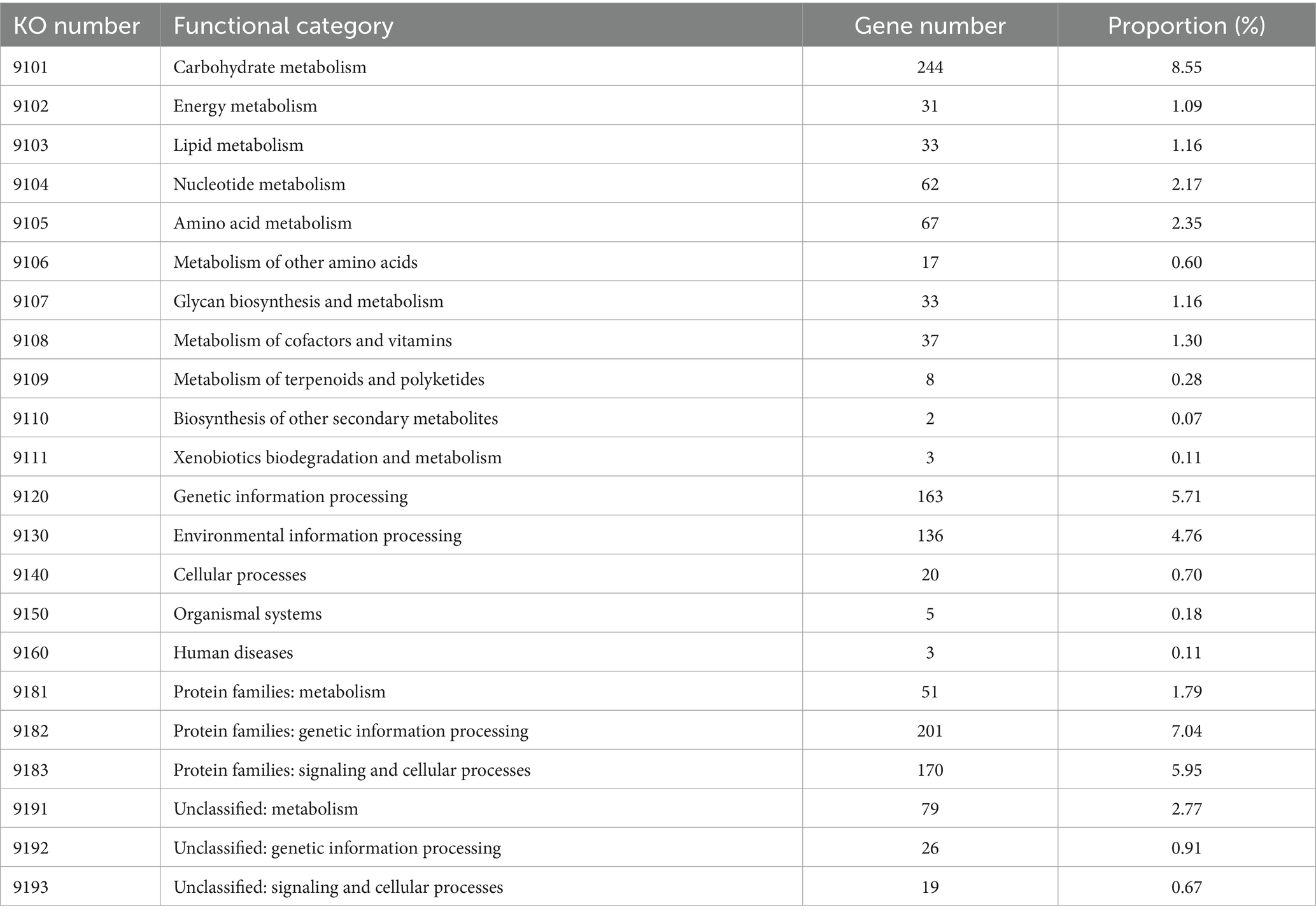
Table 5. KEGG orthology (KO) categories of identified protein-coding genes in the L. casei KACC92338 genome.
These findings of the functional annotation analysis revealed that the KACC92338 strain has high metabolic capacity with abundant functional categories of genes responsible for carbohydrate and amino acid transport and metabolism, followed by genetic information processing and protein families: signaling and cellular processes, which indicates their importance in conserved cellular processes in this genome to enable the strain to adapt extreme environments and probiotic functions.
The ability of probiotics to survive and establish themselves under harsh conditions (temperature, pH, bile, osmotic pressure, and oxidative stress) such as the gastrointestinal tract (GIT) and fermented food industries, and the ability of adhesion are critical parameters when choosing new probiotic strains (Hill et al., 2014; Evivie et al., 2017). Generally, lactobacilli strains have mechanisms to resist the environmental stress within the host to exert probiotic effects. In silico genome analysis of probiotic characteristics of L. casei KACC92338 unveiled several genes coding for temperature resistance, acid and bile tolerance, adhesion ability, oxidative stress, and immunomodulation, as listed in Table 6.
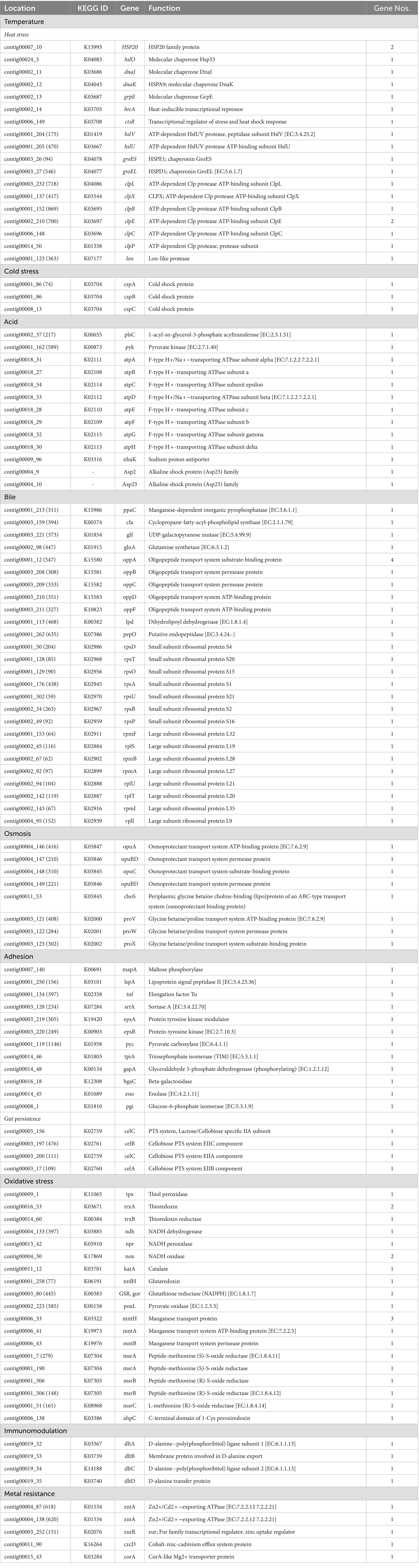
Table 6. Prediction of probiotic property-related functional genes in L. casei KACC92338 genome.
Probiotic LAB capable of surviving in the GIT tend to contain several stress response genes which are beneficial to adapt to the intestinal environment and promote health effects (Evivie et al., 2017). The strain L. casei KACC92338 contains four universal stress proteins (yugI, uspA, usp1, usp5) induced under numerous stress conditions were present in the genome. For tolerance to higher temperatures, our strain harbored 20 genes coding for heat shock proteins in its genome. These included heat shock-related regulators (hrcA, ctsR), molecular chaperones (dnaK, dnaJ, grpE, groEL, groES, hslO), and protease-encoding genes (hslU, hslV, lon, clpB, clpC, clpE, clpL, clpP, clpX). These genes have been proven for upregulated expression in Lactobacillus strains to prevent intracellular protein aggregation and membrane stabilization during heat stress conditions (Kim et al., 2022; Li et al., 2023). In addition, three genes (cspA, cspB, cspC) encoding cold shock proteins for survival under low temperatures were also identified. The CSP family genes are synthesized in several Lacticaseibacillus strains (Hill et al., 2018; Kiousi et al., 2022; Liu et al., 2023) to overcome the deleterious effect under cold stress, and hence, KACC92338 May have a similar functional property. Function.
Concerning the acid tolerance mechanism, the KACC92338 genome possessed 11 genes encoding resistance at low pH levels. Out of them, the key proteins F1F0 ATP synthase (an atp operon comprising eight genes, atpA, atpB, atpC, atpD, atpE, atpF, atpG, atpH) and sodium proton antiporter (nhaK) play a crucial role in maintaining a stable intracellular pH in the bacterial cytoplasm for survival under acidic environment or acid stress (Kiousi et al., 2022; Li et al., 2023). Additionally, a nhaK gene coding for sodium-proton (Na+/H+) antiporters (maintain pH and Na + homeostasis) along with two alkaline shock protein genes (Asp2, Asp23) were present. All these genes suggest the KACC92338 strain’s ability to adapt under acidic and alkaline environments. Concerning resistance to bile salts, 29 genes namely inorganic pyrophosphatase (ppaC, maintain surface tension and keep membrane integrity), cyclopropane-fatty-acyl-phospholipid [CFP] synthase (cfa, enhance lipid synthesis), UDP-galactopyranose mutase (glf) glutamine synthetase (glnA), oligopeptide transporting proteins, and small and large subunit ribosomal proteins were identified. A similar bile-resistance mechanism was confirmed in L. petauri LZys1 through genome and phenotype analysis (Li et al., 2021). For enduring osmotic stress in the GIT, the KACC92338 genome contained eight genes (opuA, opuC, opuBD, choS, proV, proW, proX) that are responsible for uptake and accumulation of osmoprotectants such as glycine betaine, choline or proline. Strains of the former L. casei group employed as starter or nonstarter in food industries generally present high tolerance to heat and osmotic stress conditions (Kiousi et al., 2022).
The adhesion ability of probiotics to colonize the epithelial cells of the GIT can vary depending on the LAB species or strain, as their cell surface proteins play a significant role in interaction with the environment or host (Monteagudo-Mera et al., 2019). KACC92338 genome encodes 12 genes putatively coding for adhesion-related proteins, such as maltose phosphorylase (mapA), lipoprotein signal peptidase II (lspA), elongation factor Tu (tuf), sortase A (srtA), and enolase (eno), providing evidence of high adhesion ability. Besides, four genes (celC, celB, and celA) related to gut persistence were also present in the genome. These adhesions related genes can provide stability to the strain and prolong its antagonistic effect on unwanted gut microorganisms, aiding in effective colonization of the intestinal environment and inhibition of pathogens. Similar genes related to adhesion were earlier reported in Lacticaseibacillus strains (Kim et al., 2022; Kiousi et al., 2022; Rodrigo-Torres et al., 2022).
In recent years, there has been increasing attention to probiotic bacteria with oxidative stress tolerance and antioxidant properties for investigation in the treatment of various chronic human diseases (Kang et al., 2017; Vasconcelos et al., 2019). In this study, L. casei KACC92338 harbors 23 antioxidant functional genes related to oxidative stress, and out of them, 7 genes encode the complete thioredoxin (tpx, trxA, trxB) and NADH (ndh, npr, nox) antioxidant systems involved in ROS scavenging (Table 4). The thioredoxin system provides electrons to thiol-dependent peroxidases to catalyze the reduction of ROS and RNS at a fast reaction rate. The NADH oxidase cooperates with NADH peroxidase to eliminate hydrogen peroxide, enabling the degradation of ROS (Zhang et al., 2018). The presence of catalase (katA) gene can also significantly improve the antioxidant activity of our strain. These results support the findings of H2O2 tolerance, in vitro antioxidant and protective effect demonstrated by our strain in section 3.1 to 3.3. In addition, KACC92338 encoded genes for catalase (katA), pyruvate oxidase (poxL), and glutaredoxin (nrdH). Similar to the findings of L. paracasei SP5 (Kiousi et al., 2022), intracellular Mn(II) [manganese transport systems (mntA, mntB) and protein (mntH)] accumulation in KACC92338 can act as a functional replacement for superoxide dismutase. Moreover, the methionine sulfoxide reductase system (msrA, msrB, msrC) witnessed in our genome can catalyze the oxidized methionine residues formed by ROS and RNS in proteins. These results indicated that the strain KACC92338 might be a good probiotic candidate with potential antioxidant capacity that can support survival and damage repair under aerobic conditions.
Additionally, the KACC92338 genome is equipped with genes coding for immunomodulatory activities (dlt A-D). These results suggest that L. casei KACC92338 might resist multiple stress conditions and be consistent with the adaptability characteristics of the gastrointestinal tract.
Carbohydrate-active enzymes (CAZymes) are produced by the microbiome in the human intestinal gut and play a key role in the synthesis and degradation of complex polysaccharides and their derivatives (El Kaoutari et al., 2013). Using dbCAN3, the KACC92338 strain was found to harbor 201 genes classified under five CAZy classes (Supplementary Table S1). These numbers were relatively more abundant than those for L. casei FBL6 (Kim et al., 2022). The genes annotated by CAZy were dominated by the family of glycoside hydrolases (GHs, n = 170), followed by glycosyl transferases (GTs, n = 24), polysaccharide lyases (PLs, n = 4), carbohydrate-esterases (CEs, n = 2), and auxiliary activity (AA, n = 1). The genes of GHs were clustered in 53 different families, among which GH 13, GH 30, and GH 43 are dominant. GHs are key enzymes that hydrolyze the glycosidic bonds of carbohydrates and thereby release abundant energy to support various bacterial activities, indicating that the KACC92338 strain has the potential to utilize a wide range of complex carbohydrates (El Kaoutari et al., 2013). The second most abundant, GTs (clustered in 9 different families) catalyze the transfer of sugars from activated donor molecules to specific acceptors and are pivotal in forming surface structures that can be recognized by the host immune system (Mazmanian et al., 2008). The relatively high diversity of GH and GT in the genome can help the L. casei strain to utilize various sugars and promote immune stimulation and pathogen defense (Zhang et al., 2018). The ability to metabolize a large number of carbohydrates, including those that are not found in milk-based environments, supports the hypothesis that KACC92338 is capable of inhabiting in diverse ecological niches.
Moreover, a manual search in functional annotation disclosed few sugar transporter genes related to the phosphoenolpyruvate-dependent sugar phosphotransferase (PTS) system namely, beta-glucoside, cellobiose, fructose, galactitol, galactose amine, gluconate, lactose, mannitol, mannose, and sucrose indicating the strong carbon transporting ability of KACC92338, an important attribute for organisms that inhabit the gastrointestinal environment or can inhabit a variety of ecological niches. Interestingly, there were also genes encoding for enzymes such as acetaldehyde dehydrogenase, acetate kinase, alcohol dehydrogenase, D and L- lactate dehydrogenases, fructose-6-phosphate phosphoketolase, phosphate acetyltransferase, pyruvate formate-lyase, and xylulose-5-phosphate phosphoketolase. These enzymes play a crucial role in both homo- or hetero-fermentative pathways to produce acetate or lactate, which suggests that KACC92338’s inclusion in fermented foods could be an asset in the food industry.
Insertion sequences (IS) are small (<2.5 kb) segments of DNA with a simple organization, capable of inserting at multiple sites in a target molecule, and generally only encode genetic information required for transposition. IS elements have been documented in many species of LAB (Siguier et al., 2014). Fourteen insertion sequence (IS) elements from two different families (IS1202 and IS6) were predicted in the genome of L. casei KACC92338 with the set threshold E-value of 0.00001 (Supplementary Table S2). The IS1202 family contained 11 copies of ISLrh5 (L. rhamnosus) element, with only one copy exhibiting a zero E-value. However, all copies of ISLrh5 shared 70% amino acid similarity with ISPein1 (Pediococcus inopinatus). The IS6 family contained a single copy of each ISXne2 and elements. ISXne2 shared 83% amino acid similarity with ISYps1 (L. reuteri), and ISS1Z differs from ISS1S by 11 bp (1 amino acid).
In the KACC92338 genome, Island Viewer 4 predicted five specific regions ranging in length from 9,060 to 57,794 bp, which are designated as genomic islands (GIs) (Supplementary Table S3). A total of 182 genes were assigned within these GIs, among which a relatively higher number encoding for hypothetical proteins. Additionally, genes related to transposases, integrases, bacteriocin, teichoic acid synthesis, proteins involved in DNA protection and modification, and enzymes related to carbohydrate metabolism have been found. These genes could play a significant role in enhancing the adaptability and competitiveness of the organism within the environmental niche (Ho Sui et al., 2009). Importantly, no genes related to virulence factors or drug-resistance mechanisms were annotated within the GIs.
The analysis of CRISPR sequences using CRISPR-CasFinder uncovered eight CRISPR arrays in the L. casei KACC92338 genome. Among them three are located in contigs 1 and 3 with strong evidence level 4 (Table 7A). Additional CRISPR arrays detected were with evidence level 1 were disregarded as they are potentially invalid.
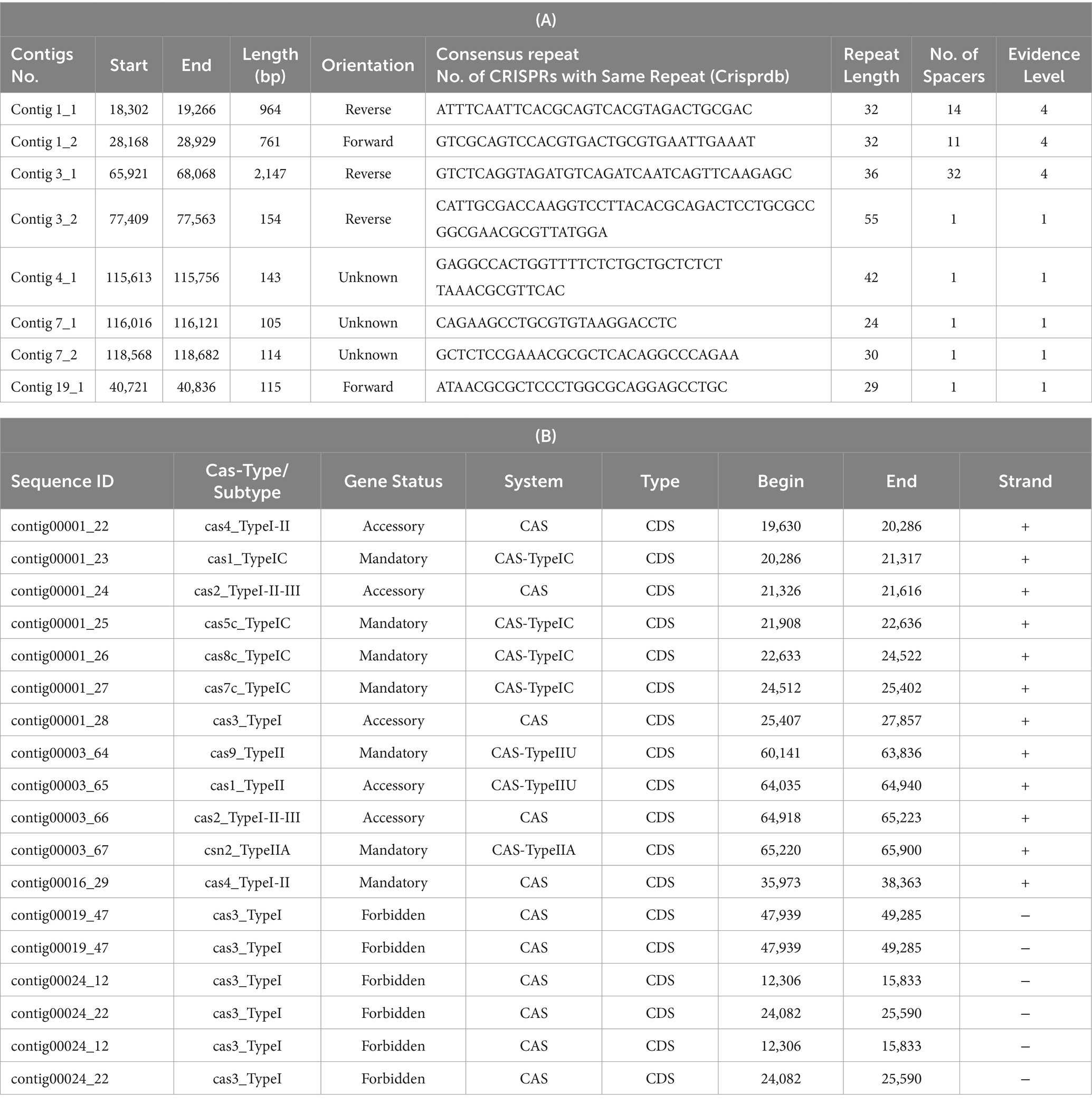
Table 7. Putative CRISPR-Cas sequences coded by CRISPR arrays (A) and Cas-related enzymes (B) in L. casei KACC92338.
Out of the total seven mandatory CRISPR-associated protein, four belonged to the Cas_TypeIC (cas1, cas5, cas8, cas7) in contig 1, two were from the Cas_TypeIIU (cas9), and Cas_Type IIA (csn2) systems in contig 3, and one from the CAS (cas4_TypeI-II) system alone in contig 16 (Table 7B). The presence of CRISPR-associated (Cas) proteins in our genome can provide a strong defense against invasive mobile genetic elements (conjugative plasmids, insertion sequences, and phages), along with restricting the dissemination of resistance genes (virulence, antibiotic) through horizontal gene transfer (Kim et al., 2022). Besides, the CRISPR functional system also confers stability to the lactic acid bacteria genomes in the dairy industry and has potential applications in gene editing for the improvement of their biotechnological applications (Song et al., 2012).
Prophages are a critical part of prokaryotic genomes and are widely distributed in several genera of LAB, including Lacticaseibacillus sp. (Pei et al., 2021; Rodrigo-Torres et al., 2022). Furthermore, prophages serve as a source of new genes added to the genome that provide various beneficial traits, such as the acquisition of antibiotic resistance genes, adaptation in new environmental niches, enhanced adhesion ability, or even turning the bacteria pathogenic (Pei et al., 2021).
A total of six regions (Table 8) were identified in the KACC92338 genome by the PHASTER webtool. These regions include two intact (regions 3 and 5), one questionable (region 2), and three incomplete (regions 1, 4 and 6) prophages. The phage regions mainly consist of hypothetical proteins and phage-related proteins from Lactobacillus, Staphylococcus, and Bacillus. The largest (35.9 kb) intact phage in region 3 (1–35,929 bp) showed a maximum (51) protein matching and resembled Lactob_PLE3_NC_031125(19), which is the most encountered prophage in L. casei strains (Xin et al., 2017). The next intact phage at region 5 (236–17,284 bp, 17 proteins; 17 kb) resembled Lactob_Lc_Nu_NC_007501(16). The questionable phage in region 2 (126909–145,470 bp, 19 proteins; 18.5 kb) corresponded to Staphy_SA7_NC_048658(2). Regarding the incomplete prophages, region 1 (153336–169,757 bp, 9 proteins; 16.4 kb), 4 (269–17,980 bp, 18 proteins; 17.7 kb) and 6 (1371–8,004 bp, 12 proteins, 6.6 kb) were consistent with Lactob_J_1_NC_022756(4), Lactob_BH1_NC_048737(17) and Lactob_phiAT3_NC_005893(5), respectively.
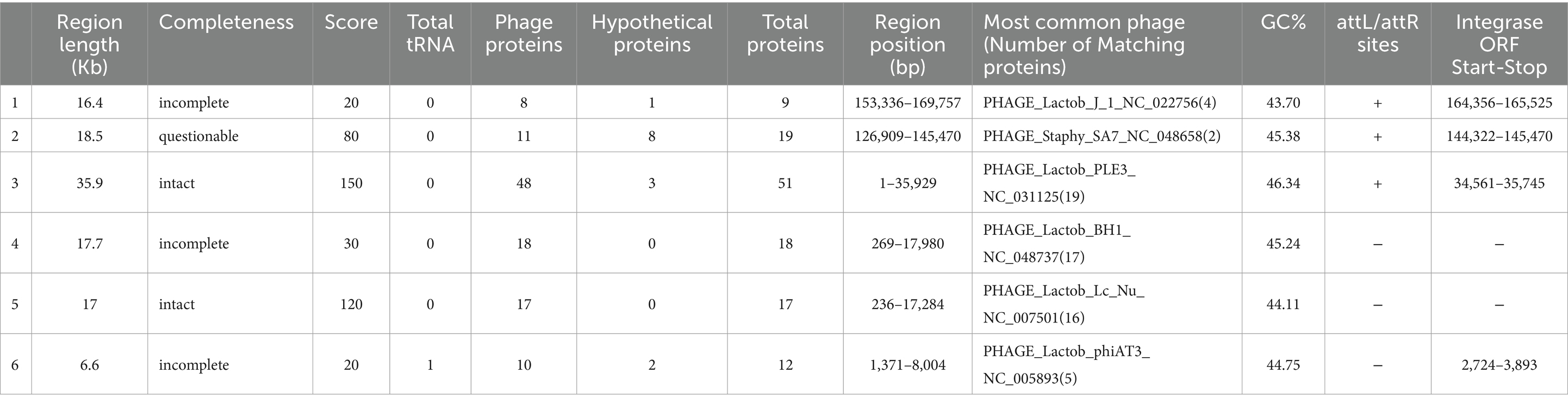
Table 8. Detection of prophage regions in L. casei KACC92338 genome using PHASTER tool.
Integrases were identified in four [PP_01651 (region 1), PP_01809 (region 2), PP_02851 (region 3), and PP_03065 (region 6)] out of the six regions. Integrases are well-recognized diagnostic markers for mobile DNA elements (prophages, pathogenicity islands, and integrative plasmids) in bacterial genomes (Xin et al., 2017). The sequences of attachment sites (attL/attR) were homologous in each intact phage. No virulence or AMR genes have been found within the intact prophages.
The in-silico prediction of ARG in the genome of KACC92338 using the CARD database under default settings (perfect/strict option) did not return any hits. However, changing the parameter to a less stringent criterion (perfect/strict/loose option), resulted in 211 loose hits (Supplementary Table S4) related to ARG associated with antibiotic target alteration (64), antibiotic target protection (13), antibiotic efflux (179), reduced permeability to antibiotic (1), antibiotic inactivation (16) and antibiotic target replacement (4). A genome map depicting the location of both ARG and MGE visualized in the Proksee server (Grant et al., 2023) (Supplementary Figure S1) showed that there is no co-localization between them, indicating there is no risks of transferability. These findings were already confirmed in the earlier section on MGE.
Furthermore, KACC92338 did not yield a significant match for any resistance genes associated with known antibiotics available in the Resfinder 4.1 database (90% threshold and 60% minimum length). The MobileElementFinder service could not find any mobile genetic elements in relation to antimicrobial resistance genes and virulence factors. It’s important to note that both the CARD and Resfinder databases mainly focus on ARG of pathogenic bacteria, therefore the ARG of non-pathogenic bacteria (eg. Lactobacillus) are usually not included.
Moreover, a search using BlastKOALA in the KACC92338 genome revealed genes related to resistance against tetracycline, macrolide, beta-lactam, and cationic antimicrobial peptides (Table 9). However, the presence of these genes does not warrant that KACC92338 surely expresses this resistance. Notably, despite the possession of macrolide and beta-lactamase resistance genes, the strain L. plantarum BCC 9546 was sensitive to these antibiotics. This May be related to various factors such as the gene expression level and substrate specificity of the expressed product (Chokesajjawatee et al., 2020). Earlier studies have shown that many LAB genera exhibit intrinsic resistance to tetracycline and macrolide due to ribosome protection, antibiotic efflux, and associated efflux pump formation. In agreement with these findings, the resistance genes in the KACC92338 code for transporters and efflux pumps, mainly related to non-specific antimicrobial resistance mechanisms. It’s also worth noting that our genome does not contain plasmids, making the transfer of antibiotic resistance genes by plasmid impossible. However, additional phenotypic testing in terms of clinical decision-making is necessary to clarify whether ARG encodes active proteins or plays different roles. These results can serve as an initial screening of genotypic ARG profiles indicating that L. casei KACC92338 is safe and unlikely to transfer any ARG to other commensals.
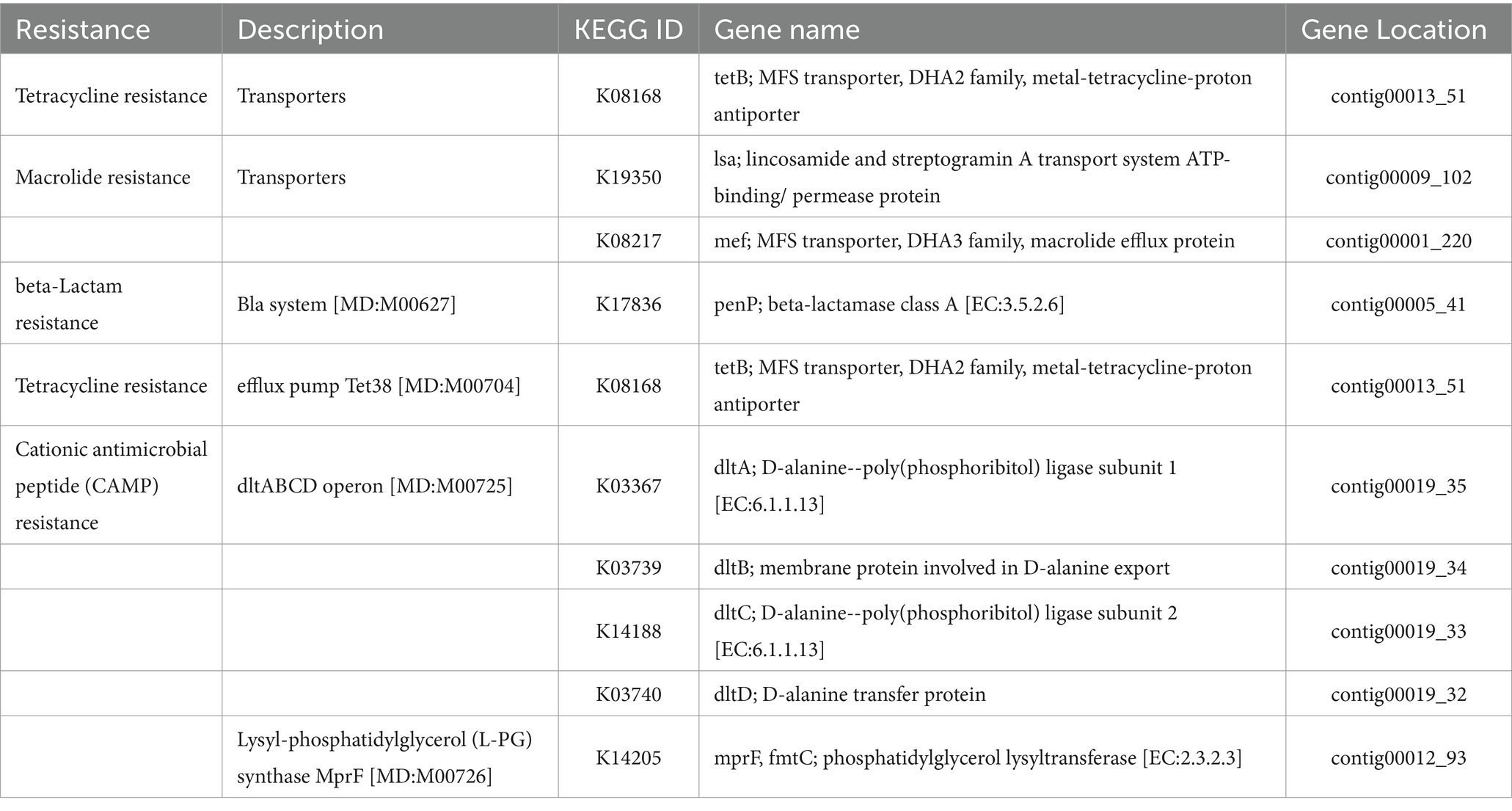
Table 9. List of antimicrobial resistance genes identified by BlastKOALA and their locations in the genome of L. casei KACC92338.
In KACC92338, no virulent genes were found using a BLASTn search in VirulenceFinder. However, a total of 22 hits were predicted through comparison with VFDB, which mainly included ABC transport proteins, stress survival, immune modulation, and adherence. It’s worth noting that these factors contributing to virulence in pathogens are also beneficial in probiotic strains as they help to increase their fitness for survival in the gut under several physiological stress conditions (Ho Sui et al., 2009).
The search for genes related to undesirable metabolites using BlastKoala identified hemolysin (hlyIII – K11068) and D-lactate dehydrogenase (K03778) in the KACC92338 genome. Further analysis revealed that the toxin gene (hlyIII) has 100% sequence identity with a predicted membrane channel-forming protein YqfA, hemolysin III family. The widespread of the hlyIII gene in commercial probiotic Lactobacillus spp. (Zafar and Saier, 2020) imply the strain harboring the gene is not considered a safety concern.
The presence of D-lactate dehydrogenase gene in the genome implies the production of D-lactic acid, which is an essential component in the cell wall peptidoglycan of several gram-positive bacteria, including L. plantarum. However, caution is necessary on the consumption of foods containing LAB with D-lactic producing ability as it can lead to acidosis in patients and infants with short-bowel syndrome and carbohydrate malabsorption (Bianchetti et al., 2017).
An analysis of genes related to the enzymes involved in biogenic amines (BA) synthesis is another essential probiotic attribute related to consumer safety issues. Based on the KEGG database search, there were no genes related to BA synthesis in our genome, indicating that the strain is a BA-non-producer and deemed safe.
The results from the Pathogen Finder (Cosentino et al., 2013) indicated our strain is a non-human pathogen with zero matches against known pathogenic gene families and a very low probability (0.179) of being a human pathogen. In conclusion, the results on the genetical analyses related to safety aspects highlighted the absence of negative characteristics and confirms that L. casei KACC92338 is a safe strain and suitable for use as a probiotic and in different food industry applications.
The antimicrobial activity of probiotics has a significant role in competing against GIT microbial pathogens by producing certain inhibitory substances, such as bacteriocins and lactic acid (Hernández-González et al., 2021). The biosynthetic gene clusters (BGC) in L. casei KACC92338 May have the ability to produce class II bacteriocins, sakacin-P, Enterolysin_A, sactipeptides and Enterocin X (Figure 5). These bacteriocins were predicted at six different locations (areas of interest (AOIs)) as follows: (i) contig 7.10 (AOI_01) (start at 145664, end at 165850), (ii) contig 22.21 (AOI_02) (start at 1, end at 11776), (iii) contig 31.22 (AOI_03) (start at 1, end at 6167), (iv) contig 4.30 (AOI_04) (start at 8053, end at 11947), (v) contig 6.32 (AOI_05) (start at 8740, end at 32412), and (vi) contig 12.6 (AOI_06) (start at 39605, end at 59758) suggesting the potential antimicrobial capacity of the strain.
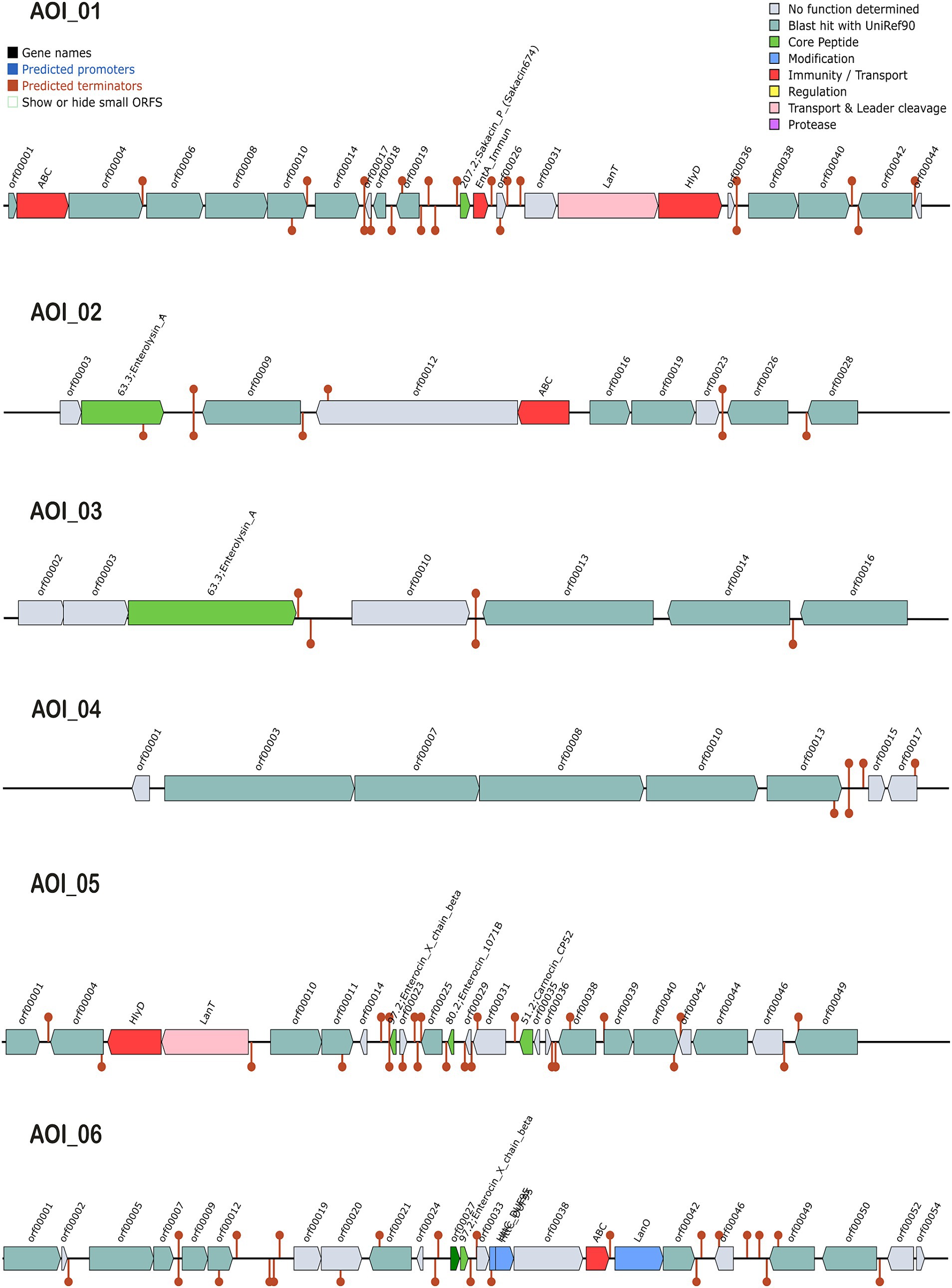
Figure 5. Graphical representation of biosynthetic gene clusters (BGCs) in L. casei KACC92338 genome predicted through the BAGEL4 webserver. Six contigs viz., contigs 7.10 (AOI_01), 22.21 (AOI_02), 31.22 (AOI_03), 4.30 (AOI_04), 6.32 (AOI_05) and 12.6 (AOI_06) encoding genes potentially related to the biosynthesis of sakacin-P, Enterolysin_A, Enterolysin_A, sactipeptides, carnocin_CP52 and Enterocin X beta chain were identified. The BCGs are represented by arrows with different colors corresponding to the operons of different functions.
The AOI_01 is comprised of 20,186 bp and contains 21 ORFs. The ORF 11 in this cluster codes for sakacin-P. Other genes in this cluster includes an ABC-type transporter, a putative immunity protein, a transport/processing ATP-binding protein (LanT), and an accessory factor for ABC-transporter PlnH (Hlyd). Protein sequence analysis of ORF11 by BLASTP showed high similarity to a class II leucocin A/sakacin P family bacteriocin (WP_070651009) from Lacticaseibacillus. A putative leucocin A/sakacin P was predicted previously in the genome of L. casei FBL6 (Kim et al., 2022). Sakacin P inhibits the growth of various gram-postive pathogens, especially Listeria monocytogenes, a common and persistent pathogen in the food industry (Katla et al., 2001).
The clusters AOI_02 (11,775 bp, 10 ORFs) and AOI_03 (6,166 bp, 7 ORFs) produce Enterolysin_A (Class III bacteriocin), a heat-resistant, cell-wall-degrading bacteriocin broadly dispensed among enterococcal strains. The bacteriocin was initially reported from Enterococcus faecalis LMG2333 (Nilsen et al., 2003) and has been found to be active against Gram-positive bacteria such as enterococci, pediococci, and lactococci. The AOI_02 includes a bacteriocin gene (ORF 2) and an ABC transporter (ORF 5), while AOI_03 contains only one bacteriocin gene (ORF3). The BLASTP analysis of enterolysin_A from AOI_02 correspond to N-acetylmuramoyl-L-alanine amidase (WP_093997694.1) of Lacticaseibacillus, while enterolysin_A from AOI_03 showed specific hits with lysozyme (WP_306402816.1) and N-acetylmuramoyl-L-alanine amidase (OFR73919.1) from Lactobacillus sp. HMSC061B07. N-acetylmuramoyl-L-alanine amidase destroys microbial cell walls leading to cell rupture and death.
The AOI_4 (3,894 bp, 8 ORFs) codes for the bacteriocin of sactipeptides (ribosomally synthesized and post-translationally modified peptides), while AOI_5 (23,672 bp, 23 ORFs) encodes carnocin_CP52 immunity protein (class II bacteriocin, ORF 14) that contain five genes including a bacteriocin secretion accessory protein (Hlyd), a peptide cleavage/export ABC transporter (LanT) and three predicted core peptides, including Enterocin X beta chain (ORF 8), Enterocin (ORF 11), and carnocin_CP52. Identification by BLASTp showed the gene sequence of Enterocin X beta chain showed 100% identity to a bacteriocin leader domain-containing protein reported in many strains of Lacticaseibacillus zeae, while Enterocin showed hits with lactococcin G-beta/enterocin 1071B family bacteriocin from Weissella cibaria and carnocin_CP52 showed 100% QC for bacteriocin immunity protein from several Lacticaseibacillus strains.
The AOI_6 (20,153 bp, 24 ORFs) encodes Enterocin X beta chain (Class IIc bacteriocin, circular peptide) which includes two class II bacteriocins (ORF 11, ORF 12), two uncharacterized proteins (ORF 14, ORF 15), ABC (ORF 17), and LanO (ORF 18). Based on BLASTp results, the bacteriocin at ORF11 was identified as a ComC/BlpC family leader-containing pheromone/ bacteriocin (WP_010492210.1) from Lacticaseibacillus, and ORF12 coding Enterocin X beta chain was found as a bacteriocin leader domain-containing protein reported in many strains of L. zeae.
The analysis concludes the ability of our strain to produce class II bacteriocins, that can contribute to the inhibitory effect against pathogens. The information presented here about the bacteriocins produced by this strain relies on the information available in the bacteriocin database and search programs employed. Further in vitro studies are necessary to confirm the exact bacteriocins or any additional proteins produced by the strain and under the conditions which they are produced.
The current study highlights the antioxidant property, whole genome sequence and in silico mining to retrieve genes related to probiotic characteristics, bacteriocin production, and genome stability of the L. casei KACC92338 strain. To our knowledge, this is the first whole-genome sequence of L. casei isolated from Korean raw milk and the first report on higher antioxidant activity by L. casei strain than reported earlier. The ability of tolerance towards H2O2, with potential in vitro scavenging activities and cellular protective effects on yeast cells indicated that L. casei KACC92338 possess good antioxidant capacity and the potential to reduce cellular damage caused by excessive oxidative stress in the host. In silico analysis of this strain showed various probiotic-associated genes that confer tolerance towards stress conditions of high and low temperatures, acid, bile, and oxidative stress, as well as abilities for adhesion, carbohydrate metabolism, and bacteriocin production. Moreover, safety assessments studies showed the absence of acquired antibiotic resistance genes, virulence factors, pathogenicity, or plasmids. The presence of prophage regions, CRISPR-Cas system, and insertion sequences further confirm genome stability. Overall, the results of genotypic validation serves as only the first step in safety assessment of L. casei KACC92338 and provides a direction for subsequent application. However, further confirmation through in vivo assessments are necessary to guarantee the safety and overall industrial stability of the strain.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/search/all/?term=PRJNA731289.
SK: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Visualization, Writing – original draft, Writing – review & editing. K-HL: Formal analysis, Methodology, Writing – review & editing. JaY: Formal analysis, Resources, Supervision, Writing – review & editing. JeY: Data curation, Formal analysis, Supervision, Writing – review & editing. HK: Data curation, Formal analysis, Methodology, Writing – review & editing. JK: Data curation, Investigation, Writing – review & editing. M-HO: Conceptualization, Supervision, Writing – review & editing. J-SH: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Validation, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by (2024) the RDA fellowship program of the National Institute of Animal Science, Rural Development and Administration, Republic of Korea (Project No: PJ01480301).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1458221/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | Antibiotic resistance genes (ARG) and Mobile genetic elements (MGE) in L. casei KACC92338 genome (visualized using Proksee genome visualization tool).
Alcock, B. P., Raphenya, A. R., Lau, T. T. Y., Tsang, K. K., Bouchard, M., Edalatmand, A., et al. (2020). CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 48, D517–D525. doi: 10.1093/nar/gkz935
Amaretti, A., Di Nunzio, M., Pompei, A., Raimondi, S., Rossi, M., and Bordoni, A. (2013). Antioxidant properties of potentially probiotic bacteria: in vitro and in vivo activities. Appl. Microbiol. Biotechnol. 97, 809–817. doi: 10.1007/S00253-012-4241-7
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/NAR/GKW387
Aziz, R. K., Bartels, D., Best, A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9, 1–15. doi: 10.1186/1471-2164-9-75/TABLES/3
Bertelli, C., Laird, M. R., Williams, K. P., Lau, B. Y., Hoad, G., Winsor, G. L., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35. doi: 10.1093/nar/gkx343
Bianchetti, D. G. A. M., Amelio, G. S., Lava, S. A. G., Bianchetti, M. G., Simonetti, G. D., Agostoni, C., et al. (2017). D-lactic acidosis in humans: systematic literature review. Pediatr. Nephrol. 33, 673–681. doi: 10.1007/S00467-017-3844-8
Bortolaia, V., Kaas, R. S., Ruppe, E., Roberts, M. C., Schwarz, S., Cattoir, V., et al. (2020). ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 75, 3491–3500. doi: 10.1093/jac/dkaa345
Cai, H., Thompson, R., Budinich, M. F., Broadbent, J. R., and Steele, J. L. (2009). Genome sequence and comparative genome analysis of Lactobacillus casei: insights into their niche-associated evolution. Genome Biol. Evol. 1, 239–257. doi: 10.1093/gbe/evp019
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P., and Huerta-Cepas, J. (2021). eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 38, 5825–5829. doi: 10.1093/MOLBEV/MSAB293
Carattoli, A., Zankari, E., Garciá-Fernández, A., Larsen, M. V., Lund, O., Villa, L., et al. (2014). In Silico detection and typing of plasmids using plasmidfinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 58, 3895–3903. doi: 10.1128/AAC.02412-14/SUPPL_FILE/ZAC007143013SO1.PDF
Chokesajjawatee, N., Santiyanont, P., Chantarasakha, K., Kocharin, K., Thammarongtham, C., Lertampaiporn, S., et al. (2020). Safety assessment of a Nham starter culture Lactobacillus plantarum BCC9546 via whole-genome analysis. Sci. Rep. 10, 10241–10212. doi: 10.1038/s41598-020-66857-2
Cosentino, S., Voldby Larsen, M., Møller Aarestrup, F., and Lund, O. (2013). PathogenFinder - distinguishing friend from foe using bacterial whole genome sequence data. PLoS One 8:e77302. doi: 10.1371/JOURNAL.PONE.0077302
Couvin, D., Bernheim, A., Toffano-Nioche, C., Touchon, M., Michalik, J., Néron, B., et al. (2018). CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 46, W246–W251. doi: 10.1093/nar/gky425
Dianin, I. B., Oliveira, A. G., Pimentel, T. C., Hernandes, N. F., and Costa, G. N. (2019). Edible biofilms formulated with whey protein isolate and L. casei probiotic culture: characterization and application in tomatoes and grapes. Chem. Eng. Trans. 75, 469–474. doi: 10.3303/CET1975079
El Kaoutari, A., Armougom, F., Gordon, J. I., Raoult, D., and Henrissat, B. (2013). The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat. Rev. Microbiol. 11, 497–504. doi: 10.1038/nrmicro3050
Evivie, S. E., Huo, G. C., Igene, J. O., and Bian, X. (2017). Some current applications, limitations and future perspectives of lactic acid bacteria as probiotics. Food Nutr. Res. 61:1318034. doi: 10.1080/16546628.2017.1318034
Feng, T., and Wang, J. (2020). Oxidative stress tolerance and antioxidant capacity of lactic acid bacteria as probiotic: a systematic review. Gut Microbes 12, 1801944–1801924. doi: 10.1080/19490976.2020.1801944
Fontana, A., Zacconi, C., and Morelli, L. (2018). Genetic signatures of dairy Lactobacillus casei group. Front. Microbiol. 9, 1–9. doi: 10.3389/fmicb.2018.02611
Grant, J. R., Enns, E., Marinier, E., Mandal, A., Herman, E. K., Chen, C. Y., et al. (2023). Proksee: In-depth characterization and visualization of bacterial genomes. Nucleic Acids Res. 51, W484–W492. doi: 10.1093/nar/gkad326
Guinane, C. M., Crispie, F., and Cotter, P. D. (2016). “Chapter 4 - value of microbial genome sequencing for probiotic strain identification and characterization: promises and pitfalls” in eds. N. Hyland and C. B. T.-T. G.-B. A. Stanton (The Gut-Brain Axis: Academic Press), 45–60.
Gulcin, İ. (2020). Antioxidants and antioxidant methods: an updated overview. Arch. Toxicol. 94, 651–715. doi: 10.1007/S00204-020-02689-3
Hernández-González, J. C., Martínez-Tapia, A., Lazcano-Hernández, G., García-Pérez, B. E., and Castrejón-Jiménez, N. S. (2021). Bacteriocins from lactic acid bacteria. A powerful alternative as antimicrobials, probiotics, and immunomodulators in veterinary medicine. Animals 11:979. doi: 10.3390/ani11040979
Hill, C., Guarner, F., Reid, G., Gibson, G. R., Merenstein, D. J., Pot, B., et al. (2014). Expert consensus document: the international scientific association for probiotics and prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat. Rev. Gastroenterol. Hepatol. 11, 506–514. doi: 10.1038/nrgastro.2014.66
Hill, D., Sugrue, I., Tobin, C., Hill, C., Stanton, C., and Ross, R. P. (2018). The Lactobacillus casei group: history and health related applications. Front. Microbiol. 9, 1–12. doi: 10.3389/fmicb.2018.02107
Ho Sui, S. J., Fedynak, A., Hsiao, W. W. L., Langille, M. G. I., and Brinkman, F. S. L. (2009). The Association of Virulence Factors with Genomic Islands. PLoS One 4:e8094. doi: 10.1371/JOURNAL.PONE.0008094
Huang, L., Zhang, H., Wu, P., Entwistle, S., Li, X., Yohe, T., et al. (2018). DbCAN-seq: a database of carbohydrate-active enzyme (CAZyme) sequence and annotation. Nucleic Acids Res. 46, D516–D521. doi: 10.1093/nar/gkx894
Joensen, K. G., Scheutz, F., Lund, O., Hasman, H., Kaas, R. S., Nielsen, E. M., et al. (2014). Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 52, 1501–1510. doi: 10.1128/JCM.03617-13
Johansson, M. H. K., Bortolaia, V., Tansirichaiya, S., Aarestrup, F. M., Roberts, A. P., and Petersen, T. N. (2021). Detection of mobile genetic elements associated with antibiotic resistance in Salmonella enterica using a newly developed web tool: MobileElementFinder. J. Antimicrob. Chemother. 76, 101–109. doi: 10.1093/JAC/DKAA390
Kandasamy, S., Yoo, J., Yun, J., Lee, K. H., Kang, H. B., Kim, J. E., et al. (2022). Probiogenomic in-silico analysis and safety assessment of Lactiplantibacillus plantarum DJF10 strain isolated from Korean raw Milk. Int. J. Mol. Sci. 23:14494. doi: 10.3390/ijms232214494
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2016). KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462. doi: 10.1093/NAR/GKV1070
Kang, J., Chung, W. H., Lim, T. J., Whon, T. W., Lim, S., and Do Nam, Y. (2017). Complete genome sequence of Lactobacillus casei LC5, a potential probiotics for atopic dermatitis. Front. Immunol. 8:239796. doi: 10.3389/FIMMU.2017.00413/BIBTEX
Kao, T. H., and Chen, B. H. (2006). Functional components in soybean cake and their effects on antioxidant activity. J. Agric. Food Chem. 54, 7544–7555. doi: 10.1021/jf061586x
Katla, T., Møretrø, T., Aasen, I. M., Holck, A., Axelsson, L., and Naterstad, K. (2001). Inhibition of Listeria monocytogenes in cold smoked salmon by addition of sakacin P and/or live Lactobacillus sakei cultures. Food Microbiol. 18, 431–439. doi: 10.1006/fmic.2001.0420
Kim, H., Kim, S., Park, S. J., Park, G., Shin, H., Park, M. S., et al. (2021). Administration of Bifidobacterium bifidum BGN4 and Bifidobacterium longum BORI improves cognitive and memory function in the mouse model of Alzheimer’s disease. Front. Aging Neurosci. 13, 1–13. doi: 10.3389/fnagi.2021.709091
Kim, E., Yang, S. M., Kim, D., and Kim, H. Y. (2022). Complete genome sequencing and comparative genomics of three potential probiotic strains, Lacticaseibacillus casei FBL6, Lacticaseibacillus chiayiensis FBL7, and Lacticaseibacillus zeae FBL8. Front. Microbiol. 12:794315. doi: 10.3389/fmicb.2021.794315
Kiousi, D. E., Efstathiou, C., Tegopoulos, K., Mantzourani, I., Alexopoulos, A., Plessas, S., et al. (2022). Genomic insight into Lacticaseibacillus paracasei SP5, reveals genes and gene clusters of probiotic interest and biotechnological potential. Front. Microbiol. 13, 1–21. doi: 10.3389/fmicb.2022.922689
Lee, J., Hwang, K. T., Heo, M. S., Lee, J. H., and Park, K. Y. (2005). Resistance of Lactobacillus plantarum KCTC 3099 from kimchi to oxidative. Stress 8, 299–304. doi: 10.1089/JMF.2005.8.299
Li, J. Y., Jin, M. M., Meng, J., Gao, S. M., and Lu, R. R. (2013). Exopolysaccharide from Lactobacillus planterum LP6: Antioxidation and the effect on oxidative stress. Carbohydr. Polym. 98, 1147–1152. doi: 10.1016/j.carbpol.2013.07.027
Li, J., Zhang, L., Mu, G., and Tuo, Y. (2023). Interpretation of safety and potential probiotic properties of Lactiplantibacillus plantarum Y42 based on genome-wide sequencing. Food Biosci. 56:103249. doi: 10.1016/j.fbio.2023.103249
Li, O., Zhang, H., Wang, W., Liang, Y., Chen, W., Ud Din, A., et al. (2021). Complete genome sequence and probiotic properties of Lactococcus petauri LZys1 isolated from healthy human gut. J. Med. Microbiol. 70. doi: 10.1099/JMM.0.001397
Li, S., Zhao, Y., Zhang, L., Zhang, X., Huang, L., Li, D., et al. (2012). Antioxidant activity of Lactobacillus plantarum strains isolated from traditional Chinese fermented foods. Food Chem. 135, 1914–1919. doi: 10.1016/j.foodchem.2012.06.048
Lin, M. Y., and Yen, C. L. (1999). Antioxidative ability of lactic acid bacteria. J. Agric. Food Chem. 47, 1460–1466. doi: 10.1021/JF981149L/ASSET/IMAGES/LARGE/JF981149LF00001.JPEG
Liu, X., Lv, X., Sun, Y., Liu, C., Wang, R., Liu, R., et al. (2023). Probiotic properties of Lacticaseibacillus rhamnosus grx10 revolved with complete genome. Food Biosci. 51:102219. doi: 10.1016/j.fbio.2022.102219
Liu, B., Zheng, D., Zhou, S., Chen, L., and Yang, J. (2022). VFDB 2022: a general classification scheme for bacterial virulence factors. Nucleic Acids Res. 50, D912–D917. doi: 10.1093/nar/gkab1107
Mazmanian, S. K., Round, J. L., and Kasper, D. L. (2008). A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 453, 620–625. doi: 10.1038/nature07008
Meier-Kolthoff, J. P., and Göker, M. (2019). TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 10, 1–10. doi: 10.1038/s41467-019-10210-3
Mikelsaar, M., and Zilmer, M. (2009). Lactobacillus fermentum ME-3 - an antimicrobial and antioxidative probiotic. Microb. Ecol. Health Dis. 21, 1–27. doi: 10.1080/08910600902815561
Mishra, V., Shah, C., Mokashe, N., Chavan, R., Yadav, H., and Prajapati, J. (2015). Probiotics as potential antioxidants: a systematic review. J. Agric. Food Chem. 63, 3615–3626. doi: 10.1021/jf506326t
Monteagudo-Mera, A., Rastall, R. A., Gibson, G. R., Charalampopoulos, D., and Chatzifragkou, A. (2019). Adhesion mechanisms mediated by probiotics and prebiotics and their potential impact on human health. Appl. Microbiol. Biotechnol. 103, 6463–6472. doi: 10.1007/s00253-019-09978-7
Mu, G., Gao, Y., Tuo, Y., Li, H., Zhang, Y., Qian, F., et al. (2018). Assessing and comparing antioxidant activities of lactobacilli strains by using different chemical and cellular antioxidant methods. J. Dairy Sci. 101, 10792–10806. doi: 10.3168/jds.2018-14989
Nilsen, T., Nes, I. F., and Holo, H. (2003). Enterolysin a, a cell wall-degrading bacteriocin from Enterococcus faecalis LMG 2333. Appl. Environ. Microbiol. 69, 2975–2984. doi: 10.1128/AEM.69.5.2975-2984.2003
Odila Pereira, J., Soares, J., Monteiro, J. P., Gomes, A., and Pintado, M. (2018). Impact of whey protein coating incorporated with Bifidobacterium and Lactobacillus on sliced ham properties. Meat Sci. 139, 125–133. doi: 10.1016/J.MEATSCI.2018.01.016
Oyaizu, M. (1986). Studies on products of Browning reaction. Japanese J. Nutr. Diet. 44, 307–315. doi: 10.5264/eiyogakuzashi.44.307
Pei, Z., Sadiq, F. A., Han, X., Zhao, J., Zhang, H., Ross, R. P., et al. (2021). Comprehensive scanning of prophages in Lactobacillus: distribution, diversity, antibiotic resistance genes, and linkages with CRISPR-Cas systems. mSystems 6:e01211-20. doi: 10.1128/msystems.01211-20
Pruksarojanakul, P., Prakitchaiwattana, C., Settachaimongkon, S., and Borompichaichartkul, C. (2020). Synbiotic edible film from konjac glucomannan composed of Lactobacillus casei-01® and Orafti®GR, and its application as coating on bread buns. J. Sci. Food Agric. 100, 2610–2617. doi: 10.1002/jsfa.10287
Richter, M., Rosselló-Móra, R., Oliver Glöckner, F., and Peplies, J. (2016). JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 32, 929–931. doi: 10.1093/BIOINFORMATICS/BTV681
Rodrigo-Torres, L., María Landete, J., Huedo, P., Peirotén, Á., Langa, S., Rodríguez-Minguez, E., et al. (2022). Complete genome sequences of Lacticaseibacillus paracasei INIA P272 (CECT 8315) and Lacticaseibacillus rhamnosus INIA P344 (CECT 8316) isolated from breast-fed infants reveal probiotic determinants. Gene 840:146743. doi: 10.1016/j.gene.2022.146743
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/BIOINFORMATICS/BTU153
Sharifpanah, F., and Sauer, H. (2016). Reactive oxygen species, oxidative stress, and cardiovascular diseases. Oxidative Stress Antioxid. Prot., The Science of Free Radical Biology and Disease, (ed.) R. D. S. Donald Armstrong. (Hoboken, NJ: John Wiley & Sons, Inc.) 281–306. doi: 10.1002/9781118832431.CH17
Siguier, P., Gourbeyre, E., and Chandler, M. (2014). Bacterial insertion sequences: their genomic impact and diversity. FEMS Microbiol. Rev. 38, 865–891. doi: 10.1111/1574-6976.12067
Siguier, P., Perochon, J., Lestrade, L., Mahillon, J., and Chandler, M. (2006). ISfinder: the reference Centre for bacterial insertion sequences. Nucleic Acids Res. 34, D32–D36. doi: 10.1093/NAR/GKJ014
Song, D., Ibrahim, S., and Hayek, S. (2012). “Recent application of probiotics in food and agricultural science” in Probiotics. ed. E. C. Rigobelo (Rijeka: IntechOpen).
Tang, W., Xing, Z., Li, C., Wang, J., and Wang, Y. (2017). Molecular mechanisms and in vitro antioxidant effects of Lactobacillus plantarum MA2. Food Chem. 221, 1642–1649. doi: 10.1016/j.foodchem.2016.10.124
Tian, Y., Wang, Y., Zhang, N., Xiao, M., Zhang, J., Xing, X., et al. (2022). Antioxidant mechanism of Lactiplantibacillus plantarum KM1 under H2O2 stress by proteomics analysis. Front. Microbiol. 13, 1–15. doi: 10.3389/fmicb.2022.897387
Toh, H., Oshima, K., Nakano, A., Takahata, M., Murakami, M., Takaki, T., et al. (2013). Genomic adaptation of the Lactobacillus casei group. PLoS One 8:e75073. doi: 10.1371/journal.pone.0075073
Valko, M., Leibfritz, D., Moncol, J., Cronin, M. T. D., Mazur, M., and Telser, J. (2007). Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 39, 44–84. doi: 10.1016/J.BIOCEL.2006.07.001
Van Heel, A. J., De Jong, A., Song, C., Viel, J. H., Kok, J., and Kuipers, O. P. (2018). BAGEL4: a user-friendly web server to thoroughly mine RiPPs and bacteriocins. Nucleic Acids Res. 46, W278–W281. doi: 10.1093/nar/gky383
Vasconcelos, F. M., Silva, H. L. A., Poso, S. M. V., Barroso, M. V., Lanzetti, M., Rocha, R. S., et al. (2019). Probiotic Prato cheese attenuates cigarette smoke-induced injuries in mice. Food Res. Int. 123, 697–703. doi: 10.1016/J.FOODRES.2019.06.001
Vijayakumar, M., Ilavenil, S., Kim, D. H., Arasu, M. V., Priya, K., and Choi, K. C. (2015). In-vitro assessment of the probiotic potential of Lactobacillus plantarum KCC-24 isolated from Italian rye-grass (Lolium multiflorum) forage. Anaerobe 32, 90–97. doi: 10.1016/J.ANAEROBE.2015.01.003
Wuyts, S., Wittouck, S., De Boeck, I., Allonsius, C. N., Pasolli, E., Segata, N., et al. (2017). Large-scale Phylogenomics of the Lactobacillus casei group highlights taxonomic inconsistencies and reveals novel clade-associated features. mSystems. 2:e00061–17. doi: 10.1128/msystems.00061-17
Xin, Y., Guo, T., Mu, Y., and Kong, J. (2017). Identification and functional analysis of potential prophage-derived recombinases for genome editing in Lactobacillus casei. FEMS Microbiol. Lett. 364. doi: 10.1093/femsle/fnx243
Zafar, H., and Saier, M. H. (2020). Comparative genomics of the transport proteins of ten lactobacillus strains. Genes (Basel) 11, 1–20. doi: 10.3390/genes11101234
Zhang, W., Ji, H., Zhang, D., Liu, H., Wang, S., Wang, J., et al. (2018). Complete genome sequencing of Lactobacillus plantarum ZLP001, a potential probiotic that enhances intestinal epithelial barrier function and defense against pathogens in pigs. Front. Physiol. 9, 1–7. doi: 10.3389/fphys.2018.01689
Zheng, J., Wittouck, S., Salvetti, E., Franz, C. M. A. P., Harris, H. M. B., Mattarelli, P., et al. (2020). A taxonomic note on the genus Lactobacillus: description of 23 novel genera, emended description of the genus Lactobacillus beijerinck 1901, and union of Lactobacillaceae and Leuconostocaceae. Int. J. Syst. Evol. Microbiol. 70, 2782–2858. doi: 10.1099/ijsem.0.004107
Keywords: Lacticaseibacillus, antioxidant, probiotics, whole genome sequencing, bacteriocin, stress-related proteins, mobile genetic elements
Citation: Kandasamy S, Lee K-H, Yoo J, Yun J, Kang HB, Kim JE, Oh M-H and Ham J-S (2024) Whole genome sequencing of Lacticaseibacillus casei KACC92338 strain with strong antioxidant activity, reveals genes and gene clusters of probiotic and antimicrobial potential. Front. Microbiol. 15:1458221. doi: 10.3389/fmicb.2024.1458221
Edited by:
Arun K. B., Christ University, IndiaReviewed by:
Corina-Diana Ceapă, National Autonomous University of Mexico, MexicoCopyright © 2024 Kandasamy, Lee, Yoo, Yun, Kang, Kim, Oh and Ham. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun-Sang Ham, aGFtanNAa29yZWEua3I=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.