 Taranjot Kaur
Taranjot Kaur Amanda M.V. Brown
Amanda M.V. Brown- Department of Biological Sciences, Texas Tech University, Lubbock, TX, United States
Bioinformatics sequence data mining can reveal hidden microbial symbionts that might normally be filtered and removed as contaminants. Data mining can be helpful to detect Wolbachia, a widespread bacterial endosymbiont in insects and filarial nematodes whose distribution in plant-parasitic nematodes (PPNs) remains underexplored. To date, Wolbachia has only been reported a few PPNs, yet nematode-infecting Wolbachia may have been widespread in the evolutionary history of the phylum based on evidence of horizontal gene transfers, suggesting there may be undiscovered Wolbachia infections in PPNs. The goal of this study was to more broadly sample PPN Wolbachia strains in tylenchid nematodes to enable further comparative genomic analyses that may reveal Wolbachia’s role and identify targets for biocontrol. Published whole-genome shotgun assemblies and their raw sequence data from 33 Meloidogyne spp. assemblies, seven Globodera spp. assemblies, and seven Heterodera spp. assemblies were analyzed to look for Wolbachia. No Wolbachia was found in Meloidogyne spp. and Globodera spp., but among seven genome assemblies for Heterodera spp., an H. schachtii assembly from the Netherlands was found to have a large Wolbachia-like sequence that, when re-assembled from reads, formed a complete, circular genome. Detailed analyses comparing read coverage, GC content, pseudogenes, and phylogenomic patterns clearly demonstrated that the H. schachtii Wolbachia represented a novel strain (hereafter, denoted wHet). Phylogenomic tree construction with PhyloBayes showed wHet was most closely related to another PPN Wolbachia, wTex, while 16S rRNA gene analysis showed it clustered with other Heterodera Wolbachia assembled from sequence databases. Pseudogenes in wHet suggested relatedness to the PPN clade, as did the lack of significantly enriched GO terms compared to PPN Wolbachia strains. It remains unclear whether the lack of Wolbachia in other published H. schachtii isolates represents the true absence of the endosymbiont from some hosts.
Introduction
Microbial symbioses are essential components of ecosystems that play major roles in evolution and ecology. These symbiotic interactions include mutualism, parasitism, and commensalism (Fransolet et al., 2012; Goffredi, 2010; Lamelas et al., 2011; Lefoulon et al., 2016; Moya et al., 2014). The bacterial endosymbiont Wolbachia is widespread in terrestrial arthropods, infecting approximately 40% of terrestrial arthropod species (Zug and Hammerstein, 2012), and is almost universally present in filarial nematodes as an obligate mutualist (Lefoulon et al., 2016; Slatko et al., 2010; Taylor et al., 2013). It has been discovered in several plant-parasitic nematodes (hereafter, PPNs) (Brown, 2018; Brown et al., 2016; Haegeman et al., 2009; Kaur and Brown, 2024). Wolbachia strains vary in role and can range from parasitic to mutualistic with their hosts (Gill et al., 2014). Except for filarial nematodes, few other nematodes have been reported to harbor Wolbachia, with only a few tylenchid reported as Wolbachia hosts: Radopholus similis (Haegeman et al., 2009), Radopholus arabocoffeae (Haegeman et al., 2009), Pratylenchus penetrans (Brown, 2018; Denver et al., 2016), a fourth PPN nematode (Weyandt et al., 2022), and an entomopathogenic tylenchid, Howardula sp. (Dudzic et al., 2022). However, it is unclear whether the apparent rarity of Wolbachia in non-filarial nematodes, including PPNs, is just a result of undersampling. Interestingly, a recent highly sensitive PCR-based survey suggests that Wolbachia may be widespread in other nematodes (Kaur and Brown, 2024).
It has been hypothesized that nematode-infecting Wolbachia may have been widespread at one point as there is evidence of horizontal gene transfers leaving Wolbachia-like fragments in nematode genomes across the nematode phylogeny (Dunning Hotopp, 2011; Husnik and McCutcheon, 2017; Koutsovoulos et al., 2014; McNulty et al., 2010). Furthermore, past phylogenomic analyses placed PPN Wolbachia strains at the origin of the Wolbachia phylogeny, at the base of other supergroups (Brown et al., 2016; Scholz et al., 2020) and with long branches suggesting longstanding symbioses in the order Tylenchida (Brown et al., 2016; Dudzic et al., 2022) and supporting Wolbachia’s emergence early in the diversification of terrestrial Ecdysozoa hosts, during a time of expanding plant-diet specialization. The conservation of iron/heme biosynthesis and other functions at the root of the clade also suggests a potential facultative nutritional mutualism (Brown et al., 2016; Weyandt et al., 2022). Whereas extant Wolbachia strains from arthropods can cause host reproductive manipulations such as cytoplasmic incompatibility (CI), parthenogenesis induction, male-killing, or genetic male feminization (Bing et al., 2023; Correa and Ballard, 2016; Sanaei et al., 2020; Werren et al., 2008; Werren, 1997; Zhu et al., 2020), Wolbachia strains from filarial nematodes and several other hosts act as beneficial or obligate mutualists (Hosokawa et al., 2010; Lefoulon et al., 2016). To date, however, the role of PPN Wolbachia remains unclear.
Among the PPNs, which are estimated to cost approximately ~$100 billion in global crop losses annually (Nicol et al., 2011), some of the most devastating species (Jones et al., 2013) have been found to host Wolbachia, raising interest in potential future Wolbachia-based controls. To evaluate this potential, it is important to understand the prevalence, diversity, and distribution of PPN Wolbachia. An early study of Wolbachia in Radopholus similis showed 100% prevalence, which might indicate a mutualist role for this strain (Haegeman et al., 2009). However, a more recent genome data mining study showed a discontinuous distribution of Wolbachia in Radopholus sp. and Pratylenchus spp. across North America, South America, and Africa, suggesting a non-obligatory role in these hosts (Wasala et al., 2023). The presence of the Wolbachia strain wPpe in Pratylenchus spp. was positively correlated with female-biased sex ratios, suggesting a potential role in host sex ratio modulation via an unknown mechanism (Wasala et al., 2019). Yet, with few PPN Wolbachia characterized to date, it is difficult to interpret these patterns.
Therefore, to understand the prevalence, diversity, and role of Wolbachia in PPNs, we searched genomic sequence data from public databases for hidden Wolbachia sequences, focusing on the top three most damaging PPNs, namely, Meloidogyne spp., Globodera spp., and Heterodera spp. (Jones et al., 2013). We screened whole-genome shotgun (WGS) assemblies and their sequence read archive (SRA) and recovered a new Wolbachia genome from a Heterodera schachtii WGS assembly (designated as wHet). We analyzed its phylogenetic place and annotated its genomic features and found functional enrichment suggesting it may be a facultative mutualist with a role in heme homeostasis.
Materials and methods
SRA data screening
We downloaded WGS assemblies and their SRA reads for Meloidogyne spp. (33 WGS assemblies), Globodera spp. (seven WGS assemblies), and Heterodera spp. (seven WGS assemblies). The SRA data and the assemblies were analyzed for the presence of Wolbachia-like sequences using a two-step approach with blastn in Blast+ v2.10.1 (Camacho et al., 2009). In the first step, SRA data and the assemblies were compared to a custom database of Wolbachia genome sequences using blastn. This custom database was made by compiling known PPN Wolbachia genomic sequences into a single file and using makeblastdb to make these a nucleotide sequence reference database to search against using blastn. In the second step, the blast hits from first step were used as queries for the second blastn analysis against the complete nt reference database from NCBI. Contigs were classified as plant-parasitic nematode (PPN) Wolbachia if their top blast hits showed a closer match to Wolbachia strains from PPN (wPpe, wTex, Pp_GH2, Pp_Cr, Rs_N1, Rs_14, Rs_5) compared to other non-plant-parasitic nematode strains.
Detection of nematode hosts
To confirm the host nematode species and look for contaminating nematodes or cryptic species in the assembly, blastn against a custom nematode COI gene database was performed.
Genome annotation and polishing by read mapping
The coverage of the Wolbachia-matching regions and flanking regions was assessed using the pileup.sh script1 from SRA Nanopore DNA reads derived from the same project as the Wolbachia-containing WGS assembly (NCBI accession PRJNA767548; reads SRR16146220-SRR16146534 and SRR16675965-SRR16675966 and SRR28675229-SRR28675543) mapped to the WGS Wolbachia scaffold using the BWA software package with the BWA-MEM algorithm (Li and Durbin, 2009). Raw reads that mapped to Wolbachia (as a sam file) were visualized in Geneious Prime and then, removing the WGS-derived Wolbachia reference, the reads spanning gap regions were assembled de novo and the final consensus contig was used to extend and fill-in the ends of the sequence containing uncalled bases (N’s) in the junctions between Wolbachia and non-Wolbachia regions of the contigs to assess a possible overlap that would create a circular genome with overlapping ends. CheckM, which looks for the genes in the genome using hmmer (Finn et al., 2011; Parks et al., 2015), was used to analyze the completeness and contamination of the assembled genome. In addition, BUSCO was also used to compute the completeness of the assembled genome (Manni et al., 2021). Prokka 1.14.6 was used to annotate the genes in the endosymbiont assembly as well as the genomes used for comparative genomics (Seemann, 2014).
Comparative genomics and pangenome analysis
Genome assemblies of Wolbachia strains from insects, filarial nematodes, and PPNs were downloaded from NCBI, for comparison to the newly discovered Wolbachia strain. Representatives included published Wolbachia genomes from supergroups A, B, C, D, E, F, J, M, S, T, and V and supergroup L (Table 1) and an out-group Candidatus Mesenet longicola.

Table 1. Description of genomes used for comparative genomics and NCBI accession numbers.
To detect orthologs, we used Roary (Page et al., 2015), and pangenome and core genome comparisons were performed based on the gene_presence_absence.csv file obtained in Roary outputs, depicted using the online Venn drawing tool.2 Venn diagrams were made to identify the overlap and differences between the gene sets and infer the biological significance of shared and unique genes in the new Wolbachia strain compared to other PPN Wolbachia genomes and Wolbachia genomes from filarial nematodes and insects.
Genomic features, such as genome size, number of proteins versus hypothetical proteins, number of tRNA and rRNA genes, % GC content, number of pseudogenes, number of ankyrin genes, transposases, phage-related genes, and coding density percent, of new Wolbachia strain were compared to members of supergroup L, including wPpe (from Pratylenchus penetrans, Oregon, USA), Pp_Cr (from Pratylenchus penetrans, Costa Rica), Pp_GH2 (from Pratylenchus penetrans, Oregon, USA), Rs_N1 (from Radopholus similis, Nigeria), Rs_14 (from Radopholus similis, Colombia), Rs_5 (from Radopholus similis, Uganda), wTex (from unknown PPN, predicted Helicotylenchus sp.), and wMel (from Drosophila melanogaster). Pseudofinder was used to detect the pseudogenes and evolutionary interference in endosymbiont genomes (Syberg-Olsen et al., 2022). The number of pseudogenes was calculated using wMel as a reference genome as it had the highest completeness score among strains (CheckM 99.79%, BUSCO 99.5%). Pseudogene indicators were classified as frameshifts, missing stop codons, and internal stop codons.
Phylogenomic analysis
The core gene alignment file (core_gene_alignment.aln) from Roary was converted to phylip format for phylogenomic analysis with PhyloBayes MPI, which uses the CAT-GTR model to predict long-branch interactions (Lartillot et al., 2013). Two independent chains were run parallelly, and the consensus was obtained by pooling all the trees of the independent chains using the bpcomp package in PhyloBayes MPI. Using the burn-in of 1,000, and sub-sampling every 10 trees, the bpcomp program calculated the largest (maxdiff) and mean (meandiff) discrepancy observed across all bipartitions. The core gene alignment file (core_gene_alignment.aln) from Roary was also imported to the Geneious Prime software for phylogeny construction with MrBayes (Ronquist et al., 2012). For MrBayes, posterior probabilities were reported for supported nodes from Bayesian 50% majority rule, with the GTR+G model with four rate categories. Additional phylogenomic comparisons were performed with RAxML using the GTR+Gamma model with support shown for 100 bootstrap replicates (Stamatakis, 2014). To test the effects of fragmented sequences and sequences with long branches, additional analyses were performed with these sequences removed.
Phylogenetic analysis of 16S rRNA gene sequences from different Wolbachia strains and out-groups was performed in RAxML using the GTR + Gamma model with support from 100 bootstrap replicates (Stamatakis, 2014). In addition, based on previous PCR assays that screened Wolbachia from different nematode populations, a Wolbachia 16S rRNA gene from Helicotylenchus sp. from Florida (Kaur and Brown, 2024) is also included in the phylogenetic analysis. A further 16S rRNA gene was included from a separate project in our laboratory (unpublished) based on an assembly of reads from Heterodera sojae from soybean roots from South Korea (SRR25626476). Phylogenetic analysis of COI genes from Heterodera spp., including COI genes recovered from Nanopore-assembled H. schachtii IRS assembly, was also performed in RAxML using the GTR + Gamma model with support from 100 bootstrap replicates (Stamatakis, 2014).
Comparison with RNA-seq datasets
Based on additional blastn screens of transcriptomic (RNA-seq) SRA datasets from H. schachtii in NCBI, several additional samples with Wolbachia-like matches were analyzed. For these samples, Illumina reads were downloaded and de novo assembled using rnaSPAdes (Bushmanova et al., 2019), and assembled transcripts matching the Wolbachia 16S rRNA gene were aligned and analyzed as described above for phylogenetic analysis.
Gene ontology enrichment analysis
Gene ontology enrichment analysis was performed to interpret the enriched GO terms for new Wolbachia strain against different Wolbachia species from supergroups A, B, C, D, E, F, J, M, S, and T and supergroup L. Functional GO enrichment was assessed using topGO v2.4.0 (Alexa and Rahnenfuhrer, 2024) which evaluates GO term graph topology and uses the ‘weight01.fisher’ algorithm to create test statistics, returning corrected p-values not affected by multiple testing. TopGO was performed in R using the script aip_topgo_usage.consider_universe.R3 for multiple gene subsets depicted in Venn diagrams using the ‘diff’ and ‘universe’ sets of genes. GO-figure was used to visualize gene ontology enrichment, summarizing the list of GO terms based on their semantic similarity and producing scatterplots with clustered GO terms of similar functions (Reijnders et al., 2021).
Results
Wolbachia found in a Heterodera schachtii but absent in Meloidogyne spp., Globodera spp., and other Heterodera spp.
No Wolbachia was detected in DNA read (SRA) data and WGS assemblies for Meloidogyne spp. and Globodera spp. (Supplementary Tables 1, 2), but among the seven Heterodera spp. WGS assemblies, one assembly from Heterodera schachtii (an isolate named ‘IRS’ from the Netherlands, sequenced using Oxford Nanopore Technologies, with NCBI accession GCA_020449115.1) was positive for Wolbachia (Table 2). Analysis of this assembly (previously assembled using Wtdgb2 v.2.3) revealed a large (1.5 Mbp) scaffold (JAIZDD010000066.1) containing a large Wolbachia region (1.1 Mbp) between scaffold positions 270,235 and 1,348,240 flanked by nematode genes. The scaffold’s upstream flanking region matched nematodes, based on blastn, and was 0.16 Mbp. The downstream flanking region also matched nematodes and was 0.25 Mbp (Figure 1). Based on the mapping of the mapped Nanopore reads from the same sample (SRA accessions SRR16146220-SRR16146534 and SRR16675965-SRR16675966 and SRR28675229-SRR28675543 from the same project) to this scaffold, the flanking nematode regions had an average fold coverage of 15,244X (upstream) and 18,099X (downstream), whereas the Wolbachia-matching region had an average fold coverage of 829X (Figure 2).
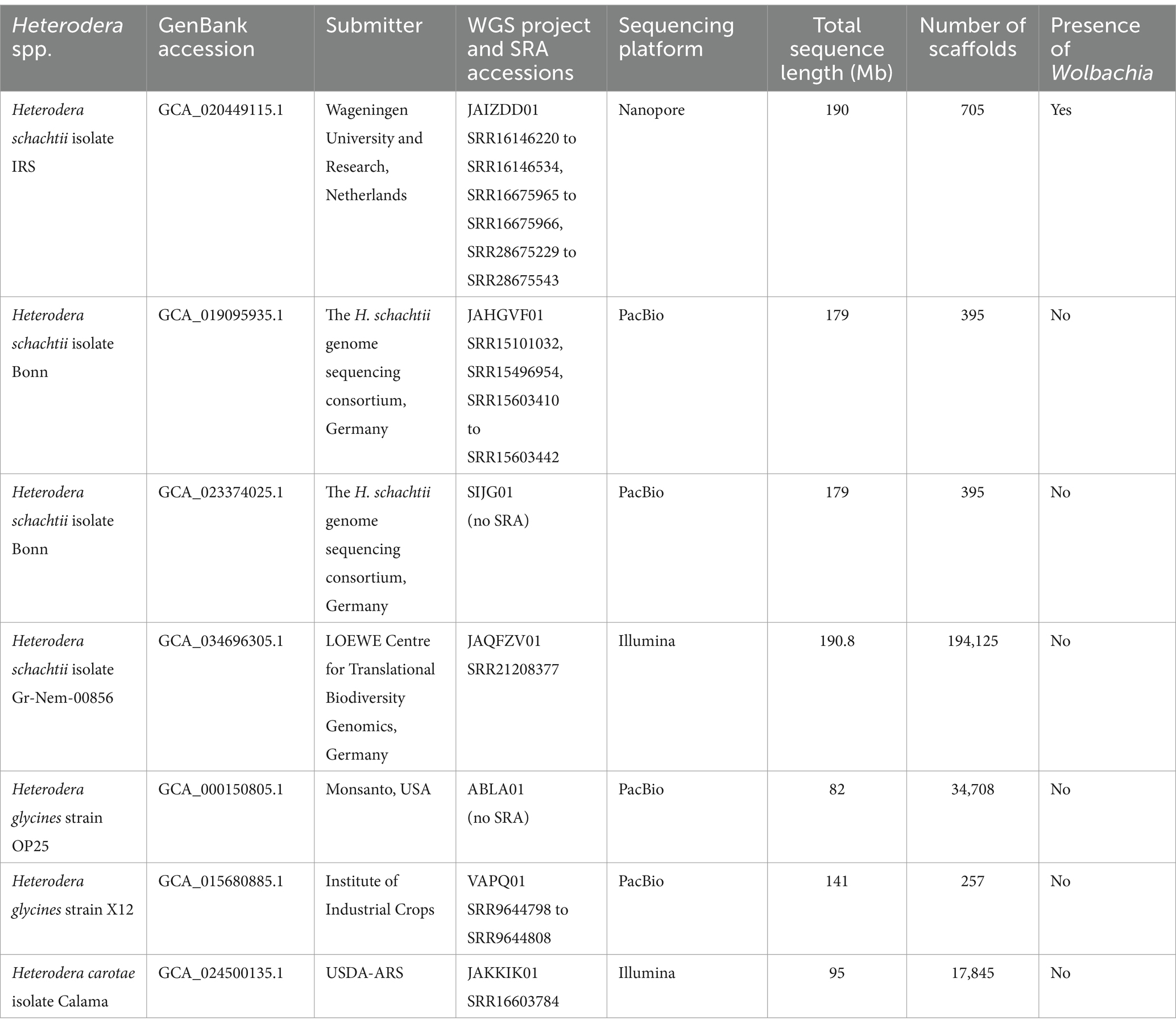
Table 2. Whole-genome sequencing (WGS) assemblies available for Heterodera spp. in NCBI with accompanying DNA reads’ SRA accessions.
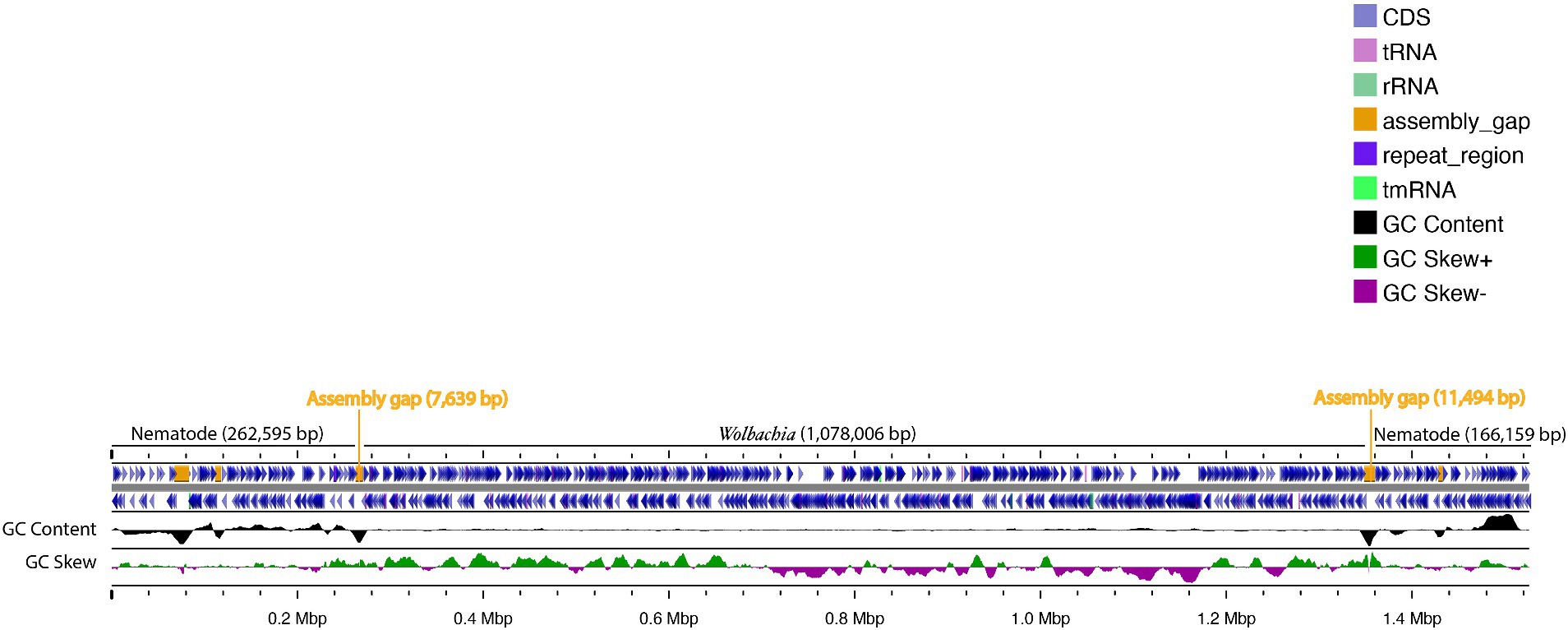
Figure 1. Representation of the Heterodera schachtii isolate IRS assembly 1.5 Mbp scaffold (JAIZDD010000066.1) harboring a Wolbachia-like sequence (1.1 Mbp) flanked by assembly gaps (Ns) and nematode-matching regions.
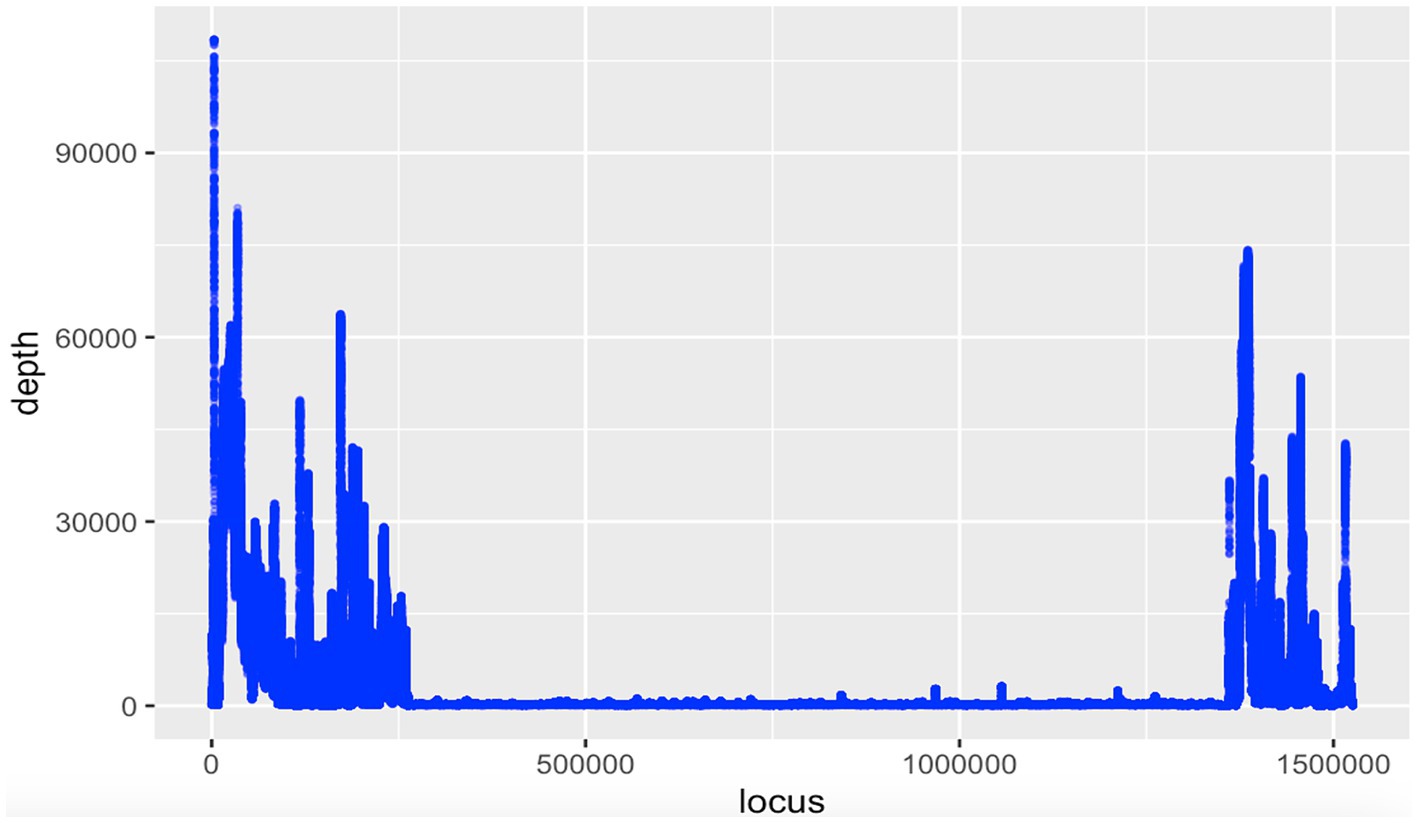
Figure 2. Coverage plot of the 1.5 Mbp scaffold (JAIZDD010000066.1 from NCBI project accession PRJNA767548) for Heterodera schachtii isolate IRS harboring a Wolbachia-like sequence (1.1 Mbp) flanked by assembly gaps (Ns) and nematode-matching regions. Mapped nanopore reads were derived from the same project (SRR16146220-SRR16146534 and SRR16675965-SRR16675966 and SRR28675229-SRR28675543).
Based on a blastn search against a custom nematode COI gene database, this Nanopore assembly for H. schachtii isolate IRS had two COI genes present in the assembly; one was most similar to H. medicaginis (382 bp hit with 90.576 percent identity to the COI gene), and other was H. schachtii (382 bp hit with 99.738 percent identity to the COI gene). The H. medicaginis-like COI region had an average fold coverage of 55,196X, whereas the H. schachtii-like COI region had an average fold coverage of 11,052X.
Upon visualization of the Wolbachia-containing scaffold (JAIZDD010000066.1) in Geneious Prime, the Wolbachia-like region was flanked by a number of non-called bases (Ns) in upstream and downstream regions in the connecting junctions between Wolbachia-like and nematode regions (Figure 1). However, mapping and re-assembly of SRA raw reads of this WGS project to this Wolbachia-containing scaffold revealed an additional 1,802 bp region downstream of this Wolbachia-like region that extended the main scaffold sequence across the downstream Ns in the assembly (i.e., filling the gap) and formed a 262 bp overlap with forward end of the main scaffold at the upstream junction with Ns, such that assembling the 1,802 bp region and the original scaffold created a complete Wolbachia circular genome (Figure 3) (denoted wHet, hereafter). Quast assembly statistics revealed this 1,079,546 bp (1.1 Mbp) sequence as a genome in single contig with GC content 32.59%, which is in the range of other Wolbachia strains (Supplementary Table 3).
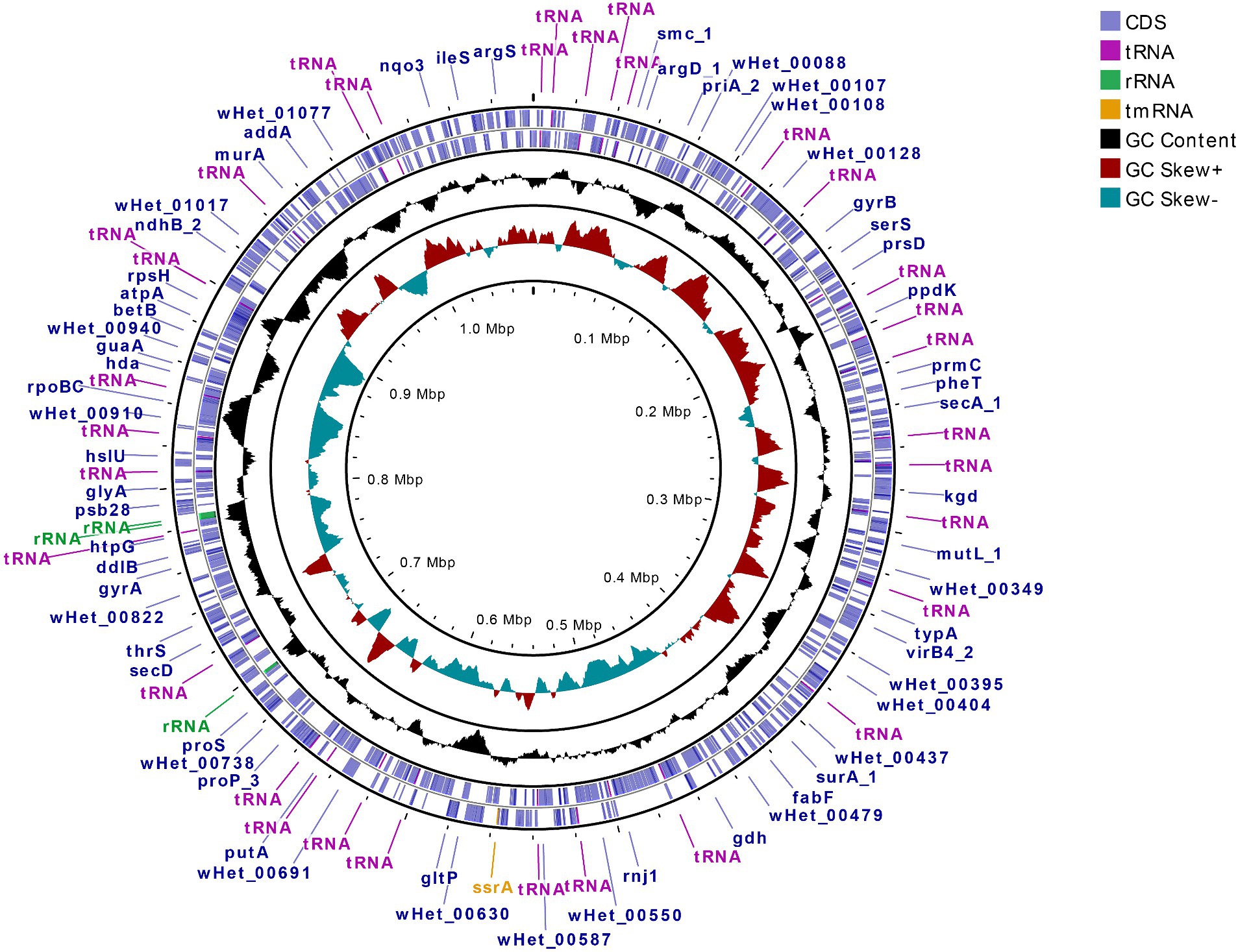
Figure 3. Circular genome representation of new Wolbachia strain wHet derived from re-assembly and circularization of overlapping scaffold ends, showing CDS, tRNA, rRNA, tmRNA, GC content, and GC skewness. Outermost circle represents the position of coding sequences (CDS) and RNA genes on the forward and reverse strand. Inner circles display plots of GC content and GC skew, respectively, showing their deviation from the average for the entire sequence.
Phylogenetic analysis of wHet with other Wolbachia strains and out-groups
Ortholog analysis of the novel Wolbachia strain (wHet) against published Wolbachia genomes from supergroups A (insects), B (insects), C (filarial nematodes), D (filarial nematodes), E, F, J, M, S, T, V, and W and supergroup L (plant-parasitic nematodes) and out-group Ca. Mesenet longicola revealed 37 core genes (orthologs) shared across all the genomes. Phylogenetic analysis of the 37-gene block (32,748 nucleotide positions) with the PhyloBayes package produced bpcomp results with the largest discrepancy results of maxdiff = 0.0414118 across all bipartitions, which indicated a good run (if maxdiff < 0.1) for independent chain runs. The consensus tree obtained using PhyloBayes MPI (Figure 4A) and MrBayes (Figure 4B) confirmed that this novel Wolbachia genome belonged to supergroup L, comprising Wolbachia from PPNs. The closest relative to wHet was wTex, followed by wPpe and Pp_GH2, with evidence supporting for the L monophyly and relationships in this clade. Pp_Cr was highly divergent from P. penetrans Wolbachia strains as its assembly was highly fragmented with a coding density of 57.47% (Table 3). Wolbachia strains from insects were grouped together for supergroup A and supergroup B. Wolbachia strains from filarial nematodes, supergroup C, and supergroup D formed separate groups (Figure 4). The results for all groups for the full alignment, and additional alignments with gaps removed, fragmented sequences removed, or long-branch sequences (wHow) removed, and analyses using RAxML produced identical phylogenies with similar node support in all cases (Supplementary Figure 1), except for the relative position of the long branch wHow. Strain wHow grouped as sister to supergroup A in PhyloBayes CAT analysis (Figure 4A), as sister to supergroup V (wCfeT) in MrBayes analysis (Figure 4B), and as sister to supergroup L with RAxML analysis.
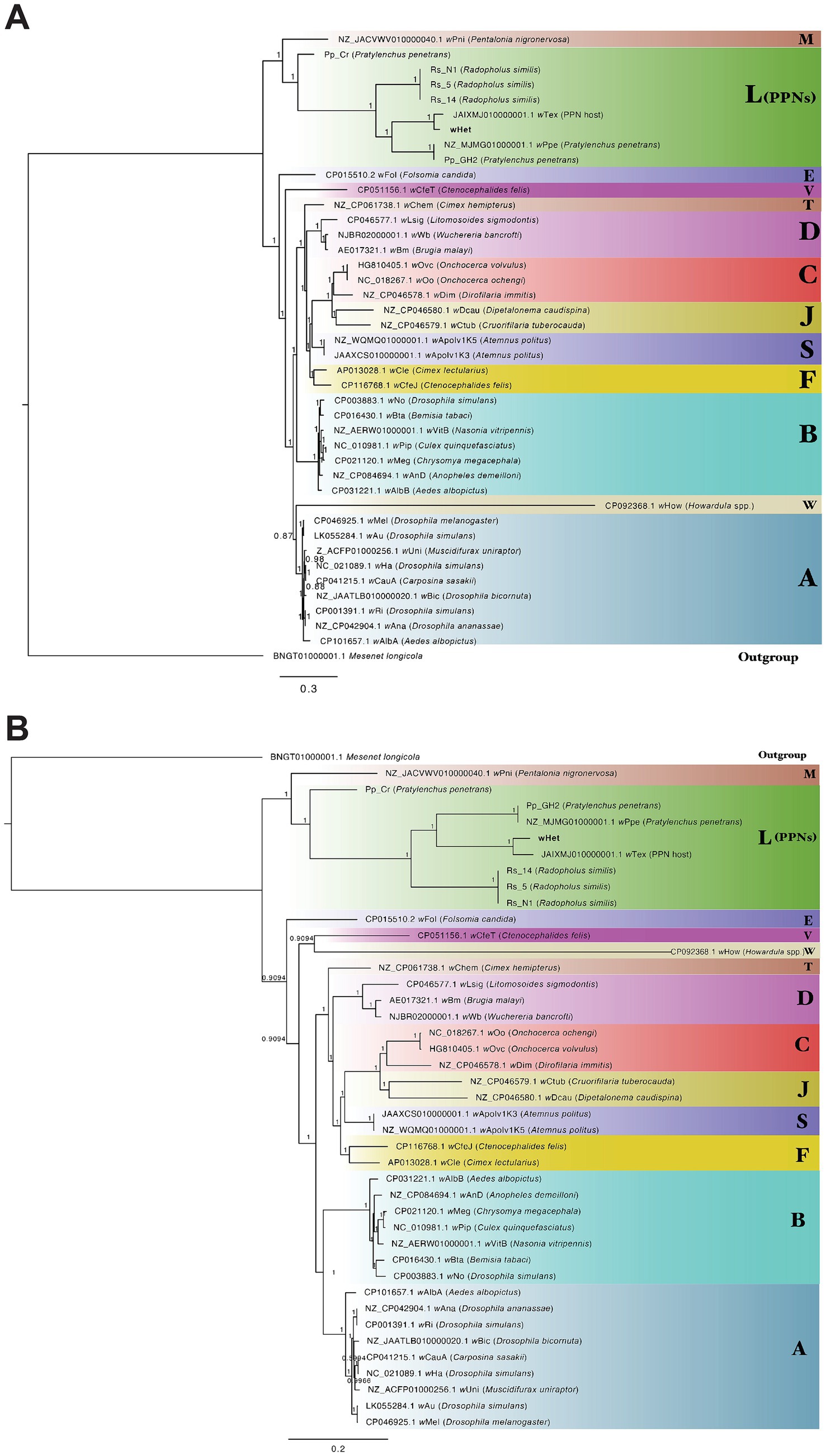
Figure 4. Phylogenetic trees of Wolbachia strains and out-group generated from an alignment of 37 core genes (32,748 nucleotide positions) (A) analyzed with PhyloBayes using the CAT-GTR model with posterior probabilities shown on branches (B) analyzed with MrBayes using the CAT-GTR model with posterior probabilities shown on branches. Strain wHet is shown in bold. Letters represent supergroups A (insects), B (insects), C (filarial nematodes), D (filarial nematodes), F, J, M, S, T, V, and W and supergroup L (plant-parasitic nematodes).
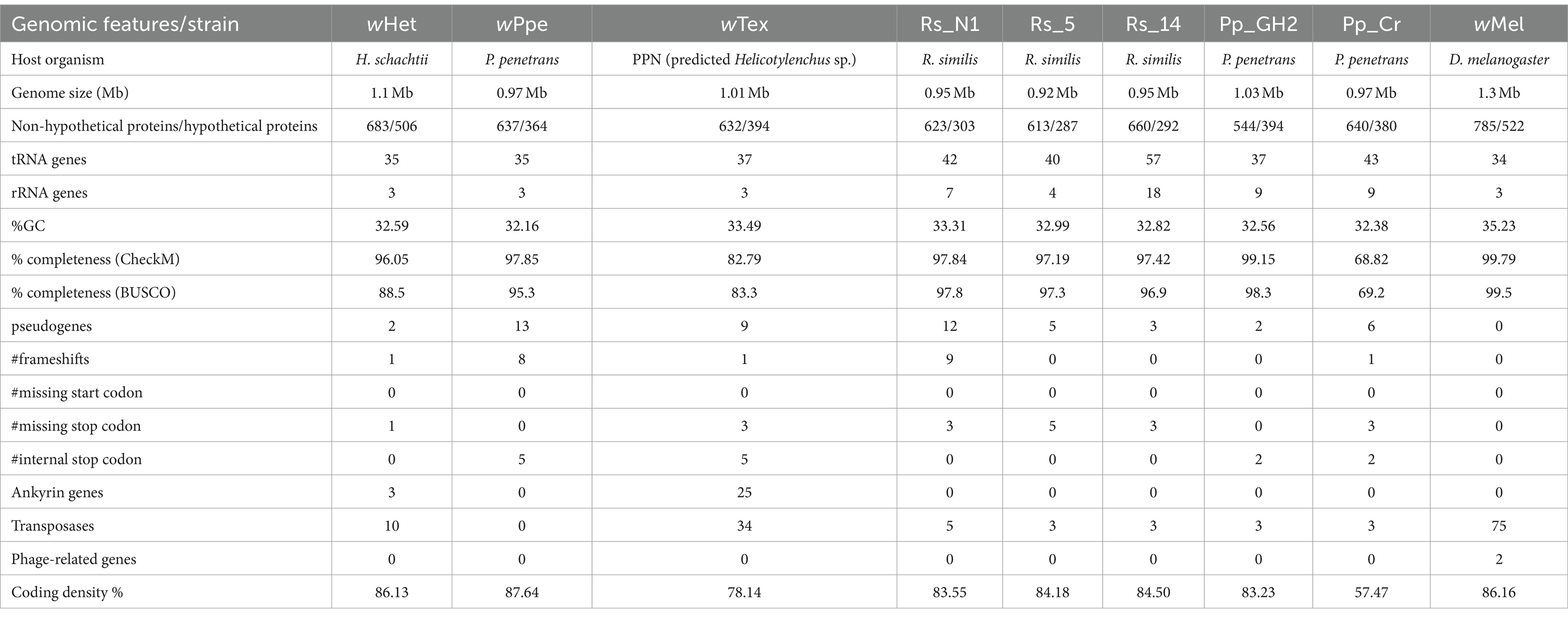
Table 3. Genomic features of wHet compared to closely related Wolbachia strains and wMel (which was included in the comparison as it was used as reference to calculate the pseudogenes).
Phylogenetic analysis of the 16S rRNA gene performed in RAxML using the GTR + Gamma model and in MrBayes revealed similar results to the core gene phylogeny, but here, wHet was shown also to be closely related to a Wolbachia strain isolated from H. sojae (soybean) roots from South Korea and a Wolbachia strain isolated from a Helicotylenchus sp. from Florida. Strain wHet was also closely related to an isolate of Heterodera humuli from Oregon (reads assembled from SRR27256751), and wTex. Evidence supported the monophyly and relationships in this clade (Figure 5; Supplementary Figures 2, 3), regardless of phylogenetic method or choice of out-groups. In addition, two transcriptomic datasets from H. schachtii RNA-seq projects on NCBI generated close matches to the 16S rRNA gene sequence of wHet (project PRJEB71637 from University of Cambridge, and a project examining roots of Arabidopsis thaliana infected with H. schachtii from Wageningen University with sample accession SAMEA14093318) (Figure 5; Supplementary Figure 2).
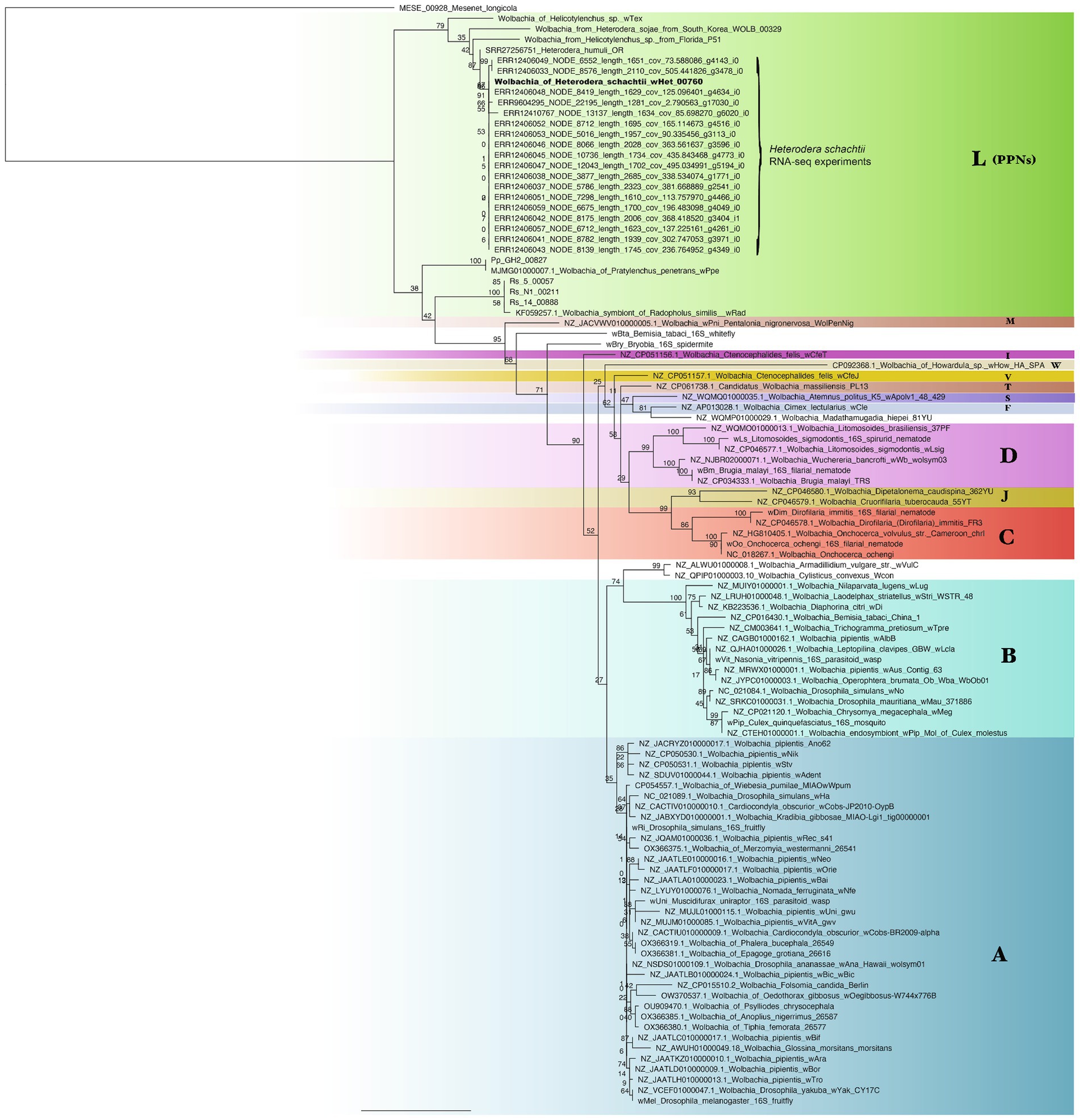
Figure 5. Phylogenetic tree of Wolbachia strains and out-groups generated by RAxML with the GTR + Gamma model from a 1,530 bp alignment of the 16S rRNA gene, with support from 100 bootstrap replicates shown on branches. The clade including wHet is shown in bold. Samples from SRA (SRR/ERR) datasets were assembled from publicly available reads on NCBI. Letters represent supergroups A (insects), B (insects), C (filarial nematodes), D (filarial nematodes), F, M, S, and T and supergroup L (plant-parasitic nematodes).
Phylogenetic analysis of the COI gene from Heterodera spp. including two COI genes recovered from the H. schachtii isolate IRS assembly showed that H. medicaginis COI and H. schachtii COI from this assembly were divergent in the tree (Supplementary Figure 4).
Comparative genome features of wHet against other Wolbachia strains
The final predicted circular genome from wHet had 96.05% estimated completeness (based on the marker gene sets present/absent that are specific to a genome’s inferred lineage within a reference genome tree in reference databases in checkM), 88.5% estimated completeness (based on BUSCO analysis), 683 predicted proteins with known functions, 506 predicted proteins with unknown functions, 35 tRNA genes, three rRNA genes, three ankyrin region containing genes, and 10 transposases (Table 3). All the PPN supergroup Wolbachia strains (wHet, wPpe, wTex, Rs_N1, Rs_14, Rs_5, Pp_Cr, and Pp_GH2) had no predicted prophage-related genes, unlike Wolbachia strains from other clades except for supergroups C and J. Compared to wMel, wHet had two pseudogenes, one frameshift, and one gene with a missing stop codon. Further examination revealed that wHet, such as wPpe, wTex, Rs_N1, Rs_14, Rs_5, Pp_Cr, and Pp_GH2, lacked homologs to known CI (cytoplasmic incompatibility) associated genes, namely, cifA and cifB, plasmid-associated genes, and WO prophages. wHet also had thiE (encoding thiamine phosphate synthase), such as others in the L (PPN) supergroup except for wTex. Various genes related to heme pathways were found in wHet, for example, hemA (encoding 5-aminolevulinate synthase), hemB (encoding delta-aminolevulinic acid dehydratase), hemC (encoding porphobilinogen deaminase), hemE (encoding uroporphyrinogen decarboxylase), hemF (encoding oxygen-dependent coproporphyrinogen-III oxidase), hemH (a ferrochelatase), ctaA (encoding heme A synthase), and ctaB (encoding protoheme IX farnesyltransferase), such as all others in L supergroup, except for hemH missing in wTex, ctaB missing in Rs_14, and catA and hemE missing in Pp_Cr.
Orthologs among wHet and related Wolbachia strains
Although wHet was closest to wTex in phylogenomic analyses, genome content similarity among this strain and others was less clear, likely due to the incompleteness of several other PPN Wolbachia genomes. Orthologous gene cluster comparisons showed wHet shared the most genes with Pp_GH2, Rs_5, and Rs_14 (549 genes shared with each) and similarly high numbers of genes with Rs_N1 and wPpe (547 and 546, respectively). wHet and wTex shared 429 genes, with 110 genes unique to wHet and 32 genes unique to wTex, likely a lower number because the wTex genome was incomplete (Supplementary Figure 5). Another Wolbachia, wHow, from an entomoparasitic tylenchid nematode and having a highly reduced genome, shared 401 genes with wHet, with 209 genes unique to wHet and 25 genes unique to wHow.
Across members of supergroup L, combined analysis of Wolbachia strains from Pratylenchus penetrans as well as wTex (unknown host) showed 365 genes shared with wHet and 32 genes unique to wHet, whereas Wolbachia strains from Radopholus similis shared 546 genes with wHet, 60 genes being unique to wHet (Supplementary Figure 6). Supergroup C Wolbachia strains wOo (Wolbachia endosymbiont of Onchocerca ochengi), wOvc (Wolbachia endosymbiont of Onchocerca volvulus), and wDim (Wolbachia endosymbiont of Dirofilaria immitis) shared 484 genes with wHet, with 97 unique genes in wHet. Supergroup D Wolbachia strains, wBm (Wolbachia endosymbiont of Brugia malayi), wLsig (Wolbachia endosymbiont of Litomosoides sigmodontis), and wWb (Wolbachia endosymbiont of Wuchereria bancrofti) shared 506 genes with wHet. Supergroup J Wolbachia strains wDcau (Wolbachia endosymbiont of Dipetalonema caudispina) and wCtub (Wolbachia endosymbiont of Cruorifilaria tuberocauda) shared 473 genes with wHet. Another close relative wPni (Wolbachia endosymbiont of Pentalonia nigronervosa) shared 538 genes with wHet (Supplementary Figure 6).
Genome length versus GC content comparing wHet to other Wolbachia strains
Comparing assembly length versus GC content showed that wHet followed the trend for all Wolbachia strains in which lower GC content is associated with smaller genomes (Figure 6). Strain wHet had a genome size and GC content similar to that of other supergroup L members, which was closer to that of supergroups D and C than to strains from groups A and B.
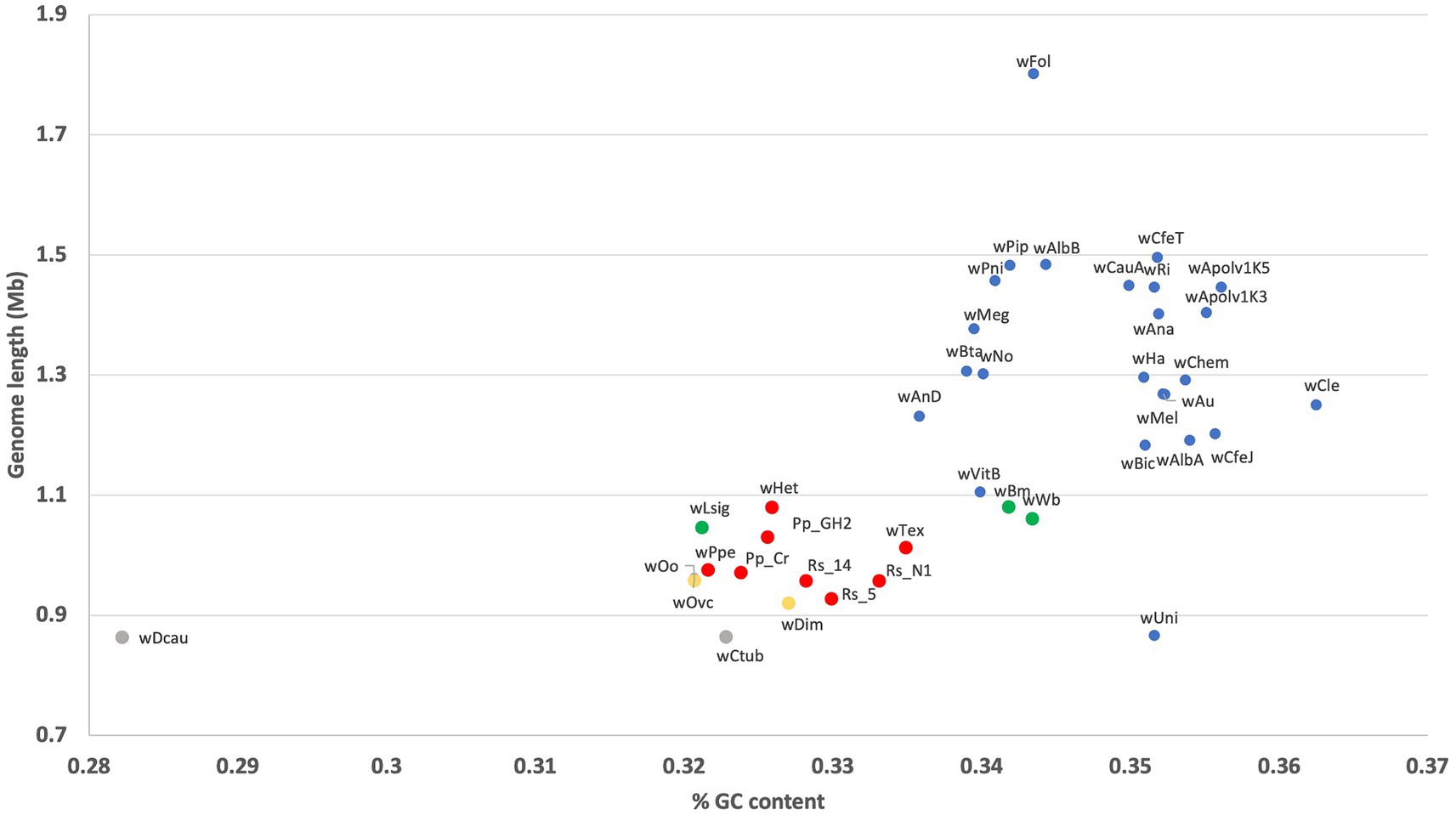
Figure 6. GC content versus genome length for different Wolbachia strains including the new Wolbachia strain wHet. Red color represents supergroup L (PPNs), yellow color represents supergroup C (filarial nematodes), green color represents supergroup D (filarial nematodes), and blue color represents supergroups A and B (insects).
Gene ontology enrichment analysis
GO-figure plots showed significant differences in enriched GO terms in wHet compared to relatives and out-groups (Supplementary Figure 7). These plots depict GO term semantic similarity in predicted gene products, where the size of the node reflects the number of genes annotated to that term. wHet enriched GO terms showed more overlapping semantic space with supergroups C, D, and L, as compared to supergroups A and B, depicting similar functional relatedness in biological processes with nematode Wolbachia supergroups. However, there were significant overlaps with supergroup S. When wHet was compared individually to supergroup L Wolbachia strains from P. penetrans, R. similis, and wTex, wTex shared the most semantic space overlap with wHet (Supplementary Figure 7).
From topGO analysis, wHet did not show any significant differences compared to wTex and supergroup B Wolbachia strains in significantly enriched GO terms, but as compared to Wolbachia strains from other supergroups (A, B, C, D, F, S, T, and L), a few significant differences were observed. The results showed that wHet was enriched for nitrogen metabolic processes compared to supergroups C, D, J, S, and T, as well as lipid catabolic processes compared to supergroup T (Table 4).
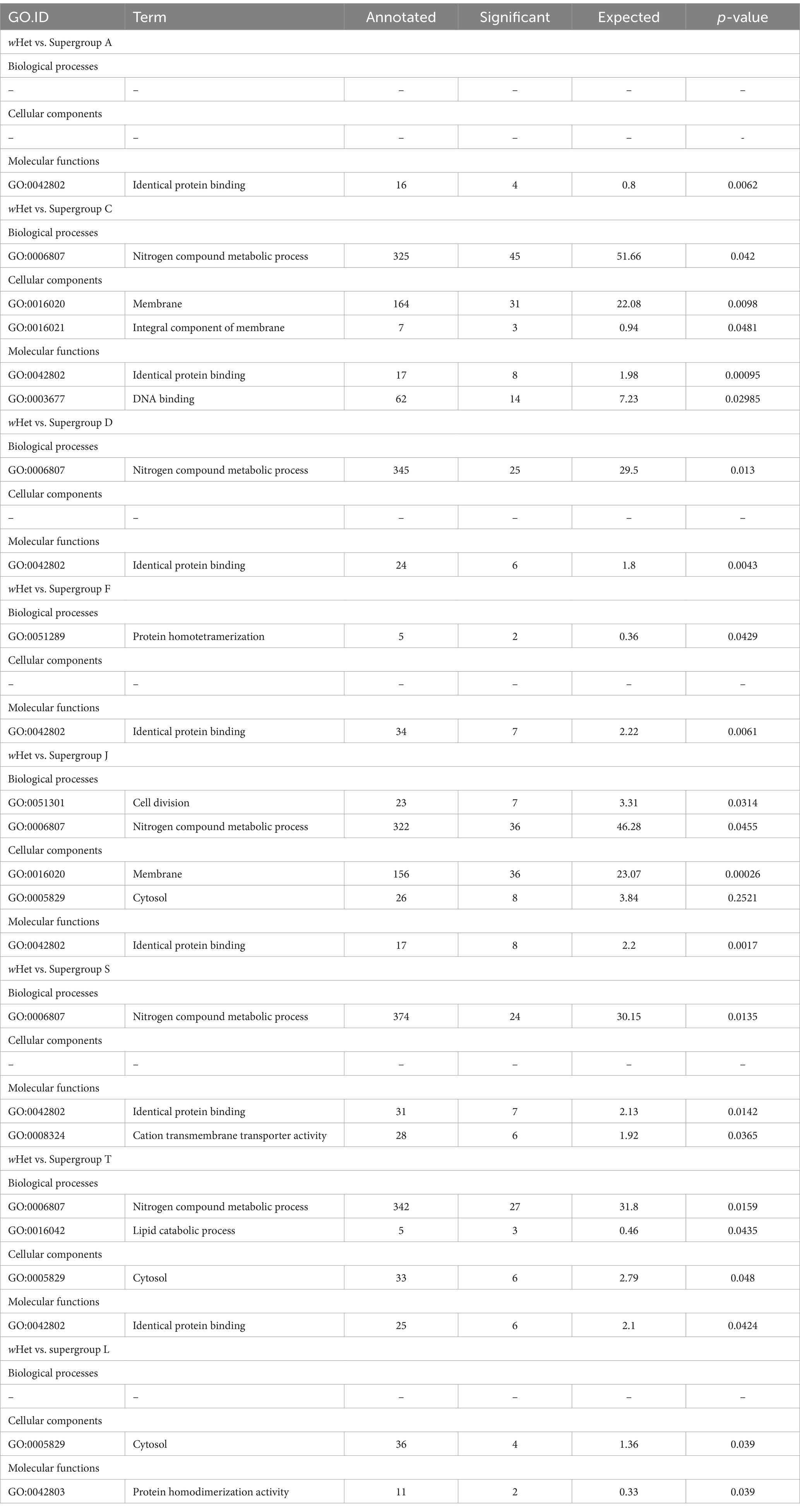
Table 4. Significantly enriched gene ontology (GO) categories based on topGO analysis for the pangenome of Wolbachia wHet compared to the pangenome Wolbachia strains from different supergroups, including genes shared between pangenomes.
Compared to supergroup L strains, wHet had significantly enriched GO terms for cytosol (cellular component) and protein homodimerization activity (molecular function) against Wolbachia strains from P. penetrans and significantly enriched GO terms for regulation of cell shape (biological process) and protein homodimerization activity (molecular function), against Wolbachia strains from R. similis (Table 5). There were similar differences as compared to supergroup A and F strains (Table 4). There were also similar differences observed as compared to supergroup D and supergroup S strains, where wHet had enriched GO terms for nitrogen compound metabolic process (biological process), identical protein binding (molecular function) against both supergroups, and enriched cation transmembrane transporter activity (molecular function) specifically against supergroup S (Table 4).
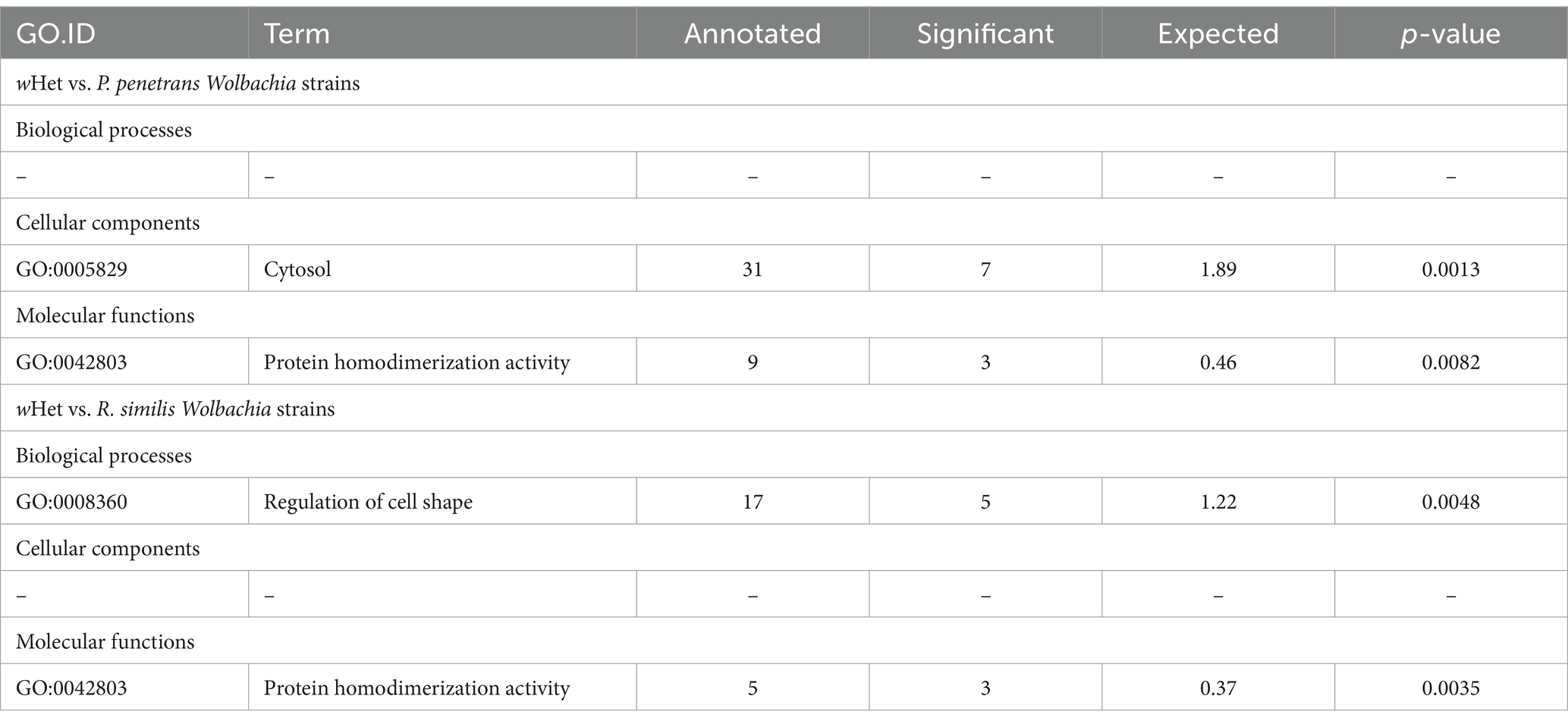
Table 5. Significantly enriched gene ontology (GO) categories based on topGO analysis for the pangenome of Wolbachia wHet compared to the pangenome of plant-parasitic nematode-associated Wolbachia (supergroup L), including genes shared between pangenomes.
Discussion
Despite Wolbachia’s widespread distribution and biological importance in arthropods and filarial nematodes, its distribution and role in plant-parasitic nematodes (PPNs) are inadequately known. Uncovering the potential roles of PPN Wolbachia is of great interest to the agricultural sector as these endosymbionts could be explored as possible candidates for developing biocontrol measures to reduce PPNs. This study sought to find PPN Wolbachia from public databases and resulted in the discovery of a new hidden Wolbachia strain, designated wHet, from long-read sequences from NCBI’s SRA database from a Heterodera schachtii assembly from the Netherlands. Previous studies have reported the occurrence of Wolbachia in PPNs across South America, North America, Africa, and Asia (Haegeman et al., 2009; Wasala et al., 2019; Wasala et al., 2023), but this is the first genome study demonstrating the occurrence of Wolbachia from a species of Heterodera in Europe.
Initially, it appeared that this new PPN Wolbachia strain, wHet, might be a large putative HGT (horizontal gene transfer) from Wolbachia to the host, perhaps similar to the whole-genome Wolbachia HGT found in Drosophila ananassae (Hotopp et al., 2007), but our analyses suggest this is not an HGT but instead represents a Wolbachia symbiont. In support of this argument, we discuss four types of evidence. First, the presence of long string of Ns directly flanking both ends of the Wolbachia region with few to no Ns in the Wolbachia region itself or in the flanking nematode regions suggests an assembly artifact. Second, our analyses showed a substantial difference in the average fold coverage of the Wolbachia-like region and flanking nematode-like regions, which suggest, again, that the apparent integration of the Wolbachia region is an assembly artifact. Third, our mapping and assembly that recovered the downstream (1,802 bp) end of the Wolbachia genome from SRA reads clearly gap-filled the Ns flanking the Wolbachia scaffold and uncovered a significant overlap (262 bp) between the upstream and downstream ends of the new combined assembly, to form a complete circular genome assembly. Finally, additional analysis of other H. schachtii projects based on RNA, rather than DNA, revealed a high-coverage 16S rRNA sequence identical with that of wHet suggesting the symbiont itself expresses its ribosomal RNA.
While our genomic analyses suggested the absence of Wolbachia in all other H. schachtii DNA assemblies from Germany (Collins et al., 2023; Siddique et al., 2022), we did detect Wolbachia wHet in other H. schachtii SRA data from transcriptome projects. Specifically, our analysis of H. schachtii RNA transcriptome assemblies revealed two separate sequencing projects with clear matches to wHet, supporting the hypothesis that the WGS-recovered Wolbachia sequence from the Nanopore ‘IRS’ project likely represents a real Wolbachia symbiont rather than a nematode host genome integration, as discussed above. Particularly, the discovery of high-coverage 16S rRNA sequences matching wHet from these transcriptome projects would be unexpected from a mere genome integration. Instead, recovery of Wolbachia 16S rRNA from these projects which used polydT library selection is a strong indicator that the ribosomal RNA itself is expressed and abundant enough to ‘contaminate’ the mature mRNA pulled down in the library preparation. To our knowledge, there is no study demonstrating that eukaryote nuclear-encoded Wolbachia rRNA genes would be expressed effectively enough to produce this result. Nevertheless, the apparent absence of Wolbachia from some DNA and RNA experiments would appear to indicate a result of biological importance: wHet may have variable presence or absence across isolates of this host. This pattern is consistent with that of another PPN Wolbachia, wPpe (Wasala et al., 2019), possibly indicating the strain has a facultative, rather than obligate role.
Steps to determine the host of wHet in this assembly revealed the presence of another nematode COI gene in the H. schachtii (sugarbeet nematode) assembly from the Netherlands, which was related at ~90% sequence identity to H. medicaginis (alfalfa cyst nematode), suggesting that this recovered sequence might reflect a cryptic and/or unnamed species present in the sample along with true H. schachtii. We note that our analyses suggest that NCBI records for H. medicaginis COI genes may reflect taxonomic issues or a polyphyletic species (see Supplementary Figure 2). To date, no SRA data were available on NCBI for H. medicaginis to search for Wolbachia in this nematode species. However, since no other species contamination was detected except for these two Heterodera species, the formation of a long branch in the PPN Wolbachia supergroup from tylenchids for this symbiont suggests a longstanding relationship of Wolbachia within this H. schachtii-like host clade, adding Heteroderidae to the short list of families of tylenchid nematode hosting Wolbachia along with Pratylenchidae (Brown, 2018; Brown et al., 2016; Haegeman et al., 2009; Kaur and Brown, 2024).
Our assembled wHet genome of 1.1 Mbp represents the most complete PPN Wolbachia discovered to date: Other PPN Wolbachia draft genomes (wPpe, wTex, Pp_Cr, Pp_GH2, Rs_14, Rs_5, and Rs_N1) are fragmented. Hence, this wHet genome provides a useful resource for improved genomic analyses. Genomic features and phylogenetic analysis of wHet compared to other Wolbachia strains confirmed that this strain belongs to supergroup L, a PPN-type Wolbachia clade. Supergroup L is a monophyletic supergroup in the Wolbachia phylogeny, including wTex, wPpe, Rs_N1, Rs_14, Rs_5, Pp_Cr, Pp_GH2, and wRad, and most analyses placed wHet and group L closet to the nearest out-groups of genus Wolbachia (i.e., Ca. Mesenet longicola and other Anaplasmataceae). Despite consistency of phylogenetic results for most Wolbachia in this study, the long branch of wHow changed position depending on the phylogenetic method used. Although it was not the focus of this study, the varying position of wHow here, and compared to a previous study (Dudzic et al., 2022), may reflect long-branch attraction (LBA) effects. Notably, the latter study using the maximum likelihood-based program IQ-TREE was more similar to our RAxML results, whereas our PhyloBayes MPI (CAT-GTR model) which reportedly corrects for some LBA (Lartillot et al., 2013) placed wHow further from out-groups. We suggest further taxonomic sampling of W and L supergroups and refinement of the phylogenetic analysis models and methods will be required to overcome possible long-branch attraction artifacts.
Our phylogenetic analysis consistently placed wHet closer to wTex than to other Wolbachia strains, whereas its gene repertoire was closer to Pp_GH2, Rs_5, and Rs_14. This gene repertoire result may be due to an artifact of the incompleteness of the wTex draft assembly. We also found the PPN Wolbachia as a group (including wHet) had a core gene repertoire more similar to that of wPni (Wolbachia endosymbiont of banana aphid, Pentalonia nigronervosa) than to other Wolbachia characterized thus far. This finding is consistent with previous analyses (Weyandt et al., 2022), suggesting evolutionary relatedness and potentially similar biological processes of the host, such as plant feeding. In terms of gene sharing with supergroup L, wHet had 32 unique genes as compared to Wolbachia strains from P. penetrans and 60 unique genes as compared to Wolbachia strains from R. similis, suggesting that these genes may be strain-specific or play a role in the biology of each strain. However, most of these accessory genes were annotated as ‘hypothetical protein,’ as for other Wolbachia strains, making it difficult to infer functional differences.
From analysis of the remaining wHet genes that could be annotated to function, it appeared that this strain was very similar to wTex, Pp_GH2, Rs_N1, Rs_14, Rs_5, Pp_Cr, and wPpe in terms of essential features, including the absence of CI (cytoplasmic incompatibility) genes cifA and cifB, plasmid-associated genes, and WO prophages or phage-like proteins, that are linked with Wolbachia phenotypes, such as CI (Beckmann and Fallon, 2013; Metcalf et al., 2014; Papafotiou et al., 2011; Siozios et al., 2013). Unlike other Wolbachia from the L supergroup, wHet had the gene thiE encoding thiamine phosphate synthase. Strain wHet harbored several genes associated with heme pathways, including hemA, hemB, hemC, hemE, hemF, hemH, ctaA, and ctaB, which were also present in wPpe and wTex, except for hemH being absent in wTex. These findings suggest that wHet may serve as an exogenous source for heme in the host nematode as nematodes are thought to be unable to synthesize heme (Rao et al., 2005). Among several nutrients (thiamine, biotin riboflavin, and heme) that were proposed as candidate nutrients supplied by other Wolbachia strains as a means to maintain Wolbachia through a weak or facultative mutualism (Brown et al., 2016; Foster et al., 2005; Gill et al., 2014; Moriyama et al., 2015), only the heme pathway appeared to be complete in wHet. However, uncertainty remains about the major functions of wHet with respect to its host, given that our genomic analysis showed that wHet overall had a large number (506) of predicted proteins with unknown functions.
At the pathway level, analysis showed some differences in functional enrichment. For example, GO-figure plots, based on semantic similarity, depicted similar functional patterns of wHet’s biological process enrichment with nematode Wolbachia supergroups (C and L). However, when compared individually to each known member of the PPN supergroup from each host (Wolbachia strains from P. penetrans and R. similis, and wTex), wHet showed more relatedness in gene repertoire and associated biological processes to wTex, which comprises a sample collected from a nematode community about which there remains little host or symbiont biological information (Weyandt et al., 2022). Notably, the gene ontology enrichment analysis showed differences in significantly enriched GO terms in wHet compared to supergroups from filarial nematodes (C, D, and J) with a notable enrichment in nitrogen metabolic processes. We speculate that this enrichment could reflect a biological role for wHet related to its nematode lifestyle in nitrogen-limited conditions of root feeding; however, this hypothesis would require experimental investigation.
Strain wHet’s GC content and genome size were similar to that of other PPN Wolbachia, as expected. Furthermore, the plot GC content and genome size, including strain wHet, shows a positive correlation between GC content and genome size across Wolbachia strains. Although endosymbionts usually have reduced genome size and metabolic capabilities compared to their free-living relatives (Newton et al., 2016; Newton and Bordenstein, 2011), here, the reduced GC content in wHet and PPN-type Wolbachia compared to most A and B group strains may be due to a stronger pattern of vertical transmission in PPN-type Wolbachia leading to higher levels of bottleneck and drift with an underlying AT-mutational bias and the inability to purge mutations under strong bottleneck (Brown et al., 2015; Moran and Bennett, 2014; Moran and Plague, 2004; Renoz et al., 2022). Phylogenetic analyses thus far showing longer branches in this clade seem to support this stronger vertical transmission hypothesis.
Acknowledging that some of the PPN Wolbachia genome lengths included in this study may be underestimated due to varying degrees of incompleteness, our data suggest clear differences in length between the complete wHet genome and those of other supergroups. The reduced genome length in wHet and PPN-type Wolbachia compared to A and B group strains may be due to the gradual erosion and elimination of non-functional sequences that become redundant within the intracellular environment where the host provides many metabolites directly to the symbiont, accelerating genome size loss due to genetic drift causing fixation of irreversible deleterious mutations (Bobay and Ochman, 2017). Although the genome repertoire and GC content of wHet and others in this supergroup were more similar to supergroups C and D, which are obligate mutualists from filarial nematodes, compared to supergroups A and B, which are mostly CI-inducing Wolbachia strains, the scant data on the PPN Wolbachia prevalence thus far suggest some supergroup L strains are not at 100% prevalence (Wasala et al., 2019) while others may be at 100% prevalence (Haegeman et al., 2009). However, the lack of previous reports of Wolbachia-like endosymbionts in Heteroderidae nematodes suggests wHet, such as wPpe, is likely not an obligate mutualist. Nevertheless, based on the trend toward shorter genome length and lower GC content similar to C and D supergroup Wolbachia strains, it is possible that PPN Wolbachia strains could be facultative mutualists.
Although we found a high-quality genome supporting the new strain wHet in H. schachtii, confirmation of the host will require additional fluorescent in situ hybridization (FISH) for isolates of sugarbeet nematode or alfalfa cyst nematode. Furthermore, to assess this symbiont’s distribution, a broad sampling of H. schachtii and H. medicaginis-like relatives will be important. Predicted genes with unknown functions in wHet, which were abundant in its accessory genome, limited the ability of GO and pathway analysis to distinguish potential functions. Future annotation of such genes may uncover important aspects of this host–symbiont relationship, but investigating the function of wHet in the host will require future experiments, such as symbiont-clearing assays and RNA-seq analysis to examine how specific gene pathways are altered in response to wHet infection.
Data availability statement
SRA data and whole genome sequencing assemblies analyzed in this study were accessed through NCBI Accessions (https://www.ncbi.nlm.nih.gov). The datasets supporting the conclusions of this article are available in the NCBI repository, BioProject PRJNA767548, BioSamples SAMN21928827, and the final assembled genome was deposited under BioProject PRJNA1148971.
Ethics statement
The manuscript presents research on animals that do not require ethical approval for their study.
Author contributions
TK: Formal analysis, Investigation, Methodology, Visualization, Writing – original draft, Writing – review & editing. AB: Conceptualization, Funding acquisition, Investigation, Methodology, Supervision, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was supported through funding to A.M.V.B. from the National Science Foundation (Award 2047684) and the United States Department of Agriculture NIFA (Award 20216701335757). Graduate student support to T.K. was provided by the Texas Tech Graduate School Study Abroad program and the Texas Tech Graduate School Water Conservation Research Scholarship.
Acknowledgments
We acknowledge support for this research from the Texas Tech Graduate School.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1446506/full#supplementary-material
Footnotes
1. ^https://github.com/BioInfoTools/BBMap/blob/master/sh/pileup.sh
References
Alexa, A., and Rahnenfuhrer, J. (2024). topGO: enrichment analysis for gene ontology. R package version 2.56.0.
Beckmann, J., and Fallon, A. M. (2013). Detection of the Wolbachia protein WPIP0282 in mosquito spermathecae: implications for cytoplasmic incompatibility. Insect. Biochem. Mol. Biol. 43, 867–878. doi: 10.1016/j.ibmb.2013.07.002
Bing, X.-L., Xia, C.-B., Ye, Q.-T., Gong, X., Cui, J.-R., Peng, C.-W., et al. (2023). Wolbachia manipulates reproduction of spider mites by influencing herbivore salivary proteins. Pest Manag. Sci. 79, 315–323. doi: 10.1002/ps.7201
Bobay, L. M., and Ochman, H. (2017). The evolution of bacterial genome architecture. Front. Genet. 8:72. doi: 10.3389/FGENE.2017.00072
Brown, A. M. V. (2018). Endosymbionts of plant-parasitic nematodes. Annu. Rev. Phytopathol. 56, 225–242. doi: 10.1146/annurev-phyto-080417-045824
Brown, A. M. V., Howe, D. K., Wasala, S. K., Peetz, A. B., Zasada, I. A., and Denver, D. R. (2015). Comparative genomics of a plant-parasitic nematode endosymbiont suggest a role in nutritional symbiosis. Genome Biol. Evol. 7, 2727–2746. doi: 10.1093/GBE/EVV176
Brown, A. M. V., Wasala, S. K., Howe, D. K., Peetz, A. B., Zasada, I. A., and Denver, D. R. (2016). Genomic evidence for plant-parasitic nematodes as the earliest Wolbachia hosts. Sci. Rep. 6:34955. doi: 10.1038/SREP34955
Bushmanova, E., Antipov, D., Lapidus, A., and Prjibelski, A. D. (2019). rnaSPAdes: a de novo transcriptome assembler and its application to RNA-Seq data. GigaScience 8, 1–13. doi: 10.1093/GIGASCIENCE/GIZ100
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Collins, G., Schneider, C., Boštjančić, L. L., Burkhardt, U., Christian, A., Decker, P., et al. (2023). The MetaInvert soil invertebrate genome resource provides insights into below-ground biodiversity and evolution. Commun. Biol. 6, 1241–1212. doi: 10.1038/s42003-023-05621-4
Correa, C. C., and Ballard, J. W. O. (2016). Wolbachia associations with insects: winning or losing against a master manipulator. Front. Ecol. Evol. 3:153. doi: 10.3389/fevo.2015.00153
Denver, D. R., Brown, A. M. V., Howe, D. K., Peetz, A. B., and Zasada, I. A. (2016). Genome skimming: a rapid approach to gaining diverse biological insights into multicellular pathogens. PLoS Pathog. 12:e1005713. doi: 10.1371/JOURNAL.PPAT.1005713
Dudzic, J. P., Curtis, C. I., Gowen, B. E., and Perlman, S. J. (2022). A highly divergent Wolbachia with a tiny genome in an insect-parasitic tylenchid nematode. Proc. R. Soc. B 289:20221518. doi: 10.1098/RSPB.2022.1518
Dunning Hotopp, J. C. (2011). Horizontal gene transfer between bacteria and animals. Trends Genet. 27, 157–163. doi: 10.1016/J.TIG.2011.01.005
Finn, R. D., Clements, J., and Eddy, S. R. (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. doi: 10.1093/NAR/GKR367
Foster, J., Ganatra, M., Kamal, I., Ware, J., Makarova, K., Ivanova, N., et al. (2005). The Wolbachia genome of Brugia malayi: endosymbiont evolution within a human pathogenic nematode. PLoS Biol. 3:e121. doi: 10.1371/JOURNAL.PBIO.0030121
Fransolet, D., Roberty, S., and Plumier, J. C. (2012). Establishment of endosymbiosis: the case of cnidarians and Symbiodinium. J. Exp. Mar. Biol. Ecol. 420-421, 1–7. doi: 10.1016/J.JEMBE.2012.03.015
Gill, A. C., Darby, A. C., and Makepeace, B. L. (2014). Iron necessity: the secret of Wolbachia’s success? PLoS Negl. Trop. Dis. 8:e3224. doi: 10.1371/JOURNAL.PNTD.0003224
Goffredi, S. K. (2010). Indigenous ectosymbiotic bacteria associated with diverse hydrothermal vent invertebrates. Environ. Microbiol. Rep. 2, 479–488. doi: 10.1111/J.1758-2229.2010.00136.X
Haegeman, A., Vanholme, B., Jacob, J., Vandekerckhove, T. T. M., Claeys, M., Borgonie, G., et al. (2009). An endosymbiotic bacterium in a plant-parasitic nematode: member of a new Wolbachia supergroup. Int. J. Parasitol. 39, 1045–1054. doi: 10.1016/J.IJPARA.2009.01.006
Hosokawa, T., Koga, R., Kikuchi, Y., Meng, X. Y., and Fukatsu, T. (2010). Wolbachia as a bacteriocyte-associated nutritional mutualist. Proc. Natl. Acad. Sci. USA 107, 769–774. doi: 10.1073/pnas.0911476107
Hotopp, J., Clark, M., Oliveira, D., Foster, J. M., Fischer, P., Muñoz Torres, M. C., et al. (2007). Widespread lateral gene transfer from intracellular bacteria to multicellular eukaryotes. Science 317, 1753–1756. doi: 10.1126/science.1142490
Husnik, F., and McCutcheon, J. P. (2017). Functional horizontal gene transfer from bacteria to eukaryotes. Nat. Rev. Microbiol. 16, 67–79. doi: 10.1038/nrmicro.2017.137
Jones, J. T., Haegeman, A., Danchin, E. G. J., Gaur, H. S., Helder, J., Jones, M. G. K., et al. (2013). Top 10 plant-parasitic nematodes in molecular plant pathology. Mol. Plant Pathol. 14, 946–961. doi: 10.1111/MPP.12057
Kaur, A., and Brown, A. M. V. (2024). Detection and analysis of Wolbachia in plant-parasitic nematodes and insights into Wolbachia evolution. Methods Mol. Biol. 2739, 115–134. doi: 10.1007/978-1-0716-3553-7_7
Koutsovoulos, G., Makepeace, B., Tanya, V. N., and Blaxter, M. (2014). Palaeosymbiosis revealed by genomic fossils of Wolbachia in a Strongyloidean nematode. PLoS Genet. 10:e1004397. doi: 10.1371/JOURNAL.PGEN.1004397
Lamelas, A., Gosalbes, M. J., Manzano-Marín, A., Peretó, J., Moya, A., and Latorre, A. (2011). Serratia symbiotica from the aphid Cinara cedri: a missing link from facultative to obligate insect endosymbiont. PLoS Genet. 7:e1002357. doi: 10.1371/JOURNAL.PGEN.1002357
Lartillot, N., Rodrigue, N., Stubbs, D., and Richer, J. (2013). PhyloBayes MPI: phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst. Biol. 62, 611–615. doi: 10.1093/SYSBIO/SYT022
Lefoulon, E., Bain, O., Makepeace, B. L., D’Haese, C., Uni, S., Martin, C., et al. (2016). Breakdown of coevolution between symbiotic bacteria Wolbachia and their filarial hosts. PeerJ 2016:e1840. doi: 10.7717/PEERJ.1840/SUPP-7
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A., and Zdobnov, E. M. (2021). BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654. doi: 10.1093/MOLBEV/MSAB199
McNulty, S. N., Foster, J. M., Mitreva, M., Hotopp, J. C. D., Martin, J., Fischer, K., et al. (2010). Endosymbiont DNA in endobacteria-free filarial nematodes indicates ancient horizontal genetic transfer. PLoS One 5:e11029. doi: 10.1371/JOURNAL.PONE.0011029
Metcalf, J., Jo, M., Bordenstein, S., Jaenike, J., and Bordenstein, S. R. (2014). Recent genome reduction of Wolbachia in Drosophila recens targets phage WO and narrows candidates for reproductive parasitism. PeerJ 2:e529. doi: 10.7717/peerj.529
Moran, N. A., and Bennett, G. M. (2014). The tiniest tiny genomes. Ann. Rev. Microbiol. 68, 195–215. doi: 10.1146/ANNUREV-MICRO-091213-112901
Moran, N. A., and Plague, G. R. (2004). Genomic changes following host restriction in bacteria. Curr. Opin. Genet. Dev. 14, 627–633. doi: 10.1016/J.GDE.2004.09.003
Moriyama, M., Nikoh, N., Hosokawa, T., and Fukatsu, T. (2015). Riboflavin provisioning underlies Wolbachia’s fitness contribution to its insect host. MBio 6, e01732–e01715. doi: 10.1128/mBio.01732-15
Moya, A., Peretó, J., Gil, R., and Latorre, A. (2014). Learning how to live together: genomic insights into prokaryote–animal symbioses. Nat. Rev. Genet. 9, 218–229. doi: 10.1038/nrg2319
Newton, I. L. G., and Bordenstein, S. R. (2011). Correlations between bacterial ecology and mobile DNA. Curr. Microbiol. 62, 198–208. doi: 10.1007/s00284-010-9693-3
Newton, I. L. G., Clark, M. E., Kent, B. N., Bordenstein, S. R., Qu, J., Richards, S., et al. (2016). Comparative genomics of two closely related Wolbachia with different reproductive effects on hosts. Genome Biol. Evol. 8, 1526–1542. doi: 10.1093/GBE/EVW096
Nicol, J. M., Turner, S. J., Coyne, D. L., den Nijs, L., Hockland, S., and Maafi, Z. T. (2011). “Current nematode threats to world agriculture” in Genomics and molecular genetics of plant-nematode interactions. eds. J. Jones, G. Gheysen, and C. Fenoll (Heidelberg, Germany: Springer), 21–43.
Page, A., Cummins, C., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Papafotiou, G., Oehler, S., Savakis, C., and Bourtzis, K. (2011). Regulation of Wolbachia ankyrin domain encoding genes in Drosophila gonads. Res. Microbiol. 162, 764–772. doi: 10.1016/j.resmic.2011.06.012
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/GR.186072.114
Rao, A. U., Carta, L. K., Lesuisse, E., and Hamza, I. (2005). Lack of heme synthesis in a free-living eukaryote. Proc. Natl. Acad. Sci. USA 102, 4270–4275. doi: 10.1073/PNAS.0500877102
Reijnders, M. J. M. F., and Waterhouse, R. M. (2021). Summary visualizations of gene ontology terms with GO-figure! Front. Bioinform. 1:638255:6. doi: 10.3389/FBINF.2021.638255
Renoz, F., Lopes, M. R., Gaget, K., Duport, G., Eloy, M.-C., Geelhand de Merxem, B., et al. (2022). Compartmentalized into Bacteriocytes but highly invasive: the puzzling case of the co-obligate symbiont Serratia symbiotica in the aphid Periphyllus lyropictus. Microbiol. Spectr. 10:e0045722. doi: 10.1128/SPECTRUM.00457-22
Ronquist, F., Teslenko, M., Van Der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/SYSBIO/SYS029
Sanaei, E., Charlat, S., and Engelstädter, J. (2020). Wolbachia host shifts: routes, mechanisms, constraints and evolutionary consequences. Biol. Rev. Camb. Philos. Soc. 96, 433–453. doi: 10.1111/brv.12663
Scholz, M., Albanese, D., Tuohy, K., Donati, C., Segata, N., and Rota-Stabelli, O. (2020). Large scale genome reconstructions illuminate Wolbachia evolution. Nat. Commun. 11, 5235–5211. doi: 10.1038/s41467-020-19016-0
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/BIOINFORMATICS/BTU153
Siddique, S., Radakovic, Z. S., Hiltl, C., Pellegrin, C., Baum, T. J., Beasley, H., et al. (2022). The genome and lifestage-specific transcriptomes of a plant-parasitic nematode and its host reveal susceptibility genes involved in trans-kingdom synthesis of vitamin B5. Nat. Commun. 13, 6190–6119. doi: 10.1038/s41467-022-33769-w
Siozios, S., Ioannidis, P., Klasson, L., Andersson, S. G. E., Braig, H. R., and Bourtzis, K. (2013). The diversity and evolution of Wolbachia Ankyrin repeat domain genes. PLoS One 8:e55390. doi: 10.1371/JOURNAL.PONE.0055390
Slatko, B. E., Taylor, M. J., and Foster, J. M. (2010). The Wolbachia endosymbiont as an anti-filarial nematode target. Symbiosis 51, 55–65. doi: 10.1007/S13199-010-0067-1
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/BIOINFORMATICS/BTU033
Syberg-Olsen, M., Garber, A., Keeling, P. J., JP, M. C., and Husnik, F. (2022). Pseudofinder: detection of pseudogenes in prokaryotic genomes. Mol. Biol. Evol. 39:msac153. doi: 10.1093/molbev/msac153
Taylor, M. J., Voronin, D., Johnston, K. L., and Ford, L. (2013). Wolbachia filarial interactions. Cell. Microbiol. 15, 520–526. doi: 10.1111/CMI.12084
Wasala, S. K., Brown, A. M. V., Kang, J., Howe, D. K., Peetz, A. B., Zasada, I. A., et al. (2019). Variable abundance and distribution of wolbachiaand cardinium endosymbionts in plant-parasitic nematode field populations. Front. Microbiol. 10:964. doi: 10.3389/fmicb.2019.00964
Wasala, S. K., Hesse, C., Wram, C. L., Howe, D. K., Zasada, I. A., and Denver, D. R. (2023). Unraveling microbial endosymbiosis dynamics in plant-parasitic nematodes with a genome skimming strategy. Appl. Microbiol. 3, 1229–1248. doi: 10.3390/APPLMICROBIOL3040085
Werren, J. H. (1997). Biology of Wolbachia. Annu. Rev. Entomol. 42, 587–609. doi: 10.1146/ANNUREV.ENTO.42.1.587
Werren, J., Baldo, L., and Clark, M. E. (2008). Wolbachia: master manipulators of invertebrate biology. Nat. Rev. Microbiol. 6, 741–751. doi: 10.1038/nrmicro1969
Weyandt, N., Aghdam, S. A., and Brown, A. M. V. (2022). Discovery of early-branching Wolbachia reveals functional enrichment on horizontally transferred genes. Front. Microbiol. 13:1274. doi: 10.3389/FMICB.2022.867392/BIBTEX
Zhu, X., You, S., Liu, T., Zhang, L., and You, M. (2020). Research progress on Wolbachia endosymbionts in arthropods. Acta Entomol Sinica. 63, 889–901. Available at: https://www.cabidigitallibrary.org/doi/full/10.5555/20203530330
Keywords: bioinformatics data mining, tylenchid, Heterodera , plant-parasitic nematode, endosymbiont, Wolbachia , comparative genomics, phylogenomics
Citation: Kaur T and Brown AMV (2024) Discovery of a novel Wolbachia in Heterodera expands nematode host distribution. Front. Microbiol. 15:1446506. doi: 10.3389/fmicb.2024.1446506
Edited by:
Steve Perlman, University of Victoria, CanadaReviewed by:
Emilie Lefoulon, The Pennsylvania State University (PSU), United StatesMichael Gerth, iDiv, Germany
Copyright © 2024 Kaur and Brown. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amanda M.V. Brown, YW1hbmRhLm12LmJyb3duQHR0dS5lZHU=