 Jinliang Hou
Jinliang Hou Yu Tan2
Yu Tan2 Yazhou Hu
Yazhou Hu Jianguo Xiang
Jianguo Xiang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 14 November 2024
Sec. Microbial Symbioses
Volume 15 - 2024 | https://doi.org/10.3389/fmicb.2024.1453617
The skin microbiota plays an essential role in helping the host adapt to different environments and maintain health. By examining the characteristics of amphibian skin flora alongside ontogenetic traits, we can gain insights into the adaptation mechanisms of amphibian skin flora to environmental changes during development. In this study, we analyzed the skin microbiota of Quasipaa spinosa during metamorphosis using Illumina sequencing. Venn diagrams and UpSet analysis revealed that the LTS (hindlimb tadpoles’ skin, aquatic habitat) and FTS (forelimb tadpoles’ skin, shift from aquatic to amphibious habitats) groups exhibited a higher number of unique amplicon sequence variants (ASVs), while the TS (tadpoles’ skin, aquatic habitat) and LFS (land frogs’ skin, amphibious habitats) groups displayed a lower abundance of ASVs. Diversity analysis indicated similarities in the microorganisms between the LTS and the FTS groups, with higher microbial diversity compared to the TS and the LFS groups. Additionally, microbial co-occurrence network analysis indicated a more stable microecology in the LTS group and FTS group. Proteobacteria, Firmicutes, and Bacteroidota were identified as the dominant phyla, although their relative abundances varied widely among groups. LEfSe (Linear discriminant analysis effect size) showed significant enrichment of beneficial bacteria at various developmental stages, including Bacteroides, Bacillus, and Lactobacillus. Functional prediction analysis shows significant differences in skin microorganism functions across various developmental stages, with a primary focus on metabolic functions. This study provides valuable insights into the compositional dynamics of skin microbiota in Q. spinosa at various developmental stages.
• Significant changes in skin microbial diversity in response to changes in developmental and perching habitats.
• Changes in microbial communities at different stages to facilitate host’s adaptation to the changes stemming from both development and environment.
• Several sensitive indicator taxa were identified for predicting host health trends.
• Critical stages determining the success of metamorphic development were identified: forelimb tadpoles to landing frogs.
The skin forms a protective barrier against the external environment and is the first line of defense against external aggressors (Kueneman et al., 2014). While the skin’s barrier function is critically dependent on its colonizing microbiota, which provides protection from pathogens, modulates immune responses, and enhances the epithelium (Harris-Tryon and Grice, 2022; Hu et al., 2021). The skin microbiota of healthy individuals maintains a stable and dynamic equilibrium (Wei et al., 2023). Studies have shown that a number of skin diseases are closely associated with altered microbial status, and these changes are also referred to as microbial dysbiosis (Christensen and Bruggemann, 2014). In recent years, the skin microbial community of aquatic organisms has gradually received more and more attention, but most of it has concentrated on disease-related microbes and the effects of purely environmental changes on the microbes (Chen et al., 2022; Fieschi-Meric et al., 2023; Jiang et al., 2022; Mulla and Hernandez-Gomez, 2023; Sadeghi et al., 2023). In contrast, skin microbial communities are affected by a variety of factors and further research is needed to find the key factors that maintain the stability of the flora.
A very special group of organisms is the tailless amphibians. During metamorphosis, amphibians develop specialized habits as they transition from aquatic life to co-inhabitation of water bodies and land (Hou et al., 2022; Tong et al., 2020). This transition presents a serious challenge to their survival, as exposure-induced habitat modification could disrupt the microecological balance of skin surface microorganisms, posing a serious threat to species health (Martinez-Ugalde et al., 2022). The second reason is that during metamorphosis amphibians in larval stages undergo physiological rearrangements (Brown and Cai, 2007), such as the skin of the organism gains a new function: assisted respiration after completing metamorphosis (Martinez-Ugalde et al., 2022), which cause immunosuppression of hormonal signaling in the organism (Rollins-Smith, 1998), and would allow for a smoother colonization by pathogenic bacteria. An increasing number of studies are investigating the infestation of microbial pathogens as a leading cause of amphibian mortality such as the pathogenic microorganisms causing “chytridiomycosis”(Bletz et al., 2018; Muletz-Wolz et al., 2019; Scheele et al., 2019), rotten-skin (Jiang et al., 2022; Xu et al., 2024a), cataracts (Knutie et al., 2017; Xu et al., 2024a) and torticollis (Xu et al., 2024b), which has been responsible for widespread amphibian deaths. Understanding the skin microbial community of healthy amphibians is important for preventing and controlling skin diseases to promote healthy farming.
The Chinese spiny frog (Quasipaa spinosa) belongs to the Dicroglossidae family and the Quasipaa genus, which belongs to the Anura order and Amphibia of chordates (Hou et al., 2023). Q. spinosa is an economically important amphibian species with a high nutritional and medicinal value (Hou et al., 2022). However, the vulnerability of the metamorphosis period and the harsh environmental requirements contribute to a high mortality rate. Previous studies have suggested a potential link between changes in gut flora and this phenomenon (Yu et al., 2014). However, the occurrence of disease phenomena like rotten-skin during metamorphosis raises questions about the role of skin microbiota in ensuring healthy survival (Hou et al., 2022). Although some studies have shown that alterations in the skin microflora of Echinodermata can lead to diseases, there is a lack of research on how developmental habitat changes relate to the dynamics of skin microflora.
In this study, skin samples were collected from spiny-breasted frogs at four key points during metamorphosis: complete tadpoles, tadpoles with hindlimbs, tadpoles with forelimbs, and amphibious living frogs. The 16S RNA and Illumina high-throughput sequencing technology were deployed to systematically compare differences in the composition, diversity, and biological function of the skin microbiota during the stages of metamorphosis. Our work sought to (1) elucidate modifications in the composition and diversity of the skin microbiota during the stages of metamorphosis; (2) predict skin microbiota-mediated functional alterations; (3) screen the indicator microorganism taxa that represent the four key stages of health. The study results emphasize the potential relevance of skin microbiota to the developmental stages of host metamorphosis, and highlight how the skin’s symbiotic microbial community responds to physiological and habitat changes caused by development in healthy hosts, with the goal of providing valuable information for healthy culture and improved survival of Q. spinosa from a microbial perspective.
The Chinese spiny frogs and tadpoles used in this study were captured from a breeding farm (111°02′40′′ E, 29°47′54′′ N, 150 m asl) in Shimen County, Changde City, Hunan Province, China. The experiment was carried out in a breeding pond for 5 months. The water was cultivated spring water at 19 ± 0.5°C, and its quality was deemed to be first class according to the Environmental Quality Standards for Surface Water in China (GB3838-2002). All frogs and tadpoles shared the same genetic background and were at the different critical stages of metamorphosis. Selective sampling was performed in order to make each stage sample representative. The tadpoles/frogs at a stage were of similar size and developmental period, and all samples were collected from healthy individuals. However, we ensured that all tadpoles were fertilized and hatched at the same time period, and that other external factors, such as the breeding environment, were consistent. Throughout the growth phase, these frogs were given the same feed and feed amount. No antibiotics were used during this experiment. According to Gosner (1960) and Kimball (2016), the developmental stage of the tadpole was determined. The TS group were tadpoles that had just started to feed after demetamorphosis (GS 25). The LTS group were tadpoles that had grown hind limbs but had not yet formed forelimbs and were in the GS 31 ~ GS 37. The FTS group were tadpoles that had already extended their forelimbs (GS 42 ~ GS 45). The LFS group were a developmental stage of frogs that had recently undergone metamorphosis and had not yet consumed food (GS 46). Six samples were collected from each group (Table 1). The weight and length of tadpoles/frogs were recorded at the same time of sampling (Table 1).
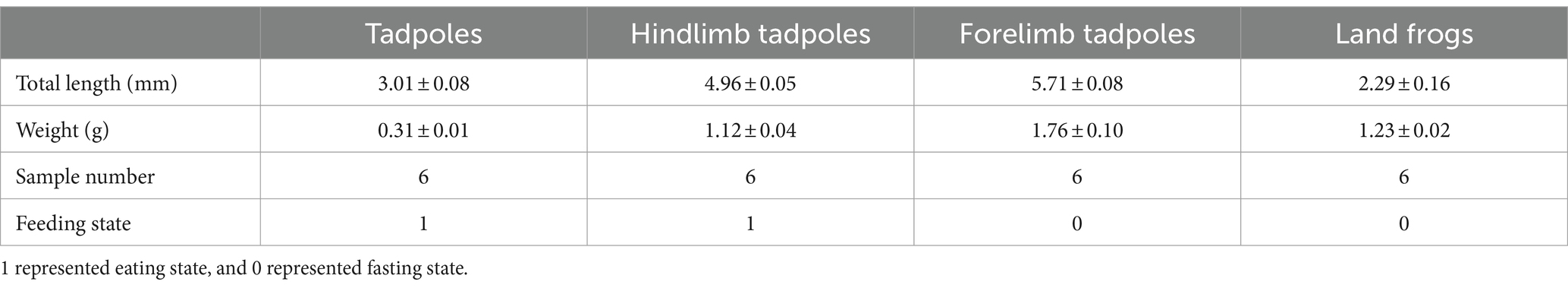
Table 1. Host information of 24 skin swab samples of Q. spinosa.
Prior to sampling, each spiny frog was rinsed with 1 × PBS (phosphate buffered saline; cytiva, United States). The skin microbiota was harvested using cotton swabs according to a previously described protocol (Costa et al., 2016). Swabs were then transferred into lyophilization tubes and immediately preserved in liquid nitrogen until DNA extraction. To ensure clean samples, the entire sampling process was carried out in ultra-clean bench with aseptic manipulation. To avoid cross contamination between samples, the instruments were replaced with new sterile cotton swabs when sampling different individuals.
The swabs were transferred into sterile centrifuge tubes with 1 × PBS (cytiva, United States), and the liquid was retained for DNA extraction after ultrasonic elution treatment. Total microbial genomic DNA was extracted from each sample with a MgaPure Stool DNA LQ Kit (Magen, Guangzhou, China) according to the manufacturer’s protocol.
Amplification of the V3-V4 regions of the 16S rRNA genes was conducted with universal primers 341F and 805R (Table 2). The KAPA HiFi HotStart ReadyMix PCR kit was used for PCR amplification according to the instructions. The system consisted of 15 μL 2 × KAPA HiFi Mix, 12.5 μL of template, 1 μL of forward primer, 1 μL of reverse primer and then add dd H2O to 30 μL, and the reaction conditions were as follows: pre-denaturation at 95°C for 3 min, denaturation at 95°C for 30 s, annealing at 55°C for 30 s, extension at 72°C for 15 s for 25 cycles, and finally extension at 72°C for 5 min. The PCR products were separated and purified from a 1% agarose gel by agarose gel electrophoresis using SanPrep Column DNA Gel Extraction Kit (Sangon Biotech, China) according to the instructions. The recovered products were quantified by Qubit Fluorometer (Thermo Fisher Scientific, United States) and mixed accordingly. Sequencing libraries were prepared using the TruSeq Nano DNA LT Library Prep Kit according to the instructions. The raw reads of skin microbial sequences were deposited in the NCBI SRA database under accession numbers PRJNA1058082.

Table 2. Primers used for this study.
Following amplicon level standardization, all samples were submitted to Benagen Technology Company Limited (Wuhan, China) for high-throughput sequencing using the Novaseq platform (Illumina, United States).
The library containing mixed samples was identified by barcode to obtain single sample sequence data. Low-quality filtering was performed using Trimmomatic v0.39 (Bolger et al., 2014) software with a sliding window size of 20 bp and an average quality score threshold within the window set to greater than 20. The primer sequences were then identified and removed using cutadapt (version 3.4) software to obtain clean sequences without primers (Martin, 2011). The obtained clean reads were filtered, denoised, and merged into amplicon sequence variants (ASVs) using DADA2 (Callahan et al., 2016).
Taxonomic classification of each ASV was assigned against the Silva database (Release138) using QIIME2 (2021.4) (Bokulich et al., 2018; Quast et al., 2013). Venn and UpSet analyses were conducted using TBtools (2.012) to the determine exclusive and shared ASVs among groups. Metrics of alpha-diversity, including richness indexes (ACE (Abundance-based Coverage Estimator) and Chao1), diversity indexes (Shannon), and evenness index (Simpson), were calculated based on vegan (2.6–4) and ggplot2 (3.4.4) package of R, respectively. The significance was performed through one-way analysis of variance (ANOVA) followed by Turkey’s post-hoc multiple comparisons. For beta-diversity analysis, Bray–curtis dissimilarity matrices were calculated based on microeco (1.5.0), and visualized by NMDS (Non-metric multidimensional scaling) using ggplot2 (Liu et al., 2021). Ternary plot was calculated and visualized based on ggtern (3.4.2) package of R. Sequences with the highest abundance of features at the genus level were chosen as representative sequences. Multiple sequence alignment was conducted using QIIME2, followed by construction of a phylogenetic tree. The tree was then visualized using the R package ggtree. LEfSe analysis was conducted to identify significant correlations between bacterial taxa and different groups (Segata et al., 2011). In this study, LDA (Linear discriminant analysis) > 4 was used as the criterion for LEfSe. Microbial networks were visualized based on Gephi (0.10). Zi-Pi was calculated and visualized based on ggClusterNet (0.1.0) and ggplot2 (3.4.4) package of R, respectively. The roles of the nodes generally fell into four categories, as previously defined (Zhao et al., 2021): (i) peripheral nodes (Zi ≤ 2.5 and Pi ≤ 0.62); (ii) connectors (Zi ≤ 2.5 and Pi > 0.62); (iii) module hubs (Zi > 2.5 and Pi ≤ 0.62); and (iv) network hubs (Zi > 2.5 and Pi > 0.62). PICRUSt2 analysis was conducted to predict functional changes in the microbiota among groups (Douglas et al., 2020). The relative abundances between the different groups were compared using Wilcoxon and multiple test corrections (Benjamini-Hochberg FDR).
A total of 1,844,167 high-quality sequences were obtained from 24 skin samples, of which the average length of the sequences was 225 bp (Supplementary Table S1). These sequences were then clustered into ASVs, and 8,593 ASVs were obtained. ASVs among skin samples ranged from 156 (TS3) to 2,256 (LTS4), with a mean of 416 (Figure 1B). The rarefaction curves, Shannon index curves and species accumulation curves were constructed based on ASVs and showed that the depth of the sequencing results was sufficient (Supplementary Figures S1A,B,D). The milder status of the rank abundance curves in the LTS and FTS groups suggested a more uniform distribution of bacteria (Supplementary Figure S1C). In this study, 491 ASVs were unique to tadpoles, 3,145 ASVs were specific to tadpoles during hindlimb development, 2,886 ASVs were specific to tadpoles during forelimb development, 492 ASVs were specific to land frogs, and 334 ASVs were common to all groups (Figure 1A). Notably, the fewest unique ASVs were observed in the TS and LFS group for microbiota samples.
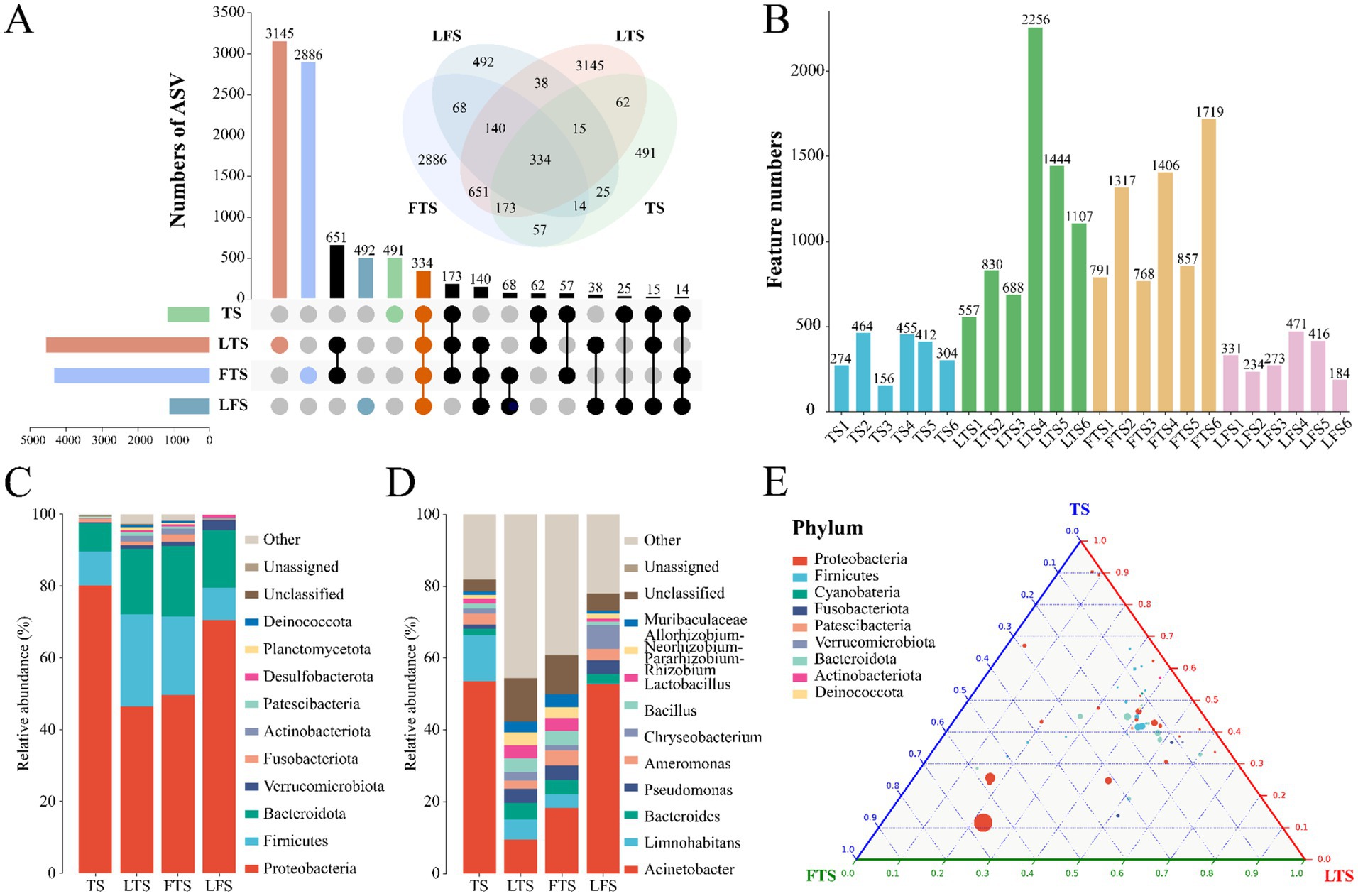
Figure 1. (A) Venn and upset diagrams show the shared and unique ASVs of skin microbiota among Q. spinosa. (B) Statistics for the number of ASVs in each sample. Relative abundances of skin microbiota at phylum (C) and genus (D) among Q. spinosa. (E) Analysis of skin microorganisms in aquatic life stage tadpoles (TS, LTS and FTS groups) using Ternary plot at the phylum level.
Overall, the coverage of skin microbiota was higher than 99.7%, indicating the reliability of the sequencing and data processing results. The ACE, Chao1, Shannon, and Simpson indexes of the skin microbiota in the LTS and FTS group were dramatically higher than those in the TS and LFS groups (Figures 2A–D; Supplementary Tables S2, S5). However, there was no significant difference between TS with LFS and LTS with FTS. Significant separation in skin microbiota was observed in all comparison sets except between the LTS and FTS groups (Figures 2E–H).
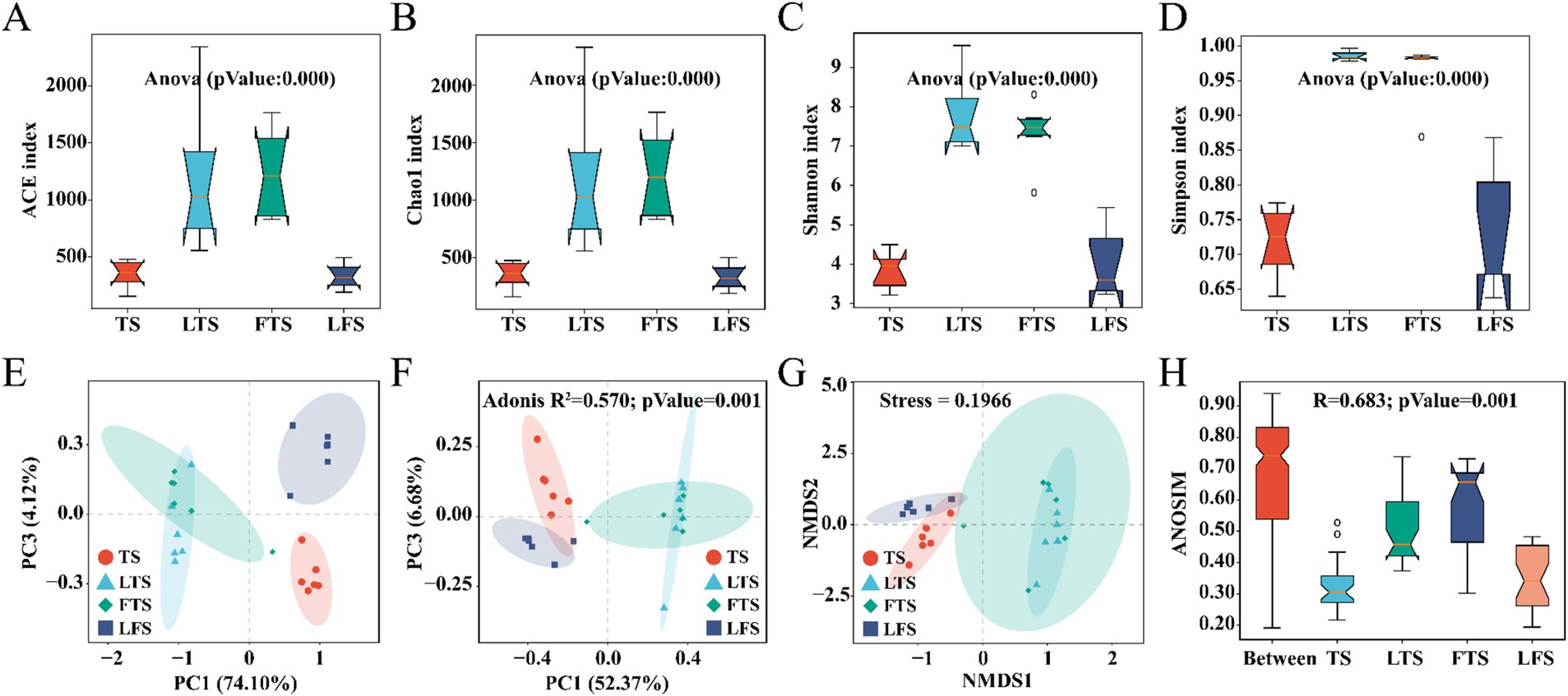
Figure 2. (A–D) Comparison of alpha-diversity in the skin microbiota among different Q. spinosa development stages. (E–H) One-way ANOVA and Waller-Duncan test were used to compare the differences among groups. Comparison of Beta-diversity in the skin microbiota among different Q. spinosa development stages. All Beta-diversity analysis based on Bray-Curtis distances reflected the degree of variation in skin microbiota among groups. Stress was used to assess the reasonableness and credibility of NMDS (G) analysis. ANOSIM (H) and PCoA (F) based on Bray-Curtis distances were conducted to evaluate the dissimilarity of NMDS analysis with adjusted p < 0.05, which was considered significant.
The dominant phyla (>1%) common to all groups of skin microbiota were Proteobacteria (TS: 80.20%, LTS: 46.35%, FTS: 49.66%, LFS: 70.43%), Firmicutes (TS: 9.45%, LTS: 25.84%, FTS: 21.88%, LFS: 9.03%), and Bacteroidota (TS: 7.83%, LTS: 18.17%, FTS: 19.61%, LFS: 16.15%) (Figure 1C). Verrucomicrobiota was the dominant phylum shared by the LTS, FTS and LFS groups, and Fusobacteriota was the dominant phylum shared by the TS and FTS groups (Figure 1C). Whereas Actinobacteriota was the dominant phylum specific to the LTS and FTS groups (Figure 1C). The dominant genera (>5%) common to all groups of skin microbiota were Acinetobacter (TS: 53.60%, LTS: 9.38%, FTS: 18.30%, LFS: 52.80%) and Limnohabitans (TS: 12.83%, LTS: 5.72%, FTS: 3.81%, RD: 0.03%) (Figure 1D). Chryseobacterium (6.61%) was the dominant genus specific to the LFS group (Figure 1D). The ternary plot illustrated the transition of skin microbial taxa from predominantly Proteobacteria to a diverse range of microorganisms across multiple clades as the aquatic phase progressed (Figure 1E). This indicates that the skin microbial community undergoes a shift towards a more stable trend as development progresses.
Molecular phylogenetic analysis of bacteria can elucidate the evolutionary processes of the species in question, shedding light on the history and mechanisms of bacterial evolution. Representative sequences with the highest abundance of features at the genus taxonomic level were chosen, and a phylogenetic evolutionary tree was generated using Fast Tree software based on the maximum likelihood method. The resulting cyclorama visually presented the phylogenetic evolutionary relationships of skin bacteria in Q. spinosa during the developmental stages (Figure 3). For all periods, Proteobacteria, Firmicutes and Bacteroidota still predominate as the main three taxa in the evolution of skin microorganisms (Figure 3).
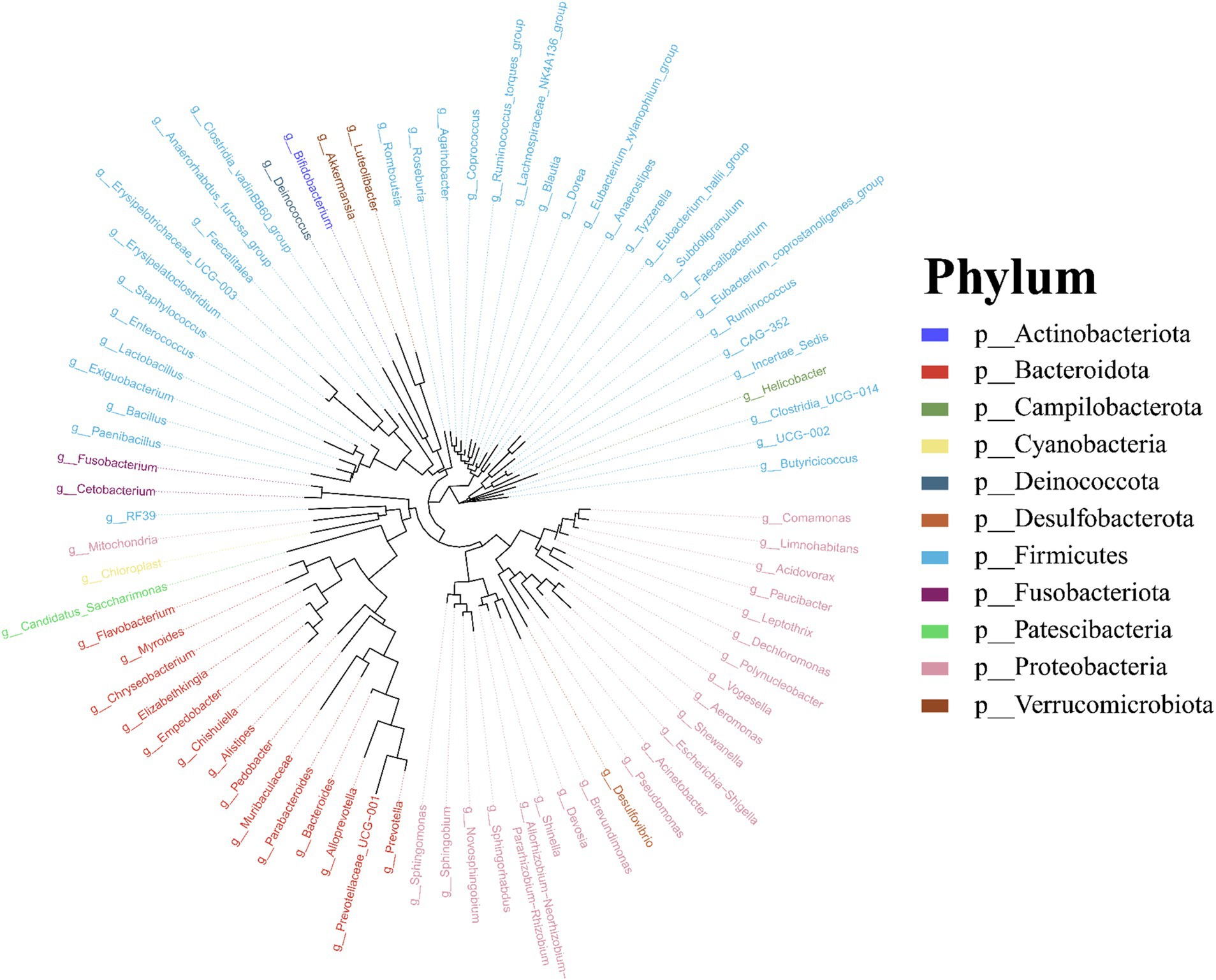
Figure 3. The phylogenetic tree of skin microbiota in Q. spinosa during metamorphosis.
The four-step rank sum test for skin microbial phylum level revealed a total of 23 phylum abundances that were significantly different (p < 0.05), and 17 phyla were significantly different after P-correction (Supplementary Table S4). LEfSe analysis revealed that Proteobacteria was significantly enriched in tadpoles’ skin, Firmicutes was significantly enriched in hindlimb tadpoles’ skin, Bacteroidota and Fusobacteriota were abundant in forelimb tadpoles’ skin, and Verrucomicrobiota was significantly enriched in land frog’s skin (Figure 4). At the genus level, Acinetobacter, Novosphingobium and Limnohabitans were enriched in the TS group, Bacteroides and Allorhizobium_Neorhizobium_Pararhizobium_Rhizobium were enriched in the LTS group, Lactobacillus, Bacillus, Muribaculaceae and Pseudomonas were enriched in the FTS group, and Luteolibacter, Comamonas and Chryseobacterium were enriched in the LFS group (Figure 4). Notably, certain microbial species, such as alpha_proteobacterium in TS group and Acinetobacter_junii in FTS group, were significantly enriched in the skin (Figure 4).
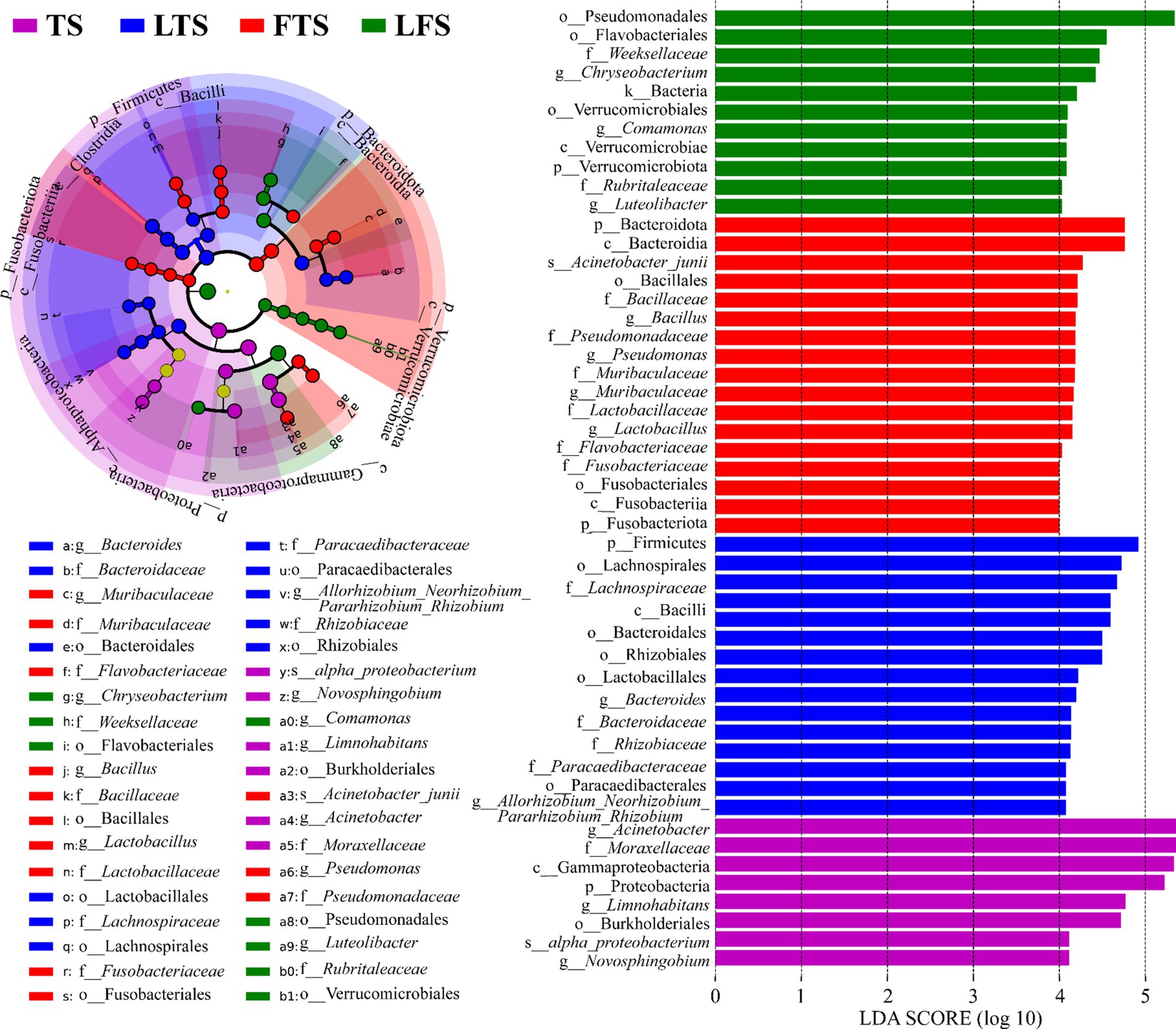
Figure 4. Differences in skin microbiota of Q. spinosa among developmental stages. The results were derived using LEfSe analysis with LDA > 4 and p < 0.05 considered significant.
To investigate the interactions between microorganisms at different developmental stages, ecological networks were constructed for each of the four stages of microbial communities. Significant differences in microbial co-occurrence relationships on the skin surface at different stages are evident (Figure 5). Based on the negative correlation connection and clustering coefficient, the stability of microbial communities in each group was ranked as LTS, FTS, LFS, TS (Table 3). The results indicated that the skin microorganisms underwent notable changes throughout the development process. The microbial community showed a progression from a low-equilibrium state to a high-equilibrium state, followed by a return to a low-equilibrium state after habitat alterations.
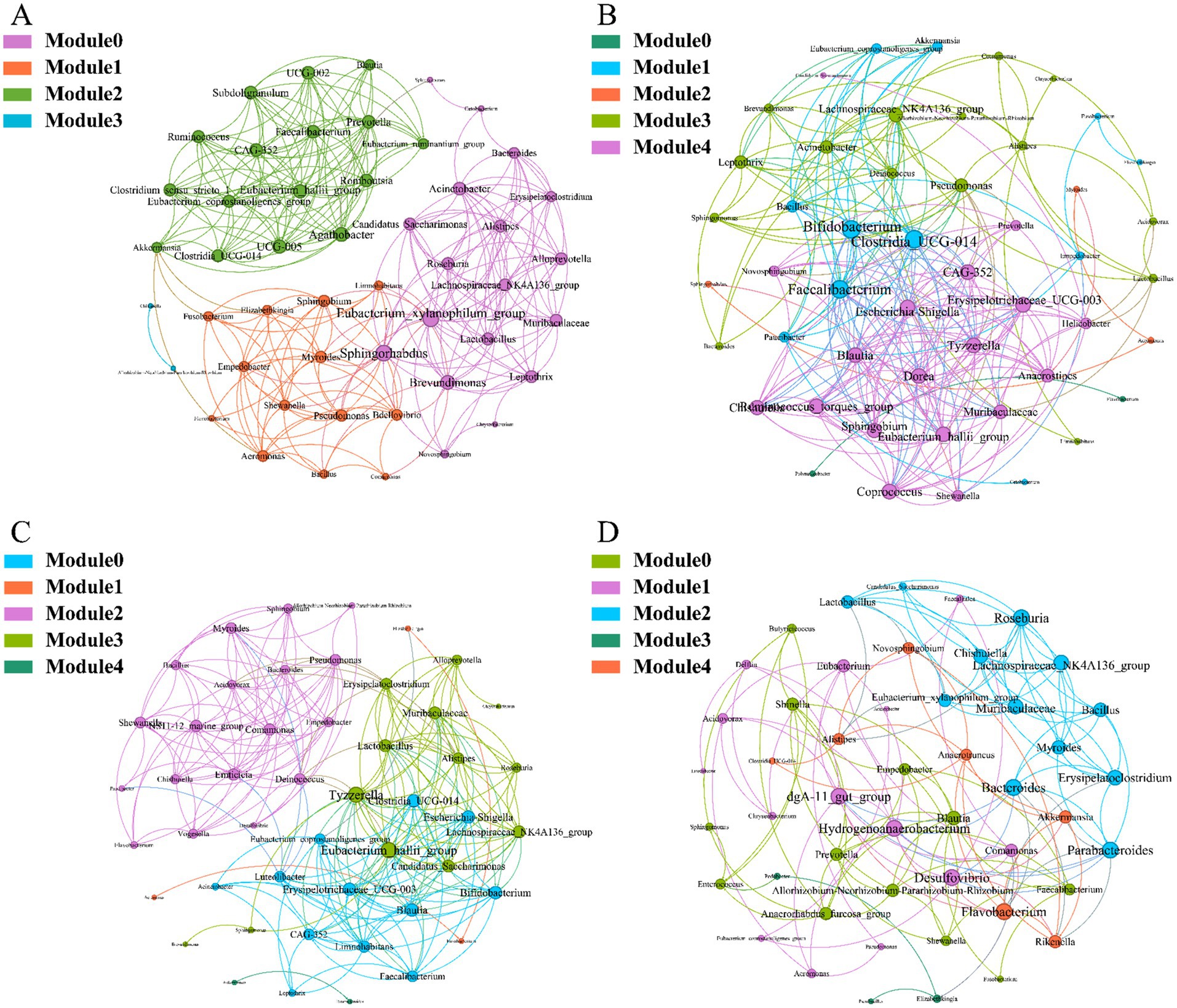
Figure 5. Co-occurrence network of Q. spinosa during metamorphosis. (A) TS group; (B) LTS group; (C) FTS group; (D) LFS group.
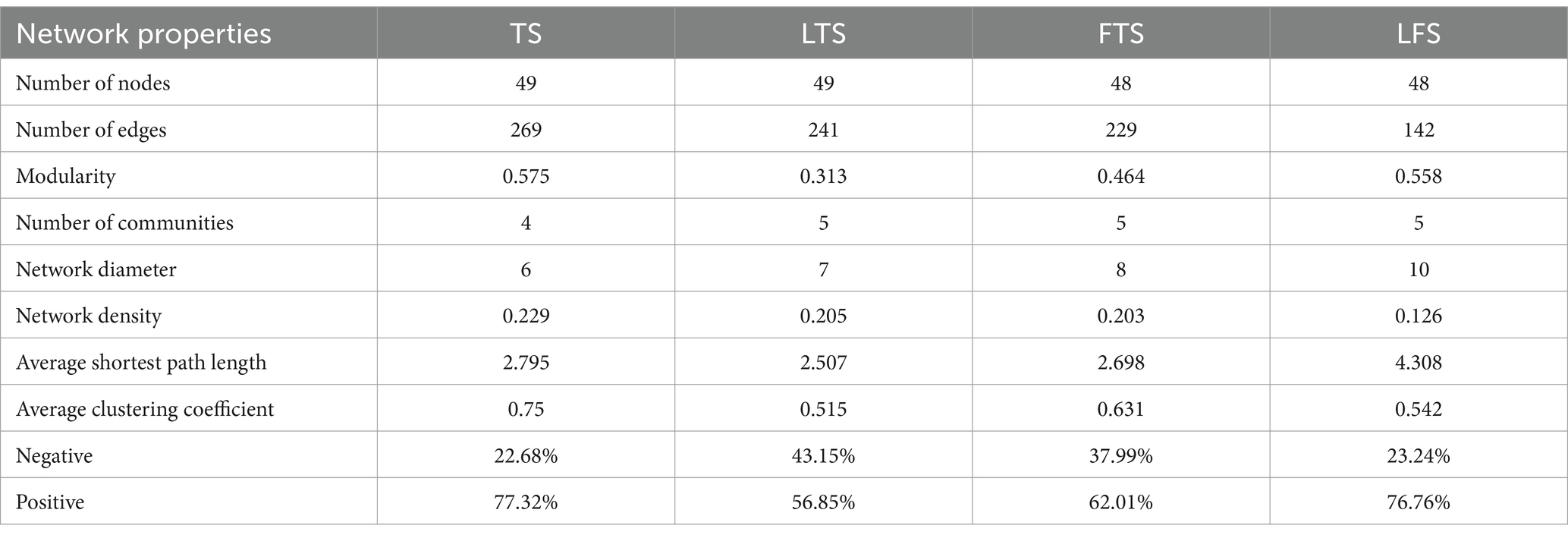
Table 3. Topological characteristics of networks.
The possible topological roles of nodes in the skin were determined by their within-module connectivity (Zi) and among-module connectivity (Pi). Most nodes were classified as peripherals, with most of their connections made within their own modules. Four nodes were identified as connectors, being highly connected to several modules. Three connecters belonged to LFS group and were affiliated with Bacteroides (0.0272), Parabacteroides (0.0094) and Flavobacterium (0.0158) (Supplementary Table S3). Pseudomonas (0.0385) belonged to LTS group (Supplementary Table S3).
PICRUSt2 analysis revealed 17 dominant KEGG leve_2 pathways (>1%) associated with the skin microbiota and remarkable divergence was mainly identified in the metabolism-related KEGG pathway (Figure 6A). A total of seven pathways were significantly enriched in three comparison sets, including amino acid metabolism, carbohydrate metabolism, cell motility, lipid metabolism, membrane transport, metabolism of terpenoids and polyketides and xenobiotics biodegradation and metabolism (Figures 6B–D). The functional pathways in the LTS and FTS groups showed great similarity with no significant differences between the two groups.
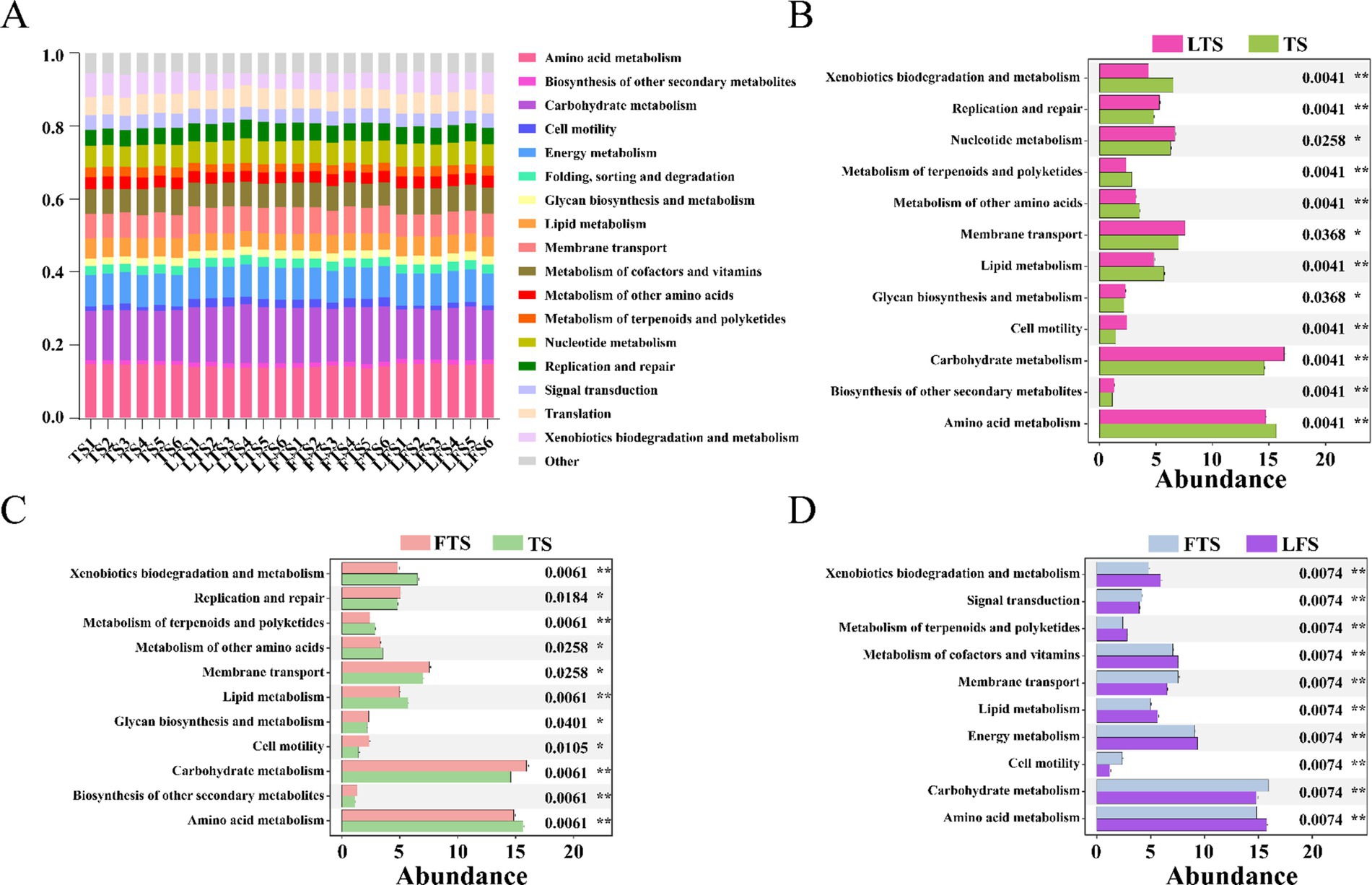
Figure 6. Relative abundances of skin ASVs assigned to level-2 KEGG pathways of Q. spinosa. PICRUSt2 was conducted to determine the predicted function. Wilcoxon and multiple test correction (Benjamini-Hochberg FDR) were used to compare the differences among groups. Data are presented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001.
The alpha diversity of microbiota serves as an indicator of species abundance and diversity, commonly used to assess animal health (Longo and Zamudio, 2017). In ecological perception, higher diversity can be considered more resilient ecosystems (Schommer and Gallo, 2013). Changes in an animal’s health often correlate with alterations in the alpha diversity of microbiota (Jiang et al., 2022). In addition, changes in the alpha diversity of the skin microbiota often occur during development (Fieschi-Meric et al., 2023; Tong et al., 2020). This study revealed that the alpha diversity of skin microbiota in tadpoles and landing frogs was significantly lower than hindlimb tadpoles and forelimb tadpoles, with no significant differences observed between tadpoles and shore frogs, and between hind-limbed tadpoles and fore-limbed tadpoles, respectively. Furthermore, the alpha diversity of skin microbiota in tadpole stages (TS, LTS, and FTS groups) showed a significant increase with developmental maturity, a trend supported by findings in amphibians like Hynobius maoershanensis (Ning, 2022) and Rana cascadae (Kueneman et al., 2014). Similarly, the co-occurrence network results showed higher ecological stability of microflora in the LTS and FTS groups, and significantly lower in the LFS group. However, the skin surface microorganisms are undergoing adaptive changes due to habitat variations and increased complexity in skin structural functions, potentially leading to a notable reduction in alpha diversity. Conversely, the diversity of skin microorganisms was higher in Lithobates vibicarius (Jimenez et al., 2019) tadpoles during development compared to juvenile and adult frogs, with no significant diversity differences between juvenile and adult frogs. These distinctions may be attributed to genetic variations among organisms and the diverse environmental requirements they must adapt to.
Similar outcomes have been observed in the microbial alpha diversity changes among other amphibian species. The skin microbial communities of Salamandra differ significantly before and after metamorphosis, and the host itself consistently influences these skin microbial communities (Sanchez et al., 2017). The phenomenon of gradually increasing microbial diversity during metamorphosis aligns with that seen in Salamanders (Hartmann et al., 2023). Additionally, a decrease in skin microbiota after the completion of metamorphosis has been observed in Anaxyrus boreas, demonstrating a consistent pattern (Kimball, 2016). After comprehensive analysis, the primary reasons for the changes in diversity can be summarized as follows. Firstly, there is a significant change in the habitat of amphibians from aquatic to terrestrial environments during metamorphosis. Secondly, during this developmental period, there are proven to be significant alterations in the skin tissue structure of these organisms (Kinoshita et al., 1986). As development progresses, the number of glands in the skin increases significantly (Fox, 1987; Rosenberg and Warburg, 1978), and the phagocytes and fibroblasts in the skin exhibit higher activity (Takahama et al., 1992). Furthermore, there are substantial changes in the immune system during amphibian development (Pollet, 2010). Studies have also demonstrated that the immune system of amphibian tadpoles differs from that of adults (Rollins-Smith, 1998), with tadpoles only being able to reject a limited number of transplanted antigens (Pasquier et al., 2000). Following the completion of metamorphosis, the number of lymphocytes in young frogs decreases by approximately 40% (Pasquier et al., 2000), but ultimately, the transformation from lymphoid tissue to lymphoid organs is completed (Manning and Horton, 1969). The antimicrobial peptides secreted by the skin of Litoria ewingii gradually increase in antibacterial activity as they develop, and their composition tends to diversify (Schadich et al., 2009).
Beta diversity statistics under the Bray-Curtis distance are based on shared species for comparison and are sensitive to changes in taxonomic abundance (Ricotta and Pavoine, 2022). The findings of this research demonstrate significant differentiation in skin microbiota across various developmental stages, but the LTS and FTS groups showing high similarity. It indicates that skin microbes evolve with the development of the organisms. Despite disturbances faced by amphibian skin microorganisms from factors like the environment, pathogenic microorganisms, and ecological imbalances (Lopez et al., 2017; Tong et al., 2023), it is the interplay between the host’s immune system and symbiotic flora that safeguards the host’s health (Hou et al., 2022). The symbiotic flora aids in repelling external pathogenic bacteria, while the immune system ensures the survival dominance of the symbiotic flora (Lopez et al., 2017; Woodhams et al., 2020). The early developmental stage of Q. spinosa is a critical period for the development and refinement of various physiological functions of the body (such as digestion, immunity, respiration, and blood circulation) (Hou et al., 2022; Xu et al., 2024a). This period is when microorganisms and hosts establish symbiotic relationships, but it is also vulnerable to bacterial imbalances and invasion by pathogenic bacteria, particularly during the critical landing period (LFS) (Long et al., 2020). This study underscores the significance of comprehending the evolution of skin commensal flora throughout development to inform healthy farming practices, health evaluations, and amphibian conservation efforts.
Similar to other amphibians (Hou et al., 2022; Jiang et al., 2022; Karlsen et al., 2017; Long et al., 2020; Zhu et al., 2024), Proteobacteria, Firmicutes, and Bacteroidota are the predominant microbiota of the skin, albeit with varying abundance across different phyla. Proteobacteria harbor many opportunistic pathogens, while most bacterial genera in Firmicutes are considered beneficial, helping maintain the acidic environment of the host skin and producing active substances through fermentation to resist pathogenic microorganisms (Huang et al., 2020; Xu et al., 2024a,b). Bacteroidota members are closely associated with the host’s immune and metabolic processes (Federici et al., 2015). This study found that the abundance of Proteobacteria significantly increased with development in Q. spinosa, indicating heightened metabolism and immunity in the early stages. The abundance of Proteobacteria among skin microorganisms decreased notably during the tadpole stage (TS, LTS and FTS group) and increased significantly post-landing (LFS group), while the abundance of Firmicutes exhibited the opposite trend. Similar patterns were observed in the development of other amphibians (Fieschi-Meric et al., 2023). We hypothesize that continuous adaptation to the aqueous environment leads to the colonization of beneficial symbiotic microorganisms on the skin. However, changes in habitat and skin functions alter the original ecology, resulting in a re-selection of host-microorganism interactions.
Interestingly, our results show that Acinetobacter sp. increased rapidly between the forelimb tadpoles and the landing frogs. In cases of disease, Acinetobacter sp. is typically associated with skin infections, ulcers, and necrosis, among other symptoms (Zhu et al., 2024). It is also commonly one of the primary causative agents of pneumonia, meningitis, and septicemia in frogs (Lee et al., 2017). In contrast, Acinetobacter sp. have a significant inhibitory effect on the pathogenic fungi of amphibian “chytridiomycosis” (Fieschi-Meric et al., 2023), and also support and protect microbial communities in biological systems, which are important for biofilm development (Zhu et al., 2024). We speculate that the enrichment of Acinetobacter sp. in the TS group may be for pre-skin development and colonization of symbiotic microbial communities. Conversely, the enrichment in the LFS group may be to resist colonization by Batrachochytrium dendrobatidis and to reorganize the microbial community. Thus, the developmental window between forelimb tadpoles and land frog could be key in establishing host defences against pathogens.
LEfSe analysis revealed several genera with significant differences in abundance among groups, including Acinetobacter, Novosphingobium, Limnohabitans, Bacteroides, Bacillus, Lactobacillus, Muribaculaceae, Pseudomonas, Luteolibacter, Comamonas, Chryseobacterium and Allorhizobium_Neorhizobium_Pararhizobium_Rhizobium. In aquaculture production, Bacillus, Bacteroides and Lactobacillus were commonly used as probiotics to maintain the health of farmed animals (Liu et al., 2023; Xu et al., 2024a; Zhu et al., 2024). For instance, B. subtilis can inhibit the growth of Vibro alginolyticus and A. bouvetii (Qi et al., 2020; Song et al., 2009). B. velezensis has an antagonistic effect on the pathogenic bacteria of aquatic conditions, including Edwardsiella tarda, A. veronii and A. hydrophila (Gao, 2019). The feed adding B. amyloliquefaciens and B. subtilis also have the role of enhancing the immunity of breeding objects and promoting growth (Lai et al., 2020; Lu et al., 2021). Bacteroides can inhibit harmful bacteria, provide more active protease and lipase, and promote feed utilization (Christensen and Bruggemann, 2014). Lactobacillus isolated from bullfrogs have been found to regulate host immune function and combat pathogens (Pereira et al., 2018). The co-occurrence network analysis revealed variations in skin microbial ecology at different stages, influencing the evaluation of host status through the identification of beneficial bacteria indicated by LEfSe. This information can be valuable for guiding aquaculture and preventive measures.
Existing reports indicate that bacterial diseases in the Chinses spiny frog often result from the synergistic action of multiple pathogenic bacteria, manifesting as a single disease with multiple symptoms or multiple diseases with a single symptom (Jiang, 2022). Based on outbreak causes and prevention and control experiences in breeding farms, the following points should be taken into consideration for the prevention and control of opportunistic pathogens during development: (1) Strict epidemic prevention measures should be implemented for the entry and exit of breeding personnel and animals into and out of the breeding area, even extending to the specific breeding ponds to prevent cross-infection of microorganisms. At the same time, mixing of breeding animals from different ponds should be prohibited. (2) Regular microbial prevention and control measures should be taken during the metamorphosis stages (LTS, FTS, and LFS), including the breeding area, related facilities and equipment, and regular incorporation of beneficial bacteria into feed. (3) Individuals or groups showing signs of illness should be promptly isolated, and the isolation ponds should be strictly disinfected.
Symbiotic microorganisms residing on the host skin are vital components that play a crucial role in defending against pathogen invasion, regulating pH levels, influencing metabolism, and supporting immune system development (Abarca et al., 2018; Harris-Tryon and Grice, 2022; Pereira et al., 2018). In this study, PICRUSt was utilized to evaluate the predicted functions of the skin microbiota. Variations in skin microbial function are observed over develop, with a particular emphasis on amino acid metabolism, carbohydrate metabolism, cell motility, lipid metabolism, membrane transport, metabolism of terpenoids and polyketides, and xenobiotics biodegradation and metabolism. The results indicate that skin microbes play a crucial role in metabolic functions, while the enrichment of cell motility and membrane transport pathways suggests that the skin symbiotic flora also aids in the developmental refinement of the host’s skin.
In this study, we systematically examined the skin microorganisms present during metamorphosis in Q. spinosa, Echinococcus auratus. Our findings demonstrate that the composition and diversity of skin microorganisms undergo changes in response to developmental and environmental factors. The dominant phyla of skin microorganisms identified were Proteobacteria, Firmicutes and Bacteroidota, with their abundance being influenced by the host’s developmental stage. Key bacterial indicators highlighted by LEfSe included Bacteroides, Bacillus, Lactobacillus, and Acinetobacter. Moreover, the functions performed by skin microbes varied significantly during development, primarily focusing on metabolic activities. The comprehensive analysis suggests that the transition from forelimb tadpole to frog landing poses a high risk of pathogenic microorganism invasion, making it a critical period for successful metamorphosis in Q. spinosa. This study underscores the importance of host skin microbes in amphibian development and habitat, offering potential implications for managing amphibian metamorphosis in relation to microbial communities.
The merged sequences are deposited in the NCBI SRA database under accession number PRJNA1058082.
The animal study was approved by Institutional Animal Care and Use Committee (IACUC) ethical guidelines of Hunan Agricultural University. The study was conducted in accordance with the local legislation and institutional requirements.
JH: Formal analysis, Investigation, Writing – original draft. YT: Software, Writing – review & editing. YFH: Data curation, Writing – review & editing. HL: Methodology, Writing – review & editing. DL: Resources, Supervision, Writing – review & editing. XL: Validation, Writing – review & editing. JL: Visualization, Writing – review & editing. YH: Conceptualization, Writing – review & editing. JX: Conceptualization, Funding acquisition, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by National Natural Science Foundation of China grant/award number [31772832]; National Key Research and Development Program of China grant/award number [2019YFD0900603]; Hunan Provincial Modern Agricultural Research System grant/award number [2019–105].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1453617/full#supplementary-material
Abarca, J. G., Vargas, G., Zuniga, I., Whitfield, S. M., Woodhams, D. C., Kerby, J., et al. (2018). Assessment of bacterial communities associated with the skin of Costa Rican amphibians at La Selva Biological Station. Front. Microbiol. 9:2001. doi: 10.3389/fmicb.2018.02001
Bletz, M. C., Kelly, M., Sabino-Pinto, J., Bales, E., Van Praet, S., Bert, W., et al. (2018). Disruption of skin microbiota contributes to salamander disease. Proc. Biol. Sci. 285:20180758. doi: 10.1098/rspb.2018.0758
Bokulich, N. A., Kaehler, B. D., Rideout, J. R., Dillon, M., Bolyen, E., Knight, R., et al. (2018). Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2's q2-feature-classifier plugin. Microbiome 6:90. doi: 10.1186/s40168-018-0470-z
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brown, D. D., and Cai, L. (2007). Amphibian metamorphosis. Dev. Biol. 306, 20–33. doi: 10.1016/j.ydbio.2007.03.021
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Chen, L. H., Lin, C. H., Siao, R. F., and Wang, L. C. (2022). Aeromonas hydrophila induces skin disturbance through mucosal microbiota dysbiosis in striped catfish (Pangasianodon hypophthalmus). mSphere 7:e19422. doi: 10.1128/msphere.00194-22
Christensen, G. J., and Bruggemann, H. (2014). Bacterial skin commensals and their role as host guardians. Benefic. Microbes 5, 201–215. doi: 10.3920/BM2012.0062
Costa, S., Lopes, I., Proenca, D. N., Ribeiro, R., and Morais, P. V. (2016). Diversity of cutaneous microbiome of Pelophylax perezi populations inhabiting different environments. Sci. Total Environ. 572, 995–1004. doi: 10.1016/j.scitotenv.2016.07.230
Douglas, G. M., Maffei, V. J., Zaneveld, J. R., Yurgel, S. N., Brown, J. R., Taylor, C. M., et al. (2020). PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38, 685–688. doi: 10.1038/s41587-020-0548-6
Federici, E., Rossi, R., Fidati, L., Paracucchi, R., Scargetta, S., Montalbani, E., et al. (2015). Characterization of the skin microbiota in Italian stream frogs (Rana italica) infected and uninfected by a cutaneous parasitic disease. Microbes Environ. 30, 262–269. doi: 10.1264/jsme2.ME15041
Fieschi-Meric, L., van Leeuwen, P., Denoel, M., and Lesbarreres, D. (2023). Encouraging news for in situ conservation: translocation of salamander larvae has limited impacts on their skin microbiota. Mol. Ecol. 32, 3276–3289. doi: 10.1111/mec.16914
Fox, H. (1987). On the fine structure of the skin of larval, juvenile and adult Ichthyophis (Amphibia, Caecilia). Zoomorphology 107, 67–76. doi: 10.1007/BF00312115
Gao, Y. (2019). Isolation, identification and application of a Bacillus velezensis LF01 strain. Shanghai Ocean University, Shanghai, pp. 79. doi: 10.27314/d.cnki.gsscu.2019.000012
Gosner, K. L. (1960). A simplified table for staging anuran embryos and larvae with notes on identification. Herpetologica 16, 183–190. Available at: http://www.jstor.org/stable/3890061
Harris-Tryon, T. A., and Grice, E. A. (2022). Microbiota and maintenance of skin barrier function. Science 376, 940–945. doi: 10.1126/science.abo0693
Hartmann, A. M., McGrath-Blaser, S. E., Colón-Piñeiro, Z., and Longo, A. (2023). Ontogeny drives shifts in skin bacterial communities in facultatively Paedomorphic salamanders. Microbiol-Sgm 169:001399. doi: 10.1099/mic.0.001399
Hou, J., Long, J., Xiang, J., Pan, W., Li, D., and Liu, X. (2022). Ontogenetic characteristics of the intestinal microbiota of Quasipaa spinosa revealed by 16S rRNA gene sequencing. Lett. Appl. Microbiol. 75, 1182–1192. doi: 10.1111/lam.13786
Hou, J., Xiang, J., Li, D., and Liu, X. (2023). Prediction of potential suitable distribution areas of Quasipaa spinosa in China based on MaxEnt optimization model. Biology-Basel 12:366. doi: 10.3390/biology12030366
Hu, C., Huang, Z., Liu, M., Sun, B., Tang, L., and Chen, L. (2021). Shift in skin microbiota and immune functions of zebrafish after combined exposure to perfluorobutanesulfonate and probiotic Lactobacillus rhamnosus. Ecotoxicol. Environ. Saf. 218:112310. doi: 10.1016/j.ecoenv.2021.112310
Huang, H., Zhou, P., Chen, P., Xia, L., Hu, S., Yi, G., et al. (2020). Alteration of the gut microbiome and immune factors of grass carp infected with Aeromonas veronii and screening of an antagonistic bacterial strain (Streptomyces flavotricini). Microb. Pathog. 143:104092. doi: 10.1016/j.micpath.2020.104092
Jiang, Y. (2022). Characterization of symbiotic microbiota of healthy and diseased Paa spinosa at the GS46 period. Master Thesis, Hunan Agricultural University, Changsha, China. Available at: https://link.oversea.cnki.net/doi/10.27136/d.cnki.ghunu.2022.000093 (overseas); https://link.cnki.net/doi/10.27136/d.cnki.ghunu.2022.000093 (domestic).
Jiang, H., Luo, S., Zhou, J., Huang, W., Li, L., Zhang, X., et al. (2022). Skin microbiota was altered in crocodile lizards (Shinisaurus crocodilurus) with skin ulcer. Front. Vet. Sci. 9:817490. doi: 10.3389/fvets.2022.817490
Jimenez, R. R., Alvarado, G., Estrella, J., and Sommer, S. (2019). Moving beyond the host: unraveling the skin microbiome of endangered Costa Rican amphibians. Front. Microbiol. 10:2060. doi: 10.3389/fmicb.2019.02060
Karlsen, C., Ottem, K. F., Brevik, O. J., Davey, M., Sorum, H., and Winther-Larsen, H. C. (2017). The environmental and host-associated bacterial microbiota of Arctic seawater-farmed Atlantic salmon with ulcerative disorders. J. Fish Dis. 40, 1645–1663. doi: 10.1111/jfd.12632
Kimball, A. K. (2016). Skin microbiome composition and diversity across Anaxyrus boreas developmental stages. Undergraduate Honors Theses, University of Colorado, Colorado, USA, p. 1175. Available at: https://scholar.colorado.edu/honr_theses/1175.
Kinoshita, T., Sasaki, F., and Watanabe, K. (1986). Regional specificity of anuran larval skin during metamorphosis: Transdifferentiation of tadpole tail-epidermis. J. Exp. Zool. 238, 201–210. doi: 10.1002/jez.1402380210
Knutie, S. A., Wilkinson, C. L., Kohl, K. D., and Rohr, J. R. (2017). Early-life disruption of amphibian microbiota decreases later-life resistance to parasites. Nat. Commun. 8:86. doi: 10.1038/s41467-017-00119-0
Kueneman, J. G., Parfrey, L. W., Woodhams, D. C., Archer, H. M., Knight, R., and McKenzie, V. J. (2014). The amphibian skin-associated microbiome across species, space and life history stages. Mol. Ecol. 23, 1238–1250. doi: 10.1111/mec.12510
Lai, Y., Luo, M., and Zhu, F. (2020). Dietary Bacillus amyloliquefaciens enhance survival of white spot syndrome virus infected crayfish. Fish Shellfish Immunol. 102, 161–168. doi: 10.1016/j.fsi.2020.04.033
Lee, C. R., Lee, J. H., Park, M., Park, K. S., Bae, I. K., Kim, Y. B., et al. (2017). Biology of Acinetobacter baumannii: pathogenesis, antibiotic resistance mechanisms, and prospective treatment options. Front. Cell Infect. Microbiol. 7:55. doi: 10.3389/fcimb.2017.00055
Liu, C., Cui, Y., Li, X., and Yao, M. (2021). Microeco: an R package for data mining in microbial community ecology. FEMS Microbiol. Ecol. 97:fiaa255. doi: 10.1093/femsec/fiaa255
Liu, Z. Y., Yang, H. L., Wei, C. Y., Cai, G. H., Ye, J. D., Zhang, C. X., et al. (2023). Commensal Bacillus siamensis LF4 induces antimicrobial peptides expression via TLRs and NLRs signaling pathways in intestinal epithelial cells of Lateolabrax maculatus. Fish Shellfish Immunol. 134:108634. doi: 10.1016/j.fsi.2023.108634
Long, J., Xiang, J., He, T., Zhang, N., and Pan, W. (2020). Gut microbiota differences during metamorphosis in sick and healthy giant spiny frogs (Paa spinosa) tadpoles. Lett. Appl. Microbiol. 70, 109–117. doi: 10.1111/lam.13251
Longo, A. V., and Zamudio, K. R. (2017). Environmental fluctuations and host skin bacteria shift survival advantage between frogs and their fungal pathogen. ISME J. 11, 349–361. doi: 10.1038/ismej.2016.138
Lopez, M. F., Rebollar, E. A., Harris, R. N., Vredenburg, V. T., and Hero, J. (2017). Temporal variation of the skin bacterial community and Batrachochytrium dendrobatidis infection in the terrestrial cryptic frog Philoria loveridgei. Front. Microbiol. 8:2535. doi: 10.3389/fmicb.2017.02535
Lu, Z., Yang, Q., Lin, Q., Wu, J., Du, H., and Zhou, C. (2021). Effects of Bacillus spp. on growth, gut microbiota compositions and immunity of sea cucumber (Apostichopus japonicus). Aquac. Res. 52, 3978–3990. doi: 10.1111/are.15241
Manning, M. J., and Horton, J. D. (1969). Histogenesis of lymphoid organs in larvae of the South African clawed toad, Xenopus laevis (Daudin). J. Embryol. Exp. Morphol. 22, 265–277. doi: 10.1242/dev.22.2.265
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 1:2096995. doi: 10.14806/ej.17.1.200
Martinez-Ugalde, E., Avila-Akerberg, V., Gonzalez, M. T., Vazquez, T. M., Zavala, H. D., Anaya-Morales, S. L., et al. (2022). The skin microbiota of the axolotl Ambystoma altamirani is highly influenced by metamorphosis and seasonality but not by pathogen infection. Anim. Microbiome 4:63. doi: 10.1186/s42523-022-00215-7
Muletz-Wolz, C. R., Fleischer, R. C., and Lips, K. R. (2019). Fungal disease and temperature alter skin microbiome structure in an experimental salamander system. Mol. Ecol. 28, 2917–2931. doi: 10.1111/mec.15122
Mulla, L., and Hernandez-Gomez, O. (2023). Wildfires disturb the natural skin microbiota of terrestrial salamanders. Environ. Microbiol. 25, 2203–2215. doi: 10.1111/1462-2920.16452
Ning, M. (2022). Diversity of skin microbiota of Hynobius maoershanensis at different development stages. Guangxi Normal University, Guilin, pp. 100. doi: 10.27036/d.cnki.ggxsu.2022.001716
Pasquier, L., Robert, J., Courtet, M., and Mußmann, R. (2000). B-cell development in the amphibian. Immunol. Rev. 175, 201–213. doi: 10.1111/j.1600-065X.2000.imr017501.x
Pereira, S. A., Jerônimo, G. T., Marchiori, N. C., Oliveira, H. M., Jesus, G. F. A., Schmidt, E. C., et al. (2018). Tadpoles fed supplemented diet with probiotic bacterium isolated from the intestinal tract of bullfrog Lithobates catesbeianus: Haematology, cell activity and electron microscopy. Microb. Pathog. 114, 255–263. doi: 10.1016/j.micpath.2017.11.033
Pollet, N. (2010). Expression of immune genes during metamorphosis of: a survey. Front. Biosci. 15, 348–358. doi: 10.2741/3624
Qi, J., Liu, Y., Tong, W., Xiao, L., Zhao, Z., and Song, Z. (2020). Isolation and identification of the pathogen of rot skin disease in Rana catesbiana and its drug sensitive test. J. South. Agric. 51, 461–469.
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Ricotta, C., and Pavoine, S. (2022). A new parametric measure of functional dissimilarity: bridging the gap between the bray-Curtis dissimilarity and the Euclidean distance. Ecol. Model. 466:109880. doi: 10.1016/j.ecolmodel.2022.109880
Rollins-Smith, L. A. (1998). Metamorphosis and the amphibian immune system. Immunol. Rev. 166, 221–230. doi: 10.1111/j.1600-065X.1998.tb01265.x
Rosenberg, M., and Warburg, M. R. (1978). Changes in structure of ventral epidermis of Rana ridibunda during metamorphosis. Cell Tissue Res. 195, 111–122. doi: 10.1007/BF00233680
Sadeghi, J., Chaganti, S. R., Johnson, T. B., and Heath, D. D. (2023). Host species and habitat shape fish-associated bacterial communities: Phylosymbiosis between fish and their microbiome. Microbiome 11:258. doi: 10.1186/s40168-023-01697-6
Sanchez, E., Bletz, M. C., Duntsch, L., Bhuju, S., Geffers, R., Jarek, M., et al. (2017). Cutaneous bacterial communities of a poisonous salamander: a perspective from life stages, body parts and environmental conditions. Microb. Ecol. 73, 455–465. doi: 10.1007/s00248-016-0863-0
Schadich, E., Cole, A. L. J., Squire, M., and Mason, D. (2009). Skin peptides of different life stages of Ewing's tree frog. J. Exp. Zool. A Ecol. Genet. Physiol. 313A, 532–537. doi: 10.1002/jez.582
Scheele, B. C., Pasmans, F., Skerratt, L. F., Berger, L., Martel, A., Beukema, W., et al. (2019). Amphibian fungal panzootic causes catastrophic and ongoing loss of biodiversity. Science 363, 1459–1463. doi: 10.1126/science.aav0379
Schommer, N. N., and Gallo, R. L. (2013). Structure and function of the human skin microbiome. Trends Microbiol. 21, 660–668. doi: 10.1016/j.tim.2013.10.001
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, WS., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60. doi: 10.1186/gb-2011-12-6-r60
Song, Z., Xiao, M., Wu, J., and Qiu, J. (2009). Isolation, identification and selection of preventable drugs of one strain of pathogen from rotten-mouth-diseases in Rana chensinensis. J. Anhui Agric. Sci. 37, 16884–16885. doi: 10.13989/j.cnki.0517-6611.2009.34.147
Takahama, H., Kinoshita, T., and Sasaki, F. (1992). Structural and endocytotic differences of fibroblasts and macrophages in the tail fin of amphibian larvae during metamorphosis. Arch. Histol. Cytol. 55, 437–448. doi: 10.1679/aohc.55.437
Tong, Q., Cui, L. Y., Hu, Z. F., Du, X. P., Abid, H. M., and Wang, H. B. (2020). Environmental and host factors shaping the gut microbiota diversity of brown frog Rana dybowskii. Sci. Total Environ. 741:140142. doi: 10.1016/j.scitotenv.2020.140142
Tong, Q., Dong, W., Xu, M., Hu, Z., Guo, P., Han, X., et al. (2023). Characteristics and a comparison of the gut microbiota in two frog species at the beginning and end of hibernation. Front. Microbiol. 14:1057398. doi: 10.3389/fmicb.2023.1057398
Wei, D., Cheng, Y., Liao, W., Yu, Q., Shi, J., Xia, X., et al. (2023). Composition and function of the skin microbiota were altered of red leg syndrome in cultured bullfrog (Rana catesbeiana). Aquacult. Rep. 29:101487. doi: 10.1016/j.aqrep.2023.101487
Woodhams, D. C., Rollins-Smith, L. A., Reinert, L. K., Lam, B. A., Harris, R. N., Briggs, C. J., et al. (2020). Probiotics modulate a novel amphibian skin defense peptide that is antifungal and facilitates growth of antifungal Bacteria. Microb. Ecol. 79, 192–202. doi: 10.1007/s00248-019-01385-9
Xu, Y., Jiang, Y., Hou, J., Li, J., Feng, X., Liu, X., et al. (2024a). Comparative profiling of the skin and gut microbiota during metamorphosis in Chinese spiny frogs (Quasipaa spinosa) highlights microbial communities involved in disease pathogenesis. Aquaculture 585:740726. doi: 10.1016/j.aquaculture.2024.740726
Xu, Y., Jiang, Y., Hou, J., Li, J., Feng, X., Liu, X., et al. (2024b). Dysplasia and torticollis on the skin microbiota of Chinese spiny frog (Quasipaa spinosa) at the stage of complete metamorphosis. Acta Hydrobiol. Sinica, 1–10. doi: 10.7541/2024.2023.0303
Yu, X., Zheng, R., Zhang, J., Shen, B., and Dong, B. (2014). Genetic polymorphism of major histocompatibility complex class IIB alleles and pathogen resistance in the giant spiny frog Quasipaa spinosa. Infect. Genet. Evol. 28, 175–182. doi: 10.1016/j.meegid.2014.09.028
Zhao, X., Yang, Y., Feng, K., Wang, X., Liu, B., Xie, G., et al. (2021). Self-regulating microbiome networks ensure functional resilience of biofilms in sand biofilters during manganese load fluctuations. Water Res. 188:116473. doi: 10.1016/j.watres.2020.116473
Keywords: developmental stage, Quasipaa spinosa, cutaneous bacterial communities, 16S amplicon sequencing, co-occurrence network
Citation: Hou J, Tan Y, Huang Y, Li H, Li D, Liu X, Li J, Hu Y and Xiang J (2024) Skin microbiota during metamorphosis of Quasipaa spinosa: guidance for maintaining mucosal symbiotic microbial flora homeostasis in early life of frogs. Front. Microbiol. 15:1453617. doi: 10.3389/fmicb.2024.1453617
Edited by:
Lifeng Zhu, Nanjing University of Chinese Medicine, ChinaReviewed by:
Jianfei Lu, Ningbo University, ChinaCopyright © 2024 Hou, Tan, Huang, Li, Li, Liu, Li, Hu and Xiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yazhou Hu, aHV5YXpob3VAaHVuYXUuZWR1LmNu; Jianguo Xiang, MjUwNzQ2NzIxMUBxcS5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.